

Abstract
Lesions involving the osteochondral unit are difficult to treat. Biomimetic scaffolds are previously shown as promising alternative. Such devices often lack multiple functional layers that mimic bone, cartilage, and the interface. In this study, multilayered scaffolds are developed based on the use of natural extracellular matrix (ECM)‐like biopolymers. Particular attention is paid to obtain a complex matrix that mimics the native osteochondral transition. Porous, sponge‐like chitosan‐collagen‐octacalcium phosphate (OCP) scaffolds are obtained. Collagen content increases while the amount of OCP particles decreases toward the cartilage layer. The scaffolds are bioactive as a mineral layer is deposited containing hydroxyapatite at the bony side. The scaffolds stimulate proliferation of human adipose‐derived mesenchymal stem cells, but the degree of proliferation depends on the cell seeding density. The scaffolds give rise to a zone‐specific gene expression. RUNX2, COL1A1, BGLAP, and SPP1 are upregulated in the bony layer of the scaffold. SOX9 is upregulated concomitant with COL2A1 expression in the cartilage zone. Mineralization in presence of the cells is prominent in the bone area with Ca and P steadily increasing over time. These results are encouraging for the fabrication of biomimetic scaffolds using ECM‐like materials and featuring gradients that mimic native tissues and their interface.
Keywords: bones, cartilage, chitosan‐collagen‐octacalcium phosphate composites, multiphasic scaffolds, osteochondral differentiation
Lesions involving two different tissues and their interface are challenging to treat. This study demonstrates that by using biomimetic, multilayered scaffolds, zone specific osteogenic, and chondrogenic differentiation are possible even without adding growth factors. The bone zone mineralizes with bone specific mineral. Relevant for such functional scaffold fabrication is the use of composite biomaterials, gradients thereof, and adequate fabrication design.

1. Introduction
Osteochondral defects may result from primary osteoarthritis (OA) or as a consequence of fractures with joint involvement. Studies on incidence revealed that 10–15% of persons aged over 60 years suffer from clinical OA,[ 1 ] with an increasing incidence due to obesity and lack of physical activity.[ 1 , 2 ] Patients with osteochondral lesions suffer from pain and loss of function that may result in disability. Current clinical approaches to heal osteochondral defects remain highly challenging. Moreover, healing mostly occurs unsatisfactorily. Advanced OA therapy often results in joint replacement with known associated risks and side effects.[ 3 ] Other treatments, like microfracture and matrix‐associated autologous chondrocytes transplantation, still produce unsatisfactory results in long‐term cartilage regeneration.[ 4 ]
Scaffold‐based regeneration models have been reported as a highly promising approach for osteochondral lesions.[ 5 ] Indeed, this type of approach may be particularly useful for the regeneration of two distinct tissues and a stable interface.[ 6 ] The clinical application of such therapeutic scaffolds may be achieved in a one‐step procedure with clear advantages. Ideally, these scaffolding materials should be able to promote attachment of the cells, mimic the natural environment, and integrate into the surrounding tissue. In addition, cell differentiation should be supported. This is crucial to promote tissue regeneration.[ 5 , 7 ] Scaffolds for osteochondral regeneration should feature multiple layers to mimic bone, cartilage, and their interface. Several strategies have been reported to develop scaffolds for osteochondral regeneration that can be summarized as i) two single scaffolds combined at the defect site, ii) scaffold for bone and scaffold‐free approach for the cartilage layer, iii) single, homogeneous scaffold for both tissues and the interface, and iv) single, heterogeneous composite scaffold (elegantly reviewed in Nooeaid et al.[ 5b ]). As materials of choice, several natural‐ and synthetic‐origin polymers are being investigated. Examples of natural materials include collagen, hyaluronan, and chitosan.[ 8 ] Bioactive ceramics, including hydroxyapatite (HA) and calcium phosphates, are mostly used for the bony layer. These ceramics are frequently entrapped in a matrix of synthetic polymers widely used for bone engineering, including polycaprolactone and poly(lactic acid).[ 9 ]
In our study, we developed a chitosan‐collagen composite scaffold that contains octacalcium phosphate (OCP) at the top of the bony layer. The aim was to develop a novel multilayered scaffold for osteochondral regeneration based on natural extracellular matrix (ECM)‐like materials. Gradients of chitosan and collagen were used that mimic the distinct tissues and the interface. Such a material gradient may promote desired stem cell differentiation that is specific for each tissue zone. In addition, a mineral component was incorporated in the bony layer. The scaffolds were designed as a continuous, single but heterogeneous material to avoid a predetermined breaking point between the bone and the cartilage layers. Chitosan and collagen were selected as biopolymers to generate the composite used in the present study. Chitosan is a natural, non‐toxic biopolymer, which is highly biocompatible, biodegradable, and bioactive. It is structurally similar to glycosaminoglycan and offers an appropriate stimulus for stem cell differentiation.[ 10 ] Collagen is the main ECM component in both native cartilage and bone tissue.[ 11 ] The bony layer in our scaffold includes OCP as the mineral component. Calcium phosphates are widely used in bone tissue engineering and have been reported to promote bone regeneration.[ 12 ] The developed scaffolds were carefully characterized for their morphology, porosity, and pore size as well as their composition, crystallinity, and mechanical properties. The bioactivity of the scaffolds was evaluated in simulated body fluid (SBF) and their biocompatibility with human adipose‐derived mesenchymal stem cells (hAMSCs). Considerable effort was made in evaluating the differentiation of hAMSCs on each of the different layers of the scaffold.
Our hypothesis is that by using a single composite scaffold that contains gradients of natural ECM‐like materials and adequate porosity, we could guide stem cells toward zone‐specific differentiation.
2. Experimental Section
2.1. Scaffold production
2.1.1. Materials
OCP was synthesized by dripping a solution of 0.04 mol L−1 calcium acetate (Ca(CH3COO)2, VWR BDH Chemicals Ltd., Radnor, PA, USA) into a solution of 0.04 mol L−1 sodium acid phosphate (Na2HPO4, Panreac Quimica SA, Barcelona, Spain) and following a reported methodology.[ 13 ] The synthesis was performed at 60 °C for 3 h. During precipitation of the OCP, the solution was maintained under stirring. Subsequently, the precipitate was filtered and washed several times with distilled water. Finally, the product obtained was dried at room temperature. Chitosan from shrimp shells, ≈75% deacetylated was obtained from Sigma Aldrich (St. Louis, MO, USA). Fibers of collagen type I were used as collagen source (bovine origin, commercial grade, Novaprom, Lins, Brazil). Both biopolymers were used as received by the manufacturers. Solutions of chitosan and collagen were prepared at a concentration of 2%, with constant stirring maintained for 24 h. Subsequently, 1% acetic acid was added and the stirring was continued for another 24 h. All raw materials used in the study were characterized by FTIR following the procedure described earlier in Section 2.2.3.
2.1.2. Production of Graded, Composite Multilayered Scaffold
Figure 1 presents a scheme illustrating the sequential layer‐by‐layer fabrication methodology to produce the scaffolds. First, the solutions corresponding to the different layers were independently produced (Figure 1A). For the bony layer and the interface, chitosan‐collagen (1:1) was used. Two percent OCP was additionally added to the solution for the bony layer. For the cartilage layer, the collagen content was increased to obtain a ratio of chitosan‐collagen of 1:3. Under stirring, 1 m NaOH was added to each layer to neutralize the acetic acid. For the fabrication of the multilayer composite scaffolds, each solution was stirred to obtain a foam and poured into a mold followed by a freezing step. Following freezing of the first layer, the polymeric solution corresponding to the next layer was deposited on top and the next freezing cycle was performed. All freezing cycles were performed at −20 °C. Subsequently, molds containing the three layers were frozen and lyophilized (Figure 1B). Finally, the scaffolds were cut out into cylinders of 5 mm diameter using a biopsy punch (Figure S2, Supporting Information) and sterilized using ethylene oxide (inpac Medizintechnik GmbH, Birkenfeld, Germany).
Figure 1.
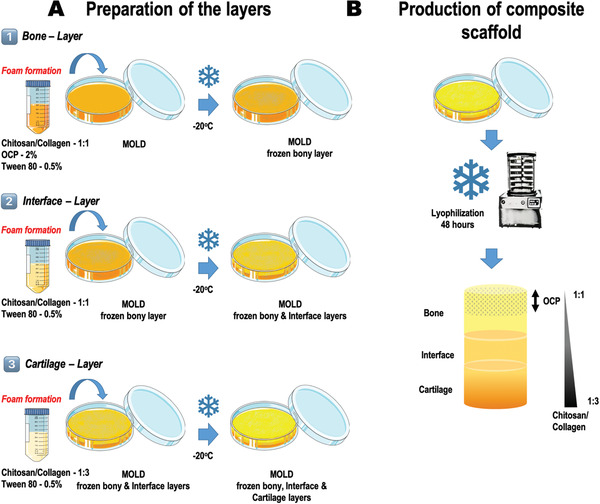
Schematic representation of the fabrication strategy followed to produce the multiphasic chitosan‐collagen‐octacalcium phosphate scaffolds. A) Material combinations used to obtain each independent layer. Consecutive freezing of the layers was performed; starting with the bony layer (1), then the interface (2), and finally the cartilage zone (3). B) Lyophilization of the material composite solutions previously deposited and frozen layer by layer into a mold. Illustration of the resulting scaffold. Different zones and material composition are identified.
2.2. Scaffold characterization
2.2.1. Morphology and Elemental Composition: Light Microscopy and Scanning Electron Microscopy—Energy Dispersive Spectroscopy
The structure and morphology (i.e., general and of the surface) of the composite scaffolds were characterized by observational light microscopy (VHX‐900F, Keyence, Osaka, Japan) and SEM (JSM‐6010LV, JEOL, Tokyo, Japan). By means of light microscopy, image depth of field and surface topography analysis were performed. For this analysis, scaffolds were bisected to obtain a plane surface. SEM was performed to analyze general morphological properties. In addition, an initial insight on porosity and pores shapes was obtained by this technique. To detect the presence of elements (i.e., Ca, P, C, and O) of different regions of interest (ROI) on the scaffolds, EDS (INCAx‐Act, PentaFET Precision, Oxford Instruments, Abingdon, UK) was performed. In addition, an element mapping of the entire scaffold was also obtained by EDS. Prior to SEM observation, the scaffolds were coated with platinum by ion sputtering. Backscattered electron imaging was used for overview images and secondary electron imaging for all detailed images (i.e., higher magnifications). For EDS analysis, scaffolds were used without coating.
2.2.2. Pore Size and Wall Thickness Analysis: Micro‐Computed Tomography
Scaffolds were characterized using µCT (SkyScan 1176, Bruker, Kontich, Belgium) for porosity, pore size, and wall thickness distribution. N = 4 scaffolds were used for the analysis. Briefly, scaffolds were stained with iodine vapor for 12 h and scanned at 40 kV and 600 µA with 9 µm resolution. The tomography was reconstructed using NRecon software (Version 1.6.9.18, Bruker). For the characterization of scaffold properties, three ROI representing the cartilage (ROI 1), interface (ROI 2), and bone (ROI 3) regions were selected and analyzed using CTAnalyser software (Version 1.17.7.2, Bruker) with adaptive thresholding between 40 and 200 (greyscale) for the collagen structure and global thresholding between 220 and 255 (greyscale) for the OCP particles. Finally, volume rendering of the scaffold was performed using CTVoxel (Version 3.3, Bruker).
2.2.3. Chemical and Crystallographic Properties: FTIR and X‐Ray Diffraction
FTIR (IR Prestige‐21 spectrometer Shimadzu, Kyoto, Japan) with attenuated total reflectance was used to analyze the chemical structure of the raw materials and scaffolds. The spectra were received in a transmittance mode of over 100 scans in a spectral range of 400–4000 cm−1. For each raw material, a standard sample preparation was followed. For each scaffold, the bone and cartilage layers were independently analyzed. The crystalline phases on the scaffolds were detected using thin‐film XRD (D8 Advance, Bruker, XRD Division, Karlsruhe, Germany). The data collection was performed by the 2θ scan method, with 1° as the incident beam angle using a Cu Kα X‐ray line and a scan speed of 0.05° min−1 over 9 h.
2.2.4. Mechanical Properties: Oscillatory Shear Measurements
Rheological measurements were performed using an AR‐2000ex rheometer (TA Instruments, New Castle, DE, USA) with the samples immersed in distilled water. In brief, swollen samples were placed between parallel plates (non‐porous, stainless steel, diameter 12 mm) and the gap was adjusted using a normal force of 0.2 N. Once the samples reached an equilibrium stage, a gap was adjusted to 1000 µm. The specimen temperature was set at 37 °C and controlled by a Peltier heating stage. Initial measurements were performed in a shear deformation mode. To start, a range of strain amplitudes was determined in which the samples exhibited a linear viscoelasticity. Dynamic strain/frequency sweep tests were performed. For the strain test, amplitudes from 0.1% to 20% at a frequency of 1 Hz were used. Thereby, the dynamic shear modulus was determined as function of strain. For the frequency test, frequencies in the range 0.01–70 Hz were used. The strain corresponding to the sample linear region was used as a fixed value. Here, the dependency of the dynamic shear modulus and loss factor on frequency was obtained. Values of the G′,
G
″, G*, and tan δ were obtained. Standard equations reported elsewhere were used for G* (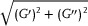 ) and tan δ (G′′/G′).[
14
]
) and tan δ (G′′/G′).[
14
]
2.3. Scaffold Bioactivity in Simulated Body Fluid
The ability of apatite to form on the scaffold surface was evaluated by in vitro bioactivity. For this test, the materials were immersed in SBF with ion concentrations nearly equal to those of human blood plasma. The test was performed by incubating the scaffolds in SBF for 1, 3, or 7 days at 37 °C with steady agitation. SBF solution was prepared according to the published protocol of Kokubo et al.[ 15 ] Briefly, NaCl, NaHCO3, KCl, KPO4.3H2O, MgCl2.6H2O, HCl, CaCl2, Na2SO4, and TRIS were dissolved in ion‐exchanged, distilled water. The final pH was adjusted to 7.40 using HCl (1 n). The solution was prepared always fresh and immediately before use. Scaffolds were immersed into 10 mL SBF solution, ensuring that all the material was fully covered. Plain SBF solution (i.e., without scaffolds) was used as a control for each observation time. After 1, 3, and 7 days, both the SBF solutions and scaffolds were collected and analyzed.
2.3.1. Analysis of SBF Solutions Recovered after Scaffolds Incubation: pH and Inductively Coupled Plasma Optical Emission Spectroscopy
pH measurements were performed on all solutions recovered. Furthermore, Ca and P concentrations were determined using inductively coupled plasma optical emission spectroscopy (JY2000‐2, HORIBA Jobin Yvon, Kyoto, Japan). The solutions were filtered using a 0.22 µm filter and diluted (1:10) in 1% nitric acid. All the treated solutions were stored at −20 °C until further use. Plain SBF solution was used as a control.
2.3.2. Analysis of the Scaffolds Harvested after SBF Incubation: XRD, SEM‐EDS, FTIR, and µCT
Scaffolds collected from the SBF solution were rinsed thoroughly with ultra‐pure water and allowed to dry overnight in a desiccator. Subsequently, the scaffolds were analyzed by thin‐film XRD, SEM‐EDS, FTIR, and µCT to evaluate chemical changes and the mineral deposits on the scaffold surface, among other relevant features. XRD, SEM‐EDS, and FTIR were used as described above. For µCT, no iodine staining was performed on the scaffolds recovered from SBF. Instead, scaffolds were scanned without any previous treatment. NRecon (Version 1.6.9.18, Bruker) software was used for reconstruction of the tomography data. The different phases were distinguished using a CTAnalyser (Version 1.17.7.2, Bruker). Briefly, a multi‐level Otsu method was used to separate the greyscale value on the entire dataset into six distinct phases. The grey scale value 25–85 was shown in white (chitosan‐collagen structure), the value 100–160 was shown in yellow (calcium phosphate deposit), and the value 200–220 was shown in red (HA deposit).
2.4. Cell Isolation and Seeding on the Scaffolds
The ethical committee of the University Hospital “Klinikum rechts der Isar” at the Technical University of Munich, Germany approved the described study. All procedures were performed in accordance with the declaration of Helsinki in its latest amendment. For all cellular experiments described hereafter, hAMSCs were isolated and used. Fresh human fat tissue harvested from abdominal fat of N = 3 patients was used. Patients provided informed consent.
Cells were isolated following the previously described protocol.[ 16 ] Briefly, harvested fat tissue was cut into small pieces, placed into 50 mL falcon tubes to which sterile Dulbecco's phosphate‐buffered saline without calcium or magnesium (DPBS, Sigma Aldrich) was added. The mixture was centrifuged at room temperature for 10 min without brakes (450 g). The resulting fat layer was collected into a new 50 mL falcon tube and washed 2–3 times with DPBS. Collagenase solution (0.8 mg mL−1 DPBS, collagenase type II, Biochrom GmbH, Berlin, Germany) was added and digestion was performed for 30 min in a 37 °C water bath. The digestion reaction was stopped by filling the tubes with pre‐warmed cell culture medium (Dulbecco's Modified Eagle's Medium—DMEM, high glucose, Sigma Aldrich) that had been supplemented with 10% fetal calf serum (Sigma Aldrich) and 1% penicillin/streptomycin (Sigma Aldrich). The solution was centrifuged for 10 min at 600 g and subsequently the pellet was resuspended in supplemented cell culture medium and filtered through a 40 µm cell strainer (BD Falcon, Franklin Lakes, NJ, USA). The resulting cell suspension was transferred into cell culture flasks. The cell seeding density was 3000 cells cm−2. Cells isolated from each individual donor were cultured independently. The cell culture medium was changed on the first day after isolation and subsequently twice weekly. For the cellular studies described in this paper, hAMSCs were used in passages 2 and 3.[ 16 ]
For cell seeding on the scaffolds, hAMSCs from 3 different donors were pooled immediately before seeding. Four different concentrations of cells were used (i.e., 104, 105, 5 × 105, and 106 cells per scaffold). The cell suspension containing the different cell concentrations were prepared in 50 µL supplemented DMEM. Sterile scaffolds were carefully placed into 48‐well plates and the 50 µL cell suspension was pipetted onto the scaffold. Care was taken to distribute the cell suspension homogeneously throughout the entire scaffold. Cell‐seeded scaffolds were placed into an incubator for 1 h at 37 °C under 5% CO2. Thereafter, 500 µL supplemented DMEM was carefully added. The constructs were cultivated for up to 35 days and media change was performed twice weekly.
2.5. Evaluation of cell attachment, toxicity, and proliferation
2.5.1. Cell Attachment, Distribution, and Morphology: SEM‐EDS
After 14, 21, and 35 days of hAMSCs culture on the scaffolds, specimens were removed from the culture media, washed twice with DPBS and fixed for SEM. Briefly, the constructs were fixed for 30 min with 2.5% glutaraldehyde in a 0.1 m sodium cacodylate buffer (pH 7.4, 2% sucrose). The fixed scaffolds were washed with sodium cacodylate buffer and dehydrated in a series of ethanol of a concentration from 30% up to 100%. Critical point drying was achieved by immersion of the samples in hexamethyldisilazane (HMDS) for 10 min. Treatment with HMDS was repeated three times. Scaffolds were used uncoated for EDS analysis and sputter coated for SEM following the same procedure as previously described in the present study.
2.5.2. Cell Viability and Potential Toxicity of the Scaffolds: Lactate Dehydrogenase and Calcein‐AM/PI Staining
To evaluate the biocompatibility of the developed scaffolds, cell viability was evaluated at 3, 7, and 14 days after seeding. In addition, the effect of the different cell concentrations used was considered. For the assay, the concentration of LDH, a marker for cell death, was evaluated in the supernatants collected after each observation time. Fluitest LDH‐L Kit (Analyticon Biotechnologies AG, Lichtenfels, Germany) was used following the manufacturer's instructions. All experiments were performed in triplicate and were normalized to a standard curve. Furthermore, live‐dead staining was performed after 21 and 35 days of cell culture on the scaffolds. Here, harvested scaffolds were washed carefully with DPBS and subsequently incubated in 500 µL staining solution (2µm calcein‐AM (Sigma Aldrich) and 1.5µm propidium iodide (Sigma Aldrich)). Following incubation at 37 °C for 20 min, the cell morphology and distribution were evaluated in each layer by confocal microscopy using an Fluoview FV10i microscope (Olympus, Tokyo, Japan). Moreover, cell infiltration through the entire scaffold was evaluated after 35 days of cell culture. For this, cell‐seeded scaffolds were horizontally sectioned, from top to bottom, to obtain 5 individual pieces. Each piece was stained with Calcein‐AM/PI and imaged using the confocal microscope. In an attempt to quantify cell morphology, ImageJ v1.53f (National Institutes of Health, MD, USA) was used. Confocal microscopy images corresponding to each zone of the scaffold after 21 days of cell culture were converted to grey‐scale, 8 bits images. Next, a fixed threshold (min. 70 and max. 255) was introduced and a binary image was created. The particle analysis tool from ImageJ was used. Events smaller than 20 µm as well as cell clusters were excluded from the analyzed population. Over 200 cells were analyzed for each scaffold zone. Shape descriptor parameters such as circularity, roundness, and aspect ratio (A/R) were considered. Cells featuring values of circularity and roundness between 0.7 and 1 were considered as round. Three independent scientists performed the analysis and results were averaged.
2.5.3. Cell Proliferation: DNA Quantification
The ability of hAMSCs to proliferate on the scaffolds was assayed by quantifying the DNA content after different observation times. Therefore, cell seeded scaffolds were cultured for 3, 7, and 14 days. At each observation time, the samples were washed using DPBS and digested using 500 µL papain solution (0.1 mg mL−1, Sigma Aldrich) per scaffold. Digestion was performed overnight at 65 °C. Subsequently, the solution was stored at −80 °C. Quantification of DNA content was performed using the Quant‐iT PicoGreen dsDNA Assay Kit (Invitrogen) following the manufacturer's instructions. Briefly, 70 µL digested cell solution and 70 µL Quant‐iT PicoGreen dsDNA working solution were pipetted into a 96‐well plate. The plate was incubated for 5 min at 37 °C in the dark. The fluorescence was quantified at an emission wavelength of 520 nm and an excitation wavelength of 485 nm using a FLUOstar Omega photometer (BMG labtech, Ortenberg, Germany). All experiments were performed in triplicate. DNA concentrations were calculated using a standard curve.
2.6. Evaluation of gene expression and cell differentiation
2.6.1. Alkaline Phosphatase Activity
Alkaline phosphatase (ALP) activity was determined after 3, 14, and 21 days of culture. ALP substrate solution was prepared with 4‐nitrophenyl phosphate (pNPP, Sigma Aldrich). In the presence of ALP, pNPP substrate will be converted to an equal amount of yellow‐colored 4‐nitrophenol (pNP). For the assay, the scaffolds were washed twice with DPBS and incubated with ALP substrate solution for 30 min at 37 °C. Following incubation, 100 µL of the solution was transferred to a 96‐well plate and the absorption was determined at 405 nm in the FLUOstar Omega photometer. All experiments were performed in triplicate. pNP concentrations were calculated from a standard curve. ALP activity was reported as a function of the pNP concentration found in each sample.
2.6.2. Expression of Apoptosis, Proliferation, Osteogenic, and Chondrogenic Genes: Quantitative Real‐Time PCR
Cell‐seeded scaffolds were used to evaluate the expression of apoptosis, proliferation, osteogenic, and chondrogenic markers. For apoptosis, the apoptosis regulator BCL2 and CASP3 were evaluated. For proliferation, MCM5 and CCND1 were analyzed. In the case of osteogenic differentiation, the expression of COL1A1, COL3A1, BGLAP (osteocalcin), SPP1 (osteopontin), and RUNX2 were evaluated. While for chondrogenesis, SOX9, COL2A1, COL10A1, and ACAN markers were analyzed. In the case of differentiation markers, the scaffolds were carefully sectioned to analyze the gene expression in the bone and cartilage layers independently. Thereby, only the upper (OCP‐rich, bone zone) or lower (cartilage zone) layer of the scaffold was used for RNA isolation and further processing. Gene expression was performed by quantitative real‐time PCR. In brief, cell‐seeded constructs were harvested using Tri Reagent (Sigma Aldrich) at 1, 3, 7, 14, and 21 days after culture. Subsequently, 100 µL chloroform was added and the samples were incubated on ice for 10 min. Following centrifugation (14 000 g for 10 min at 4 °C), the clear phase was transferred into tubes containing 250 µL isopropanol. Following another incubation step on ice, a pellet was obtained by centrifugation (14 000 g for 10 min at 4 °C). Washing was performed with 70% ethanol and the final pellet was resuspended in 30 µL ultra‐pure, DNase‐ and RNase‐free, PCR grade water. Measurement of the total RNA content was performed using a Hellma TrayCell Eppendorf BioPhotometer (Eppendorf AG, Hamburg, Germany). The cDNA transcription was performed using the First Strand cDNA Synthesis Kit (Thermo Fisher, Waltham, MA, USA) following the manufacturer's instructions. A Mastercycler nexus (Eppendorf AG) was used for the synthesis of cDNA. Following transcription, all samples were diluted with PCR grade water to a concentration of 10 ng µL−1. Quantitative real‐time PCR was performed using Sso Fast EvaGreen Super Mix (Bio‐Rad Laboratories Inc., Hercules, CA, USA) in a Bio‐Rad CFX96 thermal cycler (Bio‐Rad Laboratories Inc.). cDNA (30 ng) was used in a total reaction volume of 20 µL. Results were reported as fold expression normalized to the used housekeeper (i.e., tubulin beta class I: TUBB). Therefore, the ΔC T method using a reference gene was used.
2.6.3. Matrix Deposition on the Scaffolds: Histology
Histology examination was performed at 21 and 35 days after cell culture on the scaffolds. The cell‐seeded constructs were washed twice with DPBS and fixed with 3.7% paraformaldehyde. Dehydration was performed using gradually increasing concentrations of ethanol. Thereafter, specimens were embedded in paraffin and sectioned in 7 µm slices. The rehydration process was performed using Roti‐Histol (Carl Roth GmbH, Karlsruhe, Germany) and immersion in 100%, 95%, and 70% ethanol. Hematoxylin and eosin (H&E) staining was used to obtain an overview of the cells populating the scaffolds. Briefly, specimens were stained by 10 min immersion in hemalaun followed by a rinsing step in running tap water. The sections were immersed for 5 min in eosin solution and dehydrated using 70%, 95%, and 100% ethanol and Roti‐Histol. SafO staining was used to detect cartilage‐specific ECM. For SafO, sections were treated with hematoxylin QS solution for 5 min, washed in running tap water, and subsequently rinsed rapidly with acid ethanol. Counterstaining was performed by immersing the sections for 5 min in 0.05% fast green solution. Subsequently, the sections were rinsed with a 1% acetic acid solution and stained in 0.1% SafO solution by immersion for 5 min. The dehydration process was performed following the same procedure already described for H&E. Dried specimens were mounted and imaged with a light microscope (BZ9000 Biorevo, Keyence, Osaka, Japan). A general scan of the entire sections was obtained using the software packages BZ‐II Viewer and BZ‐II Analyzer (Keyence, Osaka, Japan). All images were taken under the same exposure time and white balance was regularly applied.
2.7. Statistical Analysis
Statistical analysis was performed using GraphPad prism version 5.00 (GraphPad Software, San Diego, CA, USA). Data are shown as mean value ± standard deviation. For multiple group comparison, either one‐way or two‐way analysis of variance (ANOVA) followed by Tukey's test was applied. Multiple t‐test corrected by Holm–Sidak was used to analyze the pH data as well as the Ca and P concentrations. All experiments were performed at least in triplicate (technical replicates = 3). p values are reported following the GraphPad style (i.e., four digits after the decimal point). A probability of p ≤ 0.05 was considered to be significant.
3. Results
3.1. Composite, Multilayered Scaffold with Smooth Bone‐to‐Cartilage Interface Mimics the Osteochondral Structure with Relevant Elements Present at the Bone Layer
Single unit scaffolds were obtained made of a chitosan‐collagen‐OCP composite (Figure 2A,B). Throughout the entire length, the chitosan‐collagen composition as well as the presence of OCP varied. Thereby, a graded scaffold was obtained. The scaffolds were fabricated in such fashion that all present zones appeared subsequently after each other in the same single 3D. Thus, the specific tissue layers were not glued, stitched, or assembled to one another post‐fabrication. Macroscopic images (Figure 2A) illustrate the fabricated scaffolds. A porous structure was observed throughout the entire scaffold length. The scaffolds consisted of three layers, that is, a bone layer, an interface, and a cartilage layer (Figure 2A). Macro‐ and microscopically, a distinction between the two main layers present in the scaffold was possible. Analysis of the scaffold surface topography (Figure 2A) showed a slightly rougher surface at the bone and interface regions compared to the cartilage region. This observation was supported by morphology analysis using scanning electron microscopy (SEM, Figure 2C). An SEM image obtained at lower magnification allowed the observation of the entire scaffold structure (Figure 2C, centered image). In this image, the transition between the different areas of the scaffold could be observed. Moreover, the gradient in mineral content, from the bony toward the cartilage layer, was also evidenced by the presence of shiny OCP particles. The OCP‐rich region was characterized by an irregular pore distribution, whereas the interface and cartilage layer were dominated by aligned pores with a smoother surface (Figure 2C, left and right images). The incorporation of OCP showed an even distribution in the entire bony layer. The incorporation of OCP crystals, solely into the bony layer, was confirmed by energy dispersive spectroscopy (EDS, Figure 2D). In the image, the presence of the elements C (turquoise), P (green), Ca (red), and O (pink) could be clearly observed for the entire scaffold. P and Ca were only detected in the zone corresponding to the bone, whereas C and O displayed a homogenous distribution throughout the entire scaffold length.
Figure 2.
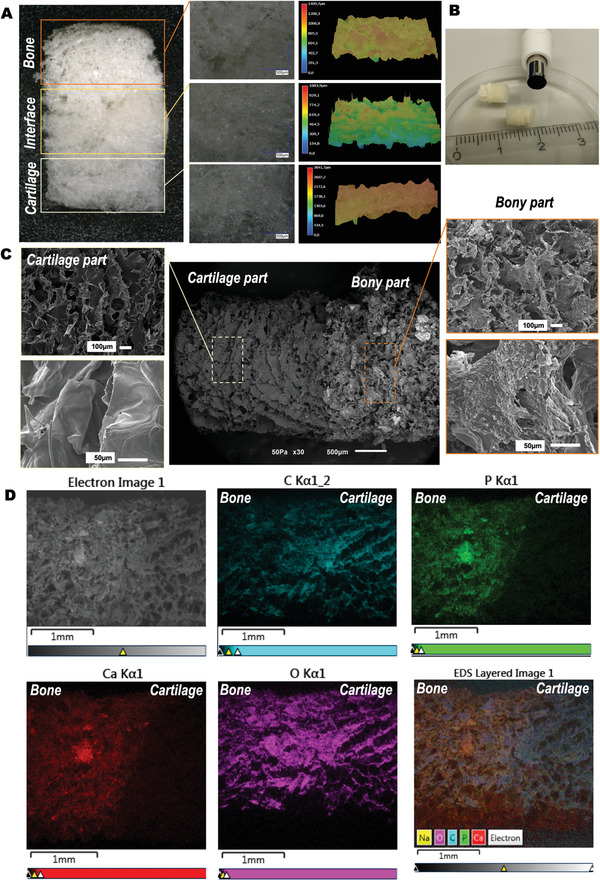
Morphological and elemental characterization of obtained chitosan‐collagen‐octacalcium phosphate scaffolds. A) Digital light microscopy images showing the different zones in the multilayered scaffold. The layers of the scaffold corresponding to the bone, interface, and cartilage regions are indicated by colored boxes added to the images. Higher magnifications of the different zones are shown. The left panel shows the surface roughness of each zone of the scaffolds. B) Macroscopic image showing the general appearance of the scaffolds. C) Scanning electron microscopy images illustrating the microstructure and morphological features of the obtained scaffolds. The central panel shows the entire scaffold. Detailed, higher magnifications images are depicted in the right and left panels for the cartilage and bone zones, respectively. D) Element mapping at the scaffolds’ surface by means of energy dispersive spectroscopy. Starting at the left upper image, images shown correspond to: SEM, C (turquoise), P (green), Ca (red), O (pink), and superposition of all elements.
Micro‐computed tomography (µCT) 3D reconstruction revealed detailed information of the scaffold's microstructure (Figure 3A). A highly porous structure was confirmed for the entire scaffold. Open and interconnected porosity characterized the scaffolds in all zones. Sagittal and axial slices generated by the µCT reconstruction (Figure 3B) indicated a rather unidirectional porosity, with a fibril‐like structure in the cartilage layer. An irregular porosity, featuring round pores, was observed in the bony layer. The incorporation of OCP into the bony layer was also corroborated in the µCT analysis (Figure 3A, lower image, OCP particles in red). The OCP particles, characterized by a high attenuation in the µCT, were displayed as having a high density with a bright appearance and were thus highly visible in the µCT scans. This clearly contrasted with the low density collagen‐chitosan composite material that constituted the overall scaffolds.
Figure 3.
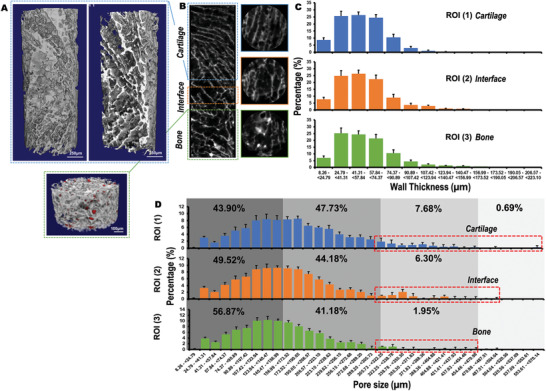
Microstructure, pore size, and wall thickness of obtained chitosan‐collagen‐octacalcium phosphate (OCP) scaffolds. A) 3D μCT reconstruction showing the cartilage (Scale bar = 250 µm) and bony layers (OCP particles in red, Scale bar = 100 µm) and B) μCT image of the different zones of the scaffold. The layers of the scaffold corresponding to the bone (green), interface (orange), and cartilage (blue) regions are indicated by colored boxes added to the images. The colors match the graphical representations for wall thickness and pore size distribution in each zone of the scaffold. C) Wall thickness and D) pore size distribution for each area of the scaffold. The pore size distribution range (µm) was divided in quarters that represent four different pore size thresholds. The quarters are indicated with different grey color shadows. Provided values indicate the % of pores that falls inside each size distribution threshold. Additionally, larger pores (i.e., >300 µm) are indicated inside a red box for each scaffold zone. For determination of both wall thickness and pore size distribution, three independent regions of interest (ROI) representing each zone were selected (i.e., ROI (1) = cartilage, ROI (2) = interface, and ROI (3) = bone). N = 4 scaffolds were used for analysis. Data are shown as mean value ± standard deviation.
Despite the varying pore morphology in the different scaffold layers, wall thickness (Figure 3C) and pore size distribution (Figure 3D) remained similar throughout the scaffold structure. Wall thickness was generally found to be <124 µm, with most of the measurements in the range of 25–50 µm. Interestingly, wall thickness increased toward the bony zone. In this region of the scaffold, 4.59% of the measurements returned values between 124 and 223 µm. By contrast, only 0.30% of the measurements in the cartilage layer showed wall thickness values <190 µm. Regarding pore size distribution, >90% of pores present in the entire scaffold had a diameter <300 µm. Most of the pores were between 75 and 240 µm, with a peak of ≈150 µm. Larger pores were found in the cartilage zone, where >8% of the pores were >300 µm in contrast to only 1.95% of such dimensions in the bony layer.
3.2. Multilayered Scaffolds Showed a Viscoelastic Behavior, with Dynamic (Complex) Shear Modulus (G*) Markedly Influenced by the Storage (Elastic) Modulus (G′)
The chitosan‐collagen‐OCP scaffolds showed a viscoelastic behavior characterized by storage modulus (G′) > loss modulus ( G ″) for viscoelastic solids. G′, G ″, and loss factor (tan δ) increased with frequency as shown in Figure 4A. This increase was at first minimal and showed a steep increase for high frequencies that are near to 10 Hz. G′ values were higher than G ″ for the studied frequency range of 0.1–10 Hz. The value of G′ was in the range of 1500–4000 Pa while G ″ was found to be <1000 Pa for the mentioned frequency interval. Figure 4C shows values obtained for G′ and G ″ at a fixed frequency of 1 Hz. For that frequency, G′ was found to be 1710 ± 20 Pa while G ″ was equal to 110 ± 10 Pa. This indicates that the storage (elastic) modulus G′ is the major contributor to the dynamic (complex) shear modulus (G*). This also reveals that our scaffolds displayed a high degree of elasticity. The loss factor tan δ was found to be equal to 0.064 (3.66°) for the chitosan‐collagen‐OCP scaffolds. This value matches well with reported ones for collagen‐based materials. G* characterizes the overall stiffness of materials, and it is considered one of the most appropriate representations of their mechanical properties. G* values are dependent on the frequency. Thus, they were determined by a frequency sweep test. Obtained results are depicted in Figure 4B, in which non‐linear as well as linear behaviors could be identified. A non‐linear relationship of G* with f ½ is found at low frequencies (i.e., <0.3 Hz), followed by a linear behavior that was lost at frequencies higher than 1 Hz. An equation that describes the dependency of G* with frequency was obtained for the chitosan‐collagen‐OCP scaffolds (Figure 4B, G* = 1556 + 163 f ½).
Figure 4.
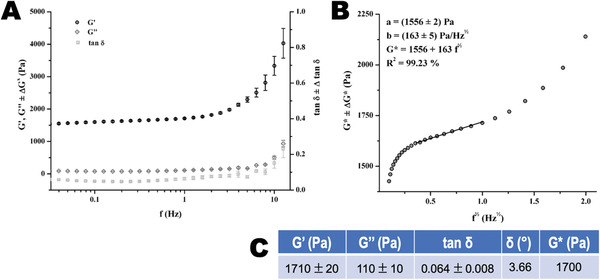
Mechanical properties of obtained chitosan‐collagen‐octacalcium phosphate (OCP) scaffolds. A) Evolution of the storage modulus (G′), loss modulus ( G ″), and loss factor (tan δ) with increasing frequency. Measurements were performed at 37 °C. Each curve corresponds to an average of three different samples analyzed. B) Dependence of the dynamic (complex) shear modulus (G*) on f 1/2 at 37 °C. Fitted line corresponds to the least‐square linear regression of the linear region. C) Average values for storage modulus (G′), loss modulus ( G ″), loss factor (tan δ), loss angle (δ), and dynamic (complex) shear modulus (G*) obtained at 1 Hz, with the corresponding standard deviation.
3.3. Multilayered Scaffolds were Bioactive with Surface Deposits Rich in Calcium and Phosphorus
Fourier‐transformed infrared spectroscopy (FTIR) spectra obtained for the raw materials and the chitosan‐collagen‐OCP composite can be found in the Figure S1, Supporting Information. The main bands on the spectra were assigned (Figure S1D, Supporting Information) and the presence of the single materials was confirmed in the composition of the blend.
Bioactive materials are expected to form an apatite‐like layer on their surface as a result of their incubation in SBF solution. FTIR and X‐Ray Diffraction (XRD) were performed to characterize the chemical and crystalline structures of the scaffold surface deposits after SBF incubation. FTIR spectra obtained for the different scaffold layers are presented in Figure 5 (A: bone and B: cartilage). The bands located at ≈1500–1650 cm−1 could be assigned to the amide I (vibration of valence of C=O) and amide II (dubbing of N—H) bands. Those chemical groups are present in the peptide bonds of collagen and in the acetyl groups of chitosan. The band located at 1057 cm−1 was attributed to the symmetric stretching vibration of the PO4 3− group of OCP and HA, while the band at ≈600 cm−1 corresponded to P—O bending vibrations in PO4 3−. A well‐defined peak, corresponding to the phosphate group, was clearly identified in the bony section of the scaffolds. With increasing incubation time in SBF, the intensity of this band notably increased. This was generally not observed in the cartilage layer of the scaffolds independently of the incubation time assayed. Noteworthy is the high intensity band at day 1, which was more likely related to a possible interference from the bony layer. XRD patterns are presented in Figure 5C. The peaks with the highest intensities have been identified with an asterisk (*) and can be observed in 2θ at 26°, 32°, and 40° and a broad peak around 50°. Several of the shown diffraction peaks could be assigned to an apatite‐like phase according to the Joint Committee on Power Diffraction Standards ASTM JCPDS 9‐432. By increasing the incubation time in SBF, the intensities of the apatite peaks increased and the single peaks became better defined.
Figure 5.
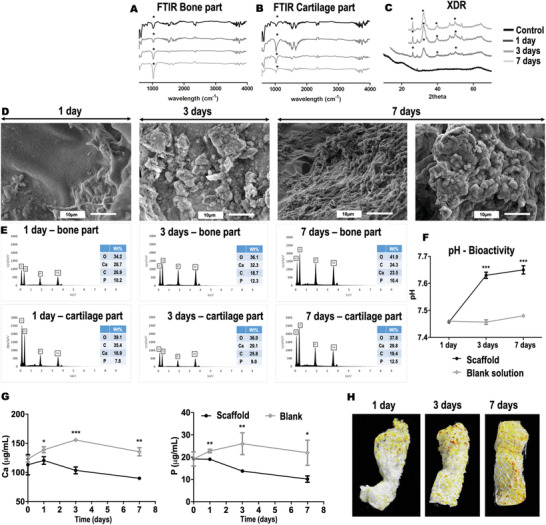
Bioactivity of the chitosan‐collagen‐octacalcium phosphate (OCP) scaffolds. FTIR spectroscopy showing the chemical composition of the scaffolds before and after immersion in simulated body fluid (SBF). Spectra obtained for A) bony and B) cartilage layers. C) X‐ray diffraction spectra showing the crystalline structure of the scaffolds before and after immersion in SBF. Relevant peaks have been identified in FTIR and XRD spectra with an asterisk (*). D) Scanning electron microscopy images of the scaffolds at 1, 3, and 7 days after SBF incubation. E) Corresponding EDS elemental analysis performed for each independent zone of the scaffolds before and after SBF incubation. The different incubation times analyzed (i.e., 1, 3, and 7 days) are depicted in the figure. F) pH of the solutions collected after scaffolds incubation in SBF. Obtained p values are indicated by *** p ≤ 0.0003. G) Ca and P concentrations in the solutions collected after scaffolds incubation in SBF as determined by ICP spectroscopy. Obtained p values are indicated by * p ≤ 0.0243, ** p ≤ 0.0038, and *** p = 0.0001. H) Mineral deposit on the scaffolds after SBF incubation as determined by μCT. Material composition have been color coded for better visualization; chitosan‐collagen in white, OCP and formed HA in orange to red coloration. Experiments were performed in triplicate (technical replicates = 3). Data are shown as mean value ± standard deviation. Multiple t‐test corrected using Holm–Sidak for multiple comparisons was used for statistical analysis of pH data as well as Ca and P determinations.
SEM‐EDS was also performed to characterize the bioactivity (Figure 5D). Because of the distinctive appearance of apatite grains and layers, SEM has been widely used—sometimes as the sole technique—to estimate apatite layer formation. In our study, a significant change in the topography of the scaffold surface was observed after incubation in SBF. Globular aggregates were observed that completely covered the surface of the scaffolds at day 3 post‐incubation. Following 7 days in SBF, a compact cauliflower‐like coating was observed (Figure 5D, right panel). The presence of Ca, P, C, and O was detected by EDS analysis (Figure 5E). At days 1 and 3 post‐incubation, higher wt% of Ca and P were identified in the bony layer of the scaffold. Unexpectedly, at day 7, these elements were slightly higher in wt% in the cartilage zone. The concentration of Ca and P in solution decreased over time (determined by ICP, Figure 5G). This decrease correlated well with a pH increase (Figure 5F) obtained at 1, 3, and 7 days post‐incubation.
Specimens collected after SBF incubation were also analyzed by µCT (Figure 5H). In the reconstructed images, the chitosan‐collagen structure is shown in white color. The OCP particles (incorporated in the bony layer) and the HA formed are shown in orange‐to‐red colors. After 1 day of incubation in SBF, only the OCP particles located in the bony zone of the scaffold were visible. By increasing the incubation time, the number of orange and red particles was elevated, indicating an increase in the amount of HA deposited on the scaffold. These deposits were mostly located in the bony area in agreement with the results obtained by the SEM/EDS analysis. Following 1 week of incubation, the entire scaffold was covered with HA deposits. This also matched the dense apatite‐like layer recognized in the SEM for the same observation time. Noteworthy is the occurrence of slight shrinkage in the scaffolds after SBF incubation. This behavior was noticeable mostly in the cartilage part of the multilayered scaffolds.
3.4. Viable Human Adipose‐Derived Mesenchymal Stem Cells Proliferated on the Multilayered Scaffolds
Cells were seeded on the scaffolds using a range of cell densities of 104–106 cells per scaffold. Cell proliferation was dependent on the cell number and culture time employed (Figure 6A). Remarkably, only a cell density of 105 cells per scaffold displayed a significant elevation of the cell proliferation with increased culture time (3 vs 14 days of culture: p = 0.0260 and 7 vs 14 days of culture: p = 0.0065). For the lowest (104 cells per scaffold) and highest (≥5 × 105 cells per scaffold) cell densities tested, no significant differences were observed for cell proliferation between 3, 7, or 14 days after cell seeding (p ≥ 0.0658). This indicated a low proliferative behavior of the cells under these culture conditions. Furthermore, 105 cells per scaffold may be optimal for hAMSCs to proliferate in the scaffolds under the culture conditions tested. The toxicity related to the scaffold materials was evaluated for the different cell densities used by quantifying the lactate dehydrogenase (LDH) activity released to the supernatant (Figure 6B). Interestingly, the highest LDH quantities were detected for the highest initial cell density, that is, 106 cells per scaffold. Even so, LDH levels still significantly decreased with culture time (p < 0.0001). Noticeably, lower cell densities resulted in significantly lower LDH production (104 vs 106 cells per scaffold showed p < 0.0001 for days 3 and 7 after culture and p = 0.0051 for day 14). Here also, LDH detection significantly decreased from day 3 to day 7 after cell seeding (p ≤ 0.05 for all cell densities except for 106 cells per scaffold) and remained low for the rest of the culture time. This indicated that the cell toxicity observed for 106 cells per scaffold was not related to the scaffold material but rather to the high cell density used per scaffold area. Based on the overall results from the cell proliferation and toxicity evaluations, a cell density of 105 cells per scaffold was used in subsequent experiments.
Figure 6.
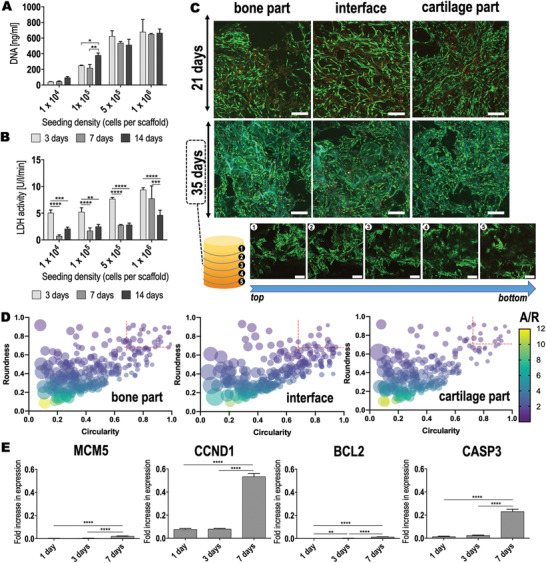
Biocompatibility of the chitosan‐collagen‐octacalcium phosphate scaffolds using hAMSCs. A) Cell proliferation and B) cell viability of hAMSCs seeded at different densities for up to 14 days of culture on the scaffolds. Two‐way ANOVA followed by a Tukey's correction for multiple comparisons was performed. Obtained p values are indicated as follows: DNA * p = 0.0260 and ** p = 0.0065; LDH activity ** p = 0.0013, *** p ≤ 0.0006, and **** p < 0.0001. C) Calcein‐AM/Propidium iodide live/dead staining performed in each specific zone of the scaffolds after 21 and 35 days of hAMSC culture (105 cells per scaffold). Calcein AM stains live cells green, while Propidium iodide stains dead cells red. Scale bar = 200 µm. After 35 days of culture, cell‐seeded scaffolds were horizontally sectioned. Each piece (numbered from 1 to 5 in the scheme) was independently imaged in the confocal microscope. Thus, cell colonization in the entire scaffold can be appreciated. Scale bar = 200 µm. D) ImageJ analysis of the cell morphology performed for each specific zone of the scaffolds after 21 days of hAMSC culture. Circularity values of 1 indicate a perfect circle. Round objects also feature high roundness and low aspect‐ratio (A/R) values. Over 200 cells were analyzed in each part of the scaffold. Cells that displayed circularity and roundness values between 0.7 and 1 were considered round. This range has been indicated in the graphs using red, discontinued lines. E) Expression of proliferation (MCM5 and CCND1) and apoptosis (BCL2 and CASP3) markers by hAMSCs seeded on the scaffolds (105 cells per scaffold) after different culture times. Results are normalized to the housekeeper TUBB. One‐way ANOVA followed by a Tukey's correction for multiple comparisons was performed. Obtained p values are indicated as ** p = 0.0013 and **** p < 0.0001. Experiments were performed in triplicate (technical replicates = 3). Data are shown as mean value ± standard deviation.
From the confocal images displayed in Figure 6C, a good cell attachment and homogenous cell distribution were visible in all layers of the scaffold. In particular, cells could be observed that colonized the entire scaffold structure independently of the tissue‐specific zone. By means of a live/dead calcein‐AM/PI staining, further insights on cell viability were obtained. All zones of the scaffold were individually observed. At 21 days after seeding, large areas of green stained cells were observed that indicated a high number of viable cells in all zones of the scaffold. Interestingly, a higher cell density was present at the cartilage zone when compared to the bony and interface layers. Cells in the cartilage area appeared rather elongated. The cellular orientation was observed in the direction of the scaffold pores. In the bony layer, the cells displayed a more roundish morphology. In the interface region, a mixture of both morphologies was observed. Cell morphology was analyzed using ImageJ and shape descriptor parameters such as circularity and roundness were calculated (Figure 6D). A value of 1 for both parameters indicates a perfect mathematical circle. Only 5.14% of the cells in the cartilage area featured values of circularity and roundness between 0.7 and 1. Conversely, 18.7% of the cells in the bony part showed values in the same range. In the interface zone, 10.6% of the cells showed circularity and roundness >0.7 in accordance to the microscopic observations. Dead cells were also identified in all three zones of the scaffold (Figure 6C, cell nuclei stained red). However, this was no longer observed at 35 days after seeding. At this observation time, a negligible number of dead cells were identified.
Further effects of the scaffolds on hAMSCs viability were analyzed by qPCR of proliferation and apoptosis markers (Figure 6E). Both minichromosome maintenance complex component 5 (MCM5) and cyclin D1 (CCND1) genes encode for proteins relevant in cell proliferation. Indeed, the protein encoded by MCM5 is involved in the initiation of DNA replication and is thus active in the regulation of the cell cycle. Similarly, CCND1 expression directly alters cell cycle progression. Following 7 days of hAMSCs culture on the scaffolds, a significant upregulation was observed for MCM5 (p < 0.0001) and CCND1 (p < 0.0001). This upregulation was more pronounced for CCND1 (24.9‐fold increased upregulation compared to MCM5). B‐cell lymphoma 2 (BCL2) and caspase 3 (CASP3) were investigated as indicators for possible apoptotic effects of the scaffolds on hAMSCs. BCL2, a gene that encodes a protein blocking the apoptotic death of cells, was significantly upregulated already at 3 days post‐culture (p = 0.0013). Interestingly, also CASP3, which plays a key role in the execution‐phase of cell apoptosis, showed a significantly higher expression at 7 days of culture (p < 0.0001).
3.5. Human Adipose‐Derived Mesenchymal Stem Cells Colonized the Entire Scaffold Surface, Displayed Multiple Intercellular Connections and Produced Extracellular Matrix
SEM was applied to evaluate cell distribution and morphology after hAMSCs culture on the scaffolds (Figure 7A–C). Interestingly, pores on the scaffold surface appeared to be occupied at 21 and 35 days after culture (overview image taken at ×30, left panel). At these observation times, more cellular material and ECM were visible (Figure 7B,C, image ×500, center panel) that may be occluding the scaffold pores. Good cell distribution and attachment were observed at the scaffold surface. Cells were visible in the bony and cartilage layers of the scaffold. Additionally, the cell density appeared to increase after longer culture periods. Interestingly, at the cartilage area, the scaffold surface had a smooth and flat appearance in which cells were well attached (Figure 7B, ×500, center panel). The morphology of the cells was elongated and abundant cell‐cell contact could be observed (Figure 7B, ×500 center panel and ×2500 right panel). At the bony layer, cells were attached surrounded by mineral deposits on a rougher surface (Figure 7A,C, ×500 center panel). Ample mineral deposition could be observed for the bony layer in images taken at ×2500 (Figure 7A,C, ×2500 right panel). After 35 days of culture on the scaffolds, cells covered the entire surface, displaying multiple intercellular connections and ECM production.
Figure 7.

hAMSCs adhesion and morphology after seeding on the chitosan‐collagen‐octacalcium phosphate scaffolds at A) 14 days, B) 21 days, and C) 35 days of cell culture. The three different panels represent different magnifications used to analyze the samples (i.e., ×30, ×500, and ×2500). From left to right, the boxed area indicates where the magnification was taken that is shown in the next panel.
3.6. Ionic Composition of the Extracellular Matrix at the Scaffold Bony and Cartilage Layers
SEM‐EDS results are depicted for scaffolds without cells (Figure 8A, control scaffolds) and for scaffolds with cells after 14, 21, and 35 days of culture (Figure 8B–D). The bone and cartilage layers were analyzed independently. On the control scaffolds, OCP incorporation in the bony region was detectable, while no Ca or P was measurable in the cartilage region (Figure 8A). With increasing cell culture time, a higher percentage of Ca and of P was detectable for both the bone and cartilage areas. This was particularly noticeable for the bone layer of the scaffolds, featuring the highest Ca and P percentages (21.9% and 9.3%, respectively, Figure 8D, left EDS spectrum).
Figure 8.
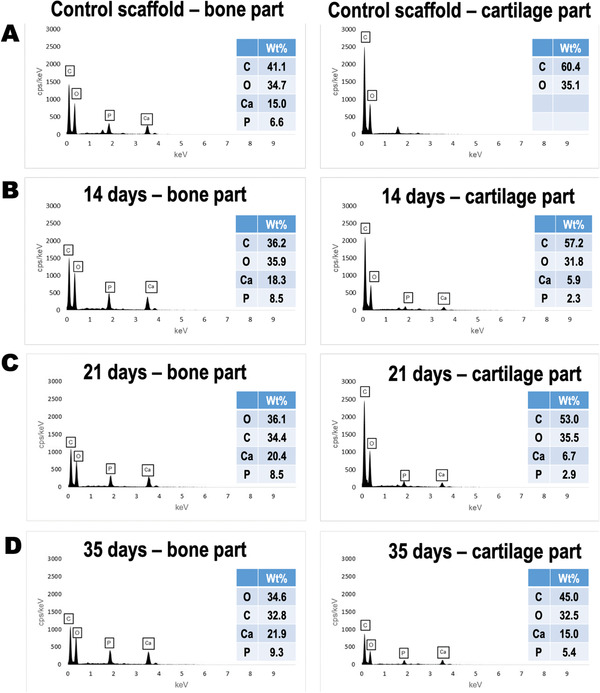
EDS elemental analysis performed for each independent zone of the scaffolds after hAMSCs culture. A) Results before cell culture. Results after B) 14 days, C) 21 days, and D) 35 days of cell culture. Values of wt% are tabulated for C, O, Ca, and P for each experimental condition analyzed.
3.7. Specific Differentiation of Human Adipose‐Derived Mesenchymal Stem Cells Occurred That was Associated with the Scaffold Zones
All analyzed osteogenic markers showed an increased expression in hAMSCs seeded on the bony layer of the scaffolds (Figure 9A). Upregulation was observed for bone gamma‐carboxyglutamate protein (BGLAP, p = 0.0022) at 14 days after culture. For the same time of observation, an increase on expression was detected for RUNX family transcription factor 2 (RUNX2) although not significant (p = 0.0520). Both, BGLAP and RUNX2 were upregulated at 21 days after culture (p < 0.05). Interestingly, collagen type I α1 chain (COL1A1) and collagen type III α1 chain (COL3A1) expression reached a maximum level at 14 days after culture. Remarkably, secreted phosphoprotein 1 (SPP1) displayed a very early expression at 3 days after culture on the scaffolds. The SPP1 expression levels further increased at 21 days after culture (p = 0.0190). ALP activity was also evaluated as an indicator for osteogenesis. ALP increased over culture time, although this increment was not statistically significant (Figure 9B, p > 0.05).
Figure 9.
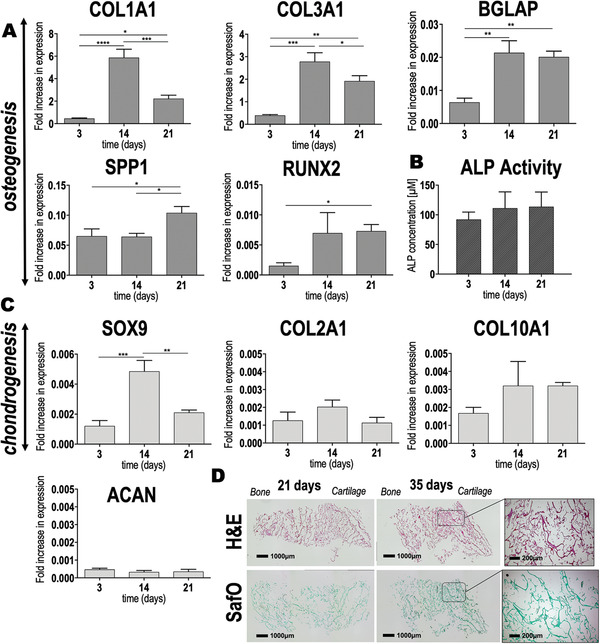
Spatial‐specific gene expression and matrix deposition of hAMSCs after seeding on the chitosan‐collagen‐octacalcium phosphate scaffolds. A) Expression of osteogenic markers (COL1A1, BGLAP, SPP1, and RUNX2) and matrix‐related fibrillar COL3A1 were assayed by qRT‐PCR after 3, 14, and 21 days of hAMSC culture on the scaffolds. Results are normalized to the housekeeper TUBB. Obtained p values are indicated as * p ≤ 0.0418, ** p ≤ 0.0034, *** p ≤ 0.0007, and **** p < 0.0001. B) ALP activity as determined by colorimetric assay (p > 0.05). C) Expression of chondrogenic markers (SOX9, COL2A1, COL10A1, and ACAN) assayed by qRT‐PCR after 3, 14, and 21 days of cell culture, was normalized to the housekeeper TUBB. Results showed statistical significance only for SOX9 (** p = 0.0022 and *** p = 0.0005). D) H&E and SafO staining of the entire scaffold after extended culture time (i.e., 21 and 35 days of cell culture). Scale bar = 1000 µm. Dashed squares represent the area of the scaffolds where higher magnification images were taken. Scale bar = 200 µm. All data are shown as mean value ± standard deviation. One‐way ANOVA followed by a Tukey's correction was performed for multiple comparisons. Experiments were performed in triplicate (technical replicates = 3).
SRY‐box transcription factor 9 (SOX9) expression was evaluated to gain an insight in early chondrogenesis. Cells seeded on the cartilage layer of the scaffolds displayed a significant upregulation of SOX9 after 14 days of culture (Figure 9C, p = 0.0005). Later, at 21 days after culture, SOX9 expression significantly decreased (p = 0.0022), such that a peak was observed at 14 days. Other markers, including collagen type II α1 chain (COL2A1) and collagen type X α1 chain, showed expression up to 21 days of hAMSCs culture on the cartilage layer of the scaffolds. Interestingly, aggrecan (ACAN) displayed same levels of expression during the entire cell culture period. Safranin O (SafO) staining after 35 days of hAMSCs culture on the scaffolds revealed a more intense staining pattern when compared to earlier culture times (Figure 9D).
4. Discussion
Functional repair of soft‐to‐hard tissue transitions remains challenging in orthopedic surgery. Clinical options feature limitations, while current research strives to develop better solutions that are rapidly translatable into patient care. In this respect, the use of tissue‐engineered scaffolds may be attractive for the treatment of osteochondral lesions. Generally, one of the problems when designing 3D scaffolds for tissue interfaces is that two different tissue types and the interface must be considered. Frequently, different materials are glued together to recreate individual layers that mimic the different tissue characteristics.[ 5b ] Such approach clearly presents the limitation of poor integration between the two layers.[ 17 ] In fact, most previous studies present two‐layer scaffolds for osteochondral repair[ 18 ] that are obtained by combining individual parts for each specific tissue. Poor integration has been reported as one of the limitations of this combinatorial fabrication strategy.[ 5 , 17 ] Gao et al. and Chen et al. introduced the use of fibrin glue to combine a bony and a cartilage scaffold.[ 19 ] Other studies recommend suturing or press fitting both layers.[ 20 ] These studies relied on the eventual formation of ECM produced by the cells initially seeded on the scaffold layers to achieve biological integration. This may be a cumbersome, hard‐to‐translate approach lacking proper integration of the bony and cartilage layers, in which the interface is barely considered. In the present study, we developed a single scaffold unit that was fabricated using biomaterial gradients. Our aim was to mimic the structure of articular cartilage and subchondral bone by tailoring the composite material and microstructure. Our scaffold design agrees with that reported by Levingstone et al.[ 8 , 21 ] The authors developed a multi‐layered collagen‐based scaffold that was obtained by an iterative layering fabrication. They also considered a cartilage, interface, and bony layer. Our work and that of Levingstone et al. demonstrate that good integration and mechanical stability can be obtained by a layering fabrication approach. In fact, similarly to Levingstone et al., our scaffold did not show any delamination of the different layers during mechanical characterization. Obtained values for G′, G ″, δ, and G* are within the range of collagen materials[ 22 ] and other biopolymers often used for cartilage regeneration (e.g., alginate gels[ 23 ] and hyaluronan[ 24 ]). Nevertheless, mechanical properties of our scaffolds were found to be lower than those reported for native articular cartilage.[ 22b ] This is a common problem in scaffolds for cartilage engineering. One possibility will be incorporating proteoglycan‐rich materials to the composite. They feature δ of 70° and may increase the typical collagen materials’ δ of 3.6° to a more cartilage‐like of ≈15°.[ 22b ] Inorganic compounds have been used with similar aim. The approach followed by Parisi et al. of introducing four different gradient zones using HA showed improved mechanics.[ 25 ] We followed these ideas in that we used a biopolymer‐ceramic composite rather than collagen alone. It may be also interesting to crosslink the matrix during fabrication. However, while crosslinking is known to increase matrix stiffness,[ 22a ] it has been well‐documented that scaffolds with minimal crosslinking and low initial stiffness result in beneficial stimulation on the synthesis of cartilage‐related molecules by cells.[ 22 , 26 ] Crosslinking has been also associated to toxicity.[ 22a ]
In addition to good integration and mechanical stability, osteochondral scaffolds should feature adequate porosity and biocompatibility.[ 5b ] Remarkably, porosity has been reported to highly impact cellular behavior in osteochondral constructs.[ 27 ] Our scaffolds were porous, with the pore size ranging mostly from 75 to 240 µm throughout the entire length of the scaffold. Microscopy analysis showed highly interconnected pores. This may have led to the good cellular distribution and extensive cell‐to‐cell connections observed from 14 days onward. Larger pores were obtained in the cartilage layer of our scaffolds. Pan et al. studied the effect of scaffold porosity on osteochondral repair in vivo.[ 27 ] The authors concluded that the scaffold with greater porosity and a larger pore size of the cartilage layer (i.e., 92% porosity, pores 200–300 µm) exhibited the best repair efficacy. Our results are in agreement with the results of Pan et al.[ 27 ] Larger pores and greater porosity at the cartilage zone of our scaffolds supported good osteochondral features in vitro. To warrant biocompatibility, our scaffolds were designed to feature a material gradient including chitosan, collagen, and OCP. Both, collagen and chitosan are natural‐origin biopolymers, highly biocompatible that present bone and cartilage ECM‐like features.[ 28 ] The chitosan‐collagen‐OCP scaffolds developed in our study were biocompatible, promoting good cell adhesion and proliferation. Our results are in agreement with Yan et al., who also demonstrated good cytocompatibility of MSCs with chitosan‐collagen porous scaffolds.[ 29 ] In vivo, their scaffolds supported differentiation of seeded MSCs toward the desired pathway. For bone, to confer additional bone‐like features to chitosan and chitosan‐blends, diverse biomimetic coating approaches have been investigated. Tuzlakoglu et al.[ 30 ] and Leonor et al.[ 31 ] reported that bioglass spraying and calcium silicate immersion, respectively, enhanced biomineralization of resulting chitosan biomaterials. Levingstone et al.[ 8 , 21 ] and Parisi et al.[ 25 ] used HA in the bony layer of their collagen‐based osteochondral scaffolds. In our study, we incorporated OCP particles into the chitosan‐collagen blend to further improve bioactivity and biomineralization at the bone layer of the scaffolds. OCP features superior in vitro and in vivo osteogenic properties compared to HA and other inorganic materials.[ 32 ] In vitro, it has been demonstrated that OCP promoted osteoblast differentiation and cell conversion to late osteocytes, while HA and beta‐tricalcium phosphate did not.[ 33 ] OCP can be highly resorbed by osteoclasts.[ 34 ] In fact, OCP can be remodeled by new bone in vivo in contrast to HA. The latter is known to be present for years at the defect site.[ 32 , 35 ] The in vivo superiority of OCP was first described in a pioneer study by Suzuki et al.[ 36 ] The authors reported superior osteoconductive properties of OCP when compared to four different inorganic materials, that is di‐calcium phosphate, amorphous calcium phosphate, Ca‐deficient HA, and HA. The OCP material had the earliest time of new bone tissue deposition (i.e., 1 week post‐implantation). In our study, by incorporating OCP in a gradual manner, bioactivity and mineral deposition increased at the bony end of the scaffolds. Interestingly, cell morphology also appeared to be impacted by the presence of OCP particles in the scaffolds. Expression of osteogenic genes, for example, RUNX2, COL1A1, BGLAP, and SPP1, also increased over time in the OCP‐rich layer of the scaffolds. Similar osteogenic features have been reported by other authors for OCP biomaterials. Anada et al. observed an enhanced expression of COL1A1, ALPL, and SP7 in mouse bone marrow stromal cells upon stimulation on OCP coatings.[ 37 ] Kouketsu et al. reported an increased level of RUNX2, COL1A1, BGLAP, and SPP1 in vivo at 4 weeks post‐OCP implantation.[ 38 ] Also relevant is the gene expression of cells at the osteochondral interface. Negligible COL2A1 and ACAN expression combined with high collagen type X α1 chain (COL10A1) expression have been reported for the interface area.[ 39 ] Our results on gene expression coincide with these reports. Our osteochondral scaffold facilitated simultaneous expression of markers for differentiated and hypertrophic chondrocyte‐like cells.
Limitations of our study are the lack of in vivo validation of the results observed in vitro. These experiments should be performed as part of the preclinical evaluation of this new biomaterial. Further composite optimization may be needed in order to achieve mechanic properties closer to that of the native tissue. Furthermore, an extended in‐depth investigation should be conducted to characterize chondrogenesis at the cartilage layer. Importantly, no growth factor or other biologic was used in our study. The chitosan‐collagen‐OCP scaffolds displayed biocompatibility and specific tissue regeneration potential based solely on microstructural features and biomaterial combinations and gradients. This is advantageous for clinical applications. Growth factor‐loaded scaffolds may pose high costs and present considerable side effects for the patients. However, it might be that such biologic is nevertheless needed to achieve robust chondrogenesis. Furthermore, the chitosan‐collagen‐OCP scaffolds are produced as soft and flexible sponges of large size (>5 cm; sponge dimensions depend on the mold used during fabrication). The sponges are easy to sterilize and manipulate. Therefore, this offers the surgeon the possibility to cut out the exact size needed with a scalpel directly prior to implantation into the patient defect. No cost‐intensive or complex patient‐individual production would be required.
5. Conclusion
Collectively, our results demonstrate the feasibility of the multiphasic chitosan‐collagen‐OCP composite scaffold for osteochondral regeneration. We demonstrated the incorporation of OCP at the top of the bony layer, forming a diminishing gradient toward the cartilage zone, to be a successful approach for osteogenic induction in the bony layer. The porosity and pore size together with the higher concentration of collagen in the cartilage layer resulted stimulative for chondrogenesis. Based on this, we conclude that multiphasic scaffolds designed as a continuous, single unit using composites of materials and gradients thereof may be promising for osteochondral repair. Relevant aspects to consider are selecting materials that mimic the natural ECM of bone and cartilage and following an adequate scaffold design that allows the development of a mechanically and chemically stable scaffold, where the tissue interface is also considered. Moreover, specific pore size for each tissue zone is highly important and this should be carefully considered.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
E.A. thanks the TUM Graduate School for funding her research stay at the 3B's Research Group, University of Minho (Guimarães, Portugal). This work was supported by the Portuguese Foundation for Science and Technology (FCT) under the project “ChimericFibre4Tendon” PTDC/BII‐BIO/28870/2017.
Amann E., Amirall A., Franco A. R., Poh P. S. P., Sola Dueñas F. J., Fuentes Estévez G., Leonor I. B., Reis R. L., van M. Griensven, Balmayor E. R., A Graded, Porous Composite of Natural Biopolymers and Octacalcium Phosphate Guides Osteochondral Differentiation of Stem Cells. Adv. Healthcare Mater. 2021, 10, 2001692. 10.1002/adhm.202001692
References
- 1. Wittenauer R., Smith L., Aden K., WHO Essential Medicines and Health Products Information Portal 2013.
- 2. Lawrence R. C., Felson D. T., Helmick C. G., Arnold L. M., Choi H., Deyo R. A., Gabriel S., Hirsch R., Hochberg M. C., Hunder G. G., Jordan J. M., Katz J. N., Kremers H. M., Wolfe F., Arthritis Rheum. 2008, 58, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Newberry S. J., FitzGerald J., SooHoo N. F., Booth M., Marks J., Motala A., Apaydin E., Chen C., Raaen L., Shanman R., Shekelle P. G., in Treatment of Osteoarthritis of the Knee: An Update Review, AHRQ Publication., Rockville, MD: 2017; [PubMed] [Google Scholar]; b) Hiligsmann M., Cooper C., Guillemin F., Hochberg M. C., Tugwell P., Arden N., Berenbaum F., Boers M., Boonen A., Branco J. C., Maria‐Luisa B., Bruyere O., Gasparik A., Kanis J. A., Kvien T. K., Martel‐Pelletier J., Pelletier J. P., Pinedo‐Villanueva R., Pinto D., Reiter‐Niesert S., Rizzoli R., Rovati L. C., Severens J. L., Silverman S., Reginster J. Y., Semin. Arthritis Rheum. 2014, 44, 271; [DOI] [PubMed] [Google Scholar]; c) Jordan K. M., Arden N. K., Doherty M., Bannwarth B., Bijlsma J. W., Dieppe P., Gunther K., Hauselmann H., Herrero‐Beaumont G., Kaklamanis P., Lohmander S., Leeb B., Lequesne M., Mazieres B., Martin‐Mola E., Pavelka K., Pendleton A., Punzi L., Serni U., Swoboda B., Verbruggen G., Zimmerman‐Gorska I., Dougados M., Ann. Rheum. Dis. 2003, 62, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camp C. L., Stuart M. J., Krych A. J., Sports Health 2014, 6, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Kon E., Filardo G., Di Matteo B., Perdisa F., Marcacci M., Bone Jt. Res. 2013, 2, 18; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nooeaid P., Salih V., Beier J. P., Boccaccini A. R., J. Cell. Mol. Med. 2012, 16, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Font Tellado S., Chiera S., Bonani W., Poh P. S. P., Migliaresi C., Motta A., Balmayor E. R., van Griensven M., Acta Biomater. 2018, 72, 150. [DOI] [PubMed] [Google Scholar]
- 7. Baiguera S., Urbani L., Del Gaudio C., Biomed Res. Int. 2014, 2014, 398069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Clearfield D., Nguyen A., Wei M., J. Biomed. Mater. Res., Part A 2018, 106, 948; [DOI] [PubMed] [Google Scholar]; b) Kumai T., Yui N., Yatabe K., Sasaki C., Fujii R., Takenaga M., Fujiya H., Niki H., Yudoh K., Int. J. Nanomed. 2019, 14, 1283; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Levingstone T. J., Thompson E., Matsiko A., Schepens A., Gleeson J. P., O'Brien F. J., Acta Biomater. 2016, 32, 149; [DOI] [PubMed] [Google Scholar]; d) Roffi A., Kon E., Perdisa F., Fini M., Di Martino A., Parrilli A., Salamanna F., Sandri M., Sartori M., Sprio S., Tampieri A., Marcacci M., Filardo G., Int. J. Mol. Sci. 2019, 20, 2227; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Seong Y. J., Kang I. G., Song E. H., Kim H. E., Jeong S. H., Adv. Healthcare Mater. 2017, 6, 1700966. [DOI] [PubMed] [Google Scholar]
- 9. Bittner S. M., Smith B. T., Diaz‐Gomez L., Hudgins C. D., Melchiorri A. J., Scott D. W., Fisher J. P., Mikos A. G., Acta Biomater. 2019, 90, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Lastra M. L., Molinuevo M. S., Cortizo A. M., Cortizo M. S., Macromol. Biosci. 2017, 17, 1600219; [DOI] [PubMed] [Google Scholar]; b) Oryan A., Sahvieh S., Int. J. Biol. Macromol. 2017, 104, 1003; [DOI] [PubMed] [Google Scholar]; c) Kim I. Y., Seo S. J., Moon H. S., Yoo M. K., Park I. Y., Kim B. C., Cho C. S., Biotechnol. Adv. 2008, 26, 1; [DOI] [PubMed] [Google Scholar]; d) Ahsan S. M., Thomas M., Reddy K. K., Sooraparaju S. G., Asthana A., Bhatnagar I., Int. J. Biol. Macromol. 2018, 110, 97. [DOI] [PubMed] [Google Scholar]
- 11.a) Lu X. L., Mow V. C., Med. Sci. Sports Exercise 2008, 40, 193; [DOI] [PubMed] [Google Scholar]; b) Muir H., BioEssays 1995, 17, 1039. [DOI] [PubMed] [Google Scholar]
- 12.a) Algul D., Sipahi H., Aydin A., Kelleci F., Ozdatli S., Yener F. G., Int. J. Biol. Macromol. 2015, 79, 363; [DOI] [PubMed] [Google Scholar]; b) Islam M. T., Felfel R. M., Abou Neel E. A., Grant D. M., Ahmed I., Hossain K. M. Z., J. Tissue Eng. 2017, 8, 10.1177/2041731417719170; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lewin S., Barba A., Persson C., Franch Serracanta J., Ginebra M. P., Ohman C., Biomed. Mater. 2017, 12, 065005; [DOI] [PubMed] [Google Scholar]; d) Thrivikraman G., Athirasala A., Twohig C., Boda S. K., Bertassoni L. E., Dent. Clin. North Am. 2017, 61, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Komlev V. S., Fadeeva I. V., Fomin A. S., Shvorneva L. I., Ferro D., Dokl. Chem. 2010, 432, 178. [Google Scholar]
- 14.a) Castro A. P., Laity P., Shariatzadeh M., Wittkowske C., Holland C., Lacroix D., J. Mater. Sci.: Mater. Med. 2016, 27, 79; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gonzalez de Torre I., Santos M., Quintanilla L., Testera A., Alonso M., Rodriguez Cabello J. C., Acta Biomater. 2014, 10, 2495. [DOI] [PubMed] [Google Scholar]
- 15. Kokubo T., Takadama H., Biomaterials 2006, 27, 2907. [DOI] [PubMed] [Google Scholar]
- 16. Schneider S., Unger M., van Griensven M., Balmayor E. R., Eur. J. Med.Res. 2017, 22, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mano J. F., Reis R. L., J. Tissue Eng. Regener. Med. 2007, 1, 261. [DOI] [PubMed] [Google Scholar]
- 18.a) Jin G. Z., Kim J. J., Park J. H., Seo S. J., Kim J. H., Lee E. J., Kim H. W., Tissue Eng., Part C 2014, 20, 895; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schutz K., Despang F., Lode A., Gelinsky M., J. Tissue Eng. Regener. Med. 2016, 10, 404. [DOI] [PubMed] [Google Scholar]
- 19.a) Chen J., Chen H., Li P., Diao H., Zhu S., Dong L., Wang R., Guo T., Zhao J., Zhang J., Biomaterials 2011, 32, 4793; [DOI] [PubMed] [Google Scholar]; b) Gao J., Dennis J. E., Solchaga L. A., Awadallah A. S., Goldberg V. M., Caplan A. I., Tissue Eng. 2001, 7, 363. [DOI] [PubMed] [Google Scholar]
- 20.a) Dormer N. H., Busaidy K., Berkland C. J., Detamore M. S., J. Oral Maxillofac. Surg. 2011, 69, e50; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schaefer D., Martin I., Jundt G., Seidel J., Heberer M., Grodzinsky A., Bergin I., Vunjak‐Novakovic G., Freed L. E., Arthritis Rheum. 2002, 46, 2524; [DOI] [PubMed] [Google Scholar]; c) Scotti C., Wirz D., Wolf F., Schaefer D. J., Burgin V., Daniels A. U., Valderrabano V., Candrian C., Jakob M., Martin I., Barbero A., Biomaterials 2010, 31, 2252. [DOI] [PubMed] [Google Scholar]
- 21. Levingstone T. J., Matsiko A., Dickson G. R., O'Brien F. J., Gleeson J. P., Acta Biomater. 2014, 10, 1996. [DOI] [PubMed] [Google Scholar]
- 22.a) Lee C. R., Grodzinsky A. J., Spector M., Biomaterials 2001, 22, 3145; [DOI] [PubMed] [Google Scholar]; b) Little C. J., Bawolin N. K., Chen X., Tissue Eng., Part B 2011, 17, 213; [DOI] [PubMed] [Google Scholar]; c) Mori H., Shimizu K., Hara M., Mater. Sci. Eng. C 2013, 33, 3230. [DOI] [PubMed] [Google Scholar]
- 23. Awad H. A., Wickham M. Q., Leddy H. A., Gimble J. M., Guilak F., Biomaterials 2004, 25, 3211. [DOI] [PubMed] [Google Scholar]
- 24. Vanderhooft J. L., Alcoutlabi M., Magda J. J., Prestwich G. D., Macromol. Biosci. 2009, 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parisi C., Salvatore L., Veschini L., Serra M. P., Hobbs C., Madaghiele M., Sannino A., Di Silvio L., J. Tissue Eng. 2020, 11, 10.1177/2041731419896068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vickers S. M., Squitieri L. S., Spector M., Tissue Eng. 2006, 12, 1345. [DOI] [PubMed] [Google Scholar]
- 27. Pan Z., Duan P., Liu X., Wang H., Cao L., He Y., Dong J., Ding J., Regener. Biomater. 2015, 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a) Liu X., Zheng C., Luo X., Wang X., Jiang H., Mater. Sci. Eng. C 2019, 99, 1509; [DOI] [PubMed] [Google Scholar]; b) Sultankulov B., Berillo D., Sultankulova K., Tokay T., Saparov A., Biomolecules 2019, 9, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan F., Yue W., Zhang Y. L., Mao G. C., Gao K., Zuo Z. X., Zhang Y. J., Lu H., Neural Regener. Res. 2015, 10, 1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tuzlakoglu K., Reis R. L., J. Mater. Sci.: Mater. Med. 2007, 18, 1279. [DOI] [PubMed] [Google Scholar]
- 31. Leonor I. B., Baran E. T., Kawashita M., Reis R. L., Kokubo T., Nakamura T., Acta Biomater. 2008, 4, 1349. [DOI] [PubMed] [Google Scholar]
- 32. Suzuki O., in Octacalcium Phosphate Biomaterials: Understanding of Bioactive Properties and Applications, (Eds: Suzuki O., Insley G.), Woodhead Publishing, Sawston, United Kingdom: 2019, Ch. 1, p. 1. [Google Scholar]
- 33. Sai Y., Shiwaku Y., Anada T., Tsuchiya K., Takahashi T., Suzuki O., Acta Biomater. 2018, 69, 362. [DOI] [PubMed] [Google Scholar]
- 34. Imaizumi H., Sakurai M., Kashimoto O., Kikawa T., Suzuki O., Calcif. Tissue Int. 2006, 78, 45. [DOI] [PubMed] [Google Scholar]
- 35. Liu B., Lun D. X., Orthop. Surg. 2012, 4, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suzuki O., Nakamura M., Miyasaka Y., Kagayama M., Sakurai M., Tohoku J. Exp. Med. 1991, 164, 37. [DOI] [PubMed] [Google Scholar]
- 37. Anada T., Kumagai T., Honda Y., Masuda T., Kamijo R., Kamakura S., Yoshihara N., Kuriyagawa T., Shimauchi H., Suzuki O., Tissue Eng., Part A 2008, 14, 965. [DOI] [PubMed] [Google Scholar]
- 38. Kouketsu A., Matsui K., Kawai T., Ezoe Y., Yanagisawa T., Yasuda A., Takahashi T., Kamakura S., J. Tissue Eng. Regener. Med. 2020, 14, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen K., Teh T. K., Ravi S., Toh S. L., Goh J. C., Tissue Eng., Part A 2012, 18, 1902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information