

Abstract
Ectoine (ECT) has recently gained considerable interest in the healthcare sector due to its promising therapeutic benefits in a variety of human disorders. This research aimed to quantify the ECT plasma level in rats by creating and optimizing a sensitive and validated UPLC-MS/MS method. Prior to analysis, ECT extraction from the plasma samples was conducted via a protein precipitation procedure, using hydroxyectoine as an internal standard (IS). A 1.7 μm UPLC C8 column (100 mm × 2.1 mm) was selected for the chromatographic separation, using a gradient mobile phase consisting of acetonitrile and 0.05% formic acid. The electrospray ionization mass spectrometry (ESI-MS) was used to detect ECT in the positive ion mode. To determine the specific precursor and the product ions of ECT, multiple reaction monitoring (MRM) methods were carried out. The selected ion pair of ECT was 143.1 > 97 and 159.1 > 113.13 for the IS. The ECT’s linearity range in rat plasma was found to be 1-1000 ng/mL, with a recovery rate of 96.48–97.37%. Consistent with FDA guidelines for bio-analytical method validation, the suggested method was validated. The method was efficiently employed to quantify the studied drug in spiked rat plasma with good accuracy and precision with no significant matrix effects. Furthermore, it was effectively used to investigate the pharmacokinetic behavior of ECT in rats after a single oral dose of 30 mg/kg.
Keywords: Ectoine, Pharmacokinetic, Rat plasma, UPLC-MS/MS, Blue applicability grade index
Introduction
Ectoine (ECT) is a naturally occurring heterocyclic compound with the IUPAC name of (4 S)-2-methyl-3, 4, 5, 6-tetrahydropyrimidine-4-carboxylic acid [1]. It is a compatible solute or extremolyte that is generated by different types of halophilic bacteria. Halophilic bacteria produce the extremolytes to survive under drastic conditions of high salinity, heat, UV irradiation, or dryness [2]. ECT was found to be able to surround the bacterial proteins, membranes, and biomolecules with water as a hydrated shield, protecting them from different stressors [3]. By the same mechanism, ECT was proved to be a great bio-functional stabilizer and human skin protector, and it has piqued an interest in the cosmetics industry [4].
Nowadays, ECT gained a significant interest in the healthcare sector due to its promising therapeutic benefits in a variety of human and animal disorders [5, 6], such as GIT inflammation disorders [7, 8], Alzheimer’s disease [9], lung syndromes [10, 11], and rhino-conjunctivitis symptoms [12, 13]. In preclinical studies, ECT was shown to guard cells against the damage induced by various stressors and prevent the subsequent activation of the inflammatory cascades, leading to reducing the inflammatory process at the membrane level [14]. This data encourages many clinical trials to harness ECT as a therapeutic or prophylactic agent for several human diseases, especially inflammatory disorders [15]. ECT is currently used as topical applications for nasal, ophthalmic, and skin disorders [13, 16]. Consequently, the requirement of validated and reliable analytical procedures is necessary to determine this bioactive drug in different preclinical or clinical studies. After reviewing the literature, a few analytical methods for quantitative analysis of ECT in different matrices were reported. Examples include HPLC methods for determining ECT in bulk powder and marine bacterial cultures [17, 18], electrochemical sensors, and a LC/MS-MS method for quantifying ECT in pharmaceutical preparations and halophilic bacterial cultures [19, 20]. But no one has been established for the determination of this drug in plasma.
This study focused on developing and optimizing a validated and sensitive UPLC/MS-MS method for quantification of ECT plasma level in rats with a simple, efficient, and low-cost extraction procedure. The proposed method was tested in a real-time situation by being employed for studying the pharmacokinetic behavior of ECT in rats after the administration of a single oral dose of 30 mg/kg. This method is expected to be useful for a large sector of scientists and researchers. Medical scientists may use this method to evaluate associated pharmacological, pharmacokinetic, or preclinical investigations. Additionally, it may also aid in the identification of infections caused by halophilic bacteria that might execrate ECT into plasma.
Experimental
Instruments
All UPLC-MS/MS experiments were performed with the Acquity UPLC chromatography system coupled to the Xevo TQD triple quadrupole mass spectrometer (Waters, Ireland). A 1.7 μm UPLC BEH C8 column (100 mm × 2.1 mm, Waters, Ireland) was used to analyze the plasma samples. A Bio-Base centrifuge model BTBK-12HRT3 (China) was used in the protein precipitation extraction procedure.
Materials
Ectoine (99.92%) and hydroxyectoine (99.65%, internal standard) were kindly obtained from Bitop AG Company (Germany) and Sigma Aldrich (Germany), respectively. Acetonitrile, methanol, and water were supplied from LiChrosolv (HPLC grade, Germany). Glacial acetic acid and formic acid were purchased from Sigma Aldrich (HPLC grade, Germany).
Standard solutions
In water, a stock standard solution of 100 µg/mL of ECT was prepared. ECT working solutions were prepared by serially diluting the stock solution with the same solvent. In addition, a standard solution of 20 µg/mL for the internal standard (IS) was also prepared in water. When not in use, all prepared solutions were refrigerated under 2 to 8 oC.
Calibration standards and QC samples
Six non-zero ECT calibration standards were prepared in the range of 1-1000 ng/mL by mixing 25 µL of each identified working standard solution of ECT with 25 µL of the IS solution in Eppendorf (EP) tubes, then mixing the resulted solution with 450 µL of blank plasma. The quality control samples (QC) were prepared in the same way as the calibration standards, but at 3 concentration levels: low, medium, and high (5, 350, and 700 ng/mL). All calibration standards and QC samples were freshly prepared during the method development, as stability data are generally not available at this time. Another set of calibration standards and QC samples were prepared in the same manner after the method development and stored at -70 oC till analysis, as they should be stored under the same conditions as the study samples [21].
Extraction procedure and sample preparation
A protein precipitation extraction procedure was applied, in which 200 µL of a mixed solution of acetonitrile, methanol, and glacial acetic acid (50:49:1, v/v/v) was mixed with 200 µL of each calibration standard or quality control sample in EP tubes. The EP tubes were vortex-mixed for 2 min before being centrifuged at 11,000 rpm for 20 min at 6 oC. The resulted supernatant of each tube was then moved to a clean glass vial for analysis.
UPLC-MS/MS parameters
The chromatographic separation was accomplished using a 1.7 μm BEH C8 column (100 mm × 2.1 mm) and a gradient elution of a mobile phase containing 0.05% formic acid (A) and acetonitrile (B) at a flow rate of 0.5 mL/min, as shown in Table 1. The column and injector temperatures were maintained at 40 oC and 4 oC, respectively, and the injection volume was 10 µL. The dynamic MRM of transitions in the positive ion mode was selected as the scanning approach for mass spectrometry. Table 2 demonstrates the ion pair and collision energy characteristics applied for the determination. The system was operated by Masslynx 3.1 software (Waters, Ireland), and the overall run time of the whole elution procedure was 8.5 min, which included one gradient elution program for 7 min with one post-equilibrium time for 1.5 min.
Table 1.
UPLC gradient elution system
| Time (min) | 0.05% formic acid (A) | Acetonitrile (B) |
|---|---|---|
| 0.0 | 55 | 45 |
| 0.5 | 65 | 35 |
| 2.5 | 75 | 25 |
| 6.5 | 65 | 35 |
| 7.0 | 55 | 45 |
| 8.5 | 55 | 45 |
Table 2.
Parameters of Mass spectrometry
| Compound | Precursor (m/z) | Product (m/z) | Cone (V) | Dwell (s) | Collision (V) |
|---|---|---|---|---|---|
| Ectoine | 143.1 | 97 | 90 | 0.165 | 30 |
| Hydroxyectoine (IS) | 159.1 | 113.13 | 22 | 0.165 | 30 |
Validation of the method
The proposed method was validated in compliance with FDA requirements for bio-analytical method validation [22].
Linearity and lower limit of quantification (LLOQ)
A set of calibration standards for ECT was prepared as mentioned in Sect. Calibration standards and QC samples. The calibration standards of ECT were 1, 10, 50, 100, 600, and 1000 ng/mL. These standard solutions were treated by the mentioned extraction procedure, and a calibration curve relating the peak area ratio of ECT to IS and the concentration of ECT in ng/mL was constructed. The FDA guidelines stated that a correlation coefficient (r2) of more than 0.99 was considered preferable. If the response of the lowest standard inside the calibration curve was five times of the blank plasma response and was within ± 20% of the nominal concentration, this standard would be accepted as the LLOQ [22].
Selectivity and specificity
In order to ascertain the extent to which the endogenous plasma substances may interact or affect the response of the target drug or the IS, six blank plasma samples were randomly chosen and treated according to Sect. Extraction procedure and sample preparation and then analyzed.
Accuracy and precision
LLOQ, low, medium, and high QC samples (1, 5, 350, 750 ng/mL) were prepared and treated by the mentioned extraction procedure. For within-run accuracy and precision (n = 6), samples were measured continuously, while for between-run accuracy and precision (n = 18), they were measured on three consecutive days. Accuracy and precision were calculated as RE% (relative error) and CV% (coefficient of variation), respectively. Accuracy should be within ± 15% of the nominal concentrations, except for LLOQ, which is ± 20%, and precision should be within ± 15% CV, except ± 20% CV at LLOQ [22].
Matrix effect, recovery, and extraction efficiency
Substances co-eluted with the analyte, but undetected, may raise or decrease the signal intensity corresponding to the mass transition of that analyte, affecting the method’s selectivity, sensitivity, accuracy, and precision. This is a phenomenon called the matrix effect, and its evaluation in the development and validation stages is essential to ensuring the reliability and selectivity of the method [23].
Consequently, to evaluate whether there was a matrix effect, QC samples (5, 350, 750 ng/mL) from six different sources (two normal plasma, two lipemic, and two hemolysates) were prepared and analyzed. The matrix effect (ME%) was measured by comparing the mean peak area ratio of QC samples prepared by adding the target analyte into extracted blank plasma samples (A) to the mean peak area ratio of pure standard solutions of equivalent concentrations (B). ME% could be calculated according to Eq. (1):
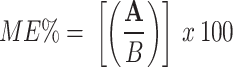 |
1 |
The recovery of the drug and IS, according to FDA recommendations, is not required to be 100.0%, but the degree of recovery should be constant, accurate, and repeatable [22]. Recovery as R% was calculated by comparing the mean peak area ratio of QC samples prepared by adding the target analyte into plasma samples before extraction (C) to the mean peak area ratio of another QC samples prepared by adding the target analyte into extracted blank plasma samples (A), as presented in Eq. (2):
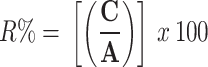 |
2 |
Extraction efficiency (EE%) of the proposed produce was also evaluated by comparing the mean peak area ratio of QC samples prepared by adding the target analyte into plasma samples before extraction (C) to the mean peak area ratio of pure standard solutions of equivalent concentrations (B), as shown in Eq. (3):
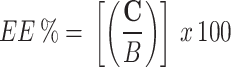 |
3 |
Carry-over
Carry-over can be defined as the presence or increase in the analyte or IS signal as a result of contamination from earlier samples. It was evaluated by injecting a calibration standard at the upper limit of quantification (ULOQ, 1000 ng/mL), then injecting blank samples. After the ULOQ standard, carry-over in blank samples should not exceed 20% and 5% of the analyte response at the LLOQ and the IS, respectively [22].
Dilution integrity
Dilution integrity is the evaluation of the sample dilution process to ensure that it does not affect the method’s accuracy or precision. It was tested on QC samples with drug concentrations higher than the ULOQ (1500 and 2000 ng/mL). Five replicates of samples after ten-fold dilution (150 and 200 ng/mL) were prepared, and their concentrations were estimated by applying the dilution factor of 10 to concentrations obtained from the calibration curve. Accuracy should be within ± 15% of the nominal concentrations, and precision should be within ± 15% CV [22].
Ruggedness
Ruggedness was studied by re-analysis of the QC samples on a different column (with the same used specifications) and also re-analysis of QC samples by a different analyst. Accuracy and precision were determined for the re-analyzed samples.
Stability
The experiment tested the storage stability of the QC samples (5, 350, 700 ng/mL) under a variety of conditions. If the deviation from the nominal concentration was within ± 15%, all stability samples were considered to be stable [22].
For long-term stability, four aliquots of each QC sample were refrigerated at -70 ± 5 °C for 30 days. Then, the samples were treated and analyzed, and the results were compared to the nominal concentrations.
Bench-top stability was also evaluated by leaving four aliquots of each QC sample at room temperature (about 23 °C) for 5 h, then were treated and analyzed, and the findings were compared to the nominal values.
Auto-sampler stability was also investigated by analyzing four aliquots of each QC sample that were treated prior to storage at 10 °C for 5 h. Samples were re-analyzed after 5 h, and the findings were compared with those of freshly prepared ones.
The plasma samples’ freeze-thaw stability was also investigated after three freeze-thaw cycles. Each QC sample was stored in four aliquots at – 70 ± 5 °C and frozen for 12 h between cycles. The samples were treated and analyzed after the end of the third cycle, and the concentrations were compared with nominal values.
Pharmacokinetic study design
The pharmacokinetic study on rats was reviewed and permitted by the ethics committee of Heliopolis University (Research No. HU.REC.A.6-2022), which strictly followed the National Institutes of Health (NIH) recommendations. Six male Sprague-Dawley rats (200 ± 30 g) were kindly obtained from the animal house of Heliopolis University (Cairo, Egypt). Before the experiment, the rats were not fed for 12 h, but the water was freely available. The rats were administered a single oral ECT dose of 30 mg/kg. This dose was selected based on different in vivo studies [7, 8]. Blood samples of 500 µL were withdrawn from each rat through the lateral tail vein. Blood samples were collected in 1.5 mL heparinized EP tubes, before (0 time) and after dosing at 0.083, 0.25, 0.5, 0.75, 1.0, 1.5, 2, 4, 6, 8, 12, and 24 h. Direct centrifugation was applied for all samples for 15 min at 10,000 rpm, then plasma samples were moved to fresh EP tubes and stored at -70 ± 5 oC till analysis. The WinNolin version 5.3 software for PK/PD analysis was used to compute the pharmacokinetic data for ECT through the non-compartmental analysis. At the end of the study, all experimental rats were euthanized by using the anesthesia method according to the AVMA Guidelines for the Euthanasia of Animals: 2020 Edition [24]. They were euthanized by intravenous pentobarbital with a dosage of 150 mg/kg. After ensuring the rats were free of life pointers, they were packaged in special plastic bags and incinerated. The entire experimental process of the animals was firmly adhered to the regulations for the care and use of laboratory animals as reviewed and permitted by the Ethics Committee of Heliopolis University (Egypt).
Results and discussion
After reviewing the published literature, no analytical method for determining ECT in plasma has been described. This encouraged the authors to create a validated UPLC-MS/MS method for quantification of this valuable bioactive molecule in plasma. The key issue was that ECT is considered to be an amino acid, as it is a cyclic derivative of aspartate [18]. This amino acid could be easily interfered with the endogenous amino acids in plasma. Consequently, the method parameters and sample processing were optimized to eliminate the potential interference of various impurities.
Tandem mass spectrometry parameters optimization
In general, compounds having a carboxyl group, such as ECT and IS, are often operated in the negative ion detection mode [25]. However, we observed that the response of the optimized positive ion mode was somewhat greater than the negative ion mode, so the positive ion mode was finally selected.
The cyclic ring in these compound structures, like ECT and hydroxyectoine, is quite stable, making it hard to produce fragments via collision-induced dissociation (CID). As illustrated in Fig. 1, CID has only cleaved the carbonyl position in the C4 side chain to produce fragment ions m/z 97 and m/z 113.13 for ECT and hydroxyectoine (IS), respectively. The m/z 143.1 > 97 and 159.1 > 113.13 were chosen as the quantitative ion pairs of MRM for ECT and IS, respectively. To enhance the mass spectrometry detection sensitivity, the collision correlation parameters and MS sources were adjusted. Cone gas was 1 L.h− 1, nitrogen de-solvation gas was 800 L.h− 1 with a temperature of 450 °C, the capillary voltage was 0.5 kV, source heat was 150 °C, and the cone voltage for ECT was 90 V and 22 V for IS.
Fig. 1.

(A) Tandem mass spectrum of ECT precursor ion m/z 143, and the major fragment m/z 97, (B) Tandem mass spectrum of Hydroxyectoine, IS, precursor ion m/z 159.1 and the major fragment m/z 113.13
Chromatographic parameters optimization
For ECT and IS, the chromatographic parameters were adjusted for optimal peak shape, resolution, and analysis time. Hydroxyectoine, the hydroxylated derivative of ECT, was chosen as the internal standard because it has a structure similarity and exhibited comparable chromatographic behavior with similar relative recovery values as ECT. Also, it was effectively operated in the positive ion mode with a high detection response.
Initially, two different columns of 1.7 μm UPLC BEH C18 and C8 stationary phases (100 mm × 2.1 mm) were examined, but the latter demonstrated a more satisfactory resolution. Generally, the use of columns with small particle sizes (1.7 μm) requires a high pumping pressure, and operating the column at high temperature reduces the pressure due to lowering the liquid viscosity [26]. In this study, it was observed that increasing column temperature up to 40 oC has an important role in shortening time of analysis without loss in the plate count, consequently increasing the column efficiency for separation. For the mobile phase, when methanol was initially used as the organic phase, the response increased by approximately two times in comparison with acetonitrile; however, the baseline noise raised by around four to five times, lowering the signal-to-noise ratio. As a result, acetonitrile was subsequently operated as the organic phase. It was observed that incorporating formic acid into the mobile phase under positive ionization conditions resulted in enhancing the ionization efficiency with sharper peak shapes and significantly higher response compared to a pure water-acetonitrile system. Different concentration levels for formic acid, 0.01%, 0.03%, 0.05%, and 0.1%, were tested, and the concentration of 0.05% was found to be the most suitable for achieving sharper peak shapes for both ECT and IS with high resolution. The gradient elution system, as shown in Table 1, was optimized by experiment to achieve high drug resolution and sensitivity. As demonstrated in Fig. 2, the retention times of both ECT and IS were 5.96 min ± 0.2 and 5.11 min ± 0.1, respectively.
Fig. 2.

UPLC-MS/MS chromatograms of (A) blank plasma sample, (B) spiked plasma sample with ECT and IS at the LLOQ = 1 ng/mL, and (C) Real rat plasma sample
Sample extraction procedure optimization
When compared to liquid-liquid extraction. Plasma protein precipitation was found to have a lower degree of matrix effect due to less endogenous interferences. It was observed that a combination of acetonitrile and methanol as the extraction solvent had a higher extraction efficiency than either methanol or acetonitrile alone. Adding the glacial acetic acid to the extraction solvent helps in acidifying and dissociating the drug from plasma proteins [27]. Different solution mixtures of acetonitrile, methanol, and glacial acetic acid with varying ratios were tested for extraction, and a solution of acetonitrile, methanol, and glacial acetic acid (50:49:1, v/v/v) was found to achieve a higher extraction efficiency with lower matrix effect. Centrifugation of samples at low temperature (6 oC for 20 min) was observed to enhance the protein precipitation process, as some of the plasma proteins precipitated only at low temperature. Besides, cooling helps to reduce the proteolytic activity of plasma enzymes and prevent unwanted degradation of the target analyte [28].
Method validation
Linearity and lower limit of quantification (LLOQ)
The peak area ratio of ECT to IS versus the concentration of ECT in ng/mL was plotted to construct the calibration curve for ECT. Over the linearity range of 1-1000 ng/mL, the calibration curve was found to be linear and precise, with an r2 value of 0.9998, and the regression equation was: Y = 0.0034 X + 0.0076, where Y was the peak area ratio and X was the corresponding concentration. According to the FDA guidelines, LLOQ represents the sensitivity of the analytical method [22]. In this study, LLOQ was found to be 1 ng/mL, which indicates the high sensitivity of the proposed method.
Selectivity and specificity
The chromatograms of a blank plasma sample, a spiked plasma sample at LLOQ, and a real rat plasma sample are shown in Fig. 2. They verified that the endogenous plasma constituents didn’t affect or impact the quantification of ECT in plasma. Consequently, the proposed method was considered to be specific and selective.
Accuracy and precision
Accuracy as RE% and precision as CV% were calculated. Mean values for all QC samples should be within ± 15% of the nominal values, except for LLOQ, which is ± 20%. The results were presented in Table 3. The within-run and between-run accuracies were between − 5.11 and − 2.16%. The within-run and between-run precisions were found to be < 12.51% and < 7.41%, respectively. These values are within the acceptable range. Therefore, the proposed method was considered to be accurate and reproducible.
Table 3.
Within- run and between run accuracy and precision of ECT in plasma samples
| Conc. (ng/mL) | Within-run | Between-run | ||||||
|---|---|---|---|---|---|---|---|---|
| n | Mean a ± SD | Accuracy (RE %) | Precision (CV %) | n | Mean b ± SD | Accuracy (RE %) | Precision (CV %) | |
| 1 | 6 | 0.97 ± 0.12 | -3.00 | 12.51 | 18 | 0.95 ± 0.07 | -5.11 | 7.41 |
| 5 | 6 | 4.81 ± 0.27 | -3.80 | 5.79 | 18 | 4.87 ± 0.30 | -2.60 | 6.29 |
| 350 | 6 | 337.53 ± 8.06 | -3.56 | 2.39 | 18 | 337.76 ± 8.61 | -3.49 | 2.55 |
| 750 | 6 | 733.74 ± 22.96 | -2.16 | 3.13 | 18 | 730.25 ± 24.68 | -2.63 | 3.38 |
a Mean of six replicate observations at each concentration
b Mean of 18 replicate observations over three different analytical runs
Matrix effect, recovery, and extraction efficiency
Table 4 demonstrated that the sample analysis was not significantly affected by the matrix effect. The extraction procedure was capable of extracting 95.23–96.26% of the studied drug from plasma samples, with a recovery rate varied from 97.66 to 98.16%.
Table 4.
Matrix effect (ME %), recovery (R %) and extraction efficiency (EE %) of the proposed method
| QCs (ng/mL) | A a (CV, %) | B b (CV, %) | C c (CV, %) | ME % d | R % e | EE % f |
|---|---|---|---|---|---|---|
| 5 | 0.0243 (3.60) | 0.0251 (1.86) | 0.02393 (0.58) | 97.01 | 98.16 | 95.23 |
| 350 | 1.1855 (1.29) | 1.2101 (0.33) | 1.16078 (0.62) | 97.98 | 97.90 | 95.93 |
| 750 | 2.5336 (0.85) | 2.5572 (1.25) | 2.47452 (1.24) | 99.07 | 97.66 | 96.26 |
a Mean peak area ratio of six replicate samples prepared by spiking ECT in extracted blank plasma
b Mean peak area ratio of six replicate of pure ECT standard solutions of equivalent concentrations
c Mean peak area ratio of six replicate samples prepared by spiking ECT in plasma before extraction
d (A/B) x 100%
e (C/A) x 100%
f (C/B) x 100%
Carry-over
Carry-over was shown to be negligible for ECT and IS (≤ 0.49%). No enhancement in the response, at the retention times of ECT and IS, was observed in the extracted blank plasma samples after the consequent injection of the ULOQ standard.
Dilution integrity
The dilution integrity experiment was performed with an aim to validate the dilution test carried out on higher analyte concentrations above the ULOQ, which may be encountered during real subject sample analysis. For ECT, the accuracy values as RE% for the ten-fold diluted QC samples (150 ng/mL and 200 ng/mL, n = 5) were found to be -2.55% and − 1.7%, respectively. While the precision values as CV% were found to be 5.2 and 6.7%, respectively. These results are within the acceptance ranges of accuracy and precision (± 15%).
Ruggedness
For re-analysis of the QC samples on a different column with the same used specifications, the precision values as CV% and accuracy values as RE% ranged from 1.8 to 3.2% and − 4.26 to -2.02%, respectively. The respective ranges for CV% and RE% for the re-analysis performed by another analyst were from 1.8 to 3.7% and − 4.22 to -1.55%, respectively.
Stability
In this work, the stability assessment was performed under a variety of experimental conditions, including long-term stability, bench-top stability, auto-sampler stability, and freeze-thaw stability. The results were summarized in Table 5. RE% and CV% values for all QC samples were within ± 15%, indicating the stability of the studied drug under different storage conditions.
Table 5.
Stability results for ECT under different conditions (n = 6)
| Stability | Storage condition | LQC (5 ng/mL) |
MQC (350 ng/mL) |
HQC (750 ng/mL) |
|||
|---|---|---|---|---|---|---|---|
| RE % | CV% | RE % | CV% | RE % | CV% | ||
| Long-term | 30 days.at − 70 ± 5 °C | -14.46 | 4.84 | -13.89 | 3.13 | -13.49 | 4.79 |
| Bench-top | 5 h. at room temp. | -12.77 | 1.41 | -11.42 | 2.31 | -11.96 | 4.39 |
| Auto-sampler | 5 h. at 10 ± 5 °C | -11.05 | 1.40 | -10.75 | 2.39 | -10.41 | 4.51 |
| Freeze-thaw | After the 3rd FT cycle | -13.99 | 2.46 | -12.39 | 1.12 | -11.89 | 3.28 |
Assessment of the method practicality
The practicality of the developed UPLC-MS/MS method was assessed by a new metric tool called BAGI, the Blue Applicability Grade Index, which was recently introduced in 2023 [29]. This tool evaluates ten main attributes that are associated with the analytical determination and sample preparation. These main attributes are: (i) type of analysis; (ii) number of analytes that are simultaneously determined; (iii) analytical technique; (iv) the number of samples that can be simultaneously treated; (v) sample preparation; (vi) the number of samples that can be analyzed per hour; (vii) type of reagents and materials; (viii) requirement of preconcentration; (ix) degree of automation; and (x) the amount of sample. The BAGI evaluation depends on four discrete scores of equal weights. Each score corresponds to a different color shade and contributes to the overall score. BAGI uses score points of 2.5, 5.0, 7.5, and 10, which correspond to white, light blue, blue, and dark blue, respectively [30]. The overall assessment is presented by an asteroid pictogram with a number in its center. This number is the assigned overall score of the analytical method and should range from 25 to 100. The method should gain at least 60 points to be considered “practical.” For more information about the criteria for assessing the scores for each attribute and overall score, readers are directed to reference [29].
Figure 3 illustrates the asteroid pictogram evaluation of our newly developed method, showing an overall BAGI score of 77.5. This score demonstrates our method’s practicality and potential to be applied in a real bioanalytical environment.
Fig. 3.

BAGI metric assessment of the newly developed UPLC-MS/MS method for determination of ECT in plasma
Pharmacokinetic results
To ascertain the validity of the proposed method in a real-time situation, the method was employed to quantify ECT plasma level in 6 rats after a single oral dosage of 30 mg/kg. Figure 4 illustrates the mean plasma concentrations (ng/mL) versus time (h) profile, and Table 6 summarizes the related pharmacokinetic parameters. After the oral administration of ECT to rats, the absorption was rapid, with a maximum plasma concentration (Cmax) of 573 ± 67.52 ng/mL. As illustrated in Fig. 4, the plasma concentration of ECT increased rapidly over time, and the time to the maximum concentration peak (Tmax) was 1.5 ± 0.61 h. A rapid decline in the plasma concentration was noticed, and the t1/2 was determined to be 6.22 ± 3.22 h. The total amount of the drug that comes into the systemic circulation after the drug administration could be expressed by AUC. AUC0–∞ and AUC0–t are the areas under the concentration-time curve from time zero extrapolated to infinite time and from time zero to the time of the last quantifiable concentration, respectively. The AUC0–∞ and AUC0–t values were found to be 4103 ± 427 and 2806 ± 234 ng h/mL, respectively. This indicates that ECT was rapidly absorbed and distributed, and also rapidly eliminated from the systemic circulation. The fraction of the drug eliminated per unit of time is called the elimination rate constant (Kel). The Kel value for ECT was 0.10 ± 0.04 h–1, this means that 10% of ECT is eliminated per hour. This high elimination rate and the rapid decline in the plasma concentration indicate that ECT might be distributed into the target tissue quickly and for a short period of time. This may lead to a rapid therapeutic effect with the need for multiple doses per day.
Fig. 4.

Mean ECT plasma concentration-time profile after 30 mg/kg single oral dose (n = 6)
Table 6.
Major pharmacokinetic parameters for ECT in rat plasma after single oral dose of 30 mg/kg (n = 6, mean ± SD)
| PK parameter | Values | Unit |
|---|---|---|
| Cmaxa | 573 ± 67.52 | ng/mL |
| Tmaxb | 1.5 ± 0.61 | h |
| t1/2c | 6.22 ± 3.22 | h |
| AUC0–td | 2806 ± 234 | ng h/mL |
| AUC0–∞e | 4103 ± 427 | ng h/mL |
| Kelf | 0.10 ± 0.04 | h–1 |
a Maximum plasma concentration
b Time to the maximum concentration peak
c Half-life of the drug
d Area under the concentration-time curve from time zero to the time of the last quantifiable concentration
e Area under the concentration-time curve from time zero extrapolated to infinite time
f Elimination rate constant
Conclusion
In this study, the proposed bioanalytical method was effectively employed to quantify the studied drug in rat plasma. It was found to be selective, sensitive, and accurate enough to be employed in preclinical bioavailability and pharmacokinetic studies. The method needed a simple and low-cost sample treatment step by a protein precipitation extraction procedure, followed by a gradient liquid chromatographic separation with tandem mass spectrometric detection. The method was capable of simultaneously estimating ECT up to 1 ng/mL in plasma with good accuracy and precision with no significant matrix effect. This study reports for the first time the pharmacokinetics of ECT in rats after a single oral dose of 30 mg/kg. Given that our study was conducted in rats and there are major differences between animals and humans, more research is needed to investigate the pharmacokinetics of ECT in humans. This might be accomplished with the help of the proposed UPLC-MS/MS method in future work.
Acknowledgements
The authors extend their appreciation to Taif University, Saudi Arabia, for supporting this work through the project number (TU-DSPP-2024-49).
Author contributions
M.R. was responsible for performing the analysis, processing the experimental data, and writing- original draft. R.A.M.S. was responsible for inspection, observation, and reviewing. I.A.N. was responsible for conceptualization, methodology, and organization. The final version of the manuscript has been completed by all authors.
Funding
This research was funded by Taif University, Saudi Arabia, Project No. (TU-DSPP-2024-49).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
The animal study was reviewed and permitted by the ethics committee of Heliopolis university- Research no. HU.REC.A.6-2022.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Su Y, Peng W, Wang T, Li Y, Zhao L, Wang X et al. Ectoine production using novel heterologous EctABCS. salarius from marine bacterium salinicola salarius. Applied Sciences (Switzerland). 2021;11(15).
- 2.Lentzen G, Schwarz T, Extremolytes. Natural compounds from extremophiles for versatile applications. Appl Microbiol Biotechnol. 2006;72(4):623–34. [DOI] [PubMed] [Google Scholar]
- 3.Hahn MB, Uhlig F, Solomun T, Smiatek J, Sturm H. Combined influence of ectoine and salt: Spectroscopic and numerical evidence for compensating effects on aqueous solutions. Phys Chem Chem Phys. 2016;18(41):28398–402. [DOI] [PubMed] [Google Scholar]
- 4.Liu M, Liu H, Shi M, Jiang M, Li L, Zheng Y. Microbial production of ectoine and hydroxyectoine as high-value chemicals. Microb Cell Fact. 2021;20(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bownik A, Stępniewska Z. Ectoine as a promising protective agent in humans and animals. Arh Hig Rada Toksikol. 2016;67(4):260–5. [DOI] [PubMed] [Google Scholar]
- 6.Merckx C, Zschüntzsch J, Meyer S, Raedt R, Verschuere H, Schmidt J et al. Exploring the therapeutic potential of Ectoine in Duchenne muscular dystrophy: comparison with taurine, a supplement with known Beneficial effects in the mdx mouse. Int J Mol Sci. 2022;23(17). [DOI] [PMC free article] [PubMed]
- 7.Abdel-Aziz H, Wadie W, Abdallah DM, Lentzen G, Khayyal MT. Novel effects of ectoine, a bacteria-derived natural tetrahydropyrimidine, in experimental colitis. Phytomedicine. 2013;20(7):585–91. [DOI] [PubMed] [Google Scholar]
- 8.Bethlehem L, van Echten-Deckert G. Ectoines as novel anti-inflammatory and tissue protective lead compounds with special focus on inflammatory bowel disease and lung inflammation. Pharmacol Res. 2021;164:105389. [DOI] [PubMed] [Google Scholar]
- 9.Kanapathipillai M, Lentzen G, Sierks M, Park CB. Ectoine and hydroxyectoine inhibit aggregation and neurotoxicity of Alzheimer’s β-amyloid. FEBS Lett. 2005;579(21):4775–80. [DOI] [PubMed] [Google Scholar]
- 10.Sydlik U, Gallitz I, Albrecht C, Abel J, Krutmann J, Unfried K. The compatible solute ectoine protects against nanoparticle-induced neutrophilic lung inflammation. Am J Respir Crit Care Med. 2009;180(1):29–35. [DOI] [PubMed] [Google Scholar]
- 11.Unfried K, Krämer U, Sydlik U, Autengruber A, Bilstein A, Stolz S, et al. Reduction of neutrophilic lung inflammation by inhalation of the compatible solute ectoine: a randomized trial with elderly individuals. Int J COPD. 2016;11(1):2573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werkhäuser N, Bilstein A, Mahlstedt K, Sonnemann U. Observational study investigating Ectoin® Rhinitis Nasal Spray as natural treatment option of acute rhinosinusitis compared to treatment with Xylometazoline. European Archives of Oto-Rhino-Laryngology; 2021. [DOI] [PMC free article] [PubMed]
- 13.Bilstein A, Heinrich A, Rybachuk A, Mösges R. Ectoine in the Treatment of Irritations and inflammations of the Eye Surface. Biomed Res Int. 2021;2021:8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buommino E, Schiraldi C, Baroni A, Paoletti I, Lamberti M, De Rosa M, et al. Ectoine from halophilic microorganisms induces the expression of hsp70 and hsp70B′ in human keratinocytes modulating the proinflammatory response. Cell Stress Chaperones. 2005;10(3):197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bilstein A, Werkhäuser N, Rybachuk A, Mösges R. The effectiveness of the Bacteria derived Extremolyte Ectoine for the treatment of allergic Rhinitis. Biomed Res Int. 2021;2021:8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kauth M, Trusova OV. Topical ectoine application in children and adults to treat inflammatory diseases associated with an impaired skin barrier: a systematic review. Dermatology Therapy. 2022;12(2):295–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Chen J, Wang S, Zhou G, Chen D, Zhang H, et al. Development and validation of polar RP-HPLC method for screening for ectoine high-yield strains in marine bacteria with green chemistry. Nat Prod Res. 2019;33(8):1122–6. [DOI] [PubMed] [Google Scholar]
- 18.Jörg Kunte H, Galinski EA, Trüper HG. A modified FMOC-method for the detection of amino acid-type osmolytes and tetrahydropyrimidines (ectoines). J Microbiol Methods. 1993;17(2):129–36. [Google Scholar]
- 19.Sattar OIA, Abuseada HHM, Emara MS, Rabee M. Green electrochemical and chromatographic quantifications of the extremolyte ectoine in halophilic bacterial cultures and related pharmaceutical preparations. J Pharm Biomed Anal. 2022;213(February):114680. [DOI] [PubMed] [Google Scholar]
- 20.He YZ, Gong J, Yu HY, Tao Y, Zhang S, Dong ZY. High production of ectoine from aspartate and glycerol by use of whole-cell biocatalysis in recombinant Escherichia coli. Microb Cell Fact. 2015;14(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redrup MJ, Igarashi H, Schaefgen J, Lin J, Geisler L, Ben M, et al. Sample Management: recommendation for best practices and Harmonization from the Global Bioanalysis Consortium Harmonization Team. AAPS J. 2016;18(2):290–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Administration F. and D. Guidance for industry. Bioanal Method Valid. 2013.
- 23.Pinto MC, Berton DC, de Oliveira AC, Lazaro CM, Carandina SAC. Method development and validation of ursodiol and its major metabolites in human plasma by hplc-tandem mass spectrometry. Clin Pharmacology: Adv Appl. 2019;11:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leary S, Pharmaceuticals F, Ridge H, Underwood W, Anthony R, Cartner S et al. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition *. 2020.
- 25.Li Xyu, Zhu S hong, Yang F, Hu G, xin, Yuan L. jing. An ultra-performance liquid chromatography-tandem mass spectrometry method for the determination of obeticholic acid in rat plasma and its application in preclinical pharmacokinetic studies. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences. 2019;1121(May):82–8. [DOI] [PubMed]
- 26.Chen LC. High-temperature liquid chromatography and the hyphenation with mass spectrometry using high-pressure electrospray ionization. Mass Spectrom. 2019;8(2):S0079–0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castro-Perez J, Prakash C. Recent advances in mass spectrometric and other analytical techniques for the identification of drug metabolites. Identif Quantification Drugs Metabolites Drug Metabolizing Enzymes Transporters. 2020;1(1):39–71.
- 28.Stone J. editor. Sample preparation techniques for mass spectrometry in the clinical laboratory. Mass spectrometry for the clinical laboratory. Elsevier; 2017. pp. 37–62.
- 29.Manousi N, Wojnowski W, Płotka-Wasylka J, Samanidou V. Blue applicability grade index (BAGI) and software: a new tool for the evaluation of method practicality. Green Chem. 2023;25(19):7598–604. [Google Scholar]
- 30.Bartosova L, Balis P, Garaj V, Kovac A, Rajtik T, Piestansky J. A simple UHPLC-MS/MS method for determination of SET2, a selective antagonist of TRPV2 receptor, in rat plasma samples. J Chromatogr B: Anal Technol Biomedical Life Sci. 2024;1235:124067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analysed during the current study.