

Abstract
Background
Stress-induced activation of ERK/Drp1 serves as a checkpoint in the segregation of damaged mitochondria for autophagic clearance (mitophagy). Elevated cytosolic calcium (Ca2+) activates ERK, which is pivotal to mitophagy initiation. This process is altered in Parkinson’s disease (PD) with mutations in leucine-rich repeat kinase 2 (LRRK2), potentially contributing to mitochondrial dysfunction. Pathogenic LRRK2 mutation is linked to dysregulated cellular Ca2+ signaling but the mechanism involved remains unclear.
Methods
Mitochondrial damages lead to membrane depolarization. To investigate how LRRK2 mutation impairs cellular response to mitochondrial damages, mitochondrial depolarization was induced by artificial uncoupler (FCCP) in wild-type (WT) and LRRK2R1441G mutant knockin (KI) mouse embryonic fibroblasts (MEFs). The resultant cytosolic Ca2+ flux was assessed using live-cell Ca2+ imaging. The role of mitochondria in FCCP-induced cytosolic Ca2+ surge was confirmed by co-treatment with the mitochondrial sodium-calcium exchanger (NCLX) inhibitor. Cellular mitochondrial quality and function were evaluated by Seahorse™ real-time cell metabolic analysis, flow cytometry, and confocal imaging. Mitochondrial morphology was visualized using transmission electron microscopy (TEM). Activation (phosphorylation) of stress response pathways were assessed by immunoblotting.
Results
Acute mitochondrial depolarization induced by FCCP resulted in an immediate cytosolic Ca2+ surge in WT MEFs, mediated predominantly via mitochondrial NCLX. However, such cytosolic Ca2+ response was abolished in LRRK2 KI MEFs. This loss of response in KI was associated with impaired activation of Ca2+/calmodulin-dependent kinase II (CaMKII) and MEK, the two upstream kinases of ERK. Treatment of LRRK2 inhibitor did not rescue this phenotype indicating that it was not caused by mutant LRRK2 kinase hyperactivity. KI MEFs exhibited swollen mitochondria with distorted cristae, depolarized mitochondrial membrane potential, and reduced mitochondrial Ca2+ store and mitochondrial calcium uniporter (MCU) expression. These mutant cells also exhibited lower cellular ATP: ADP ratio albeit higher basal respiration than WT, indicating compensation for mitochondrial dysfunction. These defects may hinder cellular stress response and signals to Drp1-mediated mitophagy, as evident by impaired mitochondrial clearance in the mutant.
Conclusions
Pathogenic LRRK2R1441G mutation abolished mitochondrial depolarization-induced Ca2+ response and impaired the basal mitochondrial clearance. Inherent defects from LRRK2 mutation have weakened the cellular ability to scavenge damaged mitochondria, which may further aggravate mitochondrial dysfunction and neurodegeneration in PD.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12964-024-01844-y.
Keywords: Parkinson disease, LRRK2 mutation, Mitophagy, Calcium-dependent pathways, Mitochondrial dysfunction, Cellular stress response, NCLX
Introduction
Mitochondrial dysfunction contributes to the pathogenesis of Parkinson’s disease (PD) [1–3] partly via mechanisms that perturb mitochondrial clearance (or mitophagy), a process by which damaged mitochondria are selectively recycled by autophagy to maintain mitochondrial quality and neuronal energy supply [4]. LRRK2 (leucine-rich repeat kinase 2) mutation is one of the most common genetic risks in both familial and sporadic PD [5], being pathologically linked to the endosomal-autophagic pathways and calcium homeostasis [6–10]. We previously showed that mutant mice carrying the pathogenic LRRK2R1441G knockin (KI) mutation [11] are more vulnerable to mitochondrial stressors, with an age-dependent accumulation of defective, ubiquitinated mitochondria in their brains compared to their age-matched wild-type (WT) littermates [12, 13]. Mouse embryonic fibroblasts (MEFs) derived from these LRRK2 mutant mice also exhibited slower rate of basal mitochondrial clearance [13]. Stressing these mutant cells by mitochondrial depolarization caused clustering of perinuclear lysosomes and accumulation of mitochondria-containing autophagosomes, which was associated with impaired phosphorylation of ERK and Drp1 (dynamin 1-like protein) [13]. The activation of ERK-Drp1 signaling axis promotes mitochondrial fission, which segregates damaged portion of mitochondria from the healthy counterpart to maintain mitochondrial quality and homeostasis [14, 15]. We hypothesize that these phenotypic changes observed in the LRRK2R1441G mutant may be due to impaired upstream signal pathway to regulate segregation of mitochondria and mitophagy in response to mitochondrial damages.
Cytosolic free calcium ions (Ca2+) play a key role in neuronal survival as an intracellular messenger to regulate a wide variety of neuronal processes in the brain [16–18]. Dysregulated Ca2+ signaling leads to mitochondrial impairment via a cascade of Ca2+-dependent cellular processes that contribute to neurodegeneration in PD [17, 19, 20]. Evidence has shown that pathogenic LRRK2 mutations are linked to dysregulated cellular Ca2+ homeostasis in PD [21, 22]. For instance, patient fibroblasts bearing LRRK2G2019S mutation exhibited impaired mitochondrial Ca2+ extrusion [23], whereas mouse cortical neurons carrying LRRK2G2019S or LRRK2R1441C mutation demonstrated impaired intracellular Ca2+ buffering and depletion of dendritic mitochondria [21]. However, the mechanism of how LRRK2 mutation impairs mitochondrial Ca2+ signaling and the subsequent cellular stress responses to maintain mitochondrial homeostasis is still unclear.
In this study, we elucidated impaired Ca2+ signaling response and a concomitant lack of CaMKII-MEK-ERK-Drp1 activation under cellular stress induced by mitochondrial depolarization in LRRK2R1441G KI MEFs. These mutant cells consistently demonstrated an accumulation of defective ubiquitinated mitochondria associated with impaired mitophagy [13]. We also demonstrated mitochondrial defects which we hypothesized to perturb cytosolic Ca2+ response under mitochondrial depolarization. Our findings demonstrate for the first time that dysregulated mitochondrial Ca2+ signaling caused by LRRK2 mutation may lead to impaired cellular stress response and mitochondrial clearance as implicated in PD.
Materials and methods
Generation of mouse embryonic fibroblasts (MEFs)
Generation of LRRK2R1441G knockin (KI) mutant mice has been previously described [12, 13, 24]. Briefly, mutant LRRK2R1441G KI mutant mice were back-crossed with wild-type (WT) C57BL/6N for over eight generations and maintained under the same genetic background. Mouse embryonic fibroblasts (MEFs) were isolated from homozygous LRRK2R1441G KI mutant mice and their WT littermates at day E12.5. Alternate clones were generated from individual WT and mutant mice littermate pairs.
Cell culture and treatments of MEFs
All MEF cell lines were cultured in Dulbecco’s Modified Eagle Medium + GlutaMAX™ (Gibco, ThermoFisher™ Scientific, 10569–010), supplemented with 10% Fetal Bovine Serum (FBS; GE Healthcare HyClone™, SH30071.03), 1% 100 units/mL penicillin, 100 µg/mL streptomycin (Gibco, ThermoFisher™ Scientific, 15140–122), 1% non-essential amino acids (Gibco, ThermoFisher™ Scientific, 11140–050). Experiments using these MEF cells were done within the passage numbers ranging from 30 to 35, in order to maintain a stable population of immortalized cell lines without the detrimental effects of long-cultures and late passaging. The same passage of the WT and KI MEF cell lines were used for each set of treatment.
All cell cultures were refreshed with the culture medium 2 h prior to FCCP treatment involving different timepoints. Cell treatments were carried out with the follow reagents: FCCP (Sigma Aldrich™, C2920), MLi-2 (Merck, SML3101), H2O2 (VWR Chemicals BDH), oligomycin A (ATP synthase inhibitor; Abcam™, ab143423), rotenone (Sigma Aldrich™, R8875) and CGP-37157 (sodium-calcium exchange inhibitor, Abcam™, ab120012).
Real-time monitoring of mitochondrial membrane potential (MMP) by TMRM
To observe the effects of MMP with 10 µM FCCP treatment in the WT and KI MEFs, cells were loaded with TMRM (ThermoFisher™ Scientific, T668), a fluorescent MMP-sensitive cationic dye, as an indicator of relative MMP based on fluorescence intensity. Briefly, cells were seeded to 80% confluency on 35 mm confocal dishes (MatTek P35G-1.5-14-C). Cells were incubated in fresh medium for 2 h prior to loading of 25 nM TMRM at 37oC, in the dark for 40 min in cell culture medium. After the incubation in TMRM, the cells were washed twice with TMRM-free medium, followed by imaging by Nikon Ti2-E Widefield Microscope using the following configurations: RFP single channel set (excitation 525 nm; emission: 600 nm), 20 × 0.75 DIC Objective, exposure time of 50 ms. TMRM fluorescence intensity was monitored via real-time acquisition of one image every 15 s for 10 min, and FCCP or DMSO (0.01% v/v) added 10–15 s prior to the 3-min time point. TMRM fluorescence intensity of individual cell tracings were quantified with the MetaMorph 7.10.2.240 software, and the data was expressed as mean intensity values with background intensity subtracted.
Live-cell imaging of cytosolic Ca2+ level by Cal-520 staining
Intracellular calcium-sensitive dye, Cal-520 (Abcam™, ab171868), was introduced to the WT and KI MEF cell lines to analyze the changes in intracellular calcium level with 10 µM FCCP. Cells were loaded with 5 µM Cal-520 at 37oC for 60 min in 1X HBSS supplemented with calcium chloride and magnesium chloride (Gibco, ThermoFisher™, 14025-076). The cells were washed twice with HBSS to remove excess dye that was non-specifically bound to the cell surface and imaged using the Nikon Ti2-E Widefield Microscope with the following configurations: GFP single channel set (excitation 475 nm; emission: 523 nm), 20 × 0.75 DIC Objective, exposure time of 80 ms, and image acquisition every 5 s. Cell treatment with FCCP on Cal-520 fluorescence intensity tracing were similarly performed as TMRM measurements above. For NCLX inhibition experiment, WT MEFs were pre-treated with CGP-37157 together with Cal-520 for 1 h prior to calcium imaging. FCCP treatment following Cal-520 dye loading was performed the same way as other FCCP treatment and calcium imaging experiments.
Mitochondrial morphology under transmission electron microscopy (TEM)
The same passage of WT and KI MEFs under normal culture condition were refreshed with new culture medium for 2 h. Cells were typsinized and pelleted for fixation with 2% paraformaldehyde with 2.5% glutaraldehyde in cacodylate buffer (0.1 M sodium cacodylate-HCl buffer pH 7.4; Electron Microscopy Sciences, 11,653) for 4 h at 8 °C, then were transferred to cacodylate buffer with 0.1 M sucrose to stop fixation. The resultant cell pellets were washed in cacodylate buffer before osmication using 1% osmium tetroxide (ThermoFisher™ Scientific, 50–332 − 14) in cacodylate buffer for 1 h at room temperature. Pellets were washed in cacodylate buffer again, then dehydrated on a rotary shaker in the following sequence: 50% ethanol – 5 min, 70% ethanol – 5 min, 90% ethanol – 5 min, 100% ethanol – 10 min each for 3 times, propylene oxide – 5 min each for 2 times. The dehydrated pellets were infiltrated and embedded in 1:1 mixture of propylene oxide and epoxy resin (Electron Microscopy Sciences, RT14040) for 1.5 h at 37 °C in form of labelled plastic capsules (Electron Microscopy Sciences, 70,000-B) or flat embedding molds. The mounts were allowed to polymerize at 60 °C overnight before sectioning. Visual examination of mitochondrial morphology was based on two-dimensional TEM photomicrographs (Philips CM100 Transmission Electron Microscope; 1:2200 magnification) taken from cross-section of cell pellets. At least 20 random micrographs were taken independently from 3 different passages of WT and KI MEFs to minimize confounding effects from variation in culture conditions. Mitochondria within a defined area of the field were identified and imaged by two independent researchers blinded from the details of each sample group.
Measurement of mitochondrial Ca2+ level by Rhod-2 staining
Rhod-2 is a calcium dye with specific sequestration to mitochondria, allowing for selective measurements of mitochondrial Ca2+ content. WT and KI MEFs were stained with 5 µM Rhod-2 at 37 °C in the dark for 45 min, in 1X HBSS supplemented with calcium chloride and magnesium chloride (Gibco, ThermoFisher™, 14025-076). Incubation at 37 °C was recommended for optimal mitochondrial compartmentalisation. The cells were washed twice with HBSS and were imaged using Nikon Ti2-E Widefield Microscope with the same configurations as TMRM staining. For flow cytometry measurements, the cells were seeded and stained in six-well plates. The red fluorescence of Rhod-2 stained cells were measured using PE-A (Excitation: 552 nm; emission: 581 nm) using the BD LSR Fortessa Cell Analyzer (PE-Texas Red Channel). Mitochondrial Ca2+ content was expressed as mean intensity of PE-A staining per cell.
Seahorse XFp Cell Mito stress test
Seahorse XFp Cell Mito Stress Tests (Seahorse XFe24 Extracellular Flux Analyser, Agilent Technologies) was performed according to the manufacturer’s instructions. The Mito Stress test involves a sequential addition of oligomycin, FCCP and rotenone with antimycin A, to generate a real-time mitochondrial bioenergetics profile of the oxygen consumption rate (OCR) in cells. Oligomycin is used to determine ATP-linked respiration by inhibiting ATP synthase. FCCP, which follows oligomycin, induces maximal respiration in attempts to restore the lost MMP from the dissipation of proton gradient. The final injection involves rotenone and antimycin A, inhibitors of complex I and III of electron transport chain, respectively, which halts respiration by stopping the electron flow in the electron transport chain, causing the oxygen consumption to drop to zero. One day prior to the assay, 30,000 WT and KI MEF cells were seeded on to Seahorse Utility plate with DMEM/GlutaMAX™ medium overnight. At day of assay, cells were incubated with XF assay medium that was supplemented with 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine, and its pH adjusted to 7.4. After the assay, the protein concentration of individual wells were measured using BCA Protein Assay kit (ThermoFisher™ Scientific, A55860).
Intracellular ATP measurements
WT MEF cells were seeded on 96-well white/clear bottom plates for ATP measurements. Intracellular ATP levels were measured with the Luminescent ATP Detection Assay kit (Abcam™, ab113849) according to manufacturer’s instructions. Cell medium was refreshed at 2 h prior to ATP measurements to minimize confounding effects from nutrient deprivation. The luminescence intensity was measured using CLARIOstar microplate reader (BMG Labtech). ATP concentration was determined through a standard curve plotted from serial dilution. The ATP concentration ranged from 1 to 3 µM ATP. The effect on intracellular ATP levels from various treatments were expressed as a percentage of baseline (at time 0 min).
Cytosolic ATP: ADP measurements
WT, and KI MEFs were seeded on 96-well white/clear bottom plates for ATP: ADP measurements using the ATP: ADP Assay Kit (Sigma Aldrich, MAK135) according to the manufacturer’s instructions. Cell medium was refreshed at 2 h prior to ATP: ADP measurements to minimize confounding effects from nutrient deprivation. The luminescence intensity was measured using CLARIOstar microplate reader (BMG Labtech). ATP: ADP was calculated using the formula provided by the manufacturer.
Real-time mitochondrial permeability transition pore (mPTP) opening assay
The opening of the mPTP was assessed by the quenching of calcein fluorescence by CoCl2 using the components from “MitoProbe™ Transition Pore Assay Kit for flow cytometry” (ThermoFisher™ Scientific, M34153). Calcein staining allows distribution of the dye into cytosol and subcellular compartments including the mitochondria. CoCl2 quenches calcein fluorescence in all cellular compartments except the mitochondria, as the latter is impermeable to CoCl2. Only upon mPTP opening can Co2+ enter the mitochondria and quench mitochondrial calcein fluorescence. Cells were loaded with calcein (1 µM) in 1X HBSS in suspension for 15 min at 37 °C in the dark, prior to the addition of 1 mM CoCl2 for another 15 min incubation. Either ionomycin (1 µM) alone or FCCP (10 µM) followed by 1 µM ionomycin was added to cells to evaluate mPTP opening by measuring the change in calcein fluorescence intensity every 30 s for 5 min using BD LSR Fortessa Cell Analyzer (FITC channel), with 10,000 cells counted for each trial. The level of mPTP opening is represented by the extent of decrease in calcein fluorescence by quenching from CoCl2.
Cell harvest and immunoblotting
At the indicated time points post-treatment, MEF cells were harvested and lysed in cold lysis buffer of 1X RIPA with SDS (Abcam™, ab156034), supplemented with 1X Halt™ protease and phosphatase inhibitor cocktail (ThermoFisher™ Scientific, 78446) and 2% phenylmethylsulfonyl fluoride (PMSF) (Pierce™, 36978). Protein concentration was determined using the Bradford Assay (Bio-Rad, Quick Start™ Bradford 1X Dye Reagent, 5000205). Equal amounts of proteins were dissolved in sample buffer (ThermoFisher™ Scientific, 39001, Pierce™ lane marker non-reducing sample buffer). Samples (15 ~ 40 µg) were electrophorized in 12% resolving SDS polyacrylamide gels at 90 V. The resulting gels were electro-transferred to PVDF membranes. The membranes were blocked in 5% Bovine Serum Albumin (Sigma Aldrich, A9647) in TBST (Santa Cruz Biotechnology, sc362311), and probed with primary antibodies overnight at 4 °C and HRP-conjugated secondary antibodies (Agilent DAKO™, P0260, polyclonal rabbit anti-mouse immunoglobulins/HRP; or Agilent DAKO™, P0448, goat anti-rabbit immunoglobulins/HRP) for 1 h. This was followed by incubation in ECL substrate solution for chemiluminescence visualization (Bio-Rad, 170–5061). The quantification of band intensity was done using the Image Lab Software (Bio-Rad). α-Tubulin served as the loading control for all immunoblots, as its molecular weight (in kilodaltons) is not close to proteins of interest. Immunoblotting was done using the following primary antibodies: Phospho-p44/42 MAPK (MAPK/ERK1/2) (Thr202/Tyr204) (1:5000; Cell Signaling Technology™, 4376 S), p44/42 MAPK (MAPK/ERK1/2) (1:5000; Cell Signaling Technology™, 4695 S), phospho-MAP2K/MEK1/2 (Ser217/221) (1:2000; Cell Signaling Technology™, 9121 S), MAP2K/MEK1/2 (1:2000; Cell Signaling Technology™, 9122 S), Phospho-RAF1 (Ser338) (1:1000; Cell Signaling Technology™, 9427 S), RAF1 (1:1000; Cell Signaling Technology™, 9422 S), phospho-Thr73 Rab10 (1:2000; Abcam™, ab230261), Rab10 (1:1000; Cell Signaling Technology™, 8127 S), Phospho-Drp1 (Ser616) (1:1000; Cell Signaling Technology™, 3455 S), Drp1 (1:1000; Cell Signaling Technology™, 8570), phospho-CaMKII (Thr287) (1:2000; ThermoFisher™ Scientific, PA5-37833), CaMKII (1:2000; Abcam™, ab52476), Phospho-CaMKI (Thr177) (1:3000; ThermoFisher™ Scientific, PA5-37833), CaMKI (1:3000; Abcam™, ab68234), MCU (1:3000; Abcam™, ab219827), COX4 (1:2000; Abcam™, ab16056), NCLX (1:1000; Abcam™, ab83551) and α-Tubulin (1:5000; Cell Signaling Technology™, 9099 S). The original western blots are available in the Supplementary file.
Cellular mitochondrial clearance assay
The mitochondrial clearance assay was performed as previously reported [13] to compare cellular clearance of mitochondria in WT and KI MEF cell lines. WT and KI MEFs were transduced by lentivirus for mito-PAmCherry protein expression, and was selected using cell sorter (BD FACSAria SORP) and puromycin (Puromycin Dihydrochloride, ThermoFisher™ Scientific, A1113803). The positively selected MEFs were refreshed with new medium for 2 h, and photoactivated for a different duration (WT MEF: 20 min; KI MEF: 18 min) under 405 nm (UV-A) exposure. Given the inconsistency of gene delivery to the genome via viral transduction, the stated duration of UV-A exposure allowed for similar levels of initial (t = 0 h) red fluorescence under unstimulated conditions. The red fluorescence at 0 and 24 h were measured by the BD LSR Fortessa Cell Analyzer (PE-Texas Red Channel). The rate of mitochondrial clearance was determined by the decline in red fluorescence (photoactivated mCherry) at 24 h compared to their initial levels at 0 h. The slopes representing relative rates of mitochondrial clearance from 0 to 24 h were plotted.
Statistical analyses
All experiments were performed based on a sufficient number of independent trials to achieve statistical significance, as indicated in figure legends. Results were expressed as means ± SEM (standard error of the mean). Conclusions were drawn based on statistical analyses using GraphPad™ PRISM software (GraphPad Inc., CA). Comparison between multiple independent groups was assessed using one-way ANOVA followed by Tukey’s multiple comparisons test. Comparison between two independent groups, and comparison of treatment effects at different time points against its respective baseline values were assessed using paired or unpaired Student’s t-test. p-values less than 0.05 (p < 0.05) were considered statistically significant.
Results
Mitochondrial uncoupler FCCP-induced cytosolic Ca2+ surge was abolished by LRRK2R1441G mutation
To understand the effect of LRRK2R1441G mutation on cellular response to mitochondrial stress, mouse embryonic fibroblasts (MEFs) carrying homozygous knockin (KI) LRRK2R1441G mutation were challenged with a mitochondrial uncoupler, FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) to mimic mitochondrial depolarization stress. To explore the possible involvement of cellular Ca2+ signaling in response to mitochondrial stress, transient changes in the cytosolic Ca2+ level in KI and wild-type (WT) control MEFs following exposure to FCCP were assessed by real-time confocal imaging of cells stained with Cal-520 (cytosolic Ca2+-sensitive green, fluorescent dye). Treatment of WT MEFs with FCCP (10 µM) caused an immediate increase in green fluorescence, indicating a rapid surge in cytosolic Ca2+ levels in response to the FCCP-induced stress (Fig. 1A). The emitted fluorescence reached a peak level within 30 s of FCCP treatment, followed by a gradual decline towards the baseline by the end of 10-min imaging duration (Fig. 1B). In contrast, similar treatment with FCCP on KI MEFs elicited no significant changes in cytosolic Ca2+ level as shown by a steady level of Cal-520 fluorescence intensity tracing (Fig. 1B). Such absence of cytosolic Ca2+ response in KI MEFs was also reflected in the area under curve (AUC) quantification from 3 to 10 min, which showed significantly smaller cumulative changes in cytosolic Ca2+ level of the KI compared to WT cells (Fig. 1C). This indicates a loss of cytosolic Ca2+ surge in response to FCCP treatment in the LRRK2R1441G mutant cells.
Fig. 1.

Depolarization of mitochondria induces transient cytosolic Ca2+ surge which is disrupted by LRRK2R1441G mutation
(A) Representative fluorescent microscopic image of WT and KI MEFs stained with Cal-520 at baseline (t = 0 min), and 1 min after the addition of FCCP (t = 4 min), and at final imaging timepoint (t = 10 min). Relative cytosolic Ca2+ level is indicated by the intensity of green fluorescence (Cal-520). Scale bar: 50 μm. (B) Real-time tracing of the Cal-520 fluorescence intensity of MEFs after addition of FCCP (10 µM), normalized by its own baseline (N = 4 experiments). (C) Area under curve (AUC) analysis of changes in cytosolic Ca2+ level after FCCP treatment from t = 3 to 10 min (N = 4 experiments). (D) Real-time tracing of the Cal-520 fluorescence intensity in WT MEFs after addition of different mitochondrial toxins: FCCP, H2O2, oligomycin and rotenone, normalized by its own baseline (N ≥ 2 experiments). (E) Real-time tracing of TMRM intensity of WT MEFs at baseline for 3 min, followed by the addition of different mitochondrial toxins (N ≥ 2 experiments). Data are presented as mean ± SEM. Statistical analyses: (C) Unpaired parametric Student’s t-test; (E) Paired parametric Student’s t-test. ***p < 0.001
To investigate whether the rapid cytosolic Ca2+ surge in response to FCCP was due to mitochondrial depolarization and not by other cellular stresses, a similar measurement of cytosolic Ca2+ transient was performed in WT MEFs following treatments with other types of toxins which caused different primary stress to the cells, including hydrogen peroxide (H2O2, oxidative stress inducer), rotenone (mitochondrial complex I inhibitor) and oligomycin (ATP synthase inhibitor). Cal-520 fluorescence intensity plots of WT MEFs showed that none of these toxins could induce a cytosolic Ca2+ surge as observed with FCCP treatment (Fig. 1D). Similarly, treatment with the drug vehicle, DMSO (0.01% v/v), had no effect on cytosolic Ca2+ levels (Fig. 1D), indicating that the cytosolic Ca2+ surge response was specific to FCCP. Based on real-time imaging of TMRM fluorescence, it was further confirmed that the other three toxins (H2O2, oligomycin and rotenone) did not cause any significant effects on MMP within the initial 10-min of treatment (Fig. 1E). This was in contrast to FCCP treatment which resulted in a rapid decline in TMRM fluorescence intensity, indicating a rapid depolarization of MMP in WT MEFs (Fig. 1E). Parallel treatment with DMSO also did not affect MMP (Fig. 1E, red tracing). The mitochondrial toxins H2O2, oligomycin or rotenone which had no acute effects on MMP did not elicit cytosolic Ca2+ surge, suggesting that the cytosolic Ca2+ surge induced by FCCP appear to be due to mitochondrial depolarization.
FCCP-induced cytosolic Ca2+ surge was predominantly mediated via mitochondrial NCLX but not mPTP opening
Given that FCCP depolarizes mitochondria, we explored whether the cytosolic Ca2+ surge induced by FCCP involved mitochondria as the origin of Ca2+ signaling. Two possible routes of mitochondrial Ca2+ efflux were investigated, namely the mitochondrial sodium-calcium exchanger (NCLX) [25] and the mitochondrial permeability transition pore (mPTP) opening [16]. To investigate the involvement of NCLX, WT MEFs loaded with Cal-520 were pre-treated with a NCLX inhibitor, CGP-37157, or drug vehicle (DMSO; 0.01% v/v) for 1 h prior to FCCP (10 µM) treatment. Since FCCP treatment in KI MEFs did not elicit any Ca2+ response, only WT MEFs were used in this experiment. As observed in the prior Ca2+ imaging experiment, WT MEFs that were pre-treated with DMSO consistently responded with an immediate Ca2+ surge within 30 s after the addition of FCCP, followed by a gradual decline back to baseline within 10 min (Fig. 2A). In contrast, pre-treatment with CGP-37157 significantly reduced the magnitude of the Ca2+ surge by 66% after the cells were exposed to FCCP [AUC analysis from 3 to 10 min of FCCP treatment] (Fig. 2B). These findings indicate that FCCP-induced cytosolic Ca2+ surge is in part due to NCLX activity (i.e. mitochondrial Ca2+ efflux).
Fig. 2.

NCLX inhibition reduces the magnitude of FCCP-induced cytosolic Ca2+ surge in WT MEFs
(A) Real-time tracing of Cal-520 fluorescence normalized by the baseline (t = 0 min) for WT MEFs pre-treated with or without CGP-37157 (NCLX inhibitor), following the addition of FCCP at 3 min timepoint (N = 4 experiments, 60 ≤ cells traced per N). (B) AUC analysis of cytosolic Ca2+ measurements in (A) from 3 to 10 min in WT MEFs (N = 4 experiments). (C) Real-time reading of percentage decrease in calcein fluorescence in WT MEFs after the addition of ionomycin for 300 s (black line, positive control) or WT MEFs treated with FCCP for the first 300 s, followed by ionomycin treatment (red line) (N = 3 experiments). Data are presented as mean ± SEM. Statistical analyses: (B) Unpaired parametric Student’s t-test between groups with and without CGP-37157 pre-treatment. **p < 0.01
Earlier study showed that FCCP can induce mPTP opening [26]. Hence, we investigated the effect of FCCP on mPTP opening in WT MEFs using an established calcein-CoCl2 quenching assay [27]. Ionomycin is a calcium ionophore that induces mitochondrial Ca2+ influx and mPTP opening, and hence was used as a positive control. In WT cells co-stained with calcein and CoCl2, addition of ionomycin resulted in an immediate decrease in calcein fluorescence intensity (Fig. 2C), indicating mPTP opening. However, unlike ionomycin, FCCP treatment did not significantly alter calcein fluorescence intensity in the treated MEFs (Fig. 2C), indicating that FCCP has no direct effect on mPTP opening. To confirm that this lack of mPTP opening after FCCP treatment was not due to confounding cross reactivity between calcein or CoCl2 with FCCP, ionomycin was added to the same cells that formerly showed no mPTP opening with FCCP. It resulted in an immediate decrease in calcein fluorescence intensity similar to when the cells were treated with ionomycin alone (Fig. 2C). These results confirmed that FCCP has no direct effect on mPTP opening, and that the impaired Ca2+ response in KI was linked to NCLX activity but not mPTP opening. These results led us to further examine how LRRK2R1441G mutation affects mitochondrial morphology and function, which can influence mitochondrial Ca2+ efflux activity.
LRRK2R1441G KI MEFs exhibited aberrant mitochondrial morphology and respiratory dysfunction
As we found that the FCCP-induced cytosolic Ca2+ surge was in part due to mitochondria NCLX activity, the mitochondrial quality and function of LRRK2 KI MEFs were assessed and compared to those of WT control. Based on morphological examination of at least 20 randomly chosen photomicrographs taken from 3 independent cell passages under transmission electron microscopy (TEM), most of the mitochondria in KI MEFs grown under normal culture condition already exhibited swollen with disorganized cristae and disintegrated, hollow structures (Fig. 3A), indicating inherent mitochondrial defects. This is in contrast to mitochondria in WT MEFs which exhibited a typical tubular and oval-shaped morphology with ordered cristae structure (Fig. 3A).
Fig. 3.

KI MEF exhibit altered unstimulated mitochondrial parameters
(A) Representative TEM photographs showing mitochondrial morphology of WT and KI MEFs under normal culture conditions. Scale bar: 500 nm. (B) Flow cytometry peaks showing mean TMRM fluorescence intensity (PE-A channel) in unstained WT (serving as blank) and TMRM-stained WT and KI MEFs. (C) Quantified mean TMRM fluorescent intensity reflecting relative basal MMP of WT and KI MEFs using flow cytometry (N = 6 experiments, 10,000 cells per N). (D) Representative fluorescent microscopic images of WT and KI MEFs stained with TMRM, a potentiometric, red fluorescent dye for mitochondrial membrane potential (MMP). Scale bar: 50 μm. (E) Quantified mean TMRM fluorescence intensity at baseline (0 min) reflecting relative basal MMP of WT and KI MEFs by microscopy (N = 4 experiments, 100 cells traced per N). (F) Real-time tracing of TMRM intensity of cells at baseline and after 3 min of FCCP (10 µM) treatment. (G) Rate of decline in absolute TMRM intensity in WT and KI MEFs as quantified by the negative slope in the first two min of FCCP treatment. (H) The magnitude of MMP depolarization in WT and KI MEFs (N = 4 experiments). (I-M) Seahorse analysis of WT and KI MEFs showing: (I) oxygen consumption rate and different mitochondrial parameters of (J) basal respiration, (K) maximal respiration, (L) spare respiratory capacity and (M) ATP production (N = 4 experiments, 10 individual wells per N). Dotted lines in (I) indicate the addition of each drug as labelled. (N) Basal cellular ATP level in WT and KI MEFs quantified using bioluminescence assay (N = 9 experiments). (O) Basal cellular ATP: ADP in WT and KI MEFs (N = 5 experiments). Data are presented as mean ± SEM. Statistical analyses by unpaired parametric Student’s t-test. *p < 0.05, **p < 0.01 and ***p < 0.001. ns, not significant
In parallel with the aberrant morphology, mitochondria in KI MEFs exhibited more depolarized basal MMP compared to those of WT cells, as indicated by significantly lower TMRM fluorescence intensity quantified by flow cytometry (Fig. 3B, C). As KI MEFs contained more depolarized mitochondria compared to WT (Fig. 3C), we compared the magnitude of decline in MMP following treatment with FCCP in the two cell lines by real-time imaging of TMRM fluorescence intensity (Fig. 3D). Cells loaded with TMRM were treated with FCCP after 3 min of baseline measurements. Subsequent changes in cellular TMRM intensity were quantitatively traced up to 10 min of imaging duration. Consistent to the flow cytometry measurements showing more depolarized mitochondria in KI MEFs under basal conditions (Fig. 3C), the baseline TMRM staining intensity in KI MEFs at time 0 min was significantly lower than that in WT MEFs (Fig. 3E). FCCP treatment resulted in a rapid and significant mitochondrial depolarization as shown by the exponential decrease in TMRM intensity in both cell lines (Fig. 3F). However, the rate of decline in TMRM intensity (within the initial two min of FCCP treatment) of KI MEFs was significantly slower than WT MEFs, by approximately 45% (Fig. 3G). AUC analysis also showed that KI MEFs responded with a significantly smaller magnitude of MMP depolarization by 35% when compared to WT (Fig. 3H).
Next, the mitochondrial respiratory function of WT and LRRK2 KI MEFs were assessed by the Seahorse™ XF Cell Mito Stress Test using the Seahorse™ analyzer (Fig. 3I). The calculated values from the cellular metabolic profile derived from the Seahorse™ analysis showed that WT and KI MEFs had a similar basal respiration level (Fig. 3J), but the maximal respiration in KI MEFs was significantly higher than that of WT (Fig. 3K). Also, the spare respiratory capacity was significantly higher in KI MEFs when compared to WT (Fig. 3L), although ATP production in KI MEFs was similar to that in WT (Fig. 3M). To verify our findings from the Seahorse analysis, total cellular ATP level and ATP-to-ADP ratio (ATP: ADP) of WT and KI MEFs were compared using a luciferase-based bioluminescence assay. Consistent with Seahorse bioenergetics profile, there were no statistical differences in total basal ATP levels between WT and KI MEFs (WT: 1.76 ± 0.088; KI: 1.69 ± 0.075 µM/µg protein) (Fig. 3N). However, KI MEFs showed significantly lower ATP: ADP when compared to WT (by 38.7%) (Fig. 3O). This implies that the cellular ADP level in KI cells was abnormally higher than WT, indicating an imbalance between cellular ATP and ADP.
LRRK2R1441G KI MEFs exhibited lower basal mitochondrial Ca2+ level and reduced mitochondrial calcium uniporter (MCU) protein expression
Mitochondria can actively buffer Ca2+ to maintain intracellular Ca2+ homeostasis [16]. Given the observation of defective mitochondria in KI MEFs, we investigated whether mitochondrial Ca2+ buffering in KI MEFs was perturbed. The relative basal mitochondrial Ca2+ store in WT and KI MEFs were compared using a mitochondrial Ca2+-sensing fluorescent dye, Rhod-2. Under basal culture condition, KI MEFs showed significantly less mitochondrial Ca2+ compared with that of WT MEFs, measured using both fluorescent microscopy (Fig. 4A) and flow cytometry (Fig. 4B, C). To explore the possible reasons for the lower mitochondrial Ca2+ level in the KI cells, protein expression levels of mitochondrial calcium channels which regulate mitochondrial Ca2+ level, namely NCLX (for mitochondrial Ca2+ efflux) and mitochondrial Ca2+ uniporter (MCU; for mitochondrial Ca2+ uptake) were assessed using western blotting (Fig. 4D). Whilst NCLX expression did not significantly differ between WT and KI cells (Fig. 4E), KI MEFs showed significantly reduced MCU expression when compared to WT MEFs (Fig. 4F). This suggests that lower basal mitochondrial Ca2+ in KI MEFs may be related to a reduced level of MCU which mediates mitochondrial Ca2+ uptake. Both MEFs showed similar COX4 levels (Fig. 4G), indicating that they had similar amounts of mitochondria, and that the observed differences in MCU expression were not due to differences in total cellular mitochondrial content.
Fig. 4.

KI MEFs exhibit reduced basal mitochondrial Ca2+ level
(A) Representative microscopy images of WT and KI MEFs stained with Rhod-2 (5 µM), a mitochondrial Ca2+ dye. Scale bar: 50 μm. (B) Representative flow cytometry histogram peaks showing Rhod-2 fluorescence readings from PE-A channel for negative control (unstained WT cells), WT and KI MEFs stained with Rhod-2. (C) Quantified mean Rhod-2 fluorescence intensity in WT and KI MEFs by flow cytometry measurement (N = 6 experiments, 10,000 cells per N). (D) Representative immunoblots showing the protein expression level of NCLX, MCU and COX4 in WT and KI MEFs. Expression level of (E) NCLX (N = 4 experiments) and (F) MCU (N = 8 experiments) were normalized using the COX4. (G) Expression level of COX4 was normalized by beta-actin (N = 8 experiments). Data are presented as mean ± SEM. Statistical analyses by unpaired parametric Student’s t-test. **p < 0.01 and ***p < 0.001. ns, not significant
CaMKII-MEK-ERK and Drp1 signals failed to be activated in LRRK2 KI MEFs under FCCP-induced mitochondrial depolarization
Having shown that LRRK2 KI MEFs failed to initiate a cytosolic Ca2+ surge under FCCP stress, potential downstream molecular events in the context of mitochondrial stress response signaling pathways in KI MEFs were investigated. Ca²⁺/calmodulin-dependent protein kinases (CaMKs) are a family of proteins that are activated by the Ca2+-binding protein, calmodulin, which themselves are activated by elevated cytosolic Ca2+ levels [28, 29]. Among the different types of CaMKs (CaMKK, CaMKI, CaMKII and CaMKIV), activation (i.e. phosphorylation) of two CaMKs which are predominantly expressed in cytosol, namely CaMKI and CaMKII, at residues of Thr177 and Thr286, respectively [30] were assessed in WT and KI MEFs after FCCP treatment. The two cell lines were treated with FCCP (10 µM) for 5, 15 and 30 min, and followed by a western blot analysis to determine the time-dependent changes in the kinase activation (phosphorylation) (Fig. 5A and Fig. S1A). In parallel with the cytosolic Ca2+ surge, FCCP induced CaMKII activation (Thr286-CaMKII phosphorylation) in WT MEFs, showing peak phosphorylation levels of 166% at 15 min from their corresponding baseline levels (Fig. 5A, B). In contrast, KI MEFs showed no significant changes in p-CaMKII level under the same FCCP treatment (Fig. 5A, B). Unlike p-CaMKII, p-CaMKI levels did not change significantly in both MEFs after FCCP treatment (Fig. S1B), indicating that CaMKI is not involved.
Fig. 5.

FCCP-induced activation of CaMKII-MEK-ERK-Drp1 is impaired in KI MEFs
(A) Left: Representative immunoblots of WT and KI MEFs treated with FCCP for 0, 5, 15 and 30 min, showing levels of phosphorylated and total CaMKII, MEK, ERK and Drp1, with α-tubulin as loading controls. Right: Diagram showing cellular events occurring in WT MEFs with exposure to FCCP: acute cytosolic Ca2+ rise, followed by activation of CaMKII, MEK, ERK and Drp1. (B-E) Quantitation of phosphorylated CaMKII, MEK, ERK and Drp1 normalized by their respective total proteins in WT and KI MEFs following FCCP treatment (N = 4 experiments). (F) Percentage clearance of mitochondria over 24 h in WT and KI MEFs is expressed as percentage decrease in mito-PAmCherry fluorescence from its own baseline fluorescence intensity at 0 h by flow cytometry (N = 5 experiments, 10,000 cells per N). (G) Calculated mean slope from 0 to 24 h showing average percentage decrease in mito-PAmCherry fluorescence per hour. (H) Representative immunoblots of KI MEFs pre-treated with either LRRK2 kinase inhibitor, MLi-2 (30 nM), or vehicle, DMSO, for 1 h prior to FCCP treatment (10 µM) for 0, 15 and 30 min (N = 4 experiments). WT MEFs were pre-treated with DMSO as a positive control, followed by FCCP treatment of same duration. (I) LRRK2 kinase inhibition by MLi-2 was confirmed by the reduction in p-Rab10 (Thr73) level. Quantitation of phosphorylated (J) MEK and (K) ERK normalized by their respective total proteins following FCCP treatment. Data are presented as mean ± SEM. Statistical analyses: (B-E) Unpaired parametric Student’s t-test, each timepoint compared to its own respective baseline value at 0 min; (C) Unpaired parametric Student’s t-test between baseline values of WT and KI MEFs, #p < 0.05; (F, G) Unpaired parametric Student’s t-test; (I) One-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01 and ***p < 0.001. ns, not significant
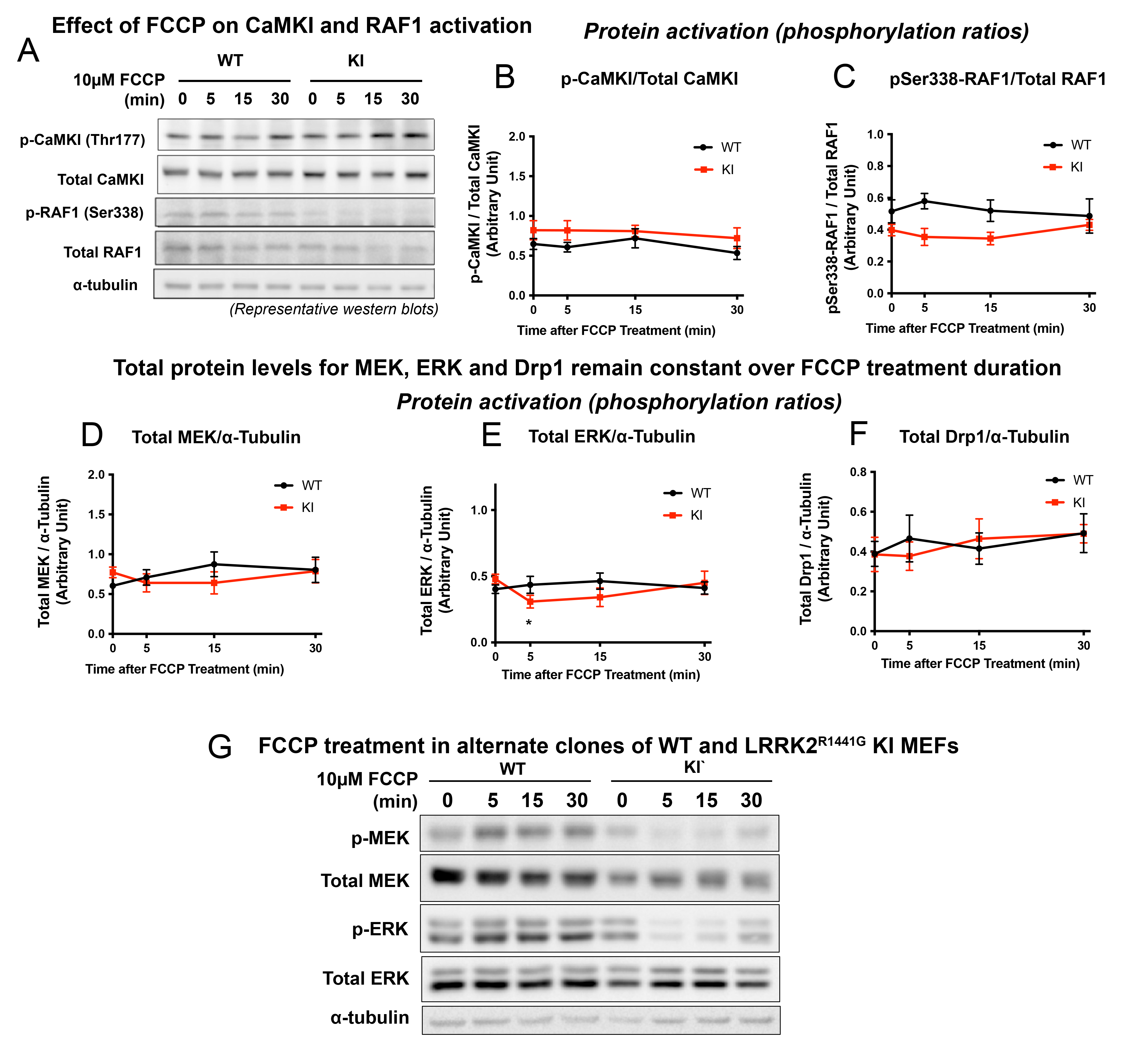
Along with CaMKII, activation of ERK is also regulated by Ca2+ [31–33]. ERK is a downstream effector of the canonical RAF-MEK-ERK cellular stress response pathway [31]. Given that both cytosolic Ca2+ surge and CaMKII activation were impaired in KI MEFs, we next explored whether the activation of RAF, MEK and ERK are also involved in FCCP-induced mitochondrial stress response, and whether such activation was impaired by LRRK2 mutation. In WT MEFs, FCCP induced phosphorylation of Ser217/221 MEK (p-MEK) at 5 min, which peaked at 15 min (245.4% of baseline), and then declined back to baseline level by 30 min (Fig. 5C). Similarly, levels of phosphorylated Thr202/Tyr204 ERK (p-ERK) were also significantly increased after FCCP treatment, and reached a peak at 15 min (308% from baseline) (Fig. 5D). Unlike MEK and ERK, levels of phosphorylated RAF1 at Ser338 (required for RAF1 activation) remained unchanged over the course of the FCCP treatment (Fig. S1A, C), indicating that FCCP-induced MEK-ERK activation was independent of the canonical RAF1-mediated pathways. In contrast to WT MEFs, KI MEFs responded with a significant reduction in levels of both p-MEK and p-ERK at 5 min after FCCP treatment, and such effect persisted even at 15 and 30 min (Fig. 5C, D), indicating an impaired MEK-ERK activation specifically in the LRRK2 mutant. In parallel with such differential responses, FCCP treatment induced Drp1 activation (i.e. phosphorylation at Ser616; pSer616-Drp1) in WT MEFs (peak activation of 138.7% from baseline) which was similarly found to be abolished in the KI (Fig. 5E). The basal phosphorylation levels of p-CaMKII, p-ERK, and pSer616-Drp1 were similar between WT and KI MEFs (Fig. 5B, D, E), whilst only the p-MEK level in KI was significantly higher compared to that of WT (Fig. 5C). Interestingly, there was a significant initial drop in total ERK protein level in KI MEFs at 5 min after FCCP treatment, but the levels were restored to its corresponding baseline level after 30 min of FCCP treatment (Fig. S1E). Total MEK and Drp1 protein levels did not change significantly over 30-min treatment (Fig. S1D, F), confirming that impaired CaMKII-MEK-ERK-Drp1 activation in KI MEFs was not due to any differences in total protein expression.
Furthermore, to verify that our findings are not confounded by clonal effects, similar measurements were repeated using alternative clones of both WT and KI MEFs which were independently generated from different mice (Fig. S1G). Consistent results in phosphorylation levels of MEK and ERK were observed in both clones in response to FCCP treatment, excluding the possibility of clonal effect.
LRRK2R1441G mutation reduced the basal mitochondrial clearance
Given the lack of Ca2+ response and CaMKII-MEK-ERK-Drp1 activation in LRRK2 KI, we compared the basal mitochondrial clearance rate between WT and KI MEFs. Based on a real-time flow cytometry assay to assess mitochondrial turnover [13], WT and KI MEFs were engineered to stably express mitochondrial-specific photoactivatable mCherry (mito-PAmCherry), a fluorescent reporter of cellular mitochondrial clearance. Cells grown under normal culture condition with refreshed medium were photoactivated by UV-A and harvested at 0 and 24 h post-photoactivation to determine the magnitude of decline in mito-PAmCherry fluorescence. Consistent with our earlier findings [13], KI MEFs exhibited a significantly slower mitochondrial clearance rate compared with that of WT MEFs by ~ 12%, as shown by the lower percentage decrease in mito-PAmCherry fluorescence over 24 h (Fig. 5F, G).
Impaired MEK-ERK activation in KI MEFs was independent of mutant LRRK2 hyperactivity
LRRK2R1441G mutation causes pathogenic kinase hyperactivity in PD [34]. Therefore, we determined whether impaired MEK-ERK activation were mediated by hyperactive kinase activity of mutant LRRK2. KI MEFs were pre-treated with MLi-2 (specific LRRK2 inhibitor; 30 nM) or drug vehicle (DMSO; 0.01% v/v) for 1 h prior to FCCP exposure (10 µM) for 15 and 30 min, and the activation of MEK-ERK was determined by western blotting (Fig. 5H). KI MEFs under basal condition exhibited increased LRRK2 kinase activity, as shown by significantly higher level of phosphorylated Rab10 at Thr73 residue (p-Rab10; a kinase phosphorylation target of LRRK2) by 486% compared with the relative level in WT cells. Therapeutic inhibition of hyperactive LRRK2 by MLi-2 treatment in KI MEFs was confirmed from p-Rab10 level in KI MEFs that was significantly reduced to level similar to DMSO pre-treated WT control MEFs (Fig. 5I). The lack of MEK-ERK activation in KI MEFs under FCCP exposure was not alleviated by therapeutic LRRK2 inhibition, as shown by a lack of difference between DMSO- and MLi-2-treated KI MEFs (Fig. 5J, K). To further validate that LRRK2 hyperactivity was not involved, KI MEFs were pre-treated again with MLi-2 (30 nM) for a longer duration of 24 h prior to FCCP challenge. Whilst LRRK2 kinase inhibition was confirmed by reduced p-Rab10 levels in KI MEFs, treating the cells with MLi-2 for longer duration did not rescue the impaired phosphorylation of MEK and ERK (Fig. S2), similar to what was observed with the 1 h MLi-2 pre-treatment. These findings indicate that the impaired MEK-ERK activation in KI MEFs was not due to hyperactivity of mutant LRRK2.
Discussion
The current work extends our previous study in which we demonstrated impaired activation of ERK and Drp1 in LRRK2R1441G mutant MEFs that was associated with abnormal accumulation of autophagosomes and impaired mitophagy [13]. In this study, we unravelled the underlying mitochondrial defects inherent to LRRK2R1441G mutant cells to elucidate how pathogenic LRRK2R1441G of PD influences mitochondrial Ca2+ signaling and a cascade of subsequent molecular events which hindered the activation of ERK-Drp1 and mitochondrial clearance under depolarization stress as mimicked by treatment with an artificial uncoupler, FCCP. We have shown that acute mitochondrial depolarization induced by FCCP caused a rapid, mitochondrial NCLX-dependent cytosolic Ca2+ surge in WT, but not in LRRK2 KI MEFs. We found that these defects in the mutant parallel a loss of CaMKII-MEK-ERK and Drp1 signaling that may be linked to mitochondrial quality control and mitophagy as implicated in the pathogenesis of PD.
Intracellular Ca2+ plays an essential role in cellular energy homeostasis and mitophagy [35–37]. In particular, ERK activation in response to elevation of cytosolic Ca2+ can affect bioenergetics and autophagy [38–40], yet whether and how pathogenic LRRK2R1441G mutation affects these processes in the context of mitochondrial homeostasis were not explored. Through treatments with different mitochondrial toxins, we showed that only FCCP which induced acute mitochondrial depolarization could elicit an immediate cytosolic Ca2+ surge in WT cells, but not those that did not induce acute mitochondrial depolarization, including rotenone, oligomycin and H2O2. Mitochondrial depolarization, also caused by exposure to environmental toxins in PD [41], serves as one of the signaling cues to initiate mitophagy as a cellular defence to replenish damaged mitochondria and to maintain energy homeostasis [42]. Therefore, we explored the molecular changes related to mitochondrial depolarization and Ca2+ signaling in the LRRK2 mutant.
Mitochondria serve as cytosolic Ca2+ buffer via transports of free Ca2+ across mitochondrial membranes [16, 43]. In particular, NCLX is a mitochondrial ion exchange channel which mediates Ca2+ efflux in neurons [44]. Cytosolic Ca2+ signal can arise from sources such as the endoplasmic reticulum and the extracellular space [45], but here we found that incubation of WT MEFs with mitochondrial NCLX inhibitor (CGP-37157) significantly suppressed the cytosolic Ca2+ surge induced by FCCP, indicating that the observed Ca2+ surge was primarily initiated by mitochondria. Given that similar treatment with FCCP did not induce a cytosolic Ca2+ surge in KI MEFs, this suggests that mitochondrial Ca2+ efflux via NCLX was repressed by LRRK2R1441G mutation. Our findings are consistent with an earlier study showing a similar defective Ca2+ efflux from NCLX in mutant LRRK2 patients’ fibroblasts, which was alleviated by upregulation of NCLX activity via protein kinase A (PKA) [23]. However, this study used permeabilized cells following stimulation with ATP or CaCl2, which may involve different pathways distinct from FCCP-induced mitochondrial stress in our study. Whether impaired NCLX activity in our LRRK2 KI MEFs also involved PKA requires further investigation. Nevertheless, our findings are also consistent with another study that reported regulation of autophagy by NCLX-mediated mitochondrial Ca2+ efflux through its modulation of cytosolic Ca2+ signaling [46]. In view of cellular response to mitochondrial depolarization, our findings may provide therapeutic insights into modulating NCLX [47] to alleviate dysregulated mitochondrial Ca2+ flux and cellular stress response in LRRK2-associated PD.
Whilst NCLX inhibition significantly reduced the magnitude of FCCP-induced cytosolic Ca2+ surge in WT cells, it did not completely abolish Ca2+ response. This suggests that additional mechanisms may also be involved. Apart from NCLX, mPTP (mitochondrial permeability transition pore) is an alternative route for mitochondrial Ca2+ efflux [48, 49]. However, our findings ruled out the involvement of mPTP in Ca2+ signaling under mitochondria depolarization stress. One possible explanation is that the toxicity level based on our current FCCP concentration, and the time of treatment may not be strong enough to induce an immediate mPTP opening. Furthermore, mPTP opening is known to be triggered by mitochondrial Ca2+ overload [50, 51]. Given that our current FCCP treatment induced mitochondrial Ca2+ efflux via NCLX as opposed to mitochondrial Ca2+ overload, it is not surprising that mPTP was not involved in this primary response. Although our current findings cannot rule out other mechanisms that may also contribute to the observed cytosolic Ca2+ surge, inhibition of NCLX repressed most of the Ca2+ response signal under mitochondrial depolarization, confirming that mitochondria have played a dominant role in FCCP-induced cellular Ca2+ response.
To explain for the defects of mitochondrial Ca2+ response in KI MEFs, our functional and morphological examination revealed a number of inherent mitochondrial abnormalities which could be linked to mitochondrial Ca2+ signaling. In particular, KI MEFs under unstressed condition already showed lower mitochondrial Ca2+ store and a concomitant reduction of MCU expression compared with those in WT MEFs. MCU is localized to the mitochondrial inner membrane where it loads Ca2+ into the matrix [52]. Hence, it is possible that the reduced MCU expression may perturb mitochondrial Ca2+ balance from inefficient ion transport. The reason for reduced MCU level in KI MEFs is unclear, but it could be due to the disruption of mitochondrial cristae where MCU is localized. Our results are in line with a transcriptomic study of dopaminergic neurons derived from mutant Drosophila model of LRRK2G2019S mutation showing reduced MCU expression [53]. However, discrepant results were also reported in another study showing increased MCU expression in LRRK2G2019S and LRRK2R1441C mutant patients’ fibroblasts [54]. The reason for such disparity is unclear, but the increased MCU expression in the latter study was associated with mitochondrial Ca2+ overload. This is in contrast with our LRRK2R1441G KI MEFs which showed a distinctive phenotype of lower MCU expression and lower mitochondrial Ca2+ store. We speculate that such reduced basal mitochondrial Ca2+ store in KI MEFs may be insufficient to initiate mitochondrial Ca2+ efflux in response to FCCP-induced depolarization [20], leading to a lack of global cytosolic Ca2+ surge in KI MEFs.
MMP (mitochondrial membrane potential) is another key factor that mobilizes mitochondrial Ca2+ by providing the electrochemical gradient across mitochondrial membrane [16, 55, 56]. Our findings showed more depolarized mitochondria in KI MEFs compared with that of WT even under unstressed conditions. As mitochondrial Ca2+ efflux depends on highly polarized MMP [57], it is reasonable to speculate that reduced basal MMP in LRRK2 mutant may render them less sensitive to FCCP-induced depolarization. This is illustrated by a significantly lower rate and magnitude of MMP depolarization in KI MEFs compared to WT after FCCP treatment. Similar observation of reduced MMP was reported in other LRRK2 mutant cell models, including PD patient fibroblasts and neuroepithelial stem cells with LRRK2G2019S and LRRK2R1441C mutations [4, 8, 58, 59]. Given that spatial organization of cristae is critical to maintain overall mitochondrial integrity and MMP [60–62], the disordered cristae structure could be a critical phenotype underlying lower MMP as observed in our KI cells. Our findings are similar to the impaired mitochondrial Ca2+ efflux seen in another cell model lacking PINK1 expression (another PD susceptible gene), where these cells also showed reduced basal MMP [47, 63]. Nevertheless, there are also contrasting findings showing impaired mitochondrial Ca2+ efflux in some LRRK2 patient fibroblasts, yet being independent of MMP [23]. This implies that although LRRK2 mutation alone may not cause mitochondrial depolarization, it could exacerbate mitochondrial dysfunction in the presence of additional cellular stress, which is consistent with the multifactorial nature of PD.
In addition to regulating Ca2+ mobilization, mitochondrial Ca2+ store is also crucial to maintain cellular energy homeostasis, e.g., via the Krebs cycle which supplies high-energy substrates for ATP synthesis [64], and efficient exchange of mitochondrial ATP for cytosolic ADP during oxidative phosphorylation [65, 66]. Although the basal ATP levels of WT and KI MEFs were similar, ATP: ADP ratio was significantly lower in KI MEFs. Despite that KI MEFs showed higher maximal respiration rate and spare respiratory capacity compared to WT MEFs, these cells did not synthesize more ATP relative to the level of WT. This explains why LRRK2 mutant cells are more susceptible to ATP depletion [13, 67]. Although the reason for higher respiratory capacity in KI MEFs is unclear, it could be due to compensation for inherent morphological and functional defects as evident by the distorted cristae (where mitochondrial Ca2+ transport takes place [16]), and a lower basal MMP in the KI cells. This is also in line with LRRK2R1441C iPSC dopaminergic neurons showing reduced MMP and oxidative phosphorylation yet with no gross energetic deficit [68]. In addition, lower ATP: ADP ratio without difference in total ATP levels also implies ADP accumulation in the mutant, which is a common phenomenon in high energy demanding cells with impaired mitochondrial function [69]. The reason for imbalance in ATP and ADP is unclear, but could be due to ineffective oxidative phosphorylation attributed to the distorted inner mitochondrial membrane, leading to accumulation of substrate ADP. ADP accumulation has been shown to affect cytosolic Ca2+ mobilization [69], which may provide clues to unveil the molecular mechanisms of impaired Ca2+ response in KI MEFs.
In the context of mitochondrial stress response, the lack of stress-induced cytosolic Ca2+ surge in KI MEFs was associated with impaired downstream Ca2+-dependent pathway that mediates ERK and Drp1 activation, which is involved in segregation of damaged mitochondria for clearance. Similar impairment in mitochondrial degradation was reported in mouse LRRK2R1441C neurons in response to induced mitochondrial damage, as demonstrated by decreased levels of mitophagy marker pS65Ub [68]. Regarding the possible mechanism(s), CaMKII plays a central role to regulate mitochondrial Ca2+ entry via transmitting cellular Ca2+ signals in response to stress [70]. CaMKII, which we found to be activated in parallel with FCCP-induced Ca2+ surge, is the upstream kinase of MEK and ERK [71–73]. KI MEFs failed to induce a cytosolic Ca2+ surge with FCCP, suggesting that such cellular stress cannot be mediated via CaMKII and transmitted to the downstream effectors. This has an important implication for neuronal resilience upon exposure to environmental stressors, in particular for how cells respond and survive from mitochondrial damages.
Mutant LRRK2 hyperactivity has been implicated in PD pathogenesis as shown by aberrant substrate phosphorylation patterns and disrupted cellular signaling [74]. One therapeutic approach to address the pathogenic effects of LRRK2R1441G mutation is to inhibit its aberrant hyperactive kinase activity [75, 76]. However, we found that the impaired MEK-ERK activation in KI MEFs was not attenuated by treatment of the LRRK2 inhibitor (MLi-2), indicating that the loss of Ca2+ response was independent of mutant LRRK2 hyperactivity. Similar findings were reported in LRRK2R1441C iPSC-derived dopaminergic neurons with impaired mitophagy and mitochondrial function that were not rescued with MLi-2 treatment [68]. Interestingly, unlike in LRRK2R1441G/C models, LRRK2 inhibition rescued impaired mitophagy and altered autophagosome transport in LRRK2G2019S KI mice [77, 78]. These divergent results suggest that the two mutations may affect distinct mitochondrial pathways, which requires further investigation.
Conclusions
Our study showed that pathogenic LRRK2R1441G mutation of PD abolishes mitochondrial depolarization-induced cytosolic Ca2+ surge and subsequent activation of CaMKII/ERK signal axis as hitherto unreported mitochondrial stress response to downstream activation of mitochondrial fission-related Drp1 and mitophagy. LRRK2 mutant MEFs exhibited inherent mitochondrial defects, including aberrant morphology and structure, ATP-ADP imbalance, and reduced basal MMP, mitochondrial Ca2+ store and MCU expression. These inherent defects may synergistically cause mitochondrial Ca2+ imbalance that failed to elicit a Ca2+ signal response under exogenous mitochondrial stress. Such lack of mitochondrial Ca2+ efflux through NCLX repressed cytosolic Ca2+ surge and subsequent activation of the CaMKII/ERK and Drp1 signaling axis in LRRK2 mutant cells. These inherent defects from LRRK2 mutation may weaken the cellular defence to scavenge damaged mitochondria via mitophagy and rejuvenate mitochondrial function. It may potentially perturb other downstream ERK-dependent stress response pathways that support neuronal survival [79, 80]. The same rationale applies to our earlier findings that KI MEFs exhibited impaired autophagic degradation with increased accumulation of oligomeric α-synuclein [24], the pathogenic species that underlies synucleinopathies in PD [81]. Our results suggest that the accumulation of dysfunctional mitochondria in LRRK2 mutant cells is a possible consequence of impaired mitochondrial Ca2+ response and loss of CaMKII/ERK and Drp1 activation, eventually forming a “death-loop” which further aggravates mitochondrial dysfunction and neuronal cell death (Fig. 6). Our findings shed light on targeting mitochondrial Ca2+ pathways as a novel therapeutic strategy to enhancing mitochondrial quality and function in LRRK2-associated PD.
Fig. 6.

Hypothetical diagram comparing differential cellular responses under FCCP-induced mitochondrial depolarization in WT (Left) and KI cells (Right)
Under FCCP-induced mitochondrial damage, WT MEFs undergoes rapid mitochondrial membrane depolarization and mitochondrial Ca2+ efflux via NCLX, which leads to a cytosolic Ca2+ surge. CaMKII-MEK-ERK signaling axis is activated, followed by activation of Drp1 via Ser616 phosphorylation. Drp1 activation then initiates mitochondrial fission prior to mitophagy, a process through which defective mitochondria is cleared. KI MEFs already exhibit basal mitochondrial dysfunction even before FCCP-induced stress. Following FCCP, KI MEFs fail to exhibit cytosolic Ca2+ surge which is followed by a lack of activation of CaMKII-MEK-ERK and Drp1. Such signaling defects could impair the mitochondrial fission and mitophagy, leading to the accumulation of defective mitochondria. The accumulation of dysfunctional mitochondria thus adversely affects overall mitochondrial quality, forming a vicious cycle in LRRK2 KI MEFs
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
Acknowledgements
We would like to acknowledge Tai Hung Fai Charitable Foundation – Edwin S H Leong Research Programme for Parkinson’s Disease, and Henry G. Leong Endowed Professorship in Neurology (Fund holder: S.L.H.) for their trust and long-term funding support. We appreciate technical assistance from the University Faculty Core Facilities, The University of Hong Kong, on confocal microscopy and flow cytometry. We also thank Mr. W.S. Lee (Senior Technical Officer) from the University Electron Microscopy Unit (EMU) for his expertise support. The authors also thank for the administrative supports from (1) Department of Medicine, The University of Hong Kong; and (2) Department of Rehabilitation Sciences, The Hong Kong Polytechnic University. We would like to express our deepest gratitude to the late Prof. David Boyer Ramsden for his invaluable guidance and mentorship that laid the foundation for our research. David was an extraordinary scientist and a true visionary, whose passion for knowledge and scientific excellence inspired us all. Though no longer with us, David’s impact on our professional growth will forever be remembered and cherished.
Abbreviations
- AUC
area under curve
- CaMKII
Ca2+/calmodulin-dependent kinase II
- DMSO
dimethyl sulfoxide
- Drp1
dynamin 1-like protein
- FCCP
carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- KI
knockin
- LRRK2
leucine-rich repeat kinase 2
- ERK
extracellular signal-regulated kinase
- MEK
mitogen-activated protein kinase kinase
- MCU
mitochondrial calcium uniporter
- MEF
mouse embryonic fibroblast
- MMP
mitochondrial membrane potential
- mPTP
mitochondrial permeability transition pore
- NCLX
sodium-calcium exchanger
- OCR
oxygen consumption rate
- PD
Parkinson’s disease
- TEM
transmission electron microscopy
- WT
wild-type
Author contributions
E.E.S.C, P.W.L.H., S.L.H. and D.B.R. designed the project. P.W.L.H., E.E.S.C., H.F.L., Z.Y.K.C., Y.M. and S.X.Y.Z. performed experiments and data analysis. M.H.W.K. managed routine laboratory activities and safety. P.W.L.H., S.L.H. and D.B.R. oversaw and evaluated all experimental data. E.E.S.C. and P.W.L.H. wrote the manuscript with input of all co-authors. All authors read and approved the final manuscript.
Funding
This work was supported primarily by Tai Hung Fai Charitable Foundation—Edwin S H Leong Research Programme for Parkinson’s Disease, and Henry G. Leong Endowed Professorship in Neurology (Fund holder: S.L.H.). PhD studentship (E.E.S.C.) was funded by the Hong Kong PhD Fellowship Scheme (HKPFS), Research Grant Council, Hong Kong SAR, China. Research consumables and research staff cost were partly supported by Start-up Fund for New Recruit, The Hong Kong Polytechnic University (Fund holder: P.W.L.H.), Postdoc Matching Fund Scheme 2023/24 3rd round, The Hong Kong Polytechnic University (Fund holder: P.W.L.H.) and Better Utilization of Allocated Budget, Department of Rehabilitation Sciences, The Hong Kong Polytechnic University (PI: Prof Benjamin Yee, Co-I: P.W.L.H.).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have approved the contents of this manuscript and provided consent for publication.
Conflict of interest
The authors declare no competing interests.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shu-Leong Ho, Email: slho@hku.hk.
Philip Wing-Lok Ho, Email: philip-wl.ho@polyu.edu.hk.
References
- 1.Keeney PM, Xie J, Capaldi RA, Bennett JP Jr. Parkinson’s Disease Brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and Misassembled. J Neurosci. 2006;26:5256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grünewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson’s disease. Prog Neurobiol. 2019;177:73–93. [DOI] [PubMed] [Google Scholar]
- 3.Osellame LD, Duchen MR. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy. 2013;9:1633–5. [DOI] [PubMed] [Google Scholar]
- 4.Walter J, Bolognin S, Antony PMA, Nickels SL, Poovathingal SK, Salamanca L, Magni S, Perfeito R, Hoel F, Qing X, et al. Neural stem cells of Parkinson’s Disease patients exhibit aberrant mitochondrial morphology and functionality. Stem Cell Rep. 2019;12:878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–59. [DOI] [PubMed] [Google Scholar]
- 6.Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107:9879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh A, Zhi L, Zhang H. LRRK2 and mitochondria: recent advances and current views. Brain Res. 2019;1702:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75:2017–20. [DOI] [PubMed] [Google Scholar]
- 9.Sanders LH, Laganière J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis. 2014;62:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alegre-Abarrategui J, Christian H, Lufino MMP, Mutihac R, Venda LL, Ansorge O, Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet. 2009;18:4022–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu H-F, Lu S, Ho PW-L, Tse HM, Pang SYY, Kung MHW, Ho JWM, Ramsden DB, Zhou ZJ, Ho SL. LRRK2 R1441G mice are more liable to dopamine depletion and locomotor inactivity. Ann Clin Transl Neur. 2014;1:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H-F, Ho PW-L, Leung GC-T, Lam CS-C, Pang SY-Y, Li L, Kung MH-W, Ramsden DB, Ho S-L. Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of Parkinson’s disease. Sci Rep-UK. 2017;7:40887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu H-F, Ho PW-L, Leung C-T, Pang SY-Y, Chang EES, Choi ZY-K, Kung MH-W, Ramsden DB, Ho S-L. Aberrant mitochondrial morphology and function associated with impaired mitophagy and DNM1L-MAPK/ERK signaling are found in aged mutant parkinsonian LRRK2R1441G mice. Autophagy. 2020;17:3196–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57:537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ansari MY, Novak K, Haqqi TM. ERK1/2-mediated activation of DRP1 regulates mitochondrial dynamics and apoptosis in chondrocytes. Osteoarthr Cartil. 2022;30:315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19:713–30. [DOI] [PubMed] [Google Scholar]
- 17.La Rovere RML, Roest G, Bultynck G, Parys JB. Intracellular ca(2+) signaling and ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium. 2016;60:74–87. [DOI] [PubMed] [Google Scholar]
- 18.Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium. 2018;70:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivero-RÃos P, Gómez-Suaga P, Fdez E, Hilfiker S. Upstream deregulation of calcium signaling in Parkinson’s disease. Front Mol Neurosci 2014, 7. [DOI] [PMC free article] [PubMed]
- 20.Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Translational Neurodegeneration. 2022;11:3–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherra SJ, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol. 2013;182:474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ludtmann MHR, Abramov AY. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci Lett. 2018;663:86–90. [DOI] [PubMed] [Google Scholar]
- 23.Ludtmann MHR, Kostic M, Horne A, Gandhi S, Sekler I, Abramov AY. LRRK2 deficiency induced mitochondrial Ca2 + efflux inhibition can be rescued by Na+/Ca2+/Li + exchanger upregulation. Cell Death Dis. 2019;10:265–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho PW-L, Leung C-T, Liu H-F, Pang SY-Y, Lam CS-C, Xian J, Li L, Kung MH-W, Ramsden DB, Ho S-L. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy. 2020;2:347–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyman L, Williams GSB, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saotome M, Katoh H, Satoh H, Nagasaka S, Yoshihara S, Terada H, Hayashi H. Mitochondrial membrane potential modulates regulation of mitochondrial ca < sup > 2+ in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;288:H1820–8. [DOI] [PubMed] [Google Scholar]
- 27.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swulius MT, Waxham MN. Ca²⁺/Calmodulin-dependent protein kinases. Cell Mol Life Sci. 2008;65:2637–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wayman GA, Lee Y-S, Tokumitsu H, Silva A, Soderling TR. Calmodulin-Kinases: modulators of neuronal development and plasticity. Neuron (Cambridge Mass). 2008;59:914–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayer KU, Schulman H. CaM kinase: still inspiring at 40. Neuron. 2019;103:380–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol. 2020;21:607–32. [DOI] [PubMed] [Google Scholar]
- 32.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron (Cambridge Mass). 1994;12:1207–21. [DOI] [PubMed] [Google Scholar]
- 33.Melien O, Nilssen LS, Dajani OF, Sand KL, Iversen J-G, Sandnes DL, Christoffersen T. Ca2+-mediated activation of ERK in hepatocytes by norepinephrine and prostaglandin F2 alpha: role of calmodulin and src kinases. BMC Cell Biol. 2002;3:5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito G, Katsemonova K, Tonelli F, Lis P, Baptista MAS, Shpiro N, Duddy G, Wilson S, Ho PW-L, Ho S-L, et al. Phos-tag analysis of Rab10 phosphorylation by LRRK2: a powerful assay for assessing kinase function and inhibitors. Biochem J. 2016;473:2671–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–78. [DOI] [PubMed] [Google Scholar]
- 36.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrank S, Barrington N, Stutzmann GE. Calcium-handling defects and neurodegenerative disease. Cold Spring Harb Perspect Biol. 2020;12:a035212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010;277:22–9. [DOI] [PubMed] [Google Scholar]
- 39.Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung K-H, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2 + transfer to Mitochondria. Cell. 2010;142:270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bootman MD, Chehab T, Bultynck G, Parys JB, Rietdorf K. The regulation of autophagy by calcium signals: do we have a consensus? Cell Calcium (Edinburgh). 2018;70:32–46. [DOI] [PubMed] [Google Scholar]
- 41.Huang M, Bargues-Carot A, Riaz Z, Wickham H, Zenitsky G, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. Impact of Environmental Risk Factors on Mitochondrial Dysfunction, Neuroinflammation, Protein Misfolding, and Oxidative Stress in the Etiopathogenesis of Parkinson’s Disease. Int J Mol Sci 2022, 23. [DOI] [PMC free article] [PubMed]
- 42.Winter JM, Yadav T, Rutter J. Stressed to death: mitochondrial stress responses connect respiration and apoptosis in cancer. Mol Cell. 2022;82:3321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dey K, Bazala MA, Kuznicki J. Targeting mitochondrial calcium pathways as a potential treatment against Parkinson’s disease. Cell Calcium. 2020;89:102216–102216. [DOI] [PubMed] [Google Scholar]
- 44.Rozenfeld M, Azoulay IS, Ben Kasus Nissim T, Stavsky A, Melamed M, Stutzmann G, Hershfinkel M, Kofman O, Sekler I. Essential role of the mitochondrial Na+/Ca2 + exchanger NCLX in mediating PDE2-dependent neuronal survival and learning. Cell Rep. 2022;41:111772–111772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the Center of Cell Signaling: interplay between endoplasmic reticulum, Mitochondria, and Lysosomes. Trends Biochem Sci (Amsterdam Regul ed). 2016;41:1035–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Miranda Ramos V, Serna JD, Vilas-Boas EA, Cabral-Costa JV, Fernanda Marques Da C, Kataura T, Korolchuk VI, Kowaltowski AJ. Mitochondrial Sodium/Calcium exchanger (NCLX) regulates basal and Starvation-Induced Autophagy through Calcium Signaling. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2023. [Google Scholar]
- 47.Kostic M, Ludtmann Marthe HR, Bading H, Hershfinkel M, Steer E, Chu Charleen T, Abramov Andrey Y, Sekler I. PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-Deficient dopaminergic neurons. Cell Rep. 2015;13:376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B, et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun. 2018;9:2293–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–49. [PMC free article] [PubMed] [Google Scholar]
- 50.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a Promising Therapeutic Target in Cardiac diseases. J Pharmacol Exp Ther. 2009;330:670–8. [DOI] [PubMed] [Google Scholar]
- 51.Baumgartner HK, Gerasimenko JV, Thorne C, Ferdek P, Pozzan T, Tepikin AV, Petersen OH, Sutton R, Watson AJM, Gerasimenko OV. Calcium elevation in Mitochondria is the main Ca2 + requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem. 2009;284:20796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pallos J, Jeng S, McWeeney S, Martin I. Dopamine neuron-specific LRRK2 G2019S effects on gene expression revealed by translatome profiling. Neurobiol Dis. 2021;155:105390–105390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT. Mitochondrial calcium Dysregulation contributes to Dendrite Degeneration mediated by PD/LBD-Associated LRRK2 mutants. J Neuroscience: Official J Soc Neurosci. 2017;37:11151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–7. [DOI] [PubMed] [Google Scholar]
- 56.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4. [DOI] [PubMed] [Google Scholar]
- 57.Drago I, Pizzo P, Pozzan T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011;30:4119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wauters F, Cornelissen T, Imberechts D, Martin S, Koentjoro B, Sue C, Vangheluwe P, Vandenberghe W. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy. 2020;16:203–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papkovskaia TD, Chau K-Y, Inesta-Vaquera F, Papkovsky DB, Healy DG, Nishio K, Staddon J, Duchen MR, Hardy J, Schapira AHV, Cooper JM. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum Mol Genet. 2012;21:4201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, van der Bliek AM, Shackelford DB, Liesa M, Shirihai OS. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019;38:e101056–101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709–17. [DOI] [PubMed] [Google Scholar]
- 62.Glancy B, Kim Y, Katti P, Willingham TB. The functional impact of mitochondrial structure across subcellular scales. Front Physiol. 2020;11:541040–541040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ivannikov Maxim V, Macleod Gregory T. Mitochondrial free Ca2 + levels and their effects on Energy Metabolism in Drosophila Motor nerve terminals. Biophys J. 2013;104:2353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. [DOI] [PubMed] [Google Scholar]
- 66.Mildaziene V, Baniene R, Nauciene Z, Bakker BM, Brown GC, Westerhoff HV, Kholodenko BN. Calcium indirectly increases the control exerted by the Adenine Nucleotide Translocator over 2-Oxoglutarate oxidation in Rat Heart Mitochondria. Arch Biochem Biophys. 1995;324:130–4. [DOI] [PubMed] [Google Scholar]
- 67.Maldonado EN, DeHart DN, Patnaik J, Klatt SC, Gooz MB, Lemasters JJ. ATP/ADP turnover and Import of Glycolytic ATP into Mitochondria in Cancer cells is Independent of the Adenine Nucleotide Translocator. J Biol Chem. 2016;291:19642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williamson MG, Madureira M, McGuinness W, Heon-Roberts R, Mock ED, Naidoo K, Cramb KML, Caiazza M-C, Malpartida AB, Lavelle M, et al. Mitochondrial dysfunction and mitophagy defects in LRRK2-R1441C Parkinson’s disease models. Hum Mol Genet. 2023;32:2808–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hancock CR, Janssen E, Terjung RL. Skeletal muscle contractile performance and ADP accumulation in adenylate kinase-deficient mice. Am J Physiology: Cell Physiol. 2005;288:C1287–97. [DOI] [PubMed] [Google Scholar]
- 70.Joiner M-lA, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, et al. CaMKII determines mitochondrial stress responses in heart. Nat (London). 2012;491:269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cipolletta E, Monaco S, Maione AS, Vitiello L, Campiglia P, Pastore L, Franchini C, Novellino E, Limongelli V, Bayer KU, et al. Calmodulin-dependent kinase II mediates vascular smooth muscle cell proliferation and is potentiated by Extracellular Signal regulated kinase. Endocrinology. 2010;151:2747–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ginnan R, Singer HA. CaM kinase II-dependent activation of tyrosine kinases and ERK1/2 in vascular smooth muscle. Am J Physiology: Cell Physiol. 2002;282:C754–61. [DOI] [PubMed] [Google Scholar]
- 73.Illario M, Monaco S, Cavallo AL, Esposito I, Formisano P, D’Andrea L, Cipolletta E, Trimarco B, Fenzi G, Rossi G, Vitale M. Calcium-calmodulin-dependent kinase II (CaMKII) mediates insulin-stimulated proliferation and glucose uptake. Cell Signal. 2009;21:786–92. [DOI] [PubMed] [Google Scholar]
- 74.Rocha EM, Keeney MT, Di Maio R, De Miranda BR, Greenamyre JT. LRRK2 and idiopathic Parkinson’s disease. Trends Neurosci. 2022;45:224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang EES, Ho PW-L, Liu H-F, Pang SY-Y, Leung C-T, Malki Y, Choi ZY-K, Ramsden DB, Ho S-L. LRRK2 mutant knock-in mouse models: therapeutic relevance in Parkinson’s disease. Transl Neurodegener. 2022;11:10–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ho PW-L, Chang EE-S, Leung C-T, Liu H, Malki Y, Pang SY-Y, Choi ZY-K, Liang Y, Lai WS, Ruan Y, et al. Long-term inhibition of mutant LRRK2 hyper-kinase activity reduced mouse brain α-synuclein oligomers without adverse effects. NPJ Parkinson’s Disease. 2022;8:115–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boecker CA, Goldsmith J, Dou D, Cajka GG, Holzbaur ELF. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr Biol. 2021;31:2140–e21542146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singh F, Prescott AR, Rosewell P, Ball G, Reith AD, Ganley IG. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. ELIFE. 2021;10:e67604–67604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheung ECC, Slack RS. Emerging Role for ERK as a Key Regulator of Neuronal Apoptosis. Science’s STKE 2004, 2004. [DOI] [PubMed]
- 80.Cao Q, Qin L, Huang F, Wang X, Yang L, Shi H, Wu H, Zhang B, Chen Z, Wu X. Amentoflavone protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through PI3K/Akt and ERK signaling pathways. Toxicol Appl Pharmcol. 2017;319:80–90. [DOI] [PubMed] [Google Scholar]
- 81.Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc Natl Acad Sci USA. 2015;112:E1994–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.