

Abstract
Osteosarcoma is a rare malignant bone‐originating tumor that usually occurs in young people. Programmed cell death 1 ligand 1 (PD‐L1), an immune checkpoint protein, is highly expressed in osteosarcoma tissues. Several recent studies have indicated that the tumor‐related role of PD‐L1 in tumors, especially non‐plasma membrane (NPM)‐localized PD‐L1, is not limited to immune regulation in osteosarcoma. Here, mass spectrometry analysis is combined with RNA‐seq examination to identify the intracellular binding partners of PD‐L1 and elucidate the underlying mechanism of its action. It is found that the NPM‐localized PD‐L1 interacted with Insulin‐like growth factor binding protein‐3 (IGFBP3) to promote osteosarcoma tumor growth by activating mTOR signaling. This interaction is enforced after phosphoglyceratekinase1 (PGK1)‐mediated PD‐L1 phosphorylation. Based on these findings, a phosphorylation‐mimicking peptide is designed from PD‐L1 and it is encapsulated with a Cyclic RGD (cRGD)‐modified red blood cell membrane (RBCM) vesicle (Peptide@cRGD‐M). The Peptide@cRGD‐M precisely delivers the PD‐L1‐derived phosphorylation‐mimicking peptide into osteosarcoma lesions and significantly promotes its therapeutic effect on the tumor. Therefore, this investigation not only highlights the function of NPM‐localized PD‐L1, but also uses an engineering approach to synthesize a small molecular peptide capable of inhibiting osteosarcoma growth.
Keywords: cRGD, osteosarcoma, PD‐L1, peptides, RBCMs, tumor growth
A model depicting that NPM localized PD‐L1 interacted with IGFBP3 to promote the tumor growth of osteosarcoma by activating the mTOR signaling. This interaction is enforced after PD‐L1 phosphorylation mediated by PGK1. The phosphorylation‐mimicking peptide of PD‐L1 is synthesized and further embedded in the membrane‐coated nanovesicles to form peptide@cRGD‐M, which inhibits the tumor growth of osteosarcoma.
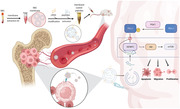
1. Introduction
Osteosarcoma is a rare malignant bone‐originating tumor that usually occurs in young people.[ 1 ] While most patients with surgically resectable osteosarcoma can survive for a long time after complete tumor resection and adjuvant chemotherapy, not every tumor is resectable. Patients with nonresectable advanced osteosarcoma have a poor outcome, with 5‐year survival rates of less than 25%.[ 2 ] Thus, identifying novel therapeutic strategies for the treatment of late‐stage osteosarcoma patients is of great necessity. Whole‐genome sequencing reveals high genomic instability in osteosarcoma, but targeting oncogenic kinases does not provide the anticipated efficiency in the treatment of this tumor type.[ 1 ]
Immunotherapy, especially immune checkpoint blockade, is a promising treatment option for multiple malignant tumor types.[ 3 ] The crosstalk between osteoblasts and monocyte‐derived osteoclasts makes the bone an immune‐related organ.[ 1 ] Therefore, because osteosarcoma originates from osteoblasts, immune‐based therapy could potentially benefit patients with the disease.[ 1 ] Some studies have shown that immune checkpoint proteins, including programmed cell death 1 ligand 1 (PD‐L1), are expressed in osteosarcoma tissues.[ 4 , 5 ] However, the poor immunogenicity of the tumor microenvironment renders the blockade of the PD‐1/PD‐L1 axis less effective in osteosarcoma.[ 6 ] Remarkably, PD‐L1 is reportedly not only involved in regulating the immune response to osteosarcoma[ 7 ] but also participates in promoting metastasis or chemotherapy resistance in osteosarcoma,[ 8 , 9 ] suggesting that the tumor‐related role of PD‐L1 in tumors is not limited to the immune regulation of osteosarcoma.
Recently, PD‐L1 has also been found to translocate from the plasma membrane to the nucleus after acetylation.[ 10 ] Nuclear PD‐L1 regulates the expression of pro‐inflammation genes,[ 10 ] while intracellular PD‐L1 competes with RNA Exosomes and modulates the response to DNA damage.[ 11 ] These findings indicate that intracellular PD‐L1 is also critical to tumor progression. However, the specific role and corresponding mechanism of non‐plasma membrane (NPM) PD‐L1 in osteosarcoma remains unclear.
Of the different circulatory cells, red blood cells (RBCs) are ideal raw materials for drug delivery because they are readily available, remarkably biocompatible, and have prolonged systematic circulation time.[ 12 ] However, the enormous sizes of intact RBCs limit their tumor retention and anti‐tumor efficiency. Red blood cell membrane (RBCM)‐modified nanoparticles could provide a feasible solution to the size issue.[ 12 ] If they can, then membrane‐camouflage technology would represent a novel drug delivery strategy for osteosarcoma patients, one that endows nanoparticles with reduced immunogenicity and anti‐phagocytosis.
Although RBCMs have been used widely as drug‐delivery carriers, they cannot be modified directly by genetic engineering because they have no nuclei.[ 13 ] Therefore, an engineering strategy must be concocted to adjust RBCMs to improve tumor retention. The surface engineering of RBCMs with a tumor‐targeting ligand is apparently useful in promoting their anti‐tumor efficiency.[ 14 , 15 ] Cyclic RGD peptide (cRGD) is an efficient tumor‐targeting ligand with a high affinity for αvβ3 integrin receptors. Reportedly, αvβ3 integrin receptors are diffusely expressed on the surface of tumor cells, including osteosarcoma.[ 16 , 17 ] Per previous inquiries, the cRGD‐modified delivery system can target tumor cells efficiently, release therapeutic agents, and improve anti‐tumor effects significantly.[ 18 ] However, whether cRGD‐modified nanoparticles can facilitate their distribution in osteosarcoma sites and contribute to efficient therapy is still unknown.
In this study, we combined mass spectrometry analysis with RNA‐seq analysis to identify the intracellular binding partners of PD‐L1 and elucidate the underlying mechanism of its action. We showed that NPM PD‐L1 binds with IGFBP3 to activate mTOR signaling and promote osteosarcoma tumor growth in a process enhanced by PGK1‐mediated phosphorylation. Based on these findings, we designed a phosphorylation‐mimicking peptide from PD‐L1 and embedded it in a cRGD‐modified RBCM vesicle to generate peptide@cRGD‐M. By combining the RBC membrane therapy with peptide therapy using an engineering approach, we developed an efficient peptide@cRGD‐M nanoparticle strategy for osteosarcoma therapy and promoted the application of RBCMs for drug delivery.
2. Results and Discussion
2.1. Non‐Plasma Membrane‐Localized PD‐L1 Binds with IGFBP3 to Impede Osteosarcoma Aggression
To determine the cancer‐related role of PD‐L1 in addition to being a membrane protein, we removed the plasma membranes of osteosarcoma cells, harvesting and subjecting the rest of the subcellular portion to mass spectrometry analysis using IgG or PD‐L1 antibodies (Figure 1A). Some proteins, including IGFBP3, PGK1, and RP‐L7a, were identified as potential binding partners of non‐plasma membrane (NPM) PD‐L1 (Figure 1A and Figure S1A,B, Supporting Information). A subsequent co‐immunoprecipitation (IP) assay revealed that PD‐L1 interacted with insulin‐like growth factor binding protein 3 (IGFBP3); particularly, NPM PD‐L1 was bound to IGFBP3 in osteosarcoma cells (Figure 1B,C).
Figure 1.
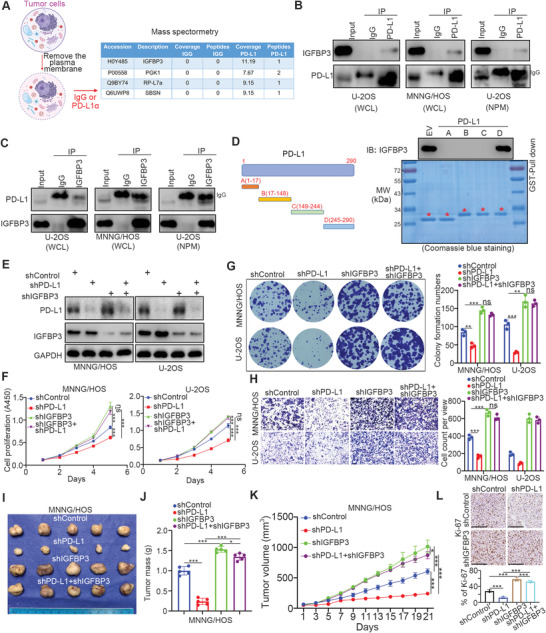
Non‐plasma membrane‐localized PD‐L1 binds with IGFBP3 to impede osteosarcoma aggression. A) The model depicting that the plasma membrane of osteosarcoma was removed and the rest of the cell compartments were subjected to mass spectrometry analysis by using the IgG or PD‐L1 antibody. The mass spectrometry of PD‐L1 was indicated in the figure. B,C) The co‐immunoprecipitation was performed in U‐2OS, MNNG/HOS and NPM U‐2OS by using the IgG, PD‐L1 or IGFBP3 antibodies. D) The diagram indicated the recombinant proteins of PD‐L1 with GST‐tagged on the left of this panel. The GST‐Pull down assay was shown on the right of this panel. E‐H. MNNG/HOS and U‐2OS cells were infected with indicated shRNAs for 72 h. These cells were harvested for western blot analysis (E), CCK‐8 assay (F), colony formation assay (G), and transwell analysis (H). Data presented as mean ± SEM with three replicates. Ns, not significant; **, P<0.01; ***, P< 0.001. I‐L. The MNNG/HOS cells were infected with indicated shRNAs for 72 h. After puromycin selection, cells were subcutaneously injected into nude mice. The tumor image was shown in panel I. The tumor mass was shown in panel J. The tumor volume was shown in panel K. Data presented as mean ± SEM with five replicates. *, P<0.05; ***, P< 0.001. The Ki‐67 staining was shown in panel L. Data presented as mean ± SEM with three replicates. ***, P< 0.001.
To explore the specific PD‐L1 region that binds with IGFBP3, we constructed recombinant proteins of PD‐L1 based on the exons of PD‐L1 (CD274) (Figure 1D). GST‐Pull down analysis showed that IGFBP3 was bound to the C‐terminal region (245‐290aa) of PD‐L1 (Figure 1D).
We previously revealed that IGFBP3 represses the progression of osteosarcoma cells by inactivating AKT signaling.[ 19 ] Thus, we sought to know whether IGFBP3 was the key mediator for the NPM PD‐L1‐induced progression of osteosarcoma cells. PD‐L1 and IGFBP3 were repressed in MNNG/HOS and U‐2OS cells, respectively, by infecting the cells with gene‐specific short‐hairpin RNA (Figure 1E). The proliferation ability of osteosarcoma tumor cells was then assessed in vitro employing the CCK‐8 assay and colony formation assay (Figure 1F,G), and the migration capacity was determined using transwell assay (Figure 1H). Tumor cell growth capability was evaluated in vivo using the xenograft tumor assay (Figure 1I–L). According to our findings, PD‐L1 silencing reduced osteosarcoma cell proliferation and migration in vitro and in vivo (Figure 1E–L). Interestingly, the downregulation of IGFBP3 alone promoted osteosarcoma progression in cells and mice, attenuating the effect of PD‐L1 in the co‐knockdown of IGFBP3 and PD‐L1 (Figure 1E–L). Therefore, these results suggest that IGFBP3 mediates the NPM PD‐L1‐induced progression of osteosarcoma.
2.2. PD‐L1 Activates the mTOR Signaling Pathway Through IGFBP3
Next, we scrutinized the underlying mechanism of the PD‐L1/IGFBP3 axis in osteosarcoma. As depicted in Figure 1E, PD‐L1, IGFBP3, or both were knocked down in MNNG/HOS cells, and the cells were evaluated using transcriptome analysis. The heat‐map of the analysis of RNA‐Seq data was constructed (Figure 2A). In view of the contrasting functions of PD‐L1 and IGFBP3 in osteosarcoma cells, we defined the genes regulated by PD‐L1 or IGFBP3 but with the opposite trend to their common target genes. 506 plus 324 genes were identified as the common target genes of PD‐L1 and IGFBP3, respectively, in osteosarcoma cells (Figure 2B,C). The gene ontology (GO) biological processes enrichment analysis and the Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis were performed on these common target genes (Figure 2D,E), revealing that the mTOR signaling pathway was the downstream pathway of the PD‐L1/IGFBP3 axis (Figure 2D,E). Western blot analysis indicated that PD‐L1 silencing decreased S6K1 T389 phosphorylation, while the co‐knockdown of PD‐L1 and IGFBP3 or the use of mTOR inhibitor treatment (RAD001) diminished the change in S6K1 T389 phosphorylation (Figure 2F). According to CCK‐8 analysis findings, the knockdown of PD‐L1 in U2‐OS cells and MNNG/HOS cells markedly reduced the IC50 value of RAD001 but knocking down both PD‐L1 and IGFBP3 resulted in a less apparent decrease (Figure 2G). We also found that knocking down PD‐L1 in U2‐OS cells and MNNG/HOS cells enhanced the growth inhibitory effect of RAD001, and treatment with RAD001 attenuated the PD‐L1/IGFBP3 axis‐induced proliferation effect change in osteosarcoma cells (Figure 2H). Hence, PD‐L1/IGFBP3 most probably modulates osteosarcoma cell growth through the mTOR pathway.
Figure 2.
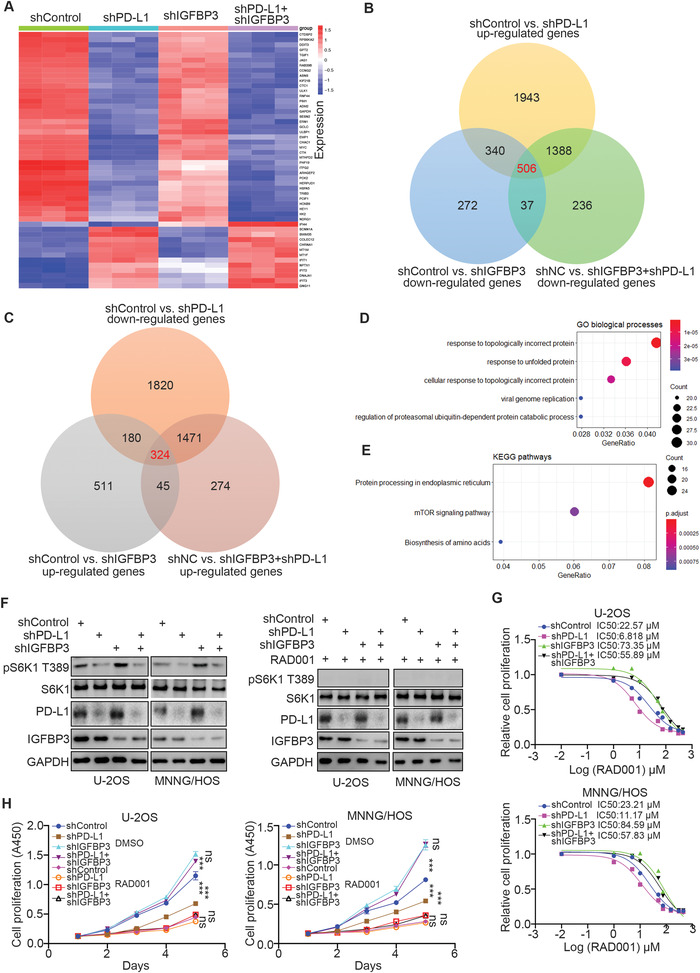
PD‐L1 activates the mTOR signaling pathway through IGFBP3. A) MNNG/HOS cells were infected with the indicated shRNAs for 72 h. Cells were used for RNA‐seq analysis. B,C) The Venn map indicated the common downstream genes of PD‐L1 and IGFBP3. D,E) GO (D) and KEGG (E) pathway enrichment analyses of the RNA‐seq analysis were performed. F–H) MNNG/HOS and U‐2OS cells were infected with indicated shRNAs for 72 h. These cells were harvested for western blot analysis (F), CCK‐8 assay with different doses of RAD001 to measure the IC50 values of RAD001 (G), or CCK‐8 assay with 20 µm RAD001. Data presented as mean ± SEM with five replicates. Ns, not significant; ***, P< 0.001.
2.3. Hyperphosphorylated PD‐L1 Enhances the Interaction between PD‐L1 and IGFBP3 in Osteosarcoma Cells
The mass spectrometry of NPM PD‐L1 also established Phosphoglycerate kinase 1 (PGK1) as a binding partner of PD‐L1 (Figure 1A). Of note, some investigations have shown that PGK1 activates the AKT/mTOR signaling pathway in non‐small cell lung cancer and tongue squamous cell carcinoma.[ 20 , 21 ] In this research, we also noted that knocking down PGK1 lessened the phosphorylation level of S6K1 T389 in MNNG/HOS and U‐2OS cells (Figure 3A). Using the co‐IP assay, we found that PGK1 interacted with PD‐L1 or NPM PD‐L1 in osteosarcoma cells (Figure 3B). Therefore, we assessed whether PGK1 regulates the mTOR signaling pathway through the PD‐L1/IGFBP3 signaling axis. First, we demonstrated that PGK1 knockdown reduced the degree of interaction between PD‐L1 and IGFBP3 in MNNG/HOS and U‐2OS cells (Figure 3C). In contrast, PGK1 overexpression enhanced PD‐L1 binding to IGFBP3 in osteosarcoma cells (Figure 3D), but this effect decreased after treatment with λ‐phosphatase (Figure 3E). Transfection with a kinase‐dead mutant of PGK1 (deletion of the K191 site of PGK1) did not increase the degree of binding between PD‐L1 and IGFBP3 (Figure 3F). These data suggest that the phosphorylation modification of PD‐L1 possibly impacts the interaction of PD‐L1 with IGFBP3.
Figure 3.
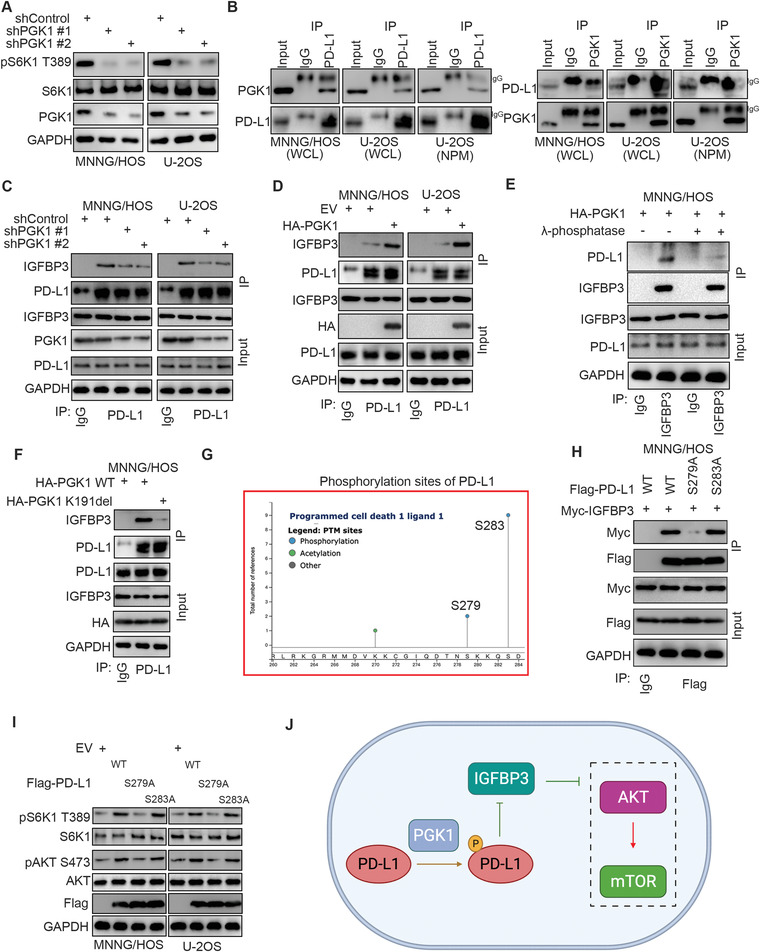
Hyperphosphorylated PD‐L1 enhances the interaction between PD‐L1 and IGFBP3 in osteosarcoma cells. A) MNNG/HOS and U‐2OS cells were infected with the indicated shRNAs for 72 h. Cells were harvested for western blot analysis. B) The co‐immunoprecipitation was performed in U‐2OS, MNNG/HOS, and NPM U‐2OS by using the IgG, PD‐L1 or PGK1 antibodies. C) MNNG/HOS and U‐2OS cells were infected with the indicated shRNAs for 72 h. Cells were harvested for immunoprecipitation by using the IgG or PD‐L1 antibodies. D) MNNG/HOS and U‐2OS cells were transfected with the indicated plasmids for 48 h. Cells were harvested for immunoprecipitation by using the IgG or PD‐L1 antibodies. E) MNNG/HOS cells were transfected with the indicated plasmids for 48 h. Cells were lysised with RIPA buffer and treated with or without λ‐phosphatase. The whole cell lysates were used for immunoprecipitation by IgG or IGFBP3 antibodies. F) MNNG/HOS cells were transfected with the indicated plasmids for 48 h. Cells were harvested for immunoprecipitation by using the IgG or PD‐L1 antibodies. G) The diagram indicated, the phosphorylation sites of c‐terminus PD‐L1 by analyzing the PhosphoSitePlus® dataset. H) MNNG/HOS cells were transfected with the indicated plasmids for 48 h. Cells were harvested for immunoprecipitation by using the IgG or Flag‐tagged antibodies. I) MNNG/HOS cells were transfected with the indicated plasmids for 48 h. Cells were harvested for western blot analysis. J) A model directing that phosphorylation of PD‐L1 mediated by PGK1 strengthens the interaction between PD‐L1 and IGFBP3 to activate the mTOR signaling pathway in osteosarcoma cells.
Specifically, these results uncovered a binding relationship between the C‐terminal region (245–290 aa) of PD‐L1 and IGFBP3 (Figure 1D). Therefore, we next employed the PhosphoSitePlus® dataset to identify the phosphorylation sites of the C‐terminal region of PD‐L1 and found two potential ones (S279 and S283) (Figure 3G). The S279A and S283A mutants of PD‐L1 were constructed to mimic the de‐phosphorylation status in the corresponding sites. Our findings showed that the PD‐L1 S279A mutant but not the S283A mutant decreased the level of interaction between PD‐L1 and IGFBP3 in MNNG/HOS cells (Figure 3H). Additionally, the PD‐L1 S279A mutant could not increase the extent of the phosphorylation of S6K1 T389 in MNNG/HOS and U‐2OS cells compared to the PD‐L1 wild type (WT) or S283A mutant (Figure 3I). Together, our results suggest that the PGK1‐mediated phosphorylation of PD‐L1 strengthens the interaction between PD‐L1 and IGFBP3 to activate the mTOR signaling pathway in osteosarcoma cells.
2.4. The PD‐L1‐Derived Phosphorylation‐Mimicking Peptide Inhibits Osteosarcoma Cell Progression
Since the phosphorylation of S279 in PD‐L1 was crucial to inhibiting IGFBP3 to activate mTOR signaling in osteosarcoma cells, we synthesized a phosphorylation‐mimicking peptide of PD‐L1, including the S279 site (Figure 4A). The purity of the synthesized peptide of PD‐L1 was tested using Mass Spectrometry (MS) and High‐ Performance Liquid Chromatograph (HPLC) (Figure 4B,C), and its ability to disrupt the PD‐L1/IGFBP3 complex was evaluated. This peptide decreased S6K1 T389 phosphorylation; however, IGFBP3 knockdown attenuated this effect, indicating that the PD‐L1 phosphorylation‐mimicking peptide repressed the mTOR pathway through IGFBP3 (Figure 4D). The peptide also reduced the proliferation and migration abilities of osteosarcoma cells in vitro (Figure 4E–G). The intratumor injection of the PD‐L1 phosphorylation‐mimicking peptide significantly lessened osteosarcoma tumor growth (Figure 4H–K). Our data, therefore, suggest that the PD‐L1‐derived phosphorylation‐mimicking peptide inhibits the progression of osteosarcoma in cells.
Figure 4.
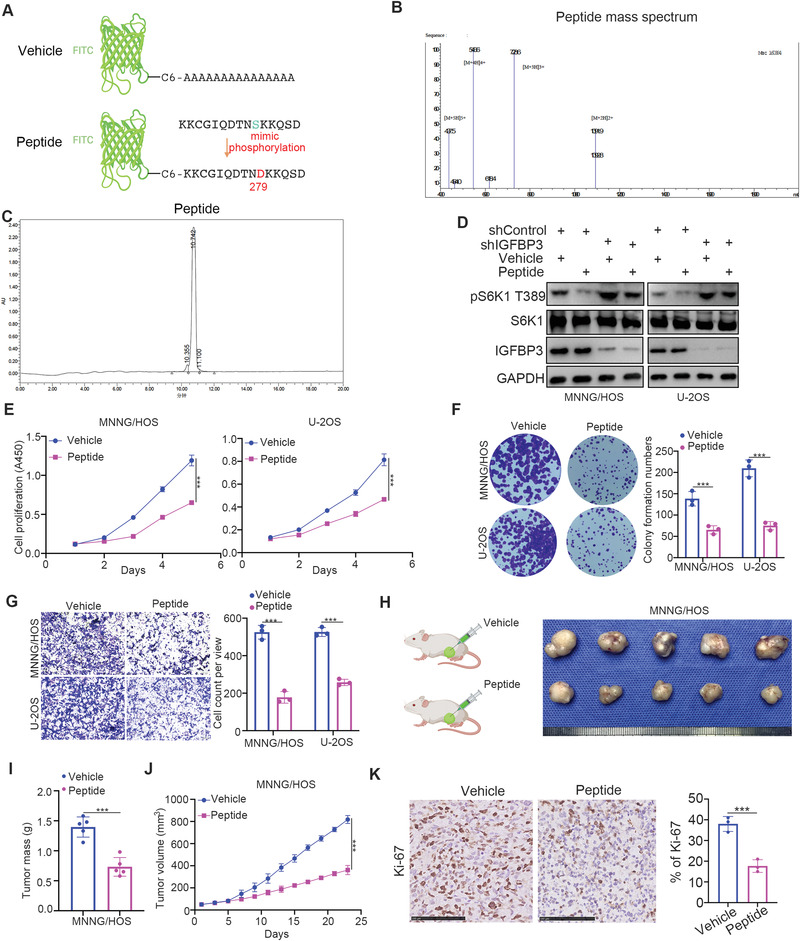
The PD‐L1‐derived phosphorylation‐mimicking peptide inhibits osteosarcoma cell progression. A) The diagram indicated the peptide of PD‐L1. B,C) the detail of synthesized peptide of PD‐L1. D) MNNG/HOS and U‐2OS cells were infected with the indicated shRNAs for 48 h. Then, cells were treated with or without peptide for 24 h. Cells were harvested for western blot analysis. (vehicle or peptide concentration: 0.05 mg mL−1) E–G) MNNG/HOS and U‐2OS cells were treated with or without peptide. These cells were subjected to CCK‐8 assay (E), colony formation assay (F), and transwell analysis (G). Data presented as mean ± SEM with three replicates. ***, P< 0.001. (vehicle or peptide concentration: 0.05 mg mL−1) H–K) The MNNG/HOS cells were subcutaneously injected into nude mice. Vehicle or peptide was injected into the tumor at day 1, 3,5, 7 (1.2 mg kg−1 every 2 h for 8 h). The tumor image was shown in panel H. The tumor mass was shown in panel I. The tumor volume was shown in panel J. Data presented as mean ± SEM with five replicates. ***, P< 0.001. The Ki‐67 staining was shown in panel K. Data presented as mean ± SEM with three replicates. ***, P< 0.001.
2.5. Characterization of peptide@cRGD‐M
Cell membranes derived from the RBCs of nude mice were used to disguise our peptide, endowing it with better biocompatibility and longer circulation time. To enhance the accumulation of peptides in tumors, cyclic RGD (cRGD) was employed to modify the RBC membrane to achieve a better tumor‐targeting capability (Figure 5A). The morphologies of the cRGD‐modified membrane vesicle (cRGD‐M) and peptide@cRGD‐M were examined under transmission electron microscopy (TEM) (Figure 5B), which demonstrated an obvious core/shell structure. The shell thickness of both cRGD‐M and peptide@cRGD‐M was ≈10 nm, pointing to the successful coverage of peptides by the RBC membrane. The mean hydrodynamic diameters of these two mono‐dispersed, spherical nanoparticles were (193.3±3.8) and (194.7±6.1) nm (Figure 5C), with the zeta potentials of (−15.3±3.7) and (‐17.1±2.4) mV (Figure 5D), respectively, which further confirmed the successful coating of membranes.
Figure 5.
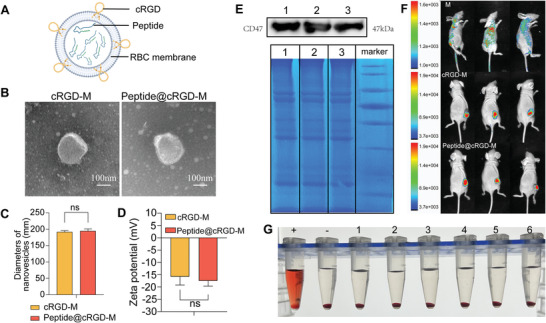
Characterization of peptide@cRGD‐M. A) Diagram of the structure of peptide@cRGD‐M. B) TEM images of cRGD‐M and peptide@cRGD‐M. (Scale bar = 100 nm) C,D) Nanovesicle diameters (C) and zeta potential (D) of cRGD‐M and peptide@cRGD‐M. E) Membrane protein characterization by Western blot analysis and SDS‐PAGE of the pure membrane of nude mice derived RBCs (lane 1); cRGD‐M (lane 2); peptide@cRGD‐M (lane 3) and marker. F) In vivo distribution of cRGD‐M and peptide@cRGD‐M in tumor‐bearing nude mice examined by fluorescence imaging. RBC membranes (M) were labeled with DiD (red) and the relative amount of membrane was detected by measuring the relative intensity of red fluorescence. G) Bio‐safety examination of peptide@cRGD‐M evaluated by RBC hemolysis test. The supernatant of RBC co‐cultured with pure water (+), PBS (‐), and peptide@cRGD‐M [peptide concentration: 0.2 mg mL−1 (lane 1), 0.1 mg mL−1 (lane 2), 0.05 mg mL−1 (lane 3), 0.025 mg mL−1 (lane 4), 0.0125 mg mL−1 (lane 5), and 0.00625 mg mL−1 (lane 6)] aqueous solutions are presented, respectively.
To find out if the membranes retained immune escape functions, Western‐blot and sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) were employed to analyze the protein ingredients of cRGD‐M and peptide@cRGD‐M. We found that CD47, one of the most important antiphagocytic proteins, and the majority of the RBC membrane proteins were preserved after co‐extrusion with peptides (Figure 5E).
To further examine the tumor‐targeting ability of cRGD presenting on the membranes, DiD‐labeled membranes (M), cRGD‐M or peptide@cRGD‐M were injected into tumor‐bearing nude mice. The captured in vivo images revealed significantly higher fluorescence intensities in the tumor specimens of both the cRGD‐M and peptide@cRGD‐M groups than those in simple membranes, demonstrating cRGD's strong targeting ability (Figure 5F). The RBC hemolysis test used to assess the different concentrations of peptide@cRGD‐M (peptide concentration ranging from 0.2 to 0.00625 mg mL−1) (Figure 5G) determined no obvious hemolysis, confirming the good biocompatibility of the nanoparticles.
2.6. The Distribution and Anti‐Tumor Efficacy of peptide@cRGD‐M In Vivo and In Vitro
To assess the intracellular uptake of the peptide with membrane coating, coated RBC membranes were fluorescently labeled with DiD (red). MNNG/HOS cells were incubated with the labeled vehicle, peptide, peptide@M, or peptide@cRGD‐M for 1 h and then examined under confocal laser scanning microscopy (CLSM). Few green (FITC) fluorescence marks were observed in MNNG/HOS cells in the first two groups (simple vehicles group and simple peptide group), while red (DiD) and green fluorescence marks were notably distributed next to the nucleus (blue, DAPI‐stained) and co‐localized in cells incubated with peptide@M and peptide@cRGD‐M, indicating that membrane‐coated peptides retained their structural integrity after they entered the cells. The average fluorescence intensity (both red and green) of peptide@cRGD‐M was higher than that of peptide@M, implying that cRGD helped intensify the intracellular uptake of peptides in vitro, an outcome that was confirmed by quantitative analysis (Figure 6A, Figure S2C, Supporting Information). The measurement of the rate of cell proliferation in the four groups after incubation for 5 days showed that peptide@cRGD‐M significantly restrained cancer cell growth in MNNG/HOS and U‐2OS cell lines compared to the other groups (Figure 6B).
Figure 6.
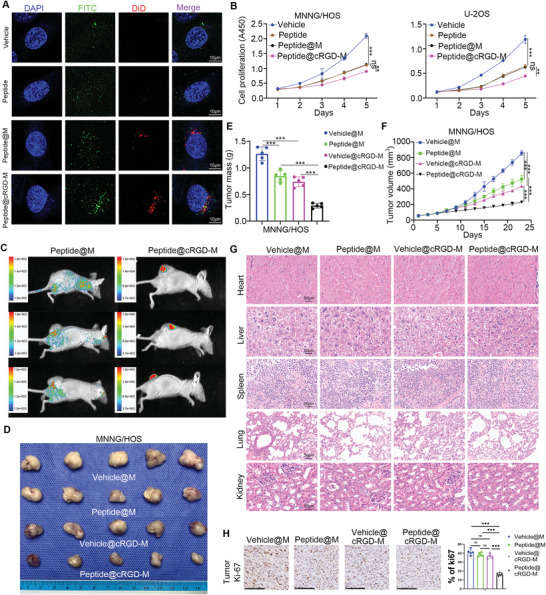
The distribution and anti‐tumor efficacy of peptide@cRGD‐M in vivo and in vitro. A) Intracellular uptake of pure vehicles, pure peptides, peptide@M, and peptide@cRGD‐M in MNNG/HOS cells after 1 h incubation were examined by CLSM. The nucleus was stained with DAPI (blue). Peptides and vehicles were labeled with FITC (green) and RBC membranes (M) were labeled with DiD (red). Scale bar = 10 µm. B) In vitro cytotoxicity of pure vehicles, pure peptides, peptide@M, and peptide@cRGD‐M (vehicle or peptide concentration: 0.05 mg mL−1) on MNNG/HOS cells and U2OS cells after 1 to 5 days incubation. Data are given as the mean ± SEM (n = 3). Ns, not significant; **, P<0.01; ***, P<0.001. C) In vivo biodistribution images after four times of injections of peptide@M or peptide@cRGD‐M in tumor‐bearing nude mice (1.2 mg kg−1 every 2 h for 8 h). The relative amount of FITC‐labeled peptide was detected by measuring the relative intensity of green fluorescence. D–F, H) Representative photographs, masses, volumes of tumors, Ki‐67 staining subjected to pure vehicles@M, pure peptides@M, vehicle@cRGD‐M, and peptide@cRGD‐M, respectively. Data are given as the mean ± SEM (n = 5). ***, P<0.001. G) Histopathologic examination of vital organs including heart, liver, spleen, lung, and kidney from nude mice after intravenous administration of pure vehicles, pure peptides, vehicle@cRGD‐M, and peptide@cRGD‐M for 23 days. Scale bar = 50 µm.
In vivo distribution of peptides was determined via scrutiny of the green (FITC) fluorescence intensity, which revealed that the accumulation of the peptides in the tumor drastically increased after camouflaging peptides with cRGD‐M (Figure 6C). In addition, tumor‐bearing nude mice received simple vehicle@M (1.2 mg kg−1 per 2 h for 8 h), simple peptides@M (1.2 mg kg−1 per 2 h for 8 h), vehicle@cRGD‐M (peptide concentration: 1.2 mg kg−1 per 2 h for 8 h) or peptide@cRGD‐M (peptide concentration: 1.2 mg kg−1 per 2 h for 8 h) intravenously via the caudal vein on Days 5, 9, 13, 17, and 21, respectively. The photos and masses of tumors on Day 23, as well as volume changes in the subcutaneously transplanted tumors in these four groups from Day 1 to Day 23, were recorded. Peptide@cRGD‐M markedly reduced tumor growth compared to the other three groups, with the difference increasing over time (Figure 6D–F,H). Hematoxylin‐eosin (HE) staining of the vehicle@M, peptide@M, vehicle@cRGD‐M, and peptide@cRGD‐M groups was used to evaluate the toxicity of nanoparticles. No obvious cardiac damage, liver injury, and/or inflammatory infiltrates were observed in the spleen, pulmonary toxicity, or renal injury (Figure 6G).
2.7. Discussion
PD‐L1, located mainly in the plasma membrane of tumors, is an immune checkpoint protein that binds with PD‐1 to inhibit the activation of T cells and regulate the immune response. PD‐L1 is expressed in the osteosarcoma tissue sample; however, its role in osteosarcoma is not limited to immune response regulation. Reports show that the LINC00657/ miR‐106a /PD‐L1 axis promotes the metastasis of osteosarcoma.[ 8 ] Additionally, the exosome carrying PD‐L1 and N‐cadherin released from osteosarcoma results in pulmonary metastasis.[ 22 ] However, the mechanism of how PD‐L1 modulates osteosarcoma metastasis is unclear. PD‐L1 also reportedly regulates cisplatin resistance in osteosarcoma.[ 23 ] Notably, intercellular PD‐L1 functions as an RNA binding protein to protect the mRNAs of NBS1 and BRCA1 from degradation and regulate DNA damage response,[ 11 ] possibly providing one explanation for how PD‐L1 leads to cisplatin resistance. Similarly, we have demonstrated that NPM PD‐L1 activates mTOR signaling to regulate osteosarcoma growth by binding with IGFBP3. mTOR signaling is crucial to tumor cell growth, proliferation and metastasis in osteosarcoma.[ 24 ] Our research has, therefore, expanded the understanding of NPM PD‐L1's function in osteosarcoma, partially explaining why PD‐L1 regulates the aggression of osteosarcoma. Furthermore, there is proof that acetylation on the c‐terminus of PD‐L1 increases its nuclear translocation.[ 10 ] In the same way, our study suggests that the phosphorylation modification of the c‐terminus of PD‐L1 enhances its binding to IGFBP3, indicating that the post‐translational modification of PD‐L1 is essential to PD‐L1's function.
IGFBP3, which belongs to the IGF binding protein family, participates in multiple types of cellular processes, such as cell proliferation, apoptosis, senescence, and differentiation.[ 25 ] While the role of IGFBP3 in cancer remains controversial, some studies have shown that it stimulates tumor growth in breast cancer via IGF‐1.[ 26 ] However, IGFBP3 is also known to repress tumor progression and metastasis through the inactivation of PI3K/AKT and RAS signaling pathways.[ 27 , 28 ] We previously reported that the E3 ligase TRAIP promotes osteosarcoma tumor growth via KANK1‐IGFBP3‐AKT signaling.[ 19 ] In this study, we further show that PD‐L1 binds with IGFBP3 to activate mTOR signaling in osteosarcoma. Moreover, we observed that the PD‐L1‐derived peptide hindered osteosarcoma proliferation through the IGFBP3‐mTOR axis. Still, the specific mechanisms of how PD‐L1 inhibits IGFBP3 and how the phosphorylation‐mimicking peptide obstructs the mTOR pathway through IGFBP3 remain unclear. Further in‐depth investigations must be carried out to answer these questions.
Biomimetic cell membrane‐coated nanocarriers are characterized by prolonged drug delivery, immunological evasion, homotypic targeting, longer blood circulation, and specific ligand/receptor recognition.[ 29 ] RBCs have attracted considerable interest as a biomaterial for nanocarrier coating. RBCs lack nuclei, which makes membrane extraction easier than when exploiting nucleus‐containing cells. For example, gradient centrifugation is often used to extract eukaryote cell membranes to separate their membranes from nuclei and other cell components. However, this technique is not required for preparing nuclei‐free membranes, such as RBCs.[ 30 ] RBCs could remove pathogens from the body via oxycytosis during blood circulation.[ 31 ] RBCs also express the “don't eat me” marker CD47, preventing them from being taken up by macrophages.[ 32 ] Accordingly, applying RBC membrane‐coated nanovesicles could improve the detoxification process, pathogen removal, and long‐term circulation. In our study, the peptide@cRGD‐M nanoparticle exhibited potent targeted anti‐tumor therapy capability in vitro and in vivo, and no significant organ toxicity was observed. Therefore, the nanoparticle is an effective and safe delivery system for osteosarcoma therapy.
However, before membrane‐coated nanovesicles can be applied clinically, several obstacles must be overcome. (a) The procedures for preparing and characterizing cell membranes are not reliable. A standardized highly repeatable method for cell membrane quality control must be developed. (b) The preparation procedures must be simplified and optimized to cut down the costs. (c) While it is relatively simple to extract and prepare cell membranes in comparison to isolating extracellular vesicles for drug transport, the targeting ability of these membranes may be reduced owing to protein loss during membrane extraction.[ 33 ] (d) RBC membrane‐coated nanocarriers lack a targeting ability. Modifying RBC membranes with specific ligands could enhance their targeting ability; however, the change might also alter the original structure of membranes and reduce the biocompatibility. Therefore, membrane hybridization could be helpful.[ 34 ] For example, Sun et al. developed a tumor cell‐RBC hybrid membrane‐coated nanovesicles characterized by the homological tumor‐targeting of the tumor cell membrane and reduced clearance by the RBC membrane.[ 35 ]
3. Conclusion
In summary, we have demonstrated that NPM‐localized PD‐L1 interacts with IGFBP3 to promote osteosarcoma tumor growth by activating mTOR signaling. We also established that this interaction is enforced by PGK1‐mediated PD‐L1 phosphorylation. Additionally, we designed and synthesized a phosphorylation‐mimicking peptide of PD‐L1 and embedded it in a membrane‐coated nanovesicle to create peptide@cRGD‐M. The peptide alone or peptide@cRGD‐M inhibited osteosarcoma tumor growth. Therefore, this study not only highlighted the non‐cell membrane function of PD‐L1, but also synthesized small molecular peptides capable of inhibiting osteosarcoma growth.
4. Experimental Section
Cell Culture
Two human osteosarcoma cell lines (MNNG/HOS and U‐2OS) were purchased from the Cell Bank of China Academy of Sciences (Shanghai, China). MNNG/HOS were cultured in MEM Medium (HyClone, USA), and U‐2OS cells were cultured in McCoy's 5a Medium (HyClone, USA) supplemented with 10% fetal bovine serum (Gibco, USA). All the cell lines were maintained at 37 °C with 5% CO2 in a humidified incubator.
Preparation of peptide@cRGD‐M
In order to prepare peptide@cRGD‐M, RBC membrane was prepared at first. Briefly, 3 mL blood from healthy nude mice was collected via orbital vein and centrifugated (600 g, 4 °C, 10 min). The bottom sediment was washed with excess PBS three times, and then the RBC cells were resuspended in 10 mL cold sterile ultrapure water containing 10 nM PMSF at 4 °C for 1 h and further homogenized 30 times using glass homogenizers. The homogenized solution was centrifuged at 700 g for 10 min at 4 °C, then the supernatant solution was centrifuged several times at 14000 × g for 30 min. Each time, the sediment was washed with cold PBS until the supernatant was clear. The sediment was collected and preserved at ‐80 °C.
Powder of phospholipid (DSPE)‐PEG2K‐cRGDyK (0.5 mg, Xi'an Ruixibio Ltd, China) was dissolved in 0.5 mL of sterile ultrapure water at 60 °C for 30 min to form micelles. And then cRGD‐modified membrane was obtained by incubating 1 mL membrane suspension with 0.5 mL cRGD micelles solution at 40 °C for 2 h, before being purified by using size‐exclusion chromatography.
In order to camouflage vehicles or peptides with membranes or cRGD‐modified membranes to obtain peptide@M or peptide@cRGD‐M, the mixture of vehicles or peptides and RBC membranes or cRGD‐modified membranes (volume ratio = 3:1) was extruded through 200 nm polycarbonate membranes (Avanti ® Mini‐Extruder, Avanti, USA) for 10 cycles or more.
Characterization of peptide@cRGD‐M
The morphologies of cRGD‐M and peptide@cRGD‐M were observed using transmission electron microscopy (TEM) (FEI Tecnai G2 F20, FEI, USA) after being stained with 1% (weight/volume) phosphotungstic acid. The particle diameter and zeta potential were measured using dynamic light scattering (DLS) (Malvern Zetasizer Nano ZS90, Malvern, UK). Western‐blot and SDS‐PAGE were utilized to evaluate the expression of RBC markers such as CD47.
Biosafety and Cytotoxicity Test
RBC hemolysis test was also used to measure the cytotoxicity. Briefly, 1 mL of blood collected from the orbital vein of healthy nude mice was washed 3 times by PBS before being centrifuged 600 g at 4 °C. The washed bottom sediment (containing RBC) was dissolved into 10 mL PBS and divided into several groups (0.2 mL solution per group). Every group of RBC solution was co‐cultured with 0.8 mL pure water (positive control), PBS (negative control), or different concentration of peptide@cRGD‐M (peptide concentration consisting 0.2 mg mL−1, 0.1 mg mL−1, 0.05 mg mL−1, 0.025 mg mL−1, 0.0125 mg mL−1, and 0.00625 mg mL−1) aqueous solutions for 4 h and then centrifuged (12,000 g, 5 min). The color of supernatant was observed.
Subcutaneous Xenograft Tumor Model
All animal procedures were performed in a specific pathogen‐free situation according to the guidelines of the ethics committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China, No. 2020IEC‐J(102)). The assigned approval number of the investigator was No. TY20190118. The osteosarcoma tumor model was established by subcutaneous injection of 5×106 cells into the inguinal region of nude mice (BALB/c, male, 6 to 8‐week‐old). After 5 days, the exogenous tumor was visible to the naked eyes. Tumor volumes were calculated by measuring the length (a, mm) and the width (b, mm): volume (mm3) = ab2/2. These mice were sacrificed after measuring the volume every other day until Day 23, whose main organs (i.e., heart, liver, spleen, lung, kidney) and tumors were collected, measured (volume, mass), and observed the cytotoxicity using Hematoxylin‐eosin (HE) staining. Immunohistochemistry for Ki67 was tested to evaluate the tumor proliferation rates.
In Vitro and In Vivo Biodistribution of Camouflaged Peptides
To investigate the biodistribution of cRGD‐M and peptide@cRGD‐M in vivo, 1 µL DiD (5 mM, D8940, Solarbio, China) was added to 1 mL cRGD‐M or peptide@cRGD‐M solution to label the membranes with red fluorescence. Tumor‐bearing mice were then separated into two groups randomly and injected with 2 mg kg−1 cRGD‐M or peptide@cRGD‐M via caudal vein, respectively. Subsequently, red fluorescence of DiD was collected after 2 h by using the in vivo fluorescence imaging system (Bruker In Vivo Xtreme, Bruker, USA). To further investigate the biodistribution of peptides, the tumor‐bearing nude mice were randomly separated into two groups and injected intravenously (i.v.) with simple vehicle (1.2 mg kg−1 per 2 h for 8 h), simple peptides (1.2 mg kg−1 per 2 h for 8 h), peptide@M (peptide concentration: 1.2 mg kg−1 per 2 h for 8 h) and peptide@cRGD‐M (peptide concentration: 1.2 mg kg−1 per 2 h for 8 h) on Day 5, 9, 13, 17, and 21, respectively. In vivo distribution of FITC‐labeled peptide was examined at 2 h after the injection of peptides or peptide@cRGD‐M by using the in vivo fluorescence imaging system (Bruker In Vivo Xtreme, Bruker, USA).
Western Blotting and SDS‐PAGE Analysis
Western blotting and SDS‐PAGE analysis were performed to identify the protein ingredients of the membrane before and after the extrusion. Briefly, SDS‐PAGE began with adding equal amounts of protein samples from the pure membrane, cRGD‐M, and peptide@cRGD‐M quantified by BCA protein assay kit (P0009, Beyotime, China) into 10% SDS‐polyacrylamide gel, which separated into different molecular weights bands by electrophoretic separation. Subsequently, the gel was treated with Coomassie brilliant blue method and imaged. For western blotting assay, the proteins were transferred to PVDF membrane from the gel, then the PVDF membrane was blocked by skim milk and incubated with primary antibodies overnight. On the next day, the membranes were washed with TBST and incubated with an HRP‐conjugated secondary antibody (BOSTER, China). Finally, the membranes were treated with ECL detection reagents, exposed using the ChemiDoc XRS imaging system (Bio‐Rad Laboratories, Inc. Hercules, CA, USA), and analyzed with the Image Lab software (Bio‐Rad Laboratories, Inc. Hercules, CA, USA).
Immunoprecipitation
The total cellular protein and the nuclear protein were extracted according to instructions of nuclear and cytoplasmic extraction reagents kit (P0027, Beyotime, Beijing, China). Cell lysis solution was incubated with an appropriate amount of antibody at 4 °C for 3 h, followed by incubation with protein A agarose (Vigorous Biotechnology, Beijing, China) for 1 h. The immune precipitates were washed three times using a lysis buffer solution, followed by elution with SDS loading buffer. The eluent was subjected to Western blot analysis.
Confocal Laser Scanning Microscopy (CLSM)
Cellular uptake of nanoparticles was evaluated by CLSM with FITC or DiD as the fluorescence indicator. MNNG/HOS cells were seeded in confocal dishes (1 × 104 cells per well), cultured for 12 h, and then incubated with 0.05 mg mL−1 labeled vehicle, peptide, peptide@M or peptide@cRGD‐M for 1 h in FBS‐free medium. Then the samples were labeled with 10 µg mL−1 DAPI. The samples were observed by CLSM after washing them several times with PBS.
Cell Viability Test
Cell viability was measured by Cell Counting Kit‐8 (CCK‐8) assay (Dojindo Laboratories Co. Ltd., Kumamoto, Japan). Briefly, MNNG/HOS cells (1×103 cells/well) were seeded onto 96‐well plates with 100 µL of α‐MEM medium (supplemented with 10% FBS) per well. The medium was replaced with 90 µL of fresh α‐MEM medium (without FBS) and 10 µL of CCK‐8 solution after 1 day. After 1 to 4 h of incubation at 37 °C, the absorbance of the medium at 450 nm in every well was recorded and analyzed.
Orthotopic Models of Tibial Injection
All animal procedures were performed in a specific pathogen‐free situation according to the guidelines of the ethics committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China, No. 2020IEC‐J(102)). The assigned approval number of the investigator was No. TY20190118. The BALB/c mice (8 weeks, male, n = 3, intact B and T cells) were purchased from Vitalriver (Beijing, China). The hind limb was prepared with a 70% ethanol swab. The lateral malleolus, medial malleolus, and lower half of the tibia were grasped and the leg bent in a combination of flexion and lateral rotation to expose the knee. Using a 0.8 mm needle and a drilling motion, the needle was inserted through the patellar ligament and into the anterior intercondylar area of the tibia. The needle was withdrawn and either a separate 0.8 mm needle filled with cell suspension was used to slowly inject K7M2 cells (1 × 106 in 10 µL PBS) into the previously drilled tibia. The volume of the grafts was measured every other day. Tumor volumes were calculated using the following formula: tumor volume (mm3) = (L × W2) ÷ 2. Mice were sacrificed on day 23 or when tumor volume reached 1000 mm3. The mass of the grafts was calculated from standard measurements.
Statistical Analysis
All the experiments were repeated at least three times and all the results were presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad 8.0 statistical software (GraphPad Software Inc., San Diego, CA). Student's t‐test was used to examine the significant differences between the two groups. One‐way analysis of variance (ANOVA) was performed to assume the statistical significance between three (or more) groups. P < 0.05 was considered to indicate a statistically significant result.
Ethical Approval
All animal experiments were approved by the Ethics Committee of the Wuhan Union Hospital, Huazhong University of Science and Technology.
Conflict of Interest
The authors declare no conflict of interest.
Authors Contribution
W.W., H.G., and D.J. contributed equally to this work. Wei Wu, Haoyu Guo, and Doudou Jing performed the experiments; Xin Jin, Zhicai Zhang, and Zhenhao Zhang collected the data; Xin Jin, Wenbo Yang, Feifei Pu, and Xin Huang wrote the paper and analyzed the data; Zengwu Shao and Xin Huang revised the manuscript. All authors read and approved the final manuscript.
Supporting information
Supporting Information
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 82072979).
Wu W., Guo H., Jing D., Zhang Z., Zhang Z., Pu F., Yang W., Jin X., Huang X., Shao Z., Targeted Delivery of PD‐L1‐Derived Phosphorylation‐Mimicking Peptides by Engineered Biomimetic Nanovesicles to Enhance Osteosarcoma Treatment. Adv. Healthcare Mater. 2022, 11, 2200955. 10.1002/adhm.202200955
Contributor Information
Xin Jin, Email: jinxinxy2@csu.edu.cn.
Xin Huang, Email: xin_huang@hust.edu.cn.
Zengwu Shao, Email: 1985XH0536@hust.edu.cn.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Kansara M., Teng M. W., Smyth M. J., Thomas D. M., Nat. Rev. Cancer 2014, 14, 722. [DOI] [PubMed] [Google Scholar]
- 2. Kager L., Tamamyan G., Bielack S., Future Oncol. 2017, 13, 357. [DOI] [PubMed] [Google Scholar]
- 3. Topalian S. L., Taube J. M., Anders R. A., Pardoll D. M., Nat. Rev. Cancer 2016, 16, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wunder J. S., Lee M. J., Nam J., Lau B. Y., Dickson B. C., Pinnaduwage D., Bull S. B., Ferguson P. C., Seto A., Gokgoz N., Andrulis I. L., Oncoimmunology 2020, 9, 1737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S., Guenther L. M., Aronhalt A., Cardillo L., Janeway K. A., Church A. J., J. Pediatr. Hematol./Oncol. 2020, 42, 482. [DOI] [PubMed] [Google Scholar]
- 6. Ge Y.‐X., Zhang T.‐W., Zhou L., Ding W., Liang H.‐F., Hu Z.‐C., Chen Q., Dong J., Xue F.‐F., Yin X.‐F., Jiang L.‐B., Biomaterials 2022, 282, 121407. [DOI] [PubMed] [Google Scholar]
- 7. Zhang M. L., Chen L., Li Y. J., Kong D. L., Oncol. Rep. 2019, 42, 2049. [DOI] [PubMed] [Google Scholar]
- 8. Zhang J., Chou X., Zhuang M., Zhu C., Hu Y., Cheng D., Liu Z., J. Cell. Biochem. 2020, 121, 4188. [DOI] [PubMed] [Google Scholar]
- 9. Liu X., He S., Wu H., Xie H., Zhang T., Deng Z., Environ. Health Prev. Med. 2019, 24, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao Y., Nihira N. T., Bu X., Chu C., Zhang J., Kolodziejczyk A., Fan Y., Chan N. T., Ma L., Liu J., Wang D., Dai X., Liu H., Ono M., Nakanishi A., Inuzuka H., North B. J., Huang Y.‐H., Sharma S., Geng Y., Xu W., Liu X. S., Li L., Miki Y., Sicinski P., Freeman G. J., Wei W., Nat. Cell Biol. 2020, 22, 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tu X., Qin B., Zhang Y., Zhang C., Kahila M., Nowsheen S., Yin P., Yuan J., Pei H., Li H., Yu J., Song Z., Zhou Q., Zhao F., Liu J., Zhang C., Dong H., Mutter R. W., Lou Z., Mol. Cell 2019, 74, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Della Pelle G., Kostevsek N., Int. J. Mol. Sci. 2021, 22, 5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou H., Fan Z., Lemons P. K., Cheng H., Theranostics 2016, 6, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding L., Wu Y., Wu M., Zhao Q., Li H., Liu J., Liu X., Zhang X., Zeng Y., ACS Appl. Mater. Interfaces 2021, 13, 52435. [DOI] [PubMed] [Google Scholar]
- 15. Zheng D., Yu P., Wei Z., Zhong C., Wu M., Liu X., Nano‐Micro Lett. 2020, 12, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan Z., Gui L., Zheng J., Chen Y., Qu S., Shen Y., Wang F., Er M., Gu Y., Chen H., ACS Appl. Mater. Interfaces 2018, 10, 30994. [DOI] [PubMed] [Google Scholar]
- 17. Lu Y., Li L., Lin Z., Li M., Hu X., Zhang Y., Peng M., Xia H., Han G., Adv. Healthcare Mater. 2018, 7, 1800602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang X., Wu W., Jing D., Yang L., Guo H., Wang L., Zhang W., Pu F., Shao Z., J. Controlled Release 2022, 343, 107. [DOI] [PubMed] [Google Scholar]
- 19. Li M., Wu W., Deng S., Shao Z., Jin X., Cell Death Dis. 2021, 12, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang Q., Wang Z., Qi Q., Li J., Xin Y., Qiu J., Mol. Ther.–Oncolytics 2022, 24, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu T., Zhao Y., Hu Z., Li J., Chu D., Zhang J., Li Z., Chen B., Zhang X., Pan H., Li S., Lin H., Liu L., Yan M., He X., Yao M., Cancer Res. 2017, 77, 5782. [DOI] [PubMed] [Google Scholar]
- 22. Wang J., Zhang H., Sun X., Wang X., Ren T., Huang Y., Zhang R., Zheng B., Guo W., J. Nanobiotechnol. 2020, 18, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J., Zhang Z., Qiu C., Wang J., Mol. Carcinog. 2022, 61, 322. [DOI] [PubMed] [Google Scholar]
- 24. Ding L., Congwei L., Bei Q., Tao Y., Ruiguo W., Heze Y., Bo D., Zhihong L., Oncotarget 2016, 7, 50805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang B., Zhang X., Du L.‐L., Wang Y., Liu D.‐B., Han C.‐Z., Jing J.‐X., Zhao X.‐W., Xu X.‐Q., World J. Gastroenterol. 2014, 20, 1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baxter R. C., Nat. Rev. Cancer 2014, 14, 329. [DOI] [PubMed] [Google Scholar]
- 27. Tu W., Yang B., Leng X., Pei X., Xu J., Liu M., Dong Q., Tao D., Lu Y., Liu Y., Yang Y., Cancer Sci. 2019, 110, 1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu D., Yu H.‐Q., Xiong H.‐J., Zhang Y.‐J., Lin X.‐T., Zhang J., Wu W., Wang T., Liu X.‐Y., Xie C.‐M., Front. Oncol. 2021, 11, 743824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lei W., Yang C., Wu Y., Ru G., He X., Tong X., Wang S., J. Nanobiotechnol. 2022, 20, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fang R. H., Kroll A. V., Gao W., Zhang L., Advanced materials (Deerfield Beach, Fla). 2018, 30, e1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Minasyan H., Immunol. Res. 2018, 66, 271. [DOI] [PubMed] [Google Scholar]
- 32. Wang H., Sun Y., Zhou X., Chen C., Jiao L., Li W., Gou S., Li Y., Du J., Chen G., Zhai W., Wu Y., Qi Y., Gao Y., J. Immunother. Cancer 2020, 8, e000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xia Y., Rao L., Yao H., Wang Z., Ning P., Chen X., Adv. Mater. 2020, 32, 2002054. [DOI] [PubMed] [Google Scholar]
- 34. Chen H.‐Y., Deng J., Wang Y., Wu C.‐Q., Li X., Dai H.‐W., Acta Biomater. 2020, 112, 1. [DOI] [PubMed] [Google Scholar]
- 35. Sun M., Duan Y., Ma Y., Zhang Q., Int. J. Nanomed. 2020, 15, 6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.