

Abstract
The therapeutic use of RNA interference is limited by the inability of siRNA molecules to reach their site of action, the cytosol of target cells. Lipid nanoparticles, including liposomes, are commonly employed as siRNA carrier systems to overcome this hurdle, although their widespread use remains limited due to a lack of delivery efficiency. More recently, nature's own carriers of RNA, extracellular vesicles (EVs), are increasingly being considered as alternative siRNA delivery vehicles due to their intrinsic properties. However, they are difficult to load with exogenous cargo. Here, EV–liposome hybrid nanoparticles (hybrids) are prepared and evaluated as an alternative delivery system combining properties of both liposomes and EVs. It is shown that hybrids are spherical particles encapsulating siRNA, contain EV‐surface makers, and functionally deliver siRNA to different cell types. The functional behavior of hybrids, in terms of cellular uptake, toxicity, and gene‐silencing efficacy, is altered as compared to liposomes and varies among recipient cell types. Moreover, hybrids produced with cardiac progenitor cell (CPC) derived‐EVs retain functional properties attributed to CPC‐EVs such as activation of endothelial signaling and migration. To conclude, hybrids combine benefits of both synthetic and biological drug delivery systems and might serve as future therapeutic carriers of siRNA.
Keywords: drug delivery, exosomes, extracellular vesicles, liposomes, nucleic acids, siRNA
The therapeutic use of RNA interference is limited by the inability of siRNA molecules to reach the site of action. In this work, extracellular vesicle (EV)–liposome hybrid nanoparticles are evaluated as siRNA drug delivery vehicle, and it is shown that these hybrid nanoparticles functionally deliver siRNA and at the same time retain important biological functions attributed to EVs.
1. Introduction
RNA interference (RNAi) is a naturally occurring process through which messenger RNA (mRNA) translation is inhibited in a sequence‐specific manner. This process is mediated by short interfering RNA (siRNA) molecules.[ 1 , 2 ] The ability of RNAi to specifically inhibit translation of (pathological) proteins makes it a powerful therapeutic agent applicable in various areas of disease.[ 3 ] However, effective delivery of siRNA molecules is limited as unmodified siRNA molecules are instable, immunogenic, and cannot reach their site of action, i.e. the cytosol of target cells.[ 4 , 5 , 6 , 7 ] In order to protect and deliver siRNA into target cells, several RNA delivery systems have been developed, including metabolically stable GalNAc‐conjugates and lipid‐based delivery systems.[ 3 ] However, these systems have limitations since their tissue distribution and cellular uptake is mainly limited to specific subsets of cells in the spleen and liver while only 1–2% of the delivered siRNA reaches the cytosol. In addition, the lipids used in the formulation of lipid‐based delivery systems can also show hepatotoxicity.[ 8 , 9 , 10 , 11 , 12 ]
The delivery of RNA molecules by naturally occurring RNA carriers, called extracellular vesicles (EVs), is an alternative to current delivery methods. EVs are small, lipid membrane vesicles secreted by a wide variety of cells, and contain biologically active complex molecules such as RNA, proteins, lipids, and sugars.[ 13 , 14 ] EVs comprise a heterogeneous group of vesicles of different intracellular origins. At least two different subtypes can be classified based on their cellular biogenesis: exosomes and ectosomes, the latter also being referred to as microvesicles.
Exosomes (30–100 nm) originate in the endosomal pathway where inward budding of the endosomal membrane results in the formation of intraluminal vesicles which upon release are referred to as exosomes. Ectosomes (50–1000 nm) are released by the cell via direct pinching of the plasma membrane at the cell surface. EVs carry different RNA molecules such as mRNA and miRNA which can be functionally transferred to recipient cells and have been suggested to play important roles in (patho)physiological processes.[ 15 , 16 , 17 ] It is also possible to load non‐naturally occurring RNA molecules, such as siRNA and sgRNA, in EVs to be functionally transferred to a recipient cell.[ 18 , 19 , 20 ]
As nature's own carriers of RNA, EVs might be an attractive alternative carrier system for therapeutic RNA as they have multiple potential benefits over current delivery vehicles in terms of delivery efficacy, intrinsic specific cell targeting properties, and toxicity/immunogenicity.[ 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 ] Interestingly, apart from the possible benefits for RNA delivery directly, EVs may, as intrinsically biologically active entities, induce additional regenerative or therapeutic effects such as induction of cell proliferation, neovascularization, immunomodulation, and prevention of cell death.[ 27 ] Opposite to the beneficial effects of EVs, some risks might be associated with the use of EVs, which originate from tumor cells as they have been implicated in cancer metastasis.[ 21 ]
Although EVs bear great potential as RNA delivery vehicles, their clinical development is hampered by a low loading efficiency of exogenous RNA molecules.[ 19 , 28 , 29 ] Multiple methods have been developed to achieve RNA loading into EVs either via loading during vesicle formation or after vesicle isolation.[ 30 , 31 ] However, for most methods, reported loading capacities are still several orders of magnitude lower compared to that of synthetic delivery systems.[ 31 ] Therefore, an alternative approach for active loading of RNA therapeutics in EVs is required to capitalize on the beneficial properties of EVs as drug delivery vehicle.
Here, we propose a biomimetic approach to generate semisynthetic hybrid nanoparticles based on EVs and liposomes termed EV–liposome hybrid nanoparticles (hybrids), thereby combining the beneficial properties of both liposomes and EVs in a single carrier of siRNA. To this end, we combined SKOV3 EVs and liposomes to produce hybrids by lipid‐film hydration followed by extrusion. We physicochemically characterized the particles and analyzed the incorporation of EV‐associated membrane proteins in the hybrids via an antibody‐based bead capture assay. Then, uptake, gene‐silencing efficacy, and toxicity of the hybrids were evaluated and compared to that of liposomes in multiple cell‐lines. Finally, we used EVs derived from cardiac progenitor cells (CPCs) to generate hybrid nanoparticles and assessed whether the functional regenerative properties of CPC EVs were retained. The data show that we successfully produced hybrid nanoparticles, which functionally deliver RNA and retain functional properties attributed to EVs.
2. Results
2.1. Hybrids Carry Physicochemical Features of Both Liposomes and EVs
First, SKOV3 EVs were isolated from conditioned medium of SKOV3 cells via an established size‐exclusion chromatography protocol.[ 32 ] The protein composition of the isolated EVs was then analyzed by western blot to verify the enrichment of specific EV‐marker proteins as compared to cell lysate. To this end, we analyzed expression of the transmembrane proteins CD81, CD63, and CD9 and luminal proteins Alix and TSG101. As a negative control, expression of endoplasmic reticulum protein Calnexin was analyzed. CD63, CD81, CD9, and Alix were enriched in EVs as compared to cell lysate (Figure 1A). Expression of TSG101 and Actin in EVs was comparable to that in cell lysate while Calnexin was clearly negatively enriched. EV purity, as determined by the number of particles per µg protein was found to be consistent among isolations (Figure S1, Supporting Information).[ 33 ] Mean and mode EV size was determined by nanoparticle tracking analysis (NTA) and found to be 100 and 75 nm, respectively. EV size, as determined by dynamic light scattering (DLS), was slightly higher at 150 nm with a polydispersity index (PDI) of ≈0.2 (Figure 1B–D). The surface charge (zeta potential) of EVs was negative, −18 mV, as measured by laser Doppler electrophoresis (Figure 1E). Cryo‐electron microscopy revealed the typical spherical, unilamellar morphology of EVs (Figure 1F). All together, these analyses confirmed successful isolation of EVs from conditioned medium of SKOV3 cells.[ 14 ]
Figure 1.
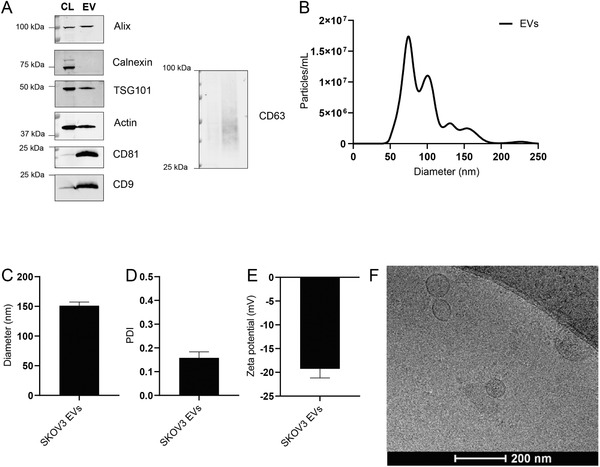
Physicochemical characterization of SKOV3 EVs. A) Western blot analysis of EV protein markers (Alix, TSG101, CD81, CD9, and CD63) and EV‐negative markers (Calnexin) in SKOV3 cell lysate (CL) and SKOV3 EVs (EV). B) Size distribution of EVs as determined by NTA. C) Average diameter of EVs as determined by dynamic light scattering. D) Polydispersity index of EVs as measured by dynamic light scattering. E) Surface charge (zeta potential) of EVs as measured by laser Doppler electrophoresis. F) Cryo‐electron microscopy image of EVs isolated from SKOV3 cells. Data are shown as mean ± SD (n = 3, technical replicates).
These EVs were then used for the production of hybrids encapsulating siRNA via lipid film hydration and subsequent extrusion. Liposomes and hybrids were prepared with DLin‐MC3‐DMA:1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphocholine (DPPC):cholesterol:18:1 Biotinyl PE:DMG‐PEG in a molar ratio of 0.3:0.3:0.355:0.015:0.03 and processed to generate a lipid film. This lipid film was hydrated with siRNA to form siRNA loaded liposomes (Figure 2A,B). For preparation of hybrids, SKOV3 EVs were added at the hydration step at two different ratios of EV‐protein to total synthetic lipid (w/w), 1:100 and 1:50, and subsequently extruded to produce hybrids (Figure 2C,D).
Figure 2.
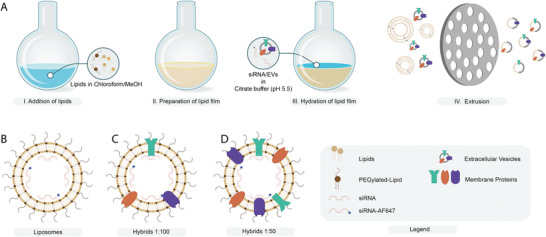
Production of liposomes and hybrids. A) Schematic illustration of hybrid production via thin‐film hydration and extrusion. B–D) Schematic illustration of liposomes and hybrids encapsulating a mixture of fluorescent and nonfluorescent siRNA. Hybrids are produced at different protein‐to‐lipid ratios (w/w): C) 1:100 and D) 1:50.
The particles were then analyzed for their physicochemical properties to evaluate the influence of increasing numbers of EVs in the formulation on size, PDI, zeta potential, siRNA encapsulation efficiency, and particle morphology (Figure 3 ). The size and PDI of liposomes and hybrids were analyzed by DLS and NTA. The average size was close to 150 nm for all formulations as measured by DLS and ≈ 100 nm as measured by NTA (Figure 3A,E). PDI seemed to increase slightly as the amount of EV material in the formulation was increased. However, this increase was not statistically significant (Figure 3B). We did observe that the zeta potential slightly decreased for hybrids as compared to liposomes but no difference was found between hybrids incorporating EVs at a ratio of 1:100 or 1:50 (Figure 3C). The decreased surface charge of hybrids could be explained by the incorporation of the negatively charged EV membrane into the newly formed hybrid nanoparticle leading to a decrease in zeta potential. The encapsulation efficiency of siRNA reduced with increasing amounts of EVs in the formulation and decreased from ≈80% for liposomes to only 50% for hybrids (1:50) (Figure 3D). Furthermore, we quantified the overall yield of each production process in terms of siRNA and cholesterol and found that while the yield for liposomes was ≈50% of both siRNA and cholesterol, the yield was slightly decreased for both hybrid formulations. Here, the influence of EV cholesterol content on the overall amount of cholesterol in the formulation was limited (0.6% and 1.2% in the 1:100 and 1:50 hybrid formulations, respectively) as EVs contained only ±0.13 µg cholesterol per µg protein (Figure S2, Supporting Information). Cryo‐electron microscopy revealed that the morphology of the nanoparticles was spherical and that all formulations consisted of unilamellar nanoparticles (Figure 3F–H).
Figure 3.
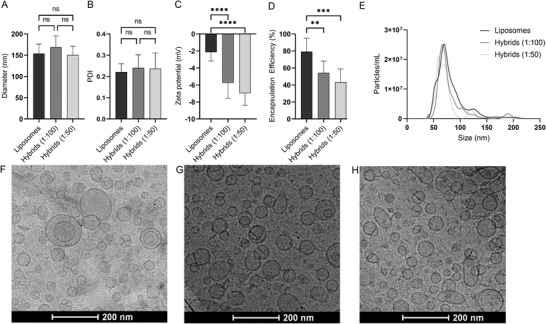
Physicochemical characterization of liposomes and hybrids. A) Nanoparticle size as determined by DLS. B) Polydispersity index of nanoparticles as determined by DLS. C) Zeta potential of nanoparticles as determined by laser Doppler electrophoresis. D) RNA encapsulation efficiency of liposomes and hybrids. E) Nanoparticle size as determine by NTA. Nanoparticle morphology as determined by cryogenic electron microscopy of F) liposomes, G) hybrids (1:100), and H) hybrids (1:50). Mean + SD are displayed. n = 8–10 (biological replicates), one‐way ANOVA with Tukey's post‐test, ns = not significant, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
In order to verify successful hybrid formation, we next evaluated the presence of siRNA and synthetic lipids in liposomes and hybrids captured using magnetic beads coated with EV‐enriched targets, including CD9, CD81, or CD63. We hypothesized that only after formation of EV–liposome hybrids, synthetic lipids and siRNA (AF647 labeled) could be detected on the beads (Figure 4A). For all beads, a clear increase in siRNA‐AF647 signal was observed for hybrid samples as compared to liposomes, which shows that only hybrids, but not liposomes, contain tetraspanins that can be captured the beads (Figure 4B). We also observed that an increase in the number of EVs used in the formulation resulted in a higher siRNA‐AF647 signal.
Figure 4.
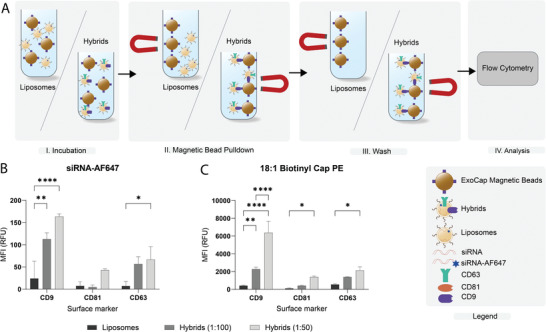
Bead capture analysis of siRNA‐AF647 and 18:1 Biotinyl Cap PE in liposomes and hybrids on beads targeting CD9, CD63, or CD81. A) Schematic illustration of bead‐capture assay. B) Flow cytometric analysis of siRNA‐AF647 on ExoCap beads. Nanoparticles were incubated with beads targeting a single epitope, CD9, CD63, or CD81, washed and analyzed. C) Flow cytometric analysis of 18:1 Biotinyl Cap PE on ExoCap beads. Data are representative of three independent experiments and expressed as mean ± SD (n = 3, technical replicates), one‐way ANOVA with Tukey's post‐hoc test, * p < 0.05, ** p < 0.01, and **** p < 0.0001.
Next, we investigated whether the incorporation of synthetic lipids in hybrids could also be detected. We prepared liposomes and hybrids containing 0.2 mol% 18:1 Biotinyl Cap PE and performed a bead‐pulldown with additional staining using streptavidin‐PE. We found that 18:1 Biotinyl Cap PE was also successfully incorporated in the hybrids given the clear increase in PE‐signal for hybrids. Moreover, this experiment confirmed that nonspecific binding of liposomes to the beads is very limited given the low PE‐signal for the liposome sample (Figure 4C). In addition, the observed trend in PE signal corresponds to that of AF647‐siRNA where the signal on CD9 beads is slightly higher compared to CD63 and CD81. All together, these results indicate that we successfully produced EV–liposome hybrid nanoparticles carrying EV surface proteins and synthetic lipids while simultaneously complexing siRNA.
2.2. Cellular Uptake of Hybrids Is Dependent on the EV‐to‐Liposome Ratio and Differs per Cell Type
Next, we evaluated the cellular internalization efficiency of liposomes and hybrids as this is an important first step in the cytosolic delivery of siRNA. We incubated three different cell types—SKOV3, HEK293T, and U87‐MG—for 4 h with liposomes and hybrids (1:100 and 1:50) and analyzed siRNA‐AF647 uptake by flow cytometry (Figure 5 ). Uptake of hybrid (1:100) nanoparticles in all three cell types was decreased as compared to liposomes. This effect was found to be statistically significant in HEK293T and U87‐MG cells. Interestingly, when more EV components were incorporated in hybrids (1:50), cellular uptake increased again, but only at statistically significant levels in HEK293T and U87 cells. Almost no nanoparticle uptake was seen at 4 °C which confirmed the effects seen are a result of active uptake processes (Figure S3, Supporting Information). The differences in cellular uptake implicate that the cellular internalization of hybrids varies per cell type and that the uptake is affected by the amount of SKOV3 EVs incorporated in the hybrid formulation.
Figure 5.
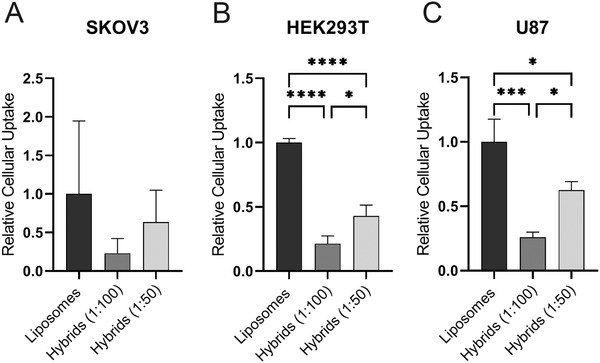
Cellular uptake of liposomes, hybrids (1:100), and hybrids (1:50) in A) SKOV3‐dluc, B) HEK293T‐dluc, and C) U87‐MG‐dluc. Cells were incubated for 4 h at 37 °C at a concentration of 25 × nm siRNA and cellular uptake was measured by flow cytometry. Data are representative of three independent experiments and expressed as mean ± SD (n = 3, technical replicates), one‐way ANOVA with Tukey's post‐hoc test, * p < 0.05, *** p < 0.001, and **** p < 0.0001.
2.3. Hybrids Show Limited Toxicity and Functionally Deliver siRNA to Multiple Cell Types
For successful application of liposomes and hybrids in RNA delivery, particles must be biocompatible and nontoxic. Therefore, we analyzed the toxicity of the nanoparticles using a cell viability assay. In SKOV3 cells, liposomes showed a dose‐dependent decrease in cell viability whereas this effect was not observed for hybrids (1:100 and 1:50) indicating increased biocompatibility of hybrids as compared to liposomes. A difference in the effect on cell viability between liposomes and hybrids was not observed in HEK293T and U87‐MG cells. In HEK293T cells, a small dose‐dependent decrease in cell viability was seen for all nanoparticles with no differences between liposomes and hybrids. In U87‐MG, administration of liposomes and hybrids did not affect the cell viability (Figure 6 ).
Figure 6.
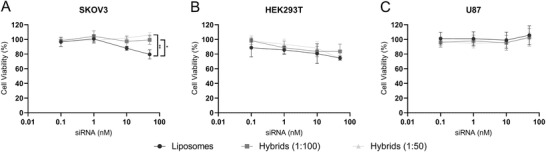
Cell viability of different cell types incubated with liposomes and hybrids as determined by an MTS assay. Cells were incubated with liposomes and hybrids at concentrations ranging from 0.1 to nm siRNA and cell‐viability was analyzed after 48 h. A) SKOV3‐dluc. B) HEK293T‐dluc. C) U87‐MG‐dluc. Data are representative of three independent experiments and expressed as mean ± SD (n = 2–3, technical replicates), one‐way ANOVA with Tukey's post‐hoc test, * p < 0.05 and ** p < 0.01.
Another possible advantageous functional characteristic of EVs as compared to liposomes could be an improved siRNA delivery efficiency.[ 19 , 20 ] Therefore, we evaluated the gene‐silencing efficacy of hybrids as compared to liposomes using a luciferase reporter assay. Liposomes or hybrids encapsulating siRNA targeting firefly luciferase or a nonspecific control siRNA were administered to SKOV3, HEK293T, and U87‐MG (Figure 7 ; Figure S4, Supporting Information). A clear dose‐dependent decrease in firefly luciferase was observed in all cell lines. In SKOV3 and HEK293T cells, the gene‐silencing effect of hybrids (1:100 and 1:50) was lower as compared to liposomes. In contrast, in U87‐MG cells, gene‐silencing efficacy of hybrids (1:100 and 1:50) was similar to that of liposomes despite a lower uptake efficiency, which may point toward more efficient cytosolic siRNA delivery. There was no difference in gene‐silencing efficacy between different EV–liposome hybrids (1:100 and 1:50). This indicates that under these conditions, gene‐silencing efficacy was not critically dependent on the EV‐protein to lipid ratio. All together, these data show that hybrids are able to functionally deliver siRNA to different cell types, although in these experiments, the potency is reduced in SKOV3 and HEK293T cells as compared to liposomes.
Figure 7.
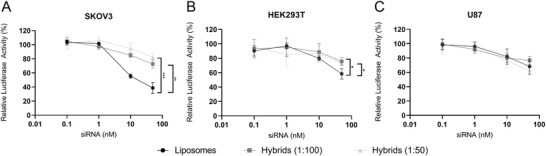
Gene silencing activity in different cell types treated with liposomes and hybrid nanoparticles encapsulating siRNA. Different cell types were incubated with liposomes and hybrids at concentrations ranging from 0.1 to 50 nm siRNA and gene‐silencing was analyzed after 48 h by measurement of luciferase expression. Data are plotted as the normalized ratio of firefly to renilla luciferase expression. A) SKOV3‐dluc. B) HEK293T‐dluc. C) U87‐MG‐dluc. Data are representative of three independent experiments and expressed as mean ± SD (n = 3, technical replicates), one‐way ANOVA with Tukey's post‐hoc test, * p < 0.05, ** p < 0.01, and *** p < 0.001.
2.4. Hybrids Based on Cardiac Progenitor Cell EVs Retain Functional Regenerative Properties
Finally, we investigated whether hybrids preserved the biological activity of EVs. To this end, we generated hybrids with CPC EVs. CPC EVs have been shown to activate endothelial signaling pathways and migration, and activate in vivo angiogenesis.[ 34 , 35 , 36 ] The physicochemical properties were analyzed in a similar manner as for SKOV3 EV derived hybrids (Figure S5, Supporting Information). Liposomes and hybrids had a size of ≈150 nm at a PDI of ≈0.2. Again, the surface charge of hybrids was lower compared to that of liposomes.
We then evaluated the functional capabilities of liposomes and CPC derived hybrids in two functional assays: an Akt phosphorylation assay and a scratch wound healing assay.
Akt is an important factor in signaling pathways involved in proliferation, angiogenesis, differentiation, adhesion, migration, and cell survival, and its phosphorylation is an indicator of functional CPC EV delivery.[ 37 , 38 ] HMEC‐1 cells were serum‐starved and subsequently incubated for 30 min with hybrids (1:100 and 1:50) and EVs as well as PBS and liposomes as negative controls. After treatment, cells were lysed and the ratio of phosphorylated Akt/Akt was analyzed by western blot analysis. PBS and liposomes did not induce phosphorylation of Akt, whereas this was observed for hybrids (1:100 and 1:50) and EVs in a dose‐dependent matter (Figure 8A). When analyzed using densitometry, hybrids (1:100 and 1:50) and EVs induced significantly more phosphorylation of Akt as compared to liposomes (Figure 8B).
Figure 8.
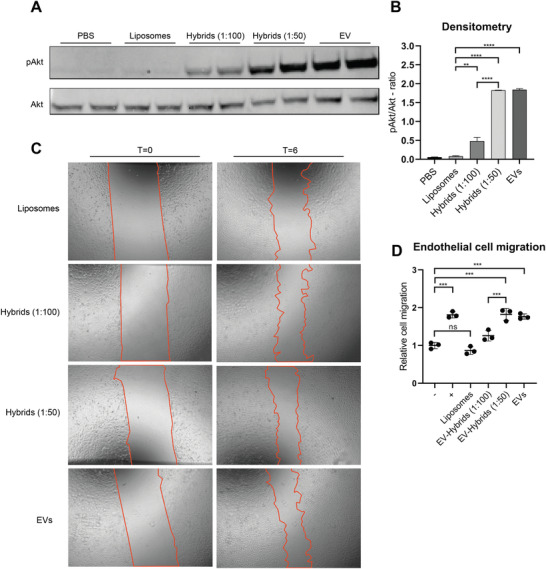
Endothelial signaling assay and scratch wound closing assay of HMEC‐1 cells treated with liposomes and hybrids. A) Representative western blot analysis of phosphorylated Akt and Akt expression levels in HMEC‐1 cells treated with liposomes, hybrids, and EVs. Liposomes and hybrids were administered at a total particle dose of 2 × 1012 and EVs at a total particle dose of 3 × 1010. B) Quantification of Akt and pAkt expression levels obtained via western blot analysis using densitometry expressed as pAkt/Akt‐ratio. C) Representative images of scratch wound healing assay before (t = 0) and after (t = 6 h) incubation with liposomes, hybrids (1:100), hybrids (1:50), and EVs. Liposomes and hybrids were administered at a total particle dose of 2 × 1012 and EVs at a total particle dose of 3 × 1010. D) Cell migration of HMEC‐1 expressed relative to the negative control. Incubation of HMEC‐1 cells with hybrids (1:50 and 1:100) and EVs increases wound closure as compared to liposomes. Data are expressed as mean + SD, one‐way ANOVA with post‐hoc test, ns = not significant, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
Second, we performed a scratch wound healing assay using a confluent monolayer of HMEC‐1 cells. Samples were normalized based on particle counts as measured by NTA and a total dose of 2 × 1012 particles for liposomes and hybrids was added as well as 3 × 1010 particles for EVs, which served as positive control. The closing of the scratch was then analyzed after 6 h. Hybrids stimulated closure of the scratch to a larger extent than liposomes and hybrids (1:50) further increased closure of the wound as compared to hybrids (1:100) (Figure 8C,D). This indicates a dose‐dependent effect of the amount of CPC EVs used in the formulation on wound closure.
These results are in good agreement with the endothelial signaling assays and indicate that hybrids produced using CPC EVs stimulate wound closure and induce phosphorylation of Akt.
3. Discussion
The delivery of RNAi therapeutics is challenging given the unfavorable characteristics of siRNA as a drug molecule. siRNA molecules are unstable in circulation, immunogenic, and are unable due to their molecular properties, to cross cellular membranes to reach their cytosolic target site.
Here, we produced EV–liposome hybrid nanoparticles, which are nanosized siRNA carriers formed through the merging of EVs and liposomes, via thin‐film hydration and extrusion. The anticipated benefits originate from the combination of liposome related properties such as high RNA loading capacity and EV related properties such as increased delivery efficacy, cell targeting properties, and possible tissue regenerative properties.
The thin‐film hydration and extrusion method has already been previously described to generate different hybrids based on the combination of EVs and liposomes.[ 39 , 40 ] For instance, Jhan et al. hydrated a lipid film in PBS and subsequently added 3T3‐ or A549‐EVs followed by sonication and sequential extrusion through 400, 200, and 100 nm membrane pores.[ 39 ] Raymajhi et al. hydrated the lipid film in the presence of J774A.1‐EVs, sonicated and subsequently extruded the lipid mixture through membrane filters with 400 nm followed by 200 nm pores.[ 40 ] The use of mechanical extrusion to generate hybrids is not limited to EVs and lipid‐based particles as it has also been used to incorporate cellular membranes, such as leukocyte membranes, in liposomes.[ 41 , 42 , 43 ] More recently, extrusion has also been used to surface coat gold nanoparticles with the membrane of EVs.[ 44 ]
An important variable in the generation of hybrids is the amount of EVs incorporated in the formulation. A useful metric to describe the amount of EVs incorporated in the formulation is the (EV) protein‐to‐(liposomal) lipid ratio. In literature, ratios can be found ranging from 1:5 to 1:1000 (protein/lipid (w/w)).[ 39 , 40 , 41 , 42 , 43 ] For instance, to incorporate the membrane of leukocytes into phosphatidylcholine liposomes, ratios varying from 1:100 to 1:300 (protein/lipid; w/w) were used, whereas for the generation of “macrophage derived hybrid exosomes,” Rayamajhi et al. used a ratio of 1:5 (protein/lipid (w/w)).[ 40 , 41 ] A potential drawback of this metric is that proteins can also be contaminants of EV isolations, which can vary from batch‐to‐batch and therefore potentially has implications for reproducibility. To account for this, we carefully monitored the number of particles per µg protein, which was found to be highly consistent among different EV isolations. Here, we generated hybrids by hydration of a lipid film with EVs at protein‐to‐lipid ratios of 1:100–1:50 (w/w) and subsequent extrusion through membranes with pores of 1000 nm followed by 100 and then 50 nm. As the majority of EVs has a size below 100 nm, based on our observation in NTA analysis where we observed a size mode value of 75 nm, a 50 nm membrane was chosen as smallest membrane. A possible limitation of the aforementioned studies regarding EV‐based hybrid nanoparticles is that samples were extruded through pores around or above the median size of EVs, which not necessarily results in deformation of the EV and subsequent reformation in a hybrid nanoparticle. In this study, we did take this into account and extruded EVs together with synthetic liposomes through a membrane with pore sizes of 50 nm.
We analyzed the yield of the production process of this formulation in terms of siRNA and synthetic lipid yield, which was found to be maximally 50% of the input siRNA and cholesterol. This could potentially be explained by the formation of siRNA/ionizable lipid aggregates, which are lost during the extrusion process. Although this effect has been shown to be overcome by the addition of 40% ethanol (v/v) combined with a rise in temperature to 65 °C, we decided not to change the production process since higher temperatures and ethanol content could potentially detrimentally affect the EV membrane proteins.[ 45 ] When we looked at the influence of EV incorporation in the formulation on several particle characteristics such as size, PDI, and zeta potential, we observed that increasing the amount of SKOV3 EVs incorporated in the formulation resulted in an increase in PDI, although this effect was not significant. For hybrids generated with CPC EVs, an increase in PDI was not observed. The surface charge, i.e., zeta potential, was decreased in hybrids, which can most likely be attributed to the incorporation of negatively charged EV membrane components. We did observe that RNA encapsulation efficiency in hybrids was decreased. This may be the results of competition for the electrostatic interaction with the ionizable lipid by negatively charged EV components such as RNA or negatively charged lipids or proteins. We also evaluated the morphology of the liposomes and hybrids. The extrusion process had no apparent detrimental effects on the morphology of hybrids as they appeared to be spherical, unilamellar membrane enclosed particles, which are comparable to liposomes and EVs. We also confirmed that hybrid particles (containing both siRNA and synthetic lipid) were captured by beads coated with antibodies against several distinctive EV marker proteins such as CD9, CD63, and CD81 indicating that surface topology of EVs is at least partly transferred to the hybrids. Nevertheless, it remains challenging to assess the efficiency of this hybridization process and fully exclude the possibility of intact EVs being present in the formulation.
Next, we quantitatively compared multiple functional characteristics, including cellular uptake, gene‐silencing efficacy, and cell viability, of liposomes and extracellular vesicle–liposome hybrids. The cellular uptake of nanoparticles via endocytosis is influenced by many variables such as size, charge, and the biomolecular corona.[ 46 , 47 , 48 , 49 ] It is known that uptake rate and route can vary between different lipid systems and EVs.[ 47 , 50 , 51 , 52 ] As the surface charge and membrane surface of hybrids differs from that of liposomes, we investigated the uptake efficiency of liposomes and hybrids. In HEK293T and U87‐MG, we observed a decrease in uptake for hybrids (1:100) as compared to liposomes. At higher concentrations of EVs in the formulation (1:50), cellular uptake increased again. The latter observation suggests that the uptake mechanism is different for hybrids as compared to liposomes, changing from predominantly liposome‐dictated to mainly EV‐dictated. This may be relevant for cell‐targeting purposes, as EVs may have intrinsic capacity to target specific cells or tissues.[ 21 , 24 , 53 ] Furthermore, this may affect endocytic routing and intracellular nanoparticle trafficking, which in turn may influence delivery efficiency.
An important drawback of the usage of liposomes or other lipid nanoparticles for RNA delivery is the dose/dose‐regimen related hepatotoxicity, which might be related to innate immune system activation.[ 8 , 54 ] In contrast to synthetic systems, EVs are generally considered to have low immunogenicity and are less toxic as observed in several preclinical studies.[ 55 , 56 ] Here, we observed a decrease in in vitro toxicity of hybrids as compared to liposomes in SKOV3 cells, which could be the results of EV component incorporation in hybrids. The effect was only observed in SKOV3 cells suggesting cell‐specific effects. These data should be interpreted with care as in vitro to in vivo translation of cell viability data for lipid‐based drug delivery systems is unclear. Moreover, the MTS‐assay as performed in this manuscript lacked an assay positive control making it more difficult to assess the value of the absolute toxicity values. However, based on the results we could still assess relative differences between the three different nanoparticle types within the same experiments, which have shown to be reproducible.
Liposomes and hybrids both functionally delivered siRNA in a dose‐dependent manner in three different cell types although the potency of hybrids was reduced in SKOV3 and HEK293T, but not in U87. Again, this is an intriguing observation as it implies that effects are cell type dependent. A possible explanation for the decrease in gene‐silencing in HEK293T might be the decreased uptake. In contrast, the combination of decreased cellular uptake and comparable gene‐silencing efficacy of liposomes and hybrids in U87‐MG might suggest different intracellular trafficking resulting in more efficient escape of siRNA from the endo‐lysosomal pathway for hybrids and is an interesting area to further explore.
Our observation that hybrids generated with SKOV3 EVs did not have a positive effect on gene‐silencing efficiency differs from others. Coating of polyethyleneimine‐based siRNA particles with SKOV3 EVs resulted in increased potency in terms of gene‐silencing efficacy of the EV‐modified particle compared to the uncoated particle.[ 57 ] This apparent discrepancy may be a result of multiple different causes, including the production method and the resulting hybrid composition.
Several groups have applied the concept of extracellular vesicle–liposome hybrids to create nanoparticles for tumor‐targeted drug delivery.[ 39 , 40 , 58 ] Here, we have shown that a similar approach can be used to convey tissue regenerative properties of CPC EVs to synthetic nanoparticles via the creation of extracellular vesicle–liposome hybrids. CPC EVs possess the ability to activate endothelial signaling and cell migration in HMEC‐1.[ 34 , 35 , 36 ] We observed that hybrids also activated endothelial signaling and cell migration whereas liposomes did not. This demonstrates that hybrids produced via thin‐film hydration and extrusion can be loaded with siRNA and retain functional properties of EVs. This implicates that hybrids potentially can be used as an efficient RNA drug delivery system while bearing intrinsic EV functionality at the same time. For instance, this can be of relevance in the salvage of myocardial tissue upon infarction where CPC EVs have shown to reduce scar size and improve ventricular function after permanent coronary occlusion.[ 59 ] Similarly, intracardiac delivery of a synthetic miRNA mimic of hsa‐miR‐590‐3p via a lipid‐based system resulted in reduced infarct size and improved cardiac output.[ 60 ] Given the results presented in this paper, hybrids might have the potential to combine both treatments in a single particle.
4. Conclusion
Currently, much is still unknown about how EV composition affects functionality and confers EVs with a potent RNA delivery capability. As long as such pivotal data are missing, the production of EV–liposome hybrids, which fully reflect the functional capabilities of EVs, remains challenging. The results presented here show that the production of hybrids via thin film hydration and subsequent extrusion results in hybrid particles with EV‐like surface topology encapsulating siRNA, which can be functionally delivered. The incorporation of EV membrane components leads to functional differences. Depending on the cell type, uptake is altered, toxicity of hybrids as compared to liposomes is reduced, and gene‐silencing effects are retained. Moreover, we also show that intrinsic functionalities of CPC EVs such as the ability to activate endothelial signaling pathways and stimulate migration of HMEC are retained in hybrids. Thus, hybrid nanoparticles could combine the functional characteristics of both liposomes and EVs and serve as a “best of both worlds” particle for therapeutic delivery of siRNA.
5. Experimental Section
Materials
Cholesterol was obtained from Sigma‐Aldrich (Saint‐Louis, USA), DPPC from Lipoid GmbH (Ludwigshafen, Germany), DMG‐PEG from NOF Corporation (Tokyo, Japan), and 18:1 Biotinyl Cap PE from Avanti Polar Lipids (Alabama, USA). DLin‐MC3‐DMA was synthesized in‐house according to a published protocol.[ 61 ] All oligonucleotides were ordered at Integrated DNA Technologies (Iowa, USA). siRNA molecules were ordered as individual strands and annealed for 5 min at 97 °C. The sequences used can be found in Table S1 in the Supporting Information.
Generation of Cells Stably Expressing Firefly and Renilla Luciferase
For the generation of stable dual luciferase cell lines, the PGK‐FFluc‐SV40‐Rluc‐NeoR_fusion cassette from the pmirGLO Dual‐Luciferase miRNA Target Expression Vector (Promega, Leiden, NL) was isolated and transferred to a pHAGE2 lentiviral vector. First, pHAGE2‐EF1a‐IRES‐NeoR‐WPRE was restricted with SpeI and XbaI restriction enzymes (New England Biolabs, Ipswich, MA, USA) and religated to remove the EF1a promoter. Then, the PGK‐FFluc‐SV40‐Rluc‐NeoR_fusion cassette was isolated from the pmirGLO plasmid using BglII and BstBI restriction enzymes (New England Biolabs, Ipswich, MA, USA) and ligated into the newly formed pHAGE2‐IRES‐NeoR‐WPRE vector digested with BamHI and ClaI restriction enzymes (New England Biolabs, Ipswich, MA, USA), generating a pHAGE2‐PGK‐FFluc‐SV40‐Rluc‐NeoR_fusion‐WPRE plasmid. All ligations were performed using a Quick Ligation Kit (all New England Biolabs, Ipswich, MA, USA) and ligation products were subsequently transformed into One Shot Stbl3 chemically competent Escherichia coli (ThermoFisher Scientific, Waltham, MA, USA). For lentiviral production, HEK293T cells were transfected overnight with psPAX2, pMD2.G, and pHAGE2‐PGK‐FFluc‐SV40‐Rluc‐NeoR_fusion‐WPRE plasmids at a 1:1:2 ratio using Lipofectamine 2000 (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturers' protocol. After 18 h, the culture medium was replaced, and lentiviral supernatant was collected after 48 h. Lentiviral supernatant was cleared from any remaining cells by a 5 min 1000 × g centrifugation step and subsequent 0.45 µm syringe filter filtration, and stored at −80 °C until further use. Cells were transduced with lentiviral supernatants overnight in the presence of 8 µg mL−1 polybrene (ThermoFisher Scientific, Waltham, MA, USA). Starting 24 h after lentiviral transduction, cells were cultured with 1000 µg mL−1 G418 for 5 days, after which they were cultured at a 500–1000 µg mL−1 G418, depending on the cell line, until further use. Transduced cells are referred to by the affix ‐dluc.
General Cell Culture
SKOV3, SKOV3‐dluc, HEK293T‐dluc, and U87‐MG‐dluc were cultured in Dulbecco's modified Eagle Medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco, Corning). SKOV3‐dluc and HEK293T‐dluc were cultured in the presence of 1000 µg mL−1 G418 (BioIVT) whereas U87‐MG‐dluc was cultured in the presence of 500 µg mL−1 G418. HMEC‐1 was cultured in MCDB‐131 medium supplemented with 10% FBS (Gibco), 2 × 10−3 m l‐glutamine (Gibco), 10 ng mL−1 rhEGF (Peprotech), and 50 × 10−9 m hydrocortisone (Sigma) in flasks/plates coated with 0.1% gelatin (Sigma). CPCs were cultured in MEM 199 + Earle's Salts and l‐glutamine (Gibco), which was supplemented with 22% EGM‐2 medium (Lonza), 10% FBS, and 1% MEM NEAA nucleic acids (Gibco). All cells were cultured at 37 °C and 5% CO2 in the presence of 100 U mL−1 penicillin and 100 U mL−1 streptomycin (Gibco).
Cell Culture and Isolation of SKOV3‐EVs and CPC‐EVs
For SKOV3‐EV production, SKOV3 cells were seeded at an appropriate density and cultured for 48–72 h to a confluence of 80–90% after which the medium was replaced and cells were cultured for another 24 h in Opti‐Mem supplemented with Glutamax, 100 U mL−1 penicillin, and 100 U mL−1 streptomycin. Conditioned medium was harvested after 24 h and spun down for 5 min at 300 × g and for 15 min at 2000 × g to remove cells and cell debris, respectively. The supernatant was filtered through a 0.45 × 10−6 m PES bottle top filter and concentrated to a volume of 15 mL by tangential flow filtration (TFF) using Vivaflow 50R hydrosart casettes, with a membrane cutoff of 100 kDa. This concentrate was then further reduced to a volume of ≈5 mL using 100 kDa Amicon Ultra‐15 Centrifugal filter (Merck) and loaded on a HiPrep 16/60 Sephacryl S‐400 HR column (GE Healthcare, Uppsala, Sweden) connected to an ÄKTA Start system (GE Healtcare) containing an UV280 flow cell. For CPC‐EVs, the procedure was slightly different. CPCs were seeded at an appropriate density and when a confluency of 80–100% was reached, cells were washed with PBS and medium was replaced for basal MEM199. The supernatant was collected after 24 h and centrifuged for 15 min at 2000 × g to remove cells and cell debris and the supernatant was filtered through a 0.45 × 10−6 m PES bottle top filter. Subsequently, the filtrate was concentrated by TFF using a minimate TFF capsule with a membrane cutoff of 100 kDa. Then, EVs were isolated by size exclusion chromatography following the same procedure as described for SKOV3‐EVs. After SEC, the fractions containing EVs were pooled, filtered through a 0.45 × 10−6 m syringe filter, and concentrated using 100 kDa Amicon Ultra‐15 Centrifugal filter (Merck). Then, the buffer was exchanged to 250 × 10−3 m citrate buffer (pH 5.5) and the sample was again concentrated using Amicon Spin Filters with a membrane cutoff of 100 kDa. The protein concentration was determined via micro‐BCA protein determination kit (Thermo Scientific). EVs were stored at 4 °C until further use. EVs were used to prepare hybrids within 72 h after isolation.
Preparation and Analysis of Liposomes and EV–Liposome Hybrids (Hybrids)
Lipid were dissolved in a mixture of chloroform/methanol (9/1; v/v) and added to a round bottom flask at a molar ratio of 30:30:35.5:1.5:3 (DLin‐MC3‐DMA:DPPC:cholesterol:18:1 Biotinyl Cap PE:DMG‐PEG). The organic solvent was evaporated using a RotoVap (Büchi Labortechnik, Flawil, Switzerland) at 60 °C and the resulting lipid film was dried under a flow of nitrogen for ≈20 min. For the liposomes, the lipid film was hydrated using a mixture of siRNA targeting firefly luciferase and fluorescently labeled siRNA targeting firefly luciferase in a ratio of 1:1 (siRNA Luc:siRNA Luc‐AF488 or AF647) dissolved in 250 × 10−3 m citrate buffer (pH 5.5) for 1 h at 45 °C. After hydration, the suspension was kept at 45 °C and extruded five times through a polycarbonate filter of 1.0 µm, then five times through 0.1 µm, and finally five times through a polycarbonate filter of 0.05 µm using an Avanti Hand Extruder (Avanti Polar Lipids). Subsequently, the liposomes were dialyzed overnight at 4 °C against an excess of PBS using Slide‐A‐Lyzer G2 Dialysis Casette with a membrane cutoff of 100 kDa to change the pH to 7.4 and to remove unencapsulated siRNA. Hybrids were produced in a similar fashion, but in this case, the lipid film was hydrated using a mixture of siRNA and extracellular vesicles. Extracellular vesicles were added at different ratios of vesicle protein to total lipid: 1:100 and 1:50 (protein/total lipid; w/w).
Characterization of EVs, Liposomes, and Hybrids
Particle size of liposomes and hybrids was measured via DLS on a Zetasizer Nano S (Malvern Panalytical, Malvern, UK). Samples were diluted with Dulbecco's PBS (DPBS) to an appropriate concentration and measured in triplicate. Liposomes, hybrid, and EV size was also measured using NTA on a NanoSight NS500 (Malvern, Panalytical, Malvern, UK). For NTA, samples were diluted in DPBS to an appropriate particle concentration and loaded in the sample chamber. Camera level 16 was selected and sample was measured three times for 30 s and subsequently analyzed using Nanosight NTA 3.4 software at a sensitivity level of 5.
Particle surface potential was measured by laser Doppler electrophoresis on a Zetasizer Nano Z (Malvern Panalytical, Malvern, UK). Samples were diluted in 0.1× DPBS and sample was measured for 20 runs in triplicate.
The RNA concentration was determined based on the fluorescence emitted by the fluorescently labeled siRNA. Samples were diluted 1:1 in 2% TX‐100 in PBS. A calibration curve of fluorescent siRNA was prepared in the same medium. Sample fluorescence was measured on a Spectramax ID3 (Molecular Devices, San Jose, California, US) at an excitation/emission wavelength of 490/530 nm or 620/665 nm for siRNA‐AF488 or siRNA‐AF647, respectively. Concentrations were determined based on a reference calibration curve.
The cholesterol concentration was determined using the LabAssay Cholesterol kit (DAKO, JP) in PBS or in the presence of 50% (v/v) isopropanol. Sample concentration was determined using a reference calibration curve. Absorbance was measured at 600 nm on a Spectramax ID3 (Molecular Devices, San Jose, California, US).
The encapsulation efficiency of siRNA was calculated by the following formula
| (1) |
Cryogenic Transmission Electron Microscopy
7 µL of liposomes or hybrid suspension were added to freshly glow‐discharged quantifoils and incubated for at least 10 min in a humidified environment and then vitrified using a FEI Mark IV Vitrobot (FEI, Hillsboro OR, USA). After vitrification, samples were stored in liquid nitrogen until imaging. Samples were imaged on an FEI Tecnai G2 20 TWIN 200 kV transmission electron microscope. Vitrified quantifoils were loaded in a Gatan 70° tilt cryo‐transfer system, which was precooled using liquid nitrogen and inserted in the microscope. Samples were imaged at a magnification of 29 000× and samples images were acquired by the bottom mounted FEI High‐Sensitive 4k × 4k Eagle camera.
Western Blotting
Protein concentration was determined via a micro‐BCA assay and ≈10 µg protein was used per sample. Samples were mixed with 4× sample buffer (40% v/v glycerol, 8% w/v SDS, 8% v/v bromophenol blue, in 0.25 m Tris‐HCL) with or without dithiothreitol (DTT) for reduced or nonreduced conditions, respectively. Samples were heated to 95 °C for 5 min and separated on a 4–12 Bis‐Tris polyacrylamide gel (Thermo Scientific). Proteins were then electrotransferred to immobilon‐FLR polyvinylidene difluoride (PVDF) membranes and blocked with 50% v/v Odyssey Blocking Buffer (LI‐COR Biosciences) in Tris buffered saline (TBS). All immune‐labeling was performed with 50% v/v Odyssey Blocking Buffer in TBS containing 0.1% Tween 20 (TBS‐T). Primary antibodies were used overnight at 4 °C and included mouse anti‐CD63 (Abcam, MEM‐259; 1:1000), Mouse Anti‐CD81 (Santa Cruz, SC‐166029; 1:500), rabbit anti‐TSG101 (Abcam, ab30871, 1:1000), mouse anti‐Alix (Thermo Scientific, 3A9, 1:1000), mouse‐anti‐β‐actin (Cell Signaling Technology, clone 8H10D10, 1:1000), rat anti‐Calnexin (Tebu‐Bio, N3C2, 1:1000), rabbit anti‐AKT (Cell Signaling Technology, 9272S, 1:1000), rabbit anti‐pAKT (Cell Signaling Technology, 4060S, 1:1000), and mouse anti‐β‐actin (Sigma, A5441,1:1000).
Secondary antibodies included Alexa Fluor 680‐conjugated antirabbit antibodies (LI‐COR Biosciences, A‐21076, 1:7500–1:10 000), Alexa Fluor 680‐conjugated antimouse antibodies (LI‐COR Biosciences, A‐21057; 1:7500–1:10 000), IRDye 800CW antimouse antibodies (LI‐COR Biosciences, 926‐32212, 1:7500–1:10 000), and IRDye 800CW antirabbit antibodies (LI‐COR Biosciences, 926‐32211, 1:7500–1:10 000). Imaging was performed on an Odyssey Infrared Imager (LI‐COR Biosciences, Leusden, The Netherlands) at 700 and 800 nm.
Proof of Hybridization: Analysis Using ExoCap CD9/CD81/CD63 Beads
Samples were incubated overnight at 4 °C with 0.75 µL ExoCap beads (JSR Life Sciences, Tokyo, Japan) in a total volume of 50 µL 2% BSA in PBS (PBSA). Samples were normalized based on siRNA concentration. Samples were incubated separately with three different beads: CD9, CD81, and CD63. After incubation, beads were captured on a magnetic plate and washed three times with PBSA. Successful bead pulldown was analyzed by measurement of siRNA‐AF647 using flow cytometry.
Incorporation of a synthetic lipid was also analyzed. To this end, 1.5% 18:1 Biotinyl Cap PE (Avanti Polar Lipids, Alblaster, Alabama, USA) was incorporated in the lipid film of both the liposomes and the hybrids. The samples were incubated with ExoCap beads as described above. After the three initial washing steps, samples were incubated with PE‐Streptavidin for 20 min and then washed three times. Samples were then suspended in 200 µL PBSA and measured by flow cytometry on a BD LSRFortessa (BD, Franklin Lakes, NJ, US). Flow cytometry data analysis was performed using FlowJo v10 software.
Analysis of Cellular Uptake by Flow Cytometry
For the measurement of liposomal and hybrid cellular uptake, flow cytometry was used. Cells were seeded at an appropriate density in a 48‐well plate. SKOV3‐dluc was seeded at 40 000 cells per well 24 h prior to the assay, HEK293T‐dluc was seeded at 20 000 cells per well 72 h prior to the assay, and U87‐MG‐dluc cells were seeded at 40 000 cells per well 24 h prior to the assay. Then, cells were incubated with different nanoparticles at a total siRNA concentration of 25 × 10−9 m per well. As a vehicle control, an equal volume of PBS was used. Cells were incubated for 4 h and then cellular uptake was analyzed by flow cytometry. Cells were washed with PBS, trypsinized, taken up in full medium, and transferred to a 96 U‐Bottom well plate (Greiner). Cells were then washed with an acid wash (0.5 m NaCL, 0.2 m acetic acid), PBS, and taken up in 2% PBSA for analysis on a LSRFortessa (BD, Franklin Lakes, NJ, US). For each experiment, cellular uptake was expressed as ratio of the uptake of the liposome sample. As a control, uptake was measured at 4 °C. To this end, cells were cooled 30 min prior to incubation in a fridge at 4 °C, and samples were added and incubated for 4 h at 4 °C. After incubation, cells were kept on ice and washed with ice‐cold PBS before trypsinization and further work‐up as described earlier.
Gene‐Silencing and Cell Viability
The gene‐silencing efficacy of liposomes and hybrids was assessed in multiple cell‐lines: SKOV3‐dluc, U87‐MG‐dluc, and HEK293T‐dluc. All cells expressed a dual luciferase construct containing both firefly and renilla luciferase under G418 selection. SKOV3‐dluc and HEK293T‐dluc cells were seeded in a 96‐well plate at a density of 5000 cells per well 48 h prior to transfection or 10 000 cells per well 24 h prior to transfection. U87‐MG‐dluc were seeded at a density of 5000 cells per well 24 h prior to transfection. Samples were added at concentrations ranging from 0 × 10−9 to 50 × 10−9 m siRNA. As a positive control, cells were transfected using Lipofectamine RNAiMAX according to the manufacturer's instructions. Luciferase activity was assessed after another 48 h of culture. Luciferase activity was measured using the Stop & Glo System (Promega, Leiden, NL) according to the manufacturer's instructions. In short, medium was aspirated and replaced by 50 µL of fresh medium. 50 µL of Glo substrate was added and cells were incubated for 10 min. After 10 min, 100 µL of lysate was transferred to a white 96‐well plate and firefly luciferase activity was measured. Then, 50 µL of Stop & Glo buffer was added and, after an incubation of 10 min, renilla luciferase activity was measured. Both firefly luciferase and renilla luciferase activities were measured on a Spectramax ID3 (Molecular Devices, San Jose, CA, USA) at an integration time of 1000 ms. For data analysis, firefly luciferase activity was normalized based on renilla luciferase activity and expressed as percentage of the blank—0 × 10−9 m siRNA—sample.
Cell viability was measured using CellTiter 96 AQueous MTS Reagent Powder according to the manufacturer's instructions. As a negative control, MTS medium was added to wells, which did not contain any cells and this background value was subtracted from sample values. Samples were normalized to untreated, blank cells, whose value was set at 100%. Absorbance was measured at 490 nm using a Spectramax ID3 (Molecular Devices).
Scratch Migration Assay
For the migration assay, HMEC‐1 cells were seeded in a 48‐well plate at a density of 90 000 cells per well 48 h prior to the assay. A scratch was made by hand using a pipet tip and the detached cells were washed away with MCDB‐131 medium without any supplementation. Subsequently, the cells were incubated in the basal MCDB‐131 medium with different samples in triplicate for 6 h. PBS was used as a negative control. At t = 0 h and t = 6 h, two pictures per well were made with the EVOS microscope (Life Technologies). The closing of the scratch was measured by image analysis using Image J software. The mean width of each scratch of t = 0 h was subtracted by the mean width at t = 6 h to determine the migrated area. The relative wound closure was calculated as compared to the negative control.
Endothelial Signaling Activation Assay
For the endothelial signaling activation assay, HMEC‐1 cells were used to measure phosphorylation of AKT after incubation with liposomes, hybrids, and EVs. HMEC‐1 cells were seeded in a 48‐well plate at a concentration of 90 000 cells per well and incubated for 48 h. Then, the medium was replaced with basal medium (MCDB‐131 medium without any supplementation), and the cells were starved for 3 h in the basal medium. After 3 h, samples were added to the wells and PBS was used as vehicle control. After 30 min, the medium was aspirated and the wells were washed with PBS. To lyse the cells, 100 µL complete lysis‐M buffer (Roche, Basel, Switzerland) including protease inhibitors (Roche) and phosphatase inhibitors (Roche) was added and incubated for 5 min on ice. Every well was scraped and the lysate was transferred to an Eppendorf tube. Samples were vortexed and centrifuged for 15 min at 12 000 × g at 4 °C. Expression of AKT and phosphorylated AKT (pAKT) was analyzed by western blotting as described above in the “Western Blotting” section. Protein concentration of samples was measured by Pierce BCA Protein Assay Kit and samples were normalized based on protein concentration.
Statistical Analysis
Data were presented as mean ± SD, unless otherwise stated. Differences in terms of particle characteristics and functionality between liposomes, hybrids (1:100), and hybrids (1:50) were analyzed by one‐way ANOVA with Tukey's post‐hoc test. An outcome was considered statistically significant if a p‐value of ≤0.05 was obtained. Statistical analysis was performed using GraphPad Prism v8.3 software (GraphPad Software, San Diego, CA, USA).
Conflict of Interest
R.M.S. is the CSO and shareholder of EXCYTEX B.V., a company devoted to the development of extracellular vesicle research tools. P.V. serves on the scientific advisory board of Evox Therapeutics.
Supporting information
Supporting Information
Acknowledgements
The work of M.J.W.E., R.M.S., and P.V. was supported by the European Union's Horizon 2020 Research and Innovation program in the project B‐SMART (to P.V. and R.M.S.) under grant agreement no. 721058. O.G.d.J. was supported by a VENI Fellowship (VI.Veni.192.174) from the Dutch Research Council (NWO). S.I.v.d.W. was supported by the Van Herk Fellowship. This work was also supported by the Project EVICARE (No. 725229) of the European Research Council (ERC) to J.P.G.S., PPS grant (No. 2018B014) to J.P.G.S./P.V., the Dutch Ministry of Economic Affairs, Agriculture and Innovation and the Netherlands CardioVascular Research Initiative (CVON): the Dutch Heart Foundation to J.P.G.S., Dutch Federations of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Sciences. P.V. acknowledges support from the Dutch Heart Foundation (Dr. E. Dekker Senior Scientist grant, no. 2019T049).
Evers M. J. W., van de S. I. Wakker, de E. M. Groot, de O. G. Jong, Gitz‐François J. J. J., Seinen C. S., Sluijter J. P. G., Schiffelers R. M., Vader P., Functional siRNA Delivery by Extracellular Vesicle–Liposome Hybrid Nanoparticles. Adv. Healthcare Mater. 2022, 11, 2101202. 10.1002/adhm.202101202
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Elbashir S. M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T., Nature 2001, 411, 494. [DOI] [PubMed] [Google Scholar]
- 2. Fire A., Xu S., Montgomery M. K., Kostas S. A., Driver S. E., Mello C. C., Nature 1998, 391, 806. [DOI] [PubMed] [Google Scholar]
- 3. Setten R. L., Rossi J. J., Han S., Nat. Rev. Drug Discovery 2019, 18, 421. [DOI] [PubMed] [Google Scholar]
- 4. Morrissey D. V., Blanchard K., Shaw L., Jensen K., Lockridge J. A., Dickinson B., McSwiggen J. A., Vargeese C., Bowman K., Shaffer C. S., Polisky B. A., Zinnen S., Hepatology 2005, 41, 1349. [DOI] [PubMed] [Google Scholar]
- 5. Kleinschmidt W. J., Ellis L. F., Van Fbank R. M., Murphy E. B., Nature 1968, 220, 167. [DOI] [PubMed] [Google Scholar]
- 6. Whitehead K. A., Dahlman J. E., Langer R. S., Anderson D. G., Annu. Rev. Chem. Biomol. Eng. 2011, 2, 77. [DOI] [PubMed] [Google Scholar]
- 7. Dowdy S. F., Nat. Biotechnol. 2017, 35, 222. [DOI] [PubMed] [Google Scholar]
- 8. Sabnis S., Kumarasinghe E. S., Salerno T., Mihai C., Ketova T., Senn J. J., Lynn A., Bulychev A., McFadyen I., Chan J., Almarsson Ö., Stanton M. G., Benenato K. E., Mol. Ther. 2018, 26, 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sago C. D., Krupczak B. R., Lokugamage M. P., Gan Z., Dahlman J. E., Cell. Mol. Bioeng. 2019, 12, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamamoto N., Sato Y., Munakata T., Kakuni M., Tateno C., Sanada T., Hirata Y., Murakami S., Tanaka Y., Chayama K., Hatakeyama H., Hyodo M., Harashima H., Kohara M., J. Hepatol. 2016, 64, 547. [DOI] [PubMed] [Google Scholar]
- 11. Sato Y., Hatakeyama H., Hyodo M., Harashima H., Mol. Ther. 2016, 24, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mui B. L., Tam Y. K., Jayaraman M., Ansell S. M., Du X., Tam Y. Y. C., Lin P. J., Chen S., Narayanannair J. K., Rajeev K. G., Manoharan M., Akinc A., Maier M. A., Cullis P., Madden T. D., Hope M. J., Mol. Ther.–Nucleic Acids 2013, 2, e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yáñez‐Mó M., J. Extracell. Vesicles 2015, 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Théry C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin‐Smith G. K., Ayre D. C., Bach J.‐M., Bachurski D., Baharvand H., Balaj L., Baldacchino S., Bauer N. N., Baxter A. A., Bebawy M., Beckham C., Bedina Zavec A., Benmoussa A., Berardi A. C., Bergese P., Bielska E., Blenkiron C., Bobis‐Wozowicz S., Boilard E., Boireau W., Bongiovanni A., et al., J. Extracell. Vesicles 2018, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skog J., Wurdinger T., van Rijn S., Meijer D., Gainche L., Sena‐Esteves M., Curry W. T. Jr., Carter R. S., Krichevsky A. M., Breakefield X. O., Nat. Cell Biol. 2008, 10, 1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pegtel D. M., Cosmopoulos K., Thorley‐Lawson D. A., van Eijndhoven M. A. J., Hopmans E. S., Lindenberg J. L., de Gruijl T. D., Wurdinger T., Middeldorp J. M., Proc. Natl. Acad. Sci. USA 2010, 107, 6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J. J., Lötvall J. O., Nat. Cell Biol. 2007, 9, 654. [DOI] [PubMed] [Google Scholar]
- 18. de Jong O. G., Murphy D. E., Mäger I., Willms E., Garcia‐Guerra A., Gitz‐Francois J. J., Lefferts J., Gupta D., Steenbeek S. C., van Rheenen J., El Andaloussi S., Schiffelers R. M., Wood M. J. A., Vader P., Nat. Commun. 2020, 11, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reshke R., Taylor J. A., Savard A., Guo H., Rhym L. H., Kowalski P. S., Trung M. T., Campbell C., Little W., Anderson D. G., Gibbings D., Nat. Biomed. Eng. 2020, 4, 52. [DOI] [PubMed] [Google Scholar]
- 20. Murphy D. E., de Jong O. G., Evers M. J. W., Nurazizah M., Schiffelers R. M., Vader P., Nano Lett. 2021, 21, 1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoshino A., Costa‐Silva B., Shen T. L., Rodrigues G., Hashimoto A., Tesic Mark M., Molina H., Kohsaka S., Di Giannatale A., Ceder S., Singh S., Williams C., Soplop N., Uryu K., Pharmer L., King T., Bojmar L., Davies A. E., Ararso Y., Zhang T., Zhang H., Hernandez J., Weiss J. M., Dumont‐Cole V. D., Kramer K., Wexler L. H., Narendran A., Schwartz G. K., Healey J. H., Sandstrom P., et al., Nature 2015, 527, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rana S., Yue S., Stadel D., Zöller M., Int. J. Biochem. Cell Biol. 2012, 44, 1574. [DOI] [PubMed] [Google Scholar]
- 23. Alvarez‐Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. J. A., Nat. Biotechnol. 2011, 29, 341. [DOI] [PubMed] [Google Scholar]
- 24. Kamerkar S., LeBlue V. S., Kalluri R., Nature 2017, 546, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grapp M., Wrede A., Schweizer M., Hüwel S., Galla H. J., Snaidero N., Simons M., Bückers J., Low P. S., Urlaub H., Gärtner J., Steinfeld R., Nat. Commun. 2013, 4, 2123. [DOI] [PubMed] [Google Scholar]
- 26. Elsharkasy O. M., Nordin J. Z., Hagey D. W., de Jong O. G., Schiffelers R. M., Andaloussi S. EL, Vader P., Adv. Drug Deliv. Rev. 2020, 159, 332. [DOI] [PubMed] [Google Scholar]
- 27. Roefs M. T., Sluijter J. P. G., Vader P., Trends Cell Biol. 2020, 30, 990. [DOI] [PubMed] [Google Scholar]
- 28. Vader P., Mol E. A., Pasterkamp G., Schiffelers R. M., Adv. Drug Delivery Rev. 2016, 106, 148. [DOI] [PubMed] [Google Scholar]
- 29. Haraszti R. A., Miller R., Stoppato M., Sere Y. Y., Coles A., Didiot M. C., Wollacott R., Sapp E., Dubuke M. L., Li X., Shaffer S. A., DiFiglia M., Wang Y., Aronin N., Khvorova A., Mol. Ther. 2018, 26, 2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vader P., Kooijmans S. A., Stremersch S., Raemdonck K., Ther. Delivery 2014, 5, 105. [DOI] [PubMed] [Google Scholar]
- 31. de Jong O. G., Kooijmans S. A. A., Murphy D. E., Jiang L., Evers M. J. W., Sluijter J. P. G., Vader P., Schiffelers R. M., Acc. Chem. Res. 2019, 52, 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nordin J. Z., Lee Y., Vader P., Mäger I., Johansson H. J., Heusermann W., Wiklander O. P. B., Hällbrink M., Seow Y., Bultema J. J., Gilthorpe J., Davies T., Fairchild P. J., Gabrielsson S., Meisner‐Kober N. C., Lehtiö J., Smith C. I. E., Wood M. J. A., Andaloussi S. E. L., Nanomedicine 2015, 11, 879. [DOI] [PubMed] [Google Scholar]
- 33. Webber J., Clayton A., J. Extracell. Vesicles 2013, 2, 19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vrijsen K. R., Maring J. A., Chamuleau S. A. J., Verhage V., Mol E. A., Deddens J. C., Metz C. H. G., Lodder K., van Eeuwijk E. C. M., van Dommelen S. M., Doevendans P. A., Smits A. M., Goumans M.‐J., Sluijter J. P. G., Adv. Healthcare Mater. 2016, 5, 2555. [DOI] [PubMed] [Google Scholar]
- 35. Vrijsen K. R., Sluijter J. P. G., Schuchardt M. W. L., van Balkom B. W. M., Noort W. A., Chamuleau S. A. J., Doevendans P. A. F. M., J. Cell. Mol. Med. 2010, 14, 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mol E. A., Goumans M., Doevendans P. A., Sluijter J. P. G., Vader P., Nanomedicine 2017, 13, 2061. [DOI] [PubMed] [Google Scholar]
- 37. Karar J., Maity A., Front. Mol. Neurosci. 2011, 4, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y., You B., Liu X., Chen J., Peng Y., Yuan Z., Med. Sci. Monit. 2019, 25, 6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jhan Y., Prasca‐chamorro D., Zuniga G. P., Moore D. M., Kumar S. A., Gaharwar A. K., Bishop C. J., Int. J. Pharm. 2019, 573, 118802. [DOI] [PubMed] [Google Scholar]
- 40. Rayamajhi S., Duong T., Nguyen T., Marasini R., Aryal S., Acta Biomater. 2019, 94, 482. [DOI] [PubMed] [Google Scholar]
- 41. Molinaro R., Corbo C., Martinez J. O., Taraballi F., Evangelopoulos M., Minardi S., Yazdi I. K., Zhao P., De Rosa E., Sherman M. B., De Vita A., Toledano Furman N. E., Wang X., Parodi A., Tasciotti E., Nat. Mater. 2016, 15, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goh W. J., Zou S., Lee C. K., Ou Y.‐H., Wang J.‐W., Czarny B., Pastorin G., Biomacromolecules 2018, 19, 22. [DOI] [PubMed] [Google Scholar]
- 43. Pitchaimani A., Duong T., Nguyen T., Aryal S., Biomaterials 2018, 160, 124. [DOI] [PubMed] [Google Scholar]
- 44. Van Deun J., Roux Q., Deville S., Van Acker T., Rappu P., Miinalainen I., Heino J., Vanhaecke F., De Geest B. G., De Wever O., Hendrix A., Cells 2020, 9, 1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Semple S. C., Klimuk S. K., Harasym T. O., Dos Santos N., Ansell S. M., Wong K. F., Maurer N., Stark H., Cullis P. R., Hope M. J., Scherrer P., Biochim. Biophys. Acta 2001, 1510, 152. [DOI] [PubMed] [Google Scholar]
- 46. Miller C. R., Bondurant B., McLean S. D., McGovern K. A., O'Brien D. F., Biochemistry 1998, 37, 12875. [DOI] [PubMed] [Google Scholar]
- 47. Miao L., Lin J., Huang Y., Li L., Delcassian D., Ge Y., Shi Y., Anderson D. G., Nat. Commun. 2020, 11, 2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang W., Kim B. Y. S., Rutka J. T., Chan W. C. W., Nat. Nanotechnol. 2008, 3, 145. [DOI] [PubMed] [Google Scholar]
- 49. Francia V., Schiffelers R. M., Cullis P. R., Witzigmann D., Bioconjugate Chem. 2020, 31, 2046. [DOI] [PubMed] [Google Scholar]
- 50. Gilleron J., Querbes W., Zeigerer A., Borodovsky A., Marsico G., Schubert U., Manygoats K., Seifert S., Andree C., Stoter M., Epstein‐Barash H., Zhang L., Koteliansky V., Fitzgerald K., Fava E., Bickle M., Kalaidzidis Y., Akinc A., Maier M., Zerial M., Nat. Biotechnol. 2013, 31, 638. [DOI] [PubMed] [Google Scholar]
- 51. Sahay G., Querbes W., Alabi C., Eltoukhy A., Sarkar S., Zurenko C., Karagiannis E., Love K., Chen D., Zoncu R., Buganim Y., Schroeder A., Langer R., Anderson D. G., Nat. Biotechnol. 2013, 31, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mulcahy L. A., Pink R. C., Carter D. R. F., J. Extracell. Vesicles 2014, 3, 24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wiklander O. P. B., Nordin J. Z., O'Loughlin A., Gustafsson Y., Corso G., Mäger I., Vader P., Lee Y., Sork H., Seow Y., Heldring N., Alvarez‐Erviti L., Smith C. I. E., Blanc K. L.e, Macchiarini P., Jungebluth P., Wood M. J. A., El Andaloussi S., J. Extracell. Vesicles 2015, 4, 26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sato Y., Matsui H., Yamamoto N., Sato R., Munakata T., Kohara M., Harashima H., J. Controlled Release 2017, 266, 216. [DOI] [PubMed] [Google Scholar]
- 55. Saleh A. F., Lázaro‐Ibáñez E., Forsgard M. A.‐M., Shatnyeva O., Osteikoetxea X., Karlsson F., Heath N., Ingelsten M., Rose J., Harris J., Mairesse M., Bates S. M., Clausen M., Etal D., Leonard E., Fellows M. D., Dekker N., Edmunds N., Nanoscale 2019, 11, 6990. [DOI] [PubMed] [Google Scholar]
- 56. Zhu X., Badawi M., Pomeroy S., Sutaria D. S., Xie Z., Baek A., Jiang J., Elgamal O. A., Mo X., La Perle K., Chalmers J., Schmittgen T. D., Phelps M. A., J. Extracell. Vesicles 2017, 6, 1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhupanyn P., Ewe A., Büch T., Malek A., Rademacher P., J. Controlled Release 2020, 319, 63. [DOI] [PubMed] [Google Scholar]
- 58. Sato Y. T., Umezaki K., Sawada S., Mukai S., Sasaki Y., Harada N., Shiku H., Akiyoshi K., Sci. Rep. 2016, 6, 21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barile L., Cervio E., Lionetti V., Milano G., Ciullo A., Biemmi V., Bolis S., Altomare C., Matteucci M., Di Silvestre D., Brambilla F., Fertig T. E., Torre T., Demertzis S., Mauri P., Moccetti T., Vassalli G., Cardiovasc. Res. 2018, 114, 992. [DOI] [PubMed] [Google Scholar]
- 60. Lesizza P., Prosdocimo G., Martinelli V., Sinagra G., Zacchigna S., Giacca M., Circ. Res. 2017, 120, 1298. [DOI] [PubMed] [Google Scholar]
- 61. Jayaraman M., Ansell S. M., Mui B. L., Tam Y. K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J. K., Rajeev K. G., Hafez I. M., Akinc A., Maier M. A., Tracy M. A., Cullis P. R., Madden T. D., Manoharan M., Hope M. J., Angew. Chem., Int. Ed. 2012, 51, 8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.