

Summary
Aberrant preterm infant gut microbiota assembly predisposes to early life disorders and persistent health problems. Here, we characterize gut microbiome dynamics over the first three months of life in 236 preterm infants hospitalized in three neonatal intensive care units using shotgun metagenomics of 2,512 stools and metatranscriptomics of 1,381 stools. Strain tracking, taxonomic and functional profiling, and comprehensive clinical metadata identify Enterobacteriaceae, Enterococci, and Staphylococci, as primarily exploiting available niches to populate the gut microbiome. Clostridioides difficile lineages persist between individuals in single centers, and Staphylococcus epidermidis lineages persist within and, unexpectedly, between centers. Collectively, antibiotic and non-antibiotic medications influence gut microbiome composition to greater extents than maternal or baseline variables. Finally, we identify a persistent low diversity gut microbiome in neonates who develop necrotizing enterocolitis after day of life 40. Overall, we comprehensively describe gut microbiome dynamics in response to medical interventions in preterm, hospitalized neonates.
Graphical Abstract
eTOC
Preterm neonates undergo numerous interventions. Thänert, Schwartz, and Keen et al. use multi-omics to show that antibiotics and other drugs alter gut microbiome development, more so than prenatal factors. Microbiome development stagnates before necrotizing enterocolitis ensues, but only among infants who experience this devastating complication after 40 days of life.
Introduction
Newborns acquire microbes during and after delivery in all body sites from their mothers and the environment1. Neonates continue to accrue microbes in their gut microbiome, an organ essential for immune and nutritional development2. Microbiome development is shaped by diverse factors, including environment, mode of delivery, antibiotic exposures, diet, and gestational age3–8. Worldwide, 10.6% of all infants are born prematurely9 (defined as birth before the 37th week of gestation) and many require months-long initial hospital care in neonatal intensive care units (NICU)10. NICU hospitalization and accompanying necessary interventions, including antibiotics and altered feeding interventions that differ from the home environment, can dramatically influence the developing gut microbiome4,11,12. In contrast to the rapid acquisition of commensal anaerobes observed in their term counterparts, the preterm infant gut is initially seeded by bacteria that include nosocomial pathobionts of hospital origin including Staphylococci, Enterococci, and Enterobacteriaceae4,13,14. These nosocomial pathobionts are abundant in the stool of infants born preterm, frequently dominate their host’s gut microbiome (to >50% of the overall community), and are transmitted between infants in the NICU environment11,13–17. The specific clinical variables and exposures governing pathobiont and commensal entrance and exclusion into the preterm microbiome remain incompletely understood.
The hospitalized preterm neonatal gut microbiome undergoes dynamic, choreographed development with species acquisition, in vivo evolution of individual microbes and the community collectively, and species loss, which are each governed by microbe-microbe, microbe-host, and exogenous factors2,4,11,18–20. Frequent antibiotic administration exacerbates this dynamicity by rapidly reshaping the content of the developing gut microbiome11,15. These rapid microbiome shifts have been well characterized in other dynamic populations such as adults with inflammatory bowel disease and traveler’s diarrhea21,22, but the relative contributions of each factor that dictate how the microbiome changes in hospitalized preterm neonates are largely undetermined. Much is known about the impact of antibiotics on the preterm gut microbiome11,12,19,20. However, we understand little about the early-life impact of other factors, together referred to as the exposome23, including comorbidities and non-antibiotic medications, many of which are known to have in vitro effects on human gut bacteria24. This is an important knowledge gap, because extremely low birthweight infants are routinely exposed to more medications, and for longer durations, than their higher birthweight counterparts in the same neonatal units25. The exposome varies in isolation and combination across intervals in neonatal care25, and the effect on microbiome composition and function is poorly understood. Importantly, alterations in specific functional units (e.g., genes, pathways) encoded in bacterial genomes, including those affecting immune homeostasis, can explain microbiome responses to perturbations not captured by taxonomic composition alone26–28. Thus, it is critical to study the presence or absence of both specific taxa and encoded microbiome genetic content to determine an infant’s susceptibility to infections and pathologies, and, more importantly, identify the precipitants of changes in community composition26,29.
In addition to unique medications and exposures, preterm neonates are at immense risk for devastating pathologies influenced by the gut microbiome, most notably necrotizing enterocolitis (NEC)30–34 and bloodstream infections caused by gut-resident pathobionts15,35,36. NEC is a necroinflammatory gastrointestinal disorder affecting ~5–10% of all very low birthweight infants in the US and remains fatal in 15–40% of cases37. Prediction of NEC prior to onset has been elusive, perhaps because a wide spectrum of disease presentation exists, and variable times to onset in the first several months of life33,38. Failure of the immature preterm neonatal immune system to control pro-inflammatory responses to an inciting gut microbiome is thought to be at the heart of NEC etiology33,39. Studies have variably reported reduced microbiota diversity, increased abundance of Enterobacteriacae, and under-representation of obligate anaerobes in the prelude to disease onset30,32,34,39. However, consistent compositional and functional signatures of microbiome communities preceding NEC, the identification of which would be essential to establish reliable biomarkers, remain elusive30–33,38–40.
Here, we sequence 1479 stools from 96 very low birthweight preterm infants, and analyze them with 409 previously sequenced stools from 92 additional preterm infants from the same cohort11,12,34,40 to comprehensively and precisely define gut microbiota assembly in preterm infants hospitalized in three NICUs in the central United States. We interrogate independent variables that are components of the NICU exposome, including antibiotic and non-antibiotic treatments, along with dependent variables of microbiome taxonomic and functional development during hospitalization. We use strain-resolved metagenomic assembled genomes (MAGs) to identify microbial dynamics within and between individuals in response to maternal factors and NICU interventions over time. Finally, we compare 96 infants without NEC using a 2:1 match to 48 infants with NEC, interrogating an additional 624 stools using shotgun metagenomics and metatranscriptomics to investigate taxonomic and functional microbiome signatures which may precede NEC onset.
Results
Gut microbiota assembly in hospitalized preterm infants
To characterize bacterial succession in the preterm gut, we performed fecal shotgun-metagenomic sequencing on 1479 stool samples (Data S1) collected at near-daily frequency from 96 preterm infants without NEC hospitalized in NICUs in St. Louis, Missouri (54/96, 56%), Oklahoma City, Oklahoma (32/96, 33%), and Louisville, Kentucky (10/96, 10%). These infants were part of a prospective cohort investigating the bacterial etiology of NEC among preterm infants but who did not develop NEC during hospitalization (Figure 1A, Data S1–S2)34,40. They were born at a mean gestational age (standard deviation) (GA) of 26.4 (2.5) weeks, with a mean birthweight of 938 g (273); 48/96 (50%) were female (Data S2).
Figure 1 |. Earliest preterm gut microbiota colonization in the NICU.
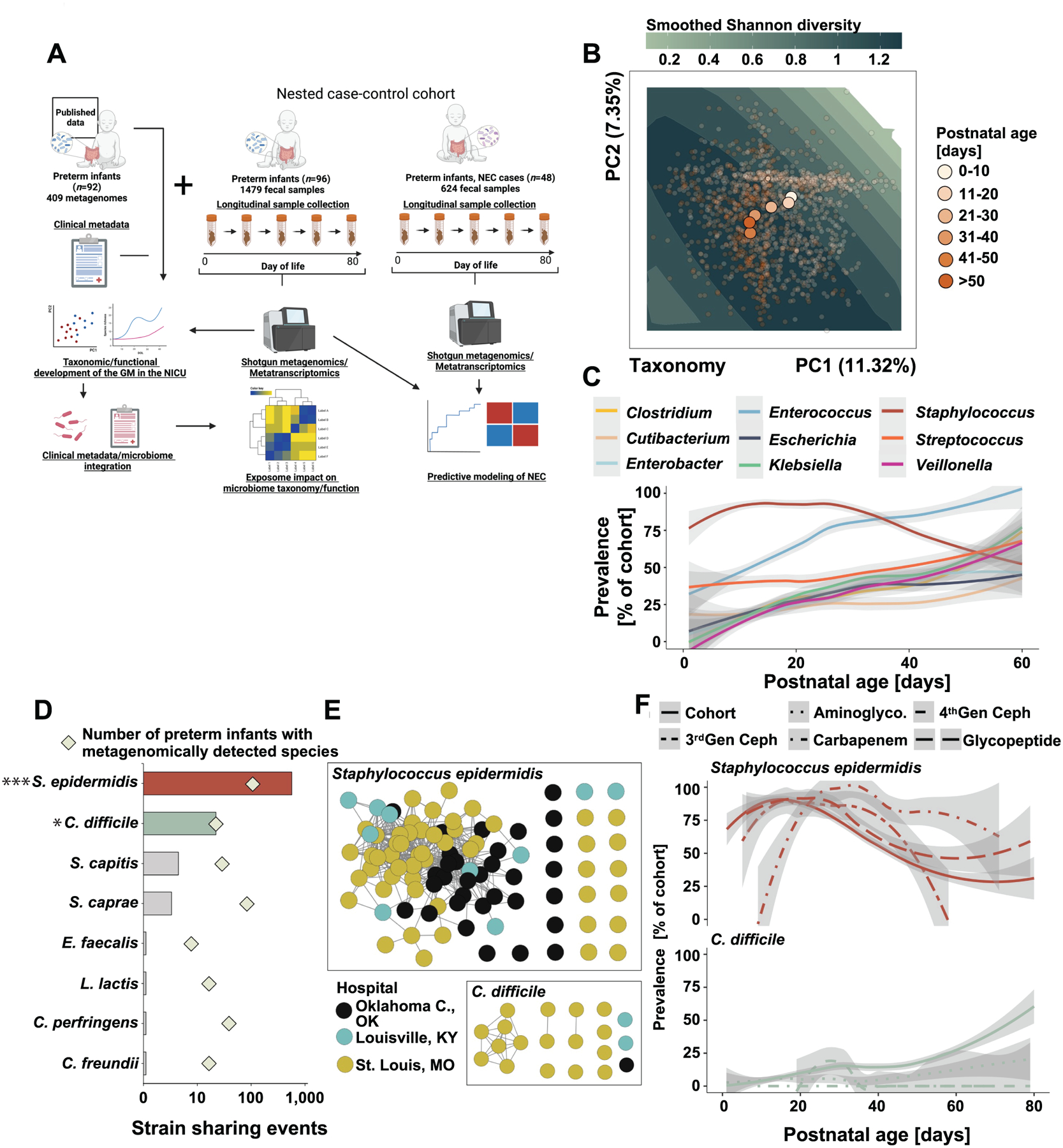
A) Schematic overview of the study design. Created with BioRender.com. B) Principal component analysis of the gut microbiota composition in 1479 samples collected from 96 of preterm infants over the first 80 days of life. Points are colored by postnatal age at sample collection and centroids of each postnatal age bin is plotted. Smoothed Shannon diversity is extrapolated based on the observed data distribution and plotted into the background. C) Prevalence over postnatal days for nine selected taxa. D) Number of pairwise strain sharing events (ANI>99.999 and breadth >0.5) across unrelated preterm infant pairs for the taxa with most such events. The number of infants metagenomically positive for the selected taxa is plotted as squares. ***, p<0.001 for S. epidermidis versus all other listed species. *, p<0.05 for C. difficile versus all species listed below it. Pairwise Chi-Square with post-hoc pairwise test with Benjamini-Hochberg adjustment. E) Network representation of strain sharing events for the two taxa with most such events across unrelated infant pairs. Each dot represents a unique preterm infant and is colored by its geographic location. F) Impact of antibiotic exposure on the prevalence of S. epidermidis (top) and C. difficile (bottom) across the entire control cohort over the first 80 days of life. Line patterns correspond to antibiotic category.
Gut microbiota development of hospitalized preterm infants has previously been described as following a common pattern, proceeding from initial dominance of Staphylococcus and other Bacilli in virtually all infants, to communities defined by pathobionts4,40. Our shotgun metagenomic data support these choreographed trajectories of the earliest microbiota assembly in preterm infants. The gut microbiota of preterm infants in our cohort diversified rapidly over the first month of life (Figure 1B, Figure S1), accruing taxa (Figure 1C, S1), and transitions from Staphylococcus epidermidis dominance to communities defined by potential pathobionts, specifically Klebsiella pneumoniae, Enterococcus faecalis, and Escherichia coli (Figure 1C, S1A). We recapitulate prior data4,11 demonstrating that pathobionts such as Enterobacteriaceae, E. faecalis, and S. epidermidis are highly prevalent and often dominate the microbiota of individual infants with >50% abundance (Figure S1B–C). Notably, we also find putatively beneficial early life colonizer microbes2,41, including Bifidobacterium spp. and Veillonella spp. (Figure S1A, S1C), demonstrating that important commensals are acquired even during hospitalization in the NICU.
Staphylococcus epidermidis and Clostridioides difficile strains are shared among infants
The earliest preterm gut microbiota is influenced by microbes acquired from the NICU environment4,13,14,17. While our study did not include environmental samples, strain-sharing between unrelated individuals is indirect evidence for environmental acquisition. Thus, to characterize species-specific microbe acquisition from environmental sources systematically, we co-assembled, binned, and taxonomically annotated infant-specific metagenome-assembled genomes (MAGs). We de-replicated a total of 1176 high- (n=864 MAGs, ≥90% completeness, ≤5% contamination) and medium- (n=312 MAGs, ≥50% completeness, ≤10% contamination) quality MAGs (Data S3), generating a set of 197 species-level representative genomes. Using this MAG database, we profiled species-specific pairwise average nucleotide identity (ANI) values for all metagenomic sample pairs using inStrain (see Methods)42 to identify instances of strain-sharing between unrelated infants.
We identified a total of 28,251 instances in which the same strain (≥99.999% popANI) was detected in multiple specimens across all species and sample pairs, of which 26,378 (93.4%) were from samples collected from the same infant over time. Eighty-two (85.4%) of the 96 infants studied shared at least one strain of at least one bacterial species with another infant. We specifically counted strain-sharing events only when ≥2 unique samples from the same infant pair shared a strain, to limit false positive associations. Strains of S. epidermidis and C. difficile were the most frequent species identified in stool samples of multiple, unrelated infants (Figure 1D, E). We did not observe a correlation between strain-sharing events and prevalence (p=0.53, Spearman). Strain-sharing events for S. epidermidis were more frequently observed than for other species with comparable cross-cohort prevalence (Figure 1D, Chi Square, p<0.001), indicating that environmental vectors may disproportionally contribute to their spread in the NICU or that these species are well-adapted to persist in the hospital environment. Strain sharing events for C. difficile were also more likely for its prevalence than other species (Figure 1D, Chi Square, p<0.05). S. epidermidis prevalence decreased with increasing day of life and was positively associated with carbapenems and glycopeptide exposure (Figure 1F). Conversely, C. difficile prevalence increased with day of life in the NICU and was negatively associated with administration of aminoglycosides, 4th generation cephalosporins, and carbapenems (Figure 1F). These observations suggest organism-specific dynamics of acquisition and spread, potentially associated with distinct environmental influences.
Surprisingly, we inferred 359 S. epidermidis strain sharing events across hospital systems (Figure 1E). To exclude the possibility that laboratory contamination explained this commonality, we randomly selected never-thawed stools from infant pairs hospitalized in three different centers (St. Louis, Louisville, and Oklahoma City) predicted to share S. epidermidis (n=21 pairs) and C. difficile (n=10 pairs) strains by sequence analysis. We then isolated single colonies from these stools and performed metagenomic sequencing, which corroborated our direct from stool sequencing that demonstrated within-hospital sharing of C. difficile and cross-hospital strain-sharing of S. epidermidis (Figure S2). Specifically, 14/21 (67%) and 7/10 (70%) of inStrain-predicted sharing events of S. epidermidis (Figure S2A–B) and C. difficile (Figure S2C–D), respectively, were validated with culturing using a relatedness cutoff of 99.999% popANI (Figure S2B,D, solid line). Strain sharing of Enterobacteriaceae, E. faecalis, and administered Bifidobacteria probiotics15,29 has been demonstrated within NICUs, and Enterobacteriaceae, Enterococcus faecium, Acinetobacter baumannii, and commercial probiotics43,44 in adults in ICUs. The identified network of S. epidermidis strains shared across large spatiotemporal distances suggests that specific strains are uniquely adapted to persist in the NICU environment.
Microbiome shifts introduce pathobionts and antibiotic resistance into the preterm gut microbiome
Changes in gut microbiota composition can rapidly alter microbial cues to the host’s immune system21. Such ‘microbiome shifts’ are thought to contribute to the pathophysiology of gastrointestinal disorders and infections in adult populations, such as inflammatory bowel disease and traveler’s diarrhea21,22. These events are characterized by interval community alterations between samplings that exceed the boundaries set by regularly observed intra-patient variation and are indistinguishable from inter-patient variation21,22.
To characterize the frequency, cause, and consequences of taxonomic microbiota shifts, we profiled Bray-Curtis dissimilarity in samples consecutively collected from the same participants. The composition of consecutive samples within a 20-day period were more similar to each other than to those from unrelated individuals (Figure S3A–B), allowing us to set a threshold for detecting microbiome shift events. We identified 131 microbiota shift events (Figure S3, 1.36/patient), defined as within-individual between-sample Bray-Curtis dissimilarity greater than the average dissimilarity of between-individual comparisons21,22 (Figure S3B). Shift events occurred significantly earlier than the average postnatal day of sample collection (Figure S3C, P=1.34e−07, two sample t-test), indicating greater instability closer to birth, and were associated with taxa appearances as well as disappearances (Data S19). Overall, shifts indicated punctuations of microbiome destabilization with dramatic changes in pathobiont abundance (Figure 2A, left) as well as discrete introductions (Figure 2A, right). Shifts were significantly more likely to introduce pathobionts (i.e., E. cloacae, E. coli, E. faecalis, and K. pneumoniae), into the preterm gut compared to non-shift sample pairs (Figure 2A, P≤0.01, permutation test). Shifts also depleted putatively beneficial taxa, including Bifidobacterium longum and Veillonella species as well as the same pathobionts introduced above (Figure S3). Common pathobionts introduced in shifts usually persisted over the subsequent 10 days (Figure 2B), suggesting that these community alterations have lasting influence on the developing preterm gut microbiome.
Figure 2 |. Antibiotic-induced microbiome shifts lead to abundance changes and introduction of pathobionts into the preterm infant gut.
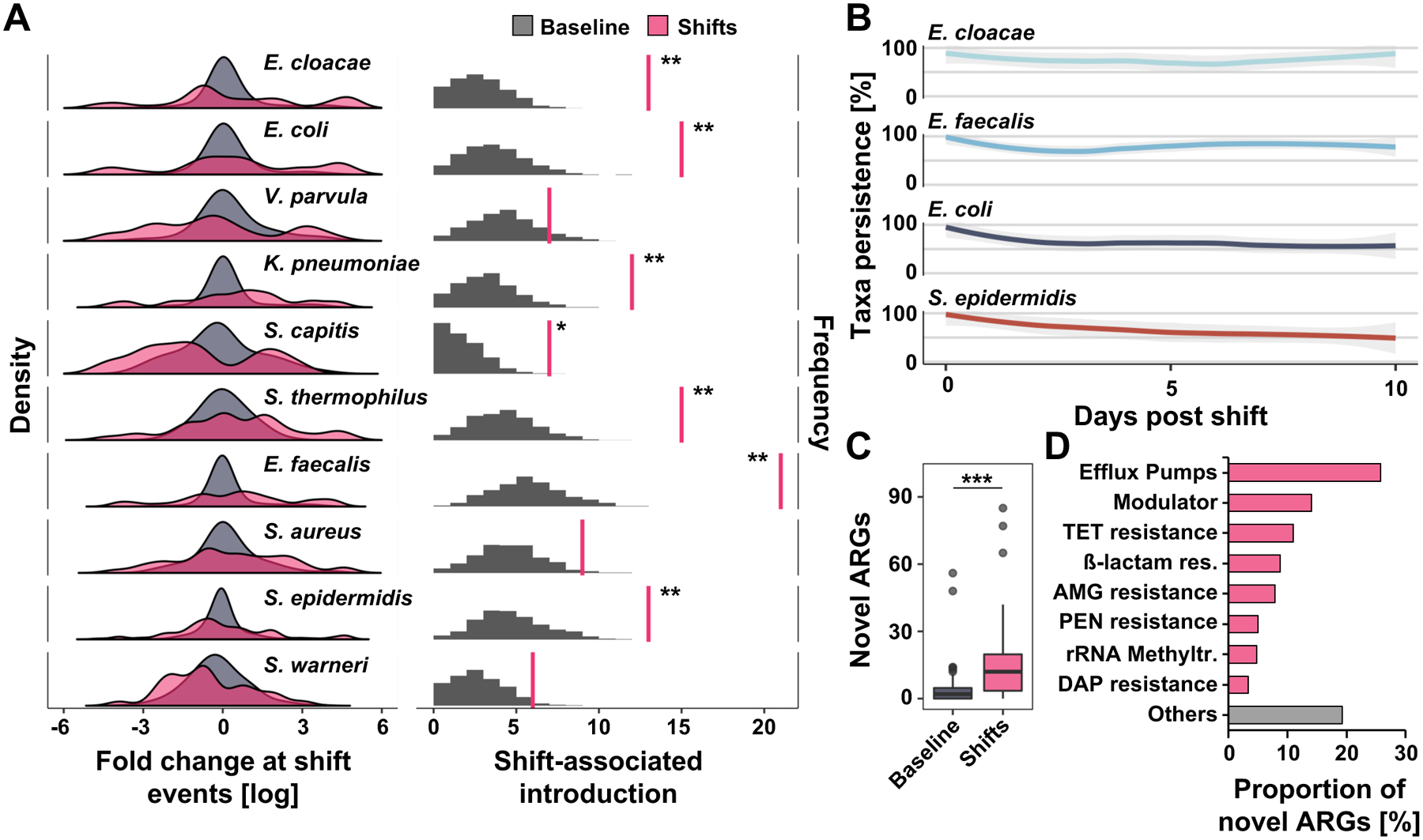
A) The left panel demonstrates density (frequency of events, Y axis) for abundance changes (X axis) of selected species associated with shift events (pink) compared to 1000 permutations of baseline non-shift events (gray). Microbiome shifts are defined as Bray-Curtis dissimilarities between consecutive samples collected from the same patient that are ≥ Bray-Curtis dissimilarities between samples from unrelated individuals collected with the same time interval (Figure S3 A and B). Right panel depicts number of shift-associated discrete introduction events when metagenomically absent prior (right pink vertical line) compared to permutation of expected introductions in non-shift baseline samples (gray, n = 262, permutation test, FDR-adjusted, *P=0.05, **P<0.01). Full taxa and statistics appear in data file S19. B) Persistence (relative abundance greater than 0%) of selected taxa introduced at shift events over ten post-shift days. C) Number of antibiotic resistance genes (ARGs) introduced at shift events and undiscovered in pre-shift samples compared to permuted non-shift samples (n=262, Wilcoxon test P=3.49e−07). D) Classification of ARGs introduced at shift events. N=1479 samples from 96 infants.
We also found that exposure to antibiotics in the prior 14 days, especially to broad-spectrum agents like 3rd and 4th generation cephalosporins, carbapenems, glycopeptides, and lincosamides, was associated with microbiome shifts (Figure S4A). Alternatively, it is also possible that the microbiome changes prompted clinical changes leading to antibiotic administration. Notably, antibiotic resistance genes not previously identified in the same infant’s gut microbiome were introduced during microbiota shifts compared to non-shift timepoints (Figure 2C, T-test P<0.001). These genes encode diverse mechanisms of antibiotic resistance (Figure 2D), including broad spectrum ß-lactamases (Figure S4B), suggesting that pathobionts introduced around shifts may proliferate because of their antimicrobial resistance gene repertoire. Consequently, our data suggest that microbiome shifts may act as a prime moment of vulnerability for clinical changes necessitating the introduction of antibiotics or antimicrobial-resistant pathobionts into the gut of hospitalized preterm infants, which further disrupt microbiome development.
The NICU exposome shapes microbiota development during hospitalization
In addition to antibiotics, hospitalized preterm infants are frequently exposed to a spectrum of host-directed medications and interventions45,46. These and other variables, such as mode and hospital of birth, maternal prenatal exposures, and infant diet, are collectively known as the ‘exposome’ and shape preterm infant gut microbiome trajectories5,11,12,47,48. To comprehensively characterize how the NICU exposome affects the preterm infant gut microbiota assembly and functional maturation during hospitalization, we integrated the 1479 metagenomic profiles of 96 infants generated above with shotgun metagenomic data from 409 stools from 92 different preterm infants from the same study population, described in previous work11,12. From this set, we synthesized 1,888 taxonomic profiles (Data S4) from 188 preterm infants collected over the first 88 days of life with extensive clinical metadata, including baseline variables, maternal information, antibiotic therapies, dietary exposures, and non-antibiotic medications (Data S5). Because of the rapid change of microbiota composition over the first weeks of life and the microbe-specific effects of some drugs4,11,24,49, we hypothesized that postnatal exposures would affect microbiome composition more individually and collectively than prenatal exposures. To address this hypothesis, we profiled the impact of metadata and the NICU exposome on Bray-Curtis dissimilarity of the gut microbiome (Figure 3A) using repeat-measures permutational analysis of variance (repeat PERMANOVA). As expected, given the longitudinal nature of our study, sample ID explained 60% of taxonomic and 59% of functional variance of the gut microbiome (Data S19, p=0.003). We divided variables into three categories: baseline/maternal/diet, antibiotic, and non-antibiotic medications, and found that only 3/27 (11%) of baseline/maternal/dietary variables were significantly (p<0.1) associated with taxonomic variance (Fig. 3B, Data S19). Postnatal age was the most explanatory feature (p=0.003), explaining 4% of taxonomic (Fig. 3A) and 6% of functional variance (Data S19). Cumulative formula and breastmilk exposure collectively contributed 1% to taxonomic variance. No other tested baseline or maternal variables, including hospital site, delivery mode, and gestational age were significantly associated with microbiome taxonomy or functional potential in the first 3 months of life (p>0.1, Data S19). In subsequent analyses, we focused on postnatal age instead of postconceptional age as we have previously done40 because gestational age was not significantly associated with metagenomic variation (p>0.05, Figure 3, Data S19). To further define the impact of postnatal age on microbiome variation, we divided our cohort into 10 DOL windows with samples from infants DOL > 50 lumped together. We found that postnatal age was significantly associated (p<0.005) with microbiome development in all DOL windows with the largest effect of 1.5% variance explained from samples at the highest DOL in our cohort (DOL 51–88, Data S19). As maternal and baseline variables have been associated with microbiome differences in term infants5,19,50, these data lead us to hypothesize that in preterm infants hospitalized in the NICU, early-life exposures contribute more to microbiome development than do prenatal exposures.
Figure 3 |. Impact of the NICU exposome on the developing preterm gut microbiome.
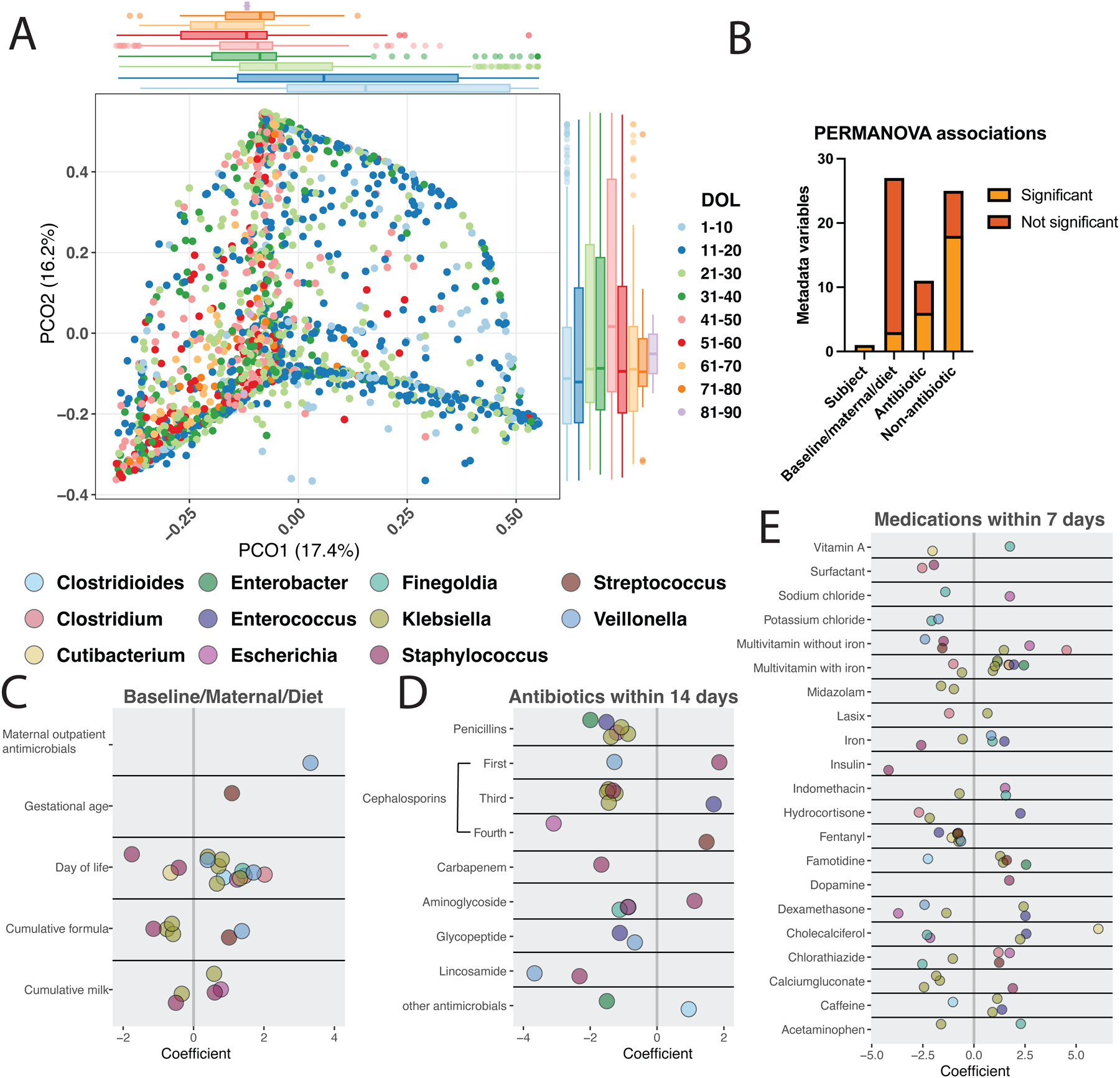
A) PCoA of Bray-Curtis dissimilarity of 1888 samples from 188 infants during the first 90 days of NICU hospitalization. Boxplots display PCO1 and PCO2 colored by postnatal age with variance explained in parentheses. B) Significant (p<0.1) and not significant (p>0.1) associations by metadata variable class are displayed as determined by repeat measures PERMANOVA. C-E) Coefficient of change of each genus corresponding to species significantly (q<0.05) changed based on the listed variable using MaAsLin2 with random effects of participant, hospital site, and study (this study, Gibson et al11, or Gasparrini et al12). Exposures affecting less than 1% of samples and fixed effects q>0.05 are not displayed, but full associations (q<0.25 per MaAsLin2 default) are listed in Data S19.
Collectively, 6/11 (55%) of antibiotic class exposures in the prior 14 days were significantly associated with microbiome taxonomic composition, and contributed 6% variance (Fig. 3B, Data S19). Surprisingly, 17/25 (68%) of non-antibiotic medication exposures in the 7 days prior to sample collection were associated with taxonomic composition, collectively contributing 10% variance (p<0.1, Fig. 3A–B, Data S19). Because medications in the NICU are often used concurrently, we determined how frequently medications were co-administered to understand the influence of non-antibiotic medications. We found that apart from the co-administration of ampicillin and gentamicin, vancomycin and gentamicin, and caffeine and gentamicin, most medication exposures did not overlap (Figure S5). Taken together, these findings suggest that the dynamics of microbiota assembly in the NICU during the first weeks of life are ordained by postnatal environmental exposures to a greater extent than by prenatal biology.
Antibiotic and non-antibiotic treatments affect early-life microbiome composition
To identify how the NICU exposome shapes taxa abundance trajectories, we implemented per-feature general linear mixed models (GLMMs) using MaAsLin251. We included participant, hospital site, and study (this study, Gibson et al11, or Gasparrini et al12) as random effects and evaluated all 62 baseline/maternal/diet, antibiotic, and non-antibiotic medication variables (Figure 3B, Data S5). This method corrects for known confounders and deconvolutes the effects of concurrent exposures but does not address the interactions between variables51. Although prenatal and maternal variables did not explain overall taxonomic variance (Figure 3B, Data S19), pre-birth maternal outpatient antimicrobials and increased gestational age were each associated with increases in single species (Figure 3C, q≤0.05). We hypothesized that prenatal and baseline variables might be more important very early in life because we did not observe that they drove microbiome composition overall (Figure 3B). We then reduced our dataset to 190 samples from 73 participants from the first 10 DOL and evaluated only the 27 baseline/maternal/diet variables and similarly found no maternal or baseline variables impacting taxonomy (q>0.05, Data S19). We conclude that maternal and baseline variables have minimal impact on early-life microbiome composition for preterm hospitalized infants.
Increasing postnatal age was associated with decreases in S. epidermidis, S. warneri, and C. avidum (Figure 3C) consistent with what we observed on the smaller cohort above (Figure S1C). DOL was also associated with increases in anaerobes such as 2 Veillonella spp., C. difficile, Finegoldia magna, and 2 Clostridium spp. as well as potentially pathogenic Enterobacteriaceae such as 5 Klebsiella spp. and E. coli (Figure 3C, Data S19). Cumulative dietary exposures, coded as fraction of days exposed to formula or breastmilk prior to a sample, were associated with alterations in key potential pathogens such as Klebsiella, E. coli, and Staphylococcus spp. (Figure 3C). Collectively, antibiotic administration within the 14 days prior to a sample was associated with abundance decreases of 24 species and increases of 5 species (q≤0.05, Figure 3D, Data S19). Aminoglycoside exposure within the prior 14 days, the most common exposure in our cohort affecting 946/1888 (50%) of samples, was associated with decreased abundance of F. magna, S. aureus, and C. difficile with increased abundance of S. epidermidis (Figure 3D). β-lactam antibiotics were associated with decreases of Enterobacteriaceae with the strongest effect of fourth generation cephalosporins in decreasing E. coli abundance (coefficient = −3.1, q=0.007). Penicillins (ampicillin, ticarcillin-clavulanate, ampicillin-sulbactam) and third generation cephalosporins were associated with decreased abundance of numerous Klebsiella species (Figure 3C). Recent third generation cephalosporins were also associated with increased E. faecalis abundance, which is likely due to their intrinsic cephalosporin resistance52. Vancomycin was associated with decreased abundance of E. faecalis consistent with known susceptibility52 and Veillonella dispar, which may reflect susceptibility given strain-level variation of Veillonella spp.53. Veillonella spp. were also depleted by recent exposure to first generation cephalosporins (coefficient = −1.3, q=0.03) and lincosamides (coefficient = −3.7, q=0.001). Thus, we demonstrate abundance decreases of many potential pathogens (Enterobacteriaceae, Staphylococcus spp., E. faecalis) as well as putatively beneficial taxa including Veillonella. While we posit that the observed changes are likely due to direct killing of susceptible bacteria from the administered antibiotic, we cannot rule out that abundance changes may be secondary to the time gap between the last dose of antibiotics and sample production with concomitant increases in other microbes or the co-administration of ampicillin or vancomycin with gentamicin (Figure S5). Indeed, we have demonstrated co-exposure of these antibiotics can alter gut microbiome abundance of Enterococcus and Enterobacteriaceae distinctly than when they are administered separately15.
We next investigated specific associations between non-antibiotic medications and gut microbiome taxonomic content. Unlike antibiotics, which were largely associated with taxonomic decreases, non-antibiotic medication exposures were associated with bi-directional changes, with 41 increases and 38 decreases of key taxa with overall prevalence greater than 10% (q≤0.05, Figure 3E, Data S19). Interestingly, the coefficient of change was often greater for non-antibiotic than antibiotic exposures (Figure 3C–E), effects that have been observed in adults with cardiometabolic disease54. 85 individual species were significantly associated (q≤0.05) with non-antibiotic exposures. Klebsiella spp. were the most frequently impacted genus accounting for 26/79 (32%) of associations with medications affecting greater than 1% of samples. Oral multivitamins and famotidine were associated with increases of 6 Klebsiella spp. and fentanyl, midazolam, and calcium gluconate were associated with decreases of 8 Klebsiella spp. (Figure 3E, Data S19). Caffeine, the most common medication overall affecting 1668/1888 (88%) of samples, was associated with decreased C. difficile, increases in two Klebsiella spp., and increased E. faecalis (Figure 3E). Oral iron, affecting nearly 1/3 of samples, was associated with decreases in S. epidermidis and K. michiganensis and increases in V. dispar and F. magna. Though some of these changes may result from the increased age of iron supplementation (43 days in exposed versus 22 days in unexposed infants, Data S5), these data also suggest that iron supplementation may lead to increases in beneficial Veillonella and Finegoldia, reflecting a more mature microbiome composition55. Fentanyl exposure in 467/1888 (25%) of samples was associated with decreased relative abundance of E. faecalis, Cutibacterium avidum, F. magna, 3 Klebsiella spp., and V. dispar. These decreases might be related to opioid-induced changes in motility or secondary to other microbial changes leading to differences in the intestinal micro-environment56. E. faecalis abundance increases were commonly associated with non-antibiotic medications including steroids (dexamethasone and hydrocortisone), iron, multivitamins with iron, and cholecalciferol. Interestingly, enterococci must acquire iron for virulence and anaerobic growth in the intestine57,58. Collectively, these results demonstrate taxonomic abundance changes associated with antibiotic and non-antibiotic exposures or the medical conditions for which they were administered for both pathobionts and key gut commensals in the preterm infant gut during NICU hospitalization.
Developmental trajectory of functions encoded by the preterm gut microbiome
Functions encoded by gut microbes are critical for infant development, because they provide vital nutrients, educate intestinal immunity, and mediate gut barrier function, important for the defense against bacterial infections of gut origin5,19,26,27. Despite its central role for infant health, our understanding of the functions encoded by the NICU-preterm gut microbiome remains rudimentary.
To better understand the kinetics of gut microbiome development, we established detailed trajectories of functional assembly of preterm gut microbiota. To do this, we mapped our shotgun metagenomic microbiome functional profiles (Data S7) to 1381 metatranscriptomes from the same stools (Data S8–S9), to capture the transcription of the encoded metabolic potential. Generally, taxonomic potential and expression corresponded to the taxonomic composition of the preterm gut microbiome, with pathway richness and transcription increasing during postnatal development (Figure S6A–E). However, we found considerable variation in transcription by functional group. Expression of core functional groups, including glycolysis and nucleoside/nucleotide biosynthesis found in virtually all microbiomes, changed less over postnatal development than did expression of genes encoding amino acid biosynthesis, carbohydrate degradation, or fatty acid and lipid biosynthesis (Figure 4A). Critically, amino acid and fatty acid biosynthesis by the microbiome are considered important for host immune modulation and development3,59,60, highlighting that the transcriptional trajectories of these functional groups during postnatal maturation may directly affect infant health. Enterobacteriaceae dominated the expression of pathways changing during postnatal development, with genomes of Escherichia spp., Klebsiella spp., and Enterobacter spp. responsible for their transcription (Figure 4B, C). Conversely, the transcriptional contribution of key commensals, including Veillonella and Clostridium species (Figure S6G), was less pronounced than the dominant effect of pathobionts on the transcriptional landscape during the first weeks of life.
Figure 4 |. Transcriptional trajectories of the gut microbiome are shaped by Enterobacteriaceae and NICU exposures.
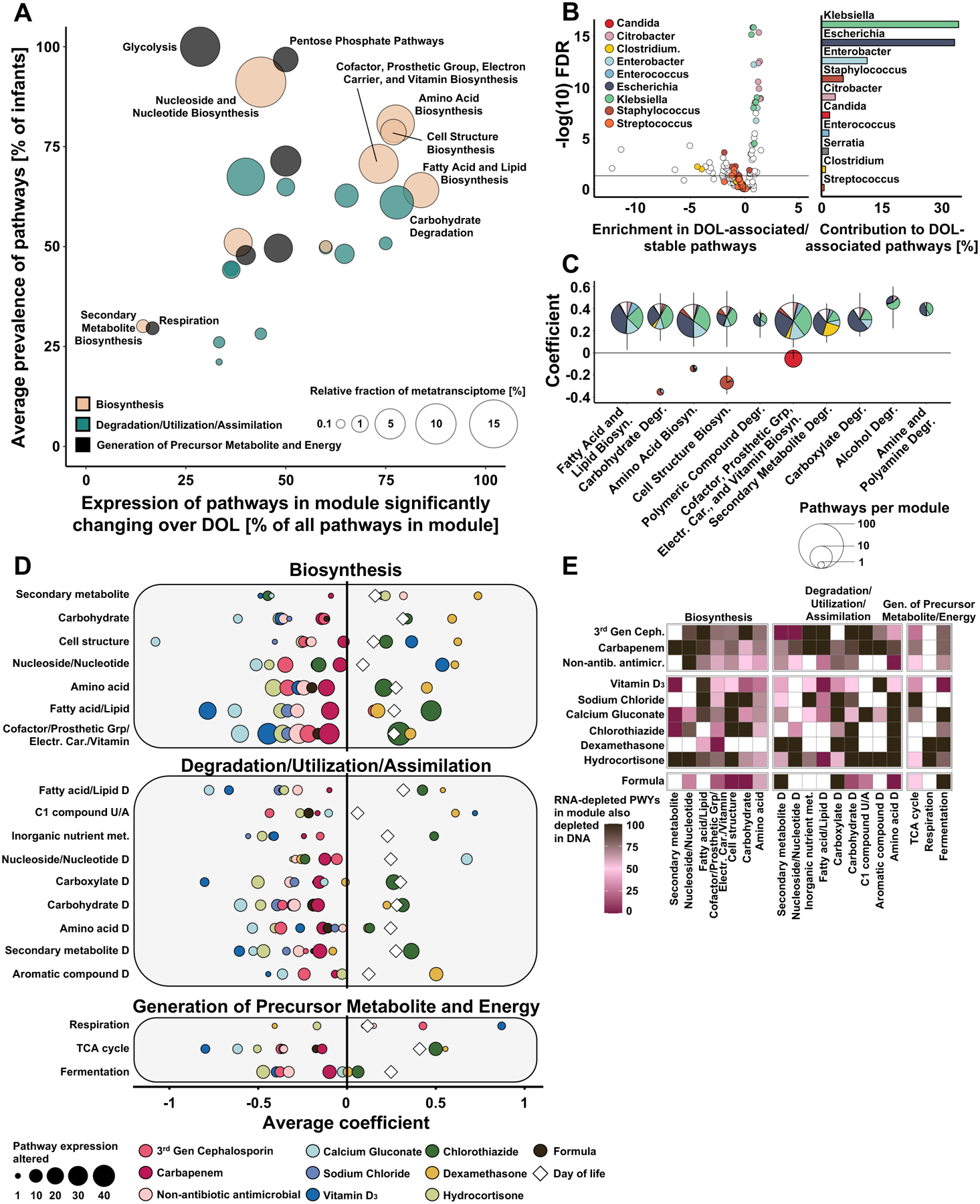
A) Characterization of transcriptional activity changes during NICU hospitalization. Pathways are colored by highest level functional categories and significant change over postnatal days is determined with MaAsLin2 (q<0.25). B) Species enrichment in pathways with stable or fold change in expression over postnatal days. C) Species association with functional categories with significant expression changes over postnatal days of life. D) Significant association of NICU exposures with changes in pathway expression. Exposures are indicated by color. Bubble size corresponds to the number of pathways within each functional category with significant expression changes associated with a given exposure. DataS19 has unaggregated and aggregated associations. E) Heatmap of pathway expression changes significantly associated with selected exposures indicating presence of concurrent changes in DNA abundance (MaAsLin2 q<0.25). Color scale represents percent change. N= 1381 samples from 95 infants.
Because we found evidence that the NICU exposome shapes the taxonomic composition of the preterm gut microbiota (Figure 3), we hypothesized that the environment also influences the transcription of genes encoding functional trajectories. Thus, we implemented GLMMs as described above and comprehensively characterized the impact of the NICU exposome on the expression of metabolic pathways. We identified a substantial impact of dietary, antibiotic, and non-antibiotic medications on the transcription of key metabolic units, including secondary metabolite, amino acid, vitamin, and fatty acid biosynthesis (Figure 4D). While antibiotics and most non-antibiotic medications were largely associated with reduced expression of metabolic potential, chlorothiazide and dexamethasone were associated with increased expression of multiple functional pathways. Importantly, transcriptional microbiome restructuring was not restricted to single pathways but frequently affected multiple functionalities (Figure 4D), highlighting the vast and overlooked impact of the NICU exposome on the preterm gut metatranscriptome.
We next asked if the observed transcriptional response was an indirect consequence of taxa abundance alterations or was driven by transcriptional regulation. To answer this question, we profiled concordance between taxonomic responses to NICU exposures on the DNA and RNA content using GLMMs. We found that while negative transcriptional responses to antibiotics were largely explained by taxa depletion rather than regulation, pathways were often downregulated in response to non-antibiotic medications and diet exposures (Figure 4E). Specifically, transcriptional responses to milk formula and Vitamin D3 were mostly unexplained by taxa depletion, suggesting active transcriptional regulation by the intestinal microbiome in response to early-life exposures.
Multi-omics analysis supports a variant microbial population as a risk factor for a subset of NEC
Previous work suggested that aberrant developmental progression of the preterm gut microbiome plays a fundamental role in the development of NEC30,32,34. Here, we used multi-omic characterization of the intestinal microbiota in hospitalized, non-NEC preterm infants to systematically query gut microbiome developmental trajectories of infants that developed NEC at any point during their NICU stay for differences in bacterial composition or function. We generated 624 longitudinal shotgun metagenomic and transcriptional profiles of the pre-NEC gut microbiome in 48 infants (see Methods, Data S1, S10–13). Comparisons were performed using a 1:2 matched case-control study design (Figure 1A), where controls were matched to cases based on NICU location (St. Louis, Louisville, and Oklahoma City), gestational age (within one week), and birthweight (within 100g). Using repeat measures PERMANOVA, and the entire NEC cohort specimens, neither the taxonomic composition of the pre-onset NEC microbiome or metatranscriptome, nor its functional profiles, varied significantly by NEC status (Figure 5A). We also found no significant difference in the bacterial replication rate of any taxonomic group prior to NEC onset (Figure 5B, Data S14), as has been previously reported32. Similarly, we found no difference in the community level virulence factor repertoire of cases and controls (Figure S7A–B, Data S15–17). Further, microbiota shifts were neither more frequent (Chi-square test P=0.876) nor occurred at different times relative to disease onset in the NEC case group (Figure 5C, Wilcoxon rank sum test P=0.664).
Figure 5 |. Gut microbiome features do not predict NEC onset for all preterm infants but differentiate late-onset cases and controls.
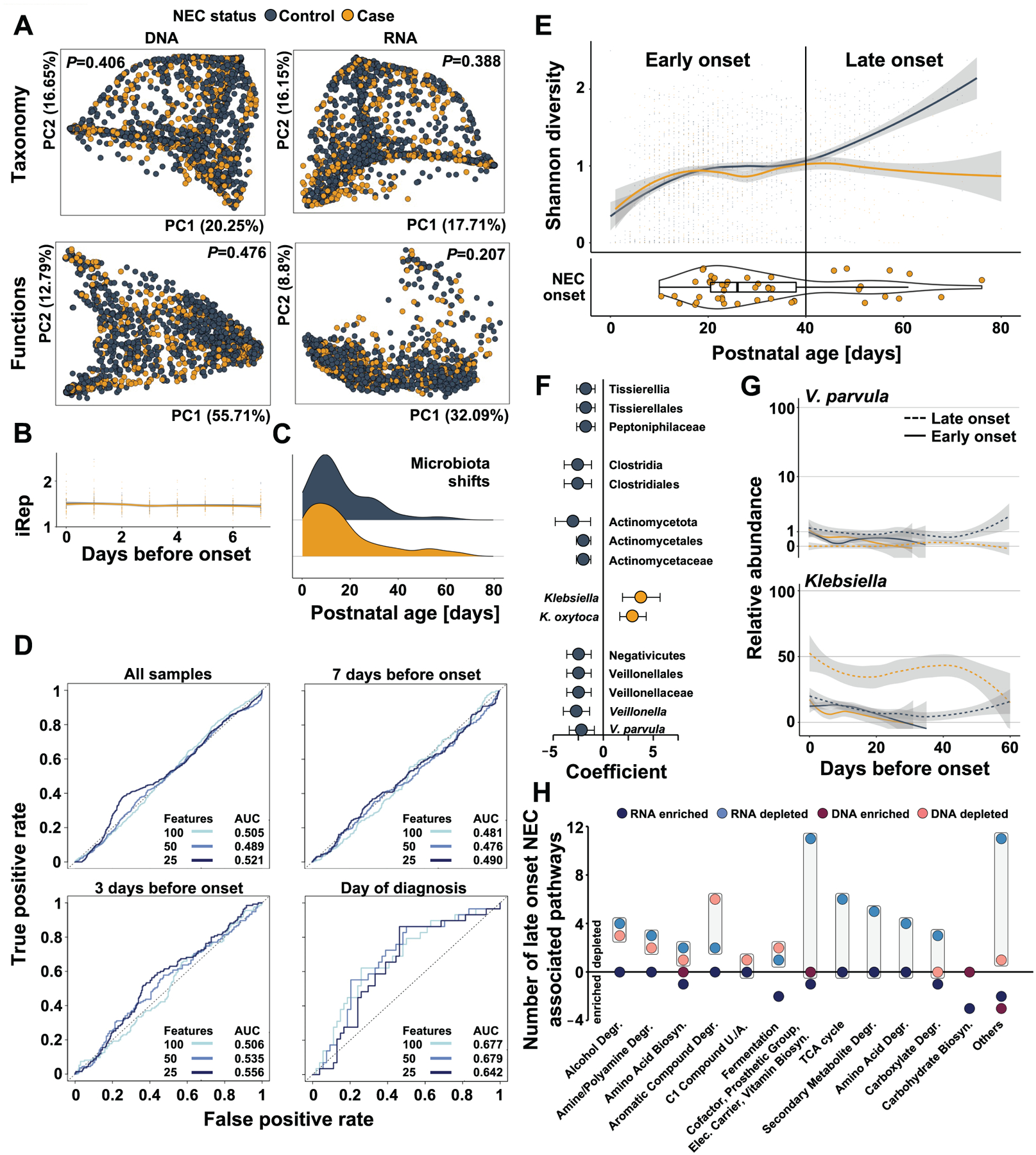
A) Principal coordinate analysis of pre-NEC gut microbiome taxonomic and functional composition in NEC cases (yellow) and matched controls (black). B) Bacterial replication rates in samples collected in the weak prior to NEC onset in cases (yellow) and matched controls (black, GLMM P>0.25). C) Distribution of microbiota shift events prior to disease onset in stool samples collected from NEC cases (yellow) and matched controls (black). D) Receiver operating characteristic curves of logistic regression models accounting for repeat measures utilizing all microbiome data collected from all samples, or samples taken in the week before, 3 days before, or at NEC onset. E) Shannon diversity of NEC cases (yellow) and matched controls (black) over postnatal days (top). Age of cases at disease onset in postnatal days (bottom). F) Taxa significantly associated with case or control status in late onset NEC when accounting for confounding exposures (MaAsLin2, q<0.25). G) Abundance of V. parvula (top) and Klebsiella (bottom) over 60 days before onset in early (solid) and late (dashed) NEC cases (yellow) and matched controls (solid). H) Functional pathway expression and abundance associated with late onset NEC. Boxes highlight the difference between the number of DNA-encoded or RNA-encoded pathways in cases vs controls. N=2103 samples from 144 infants.
As signatures of microbiome alterations preceding NEC might be more subtle, i.e., with effects limited to individual taxa or pathways, we next implemented GLMMs. Adjusting for all confounders that were associated with preterm gut microbiota development independent of disease status (i.e., in the control-only analysis), we found no association of any species, metabolic pathway, or transcriptional signature with case or control status (MaAsLin2 all q≥0.25). Further, investigating high-quality MAGs of the species K. pneumoniae, a genomically diverse species that has previously been implicated in NEC development32, we found no virulence repertoire differences between cases and controls (Figure S7C).
To account for the possibility that multi-level associations may explain individualized risk for NEC, we combined all multi-omic data (metagenomic profiles, metatranscriptomes, bacterial replication rate, community- and MAG-associated virulence profiles) using three distinct statistical modeling approaches (logistic regression, elastic net, random forest). Critically, we accounted for repeat-measures by blocking model cross-validation by individual and nesting three iterations of feature selection (25, 50, 100 features) within the cross-validation procedure, performing feature selection for each training-fold separately. Moreover, we trained models using either all pre-NEC data, a subset of data collected during the 7 or 3 days prior to onset, or on the day of clinical diagnosis. Model performance did not differ between algorithms or by number of features included in the final model (Figure 5D, Figure S8). We observed an increase in model performance peaking on the day of diagnosis, indicating that microbiome signatures are more distinguishable between cases and controls when the clinical diagnosis is made. Generally, models did not perform better than random chance, highlighting that the population-level and genome-resolved microbiome features collected in this study do not support a common microbiome imbalance that is uniformly detectable prior to the development of NEC.
While not reliably predicting NEC in all infants, we did observe a significant difference of microbiota diversity when examined by postnatal age of NEC occurrence (Figure 5E, GLMM P<0.05). Specifically, the microbiota diversity of cases and controls separated beyond postnatal day 40, when NEC case gut microbiota failed to increase in diversity in contrast to the gut microbiota in the matched case group. Twelve infants (25% of all cases) developed NEC beyond postnatal day 40, hereafter referred to as ‘late onset’ NEC. When compared to infants with onset ≤postnatal day 40 (‘early onset’), late onset cases were born at significantly younger gestational ages (24.0 (IQR 23.0–25) vs. 27.0 (IQR 25.0–28.8) weeks, Mann-Whitney p=0.0002) and lower birthweights (695g (IQR 561–798) vs 907g (IQR 773–1175) g, Mann-Whitney p=0.0006). This is consistent with reports that infants of shorter gestational age at birth develop NEC later compared to infants born following longer gestation33, and also inferred in the predecessor study34.
While no microbiome feature was significantly associated with NEC status when analyzing early onset cases and matched controls separately (MaAsLin2 all q≥0.25), several taxa, pathways, and transcriptional signatures separated the microbiomes of late onset cases from matched controls. For late onset NEC, Negativicutes, and in particular Veillonella parvula, and Clostridia, were significantly associated with control status, while Klebsiella abundance was significantly increased before the event (Figure 5F, G). When applying Dirichlet multinomial modeling to the taxonomic gut microbiota composition of cases and controls we similarly found that early onset cases and controls were indistinguishable, while the developmental enterotype trajectories of late cases and controls diverged (Figure S9). Specifically, we found that enterotypes 2–5 were over-represented among late-onset NEC cases relative to controls. Conversely, the stools of none of the late onset cases had gut microbiome compositions consistent with enterotypes 6–8 (Figure S9). Further, late-onset NEC was preceded by significant alterations in the functional maturation of the gut microbiome compared to control infants (Figure 5H). While these signatures were apparent at the level of the metagenome, they were even more pronounced, and often only detectable, in analysis of bacterial transcripts. Thus, several metabolic groups, including cofactor/prosthetic group/electron carrier/vitamin biosynthesis, TCA cycle, secondary metabolite degradation, and amino acid degradation exhibited altered transcription (vs. controls) without underlying metagenomic differences between cases and controls for several pathways (Figure 5H). Collectively, these data show that while the microbiome may not hold the key for individualized risk prediction for all infants born preterm, it may forecast disease onset after postnatal day 40, and that transcriptional analysis offers greater discriminatory information than metagenomic profiling alone.
Discussion
Intestinal host-microbe interactions educate a newborn’s immune system and affect health trajectories into adulthood50,61. Preterm infants often require months-long hospitalization in the NICU, an environment depleted of reservoirs of normal microbiota and enriched in microbiota-altering medical therapies and dietary regimens14,19. It has been proposed that preterm birth and the NICU exposome interact to individualize neonatal microbiota trajectories that affect early-life health outcomes11–13,15,19. Here, we characterized functional and taxonomic trajectories of the neonatal microbiome of 188 hospitalized preterm infants without NEC in the context of their baseline characteristics and NICU exposures, and found that the hospital environment is a major factor in seeding preterm infant bacterial gut microbiomes. Most noticeably, isogenic S. epidermidis are present in sites of care in three different states within the USA. This suggests that NICU-adapted lineages of this species could be universal. Given that S. epidermidis can be pathogenic in the NICU, including as a cause of late onset sepsis62,63, further work is warranted to validate and extend this unexpected finding. Sharing of other species previously reported to be common between infant guts and NICU surfaces13–15,17, specifically Enterobacteriaceae, Enterococcaceae, and Pseudomonas, was less frequently observed across infants and between centers in this study.
Consistent with previous reports11,12,15, we also find that antibiotic exposure is associated with profound and consequential microbiome shifts throughout the first 88 days of life. These events are not benign: they allow pathobionts to colonize hospitalized preterm infants and enable bacteria with genes encoding antibiotic resistance to enter the gut habitat. Alternatively, it is possible that gut entry of the organism leads to a clinical change prompting antibiotic administration. Teasing apart the temporal nature of these events will be important to minimize negative consequences of antibiotic prescribing64,65 while ensuring rapid treatment of potential sepsis66. Hence, our data reinforce concern that antibiotics alter bacterial community shifts that spread clinically-relevant resistance elements into and within newborn gut microbiomes, and that these organisms persist as colonizers4,11,12,36,67. Drug-resistant microbes and resistance genes enriched in preterm infant gut microbiomes during early-life hospitalization can be detected in stool long after infants have been discharged and can be sources of bloodstream infection in the NICU12,67, highlighting the durability and clinical relevance of early-life microbiome disruptions.
We also demonstrate significant microbiota alterations associated with non-antibiotic medications in preterm infants. While some effects might be explained by co-administration with antibiotics, we found that most medications that affected the microbiome were prescribed in isolation (Figure S5). Importantly, we cannot exclude that observed effects on microbial communities are caused by the pathology that obligates the medications rather than the medications per se. Our results are further supported by in vitro work that showed broad effects of non-antibiotic medications on gut microbiota using drug-by-microbe growth assays24. Intriguingly, microbial resistance mechanisms can be shared between some human-targeted drugs and antibiotics24. Therefore, our findings suggest that non-antibiotic medications may contribute to the high prevalence of drug resistance in the NICU microbiota. Further, we find that postnatal exposures explained more microbiome variation and were significantly associated with greater taxonomic changes than prenatal and baseline variables. These observations highlight the importance of high-frequency temporal sampling and integration of clinical variables and environmental exposures when profiling microbiome-associated health risks in preterm infants.
Our research adds to the literature on the role of the preterm infant gut microbiome in the multifactorial pathophysiology of NEC30,33. A current hypothesis of NEC development proposes that aberrant microbial colonization interacts with immature intestinal immunity to generate an uncontrolled inflammatory response, causing loss of barrier integrity and tissue necrosis33. At the aggregate cohort level, our results do not support this variant microbiome hypothesis, as pre-NEC multi-omic trajectories do not vary between cases and matched controls. Also, we did not identify correlates of disease onset and multi-omic integration via statistical modeling as predictors of NEC risk better than random chance before the day of onset. Nonetheless, we identified a subset of infants born after significantly shorter gestations and who developed NEC after postnatal day 40, whose microbiome trajectories differed compared to their matched controls. Despite some differences in the cases and controls studied, this finding resembles trends reported from these cohorts using 16S rRNA gene sequencing34. These infants’ microbiomes are characterized by reduced microbial diversity, increased abundance of Klebsiella, and decreased abundance of specific commensals, predominantly Veillonella. Signatures of functional microbiome disruption were most apparent in the metatranscriptome, which showed extensive functional alterations compared to matched control infants. Importantly, however, the microbiome profiles of early- and late-onset NEC cases do not differ, suggesting that NEC in infants who are born most preterm could be caused by failure of microbiome maturation, with the consequence that pathobionts persist in a more susceptible intestinal tract, i.e., a two-hit predilection. While this refined hypothesis de-emphasizes a microbiome-exclusive role in NEC causality, it extends prior work in this field that suggests fundamental biologic differences in NEC by postnatal age of onset33. Even though histologically and clinically early- and late-onset NEC are quite similar (though the latter has greater mortality37), the actual occurrence of late onset NEC (i.e., in this study after day of life 40) could relate to yet-to-be identified host responses to specific gut bacteria. It remains to be established if pre-NEC microbiome differences reflect a precipitating effect (e.g., from Enterobacteriaceae39), a lack of a protective effect (e.g., from Veillonella), or if the at-risk mucosa is more hospitable for an aberrant microbiome. Nevertheless, our data suggest that strategies to promote maturation of gut bacterial communities such as limiting unnecessary antimicrobials20,68 and possibly non-antibacterial agents might protect children born most prematurely from NEC69.
Our study has several important limitations. First, at the time of fecal sample collection, RNA transcriptomics was not planned, so preservatives were not included in storage. All samples were handled and stored for similar intervals, but we cannot exclude transcript degradation or other changes before RNA extraction. Our metagenomic associations and strain tracking are conducted on an average of 5 million reads per sample. We have previously shown this to be adequate for tracking strains between the bloodstream and gut for organisms with detectable metagenomic depth at this sequencing threshold15. Although the gut microbiome of the preterm infants studied here has low diversity, it is possible we cannot adequately track low abundance strains with inStrain or iRep42. Given these limitations, we suggest that future studies utilize appropriate preservatives and incorporate higher sequencing depth and measurements of absolute abundance to validate and extend the findings presented here. Further, our associations, like many in the microbiome field, are determined using relative abundance, which can obscure changes in absolute microbial abundance4. We would also note that breastmilk from each mother differs in composition and antibody content with similarly varied potential effects on the infant gut microbiome55,70. Additionally, the daily fraction of nutrition derived from formula versus breastmilk impacts microbiome maturation as has been previously demonstrated71,72. These personalized differences would not be addressed by our study design but are likely plausible additional drivers of individual microbiome trajectories. Our study design also prevented control over indication for use, timing, or route of antibiotic and non-antibiotic medication exposures. Thus, we cannot differentiate cause and effect with the administration of these medications versus the disease process necessitating them. Variations in timing of stool intervals could result in unequal timing of exposures, including human milk. Finally, human microbiome studies alone cannot separate cause and effect, which will require complementary systems-based mechanistic investigations, including animal models.
In summary, our integration of a vast clinical metadata database with multi-omic microbiome trajectories provides a framework for studying how gut bacterial communities ordain specific outcomes in a range of medical disorders, especially those in which there is a time series component to the variables studied. Specifically, our findings endorse identifying and systematically correcting for variables that affect the gut microbiome development in attempts to thoroughly assess microbial community-driven outcome risks.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and reasonable requests will be fulfilled by the Lead Contact, Gautam Dantas (dantas@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw sequencing data (metagenomes, metatranscriptomes, and metagenomic-assembled genomes) generated for this study was uploaded to the SRA database under BioProject PRJNA799247. Accession numbers are listed in the key resources table. Relevant raw data and metadata can be found as extended data spreadsheets. We use well-established computational and statistical analysis software and packages. Custom code for analysis and figure generation is available at https://github.com/dantaslab/NICUExposome(10.5281/zenodo.12737979) and is publicly available as of the date of publication. DOIs are listed in the key resources table. There are restrictions to the availability of infant stool raw materials due to extreme resource limitations.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Staphylococcus epidermidis from infant stool | This paper | N/A |
| Clostridioides difficile from infant stool | This paper | N/A |
| Biological samples | ||
| Fecal samples from preterm infants | This paper, Warner et al.34, La Rosa et al.40 | Neonatal Microbiome and Necrotizing Enterocolitis cohort |
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| DNeasy PowerSoil Pro Kit | Qiagen | Cat #47014 |
| Nextera XT kit | Illumina | Cat #20034197 |
| NucliSENS easyMAG system | bioMerieux | Cat #89130–520 |
| Turbo DNase | Thermo Fisher | Cat #AM2238 |
| QIAseq FastSelect – 5S/16S/23S kit | Qiagen | Cat #335925 |
| NEBNext Ultra II Directional RNA Library Prep kit for Illumina | NEB | Cat #E7760 |
| Deposited data | ||
| metagenomes, metatranscriptomes, and metagenomic-assembled genomes | This paper | BioProject PRJNA799247 |
| Fecal metagenomic DNA | Gibson et al.11 | BioProject PRJNA301903 |
| Fecal metagenomic DNA | Gasparrini et al.12 | BioProject PRJNA489090 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| Trimmomatic v0.366 | Bolger et al.74 | https://github.com/timflutre/trimmomatic |
| Deconseq v4.37 | Schmieder et al.75 | https://deconseq.sourceforge.net/ |
| MetaPhlAn3 v3.0 | Beghini et al.76 | https://github.com/biobakery/MetaPhlAn/tree/3.1.0 |
| HUMAnN3 v3.0 | Beghini et al.76 | https://github.com/biobakery/humann |
| ShortBRED | Kaminski et al.77 | https://github.com/biobakery/shortbred |
| MetaSPAdes | Nurk et al.80 | https://github.com/ablab/spades |
| Bowtie2 | Langmead et al.81 | https://github.com/BenLangmead/bowtie2 |
| samtools | Li et al.82 | https://www.htslib.org/ |
| MetaBat2 | Kang et al.83 | https://bitbucket.org/berkeleylab/metabat/src/master/ |
| checkM | Parks et al.84 | https://ecogenomics.github.io/CheckM/ |
| Quast | Gurevich et al.85 | https://github.com/ablab/quast |
| dRep | Olm et al.87 | https://github.com/MrOlm/drep |
| mash | Ondov et al.88 | https://github.com/marbl/Mash |
| R statistical software v4.0.4 - v4.4.0 | R core team | https://www.r-project.org |
| Rstudio | Rstudio | Posit.co |
| MaAslin2 | Mallick et al.51 | https://github.com/biobakery/Maaslin2 |
| DirichletMultinomial | Holmes et al.91 | https://bioconductor.org/packages/release/bioc/html/DirichletMultinomial.html |
| inStrain | Olm et al.42 | https://github.com/MrOlm/inStrain |
| pyani | Pritchard et al.89 | https://github.com/widdowquinn/pyani |
| Repeat measures PERMANOVA | Lloyd-Price et al.21 | https://bitbucket.org/biobakery/hmp2_analysis/src/master/ |
| Other | ||
| Analytic scripts and data | This paper |
https://github.com/dantaslab/NICUExposome
10.5281/zenodo.12737979 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Participants for this prospective, multi-center cohort study34,40 were recruited from among all infants born prematurely (≤37 weeks of gestation) at or below 1500 g of weight and expected to survive more than a week at St Louis Children’s Hospital (St Louis, MO, USA), Children’s Hospital at Oklahoma University Medical Center (Oklahoma City, OK, USA), and Kosair Children’s Hospital (now Norton Children’s Hospital) (Louisville, KY, USA). All infants hospitalized in the NICU were considered eligible for enrolment. Infants born with congenital noncardiac disorders were excluded. To study the impact of the gut microbiome on NEC development, we selected infants for a 1:2 case-control study from the total eligible study population. Cases were defined as infants whose clinical symptoms were consistent with NEC and whose radiographs fulfilled all criteria for Bell’s stage ≥2 NEC. Two non-NEC control infants (n=96) were matched to each NEC case (n=48), based on hospital site, gestational age at birth (±7 days) and birthweight (±100g). Demographics are provided in Data S1–S2. Metadata and samples were approved under Washington University HRPO #201105492, University of Louisville Institutional Review Board (IRB) HSPO #11.0136, and University of Oklahoma Health Sciences Center IRB 2472. The IRB at each site approved the original study, and perants were provided informed consent. Secondary analyses in the current manuscript were approved by Washington University School of Medicine HRPO #201205152.
All stools produced by infants were collected, refrigerated at 4°C, and then frozen (−80°C) daily without additives until chipped for these analyses. Samples were collected prospectively from 2009 to 2013 without prior knowledge of later NEC diagnosis. All stool samples collected prior to the day of life of clinical NEC diagnosis (day of onset, DOO) were included in the analysis for this study if quantities were sufficient for analysis. We analyzed stools from 48 infants who developed NEC, including 42 of the 46 cases of NEC in our prior publication34, 4 infants who also had congenital heart disease, 1 infant who developed NEC late in life (day of life 121, but from whom we had stool up to day of life 81), and 1 infant for whom we lowered the threshold for residual stool to include in this analysis. We used control specimens from 26 of the 94 participants without NEC from Warner et al.34, and 70 additional infants without NEC. For non-NEC controls all stool samples collected prior to DOO of their matched case were included. To robustly define the impact of the NICU exposome on gut microbiome trajectories in the NICU previously published shotgun-metagenomic data from stool specimens of an additional set of 92 non-NEC infants from the same cohort were included in the non-case-control analysis of this study11,12.
METHODS DETAILS
Metagenomic/metatranscriptomic sequencing
Metagenomic DNA was extracted from ~25mg of stool, chipped on dry ice into sterile tubes without thawing. Metagenomic DNA was extracted from stool using the DNeasy PowerSoil Pro Kit (Qiagen) following the manufacturer’s protocol, except that samples were mechanically lysed for two rounds of two minutes each using a Mini-Beadbeater-24 (Biospec Products) at 2,500 oscillations per minute. Metagenomic DNA was quantified using the PicoGreen quantitation assay (Thermo Fisher Scientific) and stored at −20°C. Genomic DNA (0.5 ng) was used as input in preparation of sequencing libraries with the Nextera XT kit (Illumina) as previously described73. Libraries were pooled and sequenced to a depth of ~2.5 million paired-end reads (2×150 bp) on a NextSeq500 High Output platform (Illumina).
Total RNA extraction for metatranscriptomic analysis was performed from ~100mg of stool, chipped on dry ice into sterile tubes without thawing using the NucliSENS easyMAG system (bioMerieux). All stool samples with ≥200 mg of remaining stool material from the NEC case-control part of this study were included in this analysis. Frozen stool was homogenized in 1 ml of easyMAG lysis buffer in a bead beating tube containing 8–10 disruption beads zirconium/silica 23mm (Research Products International Corp.). Samples were disrupted using the MP FastPrep-24 tissue homogenizer (MP Biomedicals) at 6.5 m/s for 60 sec. The lysate was centrifuged at 12,000xg for 10 minutes and the clarified supernatant was loaded into wells of the easyMAG cartridges, avoiding visual particulates. Total lysate volume of each sample was then adjusted to 2.2 ml with EasyMAG lysis buffer followed by addition of 50 μL of NucliSENS easyMAG magnetic silica beads. Samples were then loaded onto NucliSENS easyMAG system for automated nucleic acid extraction following the manufacturer’s instructions with onboard lysis “Specific A” protocol and 110 μL elution volume. Contaminating DNA was removed using the Turbo DNase (Thermo Fisher) kit and ribosomal RNA was depleted using the QIAseq FastSelect – 5S/16S/23S kit (Qiagen). Sequencing libraries with unique dual indexes were prepared using the NEBNext Ultra II Directional RNA Library Prep kit for Illumina (New England BioLabs). Libraries were sequenced to a depth of ~13 million 2×150 bp paired-end reads on the NovaSeq S4 platform (Illumina).
Sequencing data pre-processing
Sequencing adapters were removed from demultiplexed reads with Trimmomatic 0.366 (leading = 10, trailing = 10, sliding window = 4:15; minimum length = 60)74. Human reads were removed using Deconseq 4.37 with default parameters75.
QUANTIFICATION AND STATISTICAL ANALYSIS
Metagenomic/metatranscriptomic profiling of the gut microbiome
Quality-filtered short reads were profiled using MetaPhlAn3 v3.0 and HUMAnN3 v3.0 with default parameters to quantify relative abundances of bacterial species and encoded pathways, respectively76. Antibiotic resistance genes (ARGs) were quantified using ShortBRED and a marker database containing all resistance gene sequences available in the CARD database as well as additional genes confirmed to mediate resistance using functional metagenomic analysis in a previous study of this same cohort11,12,77. All marker sequences with >1 hit and >0 RPKM per sample were retained. Similarly, virulence factor abundance in both metagenomic and metatranscriptomic datasets were profiled using ShortBRED77 and a database of marker sequences built on all sequences downloaded from the Virulence Factor Database (VFDB) and Victors (September 2021)78,79. Virulence factors identified to be present in the preterm microbiome (>1 hit and >0 RPKM) were manually curated into 19 virulence categories based on literature research (e.g., Adhesion/Invasion, Capsule, Motility – see Data S15–17), allowing factors to match into multiple categories. Metatranscriptomic datasets were profiled using the MetaCYC pathway database with HUMAnN3 v3.0 with default parameters.
Metagenome-assembled genomes (MAGs) were generated for each infant using a co-assembly approach to generate data of sufficient sequencing depth for assembly. Genomes were assembled using MetaSPAdes80 using default parameters. All reads from all patient-specific samples were concatenated and aligned to indexed metagenome-assembled scaffolds using Bowtie2 (parameter: --no-mixed --very-sensitive --n-ceil 0,0.01)81, sorted using samtools (default parameter)82, and binned using MetaBat2 (parameter: --minContig 2000)83 to generate MAGs. MAG assembly quality was assessed using checkM and Quast84,85. MAGs were categorized as high-quality (completeness ≥90%, contamination ≤5%), medium-quality (completeness 50–90%, contamination 5–10%), or low-quality (completeness ≤50%, contamination ≥10%). Both high and medium quality MAGs were used in downstream analysis as supported by prior work86. These were used to generate a study-database of species-level representative genomes de-replicated across all individuals included in the case-control part of the study using dRep (gANI routine, 95% ANI)87. To taxonomically annotate all MAGs, each MAG was screened against the RefSeq genome database (downloaded July 2019) using mash-screen (default parameter)88. Subsequently, each MAG’s pairwise average nucleotide identity was profiled against the top three mash-screen hits using pyani (method: ANIm)89. Species-level taxonomical annotation was assigned to MAGs with pairwise ANI ≥94% at ≥50% reference coverage on a best-hit basis.
Bacterial replication rates were estimated based on metagenomic sequencing data using iRep as previously described32. Briefly, quality-filtered DNA short reads from each sample were mapped to all available dereplicated genomes from the corresponding patient with Bowtie2 (--very-sensitive mode). The resulting SAM files and dereplicated genomes were used as inputs for iRep with default parameters. Only iRep values passing all genome and mapping quality thresholds (min cov. = 5, min wins. = 0.98, min r^2 = 0.9, max fragments/Mbp = 175, GC correction min r^2 = 0.0) were retained for further analysis.
In silico strain-sharing prediction and validation
Metagenomic short-reads from stool samples of 96 non-NEC controls sequenced for this study were aligned to these de-replicated MAGs using inStrain42. Population ANI (popANI) values of ≥99.999% with breadth ≥0.5 at ≥25% of the reference genome being used in the comparison of two samples were considered evidence for strain-sharing. This amounts to a maximum of 50 single nucleotide polymorphisms (SNPs) for a genome with the size of 5,000,000bp. To guard against false positives resulting from contamination occurring during sample extraction or library preparation, we included infant pairs only if ≥2 unique samples from the same pair of infants indicated a strain to be shared. Strain-sharing events between unique infant-pairs were visualized as network graphs using Cytoscape90. Significance of strain-sharing by species was determined using a chi-square test followed by a pairwise nominal independence test with Benjamini-Hochberg correction using the r package rcompanion v2.4.35.
To confirm in silico predictions derived from stool metagenomes, S. epidermidis and C. difficile were selectively grown from samples of randomly selected infant pairs predicted to share the same strain. These outgrowth experiments were performed by personnel not involved in sample processing for stool-metagenomic sequencing. Approximately 10mg of fecal material was resuscitated in 0.15 ml of thioglycolate broth (Sigma). S. epidermidis was selectively cultured by inoculating and incubating TSB-CNA broth [TSB (BD) with colistin (20μg/ml), nalidixic acid (20μg/ml) and aztreonam (8μg/ml)] overnight at 37°C followed by inoculation of TSB-CNA broth with 6.5% NaCl and overnight incubation at 37°C. C. difficile was grown from stool anaerobically in reduced thioglycolate broth (Sigma) (37°C, overnight), followed by inoculation of CCMB-Tal medium (Anaerobe Systems) and incubation at 37°C overnight anaerobically. For S. epidermidis, cultures were streaked on blood agar plates (BD) and catalase tests (3% H2O2 in water) were performed following overnight incubation at 37°C. For C. difficile, cultures were streaked on selective ChromID C. difficile agar (BioMerieux Inc.) and incubated anaerobically overnight at 37°C for the appearance of black colonies typical for C. difficile. Metagenomic DNA was extracted from cultures of a single colony using the Qiagen PowerSoil Pro kit (Qiagen), libraries were prepared using a modified version of the Nextera kit (Illumina)73, and sequencing was performed on the Illumina NextSeq platform.
Metagenomic reads were assembled using MetaSPAdes80, binned using MetaBat283, annotated using mash and pyani88,89, and de-replicated using dRep87 as described above. Strain-sharing across sample pairs was investigated and confirmed using inStrain42, using the same ≥99.999% popANI and ≥25% genome-compared threshold described above.
General microbiome analysis
Statistical analyses and visualizations were conducted in R v.4.0.4, v.4.2.1, and v4.4.0 using the ggplot2, labdsv, scales, vegan, ape, ggridges, reshape2, lme4, nlme, multcomp, MuMIn, MaAsLin2, DirichletMultinomial, optparse, ggpubr, rsample, purrr, tidyr, tidyverse, rowr, metR, purrr, rsample, dplyr, permute, BiodiversityR, SIAMCAT, curatedMetagenomicData, RColorBrewer, compositions, scico, viridis, grid, glmmADMB, and rcompanion packages in Rstudio v2022-v2024.
Principal component analysis (PCA) was conducted on centered log ratio transformed relative abundance data (taxa or pathway abundances). Smoothed Shannon diversity readouts were plotted as background into the PCA space using the ordisurf function of the vegan package. For further visualizations, α-diversity was calculated with the vegan package (index = Shannon) on untransformed taxa or pathway data. Between-sample β-diversity was calculated using the vegdist function (method = Bray-Curtis). Procrustes analysis of taxonomic, functional, and transcriptional relative abundance data was performed using the procrustes function in the R vegan package.
Microbiome shift analysis
Microbiome shifts were defined as two consecutive samples collected from the same individual displaying Bray-Curtis dissimilarities equal or greater than those observed between the average of samples collected from different individuals. Bray-Curtis dissimilarity distributions were determined for consecutively collected inter- and all intra-patient sample pairings (non-NEC controls only) based on untransformed MetaPhlAn3 relative abundance data. The point at which the density distribution of interpatient dissimilarity intersects with that of intra-patient dissimilarity (Fig S3B) was set as the threshold defining a shift event. As Bray-Curtis dissimilarity between two samples increases over time (Fig S3A), shift thresholds were calculated independently for the observed range of DOL deltas between consecutively collected samples (Fig S3B).
To identify species that were significantly depleted or introduced at shift events, pre- and post-shift taxa presence was determined for all sample pairs flanking shift events (relative taxa abundance > 0%). Taxa were defined to be introduced if they were absent pre-shift but present post-shift. Conversely, depletion events were defined as taxa presence pre-shift and absence post-shift. To determine significant associations of introduction/depletion and shift events taxa-specific permutation tests were implemented. Therefore, the taxa-specific number of introduction/depletion events at shifts (n=131) was compared to the distribution of introduction/depletions observed across 1000 random permutations of an equal number of non-shift sample pairs (n=131). As shifts occurred earlier in life compared to the median sample DOL of non-shift sample pairs (Fig S3C), random permutations were restricted to generate a non-shift sample pair distribution following a similar DOL distribution as observed in the shift sample pairs using a simple bin-based density estimator in R. P-values were corrected for multiple comparisons using the Benjamini-Hochberg method (false discovery rate) in R.
Similar to introductions/depletions, permutation tests were implemented to identify clinical exposures associated with microbiome shifts. Antibiotics were aggregated at the class level. Binary recent exposure values (yes/no in the fourteen days prior to a shift) were counted for all shift events and compared to the distribution observed in 1000 permutations of non-shift sample pairs generated as described above. P-values were corrected for multiple comparisons using the Benjamini-Hochberg method (false discovery rate) in R. Significant introduction of ARGs at shifts was determined via permutation test as described above. Introduced ARGs were defined as all ARGs found directly following the shift and never seen in an individual’s gut microbiome before. ARGs were summarized by resistance mechanism for visualization purposes.
Enterotyping
Dirichlet multinomial mixture (DMM) models were implemented to generate clusters of samples of similar microbial composition (‘enterotypes’) based on relative abundances of microbial taxa transformed by 5000-fold multiplication to eliminate values below 1 for all samples in the case-control study using the R package DirichletMultinomial (k=40, iterations=1000)91. The optimal model was selected based on minimization of the model Laplace approximation. Samples were assigned cluster identity based on their highest cluster-identity probability value.
Defining the impact of the NICU exposome and baseline characteristics on gut microbiome trajectories during hospitalization
We comprehensively characterized the impact of patient baseline variables (Data S5: gestational age, birthweight, sex, delivery mode,), dietary exposures, antibiotics (aggregated on the class level), non-antibiotic medications (by route), and maternal variables (Data S5: maternal age, race, body mass index, diabetes, pre-eclampsia, chorioamnionitis, steroids, outpatient and inpatient antimicrobials, and multiple gestations) on developmental microbiota trajectories of preterm infants during hospitalization. All non-NEC controls as well as metagenomic data from 92 participants previously published11,12 were included in this analysis. Each sample was assigned patient-specific constant variables (e.g., sex, race, birthweight, gestational age at birth). We subdivided variables into three categories: maternal/baseline/dietary, antibiotic, and non-antibiotic medications. Variables changing over time were treated as follows; dietary exposures (formula, human milk) were assigned to each sample as the fraction of days of life prior to sample collection exposed. Antibiotics were assigned as binary recent exposure values if received in the 14 days and non-antibiotic medications in the 7 days prior to sample collection (Data S1, S5), previously shown to capture the acute effects of antibiotic exposures in the preterm gut microbiome12 and non-antibiotic exposures in adults92. Administration route was not available on a per-infant basis; however, many medications had predetermined routes for their use in preterm infants or as outlined in the study protocol (Data S18). Maternal exposures and comorbidities were coded as binary values and assigned to each sample on a per-patient basis.
Repeat measures permutational analysis of variance (PERMANOVA) was conducted as previously described15,21,67 to characterize the impact of the clinical exposome and baseline characteristics on the developing preterm gut microbiome. Patient identity and study source (this study, Gibson et al.11, Gasparrini et al.12) were included as a mandatory blocking factor in all repeat measure PERMANOVA analyses. Variance explained was calculated independently for each variable to avoid issues of variable ordering. Variables were considered to explain a significant portion of the observed variance of taxa or pathways if P≤0.1. P-values were corrected for multiple comparisons using the Benjamini-Hochberg method (false discovery rate) in R.
To further identify associations of the NICU exposome on specific taxa and pathway abundances and expression, generalized linear mixed models (GLMMs) were implemented using MaAsLin2 with minimum taxonomic prevalence of 10% and q-values ≤0.25 considered significant as per the default51 (Data S19). For taxonomic changes related to NICU exposures (Figure 3), all variables (Data S5) were included as fixed effects with random effects of hospital site, participant, and study source and only features q<0.05 and exposures present in >1% of samples are plotted. Subsequent analyses utilized only variables identified to explain a significant portion of the variance (P≤0.1) of a dataset (DNA pathways, RNA taxa, RNA functions) by repeat measures PERMANOVA. Day of life and gestational age at birth were included as mandatory fixed effects. Additionally, GLMMs were implemented using the R package nlme to determine significant differences in bacterial replication rates in NEC cases and controls. Significance was determined by comparing a model including NEC status against a null model using ANOVA.
Transcriptional overrepresentation analysis
To identify transcriptionally over-/under-represented taxa compared to their relative abundance in the shotgun metagenomic data, species-specific zero-inflated gaussian models were implemented on arcsine transformed relative taxa abundance data using the glmmADMB package in R. Therefore, relative abundance data for each species generated from both metagenomic and metatranscriptomic reads via MetaPhlAn3 was analyzed. Day of life was included as a fixed effect in all models. Patient ID was included as a random effect. Models took the form: abundance ~ data_type+log(day of life)+(0+log(day of life)|Patient_ID/data_type), with data_type = DNA or RNA. Significant differences between RNA and DNA relative abundance data were determined for each species individually. P-values were corrected for multiple comparisons using the Benjamini-Hochberg method (false discovery rate) in R.
Machine learning
Three classes of machine learning algorithms (logistic regression, random forest, elastic net) were trained using all microbiome features characterized in this study (metagenomic profiles, metatranscriptomes, bacterial replication rate, community- and MAG-associated virulence profiles) using the R package SiamCat v2.0.0. Features were filtered using a prevalence cut-off of 0.01 and normalized with the method ‘log.std’ (parameter: log.n0=1e−05, sd.min.q=0). To account for repeat sampling cross-validation procedure was blocked by patient. Feature selection was performed with threshold of 100, 50, or 25 features nested within-cross validation and the following parameters: methods.fs=AUC, direction=absolute. Cross-validation was performed with the following parameters: num.folds=10, num.resamples=10.
Supplementary Material
Cohort participant metadata
Metagenome-assembled genomes
Sample metadata
Expanded cohort (non-NEC) Sample-specific baseline, maternal, antibiotic, non-antibiotic, and dietary exposures
NEC case and control Sample-specific non-antibiotic medication exposure
Metagenomic species abundance controls
Metatranscriptomic species abundance controls
Expanded cohort (non-NEC) metatranscriptomic pathway abundance controls
Metagenomic species abundance NEC-cases/controls
Metatranscriptomic species abundance NEC-cases/controls
Expanded cohort (non-NEC) metagenomic pathway abundance controls
NEC-cases/controls iRep data
Virulence factor categorization
Metagenomic pathway abundance NEC-cases/controls
Metatranscriptomic pathway abundance NEC-cases/controls
Medication administration route
Select microbiome analysis data tables
Metatranscriptomic virulence factor abundance NEC-cases/controls
Metagenomic virulence factor abundance NEC-cases/controls
Highlights.
Antibiotics and non-antibiotic drugs govern infant gut microbiome development
Antibiotic-associated microbial shifts introduce pathogens and resistance
Hospitalized preterm infants share isogenic C. difficile and S. epidermidis
Microbial development stagnates before late-onset necrotizing enterocolitis
Acknowledgements
We thank the staff at the Edison Family Center for Genome Sciences and Systems Biology: Bonnie Dee, Kathleen Matheny, and Keith Page for administrative support; Jessica Hoisington-Lopez and MariaLynn Crosby for managing the high-throughput sequencing core; and Eric Martin and Brian Koebbe for computational support. This project was supported by NIH Grants UH3AI083265 and P30DK052574 (Biobank), and the CDI of Washington University and SLCH. This work was further supported in part by awards to G.D. from NIAID (grant R01AI155893) and to G.D. P.I.T., and B.B.W. from NICHD (grant R01HD092414). R.T.’s research was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation; grant 402733540). D.J.S was supported by a PIDS St. Jude Children’s Hospital Fellowship in Basic Research, a Thrasher Research Fund Early Career Award, Doris Duke Foundation Physician Scientist Fellowship 2021081, and NIH K08 KAI159384. E.C.K was supported by the NSF (DGE-1143945). J.E.S was supported by P30ES030283. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Supplementary Data
Supplementary Figures. PDF containing Figures S1–S9
Declaration of Interests
P.I.T. is a holder of equity in, a consultant to, and a member of the Scientific Advisory Board of MediBeacon Inc., which is developing a technology to noninvasively measure intestinal permeability in humans. P.I.T. is a coinventor on patents assigned to MediBeacon (U.S. patents 11,285,223 and 11,285,224 titled “Compositions and methods for assessing gut function” and U.S. patent application 2022-0326255, “Methods of monitoring mucosal healing”), which might earn royalties if the technology is commercialized. P.I.T. receives compensation for his roles as Chair, Scientific Advisory Board of the AGA Center for Microbiome Research and Education, and consultant to Temple University on waterborne enteric infections. He is a member of the Data Safety Monitoring Board of Inmunova, which is developing an immune biologic targeting Shiga toxin-producing E. coli infections, for which he receives no compensation, except for reimbursement of expenses. P.I.T. receives royalties from UpToDate from two sections on intestinal E. coli infections. G.D. is a cofounder, holder of equity in, a consultant to, and a member of the Scientific Advisory Board of Viosera Therapeutics, which is developing combination antimicrobial therapy against bacterial pathogens. G.D. is a coinventor on a patent assigned to Viosera Therapeutics (“Compositions and methods of use of antibacterial drug combinations,” U.S. Patent Application No. 62/190,588), which might earn royalties if the technology is commercialized. G.D. is a consultant to and a member of the Scientific Advisory Board of Pluton Biosciences, which is developing methods for discovering environmental microbes for commercial applications. G.D. has consulted for SNIPR Technologies Ltd. in the last 5 years, but not presently. The authors declare no other competing interests.
References
- 1.Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, and Aagaard KM (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nature medicine 23, 314–326. 10.1038/nm.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Healy DB, Ryan CA, Ross RP, Stanton C, and Dempsey EM (2022). Clinical implications of preterm infant gut microbiome development. Nat Microbiol 7, 22–33. 10.1038/s41564-021-01025-4. [DOI] [PubMed] [Google Scholar]
- 3.Robertson RC, Manges AR, Finlay BB, and Prendergast AJ (2019). The Human Microbiome and Child Growth - First 1000 Days and Beyond. Trends in microbiology 27, 131–147. 10.1016/j.tim.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Rao C, Coyte KZ, Bainter W, Geha RS, Martin CR, and Rakoff-Nahoum S (2021). Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 591, 633–638. 10.1038/s41586-021-03241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumann-Dudenhoeffer AM, D’Souza AW, Tarr PI, Warner BB, and Dantas G (2018). Infant diet and maternal gestational weight gain predict early metabolic maturation of gut microbiomes. Nature medicine 24, 1822-1829-1829. 10.1038/s41591-018-0216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reyman M, van Houten MA, Watson RL, Chu M, Arp K, de Waal WJ, Schiering I, Plotz FB, Willems RJL, van Schaik W, et al. (2022). Effects of early-life antibiotics on the developing infant gut microbiome and resistome: a randomized trial. Nature communications 13, 893. 10.1038/s41467-022-28525-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olm MR, Dahan D, Carter MM, Merrill BD, Yu FB, Jain S, Meng X, Tripathi S, Wastyk H, Neff N, et al. (2022). Robust variation in infant gut microbiome assembly across a spectrum of lifestyles. Science (New York, N.Y.) 376, 1220–1223. 10.1126/science.abj2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fouhy F, Watkins C, Hill CJ, O’Shea CA, Nagle B, Dempsey EM, O’Toole PW, Ross RP, Ryan CA, and Stanton C (2019). Perinatal factors affect the gut microbiota up to four years after birth. Nature communications 10, 1517. 10.1038/s41467-019-09252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chawanpaiboon S, Vogel JP, Moller AB, Lumbiganon P, Petzold M, Hogan D, Landoulsi S, Jampathong N, Kongwattanakul K, Laopaiboon M, et al. (2019). Global, regional, and national estimates of levels of preterm birth in 2014: a systematic review and modelling analysis. Lancet Glob Health 7, e37–e46. 10.1016/S2214-109X(18)30451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maier RF, Blondel B, Piedvache A, Misselwitz B, Petrou S, Van Reempts P, Franco F, Barros H, Gadzinowski J, Boerch K, et al. (2018). Duration and Time Trends in Hospital Stay for Very Preterm Infants Differ Across European Regions. Pediatr Crit Care Med 19, 1153–1161. 10.1097/PCC.0000000000001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibson MK, Wang B, Ahmadi S, Burnham C-AD, Tarr PI, Warner BB, and Dantas G (2016). Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nature Microbiology 1, nmicrobiol201624. 10.1038/nmicrobiol.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasparrini AJ, Wang B, Sun X, Kennedy EA, Hernandez-Leyva A, Ndao IM, Tarr PI, Warner BB, and Dantas G (2019). Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat Microbiol 4, 2285–2297. 10.1038/s41564-019-0550-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks B, Olm MR, Firek BA, Baker R, Thomas BC, Morowitz MJ, and Banfield JF (2017). Strain-resolved analysis of hospital rooms and infants reveals overlap between the human and room microbiome. Nature communications 8, 1814. 10.1038/s41467-017-02018-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks B, Olm MR, Firek BA, Baker R, Geller-McGrath D, Reimer SR, Soenjoyo KR, Yip JS, Dahan D, Thomas BC, et al. (2018). The developing premature infant gut microbiome is a major factor shaping the microbiome of neonatal intensive care unit rooms. Microbiome 6, 112. 10.1186/s40168-018-0493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz DJ, Shalon N, Wardenburg K, DeVeaux A, Wallace MA, Hall-Moore C, Ndao IM, Sullivan JE, Radmacher P, Escobedo M, et al. (2023). Gut pathogen colonization precedes bloodstream infection in the neonatal intensive care unit. Science translational medicine 15, eadg5562. 10.1126/scitranslmed.adg5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olm MR, Brown CT, Brooks B, Firek B, Baker R, Burstein D, Soenjoyo K, Thomas BC, Morowitz M, and Banfield J (2017). Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome research. 10.1101/gr.213256.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks B, Firek BA, Miller CS, Sharon I, Thomas BC, Baker R, Morowitz MJ, and Banfield JF (2014). Microbes in the neonatal intensive care unit resemble those found in the gut of premature infants. Microbiome 2, 1. 10.1186/2049-2618-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz DJ, Langdon AE, and Dantas G (2020). Understanding the impact of antibiotic perturbation on the human microbiome. Genome medicine 12, 82. 10.1186/s13073-020-00782-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thanert R, Sawhney SS, Schwartz DJ, and Dantas G (2022). The resistance within: Antibiotic disruption of the gut microbiome and resistome dynamics in infancy. Cell host & microbe 30, 675–683. 10.1016/j.chom.2022.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Milburn O, Beiersdorfer T, Du L, Akinbi H, and Haslam DB (2022). Antibiotic exposure prevents acquisition of beneficial metabolic functions in the preterm infant gut microbiome. Microbiome 10, 103. 10.1186/s40168-022-01300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boolchandani M, Blake KS, Tilley DH, Cabada MM, Schwartz DJ, Patel S, Morales ML, Meza R, Soto G, Isidean SD, et al. (2022). Impact of international travel and diarrhea on gut microbiome and resistome dynamics. Nature communications 13, 7485. 10.1038/s41467-022-34862-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayeni KI, Berry D, Wisgrill L, Warth B, and Ezekiel CN (2022). Early-life chemical exposome and gut microbiome development: African research perspectives within a global environmental health context. Trends in microbiology 30, 1084–1100. 10.1016/j.tim.2022.05.008. [DOI] [PubMed] [Google Scholar]
- 24.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, et al. (2018). Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628. 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stark A, Smith PB, Hornik CP, Zimmerman KO, Hornik CD, Pradeep S, Clark RH, Benjamin DK Jr., Laughon M, and Greenberg RG (2022). Medication Use in the Neonatal Intensive Care Unit and Changes from 2010 to 2018. The Journal of pediatrics 240, 66–71 e64. 10.1016/j.jpeds.2021.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henrick BM, Rodriguez L, Lakshmikanth T, Pou C, Henckel E, Arzoomand A, Olin A, Wang J, Mikes J, Tan Z, et al. (2021). Bifidobacteria-mediated immune system imprinting early in life. Cell 184, 3884–3898 e3811. 10.1016/j.cell.2021.05.030. [DOI] [PubMed] [Google Scholar]
- 27.Bae M, Cassilly CD, Liu X, Park SM, Tusi BK, Chen X, Kwon J, Filipcik P, Bolze AS, Liu Z, et al. (2022). Akkermansia muciniphila phospholipid induces homeostatic immune responses. Nature 608, 168–173. 10.1038/s41586-022-04985-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roodgar M, Good BH, Garud NR, Martis S, Avula M, Zhou W, Lancaster SM, Lee H, Babveyh A, Nesamoney S, et al. (2021). Longitudinal linked-read sequencing reveals ecological and evolutionary responses of a human gut microbiome during antibiotic treatment. Genome research 31, 1433–1446. 10.1101/gr.265058.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beck LC, Masi AC, Young GR, Vatanen T, Lamb CA, Smith R, Coxhead J, Butler A, Marsland BJ, Embleton ND, et al. (2022). Strain-specific impacts of probiotics are a significant driver of gut microbiome development in very preterm infants. Nat Microbiol 7, 1525–1535. 10.1038/s41564-022-01213-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pammi M, Cope J, Tarr PI, Warner BB, Morrow AL, Mai V, Gregory KE, Kroll JS, McMurtry V, Ferris MJ, et al. (2017). Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: a systematic review and meta-analysis. Microbiome 5, 31. 10.1186/s40168-017-0248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaelin EA, Rodriguez C, Hall-Moore C, Hoffmann JA, Linneman LA, Ndao IM, Warner BB, Tarr PI, Holtz LR, and Lim ES (2022). Longitudinal gut virome analysis identifies specific viral signatures that precede necrotizing enterocolitis onset in preterm infants. Nat Microbiol 7, 653–662. 10.1038/s41564-022-01096-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olm MR, Bhattacharya N, Crits-Christoph A, Firek BA, Baker R, Song YS, Morowitz MJ, and Banfield JF (2019). Necrotizing enterocolitis is preceded by increased gut bacterial replication, Klebsiella, and fimbriae-encoding bacteria. Sci Adv 5, eaax5727. 10.1126/sciadv.aax5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thanert R, Keen EC, Dantas G, Warner BB, and Tarr PI (2021). Necrotizing Enterocolitis and the Microbiome: Current Status and Future Directions. The Journal of infectious diseases 223, S257–S263. 10.1093/infdis/jiaa604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warner BB, Deych E, Zhou Y, Hall-Moore C, Weinstock GM, Sodergren E, Shaikh N, Hoffmann JA, Linneman LA, Hamvas A, et al. (2016). Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: a prospective case-control study. The Lancet 387, 1928–1936. 10.1016/s0140-6736(16)00081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carl MA, Ndao IM, Springman AC, Manning SD, Johnson JR, Johnston BD, Burnham C-AD, Weinstock ES, Weinstock GM, Wylie TN, et al. (2014). Sepsis From the Gut: The Enteric Habitat of Bacteria That Cause Late-Onset Neonatal Bloodstream Infections. Clinical Infectious Diseases 58, 1211-1218-1218. 10.1093/cid/ciu084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart CJ, Embleton ND, Marrs ECL, Smith DP, Fofanova T, Nelson A, Skeath T, Perry JD, Petrosino JF, Berrington JE, and Cummings SP (2017). Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome 5, 75. 10.1186/s40168-017-0295-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell EF, Hintz SR, Hansen NI, Bann CM, Wyckoff MH, DeMauro SB, Walsh MC, Vohr BR, Stoll BJ, Carlo WA, et al. (2022). Mortality, In-Hospital Morbidity, Care Practices, and 2-Year Outcomes for Extremely Preterm Infants in the US, 2013–2018. Jama 327, 248–263. 10.1001/jama.2021.23580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rusconi B, Good M, and Warner BB (2017). The Microbiome and Biomarkers for Necrotizing Enterocolitis: Are We Any Closer to Prediction? The Journal of pediatrics 189, 40–47 e42. 10.1016/j.jpeds.2017.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gopalakrishna KP, Macadangdang BR, Rogers MB, Tometich JT, Firek BA, Baker R, Ji J, Burr AHP, Ma C, Good M, et al. (2019). Maternal IgA protects against the development of necrotizing enterocolitis in preterm infants. Nature medicine 25, 1110–1115. 10.1038/s41591-019-0480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.La Rosa PS, Warner BB, Zhou Y, Weinstock GM, Sodergren E, Hall-Moore CM, Stevens HJ, Bennett WE Jr., Shaikh N, Linneman LA, et al. (2014). Patterned progression of bacterial populations in the premature infant gut. Proceedings of the National Academy of Sciences of the United States of America 111, 12522–12527. 10.1073/pnas.1409497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, et al. (2018). Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583-588-588. 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olm MR, Crits-Christoph A, Bouma-Gregson K, Firek BA, Morowitz MJ, and Banfield JF (2021). inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nature biotechnology 39, 727–736. 10.1038/s41587-020-00797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Souza AW, Potter RF, Wallace M, Shupe A, Patel S, Sun X, Gul D, Kwon JH, Andleeb S, Burnham CD, and Dantas G (2019). Spatiotemporal dynamics of multidrug resistant bacteria on intensive care unit surfaces. Nature communications 10, 4569. 10.1038/s41467-019-12563-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siranosian BA, Brooks EF, Andermann T, Rezvani AR, Banaei N, Tang H, and Bhatt AS (2022). Rare transmission of commensal and pathogenic bacteria in the gut microbiome of hospitalized adults. Nature communications 13, 586. 10.1038/s41467-022-28048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark RH, Bloom BT, Spitzer AR, and Gerstmann DR (2006). Reported Medication Use in the Neonatal Intensive Care Unit: Data From a Large National Data Set. Pediatrics 117, 1979-1987-1987. 10.1542/peds.2005-1707. [DOI] [PubMed] [Google Scholar]
- 46.Hsieh EM, Hornik CP, Clark RH, Laughon MM, Benjamin DK Jr., and Smith PB (2014). Medication use in the neonatal intensive care unit. American journal of perinatology 31, 811–821. 10.1055/s-0033-1361933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neumann CJ, Mahnert A, Kumpitsch C, Kiu R, Dalby MJ, Kujawska M, Madl T, Kurath-Koller S, Urlesberger B, Resch B, et al. (2023). Clinical NEC prevention practices drive different microbiome profiles and functional responses in the preterm intestine. Nature communications 14, 1349. 10.1038/s41467-023-36825-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taft DH, Ambalavanan N, Schibler KR, Yu Z, Newburg DS, Ward DV, and Morrow AL (2014). Intestinal microbiota of preterm infants differ over time and between hospitals. Microbiome 2, 36. 10.1186/2049-2618-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maier L, Goemans CV, Wirbel J, Kuhn M, Eberl C, Pruteanu M, Muller P, Garcia-Santamarina S, Cacace E, Zhang B, et al. (2021). Unravelling the collateral damage of antibiotics on gut bacteria. Nature 599, 120–124. 10.1038/s41586-021-03986-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tamburini S, Shen N, Wu HC, and Clemente JC (2016). The microbiome in early life: implications for health outcomes. Nature medicine 22, 713–722. 10.1038/nm.4142. [DOI] [PubMed] [Google Scholar]
- 51.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol 17, e1009442. 10.1371/journal.pcbi.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kristich CJ, Rice LB, and Arias CA (2014). Enterococcal Infection-Treatment and Antibiotic Resistance. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection, Gilmore MS, Clewell DB, Ike Y, and Shankar N, eds. [PubMed] [Google Scholar]
- 53.Li J, Wang H, Li N, Zhang Y, Lu X, and Liu B (2022). Antibiotic susceptibility and biofilm-forming ability of Veillonella strains. Anaerobe 78, 102667. 10.1016/j.anaerobe.2022.102667. [DOI] [PubMed] [Google Scholar]
- 54.Forslund SK, Chakaroun R, Zimmermann-Kogadeeva M, Marko L, Aron-Wisnewsky J, Nielsen T, Moitinho-Silva L, Schmidt TSB, Falony G, Vieira-Silva S, et al. (2021). Combinatorial, additive and dose-dependent drug-microbiome associations. Nature 600, 500–505. 10.1038/s41586-021-04177-9. [DOI] [PubMed] [Google Scholar]
- 55.Fehr K, Moossavi S, Sbihi H, Boutin RCT, Bode L, Robertson B, Yonemitsu C, Field CJ, Becker AB, Mandhane PJ, et al. (2020). Breastmilk Feeding Practices Are Associated with the Co-Occurrence of Bacteria in Mothers’ Milk and the Infant Gut: the CHILD Cohort Study. Cell host & microbe 28, 285–297 e284. 10.1016/j.chom.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 56.Wang F, and Roy S (2017). Gut Homeostasis, Microbial Dysbiosis, and Opioids. Toxicol Pathol 45, 150–156. 10.1177/0192623316679898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu L, Wu Y, Yang X, Pang X, Wu Y, Li X, Liu X, Zhao Y, Yu L, Wang P, et al. (2024). The Fe-S cluster biosynthesis in Enterococcus faecium is essential for anaerobic growth and gastrointestinal colonization. Gut microbes 16, 2359665. 10.1080/19490976.2024.2359665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brunson DN, Colomer-Winter C, Lam LN, and Lemos JA (2023). Identification of Multiple Iron Uptake Mechanisms in Enterococcus faecalis and Their Relationship to Virulence. Infection and immunity 91, e0049622. 10.1128/iai.00496-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strazar M, Temba GS, Vlamakis H, Kullaya VI, Lyamuya F, Mmbaga BT, Joosten LAB, van der Ven A, Netea MG, de Mast Q, and Xavier RJ (2021). Gut microbiome-mediated metabolism effects on immunity in rural and urban African populations. Nature communications 12, 4845. 10.1038/s41467-021-25213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren M, Zhang SH, Zeng XF, Liu H, and Qiao SY (2015). Branched-chain Amino Acids are Beneficial to Maintain Growth Performance and Intestinal Immune-related Function in Weaned Piglets Fed Protein Restricted Diet. Asian-Australas J Anim Sci 28, 1742–1750. 10.5713/ajas.14.0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, and Brodin P (2018). Stereotypic Immune System Development in Newborn Children. Cell 174, 1277–1292.e1214–1292.e1214. 10.1016/j.cell.2018.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong Y, and Speer CP (2015). Late-onset neonatal sepsis: recent developments. Archives of Disease in Childhood - Fetal and Neonatal Edition 100, F257. 10.1136/archdischild-2014-306213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong Y, Speer CP, and Glaser K (2018). Beyond sepsis: Staphylococcus epidermidis is an underestimated but significant contributor to neonatal morbidity. Virulence 9, 621–633. 10.1080/21505594.2017.1419117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prusakov P, Goff DA, Wozniak PS, Cassim A, Scipion CEA, Urzua S, Ronchi A, Zeng L, Ladipo-Ajayi O, Aviles-Otero N, et al. (2021). A global point prevalence survey of antimicrobial use in neonatal intensive care units: The no-more-antibiotics and resistance (NO-MAS-R) study. EClinicalMedicine 32, 100727. 10.1016/j.eclinm.2021.100727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ting JY, Synnes A, Roberts A, Deshpandey A, Dow K, Yoon EW, Lee KS, Dobson S, Lee SK, Shah PS, and Canadian Neonatal Network, I. (2016). Association Between Antibiotic Use and Neonatal Mortality and Morbidities in Very Low-Birth-Weight Infants Without Culture-Proven Sepsis or Necrotizing Enterocolitis. JAMA pediatrics 170, 1181–1187. 10.1001/jamapediatrics.2016.2132. [DOI] [PubMed] [Google Scholar]
- 66.Jiang S, Yang Z, Shan R, Zhang Y, Yan W, Yang Y, Shah PS, Lee SK, Cao Y, and Reduction of Infection in Neonatal Intensive Care Units using the Evidence-based Practice for Improving Quality Study, G. (2020). Neonatal Outcomes Following Culture-negative Late-onset Sepsis Among Preterm Infants. The Pediatric infectious disease journal 39, 232–238. 10.1097/INF.0000000000002558. [DOI] [PubMed] [Google Scholar]
- 67.Thanert R, Thanert A, Ou J, Bajinting A, Burnham CD, Engelstad HJ, Tecos ME, Ndao IM, Hall-Moore C, Rouggly-Nickless C, et al. (2021). Antibiotic-driven intestinal dysbiosis in pediatric short bowel syndrome is associated with persistently altered microbiome functions and gut-derived bloodstream infections. Gut microbes 13, 1940792. 10.1080/19490976.2021.1940792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fajardo C, Alshaikh B, and Harabor A (2019). Prolonged use of antibiotics after birth is associated with increased morbidity in preterm infants with negative cultures. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 32, 4060–4066. 10.1080/14767058.2018.1481042. [DOI] [PubMed] [Google Scholar]
- 69.DeVeaux A, Ryou J, Dantas G, Warner BB, and Tarr PI (2023). Microbiome-targeting therapies in the neonatal intensive care unit: safety and efficacy. Gut microbes 15, 2221758. 10.1080/19490976.2023.2221758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson-Hence CB, Gopalakrishna KP, Bodkin D, Coffey KE, Burr AHP, Rahman S, Rai AT, Abbott DA, Sosa YA, Tometich JT, et al. (2023). Stability and heterogeneity in the antimicrobiota reactivity of human milk-derived immunoglobulin A. J Exp Med 220. 10.1084/jem.20220839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Planer JD, Peng Y, Kau AL, Blanton LV, Ndao IM, Tarr PI, Warner BB, and Gordon JI (2016). Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 534, 263. 10.1038/nature17940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pannaraj PS, Li F, Cerini C, Bender JM, Yang S, Rollie A, Adisetiyo H, Zabih S, Lincez PJ, Bittinger K, et al. (2017). Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA pediatrics 171, 647–654. 10.1001/jamapediatrics.2017.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, and Kishony R (2015). Inexpensive multiplexed library preparation for megabase-sized genomes. PloS one 10, e0128036. 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmieder R, and Edwards R (2011). Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PloS one 6, e17288. 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beghini F, McIver LJ, Blanco-Miguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, et al. (2021). Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10. 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, and Huttenhower C (2015). High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLoS Comput Biol 11, e1004557. 10.1371/journal.pcbi.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sayers S, Li L, Ong E, Deng S, Fu G, Lin Y, Yang B, Zhang S, Fa Z, Zhao B, et al. (2019). Victors: a web-based knowledge base of virulence factors in human and animal pathogens. Nucleic acids research 47, D693–D700. 10.1093/nar/gky999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu B, Zheng D, Jin Q, Chen L, and Yang J (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic acids research 47, D687–D692. 10.1093/nar/gky1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nurk S, Meleshko D, Korobeynikov A, and Pevzner PA (2017). metaSPAdes: a new versatile metagenomic assembler. Genome research 27, 824–834. 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, and Wang Z (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome research 25, 1043–1055. 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gurevich A, Saveliev V, Vyahhi N, and Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dindhoria K, Kumar R, Bhargava B, and Kumar R (2024). Metagenomic assembled genomes indicated the potential application of hypersaline microbiome for plant growth promotion and stress alleviation in salinized soils. mSystems 9, e0105023. 10.1128/msystems.01050-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olm MR, Brown CT, Brooks B, and Banfield JF (2017). dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. The ISME journal 11, 2864–2868. 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, and Phillippy AM (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome biology 17, 132. 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pritchard L, Glover RH, Humphris S, Elphinstone JG, and Toth IK (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Analytical Methods 8, 12–24. 10.1039/c5ay02550h. [DOI] [Google Scholar]
- 90.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Holmes I, Harris K, and Quince C (2012). Dirichlet multinomial mixtures: generative models for microbial metagenomics. PloS one 7, e30126. 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nguyen CL, Markey KA, Miltiadous O, Dai A, Waters N, Sadeghi K, Fei T, Shouval R, Taylor BP, Liao C, et al. (2023). High-resolution analyses of associations between medications, microbiome, and mortality in cancer patients. Cell 186, 2705–2718 e2717. 10.1016/j.cell.2023.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cohort participant metadata
Metagenome-assembled genomes
Sample metadata
Expanded cohort (non-NEC) Sample-specific baseline, maternal, antibiotic, non-antibiotic, and dietary exposures
NEC case and control Sample-specific non-antibiotic medication exposure
Metagenomic species abundance controls
Metatranscriptomic species abundance controls
Expanded cohort (non-NEC) metatranscriptomic pathway abundance controls
Metagenomic species abundance NEC-cases/controls
Metatranscriptomic species abundance NEC-cases/controls
Expanded cohort (non-NEC) metagenomic pathway abundance controls
NEC-cases/controls iRep data
Virulence factor categorization
Metagenomic pathway abundance NEC-cases/controls
Metatranscriptomic pathway abundance NEC-cases/controls
Medication administration route
Select microbiome analysis data tables
Metatranscriptomic virulence factor abundance NEC-cases/controls
Metagenomic virulence factor abundance NEC-cases/controls
Data Availability Statement
Raw sequencing data (metagenomes, metatranscriptomes, and metagenomic-assembled genomes) generated for this study was uploaded to the SRA database under BioProject PRJNA799247. Accession numbers are listed in the key resources table. Relevant raw data and metadata can be found as extended data spreadsheets. We use well-established computational and statistical analysis software and packages. Custom code for analysis and figure generation is available at https://github.com/dantaslab/NICUExposome(10.5281/zenodo.12737979) and is publicly available as of the date of publication. DOIs are listed in the key resources table. There are restrictions to the availability of infant stool raw materials due to extreme resource limitations.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Staphylococcus epidermidis from infant stool | This paper | N/A |
| Clostridioides difficile from infant stool | This paper | N/A |
| Biological samples | ||
| Fecal samples from preterm infants | This paper, Warner et al.34, La Rosa et al.40 | Neonatal Microbiome and Necrotizing Enterocolitis cohort |
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| DNeasy PowerSoil Pro Kit | Qiagen | Cat #47014 |
| Nextera XT kit | Illumina | Cat #20034197 |
| NucliSENS easyMAG system | bioMerieux | Cat #89130–520 |
| Turbo DNase | Thermo Fisher | Cat #AM2238 |
| QIAseq FastSelect – 5S/16S/23S kit | Qiagen | Cat #335925 |
| NEBNext Ultra II Directional RNA Library Prep kit for Illumina | NEB | Cat #E7760 |
| Deposited data | ||
| metagenomes, metatranscriptomes, and metagenomic-assembled genomes | This paper | BioProject PRJNA799247 |
| Fecal metagenomic DNA | Gibson et al.11 | BioProject PRJNA301903 |
| Fecal metagenomic DNA | Gasparrini et al.12 | BioProject PRJNA489090 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| Trimmomatic v0.366 | Bolger et al.74 | https://github.com/timflutre/trimmomatic |
| Deconseq v4.37 | Schmieder et al.75 | https://deconseq.sourceforge.net/ |
| MetaPhlAn3 v3.0 | Beghini et al.76 | https://github.com/biobakery/MetaPhlAn/tree/3.1.0 |
| HUMAnN3 v3.0 | Beghini et al.76 | https://github.com/biobakery/humann |
| ShortBRED | Kaminski et al.77 | https://github.com/biobakery/shortbred |
| MetaSPAdes | Nurk et al.80 | https://github.com/ablab/spades |
| Bowtie2 | Langmead et al.81 | https://github.com/BenLangmead/bowtie2 |
| samtools | Li et al.82 | https://www.htslib.org/ |
| MetaBat2 | Kang et al.83 | https://bitbucket.org/berkeleylab/metabat/src/master/ |
| checkM | Parks et al.84 | https://ecogenomics.github.io/CheckM/ |
| Quast | Gurevich et al.85 | https://github.com/ablab/quast |
| dRep | Olm et al.87 | https://github.com/MrOlm/drep |
| mash | Ondov et al.88 | https://github.com/marbl/Mash |
| R statistical software v4.0.4 - v4.4.0 | R core team | https://www.r-project.org |
| Rstudio | Rstudio | Posit.co |
| MaAslin2 | Mallick et al.51 | https://github.com/biobakery/Maaslin2 |
| DirichletMultinomial | Holmes et al.91 | https://bioconductor.org/packages/release/bioc/html/DirichletMultinomial.html |
| inStrain | Olm et al.42 | https://github.com/MrOlm/inStrain |
| pyani | Pritchard et al.89 | https://github.com/widdowquinn/pyani |
| Repeat measures PERMANOVA | Lloyd-Price et al.21 | https://bitbucket.org/biobakery/hmp2_analysis/src/master/ |
| Other | ||
| Analytic scripts and data | This paper |
https://github.com/dantaslab/NICUExposome
10.5281/zenodo.12737979 |