

Abstract
Despite intensive research and progress in personalized medicine, pancreatic ductal adenocarcinoma remains one of the deadliest cancer entities. Pancreatic duct‐like organoids (PDLOs) derived from human pluripotent stem cells (PSCs) or pancreatic cancer patient‐derived organoids (PDOs) provide unique tools to study early and late stage dysplasia and to foster personalized medicine. However, such advanced systems are neither rapidly nor easily accessible and require an in vivo niche to study tumor formation and interaction with the stroma. Here, the establishment of the porcine urinary bladder (PUB) is revealed as an advanced organ culture model for shaping an ex vivo pancreatic niche. This model allows pancreatic progenitor cells to enter the ductal and endocrine lineages, while PDLOs further mature into duct‐like tissue. Accordingly, the PUB offers an ex vivo platform for earliest pancreatic dysplasia and cancer if PDLOs feature KRASG12D mutations. Finally, it is demonstrated that PDOs‐on‐PUB i) resemble primary pancreatic cancer, ii) preserve cancer subtypes, iii) enable the study of niche epithelial crosstalk by spiking in pancreatic stellate and immune cells into the grafts, and finally iv) allow drug testing. In summary, the PUB advances the existing pancreatic cancer models by adding feasibility, complexity, and customization at low cost and high flexibility.
Keywords: organ culture models, pancreatic cancer, stem cell differentiation, urinary bladder
The porcine urinary bladder is optimized as an easy‐to‐handle and cost‐efficient scaffold for the engraftment and maturation of various pancreatic cell types for advanced cancer modeling. Stem cell‐derived pancreatic duct‐like cells demonstrate enhanced maturation and, in addition, papillary tumor growth upon oncogene stimulation. Finally, patient‐derived organoids and pancreatic stellate cells develop into complex tumors on the porcine urinary bladder.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) represents one of the deadliest and most difficult to treat cancers among all tumor entities, with rising incidence and projection to be the second most common cause of cancer‐related death by 2030.[ 1 ] Despite intensive research and ongoing implementation of a variety of personalized medicine approaches including genome sequencing and organoid‐based decision making into treatment regimens,[ 2 , 3 , 4 ] the 5‐year overall survival of patients remains poor as it ranges between 39% in the minority of patients with localized disease to only 3% in patients with distant metastatic disease.[ 5 ]
Most recently, others and we demonstrated that the generation of human pluripotent stem cell (PSC)‐derived pancreatic duct‐like organoids (PDLOs) and pancreatic acinus‐like organoids (PALOs) enable the investigation of pancreatic disease onset and carcinogenesis in a well‐defined and easily customizable genetic background.[ 6 , 7 , 8 , 9 , 10 ] The generation of PDLOs from genetically engineered pluripotent stem cell lines or patient‐derived induced pluripotent stem cells enabled the study of pancreatic carcinogenesis in vitro and in vivo in a time‐resolved manner.[ 7 , 8 , 9 , 11 ] Complementary, patient‐derived PDAC organoids (PDOs) represent a valuable resource to study late‐stage cancer and provide the unique opportunity to reconstruct tumor biology, as well as drug response predictions with direct implications for patient treatment.[ 2 , 12 , 13 ] Thus, PSC‐based and patient‐derived pancreatic organoid systems have i) fundamentally advanced the understanding of pancreatic development and disease evolution [ 14 ] and ii) substantially extended the toolbox to investigate PDAC by a plethora of perspectives.
However, the universal hindrance of both pancreatic organoid types is their sole epithelial character. The insufficient recapitulation of their genuine in vivo pancreatic niche, containing extracellular matrix (ECM) components and cell counterparts, demands the supplementation of such cultures with niche factors.[ 13 ] Thus, the integration of the organ‐ and tumor‐specific niche into current and future ex vivo systems is imperative to elaborate suitable advanced cancer models.
To date, several approaches have been pursued to implement the niche in existing organoid cultures. They range from simple in vitro cocultures to microfluidic chips, subcutaneous, and even orthotopic in vivo xenograft models for niche exploitation.[ 15 , 16 , 17 , 18 ] Whereas coculture models of two cell types represent easy‐to‐access and ‐perform approaches, the combination with additional cellular compartments in an effort to preserve biological architecture, such as endothelial cells in tumor vasculature mimicries,[ 19 ] already presents as challenging obstacles. Innovative techniques, such as bioprinting or microfluidic chip systems, partially overcome such constraints and enable a biomimetic reconstruction of the in vivo situation.[ 19 , 20 ] While better imitating the actual in vivo niche, incorporation of bioprinting and microfluidics demands a high‐end, labor‐ and cost‐intensive technical equipment as well as thorough expertise hampering the broad application of such systems.
Until now, only xenograft models allow the direct integration into an in vivo environment. Hence, subcutaneous and orthotopic xenografts are still used as standard for more complex pancreatic tumor formation and progression models from PDOs and PDLOs/PALOs.[ 3 , 6 , 7 , 8 , 17 ] However, the absence of the immune system, its expensiveness, and the subsequent technical challenges (e.g., tumor take rate, required surgical skills) set limitations within the system.[ 17 ]
Finally, the chick chorioallantoic membrane (CAM) assay provides fast access to a relatively complex model for studying angiogenesis in a nearly in vivo setting.[ 21 , 22 ] Besides its relative low costs, CAM assays show relatively high experiment‐to‐experiment variability and a rather limited time of observation. Given the evolutionary distance, the relevance of the avian system as a universal model to study human tumor/stroma interplays is also strongly debatable.
Striving for optimization of niche‐containing culture models in terms of costs, labor intensity, accessibility and, most importantly, suitability is compelling to overcome limitations of the aforementioned models. Taking whole organs into ex vivo culture systems may seem rather evident to achieve a truthful imitation of an organ‐specific niche. In fact, generation of such scaffolds for pancreatic organ culture was reported recently.[ 23 , 24 , 25 ] Decellularization of porcine pancreata generates an ECM scaffold for a successful spontaneous differentiation of induced pluripotent stem cells toward the endodermal lineage.[ 23 ] In addition, human fetal pancreatic cells demonstrate an ECM‐dependent maturation on such scaffolds.[ 24 ] Whole organ ex vivo cultures thus provide clear benefits by creating a suitable niche for cell growth and maturation and sparing cost‐ and labor‐intensive transplantation studies. Thus, considering the body and organ size similarity between pigs and humans and the excess supply by the food industry, pig‐derived organ culture models may be an ideal source for ex vivo cultures. We and others have demonstrated that the porcine urinary bladder (PUB) is an appropriate organ culture model to study invasiveness of human urothelial cancer cells and to explore differentiation of nonmalignant cells.[ 26 , 27 , 28 ] Importantly, this model is easy to access, cost‐effective, and technically simple. Within the current study, we meticulously demonstrated the versatility of the PUB as an ex vivo organ culture system to investigate pancreatic differentiation and maturation and as a scaffold for advanced pancreatic cancer modeling.
2. Results
2.1. Transcriptomic Data Reveal High Similarity between Urinary Bladder and Pancreas Extracellular Matrices
To identify organs that may qualify as scaffolds for the recapitulation of the genuine pancreatic niche, we compared the expression of a variety of ECM genes (i.e., collagens, laminins, ELN, fibrillins, emilins, tenascins, and FN1) across multiple organs in three publicly available transcriptomic datasets (Human Protein Atlas, GTEx, FANTOM5 compiled as normalized expression in the Human Protein Atlas).[ 29 , 65 ] Focusing on ECM gene expression, principal component analysis (PCA) did not reveal any evident organ clusters, but rather showed a scattered distribution of different investigated organs, except for two main outliers (i.e., cervix/uterine and smooth muscle) (Figure 1A). As we recently established the PUB as an easy to access and handle organ culture model,[ 26 , 28 ] we performed a direct comparison of gene expression of the human pancreas and urinary bladder. Interestingly, collagens, which represent the primary structural components of the ECM, showed a comparable expression profile within the two organs (Figure 1B). This was especially observed for the most abundantly expressed COL1, COL3, COL4, and COL6 families. Laminins, another prominent group of ECM genes displayed compelling similarity including high expression of LAMB1, LAMB2, and LAMC1 in both organs (Figure 1C). As well, emilins such as EMILIN1/2/3, fibrillins such as FBN1/2/3, and tenascins such as TNN/R/C displayed similar expression patterns within the urinary bladder and pancreas. Of note, our comparative analysis also revealed several organ specificities. While laminin sub‐chain LAMA5 was strongly expressed in the pancreas, LAMA4 was exclusively present in the urinary bladder. In addition, ELN was the matrix component which was expressed at highest levels in the pancreas in the noncollagen/nonlaminin group and COL18A1 was the most abundant collagen. In contrast, EMILIN1/FN1 and COL6A1/2 were among the most abundant ECM components of the urinary bladder in their respective groups. Particularly high levels of FN1, which can be produced by smooth muscle cells,[ 30 ] and of EMILIN1 and ELN, components of elastic fibers and required to retain the original form after micturition,[ 31 ] reflect organ‐specific functions of the urinary bladder (Figure 1D). Altogether, despite certain organ specificities, pancreas and urinary bladder share comparable expression patterns of major structural components of the ECM, e.g., most collagens and many laminins among others, suggesting the latter may represent, besides other organs, a platform to host certain pancreatic cell types for ex vivo culture.
Figure 1.
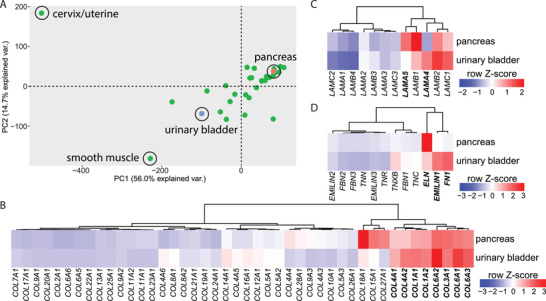
Transcriptomic analysis of the extracellular matrix of different organs reveals similarity between pancreas and urinary bladder. A) Principal component analysis of different organs based on the normalized RNA expression levels of extracellular matrix (ECM)‐filtered genes. Data were extracted from the Human Protein Atlas (https://www.proteinatlas.org/). Gene expression levels of different ECM genes including B) collagens, C) laminins, and D) tenascins, fibronectin, fibrillins, elastin, and emilins in urinary bladder and pancreas. Gene expression values depict row‐wise normalized Z‐scores and clustering was based on Ward.D2. PC, principal component; var., variance.
2.2. The Porcine Urinary Bladder Matrix Directs Pancreatic Progenitors to the Trunk Lineage
To experimentally assess the suitability of the PUB as an ex vivo organ culture model supporting pancreatic differentiation, we first employed pancreatic progenitor (PP) cells generated from human PSCs. PSC‐derived trilineage potent PPs were cultured on the de‐epithelialized porcine urinary bladder for three weeks (Figure 2A). Compellingly, tube structures with high resemblance to pancreatic ducts were identified (Figure 2B). Human origin was proven by immunostaining against human nucleoli and human‐specific CK‐19 (Figure 2C,D; Figure S1A, Supporting Information). Abundant expression of the duct marker [ 32 ] CK‐19, as well as high proportions of CK‐8‐expressing cells, and several CK‐7‐positive cells (Figure 2D) confirmed a predominant ductal identity of these tubular structures. Noteworthy, many of the detected human cells displayed a ductal phenotype albeit PPs should harbor a trilineage potential. Thus, we wondered whether the PUB predetermines a more restricted bipotent pancreatic trunk‐like fate (ductal versus endocrine identity [ 33 ]). Indeed, staining for a set of endocrine markers (chromogranin A (CHGA), glucagon (GCG), and C‐peptide (C‐pep)) demonstrated the presence of few trunk‐derived endocrine cells (Figure 2E). In contrast, tip domain‐derived acinar cells were not detected by immunostainings against MIST1 and TRY1 (Figure 2F). To exclude a pre‐restricted competence of the PPs per se toward the trunk lineage, PPs were orthotopically transplanted into murine hosts, thereby, investigating lineage specification in the genuine niche. Contrary to our observations from the on‐PUB system, ductal, endocrine, and acinar cells were present in PP‐derived xenografts (Figure S2A–G, Supporting Information). CHGA‐positive endocrine cells were only found as single scattered cells (Figure S2E, Supporting Information). Unlike in the PUB grafts, tip‐domain‐derived acinar cells were also detected, as shown by the expression of MIST1 and TRY1 (Figure S2F, Supporting Information). Of note, ductal and acinar marker expression was reflected by self‐organization in either tubular or more compact cell structures, respectively.
Figure 2.
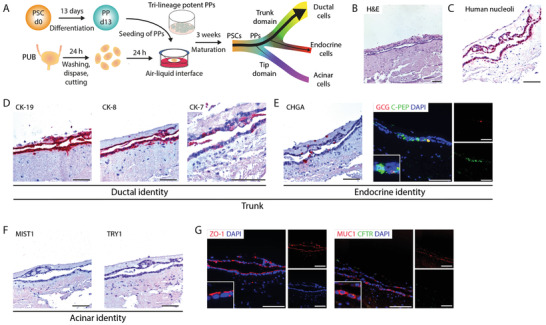
Pancreatic progenitors differentiate towards trunk lineage on the porcine urinary bladder. A) Schematic illustration of the culture of pancreatic progenitor (PP) cells on de‐epithelialized porcine urinary bladder. B) Hematoxylin and eosin (H&E) stained histological sections of PPs cultured on porcine urinary bladder (n = 2). Immunohistochemistry stainings for C) human nucleoli and D) duct‐specific cytokeratins CK‐19, CK‐8, and CK‐7 on PPs cultured on porcine urinary bladder (n = 2). E) Immunohistochemistry stainings for endocrine‐specific chromogranin A (CHGA) and immunofluorescence stainings for C‐peptide (C‐PEP, green) and glucagon (GCG, red) on PPs cultured on porcine urinary bladder (n = 2). Cells were counterstained with DAPI (blue). F) Immunohistochemistry stainings of acinar‐specific markers MIST1 and TRY1 on PPs cultured on porcine urinary bladder (n = 2). G) Immunofluorescence stainings for ZO‐1 (red) and for MUC1 (red) and CFTR (green) (n = 2). Cells were counterstained with DAPI (blue). Scale bars represent 100 µm. PSC, pluripotent stem cells; PUB, porcine urinary bladder.
Polar organization of such tubular structures on the PUB was mirrored by the strong expression of tight junction protein ZO‐1 at the luminal side of the ductal tubes (Figure 2G). Despite the strong expression of ductal markers, only a partially mature duct‐like phenotype was observed, as illustrated by moderate MUC1 but no CFTR expression (Figure 2G). In contrast, expression of MUC1 and CFTR was more abundant in murine host grafts, potentially indicative of a slightly better maturation in the genuine niche (Figure S2G, Supporting Information).
Altogether, the dominant formation of duct‐like tissue on the PUB suggests that the ex vivo culture setting provides an appropriate niche for PP engraftment and differentiation, especially to the trunk domain, and thus, may constitute a suitable scaffold for a novel organ culture‐based model of the pancreas.
2.3. Pluripotent Stem Cell‐Derived Pancreatic Duct‐Like Organoids Undergo Maturation on the Porcine Urinary Bladder
Having demonstrated the general applicability of the PUB for pancreatic engraftment and duct‐lineage commitment, we next investigated the capacity of the PUB to host already lineage committed PSC‐derived PDLOs (Figure 3A). Instead of enclosed ductal tubes, we observed the formation of large cell layers that were orientated toward the urinary bladder lumen (Figure 3B). Phenotyping revealed their ductal identity, illustrated by expression of CK‐19, CK‐7, and CK‐8 in a multitude of cells (Figure 3C) and the absence of acinar markers MIST1 and TRY1 (Figure S3A, Supporting Information), in accordance with their in vitro features (Figure S3B,G, Supporting Information). Furthermore, CHGA‐positive endocrine cells were virtually absent in the transplants (Figure S3A, Supporting Information). To gain insights if PUB‐driven PDLO maturation was enhanced compared to sole in vitro cultures, we further compared cytokeratin expression in both models. While CK‐19 was equally present, CK‐8 and CK‐7 expression was increased after on‐PUB culture. Of note, we have previously observed that CK‐7 expression is rather restricted to more mature ductal cells,[ 7 ] which is in line with observations that CK‐7 expression increases during human fetal development.[ 34 ] To investigate the maturity level in vitro, we evaluated the number of CK‐7‐expressing organoids. While only 18% of all in vitro PDLOs harbored CK‐7 expression (Figure S3C, Supporting Information), it became ubiquitously expressed in on‐PUB‐propagated cultures indicating an enhanced maturation (Figure 3C). Similarly, the maturation marker MUC1 [ 7 , 11 ] was almost absent in immunostainings of in vitro PDLOs, while it was strongly expressed in on‐PUB PDLOs (Figure 3D; Figure S3D, Supporting Information). In line, also CFTR expression increased during the ex vivo organ‐based culture as indicated by only about 2% CFTR‐positive cells after two weeks and 22% CFTR‐positive cells after four weeks of culture (Figure 3D; Figure S3D, Supporting Information). Proliferation (KI‐67) of the PUB‐based ex vivo PDLO culture was rather low compared to their in vitro counterparts in agreement with improved maturity [ 35 ] (Figure 3E; Figure S3E, Supporting Information). Functional cell polarity, a prerequisite for pancreatic duct organization, was distinctly evident in both PDLOs and ductal cells on‐PUB as indicated by the apical polarity marker ZO‐1 and the basal localization of COL4A1, which is in line with previously observed expression patterns of those markers [ 36 ] (Figure 3F; Figure S3F, Supporting Information). Apical‐basal polarity was demonstrated by the assembly of tight junctions (ZO‐1‐positive) toward the urinary bladder lumen (Figure 3F). Altogether, these data prove that the PUB constitutes a valuable scaffold for the robust propagation and differentiation of human PDLOs into more mature pancreatic ductal cells.
Figure 3.
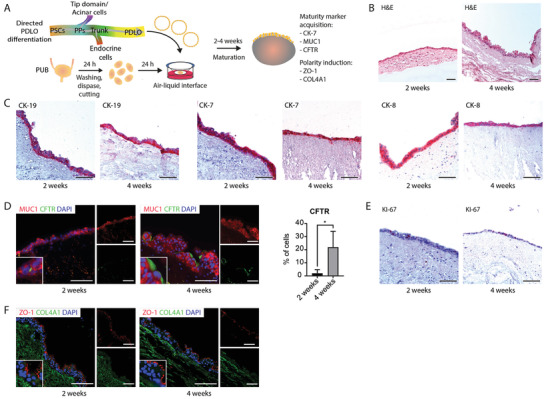
Pancreatic duct‐like organoids mature to adult ductal cells on the porcine urinary bladder. A) Schematic representation illustrating the lineage‐specific generation of pancreatic duct‐like organoids (PDLOs) originated from pluripotent stem cells and their engraftment on the porcine urinary bladder. B) Hematoxylin and eosin (H&E) stained histological sections of PDLOs cultured on porcine urinary bladder for two and four weeks (n = 4). C) Immunohistochemistry for duct‐specific cytokeratins CK‐19, CK‐7, and CK‐8 (n = 4) and D) immunofluorescence stainings for ductal maturation marker MUC1 (red) and CFTR (green) on PDLOs cultured on porcine urinary bladder for two and four weeks, with quantification of CFTR‐positive cells (n = 4). Data are shown as mean ± SD. Cells were counterstained with DAPI (blue). *, p < 0.05. E) Immunohistochemistry staining for KI‐67 on PDLOs cultured on porcine urinary bladder for two and four weeks (n = 4). F) Immunofluorescence stainings for cell polarity marker ZO‐1 (red) and COL4A1 (green) in PDLOs cultured on porcine urinary bladder for two and four weeks (n = 4). Scale bars represent 100 µm. PPs, pancreatic progenitors; PSCs, pluripotent stem cells; PUB, porcine urinary bladder.
2.4. KRASG12D‐Expressing Pancreatic Duct‐Like Organoids Form Dysplastic, Papillary, Neoplasia‐Like Lesions on Porcine Urinary Bladder
To challenge the PUB as a valuable system for human pancreatic cancer research, we next examined the tumorigenic evolution of ductal cells on‐PUB upon oncogenic stimuli. Therefore, we employed the previously described doxycycline‐inducible KRASG12D overexpression system [ 7 ] to precisely orchestrate oncogene expression in the ex vivo PDLO culture system (Figure 4A). Consistent expression of mutant KRASG12D was demonstrated by the detection of the HA‐tag and mCherry (HA‐tagged‐KRASG12D‐IRES‐mCherry construct) only in doxycycline‐treated samples (Figure 4B). Upon four weeks of doxycycline treatment and subsequent oncogenic KRASG12D expression, we observed the development of papillary lesions on the PUB (Figure 4C). In line, CK‐19 immunostaining confirmed the ductal origin of transformed and nontransformed tissue (Figure 4D). Accordingly, the papillary morphology during dysplastic lesion formation was accompanied by the expression of the epithelial tumor marker CA19‐9 (Figure 4E). In fact, we observed a trend towards increased expression of CA19‐9 and MUC1 in doxycycline‐treated samples compared to control counterparts. Such morphological and tumor marker changes illustrate the induction of early cell transformation processes [ 37 ] (Figure 4E‐G), even though the dysplastic marker MUC5AC did not increase over the course of the experiment.
Figure 4.
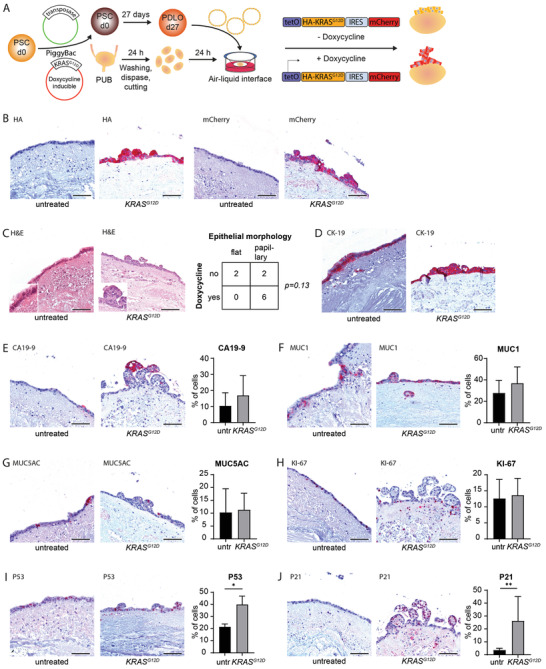
KRASG12D induces papillary neoplastic growth, dysplastic marker expression, and oncogene‐induced senescence. A) Schematic representation illustrating the propagation of pancreatic duct‐like organoids (PDLOs) harboring a Tet‐On expressing oncogenic HA‐tagged KRASG12D and mCherry on porcine urinary bladder. B) Immunohistochemistry stainings for HA‐tag and mCherry on vehicle and doxycycline‐treated Tet‐On expressing KRASG12D PDLOs cultured for four weeks on porcine urinary bladder (n = 4 for untreated (untr), n = 6 for doxycycline‐treated). C) Hematoxylin and eosin (H&E) stained histological sections of vehicle and doxycycline‐treated Tet‐On expressing KRASG12D PDLOs cultured for four weeks on porcine urinary bladder and contingency table comparing presence of papillary structures and doxycycline treatment (n = 4 for untreated, n = 6 for doxycycline‐treated). Immunohistochemistry stainings for D) CK‐19, E) CA19‐9, F) MUC1, G) MUC5AC, H) KI‐67, I) P53, and J) P21, and quantifications of respective positive cells in vehicle and doxycycline‐treated Tet‐On expressing KRASG12D PDLOs cultured for four weeks on porcine urinary bladder (n = 4 for untreated (untr), n = 6 for doxycycline‐treated). Data are shown as mean ± SD. *, p < 0.05, **, p < 0.01. Scale bars represent 100 µm. PSCs, pluripotent stem cells; PUB, porcine urinary bladder.
We previously demonstrated that oncogene‐induced growth restriction constitutes a relevant tumor barrier upon ectopic KRASG12D induction in PDLOs and in PDLO‐derived xenografts.[ 7 ] In agreement with such barrier, we did not observe an increase in proliferation (KI‐67) between doxycycline‐treated and control PUB cultures (Figure 4H). In addition, dysplastic lesions displayed a higher P53 index (Figure 4I), while in turn, the canonical downstream effector P21 was upregulated and/or stabilized upon doxycycline treatment corroborating the molecular changes during PDLO transformation on the PUB (Figure 4J). Collectively, these findings highlight the ability of our experimental system to reconstruct early preinvasive stages of pancreatic carcinogenesis and its worthiness by allowing time‐resolved and easy access to nascent malignant cells.
2.5. Patient‐Derived Pancreatic Cancer Organoids Form Stroma‐Containing Tumors on Porcine Urinary Bladder
To determine whether the PUB model implies a relevant platform to study advanced stages of PDAC as well, pancreatic cancer patient‐derived organoids were isolated and cultured on‐PUB (Figure 5A). Interestingly, PDOs formed highly proliferative (KI‐67) tumor‐gland networks with high take rate (17/18 grafts for all different approaches) (Figure 5B, C, Table 1 ). Consistent expression of CDH1 and CK‐19, along with the absence of vimentin (VIM), proved the ductal and epithelial phenotype of the malignant cells (Figure 5C). In addition, GATA6 immunostaining shows that PUB‐grown tumors preserve the classical or basal‐like subtype from their PDO ancestors (matching in 7/7 cases, 4 classical and, 3 basal‐like subtyped PDOs) [ 38 ] (Figure 5C). To further validate phenotypic stability within the PUB system, we performed a head‐to‐head comparison of three 3D‐cultured PDO lines and their on‐PUB counterparts with a carefully designed 14‐marker panel [ 39 , 40 , 41 , 42 , 43 ] quantitative PCR‐based approach. Direct comparison of basal as well as classical PDAC subtype marker genes demonstrated a high degree of similarity between in vitro and on‐PUB cultures as shown also in the PCA by forming a basal‐like clustering and a classical like cluster (Figure 5D). In fact, two basal‐like PDOs (PDO 1 and 2) retained high expression of, e.g., KRT5 and one classical PDO line (PDO 3) demonstrated high expression levels of, e.g., AGR2 both in vitro and on‐PUB. Furthermore, three PDO lines were propagated on‐PUB for 31 days to evaluate the model suitability for long‐term experiments (Figure S4A, Supporting Information). While no phenotypic alteration was observed, based on conserved respective marker expression for CDH1, CK‐19, VIM, and GATA6 (Figure S4B, Supporting Information), tumors however displayed reduced proliferation (KI‐67) and larger tumor glands, as well as necrotic areas (Figure S4A,B, Supporting Information). Thus, tumor phenotyping suggests that the PUB offers a reliable scaffold supporting PDO‐based cancer development, enabling tumor network formation, while preserving tissue differentiation and phenotype.
Figure 5.
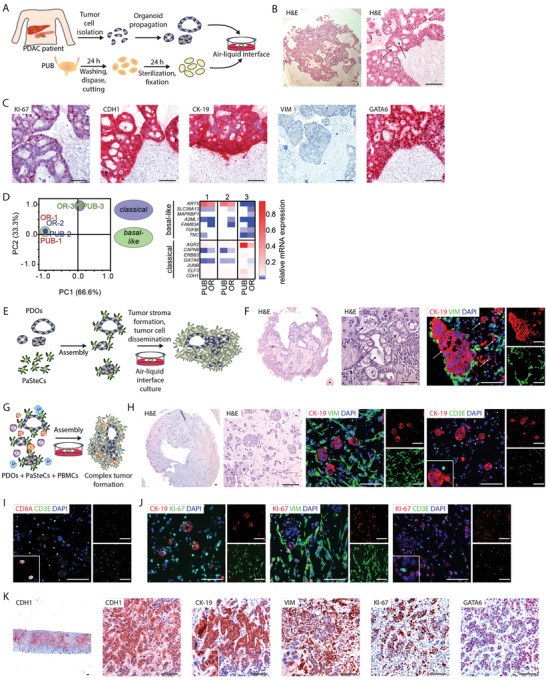
Pancreatic cancer patient‐derived organoids form stroma‐containing tumors on porcine urinary bladder. A) Schematic representation depicting the procedure for isolation, propagation, and engraftment of patient‐derived organoid (PDO) on porcine urinary bladder (PUB). B) Hematoxylin and eosin (H&E) stained histological sections of PDOs propagated on PUB for five days (n = 6). Arrows indicate tumor glands. C) Immunohistochemistry stainings for CDH1, CK‐19, VIM, KI‐67, and GATA6 of PDOs propagated on PUB for five days (n = 6). D) qRT‐PCR analysis (principal component analysis and heatmap of normalized RNA expression levels) of a relevant 14‐gene classical/basal‐like panel in PDOs propagated either on PUB or as 3D in vitro cultures (OR). E) Schematic representation illustrating the generation of assembloids consisting of PDOs and pancreatic stellate cells (PaSteCs) on PUB. F) H&E stained histological sections and immunofluorescence stainings for CK‐19 (red) and VIM (green) of PDO‐PaSteC assembloids cultured on PUB (n = 7). Cells were counterstained with DAPI (blue). Arrows indicate tumor glands/cancer cells. G) Schematic illustration depicting the generation of PDO, PaSteC, and peripheral blood mononuclear cells (PBMCs)‐containing assembloids on PUB. H) H&E stained histological sections and immunofluorescence stainings for CK‐19 (red) and VIM (green; left panel) or CD3E (green, right panel) of PDO‐PaSteC assembloids propagated on PUB for five days (n = 4). Immunofluorescence stainings for I) CD3E (green) and CD8A (red), and for J) KI‐67 (green on left panel; red on middle and right panels) and CK‐19 (red, left panel), VIM (green, middle panel), or CD3E (green, right panel) of PDO‐PaSteC‐PBMC assembloids propagated on PUB for five days (n = 4). K) Immunohistochemistry stainings for CDH1, CK‐19, VIM, KI‐67, and GATA6 of a pancreatic ductal adenocarcinoma patient liver metastasis biopsy. Scale bars represent 100 µm. PDAC, pancreatic ductal adenocarcinoma.
Table 1.
Engraftment take rates of pancreatic progenitors (PPs), pancreatic duct‐like organoids (PDLOs), and patient‐derived organoids (PDOs) on the porcine urinary bladder. PaSteCs, pancreatic stellate cells; PBMCs, peripheral blood mononuclear cells
Albeit recapitulating tumor glands, our initial approach lacked stromal counterparts that actively contribute to tumor propagation, progression, and therapy resistance.[ 44 ] To mimic a more complex tumor microenvironment on‐PUB, different nontumoral cell types were additionally incorporated to the design of the PUB‐based human PDAC model (Figure 5E, G). We first integrated a fibroblastic compartment into our system using human pancreatic stellate cells (PaSteCs). After five days, histological analysis revealed rapid development of a pancreatic cancer‐like tumor microenvironment, morphologically resembling typical pancreatic tumor‐surrounding stroma [ 45 , 46 ] (Figure 5F). Immunostainings against the fibroblast and mesenchymal marker vimentin and the ductal marker CK‐19 indeed showed tumor glands surrounded by cancer‐associated fibroblast (CAF)‐like PaSteCs, as well as scattered malignant cells indicative of potential cancer cell dissemination (Figure 5F).
To further customize the tumor microenvironment, immune cells were included in our experimental setting. Peripheral blood mononuclear cells (PBMCs) were isolated from a healthy donor and assembled with PDOs and PaSteCs onto the PUB (Figure 5G). Over the course of five days, a complex tumor rapidly developed on‐PUB (Figure 5H). Immunostainings indeed indicated a robust propagation of malignant cells and their niche, including CK‐19‐positive tumor glands and VIM‐positive CAF‐like PaSteCs. Interestingly, investigation of CD3E, in combination with CK‐19, revealed the presence of scattered T cells surrounding the tumor‐glands. Further characterization of CD3‐positive T cells demonstrated an abundance of CD8A‐positive cytotoxic T cells (Figure 5I). To assess the proliferation potential of tri‐cultured cells on the PUB, cell type‐specific KI‐67 co‐stainings were conducted (Figure 5J). Interestingly, cell proliferation was mainly observed in tumor glands and CAF‐like PaSteCs but was absent in CD3‐positive T cells. This might be due to suboptimal culture conditions and/or a tumor‐mediated immunosuppressive environment. Assembled PDAC on‐PUB was finally compared to one representative original primary patient tissue (liver metastasis biopsy) to assess cancer/niche similarities and potential differences (Figure 5K). Of note, the PUB‐assembled PDAC engraftments displayed similarities to the niche of the liver metastasis: i) morphologically with distinct tumor glands and ii) marker‐wise with epithelial (CDH1) and surrounding stromal (VIM) marker properties and iii) with a high proliferation rate (KI‐67) within PUB engraftments and the metastasis (Figure 5B,C,K). This strongly suggests that the PUB‐based ex vivo system also constitutes a relevant and accessible platform to model advanced stages of pancreatic cancer, as well as metastatic niches.
2.6. Porcine Urinary Bladder‐Based Ex Vivo System Allows Drug Testing Approaches
We finally assessed the feasibility of the PUB to serve as an ex vivo drug screening platform. Therefore, we employed Atm‐deficient malignant cells previously shown to be vulnerable upon exposure toward the PARP inhibitor olaparib and the topoisomerase 1 inhibitor irinotecan [ 47 , 48 , 49 ] (Figure S5, Supporting Information). As expected, cell proliferation (KI‐67) was blocked upon both chemotherapeutic treatments (Figure S5B,D, Supporting Information). Concomitantly, we observed an induction of DNA damage (H2AX p‐S139) leading to apoptosis (cleaved caspase‐3) in Atm knockout tumors on‐PUB (Figure S5C–E, Supporting Information), demonstrating the potential of the PUB model as an ex vivo drug testing platform.
3. Discussion
Over the last two decades, the development of a broad range of PDAC models dramatically improved our understanding of PDAC biology (e.g., genetically engineered mouse models, patient‐derived xenografts, patient‐derived organoids). Each system displays its own strengths and unfortunately limitations and constraints. Among them, PSC‐derived models are unique in their features to supply an unlimited and renewable resource of genetically defined acinar‐ and duct‐like cells.[ 6 , 7 , 8 , 11 ]They allow the study of embryonic pancreas development in vitro and, upon oncogenic hits, a robust mimicry of early steps of PDAC evolution. However, further maturation as well as final progression into cancer cells has only been observed upon time‐ and labor‐intensive orthotopic transplantation into murine hosts.[ 7 , 8 , 9 ] This indicates the importance of the ECM scaffold as trigger of signaling, maturation and transformation by driving mechanotransductional signals [ 50 , 51 , 52 ] and raises the interest for the utilization of alternative ECM scaffolds for improving pancreatic cell differentiation. In vitro cultures often rely on Matrigel, which consists mainly of laminins and collagens derived from Engelbreth–Holm–Swarm mouse sarcoma cells. Despite its state‐of‐the‐art character, other scaffolds may provide similarly suitable or even better alternatives for modeling endoderm‐derived organogenesis, pancreatic differentiation and carcinogenesis.[ 23 , 52 , 53 ] Since the ECM is the final product of dynamic modeling processes, partly orchestrated by matrix degrading metalloproteinases and their antagonists (tissue inhibitors of metalloproteinases), many components have to be taken into consideration.[ 54 ] Here, we first re‐analyzed three global datasets to identify a suitable scaffold for the elaboration of a novel tumor niche‐resembling ex vivo model. Intriguingly, the urinary bladder and pancreas extracellular matrices displayed potential similar expression patterns of highly conserved ECM components (collagens, laminins, tenascins, fibronectin, fibrillin, elastin, and emilins).[ 55 ] Albeit the mere analysis of gene expression might not allow the delineation of ECM intricate macromolecular networks following post‐translational processing,[ 54 ] the suitability of the on‐PUB ex vivo system was substantiated by a series of experimental approaches.
When propagated on‐PUB, PP cells engaged in pancreatic trunk cell fate but the presence of acinar cells was not observed. This restricted differentiation in the trunk domain raised the question of the existence of a niche‐dependent trunk‐promoting effect. While orthotopically transplanted PPs developed into ductal, endocrine, and acinar‐like structures, ductal cells were nevertheless predominant in these grafts. Hence, a lineage bias of the employed PPs cannot be excluded. Such a trunk‐promoting competence was just recently indicated by temporally‐resolved scRNA‐seq analyses demonstrating low expression of acinar markers in PPs and identifying a putative unipotent PP subcluster, whose marker expression might favor ductal lineage specification.[ 11 ]
We additionally cultured PDLOs on the ex vivo PUB model. Albeit we did not observe enclosed ductal tubular structures, such grafts highly expressed ductal epithelial markers such as CK‐19 and CK‐7. Interestingly, ductal cells were organized in an apical‐basal orientation towards the air phase of this air–liquid interphase. An increase in CK‐8, CK‐7, MUC1, and CFTRfurther gave evidence for the improved maturation of PDLOs on the PUB rendering our ex vivo system adequate to study mechanisms for ductal cell specification.
After investigation of embryonic development on‐PUB, we challenged the system to demonstrate its value for PDAC modeling. Induction of oncogenic KRASG12D expression in on PUB‐cultured PDLOs led to the formation of dysplastic, papillary, neoplastic lesions which successfully recapitulate early preneoplastic lesions in patients.[ 56 ] Albeit with lower penetrance than in acinar cells, single KRAS oncogenic hit in ductal cells can result in the development of preneoplastic lesions as pancreatic intraepithelial neoplasms (PanIN), positive for MUC1, MUC5AC, and CA19‐9 markers [ 57 , 58 , 59 ] in line with our dysplastic marker profile of KRASG12D‐expressing PDLOs cultivated on‐PUB. Accordingly, and despite this initial KRAS oncogenic insult, preneoplastic lesions did not progress to invasive tumor. The short timeframe, the absence of a second oncogenic event, and the activation of cell cycle checkpoints (P53, P21) likely accounts for the noninvasive features of the PDLO‐derived tumor formation. Given the degradation of the porcine urinary bladder in long‐term cultures, our investigations were limited to a four‐week time period. The corroboration with our recent observation that the single hit of KRASG12D expression in PDLO xenografts led to tumor formation in only one out of four grafts, after eight weeks in murine pancreata,[ 7 ] is in compliance with the absence of invasiveness in the ex vivo PUB model.
We investigated the behavior of late‐stage PDOs on the PUB. Unlike in vitro PDO cultures, organoids re‐formed complex and intricate tumor‐gland networks resembling human PDAC morphology with a complex architecture and long‐term persistence on the PUB.[ 45 ] The addition of PaSteCs in this setting allowed the formation of pancreatic cancer glands surrounded by a stromal niche of cancer‐associated fibroblast‐like cells. Finally, incorporation of PBMCs added a new level of complexity to the system with the aim to resemble the genuine pancreatic tumor niche on the PUB. Despite the intrinsic immunosuppressive nature of PDAC,[ 60 ] further optimization of culture conditions and/or prior T cell activation might allow to overcome the lack of proliferation of immune cells on‐PUB and might even enable a model to study strategies to overcome immunosuppression in PDAC.
Similarly, the meticulous reconstruction of the parental tissue might further benefit from fine‐tuning the assembled cell ratios, despite the already achieved high degree of similarity in morphology and marker landscape.
Thus, although presenting a novel organ‐based system allowing the study of complex interactions in vitro, two limitations remain: i) additional optimization steps are required to fully recapitulate tumor/stroma in vivo situation and achieve concordant morphological resemblance; ii) experimental duration is limited due to ongoing degradation of the urinary bladder and/or potential microbial contamination. Tissue fixation by peroxy acetic acid [ 27 ] was performed for studies involving PDOs and murine cells and could overcome the latter hurdle. Thus, it should be considered for long‐term experiments.
In summary, we herewith describe a novel, easy‐to‐access, and cost‐effective porcine urinary bladder‐based culture model that i) promotes pancreatic differentiation and ii) enhances cell maturation but also iii) provides a proper niche for the investigation of early and late carcinogenic events, and iv) drug response. The possibility to easily implement the system with multiple cell types, e.g., a fibroblastic and an immune compartment, makes it a unique resource to study pancreatic cancer interplays.
4. Experimental Section
Human Embryonic Stem Cells
The human embryonic stem cell (hESC) line HUES8 (Harvard University, RRID:CVCL_B207) was used in this study. The Robert Koch Institute granted the culture and differentiation of this hESC line under the application ‘‘79. Genehmigung nach dem Stammzellgesetz, AZ 3.04.02/0084.” Cells were propagated as described previously.[ 7 ] Briefly, HUES8 cells were cultured on hESC‐Matrigel (Corning)‐coated plates in mTeSR Plus (STEMCELL Technologies) at 37 °C under 5% (v/v) CO2 and 5% (v/v) O2 atmosphere with media change every second to third day.
Inducible KRASG12D Overexpression Cell Lines
To generate doxycycline‐inducible KRASG12D overexpression cell lines, an all‐in‐one vector [ 61 ] was employed as previously described.[ 7 ] Briefly, gene transfer was performed using a piggyBac (PB) transposon system with the HA‐tagged KRASG12D cDNA sequence coupled with an IRES‐mediated mCherry fluorescence reporter (described as PB‐TAC‐ERP2‐(N‐HA)‐KRAS_G12D plasmid [ 7 ]). The transfection was conducted on 2 × 105 HUES8 cells with 769 ng of all‐in‐one plasmid and 231 ng of transposase expression vector (SBI Biosciences) [ 62 ] using a 4D‐Nucleofector (Lonza) and P3 primary cell 4D‐Nucleofector Kit S (Lonza). Selection was performed by adding 1 µg mL−1 puromycin (Sigma‐Aldrich) to the culture medium.
Differentiation of Human Pluripotent Stem Cells into Pancreatic Progenitors and Pancreatic Duct‐Like Organoids
Human pluripotent stem cells were differentiated into PPs as described previously,[ 7 ] based on.[ 6 , 63 ] Pancreatic duct‐like organoids were generated from PPs as described previously.[ 7 ] Briefly, PPs were seeded into growth factor reduced (GFR)‐Matrigel and differentiated to PDLOs by adding 50 ng mL−1 FGF‐10 (R&D Systems), 50 ng mL−1 EGF (R&D Systems), 50 ng mL−1 KGF (Peprotech), 10 × 10−3 m nicotinamide (Sigma‐Aldrich), 10 × 10−6 m ZnSO4 (Sigma‐Aldrich), 10 × 10−6 m Y27632, and 50 × 10−9 m MSC2530818 (Selleckchem) to BE3 medium for the days 13–19 and 50 ng mL−1 FGF‐10, 50 ng mL−1 EGF, 10 × 10−3 m nicotinamide, and 10 × 10−6 m ZnSO4 to BE3 medium for the days 20–27 as detailed in the Supporting Information. Differentiation was performed for a minimum of 27 days, before PDLOs were subjected to further experiments.
Patient‐Derived Organoids
Pancreatic cancer patient‐derived organoids were isolated and cultured as previously described.[ 2 ] Briefly, after generation of digested tumor fragments and single cells, tumor cells were seeded in GFR‐Matrigel and cultured in RSPOI/WNT3A‐conditioned organoid medium.[ 2 , 12 ] The study was approved by the Ethics Committee of the University Hospital of Ulm (project 72/2019) and conducted in accordance with the declaration of Helsinki. Written informed consent was obtained before acquisition of any sample.
Human Pancreatic Stellate Cells
PaSteCs were isolated from a chronic pancreatitis patient, immortalized by the catalytic subunit of hTERT and SV40 large T antigen and kindly provided by Prof. Matthias Löhr (Karolinska Institute).[ 64 ] Cells were propagated in DMEM, containing 10% FBS (PanBioTech) and P/S (100 IU mL−1 penicillin and 100 µg mL−1 streptomycin sulfate) at 37 °C under 5% (v/v) CO2 atmosphere.
Peripheral Blood Mononuclear Cells
PBMCs were isolated from healthy volunteers after giving written informed consent and in accordance with the local Ethics Committee of University Hospital Ulm and in accordance with the declaration of Helsinki. PBMCs were isolated from buffy coats and bought from the local institute of transfusion medicine.
Murine Cancer Cells
Atmfl/fl; LSL‐KrasG12D/+; Ptf1aCre/+ cells were isolated and cultured as described previously.[ 47 , 48 ]
Porcine Urinary Bladders
Porcine urinary bladders were purchased from the local slaughterhouse (Ulmer Fleisch GmbH) and the following experiments were approved by the Regierungspraesidium Tuebingen (license 1406). Preparation of the urinary bladders was performed as previously described.[ 26 , 28 ] Briefly, bladders were opened, cleaned from urine and adjacent fat. After three extensive washing steps in PBS supplemented with 3% antibiotic‐antimycotic (300 IU mL−1 penicillin, 300 µg mL−1 streptomycin, 750 ng mL−1 amphotericin B; Sigma‐Aldrich), bladders were incubated with 0.5% dispase II (Sigma‐Aldrich) in PBS overnight at 4 °C. The next day, epithelial cells forming the bladder urothelium were mechanically removed. An example of successful de‐epithelialization is depicted in Figure S1A,B in the Supporting Information. Remaining bladder tissue was minced into pieces of ≈2 cm2. They were placed in 40 µm cell strainers (Corning) and incubated as air–liquid interface cultures in ROTI Cell Hanks’ BSS (Carl Roth) overnight at 37 °C under 5% (v/v) CO2 atmosphere. Bladder pieces were then transferred to new wells before being used for experiments. For co‐ and tricultures involving patient‐derived organoids, bladder pieces were sterilized by treatment with 0.1% peracetic acid (Acros Organics) overnight at 4 °C after de‐epithelialization.[ 27 ] After extensive washings in PBS, bladders were transferred to new wells for experiments.
Culture of Human Pluripotent Stem Cell‐Derived Pancreatic Cells on Porcine Urinary Bladders
On the day of cell seeding, bladder‐containing cell strainers were placed for at least 2 h in cell type specific cell culture medium which was also used for resuspending cells before seeding. Two 5 mm diameter silicon rings were then placed on each bladder piece and were allowed to settle for at least 2 h at 37 °C under 5% (v/v) CO2 atmosphere. Once harvested, pancreatic progenitors were resuspended in BE3 medium supplemented with 5% FBS and 10 × 10−6 m Y27632. An equal amount of GFR‐Matrigel was added before depositing 30 µL of cell suspension onto bladders (3 to 5 × 105 cells per ring). Supplemented BE3 medium was then used to feed air–liquid interface cultures. Cells were propagated at 37 °C under 5% (v/v) CO2 and 21% (v/v) O2 atmosphere for three weeks. Pancreatic duct‐like organoids were harvested and resuspended in ductal differentiation medium supplemented with Y27632.[ 7 ] Intact organoids were seeded onto bladders in a total volume of 30 µL of 1:1 phase II medium (as previously described;[ 7 ] Table S1, Supporting Information): GFR‐Matrigel (1.0 to 2.5 × 105 cells per ring). KRASG12D expression was induced by adding 3 µg mL−1 doxycycline hyclate (Sigma‐Aldrich) to the medium. Cells were propagated at 37 °C under 5% (v/v) CO2 and 21% (v/v) O2 atmosphere for three weeks.
Culture of Patient‐Derived Organoids, Human Pancreatic Stellate Cells, and Peripheral Blood Mononuclear Cells on Porcine Urinary Bladders
Prior to seeding, human patient‐derived organoids were harvested from GFR‐Matrigel using 1 mg mL−1 collagenase/dispase (Roche) in DMEM‐F12, GlutaMAX (Thermo Fisher Scientific). Intact organoids were then seeded onto porcine urinary bladders (600–5000 PDOs per bladder) in DMEM‐F12, GlutaMAX supplemented with 10% FBS, 10 × 10−6 m Y27632, and 50% GFR‐Matrigel in a total volume of 30 µL per bladder. For coculture experiments with PaSteCs and PDOs, cells were assembled at 100:1 ratio prior to seeding. Cells were then seeded onto porcine urinary bladders in DMEM‐F12, GlutaMAX supplemented with 10% FBS, 10 × 10−6 m Y27632 and 50% GFR‐Matrigel. For triculture experiments with PBMCs, PaSteCs, and PDOs, cells were assembled at 50:15:1 ratio prior to seeding. Cells were then seeded onto porcine urinary bladders in RPMI1640 with l‐glutamine (Gibco) supplemented with 5% FBS, 5% human serum (Sigma‐Aldrich), 1 × 10−3 m sodium pyruvate (Sigma‐Aldrich), 1 × nonessential amino acids (Sigma‐Aldrich), and 10 × 10−6 m Y27632.
Culture and Treatment of Murine Cancer Cells on Porcine Urinary Bladders
Briefly, 167 000 murine cancer cells were seeded per ring in DMEM‐F12 medium supplemented with 10% FBS and 50% GFR‐Matrigel in a total volume of 30 µL. Twenty‐four hours after seeding, cells were treated for 48 h with 3.7 × 10−6 m olaparib, 1.1 × 10−6 m irinotecan, or the vehicle control respectively. Chemotherapeutics were added to the medium beneath the bladder. Olaparib (AZD2281, KU‐00594) and irinotecan (CPT‐11) were purchased from Selleckchem.
Histology and Immunostainings
All histological experiments were performed as previously described.[ 7 ] Briefly, porcine urinary bladder samples were fixed in 4% formaldehyde for 24 h at 4 °C, subjected to automated serial dehydration series before being embedded into paraffin. Hematoxylin and eosin (H&E) stainings and immunostainings were performed as described recently.[ 7 ] Primary and secondary antibodies are listed in Table S2 in the Supporting Information. Bright‐field images were acquired using an HCX PLAN APO 20×/0.7 (Leica) objective mounted on a Leica DM5500B microscope (Leica) equipped with a Leica DMC5400 camera and Leica Application Suite software (Leica). Immunofluorescence images were obtained at room temperature using a Plan Apo 20×/0.8 objective (Carl Zeiss) mounted on a Zeiss Axioscope2 microscope (Carl Zeiss) equipped with an Axiocam 702 and ZEN3.1 imaging software (Carl Zeiss). Acquired pictures were subsequently analyzed using ImageJ software (National Institutes of Health).
RNA Extraction and Quantitative RT‐PCR
PDOs on PUB were harvested 5 days after seeding. Bladders were taken from cell strainers and briefly washed in PBS. Rings were removed from bladders and placed into accutase solution (Sigma‐Aldrich). Bladders were placed upside‐down into accutase and incubated for 1 h at 37 °C on a shaker. RNAs were extracted from harvested cells with the GeneJET RNA Purification Kit (Thermo Fisher Scientific). cDNA synthesis was performed with the iScript cDNA Synthesis Kit (Bio‐Rad). Quantitative real‐time (qRT) PCR was done using the StepOnePlus real‐time PCR System (Applied Biosystems) and iTaq Universal SYBR Green Supermix (Bio‐Rad). Data was standardized to XS13 (as housekeeping gene). Primer sequences are given in Table S6 in the Supporting Information.
Analysis of Publicly Available RNA‐Sequencing Datasets
RNA sequencing data were accessed in the human protein atlas (Protein Atlas version 20.1, https://www.proteinatlas.org/ [ 65 ]). A search within the atlas was conducted for the following terms: “collagen,” “laminin,” “emilin,” “tenascin,” “elastin,” “fibronectin,” “fibrillin.” A customized data set was downloaded for each result, including the normalized tissue RNA expression (NX) of different organs compiled from three different data‐sets. After manually checking the data sets for genes not relevant within the searching terms, a table was generated to include the RNA expression levels of all intended ECM components as indicated before. Extracted data are provided in Table S7 in the Supporting Information. The table was imported into R (Version 4.0.4) and analyzed by a principal component analysis (PCA) with the “prcomp” function. Results were plotted by using the “ggbiplot” (Version 0.55) within the “ggplot2” package (Version 3.3.3). For the generation of heatmaps, the “pheatmap” package (Version 1.0.12) was used with scaling settings to the row factor and clustering following the “ward.D2” method.
Statistical Analysis
GraphPad Prism 9.3.1 software was used for statistical analysis. For analysis of qRT‐PCR data fold expression of XS13 was calculated for each gene. mRNA fold change expression levels were plotted in GraphPad Prism to generate a heatmap. PCA was calculated in GraphPad Prism 9.3.1 based on the mean values of the respective RNA expression levels. For immunostaining evaluation, the percentage of positive cells was calculated for each marker. A minimum of 100 cells per condition were counted. Quantifications were performed on all grafts included in the study (organoids were counted as CK‐7 positive if at least one cell within expressed CK‐7. Between 31 to 122 organoids have been evaluated in five independent experiments). Bar graph data are presented as mean ± SD or SEM (indicated in figure legends). Significance was evaluated with Mann–Whitney U test. Contingency table was analyzed with Fisher's exact test.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Conceptualization was done by M.K.M., M.B., C.B., C.G., and A.K.; methodology was provided by M.K.M., M.B., F.A., A.A., J.K., J.M., L.S., V.Z., C.B., C.G., and A.K.; software and validation was provided by M.K.M. and M.B.; formal analysis was done by M.K.M., M.B., F.A., L.K., F.P., S.S. and J.G.; investigation was done by M.K.M., M.B., F.A., J.K., J.M., and S.S.; resources were provided by M.K.M., M.B., A.A., Y.R., M.Hä., M.H., C.B., C.G., and A.K.; data curation was done by M.K.M. and M.B.; original draft preparation was done by M.K.M. and J.G.; review and editing was done by M.K.M., M.B., F.A., F.W., E.R., F.Z., N.A., T.S., M.H., C.B., C.G., J.G., and A.K.; visualization was done by M.K.M.; supervision was done by C.B., C.G., J.G., and A.K.; project administration was done by M.K.M., C.G., and A.K.; funding acquisition was done by M.K.M., T.S., M.H., C.B., C.G., and A.K. All authors have read and agreed to the published version of the manuscript.
Supporting information
Supporting Information
Supplemental Table 1
Acknowledgements
Kuhn Elektro‐Technik GmbH is acknowledged for supporting the research to fight pancreatic cancer. The authors thank Katrin Köhn, Aref Saed, Ralf Köhntop, Ulrike Mayr‐Beyrle, Rashmi Bijegatte, Alica K. Beutel, and Lukas Perkhofer (DFG PE 3337/1‐1) for excellent technical support and helpful discussions. The main funding was provided by the Deutsche Forschungsgemeinschaft (DFG), Sachbeihilfe“ KL2544/7‐1, and Heisenberg‐Programm“ KL2544/6‐1. A.K. and T.S. are PIs in the HEIST RTG which was funded by the DFG GRK 2254/1. Additional funding was acquired by A.K. from the German Cancer Aid (111879), the DFG (K.L. 2544/1‐1, 1–2, 5‐1, 8‐1) and the Else Kröner‐Fresenius‐Stiftung (Excellence Grant to AK). M.H. received funding from a first application grant from Else Kröner‐Fresenius‐Stiftung (2020_EKEA.48). A.K. and T.S. are speakers in the Else Kröner‐Fresenius‐Stiftung Foschungskolleg. M.K.M., Y.R. and V.Z. are Clinician Scientists within the Else Kröner‐Fresenius‐Stiftung Forschungskolleg. M.K.M. received funding from Ulm University in the Clinician Scientist Program. F.A. received the postdoctoral start‐up funding from the DFG GRK 2254/1 (HEIST RTG). The Core Facility Organoids is funded by the Medical Faculty of Ulm University.
After initial online publication, M.K.M. was listed as a Clinician Scientist within the Else Kröner‐Fresenius‐Stiftung Forschungskolleg on April 4, 2022, due to a previous omission. The editorial office apologizes for any inconvenience caused.
Open access funding enabled and organized by Projekt DEAL.
Melzer M. K., Breunig M., Arnold F., Wezel F., Azoitei A., Roger E., Krüger J., Merkle J., Schütte L., Resheq Y., Hänle M., Zehe V., Zengerling F., Azoitei N., Klein L., Penz F., Singh S. K., Seufferlein T., Hohwieler M., Bolenz C., Günes C., Gout J., Kleger A., Organoids at the PUB: The Porcine Urinary Bladder Serves as a Pancreatic Niche for Advanced Cancer Modeling. Adv. Healthcare Mater. 2022, 11, 2102345. 10.1002/adhm.202102345
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Rahib L., Smith B. D., Aizenberg R., Rosenzweig A. B., Fleshman J. M., Matrisian L. M., Cancer Res. 2014, 74, 2913. [DOI] [PubMed] [Google Scholar]
- 2. Beutel A. K., Schütte L., Scheible J., Roger E., Müller M., Perkhofer L., Kestler A. M. T. U., Kraus J. M., Kestler H. A., Barth T. F. E., Lemke J., Kornmann M., Ettrich T. J., Gout J., Seufferlein T., Kleger A., Cancers 2021, 13, 2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frappart P. O., Walter K., Gout J., Beutel A. K., Morawe M., Arnold F., Breunig M., Barth T. F. E., Marienfeld R., Schulte L., Ettrich T., Hackert T., Svinarenko M., Rösler R., Wiese S., Wiese H., Perkhofer L., Müller M., Lechel A., Sainz B., Hermann P. C., Seufferlein T., Kleger A., United Eur. Gastroenterol. J. 2020, 8, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Driehuis E., Van Hoeck A., Moore K., Kolders S., Francies H. E., Gulersonmez M. C., Stigter E. C. A., Burgering B., Geurts V., Gracanin A., Bounova G., Morsink F. H., Vries R., Boj S., Van Es J., Offerhaus G. J. A., Kranenburg O., Garnett M. J., Wessels L., Cuppen E., Brosens L. A. A., Clevers H., Proc. Natl. Acad. Sci. USA 2019, 116, 26580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siegel R. L., Miller K. D., Fuchs H. E., Jemal A., Ca‐Cancer J. Clin. 2021, 71, 7. [DOI] [PubMed] [Google Scholar]
- 6. Hohwieler M., Illing A., Hermann P. C., Mayer T., Stockmann M., Perkhofer L., Eiseler T., Antony J. S., Müller M., Renz S., Kuo C. C., Lin Q., Sendler M., Breunig M., Kleiderman S. M., Lechel A., Zenker M., Leichsenring M., Rosendahl J., Zenke M., Sainz B., Mayerle J., Costa I. G., Seufferlein T., Kormann M., Wagner M., Liebau S., Kleger A., Gut 2017, 66, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Breunig M., Merkle J., Wagner M., Melzer M. K., Barth T. F. E., Engleitner T., Krumm J., Wiedenmann S., Cohrs C. M., Perkhofer L., Jain G., Krüger J., Hermann P. C., Schmid M., Madácsy T., Varga Á., Griger J., Azoitei N., Müller M., Wessely O., Robey P. G., Heller S., Dantes Z., Reichert M., Günes C., Bolenz C., Kuhn F., Maléth J., Speier S., Liebau S., et al., Cell Stem Cell 2021, 28, 1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang L., Desai R., Conrad D. N., Leite N. C., Akshinthala D., Lim C. M., Gonzalez R., Muthuswamy L. B., Gartner Z., Muthuswamy S. K., Cell Stem Cell 2021, 28, 1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Merkle J., Breunig M., Schmid M., Allgöwer C., Krüger J., Melzer M. K., Bens S., Siebert R., Perkhofer L., Azoitei N., Seufferlein T., Heller S., Meier M., Müller M., Kleger A., Hohwieler M., Cancers 2021, 13, 5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Breunig M., Merkle J., Melzer M. K., Heller S., Seufferlein T., Meier M., Hohwieler M., Kleger A., STAR Protoc. 2021, 2, 100913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wiedenmann S., Breunig M., Merkle J., von Toerne C., Georgiev T., Moussus M., Schulte L., Seufferlein T., Sterr M., Lickert H., Weissinger S. E., Möller P., Hauck S. M., Hohwieler M., Kleger A., Meier M., Nat. Biomed. Eng. 2021, 5, 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boj S. F., Hwang C.‐I., Baker L. A., Chio I. I. C., Engle D. D., Corbo V., Jager M., Ponz‐Sarvise M., Tiriac H., Spector M. S., Gracanin A., Oni T., Yu K. H., van Boxtel R., Huch M., Rivera K. D., Wilson J. P., Feigin M. E., Öhlund D., Handly‐Santana A., Ardito‐Abraham C. M., Ludwig M., Elyada E., Alagesan B., Biffi G., Yordanov G. N., Delcuze B., Creighton B., Wright K., Park Y., et al., Cell 2015, 160, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seino T., Kawasaki S., Shimokawa M., Tamagawa H., Toshimitsu K., Fujii M., Ohta Y., Matano M., Nanki K., Kawasaki K., Takahashi S., Sugimoto S., Iwasaki E., Takagi J., Itoi T., Kitago M., Kitagawa Y., Kanai T., Sato T., Cell Stem Cell 2018, 22, 454. [DOI] [PubMed] [Google Scholar]
- 14. Philippi A., Heller S., Costa I. G., Senée V., Breunig M., Li Z., Kwon G., Russell R., Illing A., Lin Q., Hohwieler M., Degavre A., Zalloua P., Liebau S., Schuster M., Krumm J., Zhang X., Geusz R., Benthuysen J. R., Wang A., Chiou J., Gaulton K., Neubauer H., Simon E., Klein T., Wagner M., Nair G., Besse C., Dandine‐Roulland C., Olaso R., et al., Nat. Med. 2021, 27, 1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haykal M. M., Nahmias C., Varon C., Martin O. C. B., Front. Cell Dev. Biol. 2020, 8, 606039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Byrne A. T., Alférez D. G., Amant F., Annibali D., Arribas J., Biankin A. V., Bruna A., Budinská E., Caldas C., Chang D. K., Clarke R. B., Clevers H., Coukos G., Dangles‐Marie V., Gail Eckhardt S., Gonzalez‐Suarez E., Hermans E., Hidalgo M., Jarzabek M. A., De Jong S., Jonkers J., Kemper K., Lanfrancone L., Mælandsmo G. M., Marangoni E., Marine J. C., Medico E., Norum J. H., Palmer H. G., Peeper D. S., et al., Nat. Rev. Cancer 2017, 17, 254. [DOI] [PubMed] [Google Scholar]
- 17. Garcia P. L., Miller A. L., Yoon K. J., Cancers 2020, 12, 1327.32456018 [Google Scholar]
- 18. Miyabayashi K., Baker L. A., Deschênes A., Traub B., Caligiuri G., Plenker D., Alagesan B., Belleau P., Li S., Kendall J., Jang G. H., Kawaguchi R. K., Somerville T. D. D., Tiriac H., Il Hwang C., Burkhart R. A., Roberts N. J., Wood L. D., Hruban R. H., Gillis J., Krasnitz A., Vakoc C. R., Wigler M., Notta F., Gallinger S., Park Y., Tuveson D. A., Cancer Discovery 2020, 10, 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lai B. F. L., Lu R. X. Z., Hu Y., Davenport Huyer L., Dou W., Wang E. Y., Radulovich N., Tsao M. S., Sun Y., Radisic M., Adv. Funct. Mater. 2020, 30, 2000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim E., Choi S., Kang B., Kong J. H., Kim Y., Yoon W. H., Lee H. R., Kim S. E., Kim H. M., Lee H. S., Yang C., Lee Y. J., Kang M., Roh T. Y., Jung S., Kim S., Ku J. H., Shin K., Nature 2020, 588, 664. [DOI] [PubMed] [Google Scholar]
- 21. Nowak‐Sliwinska P., Segura T., Iruela‐Arispe M. L., Anigogenesis 2014, 17, 779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen L., Wang S., Feng Y., Zhang J., Du Y., Zhang J., Van Ongeval C., Ni Y., Li Y., Cells 2021, 10, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berger C., Bjørlykke Y., Hahn L., Mühlemann M., Kress S., Walles H., Luxenhofer R., Ræder H., Metzger M., Zdzieblo D., Biomaterials 2020, 244, 119766. [DOI] [PubMed] [Google Scholar]
- 24. Elebring E., Kuna V. K., Kvarnström N., Sumitran‐Holgersson S., J. Tissue Eng. 2017, 8, 2041731417738145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuna V. K., Kvarnström N., Elebring E., Holgersson S. S., J. Vis. Exp. 2018, 140, 58302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schneider L., Liu J., Zhang C., Azoitei A., Meessen S., Zheng X., Cremer C., Gorzelanny C., Kempe‐Gonzales S., Brunner C., Wezel F., Bolenz C., Gunes C., John A., Int. J. Mol. Sci. 2021, 22, 5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rosario D. J., Reilly G. C., Salah E. A., Glover M., Bullock A. J., MacNeil S., Regener. Med. 2008, 3, 145. [DOI] [PubMed] [Google Scholar]
- 28. Wezel F., Lustig J., Azoitei A., Liu J., Meessen S., Najjar G., Zehe V., Faustmann P., Zengerling F., John A., Martini T., Bolenz C., Günes C., Int. J. Mol. Sci. 2021, 22, 2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frantz C., Stewart K. M., Weaver V. M., J. Cell Sci. 2010, 123, 4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baskin L., Howard P. S., Macarak E., J. Urol. 1993, 150, 601. [DOI] [PubMed] [Google Scholar]
- 31. Aitken K. J., Bägli D. J., Nat. Rev. Urol. 2009, 6, 596. [DOI] [PubMed] [Google Scholar]
- 32. Schussler M. H., Skoudy A., Ramaekers F., Real F. X., Am. J. Pathol. 1992, 140, 559. [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou Q., Law A. C., Rajagopal J., Anderson W. J., Gray P. A., Melton D. A., Dev. Cell 2007, 13, 103. [DOI] [PubMed] [Google Scholar]
- 34. Kasper M., von Dorsche H. H., Stosiek P., Histochemistry 1991, 96, 271. [DOI] [PubMed] [Google Scholar]
- 35. Githens S., J Pediatr Gastroenterol Nutr 1988, 7, 486. [PubMed] [Google Scholar]
- 36. Huang L., Holtzinger A., Jagan I., Begora M., Lohse I., Ngai N., Nostro C., Wang R., Muthuswamy L. B., Crawford H. C., Arrowsmith C., Kalloger S. E., Renouf D. J., Connor A. A., Cleary S., Schaeffer D. F., Roehrl M., Tsao M.‐S. S., Gallinger S., Keller G., Muthuswamy S. K., Nat. Med. 2015, 21, 1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim G. E., Bae H., Park H., Kuan S., Crawley S. C., Ho J. J. L., Kim Y. S., Gastroenterology 2002, 123, 1052. [DOI] [PubMed] [Google Scholar]
- 38. O'Kane G. M., Grunwald B. T., Jang G. H., Masoomian M., Picardo S., Grant R. C., Denroche R. E., Zhang A., Wang Y., Lam B., Krzyzanowski P. M., Lungu I. M., Bartlett J. M. S., Peralta M., Vyas F., Khokha R., Biagi J., Chadwick D., Ramotar S., Hutchinson S., Dodd A., Wilson J. M., Notta F., Zogopoulos G., Gallinger S., Knox J. J., Fischer S. E., Clin. Cancer Res. 2020, 26, 4901. [DOI] [PubMed] [Google Scholar]
- 39. Collisson E. A., Sadanandam A., Olson P., Gibb W. J., Truitt M., Gu S., Cooc J., Weinkle J., Kim G. E., Jakkula L., Feiler H. S., Ko A. H., Olshen A. B., Danenberg K. L., Tempero M. A., Spellman P. T., Hanahan D., Gray J. W., Nat. Med. 2011, 17, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maurer C., Holmstrom S. R., He J., Laise P., Su T., Ahmed A., Hibshoosh H., Chabot J. A., Oberstein P. E., Sepulveda A. R., Genkinger J. M., Zhang J., Iuga A. C., Bansal M., Califano A., Olive K. P., Gut 2019, 68, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bailey P., Chang D. K., Nones K., Johns A. L., Patch A. M., Gingras M. C., Miller D. K., Christ A. N., Bruxner T. J. C., Quinn M. C., Nourse C., Murtaugh L. C., Harliwong I., Idrisoglu S., Manning S., Nourbakhsh E., Wani S., Fink L., Holmes O., Chin V., Anderson M. J., Kazakoff S., Leonard C., Newell F., Waddell N., Wood S., Xu Q., Wilson P. J., Cloonan N., Kassahn K. S., et al., Nature 2016, 531, 47.26909576 [Google Scholar]
- 42. Moffitt R. A., Marayati R., Flate E. L., Volmar K. E., Loeza S. G. H., Hoadley K. A., Rashid N. U., Williams L. A., Eaton S. C., Chung A. H., Smyla J. K., Anderson J. M., Kim H. J., Bentrem D. J., Talamonti M. S., Iacobuzio‐Donahue C. A., Hollingsworth M. A., Yeh J. J., Nat. Genet. 2015, 47, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tu M., Klein L., Espinet E., Georgomanolis T., Wegwitz F., Li X., Urbach L., Danieli‐Mackay A., Küffer S., Bojarczuk K., Mizi A., Günesdogan U., Chapuy B., Gu Z., Neesse A., Kishore U., Ströbel P., Hessmann E., Hahn S. A., Trumpp A., Papantonis A., Ellenrieder V., Singh S. K., Nat. Cancer 2021, 2, 1185. [DOI] [PubMed] [Google Scholar]
- 44. Ho W. J., Jaffee E. M., Zheng L., Nat. Rev. Clin. Oncol. 2020, 17, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haeberle L., Esposito I., Transl. Gastroenterol. Hepatol. 2019, 4, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hosein A. N., Brekken R. A., Maitra A., Nat Rev Gastroenterol Hepatol. 2020, 17, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perkhofer L., Schmitt A., Romero Carrasco M. C., Ihle M., Hampp S., Ruess D. A., Hessmann E., Russell R., Lechel A., Azoitei N., Lin Q., Liebau S., Hohwieler M., Bohnenberger H., Lesina M., Algül H., Gieldon L., Schröck E., Gaedcke J., Wagner M., Wiesmüller L., Sipos B., Seufferlein T., Reinhardt H. C., Frappart P. O., Kleger A., Cancer Res. 2017, 77, 5576. [DOI] [PubMed] [Google Scholar]
- 48. Gout J., Perkhofer L., Morawe M., Arnold F., Ihle M., Biber S., Lange S., Roger E., Kraus J. M., Stifter K., Hahn S. A., Zamperone A., Engleitner T., Müller M., Walter K., Rodriguez‐Aznar E., Sainz B., Hermann P. C., Hessmann E., Müller S., Azoitei N., Lechel A., Liebau S., Wagner M., Simeone D. M., Kestler H. A., Seufferlein T., Wiesmüller L., Rad R., Frappart P. O., et al., Gut 2021, 70, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roger E., Gout J., Arnold F., Beutel A. K., Müller M., Abaei A., Barth T. F. E., Rasche V., Seufferlein T., Perkhofer L., Kleger A., Cells 2020, 9, 2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stanton A. E., Tong X., Yang F., Acta Biomater.. 2019, 96, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Engler A. J., Sen S., Sweeney H. L., Discher D. E., Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- 52. Wrighton P. J., Klim J. R., Hernandez B. A., Koonce C. H., Kamp T. J., Kiessling L. L., Proc. Natl. Acad. Sci. USA 2014, 111, 18126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Below C. R., Kelly J., Brown A., Humphries J. D., Hutton C., Xu J., Lee B. Y., Cintas C., Zhang X., Hernandez‐Gordillo V., Stockdale L., Goldsworthy M. A., Geraghty J., Foster L., O'Reilly D. A., Schedding B., Askari J., Burns J., Hodson N., Smith D. L., Lally C., Ashton G., Knight D., Mironov A., Banyard A., Eble J. A., Morton J. P., Humphries M. J., Griffith L. G., Jørgensen C., Nat. Mater. 2022, 21, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kular J. K., Basu S., Sharma R. I., J. Tissue Eng. 2014, 5, 2041731414557112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karamanos N. K., Theocharis A. D., Piperigkou Z., Manou D., Passi A., Skandalis S. S., Vynios D. H., Orian‐Rousseau V., Ricard‐Blum S., Schmelzer C. E. H., Duca L., Durbeej M., Afratis N. A., Troeberg L., Franchi M., Masola V., Onisto M., FEBS J. 2021, 288, 6850. [DOI] [PubMed] [Google Scholar]
- 56. Sipos B., Frank S., Gress T., Hahn S., Klöppel G., Pancreatology 2009, 9, 45. [DOI] [PubMed] [Google Scholar]
- 57. Jonckheere N., Skrypek N., Van Seuningen I., Cancers 2010, 2, 1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kopp J. L., von Figura G., Mayes E., Liu F. F., Dubois C. L., Morris J. P., Pan F. C., Akiyama H., Wright C. V. E., Jensen K., Hebrok M., Sander M., Cancer Cell 2012, 22, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Remmers N., Anderson J. M., Linde E. M., DiMaio D. J., Lazenby A. J., Wandall H. H., Mandel U., Clausen H., Yu F., Hollingsworth M. A., Clin. Cancer Res. 2013, 19, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Melzer M. K., Arnold F., Stifter K., Zengerling F., Azoitei N., Seufferlein T., Bolenz C., Kleger A., Int. J. Mol. Sci. 2020, 21, 3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim S.‐I., Oceguera‐Yanez F., Sakurai C., Nakagawa M., Yamanaka S., Woltjen K., Induced Pluripotent Stem (iPS) Cells. Methods in Molecular Biology, Humana Press, Vol. 1357, New York, NY: 2016, pp. 111–131. [DOI] [PubMed] [Google Scholar]
- 62. Rao J., Pfeiffer M. J., Frank S., Adachi K., Piccini I., Quaranta R., Araúzo‐Bravo M., Schwarz J., Schade D., Leidel S., Schöler H. R., Seebohm G., Greber B., Cell Stem Cell 2016, 18, 341. [DOI] [PubMed] [Google Scholar]
- 63. Nostro M. C., Sarangi F., Yang C., Holland A., Elefanty A. G., Stanley E. G., Greiner D. L., Keller G., Stem Cell Rep. 2015, 4, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jesnowski R., Fürst D., Ringel J., Chen Y., Schrödel A., Kleeff J., Kolb A., Schareck W. D., Löhr M., Lab. Investig. 2005, 85, 1276. [DOI] [PubMed] [Google Scholar]
- 65. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I. M., Edlund K., Lundberg E., Navani S., Szigyarto C. A. K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., Von Feilitzen K., Forsberg M., et al., Science 2015, 347, 1260419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Table 1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.