

Abstract
Dopamine neurons in the ventral tegmental area (VTA) are involved in a variety of physiological and pathological conditions, ranging from motivated behaviours to substance use disorders. While many studies have shown that these neurons can express plasticity at excitatory and inhibitory synapses, little is known about how inhibitory inputs and glial activity shape the output of DA neurons and therefore, merit greater discussion. In this review, we will attempt to fill in a bit more of the puzzle, with a focus on inhibitory transmission and astrocyte function. We summarize the findings within the VTA as well as observations made in other brain regions that have important implications for plasticity in general and should be considered in the context of DA neuron plasticity.
Keywords: astrocyte, dopamine, GABA, inhibition, plasticity, VTA
Introduction
The ventral tegmental area (VTA) is mainly populated by dopamine (DA) neurons, which are critical for reinforcing rewarding behaviours and attributing salience to important environmental cues (Schultz et al., 1997; Wise, 2004; Yamagata et al., 2015). They display a large capacity for experience-dependent plasticity (Stuber et al., 2008; Mao et al., 2011; Collo et al., 2014; Friedman et al., 2014; Gore et al., 2014), in both their synaptic connections and intrinsic excitability. These intrinsic and extrinsic cellular changes profoundly alter the output of DA neurons, and therefore, release of DA in downstream brain regions. Because changes in DA release have important consequences for motivated behaviours, learning, and memory formation (Berridge, 2006; Friedman et al., 2014; Berry et al., 2015; Popescu et al., 2016), many labs have focused on identifying ways in which DA neuron plasticity can occur.
The VTA is a rich signalling milieu that receives inputs from multiple regions and houses a heterogeneous population of cells (Watabe-Uchida et al., 2012; Barker et al., 2016). Elegant studies using modern genetic approaches and classic slice electrophysiology have unveiled numerous forms of plasticity and mechanisms that drive them (Bonci & Williams, 1996; Kalivas & Duffy, 1998). Although the different forms of excitatory synapse plasticity have been extensively reviewed (Lüscher, 2013; van Huijstee & Mansvelder, 2015; Pignatelli & Bonci, 2015), relatively little attention has been devoted to discussing the role of inhibitory inputs and glial cells in shaping the intrinsic activity and output of DA neurons (Fig. 1).
Fig. 1.
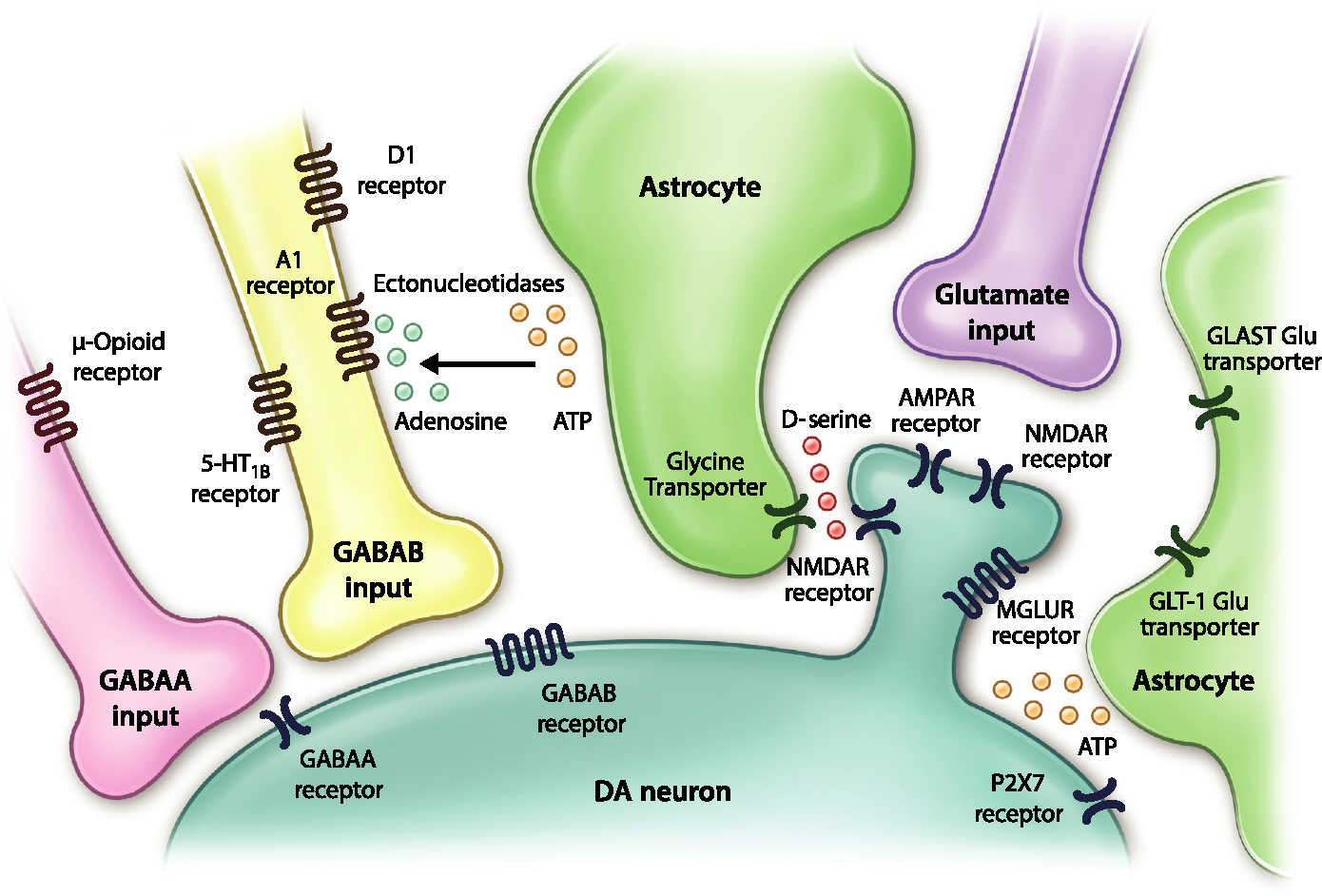
A schematic of inhibitory inputs and astrocyte processes surrounding dopamine (DA) neurons in the ventral tegmental area (VTA). As depicted, in addition to excitatory inputs, many elements are present in the local signalling milieu. Inhibitory inputs arising from multiple sources synapse onto GABAA and GABAB receptors on DA neurons. Astrocytes take up and release several signalling molecules, including glutamate, ATP, and D-serine. These processes are likely exerting important influences on the output and plasticity of DA neurons.
Excitatory synapse plasticity
Much of the existing data on plasticity in the VTA is focused on excitatory input onto DA neurons, with good reason. DA neurons can switch from tonic firing activity (1–4 Hz) to a burst firing pattern (15–30 Hz) at the presentation of behaviourally relevant stimuli (Paladini & Roeper, 2014), and this switch appears to be triggered by presynaptic excitatory inputs (Chergui et al., 1993; Floresco et al., 2003) arising from various parts of the brain (Watabe-Uchida et al., 2012; Beier et al., 2015; Tian et al., 2016). Thus, excitatory inputs critically influence DA neuron output, and consequently, DA-dependent behaviours.
Like elsewhere in the brain, excitatory synapses on DA neurons can undergo experience-dependent plasticity (Ungless et al., 2001; Chen et al., 2008; Ahn et al., 2010; Mao et al., 2011; Hausknecht et al., 2015). Changes at excitatory synapses can be elicited in vitro by high-frequency stimulation protocols and spike-timing dependent protocols (Bonci & Malenka, 1999), as well as in vivo by experiences such as social deprivation and exposure to drugs of abuse (Ungless et al., 2001; Chen et al., 2008; Mao et al., 2011; Whitaker et al., 2013; Hausknecht et al., 2015).
Long-term potentiation
Within the VTA, multiple forms of long-term potentiation (LTP) (Bellone & Lüscher, 2006; Mameli et al., 2011) and long-term depression (LTD) (Jones et al., 2000; Bellone & Lüscher, 2006; Mao et al., 2011; Labouèbe et al., 2013) of excitatory synapses have been observed. In contrast to the classic N-methyl-D-aspartate receptor (NMDAR)-dependent recruitment of additional α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) to the synapse, synaptic strength in DA neurons can increase via a switch in AMPAR subunit composition, from non-calcium permeable GluA2 subunit containing AMPARs to GluA2-lacking, calcium permeable AMPARs (Bellone & Lüscher, 2006). In the case of drug experience, changes in AMPAR subunit composition are accompanied by changes in NMDAR subunit composition, specifically a switch from NDMARs that permit calcium entry at depolarized potentials to NDMARs that are quasi-calcium impermeable (Yuan et al., 2013). NMDARs can also exhibit plasticity independent of AMPARs; in the presence of AMPAR blocker DNQX, a synaptic stimulation-burst pairing protocol was able to induce potentiation of NMDAR-mediated excitatory postsynaptic currents (EPSCs) in VTA DA neurons in animals treated with repeated amphetamine injections, but not in saline controls (Ahn et al., 2010). This facilitation of NMDAR LTP can also be observed in animals that were socially isolated during adolescence, and appears to be dependent on an increase in mGluR/IP3R mediated calcium signalling (Whitaker et al., 2013). Salient experiences like repeated drug exposure can also increase the amount of glutamate located in presynaptic terminals and extracellular glutamate levels in the VTA (Kalivas & Duffy, 1998; Kozell & Meshul, 2001).
Long-term depression
Several forms of long-term depression (LTD) have been observed at excitatory synapses onto VTA DA neurons (Jones et al., 2000; Gutlerner et al., 2002; Bellone & Lüscher, 2006; Mameli et al., 2007). The standard LTD protocol of pairing a 1-Hz presynaptic stimulation with postsynaptic depolarization produces LTD that involves internalization of GluA1 subunit containing AMPARs; but in contrast to hippocampal LTD, this requires the activation of cyclic AMP-dependent protein kinase A and is independent of NMDAR and protein phosphatase activity (Jones et al., 2000; Gutlerner et al., 2002). Stimulation of metabotropic glutamate receptors (mGluRs) can also produce LTD by causing a switch in subunit composition of AMPARs at the synapse, from GluA2-lacking AMPARs to GluA2 containing AMPARs with lower single-channel conductance (Mameli et al., 2007). On the presynaptic side, insulin and alpha-2 noradrenergic signalling can depress both AMPAR and NMDAR EPSCs in VTA DA neurons via a reduction in glutamate release probability (Jiménez-Rivera et al., 2012; Labouèbe et al., 2013; Thompson & Borgland, 2013).
Inhibitory synaptic transmission
While much work has focused on potentiation of excitatory inputs onto DA neurons, recent evidence suggests that inhibitory inputs also undergo plasticity in response to drugs of abuse. DA neurons receive dense inhibitory inputs. In the substantia nigra pars compacta (SNc), approximately 70% or more of the inputs to DA neurons are GABAergic (Ribak et al., 1976; Bolam & Smith, 1990; Henny et al., 2012). Inhibitory synaptic transmission in DA neurons is thought to underlie the extinction of learned behaviours and to control excessive DA neuron excitability. Therefore, GABAergic inputs may provide a therapeutic substrate for preventing increased DAergic activity during exposure to drugs of abuse.
GABAergic synaptic transmission onto DA neurons is mediated by both GABAA receptors (GABAARs) and GABAB receptors (GABABRs). GABAARs act by fast synaptic transmission and inhibit neurons through the activation of chloride currents, while GABABRs are slower metabotropic receptors that produce hyperpolarization via coupling to G-protein coupled inwardly rectifying potassium channels (GIRKs) (Lüscher et al., 1997). Both GABAARs and GABABRs control the tonic and burst firing of DA neurons in the VTA and SNc (Erhardt et al., 1999, 2002; Paladini & Tepper, 1999; Paladini et al., 1999). Interestingly, indirect evidence suggests that the presynaptic inputs to GABAARs and GABABRs arise from separate presynaptic sources (Sugita et al., 1992). This is demonstrated by findings showing that electrically evoked GABAA and GABAB currents undergo differential presynaptic neuromodulation. For instance, the inputs activating GABABRs are modulated by DA D1 receptors, serotonin 1B receptors, and adenosine A1 receptors, while the inputs activating GABAARs are unaffected by agonists of these receptors (Sugita et al., 1992; Cameron & Williams, 1993, 1994; Wu et al., 1995). These findings suggest that GABAAR and GABABR signalling in DA neurons are mediated by distinct inhibitory afferents and that these inputs might target separate subcellular domains within DA neurons.
In addition to the potentiation of glutamatergic inputs discussed above, many lines of evidence suggest that GABAergic inhibition is reduced by exposure to multiple drugs of abuse, potentially contributing to the maladaptive increases in DA neuron activity. Because the afferents activating GABAARs and GABABRs are thought to arise from separate sources, we will focus on the plasticity of these inputs separately.
Inputs onto GABAA receptors
DA neurons are under tonic inhibitory control by both local and long-range GABAergic inputs. Interestingly, transient blockade of GABAARs, either pharmacologically or via dynamic clamp techniques, can induce burst firing in DA neurons in slice recordings (Lobb et al., 2011). The VTA contains a large subset of GABAergic cells that form local synapses onto DA neurons and inhibit them via GABAARs (Steffensen et al., 1998; Tan et al., 2010; van Zessen et al., 2012). Additionally, recent work has shown that multiple brain regions activate GABAARs in DA neurons, including local VTA GABA neurons, the rostromedial tegmental nucleus (Jhou et al., 2009), and the lateral hypothalamus (Nieh et al., 2015). Recent studies have shown that inhibitory inputs from the nucleus accumbens and lateral hypothalamus can increase DA neuron activity by inhibiting VTA GABA neurons (Bocklisch et al., 2013; Nieh et al., 2016). These studies demonstrate the importance of local VTA GABA neurons in regulating the activity of DA neurons and suggest that modulation or plasticity of GABAergic inputs represent important mechanisms for both maladaptive processes and potential therapeutic targets.
Drugs of abuse are thought to act by increasing DA neuron activity. One of the major mechanisms by which drugs exert their effect is through the removal of tonic inhibition onto DA neurons. For example, opioids and cannabinoids are thought to produce rewarding behaviours by reducing the activity of GABAergic cells and synapses onto VTA DA neurons (Johnson & North, 1992). Recent research suggests that, similar to excitatory synapses, inhibitory synapses undergo plasticity in response to drugs of abuse. Repeated exposure to drugs of abuse show can induce long-lasting changes in GABAAR-expressing inhibitory synapses onto DA neurons. Early work showed that GABAAR-containing synapses are potentiated during withdrawal from repeated morphine exposure (Bonci & Williams, 1997). This increased GABAergic tone is due to a potentiation of presynaptic GABA release and appears to be causally related to symptoms of withdrawal (Madhavan et al., 2010).
Inhibitory synapses onto DA neurons undergo a form of LTP after high-frequency stimulation, termed LTPGABA (Nugent et al., 2007). Interestingly, LTPGABA is dependent on NMDA receptor activation and the recruitment of retrograde nitric oxide (NO) signalling (Nugent et al., 2007), suggesting an interplay between glutamatergic and GABAergic transmission in the expression of plasticity. NO-dependent potentiation of GABAAR-containing synapses does not affect GABABR-containing synapses (Nugent et al., 2009), further suggesting that the inputs onto GABAARs and GABABRs arise from separate sources and undergo differential forms of plasticity. After a single injection of morphine, DA cells no longer undergo LTPGABA (Nugent et al., 2007), due to activation of kappa opioid receptors (Graziane et al., 2013; Polter et al., 2014), revealing a potential deficit in GABAergic signalling. One possibility is that a single exposure to morphine induces LTPGABA, effectively occluding LTPGABA induction in brain slices. However, the loss of LTPGABA after morphine exposure is mirrored by a loss of LTDGABA (Dacher & Nugent, 2011; Dacher et al., 2013), suggesting that a single exposure to morphine might generally reduce GABAergic plasticity in the VTA. On the other hand, repeated exposure to morphine potentiates GABAergic transmission in the VTA (Bonci & Williams, 1997), suggesting differences between acute and chronic drug exposure. In addition to morphine, a single exposure to cocaine, nicotine, or acute stress block LTPGABA (Niehaus et al., 2010), suggesting that, similar to glutamatergic synaptic plasticity (Saal et al., 2003), each of these events triggers a common alteration in inhibitory synaptic transmission.
Little is known about how excitatory and inhibitory signalling are integrated in DA neurons. During in vitro slice recordings, synaptic blockers are usually used to isolate specific types of synaptic responses (i.e. AMPA, NMDA, or GABAA), prohibiting the ability to investigate interactions between excitatory and inhibitory plasticity. However, recent work suggests that glutamatergic and GABAergic inputs might be mutually involved in various forms of plasticity. For example, GABAergic synapses onto dopamine neurons undergo spike-timing dependent plasticity that appears to be mediated by postsynaptic activation of NMDARs (Kodangattil et al., 2013). Additionally, repeated cocaine exposure reduces GABAAR-mediated inhibition of DA neurons and allows for the induction of glutamatergic LTP (Liu et al., 2005; Pan et al., 2008a,b, 2011), indicating a role for inhibitory transmission in the expression of excitatory synaptic plasticity. However, this idea has been the subject of controversy, as other studies suggest that glutamatergic LTP is occluded by repeated drug exposure (Argilli et al., 2008; Luu & Malenka, 2008). Interestingly, benzodiazepines activate the DAergic system by inhibiting local interneurons. Recent evidence suggests that this form of disinhibition can lead to potentiation of excitatory synapses in DA neurons (Tan et al., 2010). These findings suggest a complex interplay between excitatory and inhibitory transmission that is mediated through unknown mechanisms. How might disinhibition allow for the potentiation of excitatory synapses? One possibility is that benzodiazepines increase DA release, causing the activation of D2 receptors, and preventing the occurrence of LTD (Thomas et al., 2000). Another possibility is that disinhibition might increase the efficacy of excitatory transmission by removing the shunting inhibition provided by GABAARs, lengthening the integration window for spike-timing dependent plasticity and potentially allowing for increased Ca2+ signalling. However, these hypotheses have yet to be tested. Finally, many neuromodulators transiently affect the excitatory-inhibitory balance within DA neurons by inhibiting GABAergic inputs (Johnson & North, 1992; Sugita et al., 1992). Thus, the disinhibitory role of neuromodulators could potentially shape longer lasting forms of synaptic plasticity.
Inputs onto GABAB receptors
In addition to GABAAR-mediated inhibition, GABABR-containing synapses are also affected by multiple forms of pre- and postsynaptic plasticity. Chronic exposure to either cocaine or morphine induces long-lasting changes in GABABR signalling, causing a presynaptic reduction in GABA release. These changes are mediated by increased adenosine tone, an effect that can last for a week or more (Bonci & Williams, 1996; Shoji et al., 1999), suggesting that chronic exposure to drugs of abuse can lead to prolonged disinhibition of DA neurons. However, in contrast to GABAAR-mediated inhibition, the sources of presynaptic innervation onto GABABR-containing synapses are not known. Early work suggested that nucleus accumbens inputs might inhibit VTA and SNc DA neurons via GABABRs, but this finding has been called into question by more recent studies (Brazhnik et al., 2008; Xia et al., 2011). While the monosynaptic inputs onto DA neurons have been studied extensively (Watabe-Uchida et al., 2012; Beier et al., 2015; Menegas et al., 2015), the functional contributions of many of these inputs are still unclear. Therefore, future experiments combining optogenetic manipulations of inputs with electrophysiological recordings will help determine which inputs are affected by different manipulations.
Postsynaptic GABABRs and their downstream effector proteins also undergo plasticity in response to drugs of abuse. As mentioned above, GABABRs produce inhibition through the activation of GIRKs (Lüscher et al., 1997). GIRK channels have the ability to undergo plasticity, depending on the firing activity of DA neurons (Lalive et al., 2014). Interestingly, GABA and DA neurons of the VTA express different GIRK subunits, conferring differential sensitivity to GABA, as well as other GABABR agonists (Cruz et al., 2004; Labouèbe et al., 2007). These findings suggest that GABA and DA neurons of the VTA can undergo cell-type specific modification of GABAB responses through selective plasticity of specific GIRK subunits. Indeed, a single exposure to methamphetamine causes long-term depression of GABAB signalling in VTA GABA neurons, but not DA neurons (Padgett et al., 2012). Additionally, a single cocaine injection disinhibits DA neurons through postsynaptic reduction of GABABR-GIRK signalling (Arora et al., 2011), potentially contributing to increased DA neuron activity in the early stages of addiction.
While the importance and prevalence of inhibitory plasticity is beginning to become apparent, the roles of GABABRs in DA neurons are relatively unknown. In addition to their role in inhibitory transmission, GABABRs regulate the entry of Ca2+ through NMDA receptors and through voltage-sensitive Ca2+ channels (Chalifoux & Carter, 2010, 2011). As excitatory synaptic plasticity is dependent on postsynaptic Ca2+ entry, these findings suggest that GABABRs may play a role in regulating glutamatergic LTP or LTD in DA neurons. Additionally, GABABRs within DA neurons are coupled to HCN channels which regulate the pacemaker firing of DA neurons (Schwenk et al., 2016). Therefore, plasticity of GABABR-containing synapses might be important for the control of DA neuron firing rates. Finally, GABABRs represent an important therapeutic target in the treatment of craving for cocaine and other drugs of abuse (Slattery et al., 2005; Filip et al., 2015). Future work should address the roles of DA neuron GABABRs and GABABR plasticity in addiction and other DA-dependent behaviours.
Astrocyte regulation of signalling strength and synaptic plasticity
Stimuli that trigger DA neuron plasticity can also alter astrocyte gene expression and function (Knackstedt et al., 2010; García-Pérez et al., 2014; Scofield et al., 2016). In addition, there is evidence that astrocytes can be affected by the activity level of DA neurons themselves, via dopamine receptor signalling. Blockade of D1/D2 receptor signalling can increase astrocyte calcium excitability in the substantia nigra pars reticulata (Bosson et al., 2015), and preferential deletion of D2Rs from astrocytes leads to an exaggerated inflammatory response to DA neuron injury (Shao et al., 2013). Given the limitations of existing techniques, it is essentially impossible to confirm that these effects are due to direct expression of dopamine receptors by astrocytes. Nevertheless, they indicate that dopamine signalling can significantly change the functional state of astrocytes. And regardless of whether astrocytes are undergoing changes first or reacting to neuronal changes, it would be prudent to consider how altered astrocyte function might affect neighbouring neurons. Below, we discuss just a few of the many potential ways in which astrocytes can shape DA neuron activity and plasticity.
Glutamate uptake
Perhaps the most famous role of astrocytes is the uptake of synaptically released glutamate via high levels of glutamate transporters located on their plasma membrane (Chaudhry et al., 1995). Astrocyte glutamate uptake is critical for avoiding excitotoxicity and seizures, but more interestingly, it appears to be dynamically regulated by neuronal activity (Murphy-Royal et al., 2015; Al Awabdh et al., 2016). An elegant study using quantum dot imaging showed that the astrocytic glutamate transporter GLT-1 is highly diffusible in astrocyte processes and responds to glutamate binding by diffusing away from the synapse (Murphy-Royal et al., 2015). When diffusion was impaired by crosslinking, the decay time of spontaneous EPSCs (sEPSCs) was significantly increased (Murphy-Royal et al., 2015), thus showing that astrocytic glutamate transporters can directly shape glutamatergic transmission.
In addition to modulating the time course of synaptic glutamate receptor activation, astrocyte ensheathment of synapses, or lack thereof, can determine the degree to which glutamate spillover from synapses can activate pre- and extrasynaptic glutamate receptors (Oliet et al., 2001; Oliet & Bonfardin, 2010; Wild et al., 2015). This aspect of astrocyte biology merits greater scrutiny within the VTA, as studies in other regions have shown that both the structure of astrocyte processes and the level of glutamate transporters present on these processes are altered by stimuli that impact signalling in the VTA. For example, in the nucleus accumbens (NAc), prolonged exposure to drugs of abuse downregulates astrocytic GLT-1 expression and causes a retraction of astrocyte processes from the synapse (Knackstedt et al., 2010; Scofield et al., 2016). Postmortem analysis of human tissue revealed that GLT-1 and GLAST, the other astrocytic glutamate transporter, were both downregulated in the PFC of individuals who suffered from major depressive disorder (Choudary et al., 2005). These adaptations are likely to have important behavioural consequences, as evidenced by in vivo studies using dihydrokainic acid (DHK) to block GLT-1 activity (John et al., 2012; Smith et al., 2014). A similar adaptation in the VTA could greatly impact signalling onto DA neurons. In support of this hypothesis, pharmacological blockade of GLT-1 and GLAST increases activation of extrasynaptic NMDARs in DA neurons (Wild et al., 2015), and disrupting the orthologue of GLT-1 (glt-1–6) in C. elegans produces DA neuron hyperexcitability (Hardaway et al., 2015). It is conceivable, then, that certain stimulus-triggered adaptations within the VTA may be resulting from, or at least enhanced by, a reduction in astrocyte uptake of glutamate.
Release of D-serine
Astrocyte ensheathment of synapses may also determine the amount of D-serine available to bind the glycine site of NMDARs, a necessary binding site for the activation of NMDARs (Kleckner & Dingledine, 1988). A study looking at the supraoptic nucleus of lactating rats found that depletion of D-serine, but not glycine, significantly reduced the size of NMDAR-mediated EPSCs (Panatier et al., 2006), which supports the idea that, at least in certain parts of the brain, the endogenous ligand at the NMDAR glycine site is D-serine (Mothet et al., 2000; Yang et al., 2003). The same group also made the observation that D-serine availability is correlated with the degree of synapse ensheathment by astrocytes - unsurprising, given the localization of D-serine and its synthesizing enzyme serine racemase in astrocyte processes (Schell et al., 1997; Panatier et al., 2006; Fossat et al., 2012). Slice electrophysiology experiments in the hippocampus, prefrontal cortex, and neuron astrocyte co-cultures indicate that astrocyte-derived D-serine is necessary for NMDAR-dependent induction of LTP (Yang et al., 2003; Martineau et al., 2008; Henneberger et al., 2010; Fossat et al., 2012). In addition to having the capacity to synthesize and release D-serine, astrocytes can also take up D-serine through the neutral amino acid transporter SLC1A4 (Foster et al., 2016).
Within the VTA, D-serine can participate in NMDAR activation and induction of plasticity. Intra-VTA infusion of D-serine enhances cocaine-induced behavioural sensitization and activation of CamKII, a protein kinase associated with the formation of long-term memory (Fernandez-Espejo et al., 2007). In mice lacking D-amino acid oxidase, the enzyme that degrades D-serine, VTA DA neurons exhibited in vivo burst firing more frequently than in wild-type animals (Schweimer et al., 2014). This is consistent with the known role of NMDAR activation in generating burst firing of DA neurons (Tong et al., 1996; Paladini & Roeper, 2014). Intriguingly, D-serine may also inhibit some NMDARs that are active at hyperpolarized potentials (Seif et al., 2015). Thus, astrocytes could alter the degree of NMDAR activation in VTA DA neurons - and therefore, NMDAR-dependent plasticity - by gating the availability of D-serine.
Regulation of glycine
Like D-serine, glycine is a co-agonist for NMDARs at the (aptly named) glycine site and can potentiate NMDAR responses (Johnson & Ascher, 1987; Kleckner & Dingledine, 1988). Astrocytes express high levels of the glycine transporters GlyT1 and GlyT2 (Zhang et al., 2014), and therefore have the capacity to shift extracellular concentrations of glycine through transporter-mediated uptake. Indeed, the degree of glycine uptake can be reduced by plasticity-triggering stimuli like BDNF, which promotes internalization of GlyTs in astrocyte processes (Aroeira et al., 2015). In the NAc, blocking glycine transporters pharmacologically enhances NMDA responses in vivo (Lewis & O’donnell, 2003). Intra-VTA injection of 7-chlorothiokynurenic acid, an NMDAR glycine site antagonist, impaired conditioned place preference to cocaine without affecting locomotion (Zhou et al., 2011), which suggests that co-activation of NMDARs by glycine (or D-serine) is required for NMDAR-dependent plasticity of DA neurons.
Independent of its co-agonist activity at NMDARs, glycine can also bind to glycine receptors located on both terminals and cell bodies of neurons. Within the VTA, glycine potently depressed both spontaneous and evoked IPSCs onto DA neurons by inhibiting presynaptic release of GABA (Ye et al., 2004). Thus, whether via NMDAR modulation or glycine receptor activation, changes in the degree or location of astrocytic glycine uptake can greatly impact the size, and consequence, of inputs onto DA neurons.
Release of ATP
Astrocytes can release ATP through lysosome exocytosis (Zhang et al., 2007). This ATP acts on astrocytic purinergic receptors and is responsible for the propagation of astrocytic calcium waves (Bowser & Khakh, 2007), but it can also be degraded by ectonucleotidases into adenosine and act on neuronal adenosine receptors (Pascual et al., 2005; Sun et al., 2016). Artificially elevating calcium within hypothalamic astrocytes led to a rapid decrease in the firing rate of AGRP neurons; this decrease was completely blocked by bath application of an adenosine A1 receptor antagonist (Yang et al., 2015). In the hippocampus, adenosine accumulated from astrocytic ATP release acted on presynaptic adenosine A1 receptors to suppress glutamatergic transmission (Sun et al., 2016). In addition to the effects of ATP-derived adenosine, ATP release from astrocytes can also activate postsynaptic neuronal P2X receptors and trigger dynamin-dependent endocytosis of AMPARs (Pougnet et al., 2014). There is also evidence that astrocytic ATP can regulate developmental synapse elimination via P2Y1 receptor signalling (Yang et al., 2016), although it remains to be seen whether this form of regulation remains active in adulthood.
Within the VTA, chronic exposure to cocaine and morphine produces an increased adenosine tone that reverses the effect of D1R signalling on DA neurons (Bonci & Williams, 1996). The elevation of adenosine tone could be a result of increased astrocytic ATP, as chronic drug use can impair norepinephrine clearance (Fox et al., 2015) and astrocytes respond to norepinephrine with calcium elevations that drive ATP release (Pougnet et al., 2014). Thus, whether by the actions of ATP on purinergic receptors, or by indirect activation of adenosine receptors, release of ATP by astrocytes can produce changes in both pre- and postsynaptic signalling strength, and possibly even alter the consequences of dopamine receptor signalling in DA neurons.
Concluding remarks
Landmark studies on plasticity in the hippocampus and cortex have served as phenomenal templates for studying similar adaptations in the VTA (Huganir & Nicoll, 2013). However, it is now clear that the plasticity and output of DA neurons are governed by its own set of rules – hardly surprising, as DA neurons and pyramidal neurons of the hippocampus and cortex are serving very different purposes in brain computation. Given the distinct mechanisms regulating the expression of LTP in the midbrain, it is reasonable to suspect that inhibitory transmission and glial interactions are different as well. This review highlights several ways in which inhibitory inputs and astrocytes could be involved in DA neuron signalling and plasticity, but it is by no means an exhaustive list. Modulatory inputs to the region, such as acetylcholine (Graupner et al., 2013; Solecki et al., 2013), orexin, and dynorphin (Muschamp et al., 2014), as well as microglia and oligodendrocytes (Parkhurst et al., 2013; Gibson et al., 2014), are prominent components of the VTA circuitry that we haven’t even touched on – though they deserve a full review on their own. Thus, despite our best efforts to speculate based on existing data, many more experiments need to be done within the VTA to uncover the multifaceted ways in which non-excitatory inputs and glial cells restrict and permit plasticity in DA neurons.
References
- Ahn K-C, Bernier BE, Harnett MT & Morikawa H (2010) IP3 receptor sensitization during in vivo amphetamine experience enhances NMDA receptor plasticity in dopamine neurons of the ventral tegmental area. J. Neurosci, 30, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Awabdh S, Gupta-Agarwal S, Sheehan DF, Muir J, Norkett R, Twelvetrees AE, Griffin LD & Kittler JT (2016) Neuronal activity mediated regulation of glutamate transporter GLT-1 surface diffusion in rat astrocytes in dissociated and slice cultures. Glia, 64, 1252–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argilli E, Sibley DR, Malenka RC, England PM & Bonci A (2008) Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J. Neurosci, 28, 9092–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroeira RI, Sebastião AM & Valente CA (2015) BDNF, via truncated TrkB receptor, modulates GlyT1 and GlyT2 in astrocytes. Glia, 63, 2181–2197. [DOI] [PubMed] [Google Scholar]
- Arora D, Hearing M, Haluk DM, Mirkovic K, Fajardo-Serrano A, Wessendorf MW, Watanabe M, Luján R et al. (2011) Acute cocaine exposure weakens GABA(B) receptor-dependent G-protein-gated inwardly rectifying K+ signaling in dopamine neurons of the ventral tegmental area. J. Neurosci, 31, 12251–12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Root DH, Zhang S & Morales M (2016) Multiplexed neurochemical signaling by neurons of the ventral tegmental area. J. Chem. Neuroanat, 73, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, Gao XJ, Kremer EJ et al. (2015) Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell, 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C & Lüscher C (2006) Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci, 9, 636–641. [DOI] [PubMed] [Google Scholar]
- Berridge KC (2006) The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacology, 191, 391–431. [DOI] [PubMed] [Google Scholar]
- Berry JA, Cervantes-Sandoval I, Chakraborty M & Davis RL (2015) Sleep facilitates memory by blocking dopamine neuron-mediated forgetting. Cell, 161, 1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklisch C, Pascoli V, Wong JCY, House DRC, Yvon C, de Roo M, Tan KR & Lüscher C (2013) Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science, 341, 1521–1525. [DOI] [PubMed] [Google Scholar]
- Bolam JP & Smith Y (1990) The GABA and substance P input to dopaminergic neurones in the substantia nigra of the rat. Brain Res, 529, 57–78. [DOI] [PubMed] [Google Scholar]
- Bonci A & Malenka RC (1999) Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J. Neurosci, 19, 3723–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A & Williams JT (1996) A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron, 16, 631–639. [DOI] [PubMed] [Google Scholar]
- Bonci A & Williams JT (1997) Increased probability of GABA release during withdrawal from morphine. J. Neurosci, 17, 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson A, Boisseau S, Buisson A, Savasta M & Albrieux M (2015) Disruption of dopaminergic transmission remodels tripartite synapse morphology and astrocytic calcium activity within substantia nigra pars reticulata. Glia, 63, 673–683. [DOI] [PubMed] [Google Scholar]
- Bowser DN & Khakh BS (2007) Vesicular ATP Is the predominant cause of intercellular calcium waves in astrocytes. J. Gen. Physiol, 129, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazhnik E, Shah F & Tepper JM (2008) GABAergic afferents activate both GABAA and GABAB receptors in mouse substantia nigra dopaminergic neurons in vivo. J. Neurosci, 28, 10386–10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL & Williams JT (1993) Dopamine D1 receptors facilitate transmitter release. Nature, 366, 344–347. [DOI] [PubMed] [Google Scholar]
- Cameron DL & Williams JT (1994) Cocaine inhibits GABA release in the VTA through endogenous 5-HT. J. Neurosci, 14, 6763–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR & Carter AG (2010) GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron, 66, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR & Carter AG (2011) GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J. Neurosci, 31, 4221–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC & Storm-Mathisen J (1995) Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron, 15, 711–720. [DOI] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Gilroy AM, Carelli RM, Chou JK & Bonci A (2008) Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron, 59, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chergui K, Charléty PJ, Akaoka H, Saunier CF, Brunet J-L, Buda M, Svensson TH & Chouvet G (1993) Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur. J. Neurosci, 5, 137–144. [DOI] [PubMed] [Google Scholar]
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE et al. (2005) Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. USA, 102, 15653–15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collo G, Cavalleri L & Spano P (2014) Structural plasticity in mesencephalic dopaminergic neurons produced by drugs of abuse: critical role of BDNF and dopamine. Front. Pharmacol, 5, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn M-L, Stoffel M, Slesinger PA & Lüscher C (2004) Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat. Neurosci, 7, 153–159. [DOI] [PubMed] [Google Scholar]
- Dacher M & Nugent FS (2011) Morphine-induced modulation of LTD at GABAergic synapses in the ventral tegmental area. Neuropharmacology, 61, 1166–1171. [DOI] [PubMed] [Google Scholar]
- Dacher M, Gouty S, Dash S, Cox BM & Nugent FS (2013) A-kinase anchoring protein-calcineurin signaling in long-term depression of GABAergic synapses. J. Neurosci, 33, 2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S, Nissbrandt H & Engberg G (1999) Activation of nigral dopamine neurons by the selective GABA(B)-receptor antagonist SCH 50911. J. Neural. Transm, 106, 383–394. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Mathé JM, Chergui K, Engberg G & Svensson TH (2002) GABA(B) receptor-mediated modulation of the firing pattern of ventral tegmental area dopamine neurons in vivo. N. S. Arch. Pharmacol, 365, 173–180. [DOI] [PubMed] [Google Scholar]
- Fernandez-Espejo E, Ramiro-Fuentes S, Portavella M & Moreno-Paublete R (2007) Role for D-Serine within the Ventral Tegmental Area in the Development of Cocaine’s Sensitization. Neuropsychopharmacol, 33, 995–1003. [DOI] [PubMed] [Google Scholar]
- Filip M, Frankowska M, Sadakierska-Chudy A, Suder A, Szumiec L, Mierzejewski P, Bienkowski P, Przegaliński E et al. (2015) GABAB receptors as a therapeutic strategy in substance use disorders: focus on positive allosteric modulators. Neuropharmacology, 88, 36–47. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H & Grace AA (2003) Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat. Neurosci, 6, 968–973. [DOI] [PubMed] [Google Scholar]
- Fossat P, Turpin FR, Sacchi S, Dulong J, Shi T, Rivet J-M, Sweedler JV, Pollegioni L et al. (2012) Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb. Cortex, 22, 595–606. [DOI] [PubMed] [Google Scholar]
- Foster AC, Farnsworth J, Lind GE, Li Y-X, Yang J-Y, Dang V, Penjwini M, Viswanath V et al. (2016) D-Serine is a substrate for neutral amino acid transporters ASCT1/SLC1A4 and ASCT2/SLC1A5, and is transported by both subtypes in rat hippocampal astrocyte cultures. PLoS ONE, 11, e0156551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox ME, Studebaker RI, Swofford NJ & Wightman RM (2015) Stress and drug dependence differentially modulate norepinephrine signaling in animals with varied HPA axis function. Neuropsychopharmacol, 40, 1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AK, Walsh JJ, Juarez B, Ku SM, Chaudhury D, Wang J, Li X, Dietz DM et al. (2014) Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science, 344, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Pérez D, Luisa Laorden M, Núñez C & Victoria Milanés M (2014) Glial activation and midkine and pleiotrophin transcription in the ventral tegmental area are modulated by morphine administration. J. Neuroimmunol, 274, 244–248. [DOI] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE et al. (2014) Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science, 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore BB, Soden ME & Zweifel LS (2014) Visualization of plasticity in fear-evoked calcium signals in midbrain dopamine neurons. Learn Memory, 21, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M, Maex R & Gutkin B (2013) Endogenous cholinergic inputs and local circuit mechanisms govern the phasic mesolimbic dopamine response to nicotine. PLoS Comput. Biol, 9, e1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziane NM, Polter AM, Briand LA, Pierce RC & Kauer JA (2013) Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron, 77, 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutlerner JL, Penick EC, Snyder EM & Kauer JA (2002) Novel protein kinase A-dependent long-term depression of excitatory synapses. Neuron, 36, 921–931. [DOI] [PubMed] [Google Scholar]
- Hardaway JA, Sturgeon SM, Snarrenberg CL, Li Z, Xu XZS, Bermingham DP, Odiase P, Spencer WC et al. (2015) Glial expression of the Caenorhabditis elegans gene swip-10 supports glutamate dependent control of extrasynaptic dopamine signaling. J. Neurosci, 35, 9409–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausknecht K, Haj-Dahmane S, Shen Y-L, Vezina P, Dlugos C & Shen R-Y (2015) Excitatory synaptic function and plasticity is persistently altered in ventral tegmental area dopamine neurons after prenatal ethanol exposure. Neuropsychopharmacol, 40, 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SHR & Rusakov DA (2010) Long term potentiation depends on release of D-serine from astrocytes. Nature, 463, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henny P, Brown MTC, Northrop A, Faunes M, Ungless MA, Magill PJ & Bolam JP (2012) Structural correlates of heterogeneous in vivo activity of midbrain dopaminergic neurons. Nat. Neurosci, 15, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL & Nicoll RA (2013) AMPARs and synaptic plasticity: the last 25 years. Neuron, 80, 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Huijstee AN & Mansvelder HD (2015) Glutamatergic synaptic plasticity in the mesocorticolimbic system in addiction. Front. Cell. Neurosci, 8, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Fields HL, Baxter MG, Saper CB & Holland PC (2009) The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron, 61, 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Rivera CA, Figueroa J, Vázquez R, Vélez M, Schwarz D, Velásquez-Martinez MC & Arencibia-Albite F (2012) Presynaptic inhibition of glutamate transmission by Alpha-2 receptors in the VTA. Eur. J. Neurosci, 35, 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CS, Smith KL, Van’T Veer A, Gompf HS, Carlezon WA, Cohen BM, Öngür D & Bechtholt-Gompf AJ (2012) Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacol, 37, 2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW & Ascher P (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature, 325, 529–531. [DOI] [PubMed] [Google Scholar]
- Johnson SW & North RA (1992) Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci, 12, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Kornblum JL & Kauer JA (2000) Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J. Neurosci, 20, 5575–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW & Duffy P (1998) Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. J. Neurochem, 70, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kleckner NW & Dingledine R (1988) Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science, 241, 835–837. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez RI & Kalivas PW (2010) Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine-seeking. Biol. Psychiat, 67, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodangattil JN, Dacher M, Authement ME & Nugent FS (2013) Spike timing-dependent plasticity at GABAergic synapses in the ventral tegmental area. J. Physiol, 591, 4699–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozell LB & Meshul CK (2001) The effects of acute or repeated cocaine administration on nerve terminal glutamate within the rat mesolimbic system. Neuroscience, 106, 15–25. [DOI] [PubMed] [Google Scholar]
- Labouèbe G, Lomazzi M, Cruz HG, Creton C, Luján R, Li M, Yanagawa Y, Obata K et al. (2007) RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat. Neurosci, 10, 1559–1568. [DOI] [PubMed] [Google Scholar]
- Labouèbe G, Liu S, Dias C, Zou H, Wong JCY, Karunakaran S, Clee SM, Phillips AG et al. (2013) Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat. Neurosci, 16, 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalive AL, Munoz MB, Bellone C, Slesinger PA, Lüscher C & Tan KR (2014) Firing modes of dopamine neurons drive bidirectional GIRK channel plasticity. J. Neurosci, 34, 5107–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B & O’donnell P (2003) Blockade of the GlyT1 glycine transporter prolongs response to VTA stimulation in nucleus accumbens neurons. Ann. N. Y. Acad. Sci, 1003, 431–434. [DOI] [PubMed] [Google Scholar]
- Liu Q, Pu L & Poo M (2005) Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature, 437, 1027–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb CJ, Wilson CJ & Paladini CA (2011) High-frequency, short-latency disinhibition bursting of midbrain dopaminergic neurons. J. Neurophysiol, 105, 2501–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C (2013) Cocaine-evoked synaptic plasticity of excitatory transmission intheventraltegmental area. Cold Spring Harb. Perspect Med, 3, a012013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Jan LY, Stoffel M, Malenka RC & Nicoll RA (1997) G protein-coupled inwardly rectifying K+ Channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron, 19, 687–695. [DOI] [PubMed] [Google Scholar]
- Luu P & Malenka RC (2008) Spike timing-dependent long-term potentiation in ventral tegmental area dopamine cells requires PKC. J. Neurophysiol, 100, 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan A, He L, Stuber GD, Bonci A & Whistler JL (2010) μ-Opioid receptor endocytosis prevents adaptations in ventral tegmental area GABA transmission induced during naloxone-precipitated morphine withdrawal. J. Neurosci, 30, 3276–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Balland B, Luján R & Lüscher C (2007) Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science, 317, 530–533. [DOI] [PubMed] [Google Scholar]
- Mameli M, Bellone C, Brown MTC & Lüscher C (2011) Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat. Neurosci, 14, 414–416. [DOI] [PubMed] [Google Scholar]
- Mao D, Gallagher K & McGehee DS (2011) Nicotine potentiation of excitatory inputs to ventral tegmental area dopamine neurons. J. Neurosci, 31, 6710–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau M, Galli T, Baux G & Mothet J-P (2008) Confocal imaging and tracking of the exocytotic routes for D-serine-mediated gliotransmission. Glia, 56, 1271–1284. [DOI] [PubMed] [Google Scholar]
- Menegas W, Bergan JF, Ogawa SK, Isogai Y, Umadevi Venkataraju K, Osten P, Uchida N & Watabe-Uchida M (2015) Dopamine neurons projecting to the posterior striatum form an anatomically distinct subclass. eLife, 4, e10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet J-P, Parent AT, Wolosker H, Brady RO, Linden DJ, Ferris CD, Rogawski MA & Snyder SH (2000) d-Serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. USA, 97, 4926–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L & Oliet SHR (2015) Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci, 18, 219–226. [DOI] [PubMed] [Google Scholar]
- Muschamp JW, Hollander JA, Thompson JL, Voren G, Hassinger LC, Onvani S, Kamenecka TM, Borgland SL et al. (2014) Hypocretin (orexin) facilitates reward by attenuating the antireward effects of its cotransmitter dynorphin in ventral tegmental area. Proc. Natl. Acad. Sci. USA, 111, E1648–E1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, Neve R, Wildes CP et al. (2015) Decoding neural circuits that control compulsive sucrose seeking. Cell, 160, 528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieh EH, Vander Weele CM, Matthews GA, Presbrey KN, Wichmann R, Leppla CA, Izadmehr EM & Tye KM (2016) Inhibitory input from the lateral hypothalamus to the ventral tegmental area disinhibits dopamine neurons and promotes behavioral activation. Neuron, 90, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus JL, Murali M & Kauer JA (2010) Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur. J. Neurosci, 32, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC & Kauer JA (2007) Opioids block long-term potentiation of inhibitory synapses. Nature, 446, 1086–1090. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Niehaus JL & Kauer JA (2009) PKG and PKA signaling in LTP at GABAergic synapses. Neuropsychopharmacol, 34, 1829–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR & Bonfardin VDJ (2010) Morphological plasticity of the rat supraoptic nucleus – cellular consequences. Eur. J. Neurosci, 32, 1989–1994. [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Piet R & Poulain DA (2001) Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science, 292, 923–926. [DOI] [PubMed] [Google Scholar]
- Padgett CL, Lalive AL, Tan KR, Terunuma M, Munoz MB, Pangalos MN, Martínez-Hernández J, Watanabe M et al. (2012) Methamphetamine-evoked depression of GABAB receptor signaling in GABA neurons of the VTA. Neuron, 73, 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA & Roeper J (2014) Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience, 282, 109–121. [DOI] [PubMed] [Google Scholar]
- Paladini CA & Tepper JM (1999) GABA(A) and GABA(B) antagonists differentially affect the firing pattern of substantia nigra dopaminergic neurons in vivo. Synapse, 32, 165–176. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Celada P & Tepper JM (1999) Striatal, pallidal, and pars reticulata evoked inhibition of nigrostriatal dopaminergic neurons is mediated by GABAA receptors in vivo. Neuroscience, 89, 799–812. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ & Liu Q (2008a) Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J. Neurosci, 28, 1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ & Liu Q (2008b) D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J. Neurosci, 28, 14018–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Zhong P, Sun D & Liu Q (2011) Extracellular signal-regulated kinase signaling in the ventral tegmental area mediates cocaine-induced synaptic plasticity and rewarding effects. J. Neurosci, 31, 11244–11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet J-P, Touquet B, Pollegioni L, Poulain DA & Oliet SHR (2006) Glia-derived d-Serine controls NMDA receptor activity and synaptic memory. Cell, 125, 775–784. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR III, Lafaille JJ, Hempstead BL, Littman DR et al. (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul J-Y, Takano H, Moss SJ et al. (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science, 310, 113–116. [DOI] [PubMed] [Google Scholar]
- Pignatelli M & Bonci A (2015) Role of dopamine neurons in reward and aversion: a synaptic plasticity perspective. Neuron, 86, 1145–1157. [DOI] [PubMed] [Google Scholar]
- Polter AM, Bishop RA, Briand LA, Graziane NM, Pierce RC & Kauer JA (2014) Poststress block of kappa opioid receptors rescues long-term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking. Biol. Psychiat, 76, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu AT, Zhou MR & Poo M (2016) Phasic dopamine release in the medial prefrontal cortex enhances stimulus discrimination. Proc. Natl. Acad. Sci. USA, 113, E3169–E3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pougnet J-T, Toulme E, Martinez A, Choquet D, Hosy E & Boué-Grabot E (2014) ATP P2X receptors downregulate AMPA receptor trafficking and postsynaptic efficacy in hippocampal neurons. Neuron, 83, 417–430. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Vaughn JE, Saito K, Barber R & Roberts E (1976) Immunocytochemical localization of glutamate decarboxylase in rat substantia nigra. Brain Res, 116, 287–298. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A & Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron, 37, 577–582. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Brady RO Jr, Molliver ME & Snyder SH (1997) d-Serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J. Neurosci, 17, 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P & Montague PR (1997) A neural substrate of prediction and reward. Science, 275, 1593–1599. [DOI] [PubMed] [Google Scholar]
- Schweimer JV, Coullon GSL, Betts JF, Burnet PWJ, Engle SJ, Brandon NJ, Harrison PJ & Sharp T (2014) Increased burst-firing of ventral tegmental area dopaminergic neurons in d-amino acid oxidase knockout mice in vivo. Eur. J. Neurosci, 40, 2999–3009. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Pérez-Garci E, Schneider A, Kollewe A, Gauthier-Kemper A, Fritzius T, Raveh A, Dinamarca MC et al. (2016) Modular composition and dynamics of native GABAB receptors identified by high-resolution proteomics. Nat. Neurosci, 19, 233–242. [DOI] [PubMed] [Google Scholar]
- Scofield MD, Li H, Siemsen BM, Healey KL, Tran PK, Woronoff N, Boger HA, Kalivas PW et al. (2016) Cocaine self-administration and extinction leads to reduced glial fibrillary acidic protein expression and morphometric features of astrocytes in the nucleus accumbens core. Biol. Psychiat, 80, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif T, Simms JA, Lei K, Wegner S, Bonci A, Messing RO & Hopf FW (2015) D-Serine and D-cycloserine reduce compulsive alcohol intake in rats. Neuropsychopharmacol, 40, 2357–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Zhang S, Tang M, Zhang X, Zhou Z, Yin Y, Zhou Q, Huang Y et al. (2013) Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature, 494, 90–94. [DOI] [PubMed] [Google Scholar]
- Shoji Y, Delfs J & Williams JT (1999) Presynaptic inhibition of GABA (B)-mediated synaptic potentials in the ventral tegmental area during morphine withdrawal. J. Neurosci, 19, 2347–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery DA, Markou A, Froestl W & Cryan JF (2005) The GABAB receptor-positive modulator GS39783 and the GABAB receptor agonist baclofen attenuate the reward-facilitating effects of cocaine: intracranial self-stimulation studies in the rat. Neuropsychopharmacol, 30, 2065–2072. [DOI] [PubMed] [Google Scholar]
- Smith KL, John CS, Sypek EI, Öngür D, Cohen BM, Barry SM & Bechtholt AJ (2014) Exploring the role of central astrocytic glutamate uptake in ethanol reward in mice. Alcohol. Clin. Exp. Res, 38, 1307–1314. [DOI] [PubMed] [Google Scholar]
- Solecki W, Wickham RJ, Behrens S, Wang J, Zwerling B, Mason GF & Addy NA (2013) Differential role of ventral tegmental area acetylcholine and N-Methyl-D-Aspartate receptors in cocaine-seeking. Neuropharmacology, 75, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Svingos AL, Pickel VM & Henriksen SJ (1998) Electrophysiological characterization of GABAergic neurons in the ventral tegmental area. J. Neurosci, 18, 8003–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Klanker M, de Ridder B, Bowers MS, Joosten RN, Feenstra MG & Bonci A (2008) Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science, 321, 1690–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Johnson SW & North RA (1992) Synaptic inputs to GABAA and GABAB receptors originate from discrete afferent neurons. Neurosci. Lett, 134, 207–211. [DOI] [PubMed] [Google Scholar]
- Sun X-D, Li L, Liu F, Huang Z-H, Bean JC, Jiao H-F, Barik A, Kim S-M et al. (2016) Lrp4 in astrocytes modulates glutamatergic transmission. Nat. Neurosci, 19, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KR, Brown M, Labouèbe G, Yvon C, Creton C, Fritschy J-M, Rudolph U & Lüscher C (2010) Neural bases for addictive properties of benzodiazepines. Nature, 463, 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Malenka RC & Bonci A (2000) Modulation of long-term depression by dopamine in the mesolimbic system. J. Neurosci, 20, 5581–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL & Borgland SL (2013) Presynaptic leptin action suppresses excitatory synaptic transmission onto ventral tegmental area dopamine neurons. Biol. Psychiat, 73, 860–868. [DOI] [PubMed] [Google Scholar]
- Tian J, Huang R, Cohen JY, Osakada F, Kobak D, Machens CK, Callaway EM, Uchida N et al. (2016) Distributed and mixed information in monosynaptic inputs to dopamine neurons. Neuron, 91, 1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Z-Y, Overton PG & Clark D (1996) Antagonism of NMDA receptors but not AMPA/kainate receptors blocks bursting in dopaminergic neurons induced by electrical stimulation of the prefrontal cortex. J. Neural. Transm, 103, 889–904. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC & Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature, 411, 583–587. [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A & Uchida N (2012) Whole-Brain mapping of direct inputs to midbrain dopamine neurons. Neuron, 74, 858–873. [DOI] [PubMed] [Google Scholar]
- Whitaker LR, Degoulet M & Morikawa H (2013) Social deprivation enhances VTA synaptic plasticity and drug-induced contextual learning. Neuron, 77, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild AR, Bollands M, Morris PG & Jones S (2015) Mechanisms regulating spill-over of synaptic glutamate to extrasynaptic NMDA receptors in mouse substantia nigra dopaminergic neurons. Eur. J. Neurosci, 42, 2633–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA (2004) Dopamine, learning and motivation. Nat. Rev. Neurosci, 5, 483–494. [DOI] [PubMed] [Google Scholar]
- Wu YN, Mercuri NB & Johnson SW (1995) Presynaptic inhibition of gamma-aminobutyric acidB-mediated synaptic current by adenosine recorded in vitro in midbrain dopamine neurons. J. Pharmacol. Exp. Ther, 273, 576–581. [PubMed] [Google Scholar]
- Xia Y, Driscoll JR, Wilbrecht L, Margolis EB, Fields HL & Hjelmstad GO (2011) Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J. Neurosci, 31, 7811–7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata N, Ichinose T, Aso Y, Pla,cais P-Y, Friedrich AB, Sima RJ, Preat T, Rubin GM. et al. (2015) Distinct dopamine neurons mediate reward signals for short- and long-term memories. Proc. Natl. Acad. Sci. USA, 112, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M & Duan S (2003) Contribution of astrocytes to hippocampal long-term potentiation through release of d-serine. Proc. Natl. Acad. Sci. USA, 100, 15194–15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Qi Y & Yang Y (2015) Astrocytes control food intake by inhibiting AGRP neuron activity via adenosine A1 receptors. Cell Rep., 11, 798–807. [DOI] [PubMed] [Google Scholar]
- Yang J, Yang H, Liu Y, Li X, Qin L, Lou H, Duan S & Wang H (2016) Astrocytes contribute to synapse elimination via type 2 inositol 1,4,5-trisphosphate receptor-dependent release of ATP. eLife, 5, e15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J-H, Wang F, Krnjević K, Wang W, Xiong Z-G & Zhang J (2004) Presynaptic glycine receptors on GABAergic terminals facilitate discharge of dopaminergic neurons in ventral tegmental area. J. Neurosci, 24, 8961–8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Mameli M, O’Connor EC, Dey PN, Verpelli C, Sala C, Perez-Otano I, Lüscher C et al. (2013) Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron, 80, 1025–1038. [DOI] [PubMed] [Google Scholar]
- van Zessen R, Phillips JL, Budygin EA & Stuber GD (2012) Activation of VTA GABA neurons disrupts reward consumption. Neuron, 73, 1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, Wang W, Gu X et al. (2007) Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol, 9, 945–953. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P et al. (2014) An RNA-sequencing transcriptome and splicing database of Glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci, 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Xue L, Wang X, Jiang W, Xue Y, Liu J, He Y, Luo Y et al. (2011) NMDA receptor glycine modulatory site in the ventral tegmental area regulates the acquisition, retrieval, and reconsolidation of cocaine reward memory. Psychopharmacology, 221, 79–89. [DOI] [PubMed] [Google Scholar]