

Abstract
Neuroscience research has entered a new phase of key discoveries in the realm of neurogenomics due to strong financial and intellectual support for resource-building and tool development. The previous challenge of tissue heterogeneity has been surmounted by the application of techniques that can profile individual cells at scale. Moreover, the ability to perturb genes, gene regulatory elements, and neuronal activity in a cell type-specific manner has been integrated with gene expression measures to uncover functional underpinnings of the genome at a systems level. While these insights have necessarily been grounded in model systems, now is the opportunity to apply these approaches in humans and human tissue due to further advances in human genetics, brain imaging, and tissue collection. We acknowledge that there will likely always be limits to the extent to which we can apply our genomic tools developed in model systems to human neuroscience; however, as we describe here, the neuroscience field is now primed with the optimal foundation for tackling this ambitious challenge. Importantly, the application of systems-level network analyses to these datasets will facilitate a deeper appreciation of human neurogenomics that cannot otherwise be achieved from directly observable measures.
Introduction
The human brain is considered one of the final frontiers in the biological sciences, and functional genomics and systems biology can provide unique insights into molecular mechanisms at genome and brain-wide scales. Over the last decade, the (Brain Research Through Advancing Innovative Neurotechnologies) BRAIN Initiative and the Human Brain Project (and others; see below) have spurred innovation into the study of basic mechanisms and translational approaches to understand the nervous system. These investments have aided in fast-tracking neuroscience research to catch up with, and arguably surpass, other ongoing long-term research programs such as those in cancer. Since we last discussed the pace of functional genomics and systems biology in neuroscience almost 15 years ago1, there has been remarkable progress in both tool development and discoveries. For example, deep transcriptomic mapping at the cell-type level has been carried out to generate brain atlases as well as detailed brain transcriptomic datasets from hypothesis-testing studies. These cell-type genomic datasets have been linked with circuit mapping or physiological measures and genomic tools have been harnessed to directly manipulate brain circuits or behavior (e.g.,2). However, many of these insights have been derived from and applied to model systems such as rodents or non-human primates. Because we cannot directly experiment on humans in the same manner as in model systems, the direct applicability of many of these findings to the human brain remains unknown. Thus, the neuroscience field needs to be more thoughtful about how we leverage model system data to understand human brain function and consider ways we can use summative network analysis to tease additional meaning from available human snapshot datasets.
As detailed below, our understanding of the human brain is slowly being updated and refined from cell types to circuits to behavior by embracing the windfall of technological resources that have become available in the last decade. Advances in genomics have been integrated with other approaches such as viral tools or electrophysiological measurements in single cells or circuits across brain regions. Foundational improvements in genomics have been derived from increases in the length of reads in sequencing data. For example, longer sequencing reads can be harnessed to determine the impact of gene isoform usage in cell type function in an RNA-sequencing experiment. The cost of sequencing per byte of information has also decreased, yielding greater depth of data for approachable costs viable to single labs. As another example, this has resulted in greater numbers of whole genomes being sequenced with improved insights into a number of brain disorders using genome wide association studies (GWAS)3–5. The isolation of single cells, whether by flow cytometry or droplet-based methods and followed by any number of genomic approaches, has underscored the complexity of the human brain at cellular resolution2,6. These findings include characterization of the heterogeneity of rare cell types such as vascular cells or microglia7,8, cell types or cell states related to disease8–12, the relationship between chromatin state and gene expression at cellular resolution13,14, the identification of human-specialized cell types15–17, developmental lineage information with or without cellular barcoding18–20, and the presence of somatic single nucleotide variants in single neurons21,22.
Unlike model systems or even other tissues in humans, the human brain is challenging to access in living individuals (with some exceptions). Thus, neuroscientists have gone to great lengths to 1) determine the similarities and differences between the human brain and the brains of other mammals, at multiple scales, for the purposes of interpreting modeling results and therapeutic testing; 2) develop ex vivo systems that capture a portion of living human brain tissue; and 3) examine molecular, developmental, and functional properties of human brain cells derived from pluripotent stem cells. Even though neuroscience had to initially play “catch-up” to other fields for the past decade with respect to embracing and applying functional genomics approaches, we believe neuroscience is now at the forefront of all fields with respect to developing and integrating functional genomics into most types of research questions. There is now a strong foundation in basic principles of mammalian brain development and function from a genomics and systems biology perspective that has been discovered in model systems. Moreover, the mainstay of neuroscience, electrophysiology, has been paired with measures of genetic output (optogenetics)23 or genome-wide expression (Patch-seq)24. Brain disease-focused researchers (e.g., in autism and neurodegenerative disorders) have also leaned into applying these genomic approaches in their arsenal of tools. Thus, we believe the next era of functional genomics should be focused directly on the human brain. Ongoing advances in human genetics can be coupled with our growing appreciation of human brain genomics and function. In this perspective, we outline the progress that has been made and where we think there are still exciting opportunities for expansion.
Surveying the transcriptome
In the last decade, high throughput genomics technologies have transformed the data resources that inform neuroscience research, especially with regard to human neuroscience. These technologies have become a mainstay in neuroscience research and across the community, spurred in part by the immense multi-billion dollar investments of the NIH BRAIN Initiative. These efforts across three consortium and dozens of ancillary programs have focused heavily on tool development for the characterization of single-cells within the brain25. Such efforts brought together individuals from across disciplines and enabled the marriage of multiple modalities in the characterization of the brain. Both individual and multiomic measurements have expanded our understanding of how cell type-specific properties are correlated to one another14,25–30 through studies mostly focused on mouse brain atlasing. Through these efforts, we now understand that snapshot measurements using conventional single-cell transcriptional approaches can capture much of the cell type and functional diversity that exists within the brain. Across human samples from both the normal brain and in the contexts of brain diseases and disorders, these datasets are transformation in their ability to provide a reference (Box 1) for hypothesis generation, validation of model system experiments, and development of new analytical methods. However, important details of brain function can only be captured by integrating information between modalities. The takeaways from these human and model system efforts highlight 1) electrophysiological and morphological features cannot be fully predicted by transcriptomics26, 2) spatial context allows for more heterogeneity than transcriptional analysis alone31, and 3) epigenetic profiling provides essential nuance, especially when considering state transitions or development14.
BOX 1. Relevant Human Single-Cell Dataset Repositories.
Key examples of published human brain single cell datasets together with the location and type of data.
| Dataset | Repository | Type(s) of Data | Sample Publications |
|---|---|---|---|
| Human Brain Development | NeMO, GEO, Individual Archives | sc/snRNA-seq, scATAC-seq | Nowakowski et al 201769, Fan et al 2018111, Zhong et al 2018112, Polioudakis et al. 2019113, Fan et al 2020114, Ziffra et al 202114, Eze et al 2021115, Bhaduri et al 20216, Smith et al 2021116, Ramos et al 2022117, Braun et al 2022118, Herring et al 2022119, Cameron et al 2023120 |
| Adult Human Brain | Allen Brain Atlas data portal, GEO, NeMO | snRNA-seq, snATAC-seq | Hodge et al 201972, Krienen et al 2020121, Bakken et al 2021122, Ma et al 202216, Caglayan et al 202317 |
| Neurodevelopmental and Neuropsychiatric Disorders | PsychENCODE Knowledge Portal, GEO | sc/snRNA-seq | Wang et al 201836, Velmeshev et al 201912, Gandal et al 202210 |
| Alzheimer’s Disease | Alzheimer’s Disease Knowledge Portal (subset of Synapse) | Bulk profiles and snRNA-seq | Hodes et al 2016123, Mathys et al 201911 |
| Other Neurodegenerative Disorders | dbGAP, GEO | snRNA-seq | Tryka et al 201438, Schirmer et al 2019124, Bressan et al 2023125 |
| Interactive Browsers | UCSC Cell Browser, CellXGene | sc/snRNA-seq, scATAC-seq, others | Speir et al 202135, Tabula Sapiens Consortium et al 202237 |
In human samples, beginning with bulk transcriptomics, characterizations of steady state expression have been foundational in understanding the brain and the spinal cord, highlighting important gene programs activated across brain regions and during development. While the nature of how atlases are generated has evolved, the initial RNA-based ISH and microarray atlases of the adult and developing human brain from the Allen Brain Institute32,33 are still widely and effectively used as a reference for benchmarking studies across human analyses and systems. With the advent of single-cell approaches, this concept of an atlas has expanded immensely. Efforts from the BRAIN Initiative, the Human Cell Atlas, the CZI Tabula Sapiens, and other initiatives by individual labs have begun to coalesce on a transcriptomic definition of how many cell types likely exist in the mouse and human brains, and the newest efforts are achieving these analyses in the context of spatial cell type distributions as well34. These data are collected in repositories such as dbGAP, the NeMO archives, CellxGene browsers, Synapse or the UCSC Cell Browser35–38 and are detailed further in Box 1.
Ultimately, these studies of steady state expression rely entirely on transcriptomic measurements and have not yet fully expanded to the other measurement modalities being used in rodent systems. Some of these challenges derive from more complicated methodologies being used in human samples, such as the notable phenomenon of lipofuscin in adult human brains when trying to implement spatial approaches34. Others, such as electrophysiological measurements, require access to specialized samples and long-term culture conditions39,40 to enable relevant characterizations.
As a result, existing surveys of human cells have been highly transcriptome-centric, with some measurements of open chromatin or methylation state41,42. While these measurements in the single-cell space have generally risen in quality, making broad cell typing possible, there remain limitations regarding the utility of these datasets. For example, the advent of single-nuclei sequencing43 has enabled study of hard to dissociate tissues, especially adult brain samples and banked human specimens. Yet, this approach (as well as single cell profiling) can still suffer from excessive ambient RNA content, a phenomenon that releases cytoplasmic RNA into the cell suspension prior to analysis, impeding the accuracy of the single-cell nature of the experiment44. These issues have been particularly challenging when studying glial ratios in samples, because these populations have an increased probability of mis-typing due to their naturally lower numbers of expressed genes. Additionally, even when the modalities represent samples well, gaps in terms of brain regions, developmental stages, or certain disease states exist across model organisms. These gaps are especially pronounced in the context of human dataset acquisition where sample availability and scope of existing consortia have not yet yielded comprehensive cell type atlases.
The ongoing consortia efforts pursuing atlases of the human brain present an interesting paradox to researchers working in spaces that would benefit from these atlases of the normal or diseased brain. The resources and distribution methods of consortia can be hypothesis generating and incredibly useful to individual labs, but the missing data points across regions, time points, and disease states can diminish the utility of current human datasets. However, the cost and the chance to be usurped by larger groups decreases the incentive and the opportunity for smaller groups to pursue the filling of these gaps. In the field, transparency about the planned scope of large institutes and consortium efforts paired with advances in lower cost technologies for sampling single-cell profiles would help fill these gaps more efficiently. Importantly, researchers studying human biology and disorders are the most well positioned to vet and optimize data generated from these technologically challenging approaches and are well positioned to plug the holes in terms of “missing” datasets and modalities related to human neuroscience.
Characterizing the Synapse with Omics
The nervous system is uniquely characterized by complex information flow through synapses, junctions between neurons and sometimes other cell types. The human brain is thought to contain on the order of 600 trillion to a quadrillion synapses45, and synaptic dysfunction or even minor dysregulation is implicated in numerous developmental, psychiatric, and neurodegenerative disorders. Importantly, compared to other species, humans have more synapses per neuron46. Thus, tools to study synaptic biology are necessary to truly understand brain biology. Moreover, in light of the thousands of cell types being identified from the atlas efforts described above, the characterization of cell type-specific synaptic interactions with other populations is a tantalizing link between phenotypes, behavior, and steady state measurements. Transient synaptic connections are a hallmark of key developmental processes as well, notably between the thalamocortical afferents and transient cell types and substructure of the developing cortex for example47,48, establishing the study of synapses via genomic biology a unique and essential feature of next generation neuroscience.
Notable progress has been made in this sphere. Bulk proteomic characterizations have been generated from various species and across disease contexts, especially in the context of normal brains and neurodegenerative diseases. However, recent work is also emphasizing the interesting heterogeneity to be found between cell type-specific proteomic studies of the synaptic proteome. Given the challenges of measuring electrophysiological properties accurately in human tissues, studies of synapse biology at the bulk or single cell level provide a unique glimpse into human-specialized processes from preserved tissues. For example, recent efforts exploring proteomics of synapses during development highlighted potential insight into mechanisms of neoteny in humans49. In the context of aging and neurodegeneration, profiles have identified disease associated synaptic proteins in ALS50 and Alzheimer’s disease51, as well as synaptic proteins subject to age-related changes52 or even those that seem to be resistant to these types of degradation52,53.
Synapses present a unique challenge, because not only is there a large number of synapses in the human brain, but the specific properties of these synapses are likely quite diverse. Given the size of the human brain, synapses are often located far from the cell body and associating a synapse with the originating cell is incredibly challenging. New tools to characterize single-cell gene expression of synapses with droplet-based capture approaches54 are beginning to change our ability to understand the composition and heterogeneity of synapses as well as how they are dysregulated in disease states such as Alzheimer’s. However, these approaches do not solve the question of what cell type the interacting cells are and are also somewhat controversial regarding whether they accurately measure synapses55,56. Thus, additional innovations in this space are required to fully understand the heterogeneity of synapses within the human brain. Other strategies to interrogate this problem are emerging, for example, recent innovations in capturing single dendrites highlighted key properties of local translation in rat neurons57. Importantly, these results are highlighting the opportunities to gain information from sub-cellular analyses that move beyond nuclear transcripts54, emphasizing the need to engage in more comprehensive synaptic single-cell analysis, especially because this is accessible in the human context.
Beyond the contents of what exists in a synapse specifically, understanding where this information is being shared has important implications for how individual cell types communicate with cells from different regions. This will enable a better understanding of how single cells can contribute to circuits that ultimately underlie behavior and may be dysregulated in cases of disease. Early pioneering efforts in rodents to characterize this important link between cells and function harnessed the unique properties of rabies viruses that infect the nervous system and propagates via retrograde axonal transport58. Engineered modifications of this system eliminated the extreme impacts of the virus and instead allowed for targeted tracing of an individual, genetically labeled cell, usually from the rodent brain59. As single-cell technologies have emerged, these types of approaches can now be paired with single-cell analysis enabling linkage between an individual cell and the cells it communicates with. Use of these approaches in neurons and between glial populations have expanded our understanding of how presynaptic networks are structured and what proportion of interaction partners belong to specific classes60, including identifying a role for axon guidance molecules in glial interactions61. These approaches are now being extended to the spatial context, integrating both cell type and connection location with in situ sequencing on brains previously infected with custom synaptic tracers62. Parallel tools with some cell specificity are adeno-associated viral strategies that label cell types of interest63 and can be used in ex vivo slices to map projections and morphologies64. However, an important caveat of these tracing tools in the context of human biology is that they can only be applied to slices of live tissue, limiting their utility to intra-cortical relationships, for example. These tools, while still evolving rapidly, extend the ability to fully understand synaptic function but thus far require manipulatable in vivo systems for full scale characterization, such as model organisms ranging from rodents to non-human primates. However, human neurons are characterized by a number of unique properties including decreased intrinsic excitability, increased apical dendrite length, and more circuit compartmentalization65. Given the challenges of directly assessing these properties in human explant cultures, progress in the ability to utilize xenotransplantation of human pluripotent stem cell derived developing neurons into mouse brains is an exciting approach to explore the development of human neuronal morphology and circuits66,67. Using these tools, it has been observed that human neurons mature within the rodent brain but intrinsically maintain their neoteny (prolonged developmental timeline)65; these types of experimental models are promising systems in which to apply the expanding toolkit of single-cell approaches to study synaptic biology. To derive value from these tools directly in the context of human biology, it is imperative to adapt these tools to live postmortem culture models of human tissue, explore their utility in in vitro human model systems, and connect the tracing findings from rodent models to other measures of gene expression and synaptic proteomes. These adaptations will determine whether any of these characteristics can be inferred from measures that are acceptable in the human context.
Moving from lists to networks
As detailed above, technological improvements together with consortia and initiatives have resulted in vast amounts of genomic data, much of which are relevant to the human brain. The efforts have yielded catalogs of genes that are associated with cell types, developmental trajectories, and/or disease status. However, one of the greatest challenges posed by these datasets is determining how to consolidate and prioritize the information. In other words, which genes are most crucial for a particular developmental process, phenotype, or disease? Since a single gene cannot determine a specific cell type, we need to consider genes in groups or networks. In the era of bulk tissue RNA-sequencing, this challenge was facilitated by the use of approaches to mine the relationships among patterns of gene expression in a way that was agnostic to other known genomic or biological information. Weighted gene co-expression network analysis (WGCNA) is a popular approach to systematize brain genomic datasets1. This method groups genes into modules based on their patterns of co-expression. Within each module, the genes that appear to be most linked to the pattern of co-expression can be prioritized as hub genes. It is worth noting that when starting with a bulk genomics dataset, WGCNA is useful for pulling out co-expression signatures related to cell types68. For single cell datasets, WGCNA is useful to apply to already defined major cell types or to datasets with dynamic cell type-specific expression, as in the case of a developmental dataset (Figure 1). For example, in the developing human brain where dynamic patterns of gene expression make defining subtypes of cells challenging, modules can be generated by cell type then re-clustered to obtain cleaner subclusters69. In other studies of human primary tissue and/or organoids, WGCNA has been applied to observe how defined networks change across developmental time or between species70,71. The use of WGCNA and related co-expression methods will be key to apply to the realm of underexplored human brain regions and disease states. The re-invigoration of gene networks into the human neurogenomics field could provide additional color into how biological processes are not necessarily always completely parallel to cell type. We would like to note, however, that while WGCNA is still relevant in the era of single cell modalities, application of WGCNA to single cell datasets may be limited when cell type heterogeneity is already apparent and defined. Moreover, because single cell datasets tend to be more sparse with many zeros (whether due to technical or biological reasons), the implementation of WGCNA may be challenging due to sparsity or the number of data points. Sparsity can be addressed by deeper profiling per cell type (e.g. with SMART-seq or Fluidigm)15,72, imputation of missing data points or pseudobulking values for genes across cell types73,74.
Figure 1: Mining human brain single cell datasets to infer regulatory mechanisms.
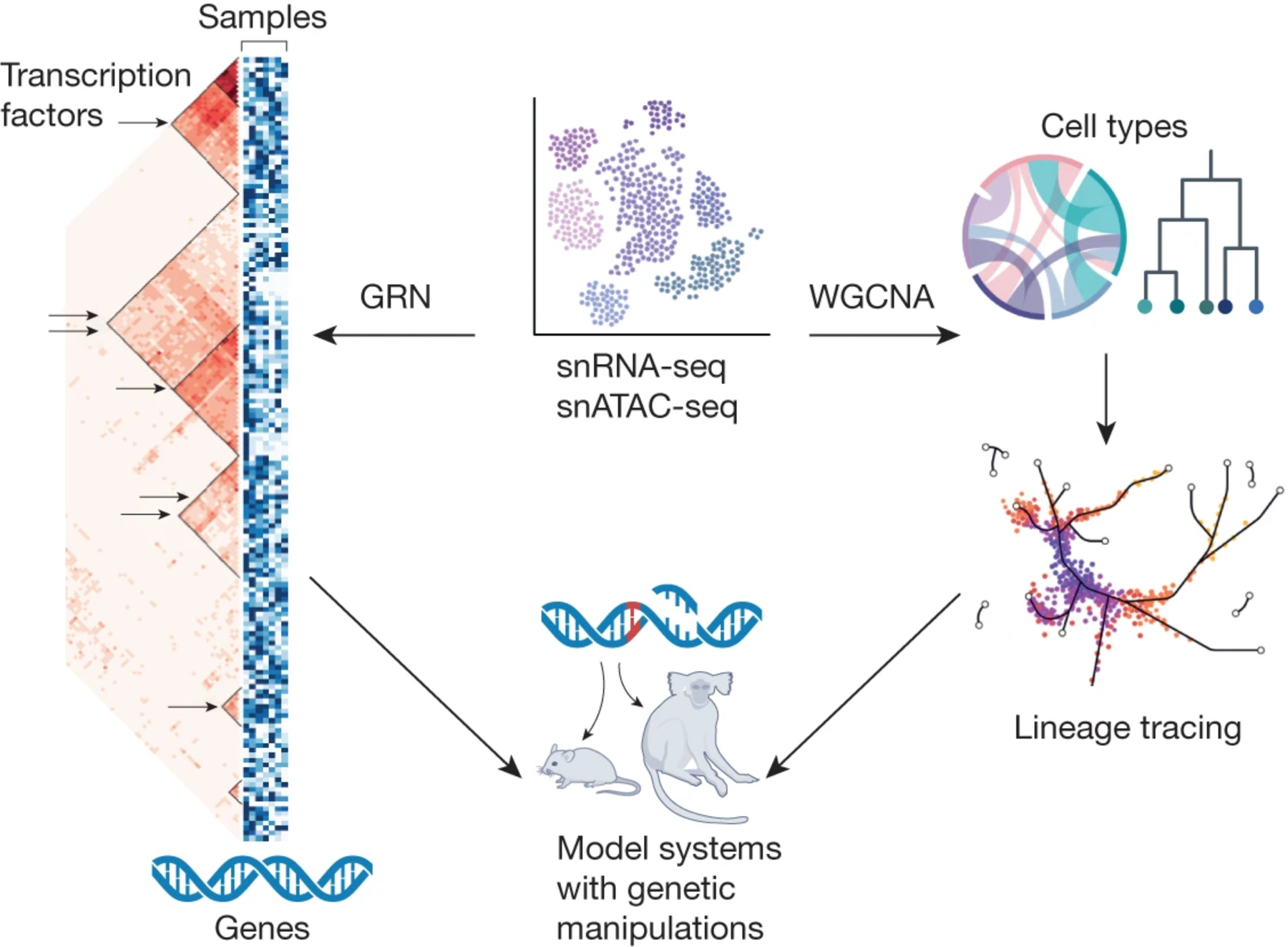
Human brain tissue can be queried for genomic information at the RNA and chromatin level. However, these datasets require further applied analysis to understand the dynamic nature of the datasets. WGCNA can be used to understand cell type composition and contributions as well as the dynamic nature of cell type development. Gene regulatory networks (GRN) can be applied to infer which transcription factors (TFs) might be important for co-regulation of sets of genes in a given cell type. Since these regulatory mechanisms cannot be directly tested in vivo in human, they can instead be tested in either human brain slice culture or model systems (e.g., rodent or monkey) using CRISPR-based tools.
Other methods such as gene regulatory networks (GRN)75 are therefore important and needed to mine single cell datasets to determine functional relevance. At its simplest level, implementation of GRN essentially identifies the genes regulated by transcription factors and builds this out at scale into a network. In a scRNA-seq dataset, this translates to determining transcription factor motifs at cell-type resolution (Figure 1). Thus, one can graph putative co-regulated (via the same transcription factor) genes in a cell type-specific manner. GRN can not only be applied to scRNA-seq but also to single cell chromatin data such as snATAC-seq. The layering and integration of both RNA and chromatin datasets is key for building functional networks, especially in human brain datasets, since perturbations to test functional outcomes are limited. The combination of RNA and chromatin at the cell type level facilitates identifying genes and regulatory mechanisms (via motif and variant mining in areas of the genome that are differential across brain states (development, disease, etc.)) and correspond to changes in gene expression. While applying some of the latest GRN methods to multimodal datasets has been shown to improve the predictive power of the GRN approach overall (e.g., MIRA, SCENIC+, and Dictys), there are still limitations even to these recent, benchmarked approaches in that some types of data such as validated TF binding sites or enhancer-promoter links are not sufficiently observed76. One of the important challenges for all these approaches with respect to human datasets, is the relatively constrained ability to confirm any of the outcomes. Both WGCNA and GRN are predictive rather than definitive of causation. In other words, these are hypothesis-generating approaches that need to be independently validated. For GRN-generated TF networks, it might be possible to validate the target genes of a specific TF using cell type specific transposase and tagmentation approaches such as single cell CUT&Tag77. We envision such approaches could adapt the use of antibodies to TFs (compared to validated antibodies for histone modifiers) and apply the approach to human brain samples.
Beyond frequently used approaches such as WGCNA and GRN, additional methods are emerging to mine multimodal genomic datasets. A causal inference approach, CoCoA-diff, has been applied to single cell RNA-seq data from Alzheimer’s disease brain tissue, facilitating the prioritization of cell type specific genes that are likely to be drivers of disease state78. One recent machine learning approach that included human brain tissue is the Geneformer tool79. Single cell RNA-seq data from human brain tissue was included within the large training dataset Geneformer used, and the networks predicted the contribution of copy number variants (i.e., dosage) of developmental disease genes with higher accuracy in neurons compared to random cells from throughout the body. There is much excitement about applying artificial intelligence approaches such as deep learning to single cell transcriptomics80. However, there are still challenges (in general81) and unknowns in how such tools might predict functional networks in a system like the human brain that is not amenable to perturbation except in select ways.
Hypothesis testing of integrated datasets can be carried in human datasets via in silico approaches such as CellOracle82 or genomic perturbation methods such as PerturbSeq83 or massively parallel reporter assays (MPRAs)84,85. These manipulations in a human context, however, require accessible models of the human brain and either immense scale in terms of cell number or limited gene sets because of the number of cells per guide RNA required for robust conclusions. These types of manipulations are the ideal scenario in which stem cell-derived models of human neurons or organoid systems of normal development or neurodevelopmental disorders can enable relevant perturbations. However, because of the in vitro nature of these models, benchmarking existing datasets from primary samples and including ample cell line and technical replicates is essential to ensuring that the phenotypes observed are not artifacts of the system.
Finally, many studies, especially those funded through BRAIN-initiative grants, are tasked with setting goals across the modalities of RNA and chromatin as already mentioned, but now also include spatial approaches due to available technology. Since single cell approaches necessarily dissociate tissue to profile individual cells, the implementation of spatial transcriptomics can be used to either independently generate or validate cellular genomic datasets across human brain subregions (e.g., a human cortical section)86. A limitation of human spatial transcriptomics is the sheer size of the human brain relative to the imaging window. However, one could imagine tiling together datasets across larger regions of the human brain to make key spatially-relevant insights. Another consideration is the sheer amount of imaging time that might be needed (and computationally intensive analysis) to cover large sections of the human brain. However, as these hurdles are surmounted, spatial transcriptomics could be scaled to slice cultures of larger areas of human brain and integrated with additional approaches that interrogate functional measures such as connectivity and viral gene manipulations, further bridging the genotype to phenotype gap.
Integrating genetic and phenotypic data
An ultimate goal of studying the human brain is to piece together the mechanisms that underlie our behavior, particularly with regard to those traits that distinguish us from other species such as language. One way to achieve this is by profiling genetic and genomic information across a wide range of individuals with varying phenotypes. This can be achieved by comparing datasets from neurotypical individuals with those who have brain disorders with distinct behavioral symptoms such as neurodevelopmental (e.g., autism spectrum disorder (ASD) or schizophrenia) or neurodegenerative (e.g., Alzheimer disease (AD)) disorders). Since it is not feasible to perturb humans genetically like we do in model systems, we can instead compare data from neurotypical individuals to those with disease to infer correlations between genes and behavior in the human brain (Figure 2A). These studies necessarily either use peripheral tissues such as blood or saliva (for DNA variants) in living individuals or postmortem tissues for brain access. While DNA polymorphism and copy number variant information have revolutionized our understanding of the contribution of both rare and common variants to risk for these brain diseases87–89, it is still not entirely clear how DNA alterations in these risk factors impact brain cells in a functional manner. In other words, how can we infer causality from DNA all the way to behavior/disease via cells and circuits? This is a significant gap in knowledge that the neurogenomics field should address over the next decade. In addition, we are challenged to identify how developmental pressures and/or brain activity intersect with genetic risk in the absence of datasets derived directly from brain tissue. Thus, researchers need to be innovative in how postmortem brain tissue can be harnessed to understand functional outcomes. One logistical challenge in leveraging postmortem tissues is that humans have diverse lifestyles and demographics, and thus available datasets are not always well matched (e.g., by age, other demographics, drug or treatment conditions, or brain region). Large scale datasets can mitigate some of these issues. For example, the UK Biobank dataset currently contains genetic data from hundreds of thousands of individuals with corresponding demographic information as well as some phenotypic information and brain imaging from tens of thousands of individuals90. These datasets have been used to make a number of important insights into how genetic variants correlate with both structural and functional brain measures and mining these datasets has identified specific genes that may underlie these phenotypes as well as link to particular brain disorders91. One disappointing outcome from these and related studies has been the inability to identify significant genetic signals that correspond to task-based fMRI measures92, in other words, the genetics underlying human behavior. It is not clear if further increases in sample size or refinement of imaging methods or behavioral tasks will alter this outcome.
Figure 2: Integrative approaches to use human brain tissue to uncover multimodal systems related to genomics.
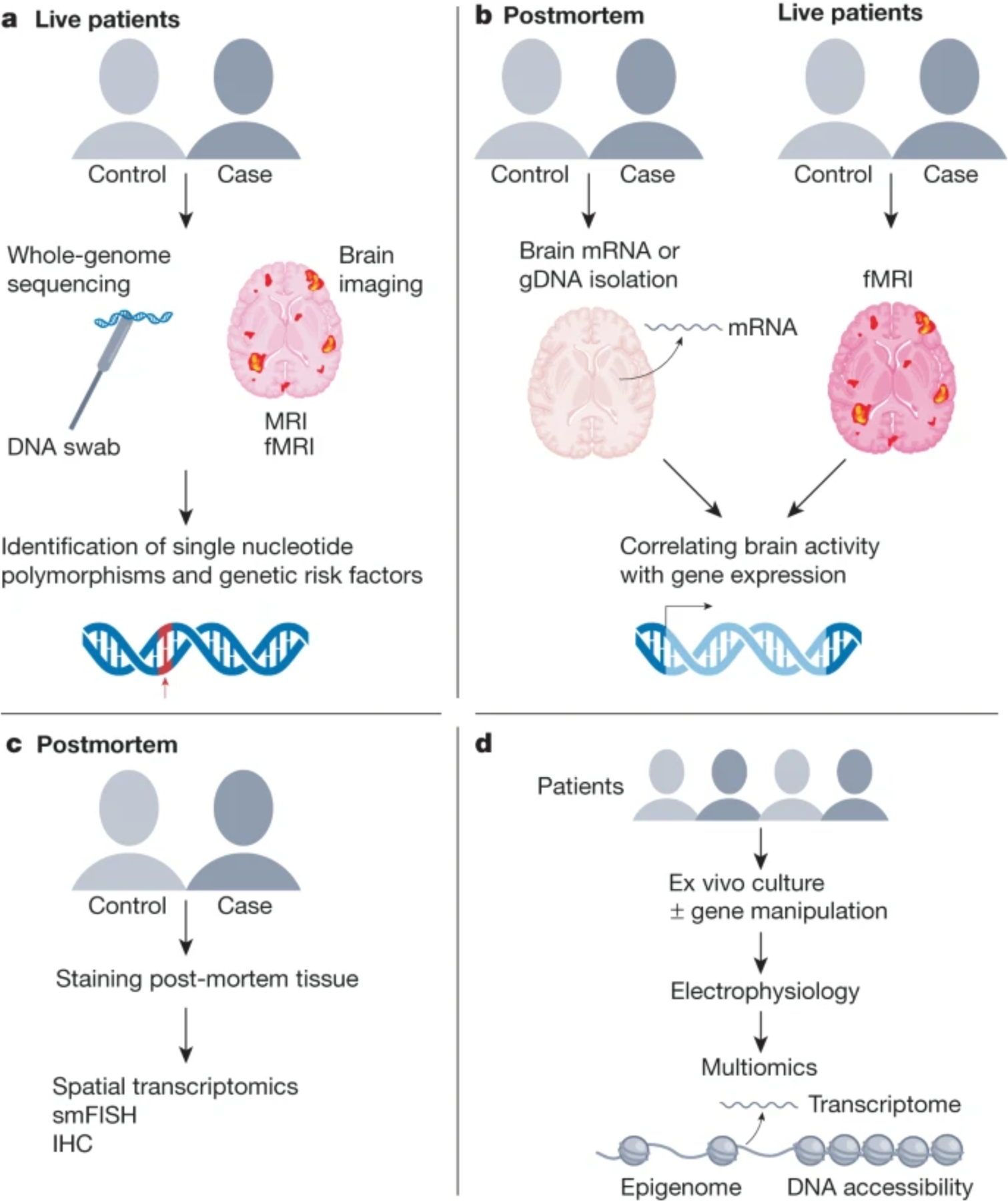
A) Brain imaging can be combined with peripheral DNA profiling to infer genetic variants that may underlie brain size or function. B) Brain gene expression and chromatin state data from post-mortem tissues can be integrated with functional MRI measures to understand how gene expression patterns may underlie brain activity. C) Human post-mortem tissues can be used to confirm findings from all other approaches. D) Ex vivo brain slice cultures can be acquired from surgical patients and use to integrate physiological and gene expression measures from the same individual.
Another recent innovation is the integration of human brain imaging with postmortem brain gene expression datasets (Figure 2B). In these comparisons, the datasets are derived from two different cohorts of people but the relatively stability of these measures across neurotypical populations has led to key insights about genomic underpinnings of brain morphology, size, lamination, and cell types93 as well as functional resting state measures94,95. These functional imaging datasets typically rely upon large repositories of imaged cohorts such as from the IMAGEN or Human Connectome Project for neurotypical individuals or disease relevant consortia such as ABIDE for ASD. Many of these integrative studies also use RNA-seq data from the human brain atlas generated by the Allen Brain Institute94, but an increasing number of studies are beginning to add additional human postmortem RNA-seq datasets95,96. Moreover, the inclusion of imaging and/or genomic datasets from individuals with a number of disorders such as depression, schizophrenia, ASD, and other neurodevelopmental disorders has resulted in the identification of genes that have patterns of expression that vary in a disease context96–99. Interestingly, these are not necessarily the same genes that are associated with genetic variant risk, suggesting a complex interplay of genetic risk and functional genomic outcomes. Beyond the use of data from disease cohorts, there are efforts to correlate genomics and brain imaging with specific behavioral repertoires relevant to humans such as language100. All these studies have laid the groundwork for human brain gene prioritization as well as insights into how groups of genes correlate together with respect to a number of brain functions or disease states. A caveat to these studies is that the postmortem tissue data are derived from separate cohorts of individuals than those who underwent imaging. Therefore, the demographics of each population can be challenging to match especially if disease or developmental datasets are considered.
Another important source of gene expression datasets (e.g., RNA-seq and/or ATAC-seq) derived from human brain tissue is surgically resected brain tissue. The use of these tissues precludes issues surrounding independent group correlations as this tissue can be obtained from individuals who undergo prior brain imaging (structural or functional), intracranial stimulation or electrophysiological measures such as electroencephalograms (EEG) (Figure 2D). For example, a within-subjects study identified the gene expression patterns associated with specific oscillatory activity during a memory recall task, uncovering specific genes that are likely important for normal human memory function101. Of course, a major caveat to the use of surgically resected tissue is that all the individuals providing tissue undergo operations for a medical condition such as medication-resistant epilepsy or cancer. The use of rapid autopsy tissue could fill a gap with respect to the availability of tissue from individuals with non-brain disease and access to the entire brain rather than where surgery dictates. Regardless, neither surgical nor rapid autopsy sources are likely to provide an abundant resource for brain tissue for genomics from individuals with cognitive disorders like ASD or AD. Therefore, between group correlations need to be made for integrating genomics with brain function in these cases.
At the cell type level, biophysical phenotypes can be integrated with genomic measures through approaches such as Patch-seq in which individual cells undergo patch clamp measurements followed by scRNA-seq24. Patch-seq has been used at scale to compare human and mouse cortical cells, identifying features that have been modified in human brain cells and might be at risk in certain disorders2. These efforts could be further expanded by manipulating disease-relevant genes or cell types in organotypic slice culture derived from surgical resections and/or rapid autopsy brain tissues. Human brain slice cultures can be kept viable for several weeks and are amenable to viral transduction40,102. Again, applying these techniques to brain tissue from individuals with specific brain disorders like ASD or AD would be limited. But high-quality gene expression and regulation measures from postmortem tissues can be obtained, and information applied to physiological studies such as key disease-relevant gene manipulation in the organotypic slice culture.
Importantly, for all these integrated studies, detailed phenotypic information related to the donor of the tissues is key in order to correlate genomic features with either demographics, disease-relevant phenotypes, or technical considerations. Coordinated brain banks that have high standards of best practices are making these tissues readily available to qualified researchers (e.g., NIH NeuroBioBank for control as well as several types of brain disorders and Simons Foundation BrainNet for ASD and control tissue). A challenge for these brain banks is to collect enough samples across the developmental and age-related epochs that are impacted by disease state. Even though the post-mortem tissue samples will reflect steady-state levels of expression, the use of Hi-C and ATAC approaches have already given insight into some aspects of molecular/chromatin features that are dynamic13,36,103–105.
An option to fill in any missing knowledge of how DNA variants lead to alterations in behavior, via changes in cell type expression patterns or brain circuits in humans, is to harness the power of non-human primate (NHP) models. NHP models that are ethically amenable to gene manipulation (e.g., marmoset or macaque) are still evolutionarily distant from humans and harbor many genomic differences at the cell type level16,17. However, their brain gene expression patterns and circuits also share many features with the human brain25. Thus, each model system such be selected based on the specific hypothesis that relates to human behavior. For example, a songbird model could be ideal for a study of vocal learning over even great apes, which do not have observable vocal learning.
Human data sharing
As the genomic tools to study neuroscience improve and expand in scale to address technical challenges associated with human samples, the number of studies utilizing donated surgical, biopsied, or postmortem tissues is expanding rapidly. Numerous organizations, including funding agencies such as the NIH are simultaneously pushing for increasingly open access to this data, including the raw sequencing reads. Scientifically this is reasonable because tools for alignment, parsing isoforms, and other applications to improve differential gene expression are improving and expanding while traditional repositories such as dbGAP or SYNAPSE are onerous to navigate bureaucratically. More streamlined access would allow speed and efficiency for tools that are yet to be conceptualized, much less developed.
However, as the tools to utilize these raw sequences improve, so also do the technologies to infer identifiable characteristics within these data. For example, while short read, low coverage 3’ enriched sequencing once was impossible to use for the identification of SNPs, tools and models now make relatively high confidence SNP calling in these sequencing datasets feasible and have been used for scientifically exciting applications such as demultiplexing identity in sequencing runs106, tracing potential lineage relationships between cells107,108, or identifying tumor subclones in single-cell studies109.
These same tools, when paired with the expanding repository of human sequencing in scientific and social contexts, are quickly approaching adaptability for identifying not only individuals but also their kin. This poses ethical risks with regards to most consent forms where deidentification is a condition of the research use of a sample and increasing societal risks to individuals and their relatives. Spelling these risk factors out directly (Figure 3) is not alarmist, but rather is specific in terms of the harms that can arise when sequencing data is not fully protected; more comprehensive access must come with legal protections against these harms. The considerations on family are particularly salient when dealing with postmortem tissues that are archived with older consent forms; not only might the research uses extend beyond those originally planned, but new technologies may expand the impact of identifying previously unknown genetic risk factors to surviving relatives who may not have been consulted during the donation process.
Thus, we as scientists and the broader community must consider what types of firewalls to protect data and community guidelines of scientific conduct must be maintained when housing, sharing, using, or publishing these sensitive data types. Additionally, when thinking about what aspects of the data or metadata will be shared, protections for future potential uses of this data must be considered in policy and scientific decision making now.
Conclusions and future directions
Data integration across modalities remains a challenging frontier in human brain genomics. However, as access to human brain tissues with associated phenotypic information increases, the gap between genes and behavior should narrow. Advances in chemistry and brain imaging may facilitate mechanisms to query gene expression in vivo. Certainly, the possibility to image the activity of a living human brain at single cell resolution might be achievable110 and resultant data could be integrated with independent genomic measures.
Most importantly, the neuroscience community has come a long way from reluctant use of genomic methods and hypothesis-generating approaches to embracing technology that profiles the brain agnostically at cellular resolution or integrates electrophysiological measures with gene expression. While significant progress has been achieved in these domains within model systems, in the next decade, neuroscience research is certain to push the boundaries of what is possible in observing and manipulating human brains. These new approaches will enable additional characterization of human biology, and existing integrative analysis approaches overviewed here can continue to drive cohesive biological understanding of existing and emerging datasets. The functional understanding of how genes influence the human brain across scales from cells to circuits will usher in a new era of neuromodulation and gene manipulations. Thus, we propose that a continuation of efforts to delve into genomic features of the human brain and how these features vary across cells, circuits, and behaviors will be rewarded with a rich appreciation of how the human brain evolved, develops, functions, and dysfunctions. Determining how these genetic features directly affect behavior via cells and circuits is a challenging feat in the human brain that may require new technological advances. These achievements will not only enhance how we understand human behavior but also have therapeutic value.
Acknowledgements
Apologies to all the authors whose work we were unable to cite due to space limitations. We would like to thank the Bhaduri and Konopka labs for feedback on the manuscript and Rachael Vollmer for assistance with Figures 1 and 2. The work in the Bhaduri lab is supported by NIMH (UM1MH130991, R00NS111731), Klingenstein-Simons Neuroscience Fellowship, Brain Behavior & Research Young Investigator Award, and The Alfred P. Sloan Foundation Fellowship. G.K. is a Jon Heighten Scholar in Autism Research and Townsend Distinguished Chair in Research on Autism Spectrum Disorders at UT Southwestern. The work in the Konopka lab is supported by NIMH (MH126481), NINDS (NS126143, NS115821), NHGRI (HG011641), the Simons Foundation (947591), and the James S. McDonnell Foundation 21st Century Science Initiative in Understanding Human Cognition – Scholar Award (220020467).
Footnotes
Competing interests: The authors declare no competing interests.
REFERENCES
- 1.Geschwind DH & Konopka G Neuroscience in the era of functional genomics and systems biology. Nature 461, 908–915 (2009). 10.1038/nature08537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg J et al. Human neocortical expansion involves glutamatergic neuron diversification. Nature 598, 151–158 (2021). 10.1038/s41586-021-03813-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halvorsen M et al. Increased burden of ultra-rare structural variants localizing to boundaries of topologically associated domains in schizophrenia. Nat Commun 11, 1842 (2020). 10.1038/s41467-020-15707-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders SJ et al. Whole genome sequencing in psychiatric disorders: the WGSPD consortium. Nat Neurosci 20, 1661–1668 (2017). 10.1038/s41593-017-0017-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trost B et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 185, 4409–4427 e4418 (2022). 10.1016/j.cell.2022.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhaduri A et al. An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204 (2021). 10.1038/s41586-021-03910-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Healy LM, Zia S & Plemel JR Towards a definition of microglia heterogeneity. Commun Biol 5, 1114 (2022). 10.1038/s42003-022-04081-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang AC et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892 (2022). 10.1038/s41586-021-04369-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batiuk MY et al. Upper cortical layer-driven network impairment in schizophrenia. Sci Adv 8, eabn8367 (2022). 10.1126/sciadv.abn8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandal MJ et al. Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD. Nature 611, 532–539 (2022). 10.1038/s41586-022-05377-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathys H et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velmeshev D et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019). 10.1126/science.aav8130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morabito S et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet 53, 1143–1155 (2021). 10.1038/s41588-021-00894-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziffra RS et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature 598, 205–213 (2021). 10.1038/s41586-021-03209-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boldog E et al. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat Neurosci 21, 1185–1195 (2018). 10.1038/s41593-018-0205-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Ma S et al. Molecular and cellular evolution of the primate dorsolateral prefrontal cortex. Science 377, eabo7257 (2022). 10.1126/science.abo7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Caglayan E et al. Molecular features driving cellular complexity of human brain evolution. Nature 620, 145–153 (2023). 10.1038/s41586-023-06338-4 [DOI] [PMC free article] [PubMed] [Google Scholar]; These studies (Ma et al., Caglayan et al.) carry out comparative single cell transcriptiomics of human and NHP brain to uncover human cellular innovations including cell type-specific changes related to FOXP2, a key gene in brain development and disease.
- 18.Allen DE et al. Fate mapping of neural stem cell niches reveals distinct origins of human cortical astrocytes. Science 376, 1441–1446 (2022). 10.1126/science.abm5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delgado RN et al. Individual human cortical progenitors can produce excitatory and inhibitory neurons. Nature 601, 397–403 (2022). 10.1038/s41586-021-04230-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You Z et al. Mapping of clonal lineages across developmental stages in human neural differentiation. Cell Stem Cell 30, 473–487 e479 (2023). 10.1016/j.stem.2023.02.007 [DOI] [PubMed] [Google Scholar]
- 21.Chung C et al. Comprehensive multi-omic profiling of somatic mutations in malformations of cortical development. Nat Genet 55, 209–220 (2023). 10.1038/s41588-022-01276-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller MB et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 604, 714–722 (2022). 10.1038/s41586-022-04640-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim CK, Adhikari A & Deisseroth K Integration of optogenetics with complementary methodologies in systems neuroscience. Nat Rev Neurosci 18, 222–235 (2017). 10.1038/nrn.2017.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24*.Cadwell CR et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat Biotechnol 34, 199–203 (2016). 10.1038/nbt.3445 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents the utility of a novel method to simultaneously profile electrophysiological activity and transcriptional identity of neurons.
- 25*.Network, B. I. C. C. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 598, 86–102 (2021). 10.1038/s41586-021-03950-0 [DOI] [PMC free article] [PubMed] [Google Scholar]; The Brain Initiative Cell Cenusus Network assembled single-cell tools and multiomic profiling to enable the study of cell types in mammalian systems, with some of the first atlas resources of the human brain.
- 26.Scala F et al. Phenotypic variation of transcriptomic cell types in mouse motor cortex. Nature 598, 144–150 (2021). 10.1038/s41586-020-2907-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang M et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature 598, 137–143 (2021). 10.1038/s41586-021-03705-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munoz-Castaneda R et al. Cellular anatomy of the mouse primary motor cortex. Nature 598, 159–166 (2021). 10.1038/s41586-021-03970-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z et al. Epigenomic diversity of cortical projection neurons in the mouse brain. Nature 598, 167–173 (2021). 10.1038/s41586-021-03223-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joglekar A et al. A spatially resolved brain region- and cell type-specific isoform atlas of the postnatal mouse brain. Nat Commun 12, 463 (2021). 10.1038/s41467-020-20343-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Codeluppi S et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods 15, 932–935 (2018). 10.1038/s41592-018-0175-z [DOI] [PubMed] [Google Scholar]
- 32.Ding SL et al. Comprehensive cellular-resolution atlas of the adult human brain. J Comp Neurol 524, 3127–3481 (2016). 10.1002/cne.24080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller JA et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). 10.1038/nature13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Fang R et al. Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science 377, 56–62 (2022). 10.1126/science.abm1741 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study compares cortical cell types and organization using novel spatial transcriptomic approaches, identifying differences in cell subtype composition and spatial arrangements.
- 35.Speir ML et al. UCSC Cell Browser: visualize your single-cell data. Bioinformatics 37, 4578–4580 (2021). 10.1093/bioinformatics/btab503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang D et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362 (2018). 10.1126/science.aat8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabula Sapiens C et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022). 10.1126/science.abl4896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tryka KA et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res 42, D975–979 (2014). 10.1093/nar/gkt1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Schwarz N et al. Long-term adult human brain slice cultures as a model system to study human CNS circuitry and disease. Elife 8 (2019). 10.7554/eLife.48417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40*.Ting JT et al. A robust ex vivo experimental platform for molecular-genetic dissection of adult human neocortical cell types and circuits. Sci Rep 8, 8407 (2018). 10.1038/s41598-018-26803-9 [DOI] [PMC free article] [PubMed] [Google Scholar]; These two papers (Schwarz et al., and Ting et al.,) describe methods for culturing human brain tissue from surgical patients and enable viral tracing of cell types and accessible intracortical connections.
- 41.Luo C et al. Single nucleus multi-omics identifies human cortical cell regulatory genome diversity. Cell Genom 2 (2022). 10.1016/j.xgen.2022.100107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li YE et al. A comparative atlas of single-cell chromatin accessibility in the human brain. bioRxiv, 2022.2011.2009.515833 (2022). 10.1101/2022.11.09.515833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakken TE et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 13, e0209648 (2018). 10.1371/journal.pone.0209648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caglayan E, Liu Y & Konopka G Neuronal ambient RNA contamination causes misinterpreted and masked cell types in brain single-nuclei datasets. Neuron 110, 4043–4056 e4045 (2022). 10.1016/j.neuron.2022.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeWeerdt S How to map the brain. Nature 571, S6–S8 (2019). 10.1038/d41586-019-02208-0 [DOI] [PubMed] [Google Scholar]
- 46.Sherwood CC et al. Invariant Synapse Density and Neuronal Connectivity Scaling in Primate Neocortical Evolution. Cereb Cortex 30, 5604–5615 (2020). 10.1093/cercor/bhaa149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alzu’bi A, Homman-Ludiye J, Bourne JA & Clowry GJ Thalamocortical Afferents Innervate the Cortical Subplate much Earlier in Development in Primate than in Rodent. Cereb Cortex 29, 1706–1718 (2019). 10.1093/cercor/bhy327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez-Bendito G Development of the Thalamocortical Interactions: Past, Present and Future. Neuroscience 385, 67–74 (2018). 10.1016/j.neuroscience.2018.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L et al. A cross-species proteomic map of synapse development reveals neoteny during human postsynaptic density maturation. bioRxiv, 2022.2010.2024.513541 (2022). 10.1101/2022.10.24.513541 [DOI] [Google Scholar]
- 50.Laszlo ZI et al. Synaptic proteomics reveal distinct molecular signatures of cognitive change and C9ORF72 repeat expansion in the human ALS cortex. Acta Neuropathol Commun 10, 156 (2022). 10.1186/s40478-022-01455-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hesse R et al. Comparative profiling of the synaptic proteome from Alzheimer’s disease patients with focus on the APOE genotype. Acta Neuropathol Commun 7, 214 (2019). 10.1186/s40478-019-0847-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlyle BC et al. Synaptic proteins associated with cognitive performance and neuropathology in older humans revealed by multiplexed fractionated proteomics. Neurobiol Aging 105, 99–114 (2021). 10.1016/j.neurobiolaging.2021.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graham LC et al. Regional Molecular Mapping of Primate Synapses during Normal Healthy Aging. Cell Rep 27, 1018–1026 e1014 (2019). 10.1016/j.celrep.2019.03.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niu M et al. Droplet-based transcriptome profiling of individual synapses. Nat Biotechnol (2023). 10.1038/s41587-022-01635-1 [DOI] [PubMed] [Google Scholar]
- 55.Hobson BD & Herzog E Methodological concerns and lack of evidence for single-synapse RNA-seq. Nat Biotechnol (2023). 10.1038/s41587-023-01877-7 [DOI] [PubMed] [Google Scholar]
- 56.Niu M & Zong C Reply to: Methodological concerns and lack of evidence for single-synapse RNA-seq. Nat Biotechnol (2023). 10.1038/s41587-023-01878-6 [DOI] [PubMed] [Google Scholar]
- 57*.Perez JD et al. Subcellular sequencing of single neurons reveals the dendritic transcriptome of GABAergic interneurons. Elife 10 (2021). 10.7554/eLife.63092 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated that subcellular, single-cell resolution transcriptional profiling provides additional information in interneurons.
- 58.Finke S & Conzelmann KK Replication strategies of rabies virus. Virus Res 111, 120–131 (2005). 10.1016/j.virusres.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 59.Wickersham IR et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647 (2007). 10.1016/j.neuron.2007.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saunders A et al. Ascertaining cells’ synaptic connections and RNA expression simultaneously with barcoded rabies virus libraries. Nat Commun 13, 6993 (2022). 10.1038/s41467-022-34334-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clark IC et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation. Science 372 (2021). 10.1126/science.abf1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan L, Chen X, Zhan H, Gilbert HL & Zador AM Massive Multiplexing of Spatially Resolved Single Neuron Projections with Axonal BARseq. bioRxiv, 2023.2002.2018.528865 (2023). 10.1101/2023.02.18.528865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Graybuck LT et al. Enhancer viruses for combinatorial cell-subclass-specific labeling. Neuron 109, 1449–1464 e1413 (2021). 10.1016/j.neuron.2021.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mich JK et al. Functional enhancer elements drive subclass-selective expression from mouse to primate neocortex. Cell Rep 34, 108754 (2021). 10.1016/j.celrep.2021.108754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanderhaeghen P & Polleux F Developmental mechanisms underlying the evolution of human cortical circuits. Nat Rev Neurosci 24, 213–232 (2023). 10.1038/s41583-023-00675-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Linaro D et al. Xenotransplanted Human Cortical Neurons Reveal Species-Specific Development and Functional Integration into Mouse Visual Circuits. Neuron 104, 972–986 e976 (2019). 10.1016/j.neuron.2019.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Revah O et al. Maturation and circuit integration of transplanted human cortical organoids. Nature 610, 319–326 (2022). 10.1038/s41586-022-05277-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kelley KW, Nakao-Inoue H, Molofsky AV & Oldham MC Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat Neurosci 21, 1171–1184 (2018). 10.1038/s41593-018-0216-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69*.Nowakowski TJ et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). 10.1126/science.aap8809 [DOI] [PMC free article] [PubMed] [Google Scholar]; This single-cell profiling of the developing human cortex serves as a reference dataset for cell types and identified differences in transcriptional profiles across cortical regions during development.
- 70.Bhaduri A et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148 (2020). 10.1038/s41586-020-1962-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pollen AA et al. Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell 176, 743–756 e717 (2019). 10.1016/j.cell.2019.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). 10.1038/s41586-019-1506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feregrino C & Tschopp P Assessing evolutionary and developmental transcriptome dynamics in homologous cell types. Dev Dyn 251, 1472–1489 (2022). 10.1002/dvdy.384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morabito S, Reese F, Rahimzadeh N, Miyoshi E & Swarup V High dimensional co-expression networks enable discovery of transcriptomic drivers in complex biological systems. bioRxiv, 2022.2009.2022.509094 (2022). 10.1101/2022.09.22.509094 [DOI] [Google Scholar]
- 75.Van de Sande B et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc 15, 2247–2276 (2020). 10.1038/s41596-020-0336-2 [DOI] [PubMed] [Google Scholar]
- 76.Costa IG Dissecting gene regulation with multimodal sequencing. Nat Methods (2023). 10.1038/s41592-023-01957-1 [DOI] [PubMed] [Google Scholar]
- 77.Wu SJ et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat Biotechnol 39, 819–824 (2021). 10.1038/s41587-021-00865-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park YP & Kellis M CoCoA-diff: counterfactual inference for single-cell gene expression analysis. Genome Biol 22, 228 (2021). 10.1186/s13059-021-02438-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Theodoris CV et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023). 10.1038/s41586-023-06139-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brendel M et al. Application of Deep Learning on Single-cell RNA Sequencing Data Analysis: A Review. Genomics Proteomics Bioinformatics 20, 814–835 (2022). 10.1016/j.gpb.2022.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma Q & Xu D Deep learning shapes single-cell data analysis. Nat Rev Mol Cell Biol 23, 303–304 (2022). 10.1038/s41580-022-00466-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamimoto K et al. Dissecting cell identity via network inference and in silico gene perturbation. Nature 614, 742–751 (2023). 10.1038/s41586-022-05688-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dixit A et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866 e1817 (2016). 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weiss CV et al. The cis-regulatory effects of modern human-specific variants. Elife 10 (2021). 10.7554/eLife.63713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whalen S et al. Machine learning dissection of human accelerated regions in primate neurodevelopment. Neuron 111, 857–873 e858 (2023). 10.1016/j.neuron.2022.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maynard KR et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci 24, 425–436 (2021). 10.1038/s41593-020-00787-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bellenguez C et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54, 412–436 (2022). 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fu JM et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet 54, 1320–1331 (2022). 10.1038/s41588-022-01104-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mollon J, Almasy L, Jacquemont S & Glahn DC The contribution of copy number variants to psychiatric symptoms and cognitive ability. Mol Psychiatry (2023). 10.1038/s41380-023-01978-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90*.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91*.Smith SM et al. An expanded set of genome-wide association studies of brain imaging phenotypes in UK Biobank. Nat Neurosci 24, 737–745 (2021). 10.1038/s41593-021-00826-4 [DOI] [PMC free article] [PubMed] [Google Scholar]; These papers (Bycroft et al., Smith et al.) harness the large UK Biobank resource to integrate genetic information with other features of the human brain including brain imaging and other phenotypes.
- 92.Bogdan R, Hatoum AS, Johnson EC & Agrawal A The Genetically Informed Neurobiology of Addiction (GINA) model. Nat Rev Neurosci 24, 40–57 (2023). 10.1038/s41583-022-00656-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Konopka G Cognitive genomics: Linking genes to behavior in the human brain. Netw Neurosci 1, 3–13 (2017). 10.1162/netn_a_00003 [DOI] [PubMed] [Google Scholar]
- 94*.Richiardi J et al. BRAIN NETWORKS. Correlated gene expression supports synchronous activity in brain networks. Science 348, 1241–1244 (2015). 10.1126/science.1255905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95*.Wang GZ et al. Correspondence between Resting-State Activity and Brain Gene Expression. Neuron 88, 659–666 (2015). 10.1016/j.neuron.2015.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]; These papers (Richiardi et al., and Wang et al.) are the first to integrate transcriptional profiling and brain imaging to begin to link gene expression and human brain function.
- 96.Berto S et al. Association between resting-state functional brain connectivity and gene expression is altered in autism spectrum disorder. Nat Commun 13, 3328 (2022). 10.1038/s41467-022-31053-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Romero-Garcia R et al. Schizotypy-Related Magnetization of Cortex in Healthy Adolescence Is Colocated With Expression of Schizophrenia-Related Genes. Biol Psychiatry 88, 248–259 (2020). 10.1016/j.biopsych.2019.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seidlitz J et al. Transcriptomic and cellular decoding of regional brain vulnerability to neurogenetic disorders. Nat Commun 11, 3358 (2020). 10.1038/s41467-020-17051-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buch AM et al. Molecular and network-level mechanisms explaining individual differences in autism spectrum disorder. Nat Neurosci 26, 650–663 (2023). 10.1038/s41593-023-01259-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kong XZ et al. Gene Expression Correlates of the Cortical Network Underlying Sentence Processing. Neurobiol Lang (Camb) 1, 77–103 (2020). 10.1162/nol_a_00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Berto S et al. Gene-expression correlates of the oscillatory signatures supporting human episodic memory encoding. Nat Neurosci 24, 554–564 (2021). 10.1038/s41593-021-00803-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eugene E et al. An organotypic brain slice preparation from adult patients with temporal lobe epilepsy. J Neurosci Methods 235, 234–244 (2014). 10.1016/j.jneumeth.2014.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103*.de la Torre-Ubieta L et al. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304 e218 (2018). 10.1016/j.cell.2017.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104*.Markenscoff-Papadimitriou E et al. A Chromatin Accessibility Atlas of the Developing Human Telencephalon. Cell 182, 754–769 e718 (2020). 10.1016/j.cell.2020.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]; These studies (de la Torre-Ubieta et al., Markenscoff-Papadimitriou et al.) utilize characterization of epigentic state to describe the regulatory programs found during human brain development.
- 105.Won H et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016). 10.1038/nature19847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94 (2018). 10.1038/nbt.4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ludwig LS et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339 e1322 (2019). 10.1016/j.cell.2019.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu J et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. Elife 8 (2019). 10.7554/eLife.45105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Meyer M et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A 112, 851–856 (2015). 10.1073/pnas.1320611111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Radecki G, Nargeot R, Jelescu IO, Le Bihan D & Ciobanu L Functional magnetic resonance microscopy at single-cell resolution in Aplysia californica. Proc Natl Acad Sci U S A 111, 8667–8672 (2014). 10.1073/pnas.1403739111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fan X et al. Spatial transcriptomic survey of human embryonic cerebral cortex by single-cell RNA-seq analysis. Cell Res 28, 730–745 (2018). 10.1038/s41422-018-0053-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhong S et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528 (2018). 10.1038/nature25980 [DOI] [PubMed] [Google Scholar]
- 113.Polioudakis D et al. A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 103, 785–801 e788 (2019). 10.1016/j.neuron.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fan X et al. Single-cell transcriptome analysis reveals cell lineage specification in temporal-spatial patterns in human cortical development. Sci Adv 6, eaaz2978 (2020). 10.1126/sciadv.aaz2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ & Kriegstein AR Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat Neurosci 24, 584–594 (2021). 10.1038/s41593-020-00794-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Smith RS et al. Early role for a Na(+),K(+)-ATPase (ATP1A3) in brain development. Proc Natl Acad Sci U S A 118 (2021). 10.1073/pnas.2023333118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ramos SI et al. An atlas of late prenatal human neurodevelopment resolved by single-nucleus transcriptomics. Nat Commun 13, 7671 (2022). 10.1038/s41467-022-34975-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Braun E et al. Comprehensive cell atlas of the first-trimester developing human brain. bioRxiv, 2022.2010.2024.513487 (2022). 10.1101/2022.10.24.513487 [DOI] [PubMed] [Google Scholar]
- 119.Herring CA et al. Human prefrontal cortex gene regulatory dynamics from gestation to adulthood at single-cell resolution. Cell 185, 4428–4447 e4428 (2022). 10.1016/j.cell.2022.09.039 [DOI] [PubMed] [Google Scholar]
- 120.Cameron D et al. Single-Nuclei RNA Sequencing of 5 Regions of the Human Prenatal Brain Implicates Developing Neuron Populations in Genetic Risk for Schizophrenia. Biol Psychiatry 93, 157–166 (2023). 10.1016/j.biopsych.2022.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krienen FM et al. Innovations present in the primate interneuron repertoire. Nature 586, 262–269 (2020). 10.1038/s41586-020-2781-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bakken TE et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature 598, 111–119 (2021). 10.1038/s41586-021-03465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hodes RJ & Buckholtz N Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) Knowledge Portal Aids Alzheimer’s Drug Discovery through Open Data Sharing. Expert Opin Ther Targets 20, 389–391 (2016). 10.1517/14728222.2016.1135132 [DOI] [PubMed] [Google Scholar]
- 124.Schirmer L et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019). 10.1038/s41586-019-1404-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bressan E et al. The Foundational Data Initiative for Parkinson Disease: Enabling efficient translation from genetic maps to mechanism. Cell Genom 3, 100261 (2023). 10.1016/j.xgen.2023.100261 [DOI] [PMC free article] [PubMed] [Google Scholar]