

SUMMARY
Chromatin priming promotes cell-type-specific gene expression, lineage differentiation, and development. The mechanism of chromatin priming has not been fully understood. Here, we report that mouse hematopoietic stem and progenitor cells (HSPCs) lacking the Baf155 subunit of the BAF (BRG1/BRM-associated factor) chromatin remodeling complex produce a significantly reduced number of mature blood cells, leading to a failure of hematopoietic regeneration upon transplantation and 5-fluorouracil (5-FU) injury. Baf155-deficient HSPCs generate particularly fewer neutrophils, B cells, and CD8+ T cells at homeostasis, supporting a more immune-suppressive tumor microenvironment and enhanced tumor growth. Single-nucleus multiomics analysis reveals that Baf155-deficient HSPCs fail to establish accessible chromatin in selected regions that are enriched for putative enhancers and binding motifs of hematopoietic lineage transcription factors. Our study provides a fundamental mechanistic understanding of the role of Baf155 in hematopoietic lineage chromatin priming and the functional consequences of Baf155 deficiency in regeneration and tumor immunity.
In brief
Wu et al. report that Baf155-deficient hematopoietic stem and progenitor cells show defects in lineage differentiation, likely due to the failure of transcription factors to open chromatin at target gene loci, leading to defects in hematopoietic regeneration and tumor control.
Graphical abstract
INTRODUCTION
Chromatin priming mechanisms allow target genes to be accessible and poised for rapid activation to ensure a timely and coordinated response during lineage-specific gene expression.1,2 The BAF (BRG1/BRM-associated factor, also known as SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complexes have been implicated in the regulation of lineage determination and differentiation in diverse tissues, including embryonic stem cells, neural development, and hematopoiesis.3–6 They interact with lineage-specific transcription factors and co-regulators to modulate the chromatin accessibility landscape required for proper lineage-specification.7 Particularly, previous studies have shown that BAF complexes are preferentially targeted to distal tissue-specific enhancers.8–11 BAF complexes interact with p300 and KDM6a/6b to activate target gene expression by modifying H3K27.9–12 Therefore, BAF targeting of the distal enhancers is essential for activating the expression of genes linked to developmental processes. Gene knockout studies have demonstrated that components of the BAF complex are required for various aspects of hematopoietic lineage development and differentiation. For example, Brg1 is required for vascular and primitive erythroid lineage development during embryogenesis.13 Baf155 regulates lineage specification of the mouse blastocyst14 and is needed for myeloid and definitive erythroid lineage differentiation from erythro-myeloid progenitors (EMPs) in the yolk sac.15 In the adult, Brg1, Baf250a, Baf200, Baf45a, Baf180, Baf60b, and Baf53a all contribute to optimal hematopoiesis.16–25 Despite these studies showing the critical role played by the BAF complex in hematopoiesis, our understanding of the underlying chromatin mechanisms of hematopoietic lineage differentiation remains incomplete.
Genes encoding BAF subunits are frequently mutated in about 20% of all human cancers.26 Consequently, there is growing interest in developing drugs that target the BAF complex for cancer therapy. However, given the critical role of the BAF complex in development, it would be essential to ensure that such drugs selectively target cancer cells without interfering with the normal functions of immune cells in the tumor microenvironment. In this study, we determined the requirement for Baf155 in homeostatic hematopoiesis, transplantation, regeneration, and tumor-mediated hematopoietic response. We demonstrate that Baf155 deficient hematopoietic stem and progenitor cells (HSPCs) show lineage differentiation defects, with neutrophil, CD8+ T, and B lineages particularly sensitive to the Baf155 deficiency. The lineage differentiation defect was manifested by failure in hematopoietic regeneration upon transplantation and regenerative stress, a more immune-suppressive tumor microenvironment, and permissive tumor growth. Chromatin accessibility in the homeostatic and regenerating Baf155-deficient HSPCs was greatly reduced compared to wild-type controls. Differentially accessible regions were marked mainly by distinct hematopoietic lineage transcription factor binding motifs, suggesting that lineage-specific transcription factors failed to establish open chromatin in the absence of Baf155. These studies suggest that BAF mediates hematopoietic lineage chromatin priming, which provides an underlying mechanism for hematopoietic differentiation, regeneration, and tumor immunity.
RESULTS
Hematopoietic Baf155 deficiency leads to homeostatic cytopenia, especially in B, CD8+ T, and neutrophil lineages
To delineate the function of Baf155 in adult hematopoiesis, we deleted Baf155 using Vav-Cre mice. Vav-Cre; Baf155f/f (hereafter called Baf155 KOVav) mice from Vav-Cre; Baf155f/+ and Baf155f/f crosses were born at a frequency lower than Mendelian expectations (~15% as opposed to 25%; Figure S1A), suggesting that some of the Baf155 KOVav animals are lost during embryogenesis. While the Vav-Cre line is believed to target mainly the hematopoietic stem cell (HSC) lineage27,28; since Baf155 deletion using Tie2-Cre mice leads to embryonic lethality due to EMP differentiation block,15 some of the loss of Baf155 KOVav mice might be due to Vav-Cre targeting of the EMP. Lineage tracing using Vav-Cre; Rosa26-floxed stop-tdTomato mice showed that, at embryonic day 8 (E8), ~41% of the endothelial (CD45−CD31+) and ~46% of the erythroid (CD45−Ter119+) cells were tdTomato+ (Figure S1B). About 35% of cKit+ (or cKit+CD41+CD16/32+) cells, enriched for EMPs, from the E9.5 yolk sac were also tdTomato+ (Figure S1C). These data indicate that Vav-Cre, while targeting the HSC lineage, also incompletely targets EMP, leading to the loss of the Baf155 KOVav mice.
Baf155 KOVav mice were born were healthy and fertile. We confirmed the Baf155 deletion in the Baf155 KOVav bone marrow (BM), including the HSC compartment (Figures S1D and S1E). Levels of other BAF components, including BRG1, BAF170, BAF57, BAF47, and BAF60a, were not altered in Baf155 KOVav BM (Figure S1F). A complete blood count (CBC) analysis of Baf155 KOVav peripheral blood (PB) showed a decrease in white blood cell counts, including lymphoid, monocytes, and neutrophils, compared to littermate wild-type controls (Figure 1A). While the platelet number was higher in Baf155 KOVav mice, red blood cell components seemed to be similar to those of control mice (Figure 1A). Fluorescence-activated cell sorting (FACS) analysis of the PB showed that the proportions of white blood cells were differentially represented. Particularly, the percentage of B cells was decreased in Baf155 KOVav mice (Figures 1B and S1G), consistent with a previous study.29 The relative frequency of the total T cells was higher, with a higher percentage of CD4+ T cells and a lower percentage of CD8+ T cells, in Baf155 KOVav compared to control mice. This bias toward CD4+ T cell over CD8+ T cell generation was seen in the thymus (Figures S1H and S1I), indicating that Baf155 deficiency in HSCs leads to more severe defects in CD8+ T cell development. The relative frequency of the myeloid compartments was higher, with higher percentages of eosinophils, Ly6Clow monocytes, and neutrophils but lower percentages of Ly6Chigh monocytes, in Baf155 KOVav PB compared to controls (Figure 1B).
Figure 1. Baf155 KO mice show cytopenia at steady-state hematopoiesis.
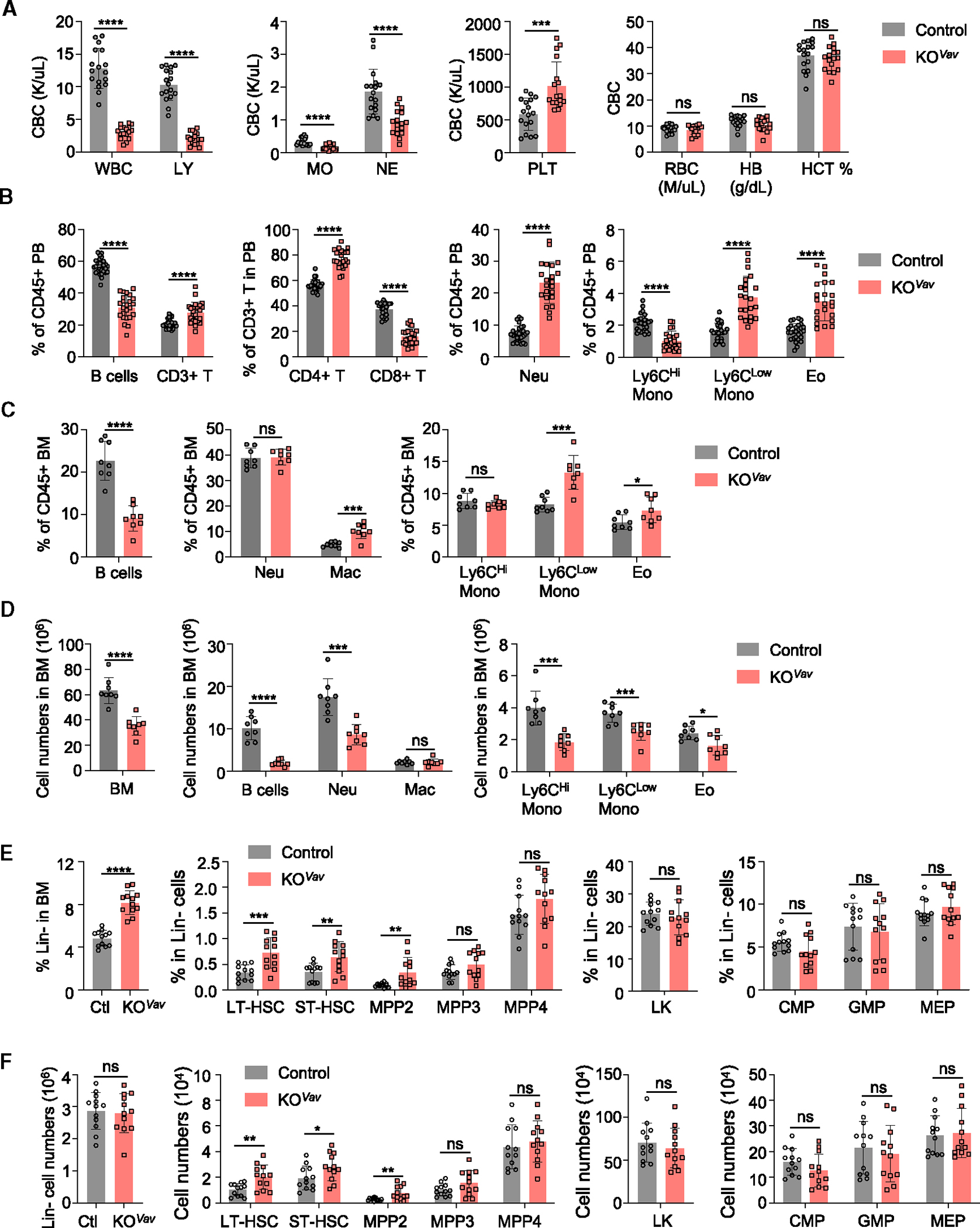
(A) Complete blood count (CBC) analysis of peripheral blood (PB) from control (n = 17) and Baf155 KOVav (n = 17) mice. WBC, whole blood cell; LY, lymphocyte; MO, monocyte; NE, neutrophil; PLT, platelet; RBC, red blood cell; HB, hemoglobin; HCT, hematocrit.
(B) Different lineage cell percentages in the PB of control and Baf155 KOVav mice were analyzed by flow cytometry. Control (n = 27–28) and Baf155 KOVav (n = 22–24). See Figure S1G for markers and gating strategy.
(C and D) Different lineage cell percentages (C) and numbers (D) in the BM of control (n = 8) and Baf155 KOVav (n = 8) mice were analyzed by flow cytometry.
(E and F) Frequency (E) and cell number (F) of different hematopoietic progenitors in BM of control and Baf155 KOVav mice as determined by flow cytometry. Control (n = 12) and Baf155 KOVav (n = 12). See Figure S1J for markers and gating strategy. Long-term hematopoietic stem cell (LT-HSC), CD150+CD48−Flk2−KSL cells; short-term hematopoietic stem cell (ST-HSC), CD150−CD48−Flk2−KSL; MPP2, CD150+CD48+Flk2−KSL; MPP3, CD150−CD48+Flk2−KSL; MPP4, CD150−Flk2+KSL; LK, Lin−Kit+Sca-1−; CMP, CD34+CD16/32−LK; GMP, CD34+CD16/32+LK; MEP, CD34−CD16/32−LK.
All experiments were performed in 8- to 10-week-old mice. Each symbol represents an individual mouse. For all graphs, data are presented as mean ± SD. Unless otherwise indicated, the p values were determined by unpaired two-tailed Student’s t test. n.s., not significant. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001.
The p values were unadjusted. See also Figure S1.
Both BM and spleen showed hypocellularity in Baf155 KOVav mice (Figures 1C, 1D, and S1J–S1L). In terms of the mature lineages, BM and PB showed similar differences between Baf155 KOVav and the control, suggesting that the differences in PB stem from BM progenitors. At the progenitor level, the percentage of Lin− cells was higher in Baf155 KOVav mice (Figure 1E), supporting the idea that mature lineage deficiency was due to differentiation defects of the progenitors. Consistent with this interpretation, long-term HSCs (LT-HSCs), short-term HSCs (ST-HSCs), and multipotent progenitor 2 (MPP2) were higher in the Baf155 KOVav BM than in controls, based on both percentages and absolute numbers (Figures 1E, 1F, and S1J).
We also generated Mx1-Cre; Baf155f/f mice to independently assess the acute deletion effect of Baf155 in adult hematopoiesis. We treated Mx1-Cre; Baf155f/f mice with poly(I:C) (hereafter called Baf155 KOMx1) and analyzed for hematopoietic compartments (Figure S1M). PB and BM analyses largely showed similar results as Vav-Cre-mediated Baf155 deletion; i.e., reduced mature blood generation while not affecting the HSPC compartments (Figures S1N–S1P). However, differences between the two models were seen in platelet reduction in the Baf155 KOMx1 mice, which could be due to poly(I:C) impairing platelet production and function30 (Figure 1A vs. Figure S1O). Additionally, the CD8+ T cell defect was less severe in Baf155 KOMx1 mice (Figures S1H and S1I vs. Figures S1Q and S1R), potentially reflecting the differences between developmental (Vav-Cre) and acute (Mx1-Cre) Baf155 deletion effects. Collectively, Baf155 deficiency did not impact the number of HSPCs in either model. However, their lineage output, especially neutrophil, B, and CD8+ T lineages, was sensitive to the Baf155 loss in homeostatic conditions.
Baf155-deficient HSPCs fail to reconstitute the hematopoietic system
To assess the effect of the overall differentiation defects of the Baf155 knockout (KO) HSPC in regeneration, we first performed competitive repopulation studies by mixing equal numbers of wild-type (WT) and Baf155 KOVav BM cells and transplanting them into lethally irradiated mice (Figure 2A). PB analysis showed that Baf155 KOVav BM contributed very little to any lineage of the recipient hematopoietic system (Figures 2B–2D and S2A–S2D). BM analysis of the recipients at 4 months showed an almost negligent contribution from Baf155 KOVav BM to LT-HSCs, ST-HSCs, MPPs, and mature hematopoietic cells (Figures 2E and 2F). However, LT-HSCs and ST-HSCs were over-represented within the recipient HSPC compartment (i.e., cKit+Sca1+Lin− [KSL] cells) derived from Baf155 KOVav, compared to WT, donors (Figure 2G).
Figure 2. Baf155-deficient BMs display defective reconstitution ability.
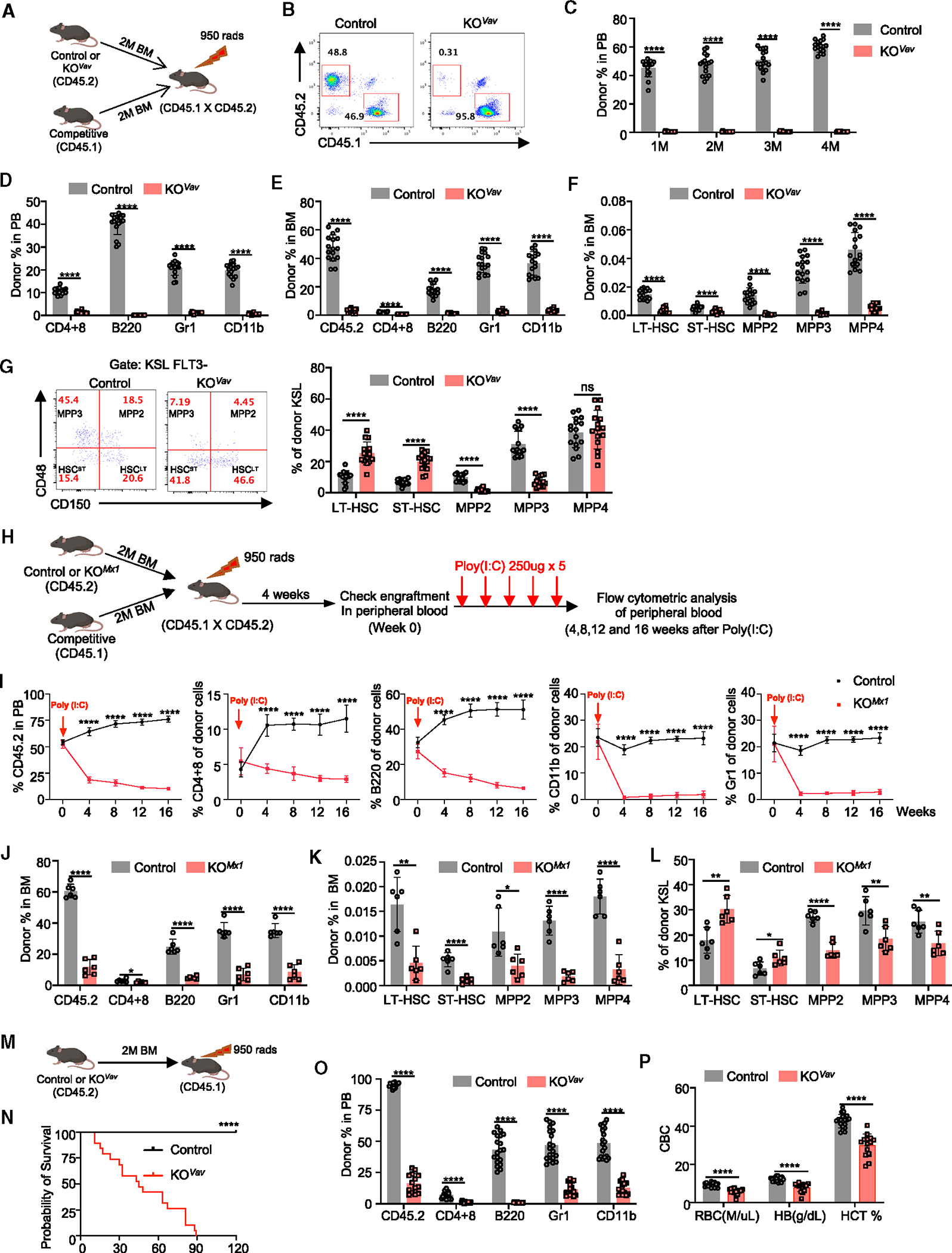
(A) Experimental design for the competitive repopulation assay with total BM cells from either control or Baf155 KOVav mice.
(B) Representative FACS plots showing donor chimerism (CD45.2) in PB of recipients transplanted with total BM from either control or Baf155 KOVav mice.
(C) Donor chimerism (CD45.2) in the PB of recipient mice was measured every month, and the results are shown in the graph. Control (n = 16) and Baf155 KOVav (n = 15).
(D) Percentage of donor-derived T cells (CD4+8), B cells (B220), granulocytes (Gr1), and Monos (CD11b) in the PB of recipient mice 4 months after transplantation. Control (n = 16) and Baf155 KOVav (n = 15).
(E) Percentage of donor chimerism (CD45.2) and donor-derived T cells (CD4+8), B cells (B220), granulocytes (Gr1), and Monos (CD11b) in the BM of recipient mice 4 months after transplantation. Control (n = 16) and Baf155 KOVav (n = 15).
(F) Percentage of donor-derived HSPC subpopulations in the BM of recipient mice 4 months after transplantation. Control (n = 16) and Baf155 KOVav (n = 15).
(G) Percentage of donor-derived HSPC subpopulations in the donor-derived KSL cells 4 months after transplantation. The representative FACS plots are shown on the left. The percentage of HSPC subpopulations in the donor-derived KSL cells are shown on the right. Control (n = 16) and Baf155 KOVav (n = 15).
(H) Experimental design for the competitive repopulation assay with total BM cells from either control or Baf155 KOMx1 mice (prior to deletion of Baf155).
(I) Percentage of donor chimerism (CD45.2) and donor-derived T cells (CD4+8), B cells (B220), granulocytes (Gr1), and Monos (CD11b) in the PB of recipient mice at different time point post poly(I:C) administration. Control (n = 6) and Baf155 KOMx1 (n = 6).
(J) Percentage of donor chimerism (CD45.2) and donor-derived T cells (CD4+8), B cells (B220), granulocytes (Gr1), and Monos (CD11b) in the BM of recipient mice at 16 weeks post poly(I:C) administration. Control (n = 6) and Baf155 KOMx1 (n = 6).
(K) Percentage of donor derived HSPC subpopulations in the BM of recipient mice at 16 weeks post poly(I:C) administration. Control (n =6) and Baf155 KOMx1 (n = 6).
(L) Percentage of donor-derived HSPC subpopulations in the donor-derived KSL cells at 16 weeks post poly(I:C) administration. Control (n = 6) and Baf155 KOMx1 (n = 6).
(M) Experimental design for the non-competitive repopulation assay with total BM cells from either control or Baf155 KOVav mice.
(N) Kaplan-Meier survival curves of recipient mice after non-competitive total BM transplantation. Control (n = 19) and Baf155 KOVav (n = 19). Mantel-Cox test, ****p < 0.0001.
(O) Percentage of donor chimerism (CD45.2) and donor-derived T cells (CD4+8), B cells (B220), granulocytes (Gr1), and Monos (CD11b) in the PB of recipient mice 30 days after non-competitive total BM transplantation. Control (n = 19) and Baf155 KOVav (n = 14).
(P) CBC analysis of PB from recipient mice 30 days after non-competitive total BM transplantation. Control (n = 19) and Baf155 KOVav (n = 14).
Each symbol represents an individual mouse. For all graphs, data are presented as mean ± SD. Unless otherwise indicated, the p values were determined by unpaired two-tailed Student’s t test. *p < 0.05, **p < 0.005, ****p < 0.0001. The p values were unadjusted. See also Figure S2.
To assess whether acute Baf155 deletion also leads to similar hematopoietic reconstitution defects, we transplanted an equal amount of Mx1-Cre; Baf155f/f and WT control BM. After engraftment, we treated mice with poly(I:C), followed by PB analysis up to 4 months (Figure 2H). In PB, Baf155 KOMx1 BM-derived myeloid progeny, CD11b+ or Gr1+, in the recipients decreased sharply even as early as 1 month after Baf155 deletion (Figure 2I). The B or T cell decrease was slower, probably reflecting their slow turnover rate (Figure 2I). At 4 months, BM showed reduced mature cells of the Baf155 KOMx1 donor origin (Figure 2J). HSPC compartments from the Baf155 KOMx1 donor were also significantly reduced (Figure 2K). Similar to Baf155 KOVav BM transplantation, LT-HSC and ST-HSC fractions were also higher in the LSK compartment derived from Baf155 KOMx1, compared to WT, donors (Figure 2L).
When transplanted without competitors, mice receiving Baf155 KOVav BM started to die after transplantation, even before 1 month (Figures 2M and 2N). PB analysis of the surviving animals at 1 or 2 months showed few mature cells of all lineages, including the erythroid lineage of the donor origin (Figures 2O, 2P, and S2E). Mice receiving Baf155 KO BM all died within 3 months (Figure 2N). These results demonstrate that Baf155-deficient HSCs, having differentiation defects, fail to reconstitute the hematopoietic system.
Baf155-deficient HSPCs fail to regenerate the hematopoietic system after 5-FU injury
Given the severe differentiation defects of the Baf155 HSPCs upon transplantation, we tested whether Baf155 KO mice also fail to regenerate the hematopoietic system in response to injury. We subjected Baf155 KOVav or Baf155 KOMx1 mice to 5-FU injection (Figures 3A and 3F). About half of the Baf155 KOVav mice died when treated with 250 mg/kg 5-FU, a highly cytotoxic dose (Figure 3B). The surviving mice showed significantly reduced HSC, MPP, KSL cells, common myeloid progenitor (CMP), and granulocyte-monocyte progenitor (GMP) numbers (Figures S3A and S3B). Although megakaryocyte-erythrocyte progenitor (MEP) numbers were similar (Figure S3B), erythroid output was impaired after 5-FU treatment (Figures S3C–S3E); Baf155 KOMx1 BM had a higher percentage of CD71+ cells with a lower percentage of more mature Ter119+ cells than controls, showing an erythroid lineage differentiation block.
Figure 3. Baf155 deficiency impairs HSC regeneration upon 5-FU injury.
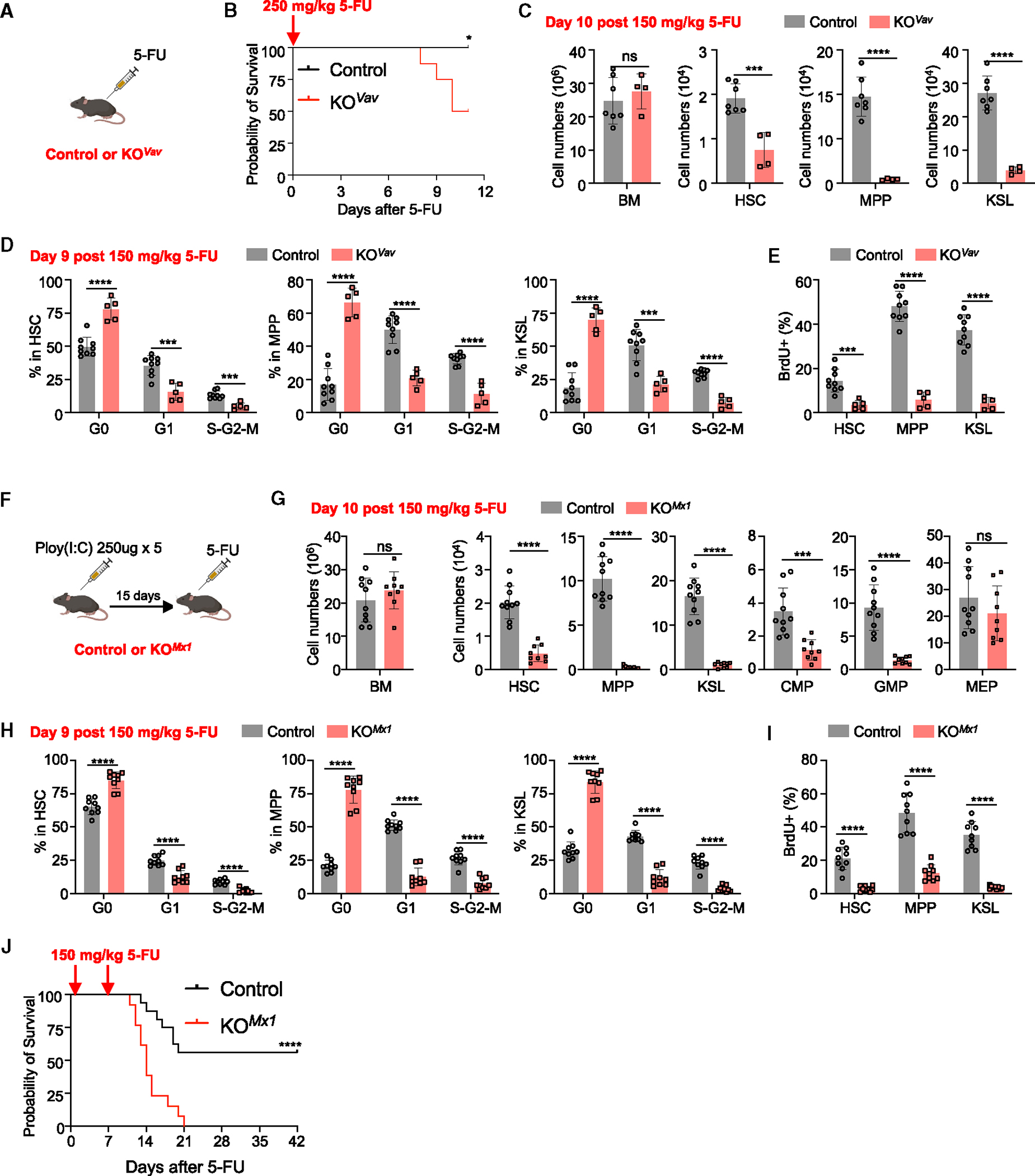
(A and B) Kaplan-Meier survival curves of control (n = 8) and Baf155 KOVav (n = 8) mice treated with one dose of 250 mg/kg 5-FU. Mantel-Cox test, *p = 0.0256.
(C) BM, HSC, MPP and KSL cell numbers from 150 mg/kg 5-FU-treated control (n = 7) and Baf155 KOVav (n = 4) mice at day 10 as determined by flow cytometry.
(D) Cell cycle analysis of HSCs, MPPs, and KSL cells from 150 mg/kg 5-FU-treated control (n = 9) and Baf155 KOVav (n = 5) mice at day 9 as determined by Ki67 and FxCycle staining.
(E) Percentage of bromodeoxyuridine (BrdU)+ cells among HSCs, MPPs, and KSL cells from 150 mg/kg 5-FU-treated control (n = 9) and Baf155 KOVav (n = 5) mice at day 9 as determined by in vivo BrdU assay.
(F) Scheme for control or Baf155 KOMx1 mice treated with 5-FU.
(G) BM cellularity and cell number of different hematopoietic progenitors from 150 mg/kg 5-FU-treated control (n = 10) and Baf155 KOMx1 (n = 9) mice at day 10 as determined by flow cytometry.
(H) Cell cycle analysis of KSL cells, MPPs, and HSCs from 150 mg/kg 5-FU-treated control (n = 9) and Baf155 KOMx1 (n = 9) mice at day 9 as determined by Ki67 and FxCycle staining.
(I) Percentage of BrdU+ cells among KSL cells, MPPs, and HSCs from 150 mg/kg 5-FU-treated control (n = 9) and Baf155 KOMx1 (n = 9) mice at day 9 as determined by in vivo BrdU assay.
(J) Kaplan-Meier survival curves of control (n = 16) and Baf155 KOMx1 (n = 13) mice treated with two doses of 150 mg/kg 5-FU at 7-day intervals. Mantel-Cox test, ****p < 0.0001.
Each symbol represents an individual mouse. For all graphs, data are presented as mean ± SD. Unless otherwise indicated, the p values were determined by unpaired two-tailed Student’s t test. *p < 0.05, ***p < 0.001, ****p < 0.0001. The p values were unadjusted. See also Figure S3.
To minimize the complications of the lethal cytotoxic effect of 5-FU, we treated mice with 150 mg/kg 5-FU, a low-cytotoxic dose, and analyzed hematopoietic compartments. Even at this low dose, where all mice survived, HSPCs and MPPs were significantly reduced in Baf155 KOVav mice, with the reduction of the HSC compartment less pronounced than that of KSL cells and MPPs (Figure 3C). When analyzed for the cell cycle status, a higher fraction of the Baf155 KOVav HSCs and MPPs was in G0 than in G1 and S cell cycle phases (Figures 3D and 3E). Baf155 KOMx1 mice also showed similar defects when treated with 5-FU (i.e., HSPC and MPP reduction) while showing less HSC reduction (Figures 3G and S3F). Similar to Baf155 KOVav, a higher percentage of Baf155 KOMx1 HSC, MPP, and KSL cells were in G0 phase in the cell cycle, suggesting that Baf155-deficient HSCs may have additional defects exiting from G0 phase, besides having differentiation defects (Figures 3H, 3I, S3G, and S3H). Importantly, all Baf155 KOMx1 mice died after two injections of 5-FU (Figure 3J). These data collectively suggest that Baf155 KO HSPCs fail to regenerate the hematopoietic system upon 5-FU injury.
Baf155-deficient HSPCs show macrophage-skewed myeloid differentiation
To better understand the role of Baf155 in hematopoietic lineage differentiation, we assessed the response of Baf155 deficient HSCs to cyclophosphamide (Cy) and granulocyte colony-stimulating factor (G-CSF). Previous studies have shown that Cy injection followed by two doses of G-CSF treatment induces HSC proliferation and differentiation (Figure 4A).31 To minimize the potential complication of developmental deletion of Baf155 using Vav-Cre, we used BaF155 KOMx1 mice. Remarkably, when we obtained a similar number of HSCs from Baf155 KOMx1 and littermate control BM, MPP or KSL cell numbers were significantly reduced in Baf155 KOMx1 BM after Cy+2 G-CSF administration (Figure 4B). Of the total CD45+ cells, the percentage of neutrophils was lower, while the percentage of macrophages was higher, in Baf155 KOMx1 BM than in controls after Cy+2 G-CSF treatment (Figures 4C and S4A). Cell cycle analysis showed that Baf155 KO HSCs cycled similarly to control HSCs (Figure 4D). However, for MPP and KSL cell compartments, Baf155 KOMx1 animals had a higher percentage of cells in G1 phase and lower percentage of cells in S-G2-M phases compared to control mice (Figure 4D). Apoptosis was similar between controls and Baf155 KOMx1 HSCs, MPPs, and KSL cells (Figures S4B and S4C). These data suggest that, when challenged with Cy+G-CSF, Baf155 KO HSCs could be maintained relatively normally, while myeloid lineage output was acutely skewed toward macrophages over neutrophils.
Figure 4. Baf155-deficient HSPCs show Mac-skewed myeloid differentiation.
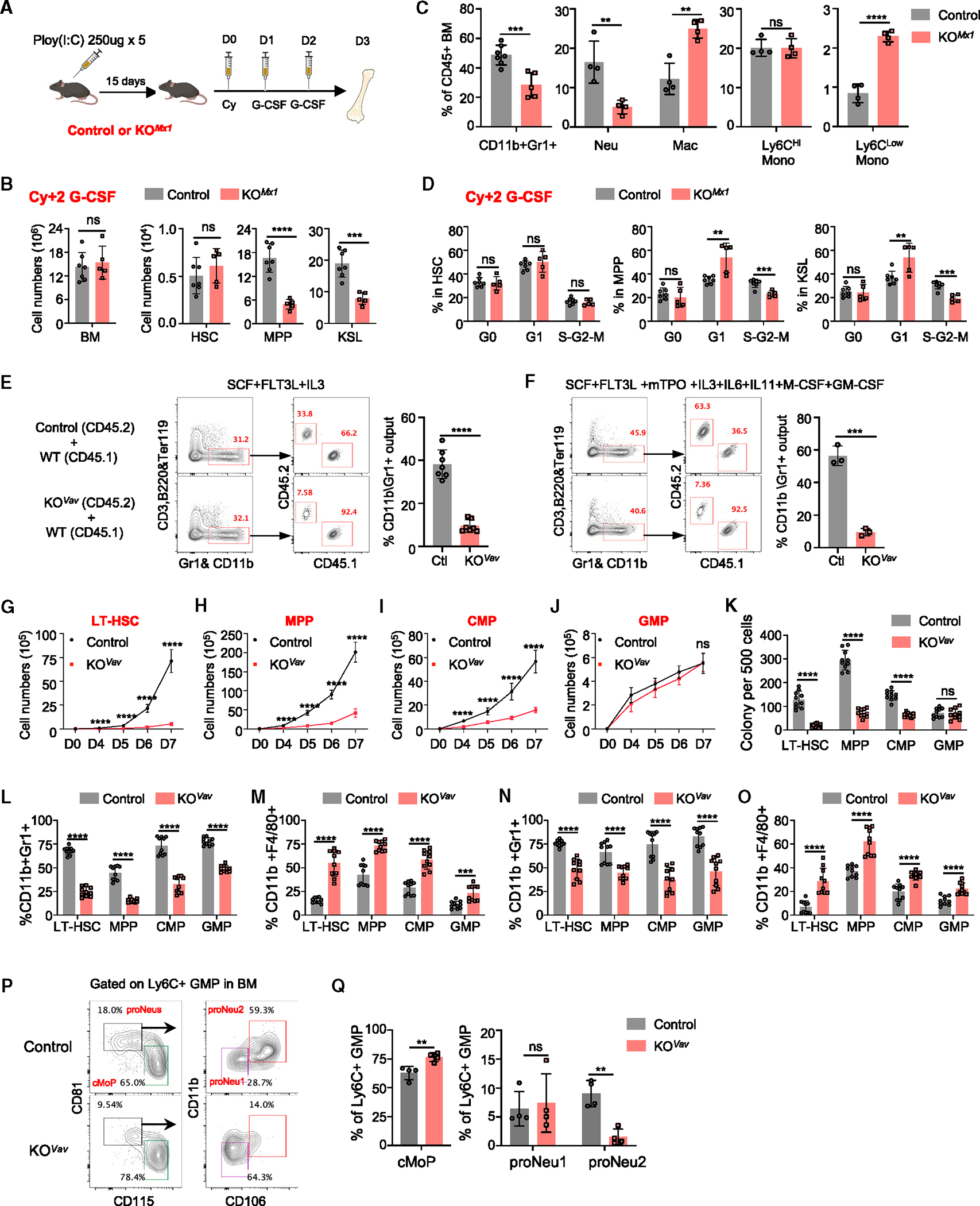
(A–D) Control or Baf155 KOMx1 mice were injected with cyclophosphamide (Cy), followed by two consecutive daily injections of G-CSF. Mice were euthanized 1 day after the last injection for analysis.
(A) Scheme for control or Baf155 KOMx1 mice treated with Cy+2G-CSF.
(B) BM cellularity; the numbers of HSCs, MPPs, and KSL cells from Cy+2G-CSF-treated control (n = 7) and Baf155 KOMx1 (n = 5) mice.
(C) The percentages of different cell lineages in the BM from Cy+2G-CSF-treated control (n = 4–7) and Baf155 KOMx1 (n = 4–5) mice.
(D) Cell cycle analysis of HSCs, MPPs, and KSL cells from Cy+2G-CSF-treated control (n = 7) and Baf155 KOMx1 (n = 5) mice as determined by Ki67 and FxCycle staining.
(E and F) KSL cells (CD45.2) from control or Baf155 KOVav mice (5,000 each) were mixed with KSL cells (CD45.1) from WT mice (5,000 each) in the same well of a 24-well plate with different combinations of cytokines: SCF+FLT3L+IL-3 (E) or SCF+FLT3L+mTPO+IL-3+IL-6+IL-11+M-CSF+GM-CSF (F) and medium containing 10% serum. Left: FACS plots depicting myeloid differentiation of KSL cells in vitro after 3.5 days of culture. Right: percentage of Mac1/Gr1+ output from KSL cells of either control or Baf155 KOVav mice. Control (n = 7) and Baf155 KOVav (n = 7) (E); control (n = 3) and Baf155 KOVav (n = 3).
(G–J) Cell numbers of ex vivo-cultured LT-HSCs (CD34−Flk2− KSL cells) (G), MPPs (Flk2hi CD34+ KSL cells) (H), CMPs (CD34+CD16/32− LK cells) (I), and GMPs (CD34+CD16/32+ LK cells) (J) at different time points in liquid medium with a combination of cytokines: SCF+FLT3L+mTPO+IL-3+IL-6+IL-11+M-CSF+GM-CSF. Control (n = 10) and Baf155 KOVav (n = 10).
(K) Colonies per 500-cell input were counted 6 days after plating in M3434 methylcellulose. Control (n = 10) and Baf155 KOVav (n = 10).
(L and M) Flow cytometry analysis of the frequency of CD11b+Gr1+ cells (L) and CD11b+F4/80+ cells (M) after 7-day culture in liquid medium. Control (n = 10) and Baf155 KOVav (n = 10).
(N and O) Flow cytometry analysis of the frequency of CD11b+Gr1+ cells (N) and CD11b+F4/80+ cells (O) after cells were replated in M3434 methylcellulose at day 6. Control (n = 10) and Baf155 KOVav (n = 10).
(P and Q) Representative flow cytometry analysis (P) and quantification (Q) of cMoP, proNeu1, and proNeu2 in control (n = 4) and Baf155 KOVav (n = 4) mice. cMoP, common Mono progenitor.
Each symbol represents an individual mouse. For all graphs, data are presented as mean ± SD. Unless otherwise indicated, the p values were determined by unpaired two-tailed Student’s t test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001. The p values were unadjusted. See also Figure S4.
To further examine the macrophage vs. neutrophil differentiation disparity, we sorted HSPCs (KSL cells) from Baf155 KO or littermate control mice and competitively cultured them with WT HSPCs.32 As expected, Baf155 KO HSPCs generated fewer myeloid cells in minimal factors (stem cell factor [SCF]+Flt3-ligand [FLT3L]+interleukin-3 [IL-3]) or in full cytokine factors (SCF+FLT3L+IL-3+Thrombopoietin [TPO]+IL-6+IL-11+macrophage colony-stimulating factor [M-CSF]+granulocyte-macrophage colony-stimulating factor [GM-CSF]) (Figures 4E and 4F). Furthermore, when we sorted HSCs, MPPs, CMPs, and GMPs and cultured them separately (1,000 cells per group), WT control MPPs showed the most robust cell output, followed by LT-HSCs, CMPs, and GMPs (Figures 4G–4J). Baf155 KO HSCs, MPPs, and CMPs generated a greatly reduced progeny output compared to WT controls (Figures 4G–4I). Similarly, MPPs generated the most colony numbers when replated in semi-solid medium, followed by CMPs and HSCs (Figure 4K). Again, Baf155 KO HSCs, MPPs, and CMPs produced reduced number of colonies (Figure 4K). Intriguingly, WT and Baf155 KO GMPs generated a similar number of progenies in culture (Figure 4J) or clonogenic assays (Figure 4K). However, the progeny output was different. Specifically, while WT GMPs produced more granulocytes, Baf155 KOVav GMPs generated more macrophages in bulk culture or in clonogenic assays (Figures 4L–4O). The lineage bias toward macrophages over granulocytes was also seen when Baf155Vav KO LT-HSC, MPP, and CMP progeny were assessed (Figures 4L–4O). Baf155 KOMx1 LT-HSCs, MPPs, CMPs, and GMPs all behaved similarly to those of Baf155 KOVav mice (Figures S4D–S4L). Since the GMP population (Lin−cKit+Sca1−CD34+CD16/32hi) has been shown to be heterogeneous and contain distinct progenitors for macrophages and neutrophils, we further analyzed the GMP population for common monocyte progenitors (cMoPs; CD115+CD81−Ly6C+GMP), pro-Neu1 (CD81+CD115−CD106−CD11bloLy6C+GMP), and a downstream proNeu2 (CD81+CD115−CD106+CD11b+Ly6C+GMP) progenitor subset.33 Baf155 KO GMPs contained a higher fraction of cMoPs and a lower fraction of the proNeu2 population (Figures 4P and 4Q). Baf155 KOMx1 GMPs showed similar trends (Figures S4M and S4N). Collectively, Baf155 deficiency leads to an increase in macrophage-committed progenitors while reducing neutrophil-committed progenitors, showing a bias toward macrophage over neutrophil lineage output.
Hematopoietic Baf155 KO mice support more robust tumor growth
We reasoned that Baf155 KOVav or Baf155 KOMx1 mice having altered hematopoietic constituents at basal level might impact tumor growth. Thus, we injected Baf155 KOVav and littermate control mice with PyMT-B6 breast cancer (Figure 5A) or 1956 sarcoma (Figure S5A) and measured tumor growth over time. Baf155 KOVav mice supported a more robust tumor growth than control mice (Figures 5B and S5B). Tumors from Baf155 KOVav mice showed a higher percentage of CD4+ T cells, including regulatory T cells (Tregs), and a lower percentage of CD8+ T cells, leading to a reduced cytotoxic T cell (CTL) to Treg ratio (Figures 5C and S5C). The percentages of tumor-associated macrophage, neutrophils, and myeloid derived suppressor cells (MDSCs) were all elevated within the tumors of the Baf155 KOVav mice (Figures 5C and S5C). Baf155 KOMx1 mice showed similar results.
Figure 5. Hematopoietic Baf155-deficient mice show more robust tumor progression.
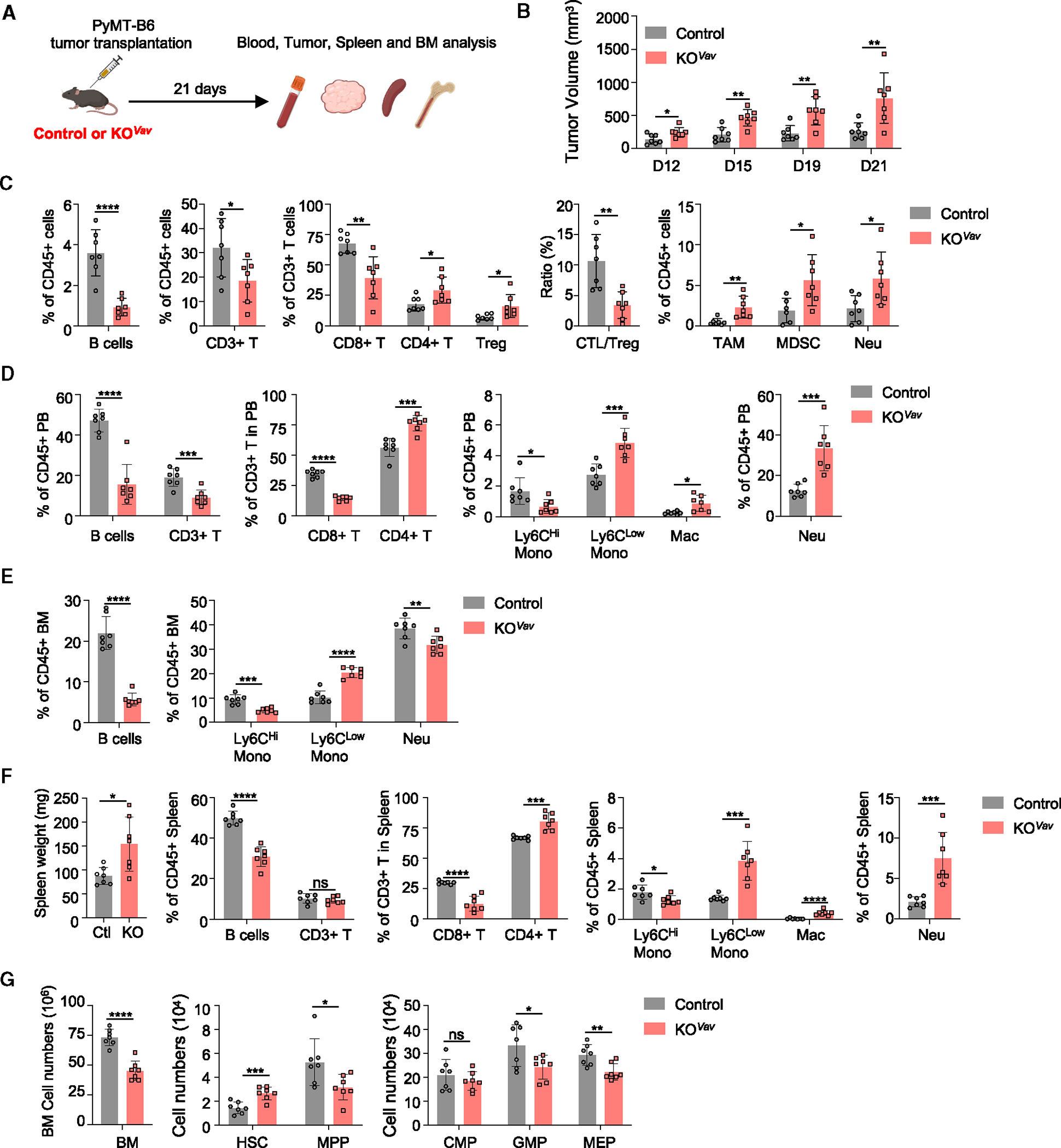
(A) Schematic of the workflow for the PyMT-B6 tumor transplantation.
(B) PyMT-B6 tumor growth in control (n = 7) and Baf155 KOVav (n = 7) mice.
(C) Different immune cell compartments in the tumors of control (n = 7) and Baf155 KOVav (n = 7) mice were analyzed by flow cytometry at the endpoints described in (B).
(D–F) Different lineage compartments in the PB (D), BM (E), and spleen (F) of PyMT-B6 tumor-bearing control (n = 7) and Baf155 KOVav (n = 7) mice were analyzed by flow cytometry at day 21.
(G) Cell numbers of different hematopoietic progenitors in the BM of PyMT-B6 tumor-bearing control (n = 7) and Baf155 KOVav (n = 7) mice at day 21.
Each symbol represents an individual mouse. For all graphs, data are presented as mean ± SD. Unless otherwise indicated, the p values were determined by unpaired two-tailed Student’s t test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001. The p values were unadjusted. See also Figure S5.
The tumor immune constituents were generally similar to those of PB (Figures 5D and S5D). BM showed similar changes in B and myeloid compartments except for neutrophils (Figures 5E and S5E), with BM neutrophils being less frequent than PB or tumor. This suggests that, while BM provides overall tumor immune cells, the spleen, assuming extramedullary hematopoiesis, might additionally supply emergency neutrophils, as often seen in tumor conditions. Indeed, both controls and Baf155 KOVav mice all showed splenomegaly (Figures 5F and S5F). Spleen analysis showed fewer B cells and CD8+ T cells with more myeloid cell presence, including neutrophils (Figures 5F and S5F). Notably, neutrophils, macrophages, and Ly6Clow monocytes significantly increased in the spleen of the tumor-bearing Baf155 KO mice. HSPC changes were less drastic between Baf155 KOVav and control tumor-bearing mice (Figures 5G and S5G). These data suggest that hematopoietic Baf155 KO mice provide an environment that is more conducive to tumor growth, characterized by the increase in immune-suppressive myeloid cells and the decrease in CD8+ T cells, which is facilitated by extramedullary hematopoiesis.
Single-nucleus multiomics analysis revealed that Baf155 is critical for chromatin priming during hematopoietic lineage differentiation
To understand how Baf155 influences hematopoietic lineage differentiation, we performed single-cell multiome ATAC+RNA sequencing on regenerating hematopoietic progenitors from littermate control (WT) and Baf155 KOVav (KO) mice. Specifically, we used sorted KSL cells enriched for HSPCs and cKit+Lin− cells representing more mature progenitors from WT and KO mice at 9 or 11 days post 5-FU injection (Figure 6A). These time points were chosen as they coincide with robust HSPC regeneration.34 Notably, the analysis of both day 9 and day 11 regenerating HSPCs revealed similar trends, supporting the robustness of our findings (Figures 6, 7, S6, and S7).
Figure 6. Single-cell multiomics characterization of WT and Baf155 KO HSPCs during regeneration.
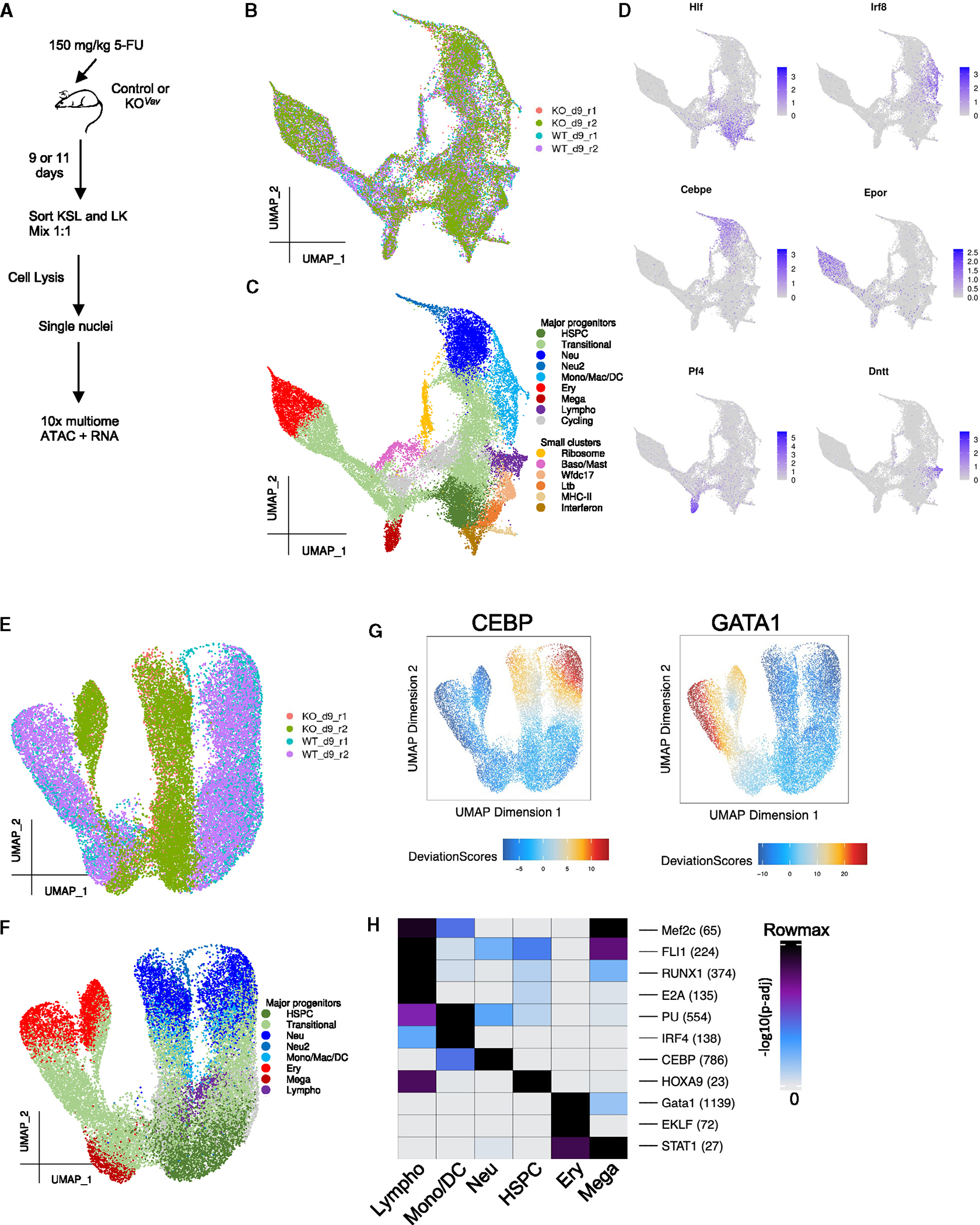
(A) Schematic view of the experimental design.
(B) UMAP based on the RNA fraction of multiomics data, colored by sample.
(C) UMAP based on the RNA fraction of the multiomics data, colored by cell type.
(D) UMAP based on the RNA fraction of the multiomics data, colored by expression of the marker genes of various hematopoietic lineages.
(E) UMAP based on the ATAC fraction of the multiomics data, colored by sample.
(F) UMAP based on the ATAC fraction of the multiomics data, colored by clusters defined in (C). Clusters with a small number of cells were removed.
(G) Smoothed chromVAR (STAR Methods) results depicting the enrichment (high deviation score) or depletion (low deviation score) of chromatin accessibility in peaks containing the CEBP or GATA1 motif on a single-cell basis.
(H) Enriched motifs in the marker peaks of 6 main clusters. Cells from all samples were used for marker peak identification. Numbers within parentheses after each motif represent the actual max(−log10(p-adj)) of that motif.
Figure 7. Baf155 regulates chromatin accessibility at hematopoietic lineage TF binding loci.
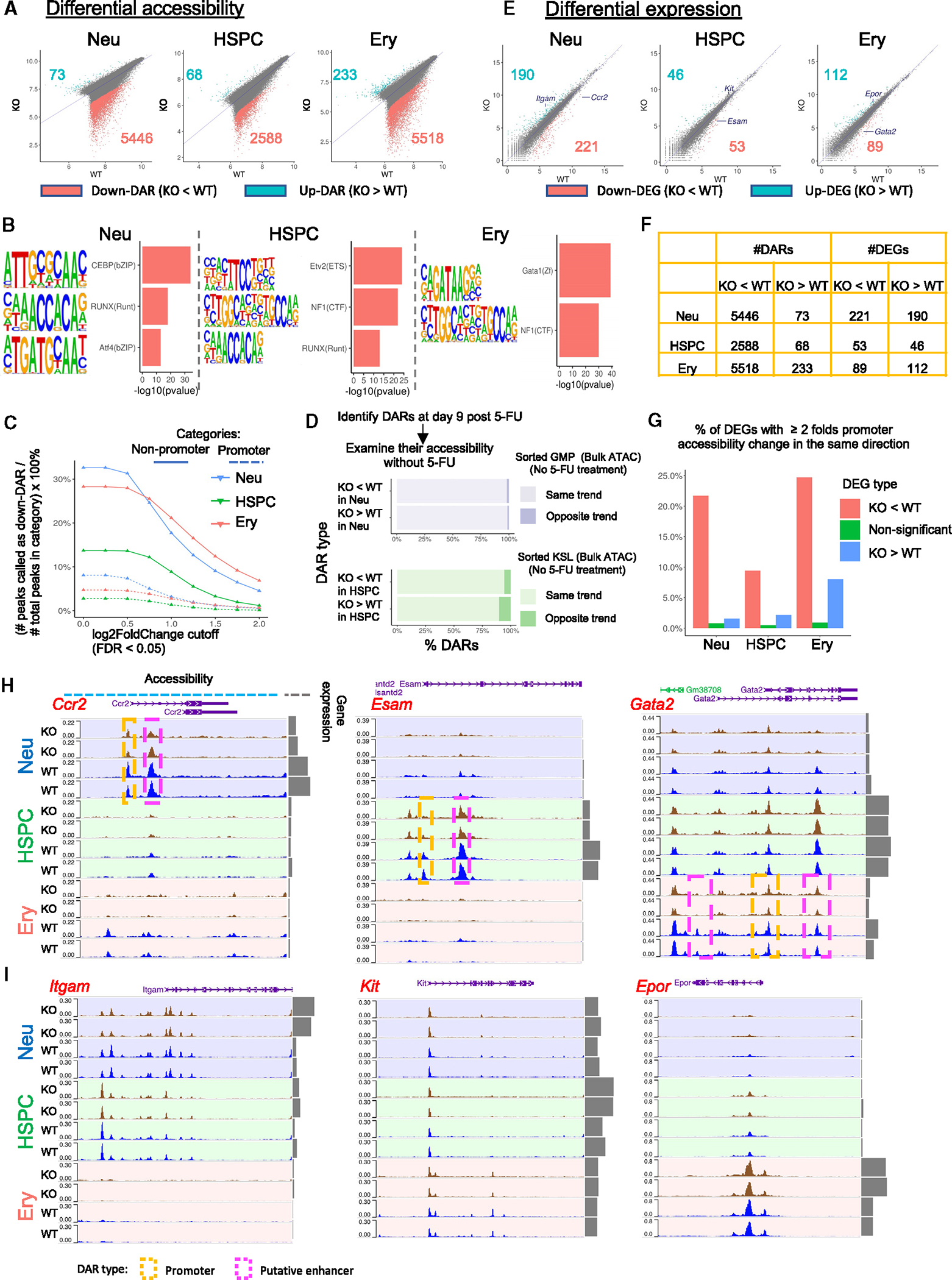
(A) Scatterplots of chromatin accessibility. Each dot represents a peak in the cluster-specific peak × sample pseudobulk matrix (STAR Methods). DARs with false discovery rate (FDR) < 0.05 and log2FoldChange > 1 were colored. The x and y axes represent the log2-transformed mean of the 2 replicates for each genotype at day 9 post 5-FU treatment.
(B) Examples of enriched motifs in the down-DARs of the 3 clusters.
(C) Solid lines: percentage of non-promoter peaks called as down-DARs, with the statistical significance cutoff of FDR < 0.05 across various log2FoldChange cutoffs as indicated by the x axis. Dotted lines: same as solid lines but calculating the percentage of promoter peaks instead.
(D) Bulk ATAC-seq on GMP and KSL cells sorted from mice without 5-FU treatment were used to approximate neutrophil progenitors and HSPC under homeostasis. For down-DARs (KO < WT at day 9 post 5-FU), “same trend” indicates those that were also less accessible in the KO compared to the WT under homeostasis, whereas “opposite trend” indicates those that were more accessible in the KO compared to the WT under homeostasis. Trends for up-DARs were determined similarly.
(E) Same as (A), with each dot representing a gene instead. Example genes are labeled. Kit is significantly upregulated in KO HSPCs (FDR = 0.008) but did not pass the log2FoldChange cutoff of 1 (log2FoldChange(Kit) = 0.6) and, therefore, is not within the list of DEGs.
(F) Table summarizing (A) and (E).
(G) Red, percentage of down-DEGs (KO < WT, defined in E) with promoter peaks showing a ≥ 2-fold decrease in accessibility; blue, percentage of up-DEGs with promoter peaks showing ≥ 2-fold increase in accessibility; green, percentage of non-significant genes with promoter peaks showing ≥ 2-fold change in accessibility in the same direction.
(H) Examples of down-DEGs and their chromatin accessibility landscapes.
(I) Examples of up-DEGs and their chromatin accessibility landscapes.
We first clustered day 9 cells by the transcriptome (Figures 6B, 6C, and S6A–S6F; Table S1). Based on known hematopoietic lineage markers,35,36 the major HSPC cluster was identified by Hlf, Ly6a (encoding Sca-1), and Procr expression; neutrophil progenitors based on Elane and Cebpe expression; and erythroid progenitors based on Epor and Gata1 expression (Figures 6D and S6E). Cells from other lineages were also recovered, including monocyte/macrophage/dendritic cell progenitors (Mono/Mac/DC) identified by Irf8, Ly86, and Csf1r; megakaryocytic progenitors (Mega) identified by Pf4 and Itga2b; lymphoid progenitors (Lympho) identified by Dntt, Flt3, and Il7r; and basophil/mast cell progenitors (Baso/Mast), identified by Ms4a2 and Cpa335 (Figure S6E). Small populations of HSPCs with the interferon response signature37 or the major histocompatibility complex class II signature38 were also detected. Additionally, minor populations of progenitors with high Ltb, Wfdc17, or ribosomal gene expression were also detected (Figures 6C and S6E), although their identities are not clear.
For the same cells, we also generated an ATAC-based UMAP, annotated using the clusters defined above (Figures 6E, 6F, S6G, and S6H). ATAC UMAP revealed that the HSPC clusters from the WT and KO were adjacent, suggesting similar chromatin accessibility landscapes. However, WT and KO samples were visibly separated in more committed erythroid and neutrophil progenitors, suggesting that differences in chromatin accessibility become more prominent as cells differentiate (Figures 6E, 6F, and S6G–S6J). Consistent with this, chromVAR39 analysis suggested that neutrophil and erythroid progenitors in KO samples showed reduced chromatin accessibility at the motifs of transcription factors (TFs) CEBP and GATA1, which are critical for the development of neutrophils40 and erythrocytes,41 respectively (Figures 6G and S6K). Last, motifs of known lineage-specific TFs were enriched in the marker peaks of each cluster (Figure 6H). For example, the motif of HOXA9, shown to be critical for HSC expansion,42 was enriched in HSPCs, suggesting that our clustering also adequately captured each lineage’s identity on the level of chromatin accessibility.
We first compared the chromatin accessibility landscape of KO vs. WT cells for neutrophil progenitors, HSPCs, and erythroid progenitors using samples from day 9 post 5-FU treatment. Among the differentially accessible regions (DARs) identified, the vast majority showed decreased accessibility in KO cells (down-DARs), and very few showed increased accessibility (up-DARs) (Figures 7A and 7F; Table S2). Down-DARs also showed a higher magnitude of accessibility changes compared to up-DARs (Figure 7A), suggesting a general loss of chromatin accessibility in KO cells. Notably, such loss was more prominent in more committed neutrophil and erythroid progenitors compared to HSPCs (Figures 7A and 7F). Down-DARs were enriched for H3K27ac signals but depleted for H3K27me3 signals in WT HSPCs, supporting the notion that BAF interacts with p300 and KDM6a/6b to activate target gene expression (Figure S7A). Neutrophil and erythroid-specific TF motifs were highly enriched in the down-DARs of corresponding clusters, suggesting that lineage-specifying TFs failed (at least partially) to initiate/maintain open chromatin in the absence of Baf155 (Figures 7B; Table S3), which is also supported by the reduced ATAC pile-up signals around the motif instances of these TFs (Figure S7B; STAR Methods). Consistent with this, chromatin immunoprecipitation (ChIP)-qPCR assay showed that down-DARs containing CEBP or GATA1 motifs also exhibited decreased CEBPb or GATA1 binding, respectively (Figure S7C). Furthermore, a higher percentage of non-promoter peaks showed downregulated chromatin accessibility compared to promoter peaks across different cutoffs (Figures 7C; Table S4). Previous studies suggest that most non-promoter ATAC peaks can be considered as putative enhancers.43–45 In addition, the DARs identified at day 9 post 5-FU showed the same trend at day 11 post 5-FU treatment (Figures S6L and S6M). To determine whether these DARs were de novo generated upon 5-FU treatment, we sorted GMP and KSL cells from WT or KO animals without 5-FU treatment for bulk ATAC-seq. With rare exceptions, DARs identified at day 9 post 5-FU showed the same trend in these bulk samples sorted from homeostatic conditions (Figure 7D). For example, almost all down-DARs (KO < WT) identified in the neutrophil progenitor cluster (Figure 7A) were also downregulated in sorted GMP cells from KO animals without 5-FU treatment (Figure 7D). These data suggest that similar chromatin accessibility defects were already present in the homeostatic conditions. Last, genes near down-DARs were enriched for development-related pathways (Table S2). Overall, Our data revealed a general loss of chromatin accessibility in the absence of Baf155 that could impair lineage TF binding and lineage differentiation under homeostasis and during regeneration.
We next identified differentially expressed genes (DEGs) in KO vs. WT cells (Figures 7E, 7F, S6N, and S6O; Table S5). More DEGs were identified in more differentiated neutrophil and erythroid progenitors compared to HSPCs, consistent with their more prominent loss of chromatin accessibility (Figures 7A and 7E). Intriguingly, similar numbers of DEGs were up- or downregulated (up-DEGs and down-DEGs), despite the general chromatin accessibility loss (Figures 7E and 7F). Down-DEGs were more likely to have consistent promoter accessibility changes, suggesting a direct effect of chromatin accessibility loss, whereas up-DEGs were more likely the result of chromatin accessibility-independent mechanisms (Figure 7G). For example, the expression of Ccr2, Esam, and Gata2, genes known to be important for neutrophils, HSPCs, and erythroid progenitors, respectively,46–48 were downregulated. Their promoters, as well as other nearby peaks, also showed decreased accessibility (Figures 7E and 7H). By contrast, Itgam, Kit, and Epor, encoding surface markers for neutrophil, HSPC, and erythroid lineages,49–51 respectively, were upregulated in KO cells. However, we did not observe any increase in chromatin accessibility at these loci (Figures 7E and 7I). Cellular differentiation-related pathways were enriched in the down-DEGs of HSPCs and neutrophil progenitors, supporting differentiation defects (Table S5). Intriguingly, immune response-related pathways were enriched in the up-DEGs of KO HSPCs and neutrophil progenitors (Table S5). Collectively, these data support that Baf155 enables lineage TFs to access and activate lineage gene expression and differentiation.
We also extended the same analyses to the Mono/Mac/DC progenitor cluster, the Lympho progenitor cluster, and the Mega progenitor cluster (Figures S7D–S7J). Similar to the neutrophil, HSPC, and erythroid data, abundant DARs were detected, most of which showed decreased accessibility in KO cells (Figure S7D; Table S2), while gene expression changes were not prominent (Figure S7E; Table S5). We note that lower numbers of DARs and DEGs were detected for lymphoid and megakaryocytic progenitors, which is probably due to the lower numbers of cells in these clusters (Figure S6F). Motifs of lineage TFs were enriched in down-DARs. For example, the E2A and FLI1 motifs, key TFs in B cell and megakaryocytic commitment, respectively,52,53 were enriched in the down-DARs of the corresponding clusters (Figure S7F; Table S3). CD4 and CD8 T cell-specific peaks showed similar levels of reduction in chromatin accessibility in the lymphoid progenitors of KO animals (Figure S7G). Interestingly, the IRF8 motif, one of the lineage-determining TFs of Mono/Mac/DC progenitors,54 was not enriched in down-DARs (Figures S7F and S7H). Since DARs only represent peaks with the most prominent changes in chromatin accessibility, we then examined the chromatin accessibility changes of all Mono/Mac/DC peaks containing the IRF8 motif. Compared to peaks with the CEBP motif, peaks with the IRF8 motif in general showed less reduction in ATAC signals (Figure S7I). Consistent with this, although the total ATAC signals around IRF8 motif instances were reduced in KO Mono/Mac/DC progenitors, such reduction was less prominent compared to the CEBP motif instances in the same cells, especially at promoters (Figure S7J). Overall, these data suggest that IRF8 activity might be less affected by the Baf155 deficiency.
To capture additional information regarding how hematopoietic differentiation might be altered in Baf155 KO animals, we performed pseudotime analyses using Slingshot55 (Figure S6P), which did not identify consistent differences in the constructed differentiation trajectories between KO and WT samples, suggesting that the general pattern of differentiation was not significantly altered in Baf155 KO animals. On the other hand, changes in hematopoietic differentiation patterns might have occurred but were missed, since pseudotime analyses only capture the direction of transition from one cell to another, not the probability of transition.
DISCUSSION
A clear understanding of lineage development would facilitate maximizing tissue regeneration in injury or disease control. Here, we investigated the role of Baf155-mediated chromatin priming in hematopoietic lineage development. Under homeostatic conditions, Baf155 deficiency led to defects in mature blood generation while maintaining HSPCs relatively normally. Despite a general decrease in the output of mature blood cells, hematopoietic lineages were affected differently. Within the myeloid compartment, we observed macrophage over neutrophil lineage output at the GMP level. Single-cell multiomics data suggest that the activity of IRF8, a lineage-determining TF of Mono/Mac/DC progenitors, might be less affected by the Baf155 deficiency compared to CEBP factors. Indeed, cMoPs were relatively increased within the GMP compartment, while proNeu2 was decreased. This interpretation is also consistent with previous observations that deficiency in Baf60b, which encodes another subunit of the BAF complex, significantly disrupted granulopoiesis, mediated by the interaction with CEBPe, a lineage-determining TF of granulocytes.24,25 Within the lymphoid lineage, we observed B cell compartment reduction, which is consistent with a previous study showing that Baf155 is required for B cell development.29 The relative frequency of CD3+ T cells was increased, probably due to B cell reduction. Of the T cells, CD4+ T cells were more represented over CD8+ T cells in Baf155 KOVav mice, suggesting that Baf155-mediated chromatin remodeling is more required for CD8+ T cell development. Consistent with this, Brg1 has been shown to control CD4+ vs. CD8+ T cell lineage output.22,23 Intriguingly, erythroid and platelet production was not impaired by the Baf155 deficiency in homeostatic conditions. Nonetheless, erythroid generation was reduced upon regenerative stress by BM transplantation and after 5-FU. Collectively, we propose that neutrophils, B cells, and CD8+ T cells are most sensitive to the Baf155 defect.
We showed that Baf155 KO HSPCs could not generate mature blood cells in a timely manner in situations where rapid HSPC expansion and lineage differentiation are needed, such as BM transplantation or 5-FU injury, leading to organismal loss. Chromatin accessibility in Baf155-deficient progenitors was greatly reduced. Regions with reduced accessibility were enriched for lineage TF binding motifs. It is conceivable that, in emergencies where blood cells need to be produced rapidly, chromatin needs to be readily accessible for lineage-specific TFs to gain access to activate their target genes. In Baf155 KO HSPCs, we envision lineage TFs failing to activate lineage genes, leading to ineffective blood cell generation in an emergency. Our data support that Baf155 establishes lineage competency through chromatin priming to allow lineage gene expression and mature blood production.
In contrast to hematopoietic regeneration conditions, where rapid production of mature blood lineages is needed, the basal hematopoietic constituents may affect disease control. Indeed, Baf155 KO mice, with altered hematopoietic compartments at the basal level, supported more robust tumor growth. As for the CD8+ T cells, recent studies have shown that the BAF complex controls CD8+ T cell exhaustion.56–58 Inhibiting the BAF complex in CD8+ T cells reduced T cell exhaustion and increased T cell persistence and anti-tumor activity. These studies suggest the distinct roles of BAF in different stages of T cell development and activation. In tumors, we also observed increased myeloid cell production, including MDSCs. Neutrophils in the periphery were also increased, although the BM production of neutrophils was compromised. We attribute this to extramedullary hematopoiesis compensating BM hematopoietic defects. Indeed, we observed splenomegaly in both control and Baf155 KO tumor-bearing mice. Recently, mice treated transiently with BRM014, a chemical BRM/BRG1 inhibitor, were shown to have similar hematopoietic changes as in our current genetic Baf155 deletion studies, which warrants further studies of the BAF inhibitors that might impact baseline hematopoiesis.59 Collectively, our data suggest that the absence of Baf155 and subsequent chromatin remodeling impairment in HSPCs leading to altered basal hematopoietic constituents affect disease control outcomes in the context of cancer. Baf155 encodes the core structural subunit of BAF and polybromo-associated BAF (PBAF). Whether the effect seen by the Baf155 deletion in this study is due to disruption of both the BAF and PBAF complex warrants further investigations in the future. In summary, our study shows the critical role of Baf155 in establishing chromatin priming to promote mature blood generation, hematopoietic regeneration, and disease control but not for maintaining hematopoietic stem and progenitor pools.
Limitations of the study
There are conflicting findings regarding the role of BAF155 in the stability of the BAF chromatin-remodeling complex. Notably, we did not observe any reduction in the BAF components when Baf155 was deleted using Vav-Cre, consistent with studies showing that Baf155 or Baf170 deletion does not lead to a decrease in the BAF components.60,61 However, Baf155 heterozygosity or siRNA treatment has been shown to reduce BRG1 or SNF5/BAF47 protein levels.62 Future extensive biochemical studies are needed to examine whether BAF155 is necessary for the structural integrity of the BAF complex. The impact of Baf155 deficiency on HSC self-renewal requires thorough investigation. Tumor studies using Vav-Cre x Baf155f/f or Mx1-Cre x Baf155f/f mice are limited to hematopoietic cell analysis. Future research needs to focus on characterizing individual immune cells and their functions. The use of bulk ATAC-seq data from homeostatic conditions for comparison with single-cell data of regenerating HSPCs may have limitations. Additionally, although we observed strong enrichments of lineage-specifying TF binding motifs in down-DARs, we do not have direct measurements of TF binding in the same cells. TFs may not bind to these regions due to a lack of chromatin accessibility or may bind but cannot open the chromatin without BAF155. The rapid development of single-cell cut&tag technology may help address these limitations soon.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author and lead contact, Dr. Kyunghee Choi (kchoi@wustl.edu).
Materials availability
This study did not generate any unique reagents.
Data and code availability
All sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Code used for sequencing data analyses has been deposited in Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| BUV395 Rat Anti-Mouse CD45 (Clone 30-F11) | BD Biosciences | Cat#564279; RRID:AB_2651134 |
| FITC anti-mouse CD45 (Clone 30-F11) | BioLegend | Cat# 103108; RRID:AB_312973 |
| Brilliant Violet 605™ anti-mouse CD45 (Clone 30-F11) | BioLegend | Cat#103155; RRID:AB_2650656 |
| PerCP-eFluor™ 710 anti-mouse CD172a (SIRP alpha) (Clone P84) | Thermo Fisher Scientific | Cat#46-1721-80; RRID:AB_10805866 |
| Alexa Fluor® 647 Rat Anti-Mouse Siglec-F (Clone E50-2440) | BD Biosciences | Cat#562680; RRID:AB_2687570 |
| Brilliant Violet 711™ anti-mouse Ly-6G (Clone 1A8) | BioLegend | Cat#127643; RRID:AB_2565971 |
| BUV805 Rat Anti-Mouse Ly-6G (Clone 1A8) | BD Biosciences | Cat#741994; RRID:AB_2871294 |
| Brilliant Violet 785™ anti-mouse Ly-6C (Clone HK1.4) | BioLegend | Cat#128041; RRID:AB_2565852 |
| Brilliant Violet 510™ anti-mouse Ly-6C (Clone HK1.4) | BioLegend | Cat#128033; RRID:AB_2562351 |
| Brilliant Violet 605™ anti-mouse CD115 (CSF-1R) (Clone AFS98) | BioLegend | Cat#135517; RRID:AB_2562760 |
| FITC anti-mouse CD4 (Clone GK1.5) | BioLegend | Cat#100405; RRID:AB_312690 |
| BUV496 Rat Anti-Mouse CD4 (Clone GK1.5) | BD Biosciences | Cat#612952; RRID:AB_2813886 |
| PE/C7 anti-mouse/human CD45R/B220 (Clone RA3-6B2) | BioLegend | Cat# 103222; RRID:AB_313005 |
| BUV395 Rat Anti-Mouse CD45R/B220 (Clone RA3-6B2) | BD Biosciences | Cat#563793; RRID:AB_2738427 |
| PE/Cy7 anti-mouse/human CD45R/B220 (Clone RA3-6B2) | BioLegend | Cat#103221; RRID:AB_313004 |
| PerCP\Cy5.5 anti-mouse/human CD45R/B220- (Clone RA3-6B2) | BioLegend | Cat#103235; RRID:AB_893356 |
| Brilliant Violet 421™ anti-mouse F4/80 (Clone BM8) | BioLegend | Cat#123137; RRID:AB_2563102 |
| PE anti-mouse F4/80 (Clone BM8) | BioLegend | Cat# 123110; RRID:AB_893486 |
| PerCP/Cy5.5 anti-mouse F4/80 (Clone BM8) | BioLegend | Cat#123127; RRID:AB_893496 |
| PE Rat Anti-Mouse CD8a (Clone 53–6.7) | BD Biosciences | Cat#553033; RRID:AB_394571 |
| PE/Cy5 anti-mouse CD8a (Clone 53–6.7) | BioLegend | Cat#100710; RRID:AB_312749 |
| FITC anti-mouse CD8a (Clone 53–6.7) | BioLegend | Cat#100706; RRID:AB_312745 |
| Brilliant Violet 650™ anti-mouse CD25 (Clone PC61) | BioLegend | Cat#102038; RRID:AB_2563060 |
| FITC anti-mouse CD3 (Clone 17A2) | BioLegend | Cat#100204; RRID:AB_312661 |
| APC/Cyanine7 anti-mouse CD3 (Clone 17A2) | BioLegend | Cat#100221; RRID:AB_2057374 |
| PE/Cy7 anti-mouse CD3 (Clone 145-2C11) | BioLegend | Cat# 100320; RRID:AB_312685 |
| Brilliant Violet 421™ anti-mouse FOXP3 (Clone MF-14) | BioLegend | Cat#126419; RRID:AB_2565933 |
| BUV737 Rat Anti-CD11b (Clone M1/70) | BD Biosciences | Cat#612800; RRID:AB_2870127 |
| PE Rat Anti-CD11b (Clone M1/70) | BD Biosciences | Cat#553311; RRID:AB_394775 |
| PE/Cy7 anti-mouse/human CD11b (Cone M1/70) | BioLegend | Cat# 101216; RRID:AB_312799 |
| Brilliant Violet 650™ anti-mouse/human CD11b (Clone M1/70) | BioLegend | Cat#101259; RRID:AB_2566568 |
| APC anti-mouse/human CD11b (Clone M1/70) | BioLegend | Cat# 101212; RRID:AB_312795 |
| APC anti-mouse Ly-6G/Ly-6C (Gr-1) (Clone RB6-8C5) | BioLegend | Cat#108412; RRID:AB_313377 |
| PE/Cy7 anti-mouse Ly-6G/Ly-6C (Gr-1) (Clone RB6-8C5) | BioLegend | Cat# 108416; RRID:AB_313381 |
| PE/Cy7 anti-mouse TER-119/Erythroid Cells (Clone TER-119) | BioLegend | Cat# 116222; RRID:AB_2281408 |
| PE anti-mouse TER-119/Erythroid Cells (Clone TER-119) | BioLegend | Cat# 116208; RRID:AB_313709 |
| APC anti-CD71 (Transferrin Receptor) (Clone R17217 (RI7 217.1.4) | Thermo Fisher Scientific | Cat# 17-0711-82; RRID:AB_1834355 |
| APC-eFluor™ 780 anti-CD117 (c-Kit) (Clone 2B8) | Thermo Fisher Scientific | Cat# 47-1171-82; RRID:AB_1272177 |
| PerCP/Cy5.5 anti-mouse Ly-6A/E (Sca-1) (Clone D7) | BioLegend | Cat# 108124; RRID:AB_893615 |
| APC anti-mouse CD135 (Clone A2F10) | BioLegend | Cat# 135310; RRID:AB_2107050 |
| PE anti-mouse CD150 (SLAM) (Clone TC15-12F12.2) | BioLegend | Cat# 115904; RRID:AB_313683 |
| Brilliant Violet 650™ anti-mouse CD150 (SLAM) (Clone TC15-12F12.2) | BioLegend | Cat# 115932; RRID:AB_2715765 |
| Brilliant Violet 785™ anti-mouse CD150 (SLAM) (Clone TC15-12F12.2) | BioLegend | Cat# 115937; RRID:AB_2565962 |
| APC anti-mouse CD48 (Clone HM48-1) | BioLegend | Cat# 103412; RRID:AB_571997 |
| BV711 anti-Mouse CD48 (Clone HM48-1) | BD Biosciences | Cat# 740687; RRID:AB_2740373 |
| FITC anti-mouse CD34 Clone RAM34) | Thermo Fisher Scientific | Cat# 11-0341-82; RRID:AB_465021 |
| Brilliant Violet 421™ anti-mouse CD16/32 (Clone 93) | BioLegend | Cat# 101332; RRID:AB_2650889 |
| Brilliant Violet 711™ anti-mouse CD16/32 (Clone 93) | BioLegend | Cat# 101337; RRID:AB_2565637 |
| PE anti-mouse/rat CD81 (Clone Eat-2) | BioLegend | Cat# 104905; RRID:AB_2076267 |
| PerCP/Cy5.5 anti-mouse CD106 (Clone 429 (MVCAM.A) | BioLegend | Cat# 105715; RRID:AB_1595594 |
| APC anti-mouse CD106 (Clone 429 (MVCAM.A) | BioLegend | Cat# 105717; RRID:AB_1877142 |
| Biotin anti-mouse Ly-6G (Clone 1A8) | BioLegend | Cat# 127603; RRID:AB_1186105 |
| Biotin anti-mouse CD90.2 (Thy1.2) (Clone 53–2.1) | BioLegend | Cat# 140313; RRID:AB_10640826 |
| Biotin Rat Anti-Mouse CD45R/B220 (Clone RA3-6B2) | BD Biosciences | Cat# 553086; RRID:AB_394616 |
| Biotin anti-mouse NK-1.1 (Clone PK136) | BioLegend | Cat# 108704; RRID:AB_313391 |
| Biotin Anti-Mouse TER-119/Erythroid Cells (Clone TER-119) | BD Biosciences | Cat# 553672; RRID:AB_394985 |
| Biotin anti-mouse Ly-6A/E (Sca-1) (Clone D7) | BioLegend | Cat# 108104; RRID:AB_313341 |
| Pacific Blue™ anti-mouse CD45.2 (Clone 104) | BioLegend | Cat# 109820; RRID:AB_492872 |
| PE/Cy7 anti-mouse CD45.1 (Clone A20) | BioLegend | Cat# 110730; RRID:AB_1134168 |
| FITC anti-mouse CD45.1 (Clone A20) | BioLegend | Cat# 110706; RRID:AB_313495 |
| SMARCC1/BAF155 (D7F8S) Rabbit mAb antibody | Cell Signaling Technology | Cat# 11956; RRID:AB_2797776 |
| BRG1 (D1Q7F) Rabbit mAb antibody | Cell Signaling Technology | Cat# 49360; RRID:AB_2728743 |
| SMARCC2/BAF170 antibody | Cell Signaling Technology | Cat# 8829; RRID:AB_11141240 |
| SMARCE1/BAF57 (E6H5J) Rabbit mAb | Cell Signaling Technology | Cat# 33360; RRID:AB_2799034 |
| SMARCB1/BAF47 (D9C2) Rabbit mAb | Cell Signaling Technology | Cat# 8745; RRID:AB_10950321 |
| SMARCD1/BAF60A (E7W9W) Rabbit mAb | Cell Signaling Technology | Cat# 35070; RRID:AB_3096176 |
| Lamin B1 (D9V6H) Rabbit mAb | Cell Signaling Technology | Cat# 13435; RRID:AB_2737428 |
| Anti-rabbit IgG, HRP-linked antibody | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID:AB_1031062 |
| CEBPB Polyclonal antibody | Proteintech | Cat# 23431-1-AP; RRID:AB_2879278 |
| GATA-1 Antibody (N6) | Santa Cruz Biotechnology | Cat# sc-265; RRID:AB_627663 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Proteinase K | Thermo Fisher Scientific | Cat# EO0491 |
| Poly(I:C) (HMW) VacciGrade™ | InvivoGen | Cat# vac-pic |
| 5-Fluorouracil | Millipore-Sigma | Cat# F6627 |
| Cyclophosphamide monohydrate | Millipore-Sigma | Cat# C0768 |
| G-CSF (Filgrastim) | Amgen | N/A |
| DMEM | Thermo Fisher Scientific | Cat# 1196509 |
| RMPI 1640 | Gibco | Cat# 11875-085 |
| FBS | Millipore-Sigma | Cat# 12103C |
| 0.25% trypsin-EDTA | Gibco | Cat#25200-056 |
| L-Glutamine | Gibco | Cat#35050061 |
| Penicillin-streptomycin | Gibco | Cat# 15140122 |
| Sodium Pyruvate | Thermo Fisher Scientific | Cat# BW17-613E |
| 2-Mercaptoethanol | Millipore-Sigma | Cat# M-6250 |
| Matrigel | Corning | Cat# 354248 |
| ACK lysing buffer | Gibco | Cat# A10492-01 |
| BSA | Millipore-Sigma | Cat# BSAV-RO |
| Collagenase-II | Worthington | Cat#LS004176 |
| Collagenase-III | Worthington | Cat#LS004182 |
| Deoxyribonuclease I | Worthington | Cat#LS002139 |
| Dispase-II | Millipore-Sigma | Cat# D6693 |
| BrdU | Millipore-Sigma | Cat# B5002 |
| FxCycle™ Violet Stain | Thermo Fisher Scientific | Cat# F10347 |
| StemSpan™ SFEM II | Stem Cell Technologies | Cat# 09655 |
| MethoCult3434 | Stem Cell Technologies | Cat# M3434 |
| Recombinant Murine SCF | PeproTech | Cat# 250-03 |
| Recombinant Murine Flt3-Ligand | PeproTech | Cat# 250-31L |
| Interleukin-3 (IL-3) supernatant | This paper | N/A |
| Recombinant Murine TPO | PeproTech | Cat# 315-14 |
| Recombinant Murine IL-6 | PeproTech | Cat# 216-16 |
| Recombinant Murine IL-11 | PeproTech | Cat# 220-11 |
| Recombinant Murine M-CSF | PeproTech | Cat# 315-02 |
| Recombinant Murine GM-CSF | PeproTech | Cat# 315-03 |
| Protease inhibitor cocktail | Sigma | Cat#11836170001 |
| ECL chemiluminescence substrate | Thermo Fisher Scientific | Cat#32106 |
| 10% solution Nonidet P-40 substitute | Abcam | Cat# ab142227 |
| Tween 20 | Millipore-Sigma | Cat# 11332465001 |
| Digitonin | Promega | Cat# G9441 |
| Dimethyl Formamide | Millipore-Sigma | Cat# D4551 |
| Tagment DNA Enzyme 1 | Illumina | Cat# 20034197 |
| 2x NEBNext Master Mix | New England Biolabs | Cat# M0541 |
| Ampure XP beads | Beckman Coulter | Cat# A63880 |
| 4200 TapeStation High Sensitivity D1000 ScreenTape | Agilent Technologies | Cat# 5067-5584 |
| 4200 TapeStation High Sensitivity D1000 Reagents | Agilent Technologies | Cat# 5067-5583 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse Direct PCR Kit | Bimake | Cat# B40013 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34961 |
| Tumor Dissociation Kit, mouse | Miltenyi Biotec | Cat# 130-096-730 |
| BD Cytofix/Cytoperm™ Fixation/Permeabilization Kit | BD Biosciences | Cat# 554714 |
| Intracellular Fixation & Permeabilization Buffer Set | Thermo Fisher Scientific | Cat# 88-8824-00 |
| Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| PE Mouse Anti- BrdU Set | BD Biosciences | Cat# 556029 |
| FITC Mouse Anti-Ki-67 Set | BD Biosciences | Cat# 556026 |
| PE Annexin V Apoptosis Detection Kit | BD Biosciences | Cat# 559763 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| SimpleChIP® Plus Enzymatic Chromatin IP Kit | Cell Signaling Technology | Cat#9005 |
| DNA Clean and Concentrator-5 Kit | Zymo Research | Cat# D4014 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32851 |
| Deposited data | ||
| All sequencing data generated in this study | This study | GEO: GSE240585 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| PyMT-B6 | (Barisas DAG et al.)63 | N/A |
| 1956 sarcoma cells | (Kabir AU et al.)64 | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Baf155f/f | (Choi J et al.)29 | N/A |
| Vav-Cre | Jackson Laboratories | RRID: IMSR_JAX:008610 |
| Mx1-Cre | Jackson Laboratories | RRID: IMSR_JAX:003556 |
| CD45.1 | Jackson Laboratories | RRID: IMSR_JAX:002014 |
| CD45.2 | Jackson Laboratories | RRID: IMSR_JAX:000664 |
| C57BI6/J Wild Type | Jackson Laboratories | RRID: IMSR_JAX:000664 |
|
| ||
| Oligonucleotides | ||
|
| ||
| ChIP-qPCR primers: see Table S6 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo software version 10.10.0 | TreeStar Inc. | https://www.flowjo.com |
| Graphpad Prism version 10.2.3 (347) | Graphpad Software, LLC. | https://www.graphpad.com/scientific-software/prism/ |
| R(v3.6.1) | R Foundation | https://www.r-project.org/ |
| Seurat (v3.2.3) | (Hao Y et al.)65 | https://satijalab.org/seurat/ |
| ArchR (v1.0.1) | (Granja JM et al.)66 | https://www.archrproject.com/ |
| Cellranger-arc (v2.0) | 10x genomics | https://www.10xgenomics.com/support/software/cell-ranger-arc/latest |
| AIAP (v1.1) | (Liu S et al.)67 | https://github.com/Zhang-lab/ATAC-seq_QC_analysis |
| deepTools (v3.5.0) | (Ramírez F et al.)68 | https://github.com/deeptools/deepTools |
| DESeq2 (v1.26.0) | (Love MI et al.)69 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| WashU Epigenome Browser (v54.0.6) | (Li D et al.)70 | https://epigenomegateway.wustl.edu/browser/ |
| Code used for sequencing data analyses | This study | https://doi.org/10.5281/zenodo.11992331 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Baf155f/f mice have been previously described.29,34 Vav-Cre (008610) mice were purchased from Jackson Labs and the line was maintained in a rodent barrier facility. Vav-Cre; Baf155 conditional knockout mice were generated by first crossing Vav-Cre positive females to Baf155f/f males. Vav-Cre; Baf155f/+ females were then crossed to Baf155f/f males to generate the conditional knockout (Baf155 KOVav). The same method was used to generate Mx1-Cre; Baf155 conditional knockout mice (Baf155 KOMx1), with either male or female carrying Cre. CD45.1 (002014) and CD45.2 (000664) mice were purchased from Jackson Labs and the lines were maintained in a rodent barrier facility. CD45.1/2 mice were generated by crossing CD45.1 and CD45.2. Animal studies were approved by the Animal Studies Committee at Washington University School of Medicine. All in vivo experiments were performed following the guidelines set by the Institutional Animal Care and Use Committee. Animals were fed standard chow diet in an ad libitum manner, and kept under an ambient temperature of 22°C and 50–60% humidity with a 12-h dark/light cycle (6 a.m. - 6 p.m.: light; 6 p.m. - 6 a.m.: dark). Both male and female mice were used in all the in vivo experiments. The influence of sex on phenotype was not observed.
Cell culture
PyMT-B663 tumor cells were cultured in DMEM high glucose (Cat:1196509, ThermoFisher Scientific) growth medium supplemented with 10% (v/v) FBS (Cat:12103C, Millipore Sigma), 100 unit/ml penicillin-streptomycin (Cat:15140122, ThermoFisher Scientific). 1956 sarcoma cells64 were cultured in RPMI 1640 growth medium supplemented with 10% (v/v) FBS, 100 unit/ml penicillin-streptomycin, 1% (v/v) L-glutamine (200mM) (Cat:BW17–605E, ThermoFisher Scientific), 1% (v/v) Sodium Pyruvate (100mM) (Cat: BW13–115E, ThermoFisher Scientific), 0.5% (v/v) Sodium Bicarbonate (7.5% w/v stock) (Cat: BW17–613E, ThermoFisher Scientific), and 0.1% (v/v) 2-Mercaptoethanol (Cat: M-6250, Millipore Sigma).
METHOD DETAILS
Genotyping
Mouse genomic DNA was isolated using the Mouse Direct PCR kit (Bimake). Genotyping was performed by PCR. To assess Vav-Cre excision efficiency in hematopoietic stem cells (HSCs), sorted HSCs (CD34−CD150+CD48−KSL) were re-suspended in 100μL lysis buffer + proteinase K, incubated at 55°C for 20 min, and then 95°C for 5 min. The lysates were subsequently used for genotyping. Sequences for genotyping primers are as follows: Vav-Cre: 5′-AGATGCCAGGAC ATCAGGAACCTG-3′ and 5′-ATCAGCCACACCA GACACAGAGATC-3’ (236bp), Mx1-Cre: 5′- GTGAGT TTCGTTTCTGAGCTCC-3′ and 5′-CGGTTATTCAACTTGCACCA-3’ (340bp), Baf155: P1: 5′-TGTCATCCATGAGGAGTGGTC-3′, P2: 5′-GGTAGCTCACAAATGCCTGT-3’ (WT = 400bp, Flox = 450bp), and P3: 5′-AACCTAGATGGCTCAGTAGGC-3’ (Deletion = 740bp). Primers for genomic quantitative PCR are as follows: (i) 5′-AGGCGGAG ATTTGATCTTCAG-3′, (ii) 5′-AGAGGAAAGATGAGGCAAAGG-3’.
In vivo treatment
For Mx1-Cre mediated deletion, four to six-week-old Mx1-Cre-posivite or -negative mice were intraperitoneally (i.p.) injected every other day for five total injections with 250 μg of poly (I:C) HMW (InvivoGen). Poly (I:C) injection was completed at least 15 days prior to each experiment.
For the induction of hematopoietic stress, mice were injected i.p. with one dose of 5-FU (Sigma, Cat#: F6627) in PBS (150 mg/kg or 250 mg/kg body weight). Total BM cell count, hematopoietic progenitors’ frequency and numbers were determined after 11 days (250 mg/kg) or 10 days (150 mg/kg) of 5-FU administration. Cell cycles were determined after 9 days (150 mg/kg) of 5-FU administration. For the evaluation of susceptibility, mice were challenged with two doses of 5-FU (150 mg/kg) at 7-day interval and survival time was recorded from the first injection until death.
For Cy/GCSF induced stress, mice were injected i.p. with one dose of cyclophosphamide (4 mg/mouse, Sigma) followed by two daily subcutaneous (s.c.) injection of 5 μg G-CSF (Neupogen, Amgen). 1 day after the last G-CSF injection, mice were euthanized for subsequent analysis.31
Tumor transplantation models
For tumor transplantation studies, 5 × 105 PyMT-B6 tumor cells or 1 × 106 1956 cells were mixed with growth factor reduced Matrigel (Cat: 354248; Corning) at a 1:1 (v/v) ratio in PBS and injected subcutaneously into the back of the mice. Tumor growth was measured accordingly, and tumors were harvested at 21 days for PyMT-B6 and 18 days for 1956.
BM transplantation assay
For competitive BM transplantation (BMT), 2 million total BM cells from 8 to 12-week-old control or Baf155 KOVav (CD45.2) mice were mixed with an equal number of competitor total BM cells (CD45.1) and transplanted into lethally irradiated (9.5Gy) recipient mice (CD45.1/2) by retro-orbital injection. Every four weeks post BMT, donor cell reconstitution in PB was evaluated by flow cytometry. Sixteen weeks after BMT, reconstituted donor stem and progenitor cells (HSPC) from BM were analyzed by flow cytometry. For competitive BMT prior to Baf155 deletion, 2 million total BM cells from 8 to 12-week-old control or Baf155 KOMx1 (CD45.2) mice were mixed with an equal number of competitor total BM cells (CD45.1) and transplanted into lethally irradiated (9.5Gy) recipient mice (CD45.1/2) by retro-orbital injection. Donor engraftment was assessed in PB 4 weeks after transplantation, Baf155 deletion was then induced in recipient mice using poly (I:C). The reconstitution of donor cell was determined as described above. For whole BMT, 2 million total BM cells from 8 to 12-week-old control or Baf155 KOVav (CD45.2) mice were transplanted into lethally irradiated (9.5Gy) recipient mice (CD45.1) by retro-orbital injection. The survival time of recipient mice was recorded from BMT until death. PB analysis was performed on surviving recipient mice at day 30 and day 60 post BMT.
Flow cytometric analysis
BM cells were harvested from femurs and tibias by centrifugation at 6000rpm for 5 min at 4°C. Spleen was collected and meshed into a single-cells suspension through a 70-μm cell strainer using the back side of a sterile 5mL syringe plunger. Peripheral blood (PB) was collected by venipuncture of the facial vein. For staining, cells from PB, BM and Spleen were lysed with ACK lysing buffer (Gibco) for 3 min at RT to remove red blood cells, followed by staining with different fluorochrome-conjugated anti-CD45-BUV395 (30-F11) (1:200), -CD11b-BV650 (M1/70) (1:200), -CD172a-PerCP/e710 (P84) (1:200), -SiglecF-AF647 (E50–2440) (1:200), -Ly6G-BV711 (1A8) (1:200), -Ly6C-BV785 (HK1.4) (1:200), -CD115-BV605 (AFS98) (1:200), -CD3-APC\Cy7 (17A2) (1:200), -CD4-FITC (RM4–5) (1:200), -CD8-PE (53–6.7) (1:200), -B220-PE\Cy7 (RA3–6B2) (1:200), -F4\80-BV421 (BM8) (1:200) antibodies, for 40 min at 4°C. Cells were washed and suspended in FACS buffer (PBS with 0.5% BSA) for flow cytometry analysis. The gating strategy of different cell lineages is shown in Figure S1G.71
Tumor tissues were harvested, minced into fine pieces, and dissociated into single-cell suspensions with an enzymatic digestion buffer consisting of Collagenase-II (For 1956 tumors) (Cat: LS004176, Worthington) or Collagenase-III (For PyMT-B6 tumors) (Cat: LS004182, Worthington), along with Dispase-II (Cat: D4693, Millipore Sigma) and Deoxyribonuclease 1 (Cat: LS002139, Worthingon). Next, the cells suspensions were incubated with LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Cat: L34961) alone with different panels of fluorophore-conjugated surface staining antibodies: CD45-BV605 (30-F11) (1:200), CD3-APC\Cy7 (17A2) (1:200), CD4-BUV496 (GK1.5) (1:200), CD8-PE\Cy5 (53–6.7) (1:200), CD25-BV650 (PC61) (1:200), CD11b-BUV737 (M1/70) (1:200), F4/80-PercP\Cy5.5 (BM8) (1:200), Ly6G-BUV805 (1A8) (1:200), Ly6C-BV510 (HK1.4) (1:200), B220-BUV395 (RA3–6B2) (1:200). For Subsequent intracellular staining, cell suspensions were fixed and permeabilized with either Foxp3/Transcription Factor Staining Buffer Set (Cat: 00–5523-00, ThermoFisher Scientific) or Intracellular Fixation & Permeabilization Buffer Set (Cat: 88–8824-00, ThermoFisher Scientific) and subsequently stained with intracellular anti-Foxp3-BV421 (1:100). The different lineages are defined as: B cells (CD45+B220+), Treg (CD45+CD3+CD4+CD25+Foxp3+), CTL (CD45+CD3+CD8+), TAM (CD45+CD11b+F4/80+), MDSC (CD45+CD11b+Ly6G + Ly6C+), Neutrophil (CD45+CD11b+Ly6G + Ly6CLow).
Samples were analyzed using either BD FACSSymphony A3 (BD Biosciences) or BD LSRFortessa X-20 (BD Biosciences) and later processed with FlowJo software (BD Life Sciences).
Hematopoietic stem and progenitor cell analysis
For lineage staining, cells from PB or BM were lysed with ACK lysing buffer (Gibco) for 3 min at RT to remove red blood cells, followed by staining with different fluorochrome-conjugated anti-Gr-1 (RB6–8C5) (granulocytes) (1:200), -CD11b (M1/70) (macrophage) (1:200), -B220 (RA3–6B2) (B cells) (1:200), -CD4 (GK1.5) (1:200) and -CD8 (53–6.7) (1:200) or -CD3 (17A2) (T cells) (1:200) antibodies, for 40 min at 4°C. Cells were washed and suspended in FACS buffer (PBS with 0.5% BSA) for flow cytometry analysis.
For HSPC and committed progenitors staining, BM cells were harvested from femurs and tibias by centrifugation at 6,000rpm for 5 min at 4°C and quickly lysed with ACK lying buffer for 3 min at RT. Cells were suspended in FACS buffer and counted on an automated Nexcelom cell counter. Cells were then stained with PE-Cy7 conjugated anti-Gr-1 (RB6–8C5) (1:400), -CD11b (M1/70) (1:200), -B220 (RA3–6B2) (1:200), -Ter119 (TER-119) (1:200) and -CD3 (145–2C11) (1:200), in combination with APC-Cy7-c-Kit (2B8) (1:100), PerCP-Cy5.5-Sca1 (E13–161.7) (1:100), APC-Flk2 (A2F10.1) (1:100), PE-CD150 (TC15–12F12.2) (1:100), BV711-CD48 (HM48–1) (1:100), FITC-CD34 (RAM34) (1:100) and BV421-CD16/32 (93) (1:200) antibodies for 40 min on ice. Flow cytometry was carried out on BD Symphony A3 machine. Data were analyzed on FlowJo software (FlowJo, LLC). Different HSPC subpopulations were defined as long-term hematopoietic stem cells (LT-HSC, Lin−c-Kit+Scal1+Flk2−CD150+CD48−), short-term hematopoietic stem cells (ST-HSC, Lin−c-Kit+Scal1+Flk2−CD150−CD48−), multiple potent progenitors (megakaryocyte/erythroid-biased MPP2, Flk2−CD150+CD48+ KSL; Myeloid-biased MPP3, Flk2−CD150−CD48+ KSL; lymphoid-biased MPP4, Flk2+CD150− KSL).72 Different committed progenitors were defined as granulocyte-monocyte progenitor (GMP, CD34+CD16/32+Lin−c-Kit+Sca1−) cells, common myeloid progenitor (CMP, CD34+CD16/32− Lin−c-Kit+Sca1−) cells and megakaryocyte-erythrocyte progenitor (MEP, CD34−CD16/32− Lin−c-Kit+Sca1−).
For the identification of BM myeloid progenitor cell subsets, BM cells were stained with BUV395-CD45 (30-F11) (1:200), APC-Cy7-c-Kit (2B8) (1:100), FITC-CD34 (RAM34) (1:100), BV711-CD16/32 (93) (1:200), APC-Flt3 (A2F10.1) (1:100), BV785-Ly6C (HK1.4) (1:200), PE-CD81 (Eat-2) (1:100), BV605-CD115 (AFS98) (1:200), BUV737-CD11b (M1/70) (1:200), PerCP-Cy5.5-CD106 (429) (1:100), together with exclusion lineage markers that include Ly6G (1A8), CD90.2 (53–2.1), B220 (RA3–6B2), NK1.1 (PK136), Ter119 (TER119), and Sca-1 (D7). After exclusion of cell doublets and dead cells with DAPI, proNeu1 were identified as CD45+ Lin−cKithiCD34+CD16/32+Flt3−Ly6C+CD115−CD81+CD11blowCD106−, proNeu2 were identified as CD45+Lin−cKithiCD34+CD16/32+ Flt3−Ly6C+CD115−CD81+CD11b+CD106+, cMoP were identified as CD45+Lin−cKithiCD34+CD16/32+Flt3−Ly6C+CD115+CD81−.
For transplantation experiments, PB chimerism and lineage distribution of donor-derived cells were assessed by cell staining with Pacific blue-CD45.2 (104), PE-Cy7-CD45.1 (A20), FITC-CD4 (GK1.5) and -CD8 (53–6.7), PerCP-Cy5.5-B220 (RA3–6B2), APC-Gr1 (RB6–8C5), PE-CD11b (M1/70).
BrdU and cell cycle assay
Mice were i.p. injected with 200μL BrdU (10 mg/kg; Sigma) for 2 h. Total BM cells were stained with cell surface markers for HSCs/MPPs, and then fixed and permeabilized with BD Cytofix/Cytoperm kit. After washing with BD Perm/Wash buffer, cells were intracellularly stained with PE-anti-BrdU (Cat#: 556029, BD Pharmingen) (20μL per test) and FITC-anti-Ki67 (Cat#: 556026, BD Pharmingen) (20μL per test) for 2 h at RT. After washing, cells were incubated with FxCycle Violet dye (ThermoFisher) for 1–2 h before acquisition.
Hematopoietic stem and progenitor cell sorting and ex vivo cultures
Single-cell suspensions from BM were prepared as described above. Cells were then stained with PE-Cy7 conjugated anti-Gr-1 (RB6–8C5), -CD11b (M1/70), -B220 (RA3–6B2), -Ter119 (TER-119) and -CD3 (145–2C11), in combination with APC-Cy7-c-Kit (2B8), PerCP-Cy5.5-Sca1 (E13–161.7), APC-Flk2 (A2F10.1), PE-CD150 (TC15–12F12.2), FITC-CD34 (RAM34) and BV421-CD16/32 (93) antibodies for 40 min on ice. KSLs (Lin−c-Kit+Scal1+), LT-HSCs (CD34−Flk2−KSL), MPPs (Flk2hiCD34+KSL), CMPs and GMPs were sorted on FACS Aria II (BD Biosciences) sorter using 85μm nozzles.
For in vitro competitive culture, sorted KSL cells from control or Baf155 KOVav (CD45.2) and WT mice (CD45.1) were mixed at a 1:1 ratio (5000 cells each) and cultured in 24-well tissue-culture plates. Culture media consisted of StemSpan serum-free base medium (StemCell Technologies), 10% serum, penicillin (50U/ml) and streptomycin (50U/ml) and different combination of cytokines, either SCF (25 ng/ml, PeproTech), FLT3L (20 ng/ml, PeproTech) and IL3 (1% supernatant), or SCF (25 ng/ml, PeproTech), FLT3L (20 ng/ml, PeproTech) and IL3 (1% supernatant), mTPO (20 ng/ml, PeproTech), IL6 (10 ng/ml, PeproTech), IL11 (10 ng/ml, PeproTech), M-CSF (10 ng/ml, PeproTech) and GM-CSF (10 ng/ml, PeproTech). Cells were analyzed after 3.5–4 days of culture by flow cytometry stained with PE-Cy7 conjugated anti-B220 (RA3–6B2), -Ter119 (TER-119) and -CD3 (145–2C11), APC-conjugated anti-Gr-1 (RB6–8C5) and -CD11b (M1/70), Pacific blue-CD45.2 (104) and FITC-CD45.1 (A20).
For myeloid differentiation and surface marker analyses, LT-HSCs, MPPs, CMPs and GMPs were sorted from BM directly into a round-bottom 96-well plate at a density of 1000 cells/well. Culture media consisted of StemSpan serum-free base medium (StemCell Technologies), 10% serum, penicillin (50U/mL) and streptomycin (50U/mL) and SCF (25 ng/ml, PeproTech), FLT3L (20 ng/ml, PeproTech), IL3 (1% supernatant), mTPO (20 ng/ml, PeproTech), IL6 (10 ng/ml, PeproTech), IL11 (10 ng/ml, PeproTech), M-CSF (10 ng/ml, PeproTech) and GM-CSF (10 ng/ml, PeproTech). Cells were counted at indicated time point and analyzed at day 7 by flow cytometry using FITC-CD45 (30-F11), BV421-CD11b (M1/70), APC-Gr1 (RB6–8C5) and PE-F4/80 (BM8).
Complete blood count (CBC)
Peripheral blood was collected by venipuncture of the facial vein and immediately transferred into EDTA-coated tubes (BD Microtainer). Blood samples were mixed and analyzed using the HV950 Hemavet (Drew Scientific, Inc.).
Colony-forming unit (CFU) assay
Unfractionated BM cells or sorted HSCs, MPPs, CMPs and GMPs from the various mouse model and their corresponding controls were plated in a 3cm Petri dish containing 1mL MethoCult M3434 methylcellulose medium (StemCell Technologies) at the indicated numbers and cultures were maintained in a humidified incubator at 37°C, 5% CO2. Colonies were counted and collected for further analysis at day 6.
Nuclear extract preparation and western blotting
Bone marrow cells were collected from control and KOVav mice. Cells were washed twice with phosphate-buffered saline (PBS) and centrifuged for 5 min at 3000 rpm. Nuclear extraction was conducted following manufacturing protocol (Abcam, ab113474). Cell pellet was resuspended in pre-extraction buffer containing dithiothreitol (DTT) and protease inhibitor cocktail (PIC), incubated on ice for 10 min, vortexed vigorously for 10s, and centrifuged at 12000 rpm for 1 min. The pellet containing nuclei was resuspended in nuclear extraction buffer containing DTT and PIC, incubated on ice for 15 min, vortexed vigorously every 3 min, then centrifuged at 14000 rpm for 10 min. Supernatant was collected and used for immunoblotting.
Western blotting was conducted following standard protocols. Primary antibodies used for western blotting are BAF155 (11956) (1:1000), BRG1 (49360) (1:1000), BAF170 (12760) (1:1000), BAF57 (33360) (1:1000), BAF47 (8745) (1:1000), BAF60A (35070) (1:1000) and Lamin B1 (13435) (1:1000) all from Cell Signaling Technology. Secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit IgG (7074, Cell Signaling Technology) (1:3000). Membranes were developed with ECL chemiluminescence substrate (ThermoFisher) and detected using ChemiDoc (Bio-Rad).
Single cell multiomic library preparation, sequencing and mapping
LK and LSK cells were FACS-isolated from the bone marrow of WT and KO animals at 9- or 11-days post 5-FU (150 mg/kg) treatment. 3 to 5 animals were pooled for each sample. After sorting, LK and LSK cells from each sample were mixed at a 1:1 ratio. Nuclei were isolated following the 10x protocol CG000365 • Rev B. Isolated nuclei were further processed using the 10x “Single Cell Multiome ATAC + Gene Expression” platform (CG000338 Rev B). The final libraries were sequenced to a depth of 50K reads per cell for the RNA fraction and 60K reads per cell for the ATAC fraction, following the sequencing recommendations in CG000338 Rev B and CG000373 • Rev B. Raw reads were aligned using cellranger-arc (v2.0).
Single cell multiomic data cleaning
First, for the RNA fraction of the multiomic data, gene x cell matrices from cellranger-arc were imported into Seurat65,73 (v3.2.3). All cells with fewer than 750 genes detected and all genes detected in fewer than 5 cells were removed from the matrices. Cells were then ranked based on the number of detected genes (nFeature, small to large) to construct a scatterplot with rank and nFeature as x and y axes, respectively. For each sample, an elbow point was identified on x, on the right side of which y increases sharply with respect to x. Cells after the elbow point were filtered out as putative doublets. Similarly, a scatterplot was constructed for the of percentage of UMIs mapped to mitochondria genes (percent.mt) against its rank, and all cells on the right side of the elbow point was removed to filter out apoptotic cells. For the remaining cells in each sample, normalization (Seurat::scTransform()), dimensionality reduction (Seurat::RunPCA(npcs = 60)), and clustering (Seurat::FindNeighbors(dims = 1:30) followed by Seurat::FindClusters(resolution = 0.7)) were performed.
For the ATAC fraction, cellranger-arc output was imported into ArchR66 (v1.0.1). Cells with fewer than 1000 ATAC fragments or TSS enrichment score < 10 were filtered out. For each sample, to remove putative doublets: 1) ArchR::addIterativeLSI(iterations = 2, dimsToUse = 1:30, clusterParams = list(resolution = 0.2, n.start = 10)) was used for dimensionality reduction; 2) ArchR::addClusters(resolution = 0.8, dimsToUse = 1:15) was used to find clusters; 3) ArchR::addUMAP(nNeighbors = 30, minDist = 0.5, dimsToUse = 1:15) was used to generate UMAP embeddings; 4) ATAC UMAP annotated by ATAC based clustering or RNA based clustering was then contrasted. If the cells in a given ATAC-based cluster resided closely to each other on the ATAC UMAP, but were from distinct lineages according to RNA based clustering, we considered this cluster as putative doublets and removed it from analyses; 5) for the remaining cells, those receiving top 3% DoubletEnrichment scores calculated by ArchR were further removed. Cells passing both RNA and ATAC based filtering were used for subsequent analyses.
Single cell multiomic data clustering and cell type identification
The scRNA data from all samples were integrated using the reciprocal PCA method in Seurat, with SCTransform based normalization and k.anchor = 20. Subsequently, dimensionality reduction (Seurat::RunPCA(npcs = 60)), clustering (Seurat::FindNeighbors(dims = 1:30) followed by Seurat::FindClusters(resolution = 0.8)), and UMAP visualization (Seurat::RunUMAP(dims = 1:30)) were performed. The known marker genes of HSPC and various blood lineages were used to assign cell types to clusters.
The ATAC fraction of all samples were merged using ArchR without batch effect correction. Dimensionality reduction and UMAP were accomplished using the same functions and parameters mentioned above for the data cleaning of single samples. Cluster-specific peaks (“marker peaks”) for each RNA based cluster were identified using ArchR::getMarkerFeatures(). Motifs enriched in marker peaks were identified using ArchR::peakAnnoEnrichment() with the HOMER motif database. chromVAR analyses were performed using ArchR::addDeviationsMatrix() with the HOMER motif database. ArchR::addImputeWeights() with default parameters was used to smooth the chromVAR results for visualization.
Single cell multiomic data differential analyses
Differential analyses were performed using samples obtained on day 9 post 5-FU treatment. However, identified DEGs and DARs showed the same trend in day 11 post 5-FU treatment samples. For differential gene expression, a pseudobulk gene expression matrix was generated for each cluster through aggregating the raw counts from all pass-filter cells in each sample. Each cluster-specific pseudobulk matrix was filtered to only retain genes that are detected in all pseudobulk samples. The rationale is that for non-detected genes, it is uncertain if they were not expressed or not detected, especially when the number of cells in the cluster is low. Additionally, it is uncertain how to normalize 0’s. DESeq269 (v1.26.0) was used to perform differential expression analyses from the filtered pseudobulk matrices. DEGs were determined based on the cutoff of FDR < 0.05 and log2FoldChange > 1. A similar method was used to identify DARs: 1) A union peakset was created containing peaks from all clusters using ArchR::addReproduciblePeakSet(). A peak x sample pseudobulk matrix was subsequently created for each cluster. Each cluster-specific pseudobulk matrix was filtered such that: 1) Only peaks with at least 1 fragment in all pseudobulk samples were retained; 2) A cpm matrix was calculated using edgeR::cpm(),74 and only peaks with cpm ≥ 6 in at least 1 sample were retained. This filter is added because the union peakset contains peaks from all clusters; 3) The maximum across all samples were calculated for each remaining peak. Peaks within the highest 2 percentiles in terms of this maximum were further filtered out. DARs were called with the same method and cutoffs as DEGs.
For motif enrichment in DARs, up (KO > WT) and down (WT > KO) DARs were separately ranked by p value and the top 1000 peaks (or all peaks when fewer than 1000 were available) in each category were used for motif enrichment. For each DAR used for motif enrichment, its 25 closest non-DARs in the Euclidean space constructed from WT samples were selected without replacement as background peaks. HOMER (v4.11.1) was used for motif enrichment in DARs relative to background peaks (findMotifsGenome.pl up_DAR.bed mm10.fa out_dir -bg up_DAR_background.bed -nomotif -size given).75
GREAT analysis was used to identify biological pathways enriched in genes in the vicinity of down-DARs.76 The background and foreground peaks used in motif enrichment were used as input for GREAT. “Basal plus extension” was used to associate genes to DARs.
ATAC signal pile-up was performed using ArchR::getFootprints(smoothWindow = 20). Motifs were taken from the HOMER motif database. No bias correction was applied. Confidence intervals (shaded areas around curves) were calculated based on the 2 replicates of each condition (KO vs. WT) collected at day 9 post 5-FU treatment. The union peak set from all clusters was partitioned into “Promoter”, “Intronic”, and “Intergenic” peak sets using ChIPseekerannotatePeak (tssRegion = c(−1000, 500), TxDb = TxDb. Mmusculus.UCSC.mm10.knownGene, annoDb = “org.Mm.eg.db”). Peaks in other categories (e.g., Exonic and UTR peaks) were not assessed since each of these categories only contained a small number of peaks (< 8% of all peaks), and since this analysis only uses peaks that contain a specific motif, an even smaller number of peaks can be used. Therefore, the pile-up signatures for these other categories are less accurate. For each peak set (“Promoter”, “Intronic”, or “Intergenic”), this approach finds all instances of a specific motif (CEBP motif, for example) across the peak set, uses the centers of these motif instances as anchors, and computes the total numbers of Tn5 insertions across all motif instances for each bp within the motif center ± 250 bp region. The resulting ATAC pile-up signature was then normalized. The ATAC pile-up signature was quantified as the mean pile-up value of the motif center ±25 bp region, normalized to the mean of the WT samples, and presented in bar plots.
ChIPseeker77 (v1.24.0) was used to annotate each peak in the peakset as “Promoter”, “5′ UTR”, “Exon”, “Intron”, “Intergenic”, etc. (ChIPseeker::annotatePeak (tssRegion = c(−1000, 500), TxDb = TxDb.Mmusculus.UCSC.mm10.knownGene, annoDb = “org.M-m.eg.db”)). Peaks whose annotation is not “Promoter” were considered as putative enhancers.
Pathway enrichment in DEGs were performed using clusterProfiler::enricher78 (v3.12.0). The database of pathways included hall-mark and GOBP gene sets from MsigDB79 (v7.2). For each cluster, all genes in the pass-filter psedubulk matrix of that cluster were used as the “universe”.
For genome browser visualization, bigwig files were generated using ArchR::getGroupBW(), and visualized using WashU Epigenome Browser.70
Single cell pseudotime analyses
Pseudotime analyses were performed using Slingshot.55 Briefly, small clusters (as indicated in figures) were removed. Lineages was then inferred using slingshot::getLineages(). The PCA space (PC1–30) was used. The HSPC cluster was indicated as starting cluster (start.clus), whereas downstream progenitors were used as end clusters (end.clus). Then, slingshot::getCurves(approx_points = 400) was used to obtain trajectory curves in the PCA space. Finally, slingshot::embedCurves was used to project the trajectory from PCA space to UMAP space.
Bulk ATAC-seq library generation and data analyses
For ATAC-seq library generation, approximately 10,000 KSL cells and 30,000 GMP cells per sample were isolated from control and Vav-Cre; Baf155 KO BM using FACS sorter as described above. ATAC-seq libraries were generated following the Omni-ATAC protocol80 with modifications.15 The reads were de-multiplexed by using sample-specific index sequences. ATAC-seq data was processed using the AIAP pipeline67 (v1.1). We used neutrophil progenitors (cluster 0) or HSPC (cluster 1) specific pass-filter peaks for the analysis of GMP or KSL bulk data, respectively. Peak x sample matrices were constructed through counting the number of Tn5 insertions (output by AIAP) in each peak for each sample. Matrices were normalized using DESeq2::estimateSizeFactors(). The trend of chromatin accessibility changes (KO > WT or KO < WT) were determined by the sign of the log2FoldChange column returned by DESeq2::results().
To obtain CD4 and CD8 T cells-specific ATAC-peaks, Immgen ATAC data were retrieved from GEO (GEO: GSM2692186, GSM2692187, GSM2692188, GSM2692343), and processed using the AIAP pipeline. For each sample, peaks were called as a part of the AIAP pipeline, and were further filtered to only retain those with −log10(FDR) ≥ 8. Pass-filter peaks from all samples were merged. DESeq2 (default parameters) were used to find CD4 and CD8 specific peaks (FDR < 0.05, log2FoldChange > 1). These peaks were intersected with the lymphoid progenitor peaks used in the pseudobulk analyses of single-nuclei data (“single nuclei peaks”; GenomicRanges::findOverlaps()). The log2FoldChange of CD4 and CD8-specific peaks in KO vs. WT animals were represented by the log2FoldChange of the corresponding single nuclei peaks in the single-nuclei data. CD4 and CD8-specific peaks that did not overlap single nuclei peaks were removed.
H3K27ac and H3K27me3 profiles from public datasets
Public histone modification datasets were retrieved from GEO DataSets (https://www.ncbi.nlm.nih.gov/gds). The data source for each heatmap is indicated in figure legends.81–87 Original bigwig files submitted by the authors were used. For each cell type, 500 randomly sampled down-DARs not overlapping with repeats were used. Repeats were retrieved from UCSC table browser (mm10 genome, updated 2021–04-08). Heatmaps were generated using deeptools (v3.5.0).68
ChIP-qPCR
All target regions of ChIP-qPCR were 500 bp ATAC-peaks (defined by ArchR) with CEBP or GATA1 motifs. Motif occurrences were downloaded from https://www.vierstra.org/resources/motif_clustering. Some targets are down-DARs close to the transcription start sites of down-DEGs (Targets 1, 2, 5, 6), while others are non-DARs close to non-down-DEGs as control regions (Targets 3, 4, 7). All targets contain the CEBP or GATA1 motif.
Chromatin immunoprecipitation (ChIP) was performed according to the manufacture’s protocol (ChIP kit, 9005, Cell Signaling Technologies) with the following modification: 4 million whole BM cells were used per preparation. After cell lysis, nuclei extracts were digested by adding 0.5 μL Micrococcal Nuclease per IP prep and incubating for 20 min at 37°C with frequent mixing to digest DNA to a size of approximately 150–900 bp. Digestion was stopped by adding 10 μL 0.5 M EDTA and samples placed on ice for 2 min. Nuclei was pelleted and resuspend in 100 μL ChIP buffer. Nuclear lysates were further subjected to sonication to break nuclear membrane using a 120 Sonic Dismembrator (Fisher Scientific) at 4°C for 3 cycles, cycling ON for 10 s and OFF for 30 s at 40% amplitude. Approximately 5 μg of digested, cross-linked chromatin and 5 mg of antibody (CEBPB, ProteinTech, 23431–1-AP; GATA1, Santa Cruz, sc-265) were used per immunoprecipitation. IP samples were incubated overnight at 4°C with rotation, followed by 30 μL of protein G Magnetic Beads per IP reaction, and incubated for an additional 2 h at 4°C with rotation. After elution of chromatin from the antibody/protein G magnetic beads, reverse cross-link performed by adding 6 μL 5 M NaCl and 2 μL proteinase K per IP, and incubating for 6 h at 65°C. Immunoprecipitated DNA fragments were isolated using spin columns provided by the kit and subjected to qPCR with appropriate primers indicated in Table S6. Rabbit IgG (Cell Signaling Technologies, 2729) was used as a negative control. Quantitative PCR was performed in duplicate, and data were normalized to input values. The fold changes over IgG were shown in the figures.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
GraphPad Prism 10 software was used for performing statistical analysis and generating graphs/plots. Data are presented as mean with standard deviation for all the measurements. All experimental data were reliably reproduced in two or more individual biological replicates. Details of the statistical tests performed are given in the respective figure legends. p < 0.05 was considered statistically significant. No methods were used to determine whether the data met assumptions of the statistical approach.
Supplementary Material
Highlights.
Baf155 is needed for lineage-specific chromatin remodeling
Baf155 is critical for hematopoietic lineage differentiation
Baf155 is indispensable for hematopoietic regeneration
Baf155 deficiency compromises anti-tumor immunity
ACKNOWLEDGMENTS
We thank the Choi lab members for constructive criticism and support. We also thank Drs. Emery Bresnick and Leonard Zon for discussions and input on the manuscript. We thank Dr. Daniel Link for GCSF. We thank the Washington University Pathology FACS core. This work was supported by NIH grants R01HG007175, U01HG009391, and U24HG012070 (to T.W.) and R01HL55337 and R01HL149954 (to K.C.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114558.
REFERENCES
- 1.Bonifer C, and Cockerill PN (2017). Chromatin priming of genes in development: Concepts, mechanisms and consequences. Exp. Hematol. 49, 1–8. 10.1016/j.exphem.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Jimenez G, Griffiths SD, Ford AM, Greaves MF, and Enver T (1992). Activation of the beta-globin locus control region precedes commitment to the erythroid lineage. Proc. Natl. Acad. Sci. USA 89, 10618–10622. 10.1073/pnas.89.22.10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hargreaves DC, and Crabtree GR (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420. 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alfert A, Moreno N, and Kerl K (2019). The BAF complex in development and disease. Epigenet. Chromatin 12, 19. 10.1186/s13072-019-0264-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centore RC, Sandoval GJ, Soares LMM, Kadoch C, and Chan HM (2020). Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 36, 936–950. 10.1016/j.tig.2020.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 1, e1500447. 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho PJ, Lloyd SM, and Bao X (2019). Unwinding chromatin at the right places: how BAF is targeted to specific genomic locations during development. Development 146, dev178780. 10.1242/dev.178780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu G, Schones DE, Cui K, Ybarra R, Northrup D, Tang Q, Gattinoni L, Restifo NP, Huang S, and Zhao K (2011). Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res. 21, 1650–1658. 10.1101/gr.121145.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, and Roberts CW (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 14648. 10.1038/ncomms14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Lee RS, Alver BH, Haswell JR, Wang S, Mieczkowski J, Drier Y, Gillespie SM, Archer TC, Wu JN, et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295. 10.1038/ng.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bossen C, Murre CS, Chang AN, Mansson R, Rodewald HR, and Murre C (2015). The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 16, 775–784. 10.1038/ni.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narayanan R, Pirouz M, Kerimoglu C, Pham L, Wagener RJ, Kiszka KA, Rosenbusch J, Seong RH, Kessel M, Fischer A, et al. (2015). Loss of BAF (mSWI/SNF) Complexes Causes Global Transcriptional and Chromatin State Changes in Forebrain Development. Cell Rep. 13, 1842–1854. 10.1016/j.celrep.2015.10.046. [DOI] [PubMed] [Google Scholar]
- 13.Griffin CT, Brennan J, and Magnuson T (2008). The chromatin-remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development 135, 493–500. 10.1242/dev.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panamarova M, Cox A, Wicher KB, Butler R, Bulgakova N, Jeon S, Rosen B, Seong RH, Skarnes W, Crabtree G, and Zernicka-Goetz M (2016). The BAF chromatin remodelling complex is an epigenetic regulator of lineage specification in the early mouse embryo. Development 143, 1271–1283. 10.1242/dev.131961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Krchma K, Lee HJ, Prabhakar S, Wang X, Zhao H, Xing X, Seong RH, Fremont DH, Artyomov MN, et al. (2020). Requisite Chromatin Remodeling for Myeloid and Erythroid Lineage Differentiation from Erythromyeloid Progenitors. Cell Rep. 33, 108395. 10.1016/j.celrep.2020.108395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tu J, Liu X, Jia H, Reilly J, Yu S, Cai C, Liu F, Lv Y, Huang Y, Lu Z, et al. (2020). The chromatin remodeler Brg1 is required for formation and maintenance of hematopoietic stem cells. Faseb. J. 34, 11997–12008. 10.1096/fj.201903168RR. [DOI] [PubMed] [Google Scholar]
- 17.Han L, Madan V, Mayakonda A, Dakle P, Woon TW, Shyamsunder P, Nordin HBM, Cao Z, Sundaresan J, Lei I, et al. (2019). Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia 33, 2291–2305. 10.1038/s41375-019-0438-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Wan X, Zhou P, Zhou X, Zhang W, Hui X, Yuan X, Ding X, Zhu R, Meng G, et al. (2018). The chromatin remodeling subunit Baf200 promotes normal hematopoiesis and inhibits leukemogenesis. J. Hematol. Oncol. 11, 27. 10.1186/s13045-018-0567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krasteva V, Buscarlet M, Diaz-Tellez A, Bernard MA, Crabtree GR, and Lessard JA (2012). The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood 120, 4720–4732. 10.1182/blood-2012-04-427047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krasteva V, Crabtree GR, and Lessard JA (2017). The BAF45a/PHF10 subunit of SWI/SNF-like chromatin remodeling complexes is essential for hematopoietic stem cell maintenance. Exp. Hematol. 48, 58–71.e15. 10.1016/j.exphem.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Lee H, Dai F, Zhuang L, Xiao ZD, Kim J, Zhang Y, Ma L, You MJ, Wang Z, and Gan B (2016). BAF180 regulates cellular senescence and hematopoietic stem cell homeostasis through p21. Oncotarget 7, 19134–19146. 10.18632/oncotarget.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chi TH, Wan M, Zhao K, Taniuchi I, Chen L, Littman DR, and Crabtree GR (2002). Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature 418, 195–199. 10.1038/nature00876. [DOI] [PubMed] [Google Scholar]
- 23.Gebuhr TC, Kovalev GI, Bultman S, Godfrey V, Su L, and Magnuson T (2003). The role of Brg1, a catalytic subunit of mammalian chromatin-remodeling complexes, in T cell development. J. Exp. Med. 198, 1937–1949. 10.1084/jem.20030714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witzel M, Petersheim D, Fan Y, Bahrami E, Racek T, Rohlfs M, Puchalka J, Mertes C, Gagneur J, Ziegenhain C, et al. (2017). Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat. Genet. 49, 742–752. 10.1038/ng.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Priam P, Krasteva V, Rousseau P, D’Angelo G, Gaboury L, Sauvageau G, and Lessard JA (2017). SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPϵ dependent mechanism. Nat. Genet. 49, 753–764. 10.1038/ng.3812. [DOI] [PubMed] [Google Scholar]
- 26.Mittal P, and Roberts CWM (2020). The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 17, 435–448. 10.1038/s41571-020-0357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, and Adams JM (1999). Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94, 1855–1863. [PubMed] [Google Scholar]
- 28.Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, and Speck NA (2009). Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457, 887–891. 10.1038/nature07619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi J, Ko M, Jeon S, Jeon Y, Park K, Lee C, Lee H, and Seong RH (2012). The SWI/SNF-like BAF complex is essential for early B cell development. J. Immunol. 188, 3791–3803. 10.4049/jimmunol.1103390. [DOI] [PubMed] [Google Scholar]
- 30.Rivadeneyra L, Pozner RG, Meiss R, Fondevila C, Gomez RM, and Schattner M (2015). Poly (I:C) downregulates platelet production and function through type I interferon. Thromb. Haemostasis 114, 982–993. 10.1160/TH14-11-0951. [DOI] [PubMed] [Google Scholar]
- 31.Morrison SJ, Wright DE, and Weissman IL (1997). Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc. Natl. Acad. Sci. USA 94, 1908–1913. 10.1073/pnas.94.5.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oduro KA Jr., Liu F, Tan Q, Kim CK, Lubman O, Fremont D, Mills JC, and Choi K (2012). Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood 120, 2203–2213. 10.1182/blood-2011-11-391342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwok I, Becht E, Xia Y, Ng M, Teh YC, Tan L, Evrard M, Li JLY, Tran HTN, Tan Y, et al. (2020). Combinatorial Single-Cell Analyses of Granulocyte-Monocyte Progenitor Heterogeneity Reveals an Early Unipotent Neutrophil Progenitor. Immunity 53, 303–318.e5. 10.1016/j.immuni.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Xu CX, Lee TJ, Sakurai N, Krchma K, Liu F, Li D, Wang T, and Choi K (2017). ETV2/ER71 regulates hematopoietic regeneration by promoting hematopoietic stem cell proliferation. J. Exp. Med. 214, 1643–1653. 10.1084/jem.20160923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dahlin JS, Hamey FK, Pijuan-Sala B, Shepherd M, Lau WWY, Nestorowa S, Weinreb C, Wolock S, Hannah R, Diamanti E, et al. (2018). A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice. Blood 131, e1–e11. 10.1182/blood-2017-12-821413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, and Gottgens B (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31. 10.1182/blood-2016-05-716480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fast EM, Sporrij A, Manning M, Rocha EL, Yang S, Zhou Y, Guo J, Baryawno N, Barkas N, Scadden D, et al. (2021). External signals regulate continuous transcriptional states in hematopoietic stem cells. Elife 10, e66512. 10.7554/eLife.66512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Williams MJ, Park HJ, Bastos HP, Wang X, Prins D, Wilson NK, Johnson C, Sham K, Wantoch M, et al. (2022). STAT1 is essential for HSC function and maintains MHCIIhi stem cells that resist myeloablation and neoplastic expansion. Blood 140, 1592–1606. 10.1182/blood.2021014009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). Chrom-VAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nerlov C (2007). The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 17, 318–324. 10.1016/j.tcb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Pevny L, Simon MC, Robertson E, Klein WH, Tsai SF, D’Agati V, Orkin SH, and Costantini F (1991). Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349, 257–260. 10.1038/349257a0. [DOI] [PubMed] [Google Scholar]
- 42.Lawrence HJ, Christensen J, Fong S, Hu YL, Weissman I, Sauvageau G, Humphries RK, and Largman C (2005). Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 106, 3988–3994. 10.1182/blood-2005-05-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shih HY, Sciume G, Mikami Y, Guo L, Sun HW, Brooks SR, Urban JF Jr., Davis FP, Kanno Y, and O’Shea JJ (2016). Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 165, 1120–1133. 10.1016/j.cell.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koues OI, Collins PL, Cella M, Robinette ML, Porter SI, Pyfrom SC, Payton JE, Colonna M, and Oltz EM (2016). Distinct Gene Regulatory Pathways for Human Innate versus Adaptive Lymphoid Cells. Cell 165, 1134–1146. 10.1016/j.cell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936. 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujimura N, Xu B, Dalman J, Deng H, Aoyama K, and Dalman RL (2015). CCR2 inhibition sequesters multiple subsets of leukocytes in the bone marrow. Sci. Rep. 5, 11664. 10.1038/srep11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokota T, Oritani K, Butz S, Kokame K, Kincade PW, Miyata T, Vestweber D, and Kanakura Y (2009). The endothelial antigen ESAM marks primitive hematopoietic progenitors throughout life in mice. Blood 113, 2914–2923. 10.1182/blood-2008-07-167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vicente C, Conchillo A, Garcia-Sanchez MA, and Odero MD (2012). The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit. Rev. Oncol. Hematol. 82, 1–17. 10.1016/j.critrevonc.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Carlos TM, and Harlan JM (1994). Leukocyte-endothelial adhesion molecules. Blood 84, 2068–2101. [PubMed] [Google Scholar]
- 50.Broxmeyer HE, Maze R, Miyazawa K, Carow C, Hendrie PC, Cooper S, Hangoc G, Vadhan-Raj S, and Lu L (1991). The kit receptor and its ligand, steel factor, as regulators of hemopoiesis. Cancer Cells (Cold Spring Harbor) 3, 480–487. [PubMed] [Google Scholar]
- 51.Wickrema A, Krantz SB, Winkelmann JC, and Bondurant MC (1992). Differentiation and erythropoietin receptor gene expression in human erythroid progenitor cells. Blood 80, 1940–1949. [PubMed] [Google Scholar]
- 52.Hart A, Melet F, Grossfeld P, Chien K, Jones C, Tunnacliffe A, Favier R, and Bernstein A (2000). Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity 13, 167–177. 10.1016/s1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- 53.Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S, and Busslinger M (2008). Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 28, 751–762. 10.1016/j.immuni.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 54.Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, and Ozato K (2000). ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity 13, 155–165. 10.1016/s1074-7613(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 55.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 19, 477. 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Battistello E, Hixon KA, Comstock DE, Collings CK, Chen X, Rodriguez Hernaez J, Lee S, Cervantes KS, Hinkley MM, Ntatsoulis K, et al. (2023). Stepwise activities of mSWI/SNF family chromatin remodeling complexes direct T cell activation and exhaustion. Mol. Cell 83, 1216–1236.e12. 10.1016/j.molcel.2023.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belk JA, Yao W, Ly N, Freitas KA, Chen YT, Shi Q, Valencia AM, Shifrut E, Kale N, Yost KE, et al. (2022). Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 40, 768–786.e7. 10.1016/j.ccell.2022.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo A, Huang H, Zhu Z, Chen MJ, Shi H, Yuan S, Sharma P, Connelly JP, Liedmann S, Dhungana Y, et al. (2022). cBAF complex components and MYC cooperate early in CD8(+) T cell fate. Nature 607, 135–141. 10.1038/s41586-022-04849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chambers C, Cermakova K, Chan YS, Kurtz K, Wohlan K, Lewis AH, Wang C, Pham A, Dejmek M, Sala M, et al. (2023). SWI/SNF Blockade Disrupts PU.1-Directed Enhancer Programs in Normal Hematopoietic Cells and Acute Myeloid Leukemia. Cancer Res. 83, 983–996. 10.1158/0008-5472.CAN-22-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tuoc TC, Boretius S, Sansom SN, Pitulescu ME, Frahm J, Livesey FJ, and Stoykova A (2013). Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell 25, 256–269. 10.1016/j.devcel.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 61.Narayanan R, Pham L, Kerimoglu C, Watanabe T, Castro Hernandez R, Sokpor G, Ulmke PA, Kiszka KA, Tonchev AB, Rosenbusch J, et al. (2018). Chromatin Remodeling BAF155 Subunit Regulates the Genesis of Basal Progenitors in Developing Cortex. iScience 4, 109–126. 10.1016/j.isci.2018.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sohn DH, Lee KY, Lee C, Oh J, Chung H, Jeon SH, and Seong RH (2007). SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J. Biol. Chem. 282, 10614–10624. 10.1074/jbc.M610563200. [DOI] [PubMed] [Google Scholar]
- 63.Barisas DAG, Kabir AU, Wu J, Krchma K, Kim M, Subramanian M, Zinselmeyer BH, Stewart CL, and Choi K (2023). Tumor-derived interleukin-1a and leukemia inhibitory factor promote extramedullary hematopoiesis. PLoS Biol. 21, e3001746. 10.1371/journal.pbio.3001746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kabir AU, Subramanian M, Lee DH, Wang X, Krchma K, Wu J, Naismith T, Halabi CM, Kim JY, Pulous FE, et al. (2021). Dual role of endothelial Myct1 in tumor angiogenesis and tumor immunity. Sci. Transl. Med. 13, eabb6731. 10.1126/scitranslmed.abb6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411. 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu S, Li D, Lyu C, Gontarz PM, Miao B, Madden PAF, Wang T, and Zhang B (2021). AIAP: A Quality Control and Integrative Analysis Package to Improve ATAC-seq Data Analysis. Dev. Reprod. Biol. 19, 641–651. 10.1016/j.gpb.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li D, Purushotham D, Harrison JK, Hsu S, Zhuo X, Fan C, Liu S, Xu V, Chen S, Xu J, et al. (2022). WashU Epigenome Browser update 2022. Nucleic Acids Res. 50, W774–W781. 10.1093/nar/gkac238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Z, Gu Y, Shin A, Zhang S, and Ginhoux F (2020). Analysis of Myeloid Cells in Mouse Tissues with Flow Cytometry. STAR Protoc. 1, 100029. 10.1016/j.xpro.2020.100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, and Passegue E (2015). Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 17, 35–46. 10.1016/j.stem.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296. 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 78.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Antoszewski M, Fournier N, Ruiz Buendia GA, Lourenco J, Liu Y, Sugrue T, Dubey C, Nkosi M, Pritchard CEJ, Huijbers IJ, et al. (2022). Tcf1 is essential for initiation of oncogenic Notch1-driven chromatin topology in T-ALL. Blood 139, 2483–2498. 10.1182/blood.2021012077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Celik H, Koh WK, Kramer AC, Ostrander EL, Mallaney C, Fisher DAC, Xiang J, Wilson WC, Martens A, Kothari A, et al. (2018). JARID2 Functions as a Tumor Suppressor in Myeloid Neoplasms by Repressing Self-Renewal in Hematopoietic Progenitor Cells. Cancer Cell 34, 741–756.e8. 10.1016/j.ccell.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu T, Morita K, Hill MC, Jiang Y, Kitano A, Saito Y, Wang F, Mao X, Hoegenauer KA, Morishita K, et al. (2019). PRDM16s transforms megakaryocyte-erythroid progenitors into myeloid leukemia-initiating cells. Blood 134, 614–625. 10.1182/blood.2018888255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ling T, Birger Y, Stankiewicz MJ, Ben-Haim N, Kalisky T, Rein A, Kugler E, Chen W, Fu C, Zhang K, et al. (2019). Chromatin occupancy and epigenetic analysis reveal new insights into the function of the GATA1 N terminus in erythropoiesis. Blood 134, 1619–1631. 10.1182/blood.2019001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Man N, Mas G, Karl DL, Sun J, Liu F, Yang Q, Torres-Martin M, Itonaga H, Martinez C, Chen S, et al. (2021). p300 suppresses the transition of myelodysplastic syndromes to acute myeloid leukemia. JCI Insight 6, e138478. 10.1172/jci.insight.138478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Poplineau M, Vernerey J, Platet N, N’Guyen L, Herault L, Esposito M, Saurin AJ, Guilouf C, Iwama A, and Duprez E (2019). PLZF limits enhancer activity during hematopoietic progenitor aging. Nucleic Acids Res. 47, 4509–4520. 10.1093/nar/gkz174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yun H, Narayan N, Vohra S, Giotopoulos G, Mupo A, Madrigal P, Sasca D, Lara-Astiaso D, Horton SJ, Agrawal-Singh S, et al. (2021). Mutational synergy during leukemia induction remodels chromatin accessibility, histone modifications and three-dimensional DNA topology to alter gene expression. Nat. Genet. 53, 1443–1455. 10.1038/s41588-021-00925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Code used for sequencing data analyses has been deposited in Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| BUV395 Rat Anti-Mouse CD45 (Clone 30-F11) | BD Biosciences | Cat#564279; RRID:AB_2651134 |
| FITC anti-mouse CD45 (Clone 30-F11) | BioLegend | Cat# 103108; RRID:AB_312973 |
| Brilliant Violet 605™ anti-mouse CD45 (Clone 30-F11) | BioLegend | Cat#103155; RRID:AB_2650656 |
| PerCP-eFluor™ 710 anti-mouse CD172a (SIRP alpha) (Clone P84) | Thermo Fisher Scientific | Cat#46-1721-80; RRID:AB_10805866 |
| Alexa Fluor® 647 Rat Anti-Mouse Siglec-F (Clone E50-2440) | BD Biosciences | Cat#562680; RRID:AB_2687570 |
| Brilliant Violet 711™ anti-mouse Ly-6G (Clone 1A8) | BioLegend | Cat#127643; RRID:AB_2565971 |
| BUV805 Rat Anti-Mouse Ly-6G (Clone 1A8) | BD Biosciences | Cat#741994; RRID:AB_2871294 |
| Brilliant Violet 785™ anti-mouse Ly-6C (Clone HK1.4) | BioLegend | Cat#128041; RRID:AB_2565852 |
| Brilliant Violet 510™ anti-mouse Ly-6C (Clone HK1.4) | BioLegend | Cat#128033; RRID:AB_2562351 |
| Brilliant Violet 605™ anti-mouse CD115 (CSF-1R) (Clone AFS98) | BioLegend | Cat#135517; RRID:AB_2562760 |
| FITC anti-mouse CD4 (Clone GK1.5) | BioLegend | Cat#100405; RRID:AB_312690 |
| BUV496 Rat Anti-Mouse CD4 (Clone GK1.5) | BD Biosciences | Cat#612952; RRID:AB_2813886 |
| PE/C7 anti-mouse/human CD45R/B220 (Clone RA3-6B2) | BioLegend | Cat# 103222; RRID:AB_313005 |
| BUV395 Rat Anti-Mouse CD45R/B220 (Clone RA3-6B2) | BD Biosciences | Cat#563793; RRID:AB_2738427 |
| PE/Cy7 anti-mouse/human CD45R/B220 (Clone RA3-6B2) | BioLegend | Cat#103221; RRID:AB_313004 |
| PerCP\Cy5.5 anti-mouse/human CD45R/B220- (Clone RA3-6B2) | BioLegend | Cat#103235; RRID:AB_893356 |
| Brilliant Violet 421™ anti-mouse F4/80 (Clone BM8) | BioLegend | Cat#123137; RRID:AB_2563102 |
| PE anti-mouse F4/80 (Clone BM8) | BioLegend | Cat# 123110; RRID:AB_893486 |
| PerCP/Cy5.5 anti-mouse F4/80 (Clone BM8) | BioLegend | Cat#123127; RRID:AB_893496 |
| PE Rat Anti-Mouse CD8a (Clone 53–6.7) | BD Biosciences | Cat#553033; RRID:AB_394571 |
| PE/Cy5 anti-mouse CD8a (Clone 53–6.7) | BioLegend | Cat#100710; RRID:AB_312749 |
| FITC anti-mouse CD8a (Clone 53–6.7) | BioLegend | Cat#100706; RRID:AB_312745 |
| Brilliant Violet 650™ anti-mouse CD25 (Clone PC61) | BioLegend | Cat#102038; RRID:AB_2563060 |
| FITC anti-mouse CD3 (Clone 17A2) | BioLegend | Cat#100204; RRID:AB_312661 |
| APC/Cyanine7 anti-mouse CD3 (Clone 17A2) | BioLegend | Cat#100221; RRID:AB_2057374 |
| PE/Cy7 anti-mouse CD3 (Clone 145-2C11) | BioLegend | Cat# 100320; RRID:AB_312685 |
| Brilliant Violet 421™ anti-mouse FOXP3 (Clone MF-14) | BioLegend | Cat#126419; RRID:AB_2565933 |
| BUV737 Rat Anti-CD11b (Clone M1/70) | BD Biosciences | Cat#612800; RRID:AB_2870127 |
| PE Rat Anti-CD11b (Clone M1/70) | BD Biosciences | Cat#553311; RRID:AB_394775 |
| PE/Cy7 anti-mouse/human CD11b (Cone M1/70) | BioLegend | Cat# 101216; RRID:AB_312799 |
| Brilliant Violet 650™ anti-mouse/human CD11b (Clone M1/70) | BioLegend | Cat#101259; RRID:AB_2566568 |
| APC anti-mouse/human CD11b (Clone M1/70) | BioLegend | Cat# 101212; RRID:AB_312795 |
| APC anti-mouse Ly-6G/Ly-6C (Gr-1) (Clone RB6-8C5) | BioLegend | Cat#108412; RRID:AB_313377 |
| PE/Cy7 anti-mouse Ly-6G/Ly-6C (Gr-1) (Clone RB6-8C5) | BioLegend | Cat# 108416; RRID:AB_313381 |
| PE/Cy7 anti-mouse TER-119/Erythroid Cells (Clone TER-119) | BioLegend | Cat# 116222; RRID:AB_2281408 |
| PE anti-mouse TER-119/Erythroid Cells (Clone TER-119) | BioLegend | Cat# 116208; RRID:AB_313709 |
| APC anti-CD71 (Transferrin Receptor) (Clone R17217 (RI7 217.1.4) | Thermo Fisher Scientific | Cat# 17-0711-82; RRID:AB_1834355 |
| APC-eFluor™ 780 anti-CD117 (c-Kit) (Clone 2B8) | Thermo Fisher Scientific | Cat# 47-1171-82; RRID:AB_1272177 |
| PerCP/Cy5.5 anti-mouse Ly-6A/E (Sca-1) (Clone D7) | BioLegend | Cat# 108124; RRID:AB_893615 |
| APC anti-mouse CD135 (Clone A2F10) | BioLegend | Cat# 135310; RRID:AB_2107050 |
| PE anti-mouse CD150 (SLAM) (Clone TC15-12F12.2) | BioLegend | Cat# 115904; RRID:AB_313683 |
| Brilliant Violet 650™ anti-mouse CD150 (SLAM) (Clone TC15-12F12.2) | BioLegend | Cat# 115932; RRID:AB_2715765 |
| Brilliant Violet 785™ anti-mouse CD150 (SLAM) (Clone TC15-12F12.2) | BioLegend | Cat# 115937; RRID:AB_2565962 |
| APC anti-mouse CD48 (Clone HM48-1) | BioLegend | Cat# 103412; RRID:AB_571997 |
| BV711 anti-Mouse CD48 (Clone HM48-1) | BD Biosciences | Cat# 740687; RRID:AB_2740373 |
| FITC anti-mouse CD34 Clone RAM34) | Thermo Fisher Scientific | Cat# 11-0341-82; RRID:AB_465021 |
| Brilliant Violet 421™ anti-mouse CD16/32 (Clone 93) | BioLegend | Cat# 101332; RRID:AB_2650889 |
| Brilliant Violet 711™ anti-mouse CD16/32 (Clone 93) | BioLegend | Cat# 101337; RRID:AB_2565637 |
| PE anti-mouse/rat CD81 (Clone Eat-2) | BioLegend | Cat# 104905; RRID:AB_2076267 |
| PerCP/Cy5.5 anti-mouse CD106 (Clone 429 (MVCAM.A) | BioLegend | Cat# 105715; RRID:AB_1595594 |
| APC anti-mouse CD106 (Clone 429 (MVCAM.A) | BioLegend | Cat# 105717; RRID:AB_1877142 |
| Biotin anti-mouse Ly-6G (Clone 1A8) | BioLegend | Cat# 127603; RRID:AB_1186105 |
| Biotin anti-mouse CD90.2 (Thy1.2) (Clone 53–2.1) | BioLegend | Cat# 140313; RRID:AB_10640826 |
| Biotin Rat Anti-Mouse CD45R/B220 (Clone RA3-6B2) | BD Biosciences | Cat# 553086; RRID:AB_394616 |
| Biotin anti-mouse NK-1.1 (Clone PK136) | BioLegend | Cat# 108704; RRID:AB_313391 |
| Biotin Anti-Mouse TER-119/Erythroid Cells (Clone TER-119) | BD Biosciences | Cat# 553672; RRID:AB_394985 |
| Biotin anti-mouse Ly-6A/E (Sca-1) (Clone D7) | BioLegend | Cat# 108104; RRID:AB_313341 |
| Pacific Blue™ anti-mouse CD45.2 (Clone 104) | BioLegend | Cat# 109820; RRID:AB_492872 |
| PE/Cy7 anti-mouse CD45.1 (Clone A20) | BioLegend | Cat# 110730; RRID:AB_1134168 |
| FITC anti-mouse CD45.1 (Clone A20) | BioLegend | Cat# 110706; RRID:AB_313495 |
| SMARCC1/BAF155 (D7F8S) Rabbit mAb antibody | Cell Signaling Technology | Cat# 11956; RRID:AB_2797776 |
| BRG1 (D1Q7F) Rabbit mAb antibody | Cell Signaling Technology | Cat# 49360; RRID:AB_2728743 |
| SMARCC2/BAF170 antibody | Cell Signaling Technology | Cat# 8829; RRID:AB_11141240 |
| SMARCE1/BAF57 (E6H5J) Rabbit mAb | Cell Signaling Technology | Cat# 33360; RRID:AB_2799034 |
| SMARCB1/BAF47 (D9C2) Rabbit mAb | Cell Signaling Technology | Cat# 8745; RRID:AB_10950321 |
| SMARCD1/BAF60A (E7W9W) Rabbit mAb | Cell Signaling Technology | Cat# 35070; RRID:AB_3096176 |
| Lamin B1 (D9V6H) Rabbit mAb | Cell Signaling Technology | Cat# 13435; RRID:AB_2737428 |
| Anti-rabbit IgG, HRP-linked antibody | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID:AB_1031062 |
| CEBPB Polyclonal antibody | Proteintech | Cat# 23431-1-AP; RRID:AB_2879278 |
| GATA-1 Antibody (N6) | Santa Cruz Biotechnology | Cat# sc-265; RRID:AB_627663 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Proteinase K | Thermo Fisher Scientific | Cat# EO0491 |
| Poly(I:C) (HMW) VacciGrade™ | InvivoGen | Cat# vac-pic |
| 5-Fluorouracil | Millipore-Sigma | Cat# F6627 |
| Cyclophosphamide monohydrate | Millipore-Sigma | Cat# C0768 |
| G-CSF (Filgrastim) | Amgen | N/A |
| DMEM | Thermo Fisher Scientific | Cat# 1196509 |
| RMPI 1640 | Gibco | Cat# 11875-085 |
| FBS | Millipore-Sigma | Cat# 12103C |
| 0.25% trypsin-EDTA | Gibco | Cat#25200-056 |
| L-Glutamine | Gibco | Cat#35050061 |
| Penicillin-streptomycin | Gibco | Cat# 15140122 |
| Sodium Pyruvate | Thermo Fisher Scientific | Cat# BW17-613E |
| 2-Mercaptoethanol | Millipore-Sigma | Cat# M-6250 |
| Matrigel | Corning | Cat# 354248 |
| ACK lysing buffer | Gibco | Cat# A10492-01 |
| BSA | Millipore-Sigma | Cat# BSAV-RO |
| Collagenase-II | Worthington | Cat#LS004176 |
| Collagenase-III | Worthington | Cat#LS004182 |
| Deoxyribonuclease I | Worthington | Cat#LS002139 |
| Dispase-II | Millipore-Sigma | Cat# D6693 |
| BrdU | Millipore-Sigma | Cat# B5002 |
| FxCycle™ Violet Stain | Thermo Fisher Scientific | Cat# F10347 |
| StemSpan™ SFEM II | Stem Cell Technologies | Cat# 09655 |
| MethoCult3434 | Stem Cell Technologies | Cat# M3434 |
| Recombinant Murine SCF | PeproTech | Cat# 250-03 |
| Recombinant Murine Flt3-Ligand | PeproTech | Cat# 250-31L |
| Interleukin-3 (IL-3) supernatant | This paper | N/A |
| Recombinant Murine TPO | PeproTech | Cat# 315-14 |
| Recombinant Murine IL-6 | PeproTech | Cat# 216-16 |
| Recombinant Murine IL-11 | PeproTech | Cat# 220-11 |
| Recombinant Murine M-CSF | PeproTech | Cat# 315-02 |
| Recombinant Murine GM-CSF | PeproTech | Cat# 315-03 |
| Protease inhibitor cocktail | Sigma | Cat#11836170001 |
| ECL chemiluminescence substrate | Thermo Fisher Scientific | Cat#32106 |
| 10% solution Nonidet P-40 substitute | Abcam | Cat# ab142227 |
| Tween 20 | Millipore-Sigma | Cat# 11332465001 |
| Digitonin | Promega | Cat# G9441 |
| Dimethyl Formamide | Millipore-Sigma | Cat# D4551 |
| Tagment DNA Enzyme 1 | Illumina | Cat# 20034197 |
| 2x NEBNext Master Mix | New England Biolabs | Cat# M0541 |
| Ampure XP beads | Beckman Coulter | Cat# A63880 |
| 4200 TapeStation High Sensitivity D1000 ScreenTape | Agilent Technologies | Cat# 5067-5584 |
| 4200 TapeStation High Sensitivity D1000 Reagents | Agilent Technologies | Cat# 5067-5583 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse Direct PCR Kit | Bimake | Cat# B40013 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34961 |
| Tumor Dissociation Kit, mouse | Miltenyi Biotec | Cat# 130-096-730 |
| BD Cytofix/Cytoperm™ Fixation/Permeabilization Kit | BD Biosciences | Cat# 554714 |
| Intracellular Fixation & Permeabilization Buffer Set | Thermo Fisher Scientific | Cat# 88-8824-00 |
| Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| PE Mouse Anti- BrdU Set | BD Biosciences | Cat# 556029 |
| FITC Mouse Anti-Ki-67 Set | BD Biosciences | Cat# 556026 |
| PE Annexin V Apoptosis Detection Kit | BD Biosciences | Cat# 559763 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| SimpleChIP® Plus Enzymatic Chromatin IP Kit | Cell Signaling Technology | Cat#9005 |
| DNA Clean and Concentrator-5 Kit | Zymo Research | Cat# D4014 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32851 |
| Deposited data | ||
| All sequencing data generated in this study | This study | GEO: GSE240585 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| PyMT-B6 | (Barisas DAG et al.)63 | N/A |
| 1956 sarcoma cells | (Kabir AU et al.)64 | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Baf155f/f | (Choi J et al.)29 | N/A |
| Vav-Cre | Jackson Laboratories | RRID: IMSR_JAX:008610 |
| Mx1-Cre | Jackson Laboratories | RRID: IMSR_JAX:003556 |
| CD45.1 | Jackson Laboratories | RRID: IMSR_JAX:002014 |
| CD45.2 | Jackson Laboratories | RRID: IMSR_JAX:000664 |
| C57BI6/J Wild Type | Jackson Laboratories | RRID: IMSR_JAX:000664 |
|
| ||
| Oligonucleotides | ||
|
| ||
| ChIP-qPCR primers: see Table S6 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo software version 10.10.0 | TreeStar Inc. | https://www.flowjo.com |
| Graphpad Prism version 10.2.3 (347) | Graphpad Software, LLC. | https://www.graphpad.com/scientific-software/prism/ |
| R(v3.6.1) | R Foundation | https://www.r-project.org/ |
| Seurat (v3.2.3) | (Hao Y et al.)65 | https://satijalab.org/seurat/ |
| ArchR (v1.0.1) | (Granja JM et al.)66 | https://www.archrproject.com/ |
| Cellranger-arc (v2.0) | 10x genomics | https://www.10xgenomics.com/support/software/cell-ranger-arc/latest |
| AIAP (v1.1) | (Liu S et al.)67 | https://github.com/Zhang-lab/ATAC-seq_QC_analysis |
| deepTools (v3.5.0) | (Ramírez F et al.)68 | https://github.com/deeptools/deepTools |
| DESeq2 (v1.26.0) | (Love MI et al.)69 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| WashU Epigenome Browser (v54.0.6) | (Li D et al.)70 | https://epigenomegateway.wustl.edu/browser/ |
| Code used for sequencing data analyses | This study | https://doi.org/10.5281/zenodo.11992331 |