

Abstract
Osteoarthritis is a degenerative joint disease characterized by cartilage deterioration and subsequent inflammatory changes in the underlying bone. Injectable hydrogels have emerged as a promising approach for controlled drug delivery in cartilage therapies. This review focuses on the latest developments in utilizing injectable hydrogels as vehicles for targeted drug delivery to promote cartilage repair and regeneration. The pathogenesis of osteoarthritis is discussed to provide a comprehensive understanding of the disease progression. Subsequently, the various types of injectable hydrogels used for intra‐articular delivery are discussed. Specifically, physically and chemically crosslinked injectable hydrogels are critically analyzed, with an emphasis on their fabrication strategies and their capacity to encapsulate and release therapeutic agents in a controlled manner. Furthermore, the potential of incorporating growth factors, anti‐inflammatory drugs, and cells within these injectable hydrogels are discussed. Overall, this review offers a comprehensive guide to navigating the landscape of hydrogel‐based therapeutics in osteoarthritis.
Keywords: biomaterials, drug delivery, hydrogels, injectable, osteoarthritis
Injectable biomaterials have emerged as a prominent approach for therapeutic delivery in cartilage repair and regeneration. This review offers brief insights into the pathogenesis of osteoarthritis and critically evaluates recent advancements in injectable biomaterials for these applications. Furthermore, it also discusses fabrication strategies for the encapsulation and release of therapeutics, including growth factors, anti‐inflammatory drugs, and stem cells.
1. Introduction
Osteoarthritis (OA) is the most common chronic joint disease, affecting 32.5 million Americans.[ 1 ] OA involves changes in the molecular, biochemical, morphological, and biomechanical components of the joints.[ 1 ] During OA, the articular hyaline cartilage, joint capsule synovium, and subchondral bone in the knee are primarily affected. As OA progresses, there is a notable degeneration of cartilage, leading to the narrowing of the joint space.[ 2 ] Additionally, subchondral bone thickening, the formation of osteophytes or bone spurs, and joint inflammation occur, often accompanied by swelling and persistent pain. Cartilage, an avascular, aneural, and alymphatic connective tissue, is enveloped by a perichondrium‐like fibrous membrane. It is characterized by a low density of chondrocytes responsible for producing the majority of the extracellular matrix components.[ 3 ] These factors contribute to the challenge of insufficient nutrient supply to cartilage and its limited regenerative ability.[ 4 ]
Current clinical approaches for OA treatment rely on pain management (nonsteroidal anti‐inflammatory drugs (NSAIDs) administration), restoration of joint function (hyaluronan injection to improve lubrication), and delaying the onset of permanent joint deformities (arthrocentesis).[ 5 ] Oral NSAIDs are used to treat OA by inhibiting cyclooxygenase enzymes that produce inflammation‐causing prostaglandins. However, NSAIDs provide palliative relief and cannot reverse the disease progression.[ 6 ] They also tend to accumulate in systemic tissues rather than the targeted cartilage, resulting in insufficient therapeutic efficacy. Therefore, intra‐articular (IA) drug delivery is preferred as it allows direct delivery to the site of injury. IA injection increases the bioavailability of the therapeutic agents at the target site, reducing systemic exposure, minimal systemic side effects, and overall cost.[ 7 ] However, rapid clearance by synovial fluid leads to rapid drug depletion in the joint cavity, necessitating frequent administration and increasing the risk of systemic toxicity.
Hydrogels offer a solution by sustainably delivering drugs, enabling long‐term treatment for osteoarthritis. Hydrogels possess desirable characteristics such as biofunctionality, biocompatibility, and tunable properties, including a porous framework, high water absorption, and mechanical stability. These attributes make hydrogels well‐suited for delivering drugs and other therapeutic agents, such as protein/peptides, and cells, to facilitate cartilage regeneration in the treatment of OA.[ 8 ] The properties of hydrogels can be adjusted based on their composition and synthesis process. The porous structure of hydrogels allows for the entrapment of the therapeutic agents, and the release kinetics can be controlled by manipulating the pore architecture. Moreover, the physical properties of hydrogels can be tailored to mimic the extracellular matrix for the differentiation and proliferation of cells. Injectable hydrogels can be designed to crosslink rapidly, enabling in situ hydrogel formation upon injection. Injectable hydrogels offer many advantages including convenient synthesis, good biocompatibility and tunable biodegradability, high drug loading capacity, encapsulation, and controlled release of therapeutics.[ 9 ] This minimally invasive approach is advantageous for targeting irregularly shaped sites affected by OA, providing precise treatment at the desired location.
This comprehensive review focuses on the latest advancements in utilizing injectable hydrogels as controlled drug delivery to facilitate cartilage repair and regeneration. A thorough discussion of the pathogenesis of osteoarthritis is provided, offering a comprehensive understanding of the disease process. Subsequently, the various types of injectable hydrogels employed for drug delivery to cartilage are explored, with a specific emphasis on physically and chemically crosslinked hydrogels. The fabrication strategies and the ability of these hydrogels to encapsulate and release therapeutic agents in a controlled manner are thoroughly discussed. Furthermore, the potential of incorporating growth factors, anti‐inflammatory drugs, and stem cells within these hydrogel systems is highlighted. The critical roles these components play in modulating cellular behavior, promoting cartilage regeneration, and reducing inflammation are extensively addressed. Overall, this review provides a comprehensive guide to navigating the promising landscape of hydrogel‐based therapeutics in osteoarthritis.
2. Overview of OA Pathogenesis
OA is characterized by the degeneration of articular cartilage and the remodeling of the underlying bone (Figure 1A). This condition commonly leads to pain and stiffness, particularly affecting the hip, knee, and thumb joints. Factors that play an important role in the development and progression of OA include aging, sex, obesity, congenital factors, joint trauma, joint overuse, infectious factors, genetic predisposition, surgical intervention, and other systemic diseases.[ 10 ] The role of the immune system in the onset and progression of OA is a crucial factor in its pathogenesis.[ 11 ] OA is caused due to the imbalance between the synthesis and degradation of the extracellular matrix components by the chondrocytes.[ 8 ] During OA, a sequence of changes occurs in the composite of articular cartilage, calcified cartilage, and subchondral bone, known as the osteochondral unit (Figure 1B).[ 12 ] A healthy osteochondral unit has a thin layer of calcified cartilage beneath the articular cartilage separated by a well‐defined tidemark.[ 13 ] Beneath the calcified cartilage lies the cortical plate and an interconnected network of osteocytes. However, in the early stages of OA, the cartilage matrix swells, and the metabolic activity of chondrocytes increases.[ 14 ] In addition, surface fibrillations are observed in the articular cartilage, and remodeling of the cortical plate with an increase in porosity is observed in the subchondral bone. During the OA progression, the loss of proteoglycans in the cartilage matrix disrupts the collagen network.[ 12 ] This leads to the development of deep fissures and delamination of the cartilage, resulting in the exposure of calcified cartilage and subchondral bone. In late‐stage OA, chondrocyte clustering and chondrocyte apoptosis are observed. Also, vascularization occurs in the calcified cartilage. This expands the calcified cartilage into the articular cartilage, leading to the duplication of the tidemark.[ 15 ] In the subchondral bone, the osteocyte canalicular network is damaged, resulting in osteocyte apoptosis.[ 12 ] Moreover, inflammation also plays a significant role in the origin and progression of OA.[ 16 ] Studies show a high correlation between the progression of cartilage degradation and the presence of inflammatory synovium.[ 16 ] Inflammatory cytokines are one of the major contributors to inflammation and, correspondingly, to the pathogenesis of OA.[ 10 ]
Figure 1.
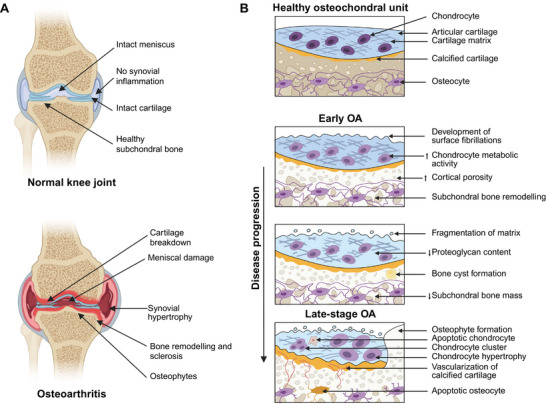
Osteoarthritis pathophysiology. A) Schematic comparing a normal knee joint with osteoarthritis (OA). B) Schematic comparing a healthy osteochondral unit with the changes in the osteochondral unit during OA progression. OA is characterized by chronic inflammation, cartilage damage due to mechanical and proteolytic degradation, and abnormal subchondral bone formation, leading to the formation of bony outgrowths into the joint capsule referred to as osteophytes. Adapted and redrawn with permission.[ 12 ] Copyright 2016, Nature.
2.1. Inflammatory Cytokines
Interleukin‐1 beta (IL‐1β) is a proinflammatory cytokine that induces catabolic effects on the articular cartilage and the joint (Figure 2A).[ 10 ] Studies show that high levels of IL‐1β are usually observed in the synovial fluid, synovial membrane, cartilage, and the subchondral bone layer of patients with OA.[ 17 ] Usually, IL‐1β is in an inactive form, as a cytosolic precursor protein (pro‐IL‐1β), and it is converted to active form when it undergoes intracellular proteolysis by Caspase 1.[ 18 ] The IL‐1β binds to the interleukin‐1 receptor type 1 (IL‐1R1) receptor and recruits an additional IL‐1 type 3 receptor (IL‐1R3) chain, forming a complex, which then recruits the adapter protein myeloid differentiation primary response protein 88 (MyD88).[ 19 ] This complex also binds to the IL‐1 receptor‐associated kinase (IRAK), which then affects tumor necrosis factor receptor associated factor 6 (TRAF6) protein, and induces additional binding of TAK1), TAB1, and TAB2.[ 20 ] This activates the transcription factors nuclear factor‐κB (NF‐κB), p38 mitogen‐activated protein kinase (p38MAPK), and c‐Jun N‐terminal kinase (JNK), which enable the production of several other cytokines, adhesion molecules, inflammatory mediators, and enzymes.[ 19 , 20 , 21 ] Through these mechanisms, IL‐1β affects the metabolism of cells and extracellular matrix.[ 22 ] Furthermore, IL‐1β affects the operation of chondrocytes via two mechanisms. One, interfering with the synthesis of proteins such as type‐II collagen and aggrecan decreases extracellular matrix production.[ 23 ] The other is by increasing the synthesis of enzymes such as interstitial collagenase (MMP‐1), stromelysin‐1 (MMP‐3), and collagenase 3 (MMP‐13), which have a destructive effect on cartilage.[ 24 ] IL‐1β present in the joint can also stimulate the synthesis of other proinflammatory cytokines such as tumor necrosis factor alpha (TNFα), interleukin‐6 (IL‐6), interleukin‐8 (IL‐8), and chemokine (C‐C motif) ligand 5 (CCL5).[ 25 ] In addition, IL‐1β also secretes other enzymes such as NO, phospholipase A2, cyclooxygenase‐2 (COX‐2), and prostaglandin E2 (PGE2), which play a significant role in the pathophysiology of OA.[ 26 ] IL‐1β generates reactive oxygen species, leading to peroxides and hydroxylated radicals formation and articular cartilage degradation.[ 27 ]
Figure 2.
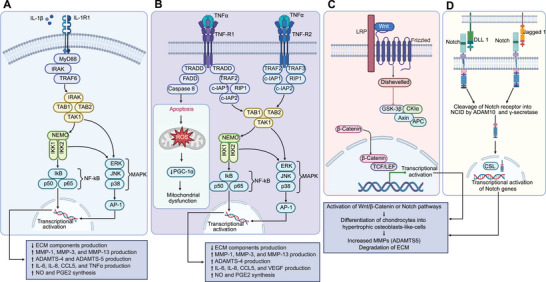
Pathogenesis of OA. A) Intracellular signaling pathways associated with the inflammatory cytokine IL‐1β and its downstream cellular targets and effects. B) Intracellular signaling pathways associated with the inflammatory cytokine TNFα and its downstream cellular targets and effects. C) Wnt–β‐catenin signaling pathway. D) Notch pathway.
TNFα, in combination with IL‐1β, is a proinflammatory cytokine involved in the pathogenesis of OA (Figure 2B).[ 10 ] Studies show that similar to IL‐1β, high levels of TNFα are also observed in the synovial fluid, synovial membrane, cartilage, and the subchondral bone layer of patients with OA.[ 17 , 28 ] Usually, macrophages in the joint can secrete TNFα in addition to that of IL‐1β.[ 17 a] This secreted TNFα has the ability to bind to membrane receptors TNF‐R1 and TNF‐R2 present on nucleated cell surfaces.[ 29 ] Binding of TNFα with TNF‐R1 enables the TRADD adapter protein to interact with the DD domain and gradually bind to other adapter proteins such as cellular inhibitor of apoptosis protein 1 (c‐IAP1), c‐IAP2, TRAF2, and receptor interacting protein 1 (RIP1).[ 30 ] This complex then enables RIP1 protein to bind with TAK1, TAB1, and TAB2, stimulating the phosphorylation of the IKK complex.[ 31 ] This results in the activation of NF‐κB, JNK, and p38MAPK transcription pathways.[ 31 ] Similarly, the binding of TNFα with TNF‐R2 will also enable mutual interactions of proteins such as c‐IAP1, c‐IAP2, TRAF2, and TRAF3, resulting in the activation of JNK kinase and the transcription factor NF‐κB.[ 32 ] In most cases, the effect of TNFα is similar to the effect of IL‐1β in the pathogenesis of OA.[ 33 ] This similarity is due to the activation of the same group of intracellular signaling pathways, which triggers inflammation and catabolism in joint tissues.[ 21 , 22 ] Like IL‐1β, TNFα also affects the chondrocytes from synthesizing proteins such as type‐II collagen and proteoglycans, resulting in decreased extracellular matrix production.[ 34 ] Moreover, TNFα enables increased production of MMP‐1, MMP‐3, MMP‐13, and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS‐4).[ 35 ] TNFα, in conjunction with IL‐1β, can induce rapid aging in chondrocytes, leading to apoptosis.[ 36 ] This affects chondrogenic progenitor cells (CPCs) migration and disables the cartilage from regeneration.[ 36 , 37 ] This is attributed to reduction in the efficiency of the respiratory chain, leading to reduced ATP production within the mitochondria located in chondrocytes.[ 38 ] Moreover, TNFα also increases the synthesis of other proinflammatory cytokines such as IL‐6 and IL‐8 and secretes enzymes such as iNOS, COX‐2, and PGE2,[ 26 , 39 ] which play a significant role in the pathophysiology of OA.[ 26 ]
Interleukin‐15 (IL‐15) is another proinflammatory cytokine involved in the pathogenesis of OA. IL‐15, a glycoprotein with a mass of 14–15 kDa,[ 40 ] contributes to OA by stimulating the differentiation and proliferation of T‐cells and NK cells.[ 41 ] Moreover, IL‐15 also stimulates the secretion of specific metalloproteinases.[ 42 ] Studies show that increased levels of IL‐15 in patients’ serum also correlate with a higher sensation of pain and severity of lesions.[ 43 ] The CD4+ T cells and mast cells that penetrate the synovial membrane can produce another proinflammatory cytokine, interleukin‐17 (IL‐17),[ 44 ] which affects chondrocytes and fibroblast‐like synoviocytes exhibiting IL‐17R expression on their surface.[ 45 ] Moreover, IL‐17 also influences the secretion of other proinflammatory cytokines such as IL‐1β, TNFα, and IL‐6.[ 46 ] Similar to IL‐15, increased levels of IL‐17 in patients’ serum and synovial fluid correlate with the severity of lesions.[ 47 ] Interleukin‐18 (IL‐18) is a precursor form of the pro‐IL‐18, which is transformed into a biologically active form after the activation of Caspase 1.[ 48 ] High levels of Caspase 1 in articular cartilage and synovium stimulate the formation of IL‐1β and IL‐18.[ 49 ] Similar to IL‐15 and IL‐17, an increased concentration of IL‐18 in patients’ serum, synovial fluid, synovium, and cartilage also correlates with a higher degree of severity.[ 50 ]
2.2. Wnt–β‐Catenin Signaling
In addition to proinflammatory cytokines and their signaling pathways, the canonical Wnt frizzled–β‐catenin pathway also plays a major role in the pathogenesis of OA (Figure 2C).[ 51 ] Studies show that increased levels of β‐catenin are observed in patients with deteriorating cartilage. This suggests that the failure to limit Wnt signaling might play a major role in OA. Generally, β‐catenin in the cytoplasm is in the form of an oligomeric complex consisting of Axin, casein kinase (CK), the adenomatous polyposis coli tumor suppressor protein (APC), and glycogen synthase kinase 3β (GSK3β).[ 52 ] However, when soluble Wnt glycoproteins bind to coreceptors LRP5, LRP6, and CRD of the frizzled receptor, a signal is transmitted through the β‐catenin‐dependent canonical Wnt pathway. This signal relocates Axin to the plasma membrane complex and releases β‐catenin from the oligomeric CK–APC–GSK3β–Axin complex.[ 51 ] This increases the levels of free cytoplasmic β‐catenin, leading to the translocation of cytoplasmic β‐catenin into the nucleus, where it interacts with transcription factors and activates a transcriptionally active complex.[ 53 ] This complex targets genes such as cyclin D1, MMP‐3, ADAMTS‐5, and CD‐44, resulting in the degradation of the extracellular matrix.
2.3. Notch Pathway
Another canonical pathway that can cause OA is the Notch signaling pathway (Figure 2D).[ 54 ] This pathway is initiated when furin convertase cleaves (first cleavage) the trans‐Golgi network into two fragments. These two fragments can then reassociate and reach the cell surface as a transmembrane receptor, consisting of a Notch tethered membrane and an extracellular domain. This complex then binds to the ligand, exposing the receptor to enzymes such as ADAM10 and ADAM17. These enzymes then cleave (second cleavage) the transmembrane receptor into a transmembrane domain and the intracellular domain.[ 55 ] Next, an enzyme, γ‐secretase cleaves (third cleavage) the transmembrane domain of the receptor and releases the intracellular domain of the Notch receptor. This can then translocate to the nucleus and interact with the transcription factor, CSL to activate the transcription of target genes, resulting in increased production of MMPs and the degradation of extracellular matrix.[ 54 ]
3. Injectable Hydrogel for Intra‐Articular Delivery of OA‐Therapeutic
A range of injectable hydrogels have been engineered for the intra‐articular delivery of therapeutics aimed at targeting OA pathogenesis. These hydrogels are capable of delivering small molecule drugs, peptides, and proteins. Owing to their shear‐thinning properties, these injectable hydrogels can be administered through minimally invasive techniques, thereby mitigating complications commonly associated with surgical interventions. Furthermore, the minimally invasive nature of these injectable hydrogels allows for targeted delivery of therapeutics directly to the site, thereby eliminating the systemic side effects commonly associated with oral or intravenous administration.
Injectable hydrogels can be synthesized using various natural and synthetic polymers. Natural polymers, including alginate, chitosan, hyaluronic acid, collagen, chondroitin sulfate, gelatin, dextrin, and fibrin offer biocompatibility and bioactivity, making them suitable for hydrogel synthesis. Conversely, synthetic polymers such as poly(ethylene) glycol (PEG), polyethylene oxide, poly(vinyl alcohol) (PVA), poly(lactic‐co‐glycolic acid) (PLGA), poly(lactic acid), and poly(N‐isopropyl acrylamide) provide tunable mechanical properties and degradation rates. In this section, we will discuss various injectable hydrogels for delivery of therapeutics.
Injectable hydrogels can be crosslinked using either physical or chemical crosslinking strategies (Figure 3 ). Physical crosslinking involves noncovalent interactions such as ionic bonds, hydrogen bonds, hydrophobic interactions, van der Waals, dipole–dipole, or London dispersion forces.[ 56 ] These physical hydrogels, being crosslinked without chemical modification, exhibit compatibility with biomolecules and living cells. However, due to the involvement of weak interactions, these gels often demonstrate poor mechanical properties and susceptibility to degradation.[ 57 ] In contrast, chemical crosslinking involves the formation of covalent bonds using strategies such as click chemistry, Micheal addition, Schiff base reaction, enzyme‐mediated reaction, or photopolymerization.[ 58 ] Unlike physical crosslinking, these hydrogels offer stability to the hydrogel against degradation. They exhibit robust mechanical strength and flexibility in terms of physicochemical properties like gelation time, pore size, chemical functionalization, and degradation characteristics.[ 57 , 59 ] However, some by‐products or chemical crosslinkers may affect the biocompatibility, stability, or functionality of the hydrogels, necessitating additional purification steps. In recent years, dual crosslinking of hydrogels has shown great promise to improve the efficacy of injectable hydrogels. Dual crosslinking utilizes a combination of physical and chemical crosslinking yielding hydrogels with improved mechanical strength, better control over swelling properties, enhanced stability, and tunable degradation rates compared to hydrogels formed via a single crosslinking method.[ 60 ]
Figure 3.
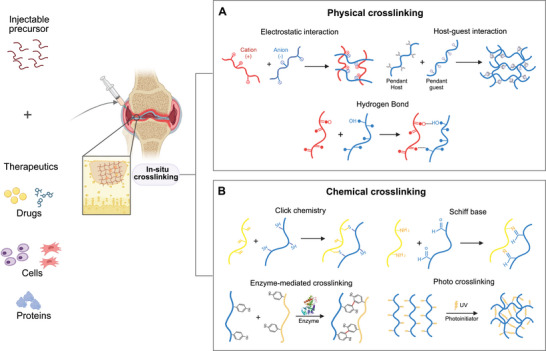
Schematic depicting the crosslinking process of an injectable hydrogel incorporating therapeutic agents such as small molecules, proteins, and cells. Injectable hydrogels can be crosslinked using two common methods: A) Physical crosslinking, and B) Chemical crosslinking. Physical crosslinking involves the formation of non‐covalent bonds, such as electrostatic interactions, hydrogen bonds, and host‐guest interactions. On the other hand, chemical crosslinking utilizes covalent bonds through various strategies, includingclick chemistry, Schiff base reactions, enzyme‐mediated reactions, and photopolymerization.
Consequently, each approach carries distinct advantages and challenges, requiring careful consideration when selecting an appropriate method for developing the desired injectable hydrogel formulation. In the following section, physical, chemical, and dual crosslinked hydrogels are investigated for intra‐articular delivery of the therapeutic.
3.1. Physically Crosslinked Injectable Hydrogels
Electrostatic interaction is a physical crosslinking method of fabricating an injectable hydrogel. The electrostatic interaction between an anionic and a cationic polymer chain can be utilized to form the physically crosslinked hydrogels. Anionic polymers include alginate, hyaluronate, pectin, heparin, chondroitin sulfate, dextran sulfate, poly‐l‐glutamates, polyacrylic acids, and poly(methyl vinyl ether‐co‐maleic anhydride), while cationic polymer includes chitosan, poly(l‐lysine), poly(ethylene amine), poly(allylamine), polyvinyl amine hydrochloride, and poly(diallyldimethylammonium chloride). In some instances, oppositely charged ions can also be used for crosslinking. For instance, alginate can undergo crosslinking upon mixing with calcium chloride and other divalent ions.[ 61 ] These physically crosslinked hydrogels can entrap therapeutic and release it as they dissolve over time under in vivo conditions.
A hybrid injectable hydrogel made from alginate has been synthesized, featuring microspheres of poly(ε‐caprolactone)‐b‐poly(ethylene glycol)‐b‐poly(ε‐caprolactone) (Figure 4A).[ 62 ] This hydrogel incorporated calcium gluconate crystals as crosslinking agents for the alginate matrix, achieving crosslinking within 3 min postinjection. By varying the concentration of porous microspheres, the mechanical strength and degradation rate of the hydrogel was controlled. A three‐day study confirmed that the scaffold facilitated chondrocyte proliferation. The hydrogel was then injected into rabbits with full‐thickness cartilage defects in their left knees. After 18 weeks, the microspheres/alginate + cells group demonstrated a 96.7% improvement in osteochondral cell regeneration compared to control groups (blank, alginate, microsphere, and cells‐only). The scaffold was fully degraded by the end of study, making way for new cartilage formation. One limitation observed was the migration of microspheres into adjoining tissues, which could be attributed to slower gelation times of alginate.
Figure 4.
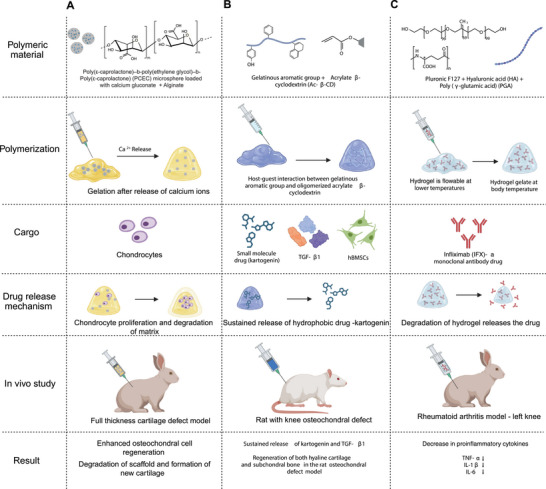
Physical crosslinking hydrogels. A) Formation of hybrid injectable alginate hydrogel with poly(ε‐caprolactone)‐b‐poly(ethylene glycol)‐b‐poly(ε‐caprolactone) (PCEC) microsphere containing calcium gluconate crystals crosslinked using the electrostatic interaction between alginate and calcium ions. The fully degradable hydrogel scaffold provided a suitable environment for proliferation and osteochondral cell regeneration of chondrocytes for repairing damaged cartilage. B) Injectable hydrogel formed by crosslinking GelMa and acrylate β‐cyclodextrin using the host–guest supramolecular interaction. The self‐healing hydrogel mediated a sustained release of kartogenin and TGF‐β1 for enhanced chondrogenesis of bone marrow‐derived mesenchymal stem cells (BMSCs) in vitro and in vivo. C) The thermoresponsive hydrogel formed by mixing PF127, HA, and PGA gelled under body temperature (37 °C). The injectabale hydrogel loaded with infliximab downregulated the expression of inflammatory cytokines, such as tumor necrosis factor α (TNFα), interleukin‐1β (IL‐1β), interleukin‐6 (IL‐6), and interleukin‐17 (IL‐17) in the synovial fluid and cartilage.
Another mechanism for physical crosslinking is host–guest chemistry, which relies on the formation of complexes between molecules or ions through noncovalent interactions, such as ionic/hydrogen bonding, hydrophobic interactions, and van der Waals forces. These interactions are pivotal in stabilizing and maintaining the 3D structure of large protein molecules. Using a host–guest macromer approach, a supramolecular gelatin hydrogel was developed that exhibited resilience, self‐healing, and injectability (Figure 4B).[ 63 ] This hydrogel was engineered to carry multiple types of cargo: the small molecule drug kartogenin, the protein TGF‐β1, and human bone marrow‐derived mesenchymal stem cells (BMSCs) encapsulated within the matrix. This hydrogel was injected into the knee of a rat osteochondral defect model, and its performance in cartilage repair was evaluated. This hydrogel provided sustained therapeutic delivery for up to 28 days and permitted cell infiltration, as well as shape adaptation for the delivered mesenchymal stem cells (MSCs) in both in vitro and in vivo environments. These features are key for enabling long‐term chondrogenesis of MSCs, essential for cartilage repair. Despite these advantages, the study identified limitations, including the hydrogel's suboptimal mechanical properties, the challenges associated with controlling therapeutic dose release, and questions about the material's suitability for the regeneration of other tissue types.
Thermoresponsive physical crosslinking is another widely‐used method for hydrogel synthesis, relying on temperature‐dependent reversible reactions for the crosslinking process. Hydrogels synthesized through this technique are designed to transition from liquid to solid or solid to liquid after injection. One study synthesized an organic hydrogel using triblock polymers and isopropyl myristate with thermoresponsive crosslinking.[ 64 ] The resulting nanoemulsion maintained relative stability and successfully transitioned from a solid to a liquid state. In another study, a thermoresponsive injectable hydrogel was created using PGA, hyaluronic acid, and a monoclonal antibody therapeutic (Figure 4C).[ 65 ] This hydrogel exhibited a reduction in proinflammatory cytokines such as TNFα, IL‐1β, and IL‐6. These cytokines are key players in the inflammatory immune response. Additionally, the hydrogel caused minimal tissue damage upon injection, demonstrated high levels of biocompatibility, and effectively prevented further cartilage deterioration.[ 65 ] Both studies highlight the potential of thermoresponsive injectable hydrogels as immunosuppressive agents. These hydrogels also demonstrated stability and other beneficial properties, particularly in the context of rheumatoid arthritis models. These findings suggest the need for further research to explore their applicability in OA therapy. Additional examples of physically crosslinked injectable hydrogels that are evaluated for OA treatment are shown in Table 1 .[ 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 ]
Table 1.
Injectable hydrogels via physical crosslinking.
| Cargo | Polymeric material | Gelation mechanism | Properties | Refs. |
|---|---|---|---|---|
| Small drug molecules | ||||
| Kartogenin and TGF‐β1 | Methacrylated gelatin with acrylated β‐cyclodextrins | Host–guest interactions | Natural compound delivery to enhance the activities of the critical protein for cell survival proliferation and MSCs for improving the regeneration of articular cartilage | [63] |
| Kartogenin | Silk–fibroin–chitosan | Lyophilization | Self‐healing, biocompatibility | [66] |
| Dexamethasone | Drug conjugated to multiarm Avidin | Electrostatic interactions | Promotion of chondrogenesis and cartilage regeneration in rabbit knees | [67] |
| Crystal violet | Heparin‐based hydrogel | Electrostatic interactions | Sustained drug release of water‐soluble steroid | [68] |
| Icaritin | Gelatin hydrogels | Host–guest interactions | Self‐assembling, tunable shear‐thinning properties of gels to maintain shape and viscoelastic properties under stress | [69] |
| Ginsenoside Rb1 with TGF‐β1 | Silk fibroin‐coated gelatin scaffold | Lyophilization | Porous microstructure, proper mechanical strength, degradation rate, and sustained release of Rb1 and TGF‐β1; scaffolds promoted the chondrogenic differentiation of rBMSCs and suppression of inflammation genes expression; implanted scaffolds in rats with osteochondral defects promoted hyaline cartilage regeneration | [70] |
| Cells | ||||
| Bone marrow‐derived mesenchymal stem cells (BMSCs), platelet‐rich‐plasma (PRP) | Sodium alginate | Electrostatic interactions | In vitro degradation of the alginate gel was accelerated by the addition of PRP, excellent cytocompatibility, chondrogenesis of BMSCs with upregulation of chondrogenic marker genes SOX9 and Aggrecan | [71] |
| (TGF)‐β3, IFP‐derived stem cells | Fibrin with cartilage microparticles | Lyophilization | Chondrogenesis of freshly isolated fat pad‐derived stromal cells in vivo, included higher levels of sulphated glycosaminoglycan (sGAG), collagen accumulation, and cartilage formation | [72] |
| Autologous nasal chondrocytes (NCs) | Alginate | Electrostatic interactions | Superior and more hyaline‐like repaired tissue both at 3 and 6 months after surgery in rabbits with knee osteochondral defects, repaired tissue possessed similar mechanical properties to the native cartilage | [73] |
| Bone marrow‐derived mesenchymal stem cells (BMSCs), platelet‐rich‐plasma (PRP) | Sodium alginate, strontium‐doped bioglass | Electrostatic interactions | Dual role in cartilage repair, by activating both differentiation of stem cells and macrophage polarization to the M2 phenotype for cartilage regeneration in mice | [74] |
| Mesenchymal stem cells (MSCs) | β‐cyclodextrin (β‐CD)‐modified HA (HA‐CD), and adamantane (Ad)‐modified HA (HA‐Ad) | Host–guest interactions | Shear‐thinning and self‐healing, high cell viability of encapsulated MSCs, notable cartilage tissue regeneration in cartilage defect model rats in 28 days | [75] |
| Immunotherapy | ||||
| Insulin‐like growth factor 1 | Gelatin/PEGDA hydrogel | Host–guest interactions | Biocompatible, sustained release, encourages MSC differentiation, promotes chondrogenesis. | [76] |
| Chitosan | Silver‐doped nanoparticles and chitosan hydrogel | Electrostatic interactions | Reduction in inflammation and activity of p38 protein, preservation of cartilage, and bone integrity | [77] |
| Chitosan and citric acid | Subchondral lamellar bone hydrogel | Lyophilization | Improvement in the strength of mechanical structure, proper flexibility, and cytokine free reaction, which would be less likely to induce an inflammatory response | [78] |
3.2. Covalently Crosslinked Injectable Hydrogels
3.2.1. Click Chemistry
Introduced by Sharpless and colleagues in 2001, “Click” chemistry has garnered significant attention for its role in synthesizing hydrogels and microgels, particularly for applications in tissue engineering and drug delivery.[ 79 ] Characterized by high selectivity, efficiency, and compatibility with a diverse array of functional groups, “Click” chemistry has become a preferred method for crafting injectable hydrogels. This approach encompasses a range of reactions including azide and alkyne cycloaddition, thiol–ene, and Diels–Alder (DA). In recent years, injectable hydrogels produced through “Click” chemistry have been successfully engineered to carry various therapeutic agents. These range from small molecule drugs and growth factors to cells and proteins/peptides. Such hydrogels have shown promise in enhancing cartilage regeneration and alleviating pain, specifically in the treatment of OA.
Researchers have employed cartilage‐mimetic hydrogels to deliver small molecules like extracellular matrix analogs (e.g., chondroitin sulfate and RGD peptides), aiming to induce mechanical stimulations that enhance chondrogenesis.[ 80 ] These mechanical cues have been found to decrease the expression of hypertrophic markers such as collagen X and MMPs, while also reducing calcium deposition in MSCs. For instance, one study utilized a photo‐clickable thiol: norbornene reaction to crosslink thiolated chondroitin sulfate, 2‐arm PEG dithiol, 8‐arm PEG norbornene, and RGD peptide into a cartilage‐mimetic hydrogel. Human MSCs encapsulated in these hydrogels received physiochemical cues native to the cartilage environment. This led to the inhibition of MSC hypertrophy and the maintenance of a stable chondrogenic population, achieved by upregulating the p38 MAPK pathway and downregulating the SMAD 1/5/8 signaling pathways (Figure 5A).[ 80 ] However, the study had its limitations. It primarily focused on two well‐known pathways responsible for chondrogenesis, ignoring the likelihood of other signaling mechanisms being involved. Additionally, the use of nondegradable, stable hydrogel matrices minimized variability in hydrogel chemistry and mechanical properties but also constrained extracellular matrix deposition to paracellular spaces. Given these constraints, future comprehensive research employing a degradable and biomimetic hydrogel that investigates additional signaling pathways would be beneficial.
Figure 5.
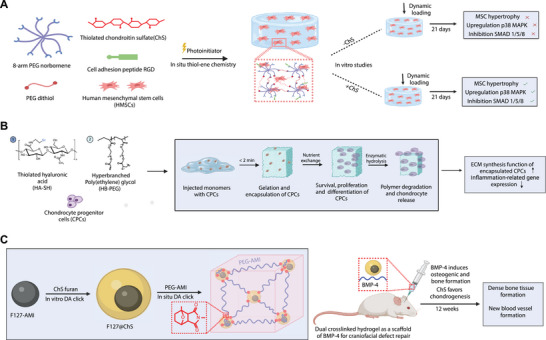
Click chemistry‐mediated crosslinked hydrogel. A) Formation of cartilage‐mimetic injectable hydrogel utilizing the photo‐clickable thiol–ene reaction between multiarm PEG norbornene monomer and crosslinker PEG dithiol, along with the incorporation of extracellular matrix mimetics of chondroitin sulfate, RGD, and HMSCs. Hydrogel with chondroitin sulfate when combined with dynamic compressive loading acted as a potent physiochemical cue to the cartilage environment upregulating p38 MAPK and downregulating SMAD 1/5/8 signaling pathways maintaining stable HMSCs chondrogenesis for cartilage repair. B) Encapsulation of chondrocytes progenitor cells in HA‐SH and HB‐PEG injectable hydrogel crosslinked via thiol–ene click chemistry for cartilage regeneration. The porous hydrogel promoted the extracellular matrix synthesis function of the encapsulated cells and downregulated gene expression related to inflammation for treating osteoarthritis. C) In situ crosslinking of chondroitin sulfate with PEG monomers using Diels–Alder reaction. Hydrogels containing ChS decreased the expression of TNFα and IL‐1β, and favored chondrogenesis of encapsulated cells.
Another research team took a similar approach, encapsulating cartilage‐derived progenitor cells (CPCs) from rat articular cartilage into an injectable hydrogel. This hydrogel was fabricated using hyperbranched poly(ethylene glycol) (HB‐PEG) and thiol‐functionalized hyaluronic acid (HA‐SH) via an in situ thiol–ene Michael reaction, aimed at cartilage regeneration (Figure 5B).[ 81 ] Remarkably, the gelation time for this HB‐PEG and HA‐SH mixture was less than 2 min. This is considerably shorter than the gelation times observed for dithiol peptides and PEGDA (≈13 min), and for HA‐SH and PEGDA (≈5–30 min). This rapid gelation was advantageous for ensuring an orderly distribution of both the hydrogel precursor and the encapsulated cells, crucial for optimal in vivo gel formation. Moreover, the chemical crosslinking between the vinyl groups of HB‐PEG and HA‐SH led to the formation of a porous network with pore sizes ranging from 5 to 30 µm. This porosity enhanced nutrient exchange and facilitated cell migration within the hydrogel. While PEGDA‐based hydrogels have previously been employed for similar applications, their photopolymerization crosslinking method—utilizing UV irradiation—poses a risk of tissue damage due to DNA fragmentation and cell injury.[ 82 ] In terms of biocompatibility, this thiol–ene‐based hydrogel demonstrated high cell compatibility for the encapsulated CPCs, with an impressive 85% cell viability after seven days in culture.[ 81 ] Gene expression analyses further showed increased expression of anabolic genes like SOX9, collagen type II, and aggrecan, suggesting enhanced extracellular matrix synthesis favorable for cartilage regeneration.
Click chemistry offers versatile applications for formulating various drug types, including those aimed at inhibiting TNFα. One recent study explored the therapeutic potential of a hydrogel made from chondroitin sulfate and glucosamine for inhibiting TNFα.[ 83 ] Being a native component of the extracellular matrix, chondroitin sulfate is highly biocompatible.[ 84 ] The study found a positive correlation between the administration of this hydrogel and decreased levels of TNFα in the synovial fluid, which corresponded to reduced inflammation and joint pain in affected subjects.[ 83 ] Another investigation utilized click chemistry to create a hydrogel comprising chondroitin sulfate and 11‐azido‐3,6,9‐trioxaundecan‐1‐amine.[ 82 ] This formulation successfully promoted chondrocyte production, further inhibiting TNFα. In addition, chondroitin sulfate hydrogels can also inhibit interleukins, specifically IL‐1β. The hydrogel achieves this by downregulating the release of MMPs, which play a role in cartilage degradation and consequently cause inflammation.[ 85 ] The DA reaction offers another avenue for creating biocompatible hydrogels, particularly those containing chondroitin sulfate, to counteract the effects of TNFα and IL‐1β. One innovative study employed this reaction to synthesize a dienophile and thermosensitive copolymer, which was then used as a component in a hydrogel aimed at bone repair applications (Figure 5C).[ 86 ] The study demonstrated that this chemical approach produced a stable, biocompatible scaffold while preserving the chondrogenic activity of the incorporated materials.[ 86 ] In a rat model, this DA‐reaction‐based hydrogel was employed as a scaffold to carry bone morphogenetic protein 4 (BMP‐4) for craniofacial defect repair. The results were promising, showing a substantial increase in osteoid tissues within an 8‐week period and the development of denser bone tissues rich in blood vessels by the 12‐week mark.[ 86 ]
3.2.2. Injectable Microgels (Granular Hydrogels)
Microscopic particles of hydrogels (microgels) are a special class of injectable hydrogels that are highly investigated for cartilage repair.[ 87 ] Click chemistry strategies and microfluidic technology play a major role in the generation of these microgels.[ 88 ] Researchers have developed injectable stem cell‐laden microgels to promote cartilage repair.[ 89 ] They generated the microgels by mixing thiolated gelatin (Gel‐SH) and vinyl sulfonated hyaluronic acid (HA‐VS) with BMSCs via the droplet‐based microfluidic approach.[ 90 ] These materials were crosslinked into hydrogels via the thiol‐Micheal addition reaction under ambient temperature and neutral pH (Figure 6A).[ 89 ] Since Gel‐SH is a protein derivative and HA‐VS is a polysaccharide derivative, it mimics the extracellular matrix and provides an excellent microenvironment for cell proliferation. Moreover, this cell proliferation induces self‐assembly of microgels into macroporous scaffolds, which enhances nutrient transport and thereby promotes stem cell chondrogenesis. These microgels were hypodermically implanted in a mouse model to observe ectopic cartilage formation. Immediately after the implantation, the implant size in the control group (pure BMSCs) was slightly larger than the microgel group. However, it reduced dramatically within 3 days, whereas the microgel group retained a stable implant size. This is due to the increased BMSC viability in the microgels. Moreover, removing the implants after 8 weeks showed that the formation of cartilage tissues with smooth surfaces in the microgel group whereas smaller morphologies with rough and irregular surfaces in the control group.[ 89 ] This shows that the BMSC‐laden microgels can facilitate cartilage repair and aid in the reconstruction of chondral defects.
Figure 6.
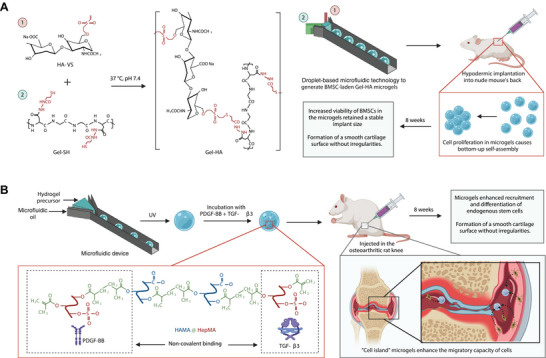
Click chemistry‐mediated crosslinked microgels. A) BMSC‐laden gelatin/hyaluronic acid hybrid (Gel‐HA) microgels generated by droplet‐based microfluidic technology and crosslinked at 37 °C and pH 7.4 via thiol‐Michael addition reaction. The proliferation of BMSC encapsulated within the microgels causes bottom‐up self‐assembly. This self‐assembly enhances the BMSC viability in microgels, which enables formation of cartilage tissues with smooth surfaces in vivo. B) Growth‐factor loaded methacrylated hyaluronic acid and heparin blend (HAMA@HepMA) “cell island” microgels generated using microfluidic technology and photopolymerization process. These microgels recruit endogenous stem cells and promote chondrogenic differentiation by releasing growth factors and aid in cartilage formation.
To further improve the microgel injectability and BMSC viability, the same research group has developed microgels using dynamic covalent bonds.[ 91 ] To generate these microgels, KGN‐loaded cyclodextrin nanoparticles (KGN@CD NPs) were included with phenylboronic acid‐grafted methacrylate hyaluronic acid (HAMA‐PBA), methacrylate gelatin (GelMA), and BMSCs via droplet‐based microfluidic approach and photocrosslinked. These microgels were then assembled via dynamic crosslinking between phenyl‐boronic acid and dopamine‐modified hyaluronic acid (HA‐DA). Furthermore, these microgels were injected into articular cartilage defects of a rabbit model, and their repair performances were observed. After 8 weeks of implantation, the newly formed cartilages were thicker, abundant, and transparent compared to the control group (normal saline).[ 91 ] Comparing the properties of the newly formed cartilages between the BMSC‐laden HA‐Gel group and KGN@CD‐HA‐Gel group shows that the KGN@CD‐HA‐Gel group had significantly higher O'Driscoll scores than the BMSC‐laden HA‐Gel group, demonstrating the importance of sustained KGN release in cartilage repair.
Another research group has developed a new approach in which injectable microgels can recruit endogenous stem cells for repairing OA (Figure 6B).[ 92 ] This approach where the microgels act as a “cell island” was inspired by a biological phenomenon where islands recruit plenty of seabirds for nesting and breeding. The microgels were generated by blending methacrylated hyaluronic acid and heparin (HAMA@HepMA) via a microfluidic device followed by photocrosslinking.[ 92 ] Furthermore, transforming growth factor‐β3 (TGF‐β3) and platelet‐derived growth factor‐BB (PDGF‐BB) were noncovalently included in the microgels. Moreover, these microgels were injected into the intraknee joints of the osteoarthritic rat model to evaluate repair performance. After 8 weeks, radiological examination showed reduced cartilage degradation in the microgel group compared to the control groups, and histological results showed the formation of a smooth cartilage surface without irregularities in the microgel group compared to the control groups.[ 92 ] This could be due to the enhanced recruitment and differentiation of endogenous stem cells. This suggests that “cell island” microgels might provide a new approach to treat cartilage damage.
3.2.3. Enzyme‐Mediated Crosslinking
Enzyme‐mediated crosslinking offers a compelling avenue for developing injectable hydrogels, particularly due to its rapid gelation capabilities and mild reaction conditions.[ 84 ] Such methods enable polymer crosslinking at neutral pH and moderate temperatures, all while avoiding the production of cytotoxic byproducts. A variety of enzymes have been employed for this purpose in the realm of cartilage tissue engineering, including but not limited to tyrosinase, transglutaminase, phosphopantetheinyl transferase, lysyl oxidase, thermolysin, β‐lactamase, phosphatase/kinase, and horseradish peroxidase.[ 93 ] Of these, tyrosinase and horseradish poeroxidase are most frequently used due to their ability to catalyze the oxidation of phenolic compounds, such as tyramine, to facilitate crosslinking.[ 94 ] This adds another dimension to the array of methodologies available for creating functional, biocompatible hydrogels suited for cartilage regeneration applications.[ 94 ]
Injectable hydrogels synthesized through enzyme‐mediated chemical crosslinking have shown promise in the treatment of OA. These hydrogels can be rapidly formed, crucially preserving the bioactive properties of their constituent materials. This unique combination of rapid synthesis and bioactivity retention enhances their therapeutic potential for OA interventions. One study utilized tyrosinase, a common chemical crosslinking agent, to modify polyphenolic compounds that inhibit natural inflammatory agents.[ 95 ] This work demonstrated that the activity of TNFα could be inhibited as the conjugation of the polyphenolic components into their respective polymers could form a stable, bioactive hydrogel.[ 95 ] The importance of these findings lies in the hydrogels’ demonstrated ability to reduce monocyte and macrophage activity, which are both modulated by TNFα and IL‐1 cytokines.[ 87 ] This suggests that enzyme‐mediated hydrogels could be an effective tool in mitigating inflammation and enhancing tissue repair in OA treatments.[ 95 ]
In another study, an injectable hydrogel that incorporated liposome anchored teriparatide (PTH(1‐34)) into gallic acid‐grafted gelatin lipo@PTH(1‐34)GGA) was obtained by crosslinking using transglutaminase (Figure 7A).[ 96 ] The hydrogel exhibited a porous structure and sustainably released PTH(1‐34) over a 20‐day period, while fully degrading in phosphate‐buffered saline (PBS) within 28 days. This formulation promoted the proliferation of ATDC5 cells, as evidenced by the upregulation of key proteins such as Ki‐67, c‐Fos, and PTH1R. Furthermore, the hydrogel had protective effects against apoptosis in IL‐1β‐induced ATDC5 cells. Specifically, the apoptosis rate decreased from 11% to 9.05% upon the introduction of the hydrogel. This protective mechanism is attributed to the upregulation of key anabolic genes like SOX9, Bcl‐2, Col2a1, and Acan, as well as the downregulation of catabolic genes such as BAX, ADAMTS5, iNOS, COX‐2, IL‐6, and TNFα. These genetic changes are thought to modulate the PI3K/AKT signaling pathway. Additionally, in a 10‐week OA mouse model, the injection of this hydrogel led to a decrease in cartilage degradation, promotion of extracellular matrix synthesis, and significant improvement in GAC content.
Figure 7.
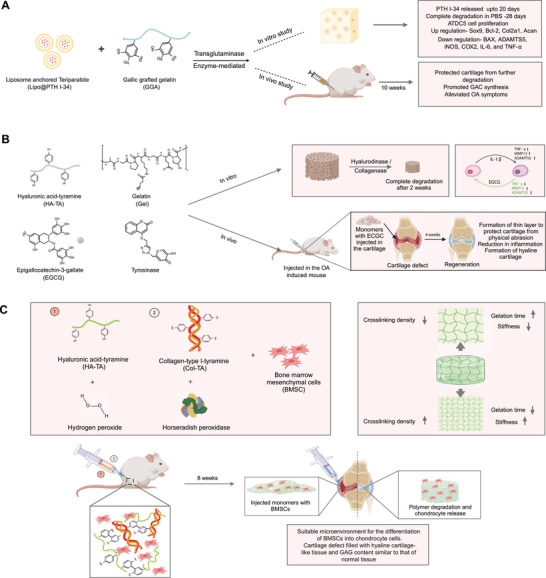
Enzyme‐mediated crosslinked hydrogel. A) Gallic acid‐grafted gelatin hydrogel incorporating liposome loaded with PTH(1‐34), crosslinked using transglutaminase enzymatic method. The injectable hydrogel upregulated Ki‐67, c‐Fos, and PTH1R resulting in ATDC5 proliferation. After 10 weeks of injection in OA model mice, cartilage degradation decreased, and extracellular matrix synthesis was promoted. B) EGCG‐loaded HA/gelatin hybrid hydrogel crosslinked via tyrosinase enzymatic method for OA therapy. EGCG inhibited proinflammatory signaling cascades by limiting nuclear translocation of nuclear factor‐kappa B (NF‐kB), inhibiting metalloproteinases (MMP‐13) and cyclooxygenase‐2 (COX‐2). The in vivo evaluation of EGCG‐laden hydrogel demonstrated synergetic anti‐inflammatory and chondroprotective effects for OA therapy. C) Encapsulation of BMSCs in hyaluronic acid and collagen functionalized with tyramine‐ crosslinked via enzyme horseradish peroxidase and hydrogen peroxide. The gelation time and stiffness varied depending on the concentration of HRP and H₂O₂. In cartilage‐defective mice, the hydrogel provided an ideal microenvironment for differentiation of BMSC promoting repair of damaged cartilage.
In a related study, an injectable hydrogel system was developed using tyramine‐conjugated hyaluronic acid and gelatin, which were enzymatically crosslinked via tyrosinase (Figure 7B).[ 97 ] This hybrid hydrogel was enhanced by the incorporation of epigallocatechin‐3 gallate (EGCG) molecules, recognized for their anti‐inflammatory properties and radical‐scavenging capabilities. Remarkably, integrating EGCG into this tyramine‐tethered hydrogel system reduced cytotoxicity compared to its freestanding form, while preserving its anti‐inflammatory effects. In addition to these attributes, the enzyme‐mediated hydrogel system demonstrated several in vivo advantages in mouse OA model, including robust crosslinking, ease of injection, high cytocompatibility, and a significant reduction in cartilage degradation.[ 97 ] Moreover, when chondrocytes were encapsulated within these hydrogels, they promoted GAG and collagen II accumulation in vivo and showed progression in the OA treatment for the mouse model.
The mild conditions of enzymatic reactions offer the advantage of crosslinking natural polymers without compromising their bioactivity.[ 93 ] For example, collagen, a natural polymer, has been shown to support the attachment, proliferation, and differentiation of MSCs in cartilage regeneration applications.[ 98 ] However, collagen‐based biomaterials have limitations such as poor water solubility, rapid degradation, extended gelation time, and weak mechanical strength.[ 99 ] To overcome these challenges, an injectable hydrogel consisting of collagen‐type I‐tyramine (Col‐TA) and hyaluronic acid‐tyramine (HA‐TA) alongside BMSCs and transforming growth factor‐β1 (TGF‐β1) was synthesized (Figure 7C).[ 100 ] This hydrogel was enzymatically crosslinked using horseradish peroxidase (HRP) and hydrogen peroxide (H₂O₂). Importantly, the gelation time could be adjusted from seconds to minutes by varying the concentrations of HRP and H₂O₂. The resulting hydrogel exhibited a favorable swelling ratio, enabling uniform BMSC proliferation throughout the material and a significant increase in cell viability by the seventh day of in vitro culture. Additionally, the hydrogel's high swelling ratio created an optimal microenvironment for BMSC survival, differentiation, and proliferation, while facilitating nutrient and metabolite exchange.[ 101 ] In vitro assays under chondrogenic differentiation conditions for 28 days revealed upregulated expression of key extracellular matrix proteins such as Col II, GAGs, and SOX9. This suggests that the hydrogel successfully provided a structured microenvironment for BMSC differentiation into chondrocyte cells. In vivo studies further supported these findings. When the hydrogel was injected into rat cartilage defects and allowed to set for eight weeks, it facilitated the formation of hyaline cartilage‐like tissue and glycosaminoglycan content comparable to that of normal cartilage. Overall, both in vitro and in vivo results indicate that this enzymatically crosslinked hydrogel offers an optimal setting for BMSC differentiation into chondrocyte cells, thereby promoting the repair of damaged cartilage.
3.2.4. Schiff Base Crosslinking
Schiff base crosslinking involves the formation of a covalent imine bond between a primary amine and a carbonyl compound, which can be either an aldehyde or a ketone. This reaction is particularly advantageous because it can occur under physiological conditions in an aqueous solution, offering fast gelation times and excellent biocompatibility.[ 102 ] Polysaccharides serve as ideal candidates for the development of injectable hydrogels through Schiff base crosslinking. This is largely due to the abundance of functional groups present in their molecular backbone, which can readily participate in Schiff base reactions. Some polysaccharides, like chitosan, contain primary amine groups that can directly engage in these reactions. Alternatively, functional groups in polysaccharides can be chemically modified into aldehydes and amines to facilitate Schiff base crosslinking. Overall, the presence of multiple reactive sites makes polysaccharides versatile building blocks for injectable hydrogels optimized through Schiff base chemistry.[ 58 , 102 ]
Chitosan has traditionally been crosslinked using small molecule dialdehydes such as glyoxal and glutaraldehyde, substances known for their toxic and mutagenic properties.[ 103 ] However, recent advancements have demonstrated the feasibility of crosslinking chitosan through Schiff base reactions without the use of toxic catalysts. For instance, an injectable hydrogel comprising multi‐benzaldehyde functionalized poly(ethylene oxide‐co‐glycidol) (poly(EO‐co‐Gly)‐CHO) and glycol chitosan have been developed (Figure 8A).[ 104 ] This hydrogel was formulated using a Schiff base reaction under physiological conditions. One of its notable features was the tunability of its physical properties, such as gelation time, water absorption, stiffness, and rate of degradation, which could be adjusted by varying the concentration of poly(EO‐co‐Gly)‐CHO. Although these hydrogels were not tested in animal models for cartilage repair, several in vitro findings suggest their ability to achieve cartilage regeneration. Encapsulated chondrocytes within this hydrogel matrix showed excellent viability, exceeding 90% even after two weeks. The hydrogel supported oxygen and nutrient transport, offering an ideal microenvironment for cell survival and proliferation. This also increased the capability of the encapsulated chondrocytes to preserve their phenotype, which is a prerequisite for hyaline cartilage regeneration.
Figure 8.
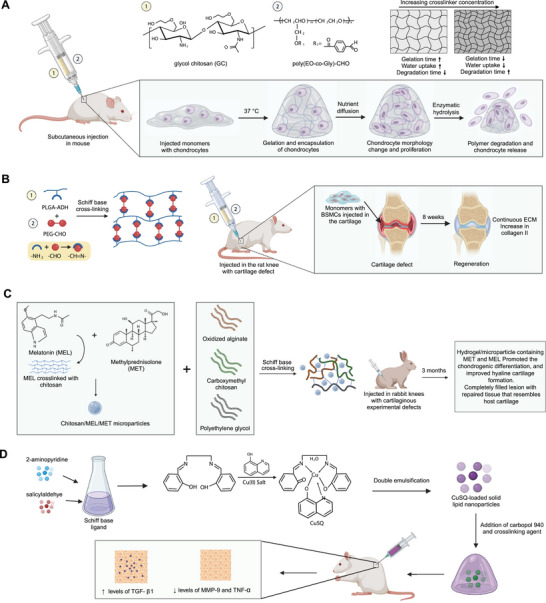
Schiff base mediated crosslinked hydrogel. A) Formation of poly(ethylene oxide‐co‐glycidol)‐CHO ((poly(EO‐co‐Gly)‐CHO)) and glycol chitosan‐based injectable hydrogel crosslinked using the Schiff base reaction. The degradable hydrogel provided a suitable microenvironment for oxygen and nutrient exchange for the proliferation of chondrocytes. B) In situ self‐crosslinking of PLGA/PEG hydrogels. Injecting hydrogels with BMSCs in the cartilage increased collagen II and promoted extracellular matrix formation. C) Injectable in situ forming microparticle/hydrogel system consisting of small molecule drugs, melatonin, and methylprednisolone crosslinked with chitosan using ionic gelation to form microparticles. The microparticles were added in the hydrogel system of oxidized alginate, carboxymethyl chitosan (CMC), and polyethylene glycol, crosslinked through the Schiff base reaction between the amine group of CMC and aldehyde group of oxidized alginates. The hydrogel injected in vivo model with cartilaginous experimental defects promoted chondrogenic differentiation, and improved hyaline cartilage formation. D) Injectable hydrogel loaded with Schiff base CuSQ solid lipid nanoparticles to express enhanced TGF‐β1 and reduced levels of MMP‐9. Schiff base CuSQ was formulated using 2‐aminopyridine, salicylaldehyde, and Cu(II) salt.
Similarly, in another study injectable hydrogels are designed by crosslinking adipic dihydrazide‐modified poly(l‐glutamic acid) (PLGA‐ADH) with benzaldehyde‐terminated poly(ethylene glycol) (PEG‐CHO) through a Schiff base mechanism (Figure 8B).[ 105 ] The degradation rate of these hydrogels could be fine‐tuned by adjusting the molar ratios of ─CHO to ─NH2 groups. Importantly, BMSCs also showed favorable survival and proliferation within these hydrogels. In an in vivo study using a rat model with cartilage defects, the injectable hydrogels facilitated cartilage regeneration over an eight‐week period. The new cartilage‐like tissue integrated well with the surrounding tissue, and the resulting extracellular matrix showed high levels of collagen II expression, which is indicative of successful cartilage repair.
Others explored the potential for small molecule delivery in rats by creating an injectable, self‐healing hydrogel.[ 106 ] This was achieved by crosslinking chondroitin sulfate with multiple aldehydes and N‐succinyl‐chitosan through a Schiff base reaction. By manipulating monomer ratios, the team could fine‐tune gel stiffness, gelation kinetics, and water content. A standout feature of this approach was the hydrogel's robust gelation under physiological conditions. Additionally, the dynamic equilibrium facilitated by the Schiff base linkages endowed the hydrogel with a self‐healing capability. While the hydrogels evoked minimal inflammatory responses and demonstrated in vivo biodegradability, it is worth noting that these hydrogels were only administered subcutaneously in rats.
In a related study, Naghizadeh et al. developed an injectable in situ‐forming hydrogel/microparticle system (Figure 8C).[ 107 ] Initially, chitosan microparticles were loaded with two drugs, melatonin and methylprednisolone. These microparticles were then integrated into a hydrogel composed of oxidized alginate, carboxymethyl chitosan, and polyethylene glycol, using Schiff base crosslinking. Upon implantation into rabbit knees with cartilaginous defects, these drug‐laden hydrogel/microparticle systems showed promising therapeutic potential. Within 14 days, the expression of key marker genes increased, leading to elevated production of chondrocytes within the damaged cartilage. Histological analyses after 14 and 21 days showed a significant increase in the amount of glycosaminoglycans. Six months postimplantation, the repaired lesion closely resembled the surrounding healthy tissue, indicating successful cartilage repair and hyaline cartilage formation.
Hydrogels synthesized through Schiff base crosslinking present promising avenues for immunotherapy, primarily due to their anti‐inflammatory and antimicrobial properties.[ 108 ] For example, a recent study formulated a hydrogel consisting of lipid nanoparticles and a copper (II) Schiff base 8‐hydroxyquinoline complex (Figure 8D).[ 109 ] Notably, this formulation led to a significant reduction in TNFα activity in treated areas. This suggests that the copper complex is effective in modulating key immune cell activities while concurrently supporting collagen synthesis. Furthermore, the hydrogel underwent tests for homogeneity, stiffness, and stability, all of which indicated no irritability or toxicity on human cell lines. Additional findings from this study revealed that the copper Schiff base complex also significantly reduced the expression of TNF‐α and MMPs.[ 109 ] In a parallel study, a hydrogel comprising tilapia gelatin, trivalent iron, and 2,3,4‐trihydroxy benzaldehyde was synthesized, also utilizing Schiff base reactions for crosslinking.[ 110 ] Their investigation found that the hydrogel effectively lowered the levels of various cytokines, including TNFα, IL‐6, IL‐10, and IL‐1β, showcasing its anti‐inflammatory capabilities. Collectively, these studies underscore the utility of Schiff base crosslinking in the development of injectable hydrogels designed to modulate the inflammatory response of immune cells. Although in the rat models, only the wound healing ability of these hydrogels were studied, these hydrogels reduced the levels of proinflammatory cytokines including TNFα, IL‐6, IL‐10, and IL‐1β. Several studies show the influence of reduced proinflammatory levels in cartilage repair.[ 111 ] Hence, further in vivo testing of these hydrogels for cartilage repair would help in understanding their ability in OA treatment. Additional examples of chemically crosslinked injectable hydrogels for OA treatment are listed in Table 2 .[ 112 , 113 , 114 , 115 , 116 , 117 , 118 , 119 , 120 , 121 , 122 , 123 , 124 , 125 , 126 , 127 , 128 , 129 , 130 , 131 , 132 , 133 , 134 , 135 , 136 , 137 , 138 , 139 ]
Table 2.
Injectable hydrogels via chemical crosslinking.
| Cargo | Polymeric material | Gelation mechanism | Properties | Refs. |
|---|---|---|---|---|
| Small drug molecules | ||||
| Resveratrol | Gelatin | NHS‐EDC chemistry | Natural compound delivery to enhance the critical proteins activities for cell survival proliferation and MSCs for improving the regeneration of articular cartilage | [112] |
| Melatonin with kartogenin | HA methacryloyl (HAMA), gelatin methacryloyl (GelMA) | Radical photoinitiated crosslinking/host–guest interaction | Self‐healing, biocompatibility | [113] |
| Melatonin with methylprednisolone | Oxidized alginate, carboxymethyl chitosan, and PEG microparticles | Schiff base chemistry | Promotion of chondrogenesis and cartilage regeneration in rabbit knees | [107] |
| Betamethasone | Poly(lactic‐co‐glycolic acid) (PLGA) nanospheres (300–490 nm) | Emulsion‐solvent evaporation | Sustained drug release of water‐soluble steroid | [114] |
| Chondroitin sulfate | Chondroitin sulfate multiple aldehyde (CSMA) and N‐succinyl‐chitosan (SC) | Schiff base chemistry | Self‐healing, biodegradable, minimal inflammatory response in rats | [106] |
| Celecoxib | PCLA (poly(‐caprolactone‐co‐lactide))–PEG (polyethylene glycol)–PCLA triblock copolymer hydrogel | Ring‐opening polymerization | Biodegradable, biocompatible, slow release of drug over time, thermo‐responsive gel | [115] |
| Rhein | PLGA microparticles | Emulsion‐solvent evaporation | Controlled release profile of drug, cytocompatibility of drug‐loaded microspheres | [116] |
| Diflunisal and etodolac | Multidomain peptide (MDP) hydrogel | Self‐assembly of hydrophobic and hydrophilic domains | In situ delivery of poorly water‐soluble drugs, prolonged drug release profiles due to intrafibrillar drug encapsulation | [117] |
| Kartogenin | N‐(β‐maleimidopropyloxy) succinimide ester‐ modified chitosan | NHS‐EDC chemistry | Thermosensitive, tunable, sustained release of drug, cytocompatibility | [118] |
| Kartogenin with curcumin | PLGA microparticles | Oil‐in‐water emulsion‐solvent evaporation | Use of drug‐ and morphogen‐loaded microparticles with stem cells for repair and regeneration | [119] |
| Kartogenin with histidine–alanine–valine and RGD | PLGA microspheres in methacrylated HA | Radical photoinitiated crosslinking | Promoted cell proliferation, adhesion, and differentiation using biomimetic peptides | [120] |
| Diclofenac sodium | GelMA microspheres coated with N,N‐dimethylacrylamide (DMA) and methacryloyloxyethyl phosphorylcholine (MPC) polymer | Water‐in‐oil emulsion polymerization then photo crosslinking, dip coating | Enhanced lubricating hydrogel microspheres, controllable drug release in in vivo rat‐knee model | [121] |
| Diclofenac sodium | Albumin microspheres | Water‐in‐oil emulsion polymerization | Biodegradability, lack of toxicity, and antigenicity of drug‐loaded microspheres | [122] |
| Diclofenac sodium | Gelatin microspheres | Water‐in‐oil emulsion polymerization | Slow drug release using microspheres | [123] |
| Diclofenac sodium and bovine serum albumin | IPN hydrogels based of poly(ethylene glycol) methacrylate, N‐isopropylacrylamide, and methacrylated alginate | Redox initiator system (APS/TEMED) | Temperature and pH‐responsive hydrogel, good mechanical strength, and stability under physiological conditions, cytocompatibility | [124] |
| Paclitaxel | PLGA microspheres (50 µm diameter) | Emulsion‐solvent evaporation | No significant effect on hemodynamic, increased trans synovial fluid forces and permeability to fluid transport; minimal early joint inflammatory reaction, and no gross or histological abnormalities in the synovium or cartilage. | [125] |
| Methotrexate | Poly(l‐lactic acid) (PLLA) microspheres (62–83 µm diameter) | Emulsion‐solvent evaporation | Biocompatibility, controlled release/delivery | [126] |
| Indomethacin | Polymeric micelles of polyphosphazene substituted with poly(N‐isopropylacrylamide) and ethyl glycinate copolymers | Thermal ring‐opening polymerization and dialysis method for micelles | Sustained therapeutic efficacy and prolonged drug concentration maintained in rats | [127] |
| Naproxen or dexamethasone | Oxidized dextran (Dex‐ox), gelatin, and hyaluronic acid | Reactive carbonyl chemistry | Drug‐dependent degradation/release profile, effective against the progression of osteoarthritis and cartilage degeneration, promoted cartilage regeneration in a rabbit model | [128] |
| Cells | ||||
| Mesenchymal stem cells (MSCs) | Poly(ethylene glycol) diacrylate (PEGDA), thiolated hyaluronic acid (HA) | Michael addition | No UV illumination for crosslinking, efficient chondrogenesis in vitro, repair of full‐thickness cartilage defects in a rat model after 8 weeks, hyaline cartilage formation | [129] |
| Adipose‐derived stem cells (ADSCs) | Thiolated chondroitin sulfate (CS‐SH), hyperbranched PEG | Thiol–ene | Reduced stem cell inflammatory response, prolonged degradation properties, improved cell viability, and chondrogenesis | [130] |
| Chondrocytes | Thiolated hyaluronic acid (HA‐SH) and maleimided hyaluronic acid (HA‐Mal) | Thiol–ene | Promoted chondrocytes adhesion, spreading, and proliferation, improved hyaline cartilage formation, and proteoglycan secretion in vitro and in vivo (nude mice) | [131] |
| Chondrocytes | Dibenzocyclooctyl (DBCO)‐modified HA, HA‐PEG4‐DBCO | NHS‐EDC chemistry | Flexible elastic modulus properties, development of cartilage lacunae in mice after 5 weeks of implantation without any signs of inflammation | [132] |
| Chondrocytes | Poly(ethylene oxide‐co‐glycidol)‐CHO (poly(EO‐co‐Gly)‐CHO), glycol chitosan | Schiff base | Lifetime in vivo was greater than 3 months, no dedifferentiation of chondrocytes was observed after 2 weeks of in vitro culture, maintenance of the chondrocyte phenotype | [104] |
| Bone marrow‐derived mesenchymal stem cells (BMSCs) | Adipic dihydrazide (ADH)‐modified poly(l‐glutamic acid) (PLGA‐ADH) and benzaldehyde‐terminated poly(ethylene glycol) (PEG‐CHO) | Schiff base | Effectively supported BMSC proliferation and deposition of glycosaminoglycans, upregulated the expression of cartilage‐specific genes, and repair of cartilage defect in mice after 8 weeks | [105] |
| Fibrochondrocyte | Tyramine (TA)‐conjugated hyaluronic acid (TA‐HA) and gelatin | Enzyme‐mediated | Enhancement of cartilage‐specific gene expression | [81b] |
| Chondrocytes | Tyrosine‐conjugated alginate sulfate | Enzyme‐mediated | Redifferentiation of chondrocytes, adhesive to cartilage tissue | [133] |
| Mesenchymal stem cells (MSCs) | Phenol‐functionalized chitosan (CHPH) and hyaluronic acid (HAPH) | Enzyme mediated | Expression of cartilage tissue markers, higher propensity of MSCs differentiation into cartilage‐like cells | [134] |
| Immunotherapy | ||||
| Betamethasone | Sodium alginate‐g‐(QCL‐co‐HEMA) hydrogels | Free radical polymerization | Biocompatible, sustained, and controlled release | [135] |
| Anti‐RANKL antibodies | HA/PEG‐based hydrogel | Michael addition | Reduction in activities of inflammatory cytokines and proteins, induce MSC maturation | [136] |
| Dexamethasone | Nitric oxide responsive hydrogel | Click cyclo‐addition | Sustained drug release, self‐healing properties, reduction in nitric oxide levels | [137] |
| Chitosan | Hydroxypropyl chitin hydrogel | Amide linkage | Efficient mechanical stability encourages MSC differentiation, encourages cell recruitment for tissue regeneration | [138] |
| Anti‐TNFα antibodies | Tyramine‐gellan gum hydrogel | Amide bond formation | Resistant to degradation, high levels of biocompatibility, successful inhibition of TNFα | [139] |
3.3. Dual Crosslinked Injectable Hydrogels
Dual crosslinking hydrogels can include structures with physical–physical, physical–chemical, and chemical–chemical networks.[ 60 ] Due to the combination of the different crosslinking methods dual crosslinked hydrogels provide a synergetic effect resulting in enhanced mechanical strength, controlled degradation properties, enhanced stability, and self‐healing properties. These properties have made dual crosslinked hydrogel a promising candidate for injectable hydrogel for biomedical applications.
Yu et al. recently developed a dual crosslinked hydrogel to overcome the poor mechanical properties and fast enzymatic degradation of HA for cartilage regeneration (Figure 9A).[ 140 ] The injectable dual‐crosslinking HA hydrogel was synthesized by integrating Diels–Alder click chemistry and phenylboronate ester bond. The Diels–Alder reaction provides mechanical strength and the phenyl boronate bonds impart injectability, adhesion, and antidegradation properties to the hydrogel. In brief, an equimolar mixture of HA–furfurylamine–3‐aminophenylboronic acid (HA–Furan–PBA) and 4‐arm‐PEG–maleimide (MAL) was mixed with HA–furan–dopamine hydrochloride (HA–furan–DA), creating ester bond between the PBA and DA groups at pH 7.4 in few seconds.[ 140 ] This was followed by a Diels–Alder click chemistry between the furan and MAL in the subsequent 1.5 h. The DC gel exhibited pH‐responsive behavior with regard to the pore size and swelling ratio. Compared to the single crosslinked hydrogels, DC gels also demonstrated excellent mechanical properties. Additionally, the gels demonstrated self‐healing ability, shear thinning behavior for injectability, tissue adhesion, shape recovery, fatigue resistance, and controlled degradation. This hydrogel matrix also supported the survival, proliferation, and morphology of the encapsulated ATDC‐5 cells.
Figure 9.
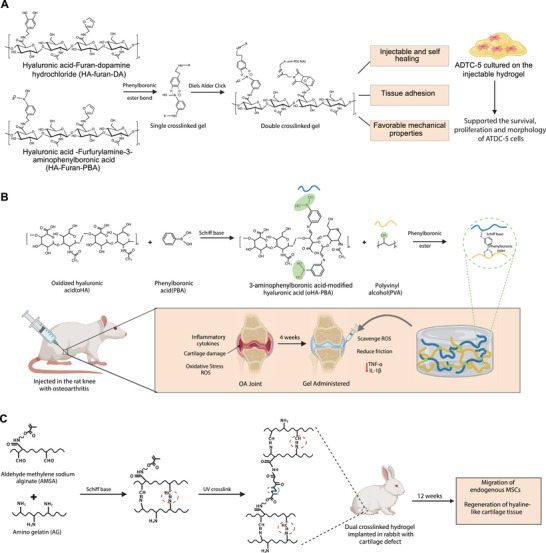
Dual crosslinked hydrogel. A) Hyaluronic acid (HA) modified dual crosslinked hydrogel formed using Diels–Alder click chemistry and phenyl boronate ester bond. Dual crosslinking enhanced mechanical strength and imparted injectability, adhesion, and antidegradation properties to the hydrogel. The hydrogel matrix supported the survival, proliferation, and morphology maintenance of the ATDC‐5 cells. B) Dual crosslinked hydrogel utilizing the dynamic covalent bonds; Schiff base, and phenylboronic ester bond to develop an injectable self‐healing hydrogel for OA therapy. In vivo study in mice with OA showed improved mobility, lower level of inflammatory cytokine expression TNFα and IL‐1β, promoting ECM decomposition in the joints. C) Aldehyde methylene sodium alginate (AMSA) and amino gelatin (AG) based dual crosslinked hydrogel utilizing Schiff base and photopolymerization for cartilage regeneration. The hydrogel loaded with KGN‐conjugated polyurethane nanoparticles (PN‐KGN) and TGF‐β3 promoted the regeneration of hyaline‐like cartilage tissue in 12 weeks in OA defective rabbits.
Similarly, in a separate study dual dynamic covalent crosslinking was employed to develop an injectable and self‐healing hydrogel for treating OA (Figure 9B).[ 141 ] These bonds were established between 3‐aminophenyl boronic acid‐modified hyaluronic acid (oHA‐PBA) and PVA. The Schiff base bonds were formed between amino groups of PBA and residual aldehyde groups of oHA. Subsequently, the phenylboronic ester bonds are formed between boronic groups of oHA‐PBA and hydroxyl groups of PVA. The dual crosslinked gel exhibited excellent injectability, seal‐healing, swelling, and stress relaxation properties along with high compressive yield strength, oscillatory shear strengthening properties, and low friction coefficient resulting in an excellent lubrication effect. This effect could potentially alleviate the wear and pain during friction of the joint surfaces. The in vitro experiments conducted on gel incubated with ATDC5 mouse chondrocytes showed low toxicity and good biocompatibility by reducing the oxidative stress and expression of proinflammatory cytokines. An in vivo study conducted on mice with OA also revealed improved mobility, and a lower level of inflammatory cytokine expression (TNFα and IL‐1β) promoting ECM decomposition in the joints within 4 weeks.
In a related study, Fan et al. developed a dual‐crosslinked hydrogel for cartilage defect repair using aldehyde methylene sodium alginate (AMSA) and amino gelatin (AG) using Irgacure 2959 as photoinitiator (Figure 9C).[ 142 ] The aldehyde group of ASMA was crosslinked with an amino group of AG via the Schiff base reaction within 120 s. Subsequently, a second crosslink was formed when the gel was exposed to UV light at 365 nm. The dual crosslinked hydrogel exhibited higher compressive modulus, and slower degradation in PBS and collagenase solution compared to the single crosslinked hydrogel. Loaded with KGN‐conjugated polyurethane nanoparticles (PN‐KGN) and TGF‐β3, it facilitated the controlled release of biomolecules, promoting the MSC migration and cartilage regeneration. Upon injection into rabbits with osteochondral cartilage defects, the hydrogel promoted the migration of endogenous MSCs and the regeneration of hyaline‐like cartilage tissue within 12 weeks.
4. Conclusions
Osteoarthritis and cartilage repair are intricate processes orchestrated by a complex interplay of biomechanical forces, inflammatory responses, and cell signaling pathways. A comprehensive understanding of these multifactorial interactions and the specific factors contributing to individual variations in disease progression remains elusive, hindering the development of targeted therapies. While hydrogel‐based approaches have laid the groundwork for drug delivery systems in OA treatment, traditional hydrogels suffer from limitations such as inadequate mechanical strength, poor tissue integration, and regeneration, restricted sustained drug release, and scalability issues. However, the advent of advanced injectable hydrogels offers a promising avenue for overcoming these limitations. Characterized by biomimetic, self‐healing, and tunable properties, these materials address many shortcomings inherent in traditional hydrogel systems. By incorporating multicompartment drug delivery systems into these advanced polymeric materials, researchers are striving to achieve sustained and targeted therapeutic release, a crucial step toward effective OA treatment.
Furthermore, the dynamic and tunable mechanical properties of some shear‐thinning biomaterials and synthetic hydrogels hold promise for facilitating easier integration with surrounding cartilage tissue, thereby promoting tissue regeneration. Nevertheless, overcoming hurdles related to fine‐tuning the degradation kinetics of these hydrogels remains crucial for their successful clinical translation. Researchers are actively exploring strategies to achieve the optimal balance between on‐demand degradation of hydrogel matrices for payload delivery of effective therapeutic dosages, tailoring the degradation profile to the in situ generated inflammatory responses. Additionally, ensuring the timely replacement of injected materials with regenerative extracellular matrix is crucial for long‐term therapeutic success. Some researchers have taken this approach one step further by codelivering stem cells, differentiation factors, or recruitment factors alongside therapeutic agents within the hydrogel matrix. This approach holds promise for achieving the next generation of OA therapies, where degraded hydrogel materials are replaced by newly formed cartilage tissue, orchestrating a regenerative healing process.
In conclusion, while challenges remain in optimizing hydrogel‐based approaches for OA treatment and cartilage repair, significant progress has been made in recent years. Advanced hydrogels with biomimetic, self‐healing, and tunable properties, coupled with innovative drug delivery systems and codelivery strategies, offer promising avenues for overcoming limitations inherent in traditional therapies. As research continues to unravel the complexities of OA and cartilage repair, these advanced materials hold immense potential for paving the way toward personalized and effective treatments.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
A.K.G. acknowledges financial support from the National Institute of Dental and Craniofacial Research (NIDCR) (R01 DE032031), the National Institute of Biomedical Imaging and Bioengineering (NIBIB) (DP2 EB026265), the National Science Foundation (NSF) Award (CBET 1705852), the Congressionally Directed Medical Research Program (CDMRP) (W81XWH2210932), and President's Excellence Fund (X‐Grants) from Texas A&M University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agency. All the images in the article were created using BioRender.com.
Biographies
Manivannan Sivaperuman Kalairaj is a Ph.D. student in biomedical engineering at Texas A&M University. He received his B.E. in mechanical engineering from Anna University, India, and his M.Sc. in Smart Product Design from Nanyang Technological University, Singapore. Before joining Texas A&M University, Manivannan worked as an Application Engineer at Mason Industries Pte Ltd, Singapore, and as a research engineer at the National University of Singapore. His current research focuses on developing implantable engineered living materials for therapeutic applications.

Ridhi Pradhan received the B.S. degree in biomedical engineering from University of Texas, Arlington, USA in 2021. She is currently pursuing the Ph.D. degree in biomedical engineering at Texas A&M University. Her research interests include developing hydrogels for implantable biosensors and tissue engineering applications.

Akhilesh K. Gaharwar is a Professor in the Department of Biomedical Engineering and Presidential Impact Fellow at Texas A&M University. He received his Ph.D. in biomedical engineering from Purdue University and completed his postdoctoral training from the Massachusetts Institute of Technology (MIT) and Harvard University. The goal of his lab is to design new biomaterials for regenerative medicine and biomanufacturing. In particular, his lab is leveraging principles from materials science, cell biology, additive biomanufacturing, and high throughput genomics to design smart and responsive biomaterials, with wide‐ranging applications in the field of bioengineering. He is fellow of the Biomedical Engineering Society (BMES), American Institute for Medical and Biological Engineering (AIMBE), and senior member of National Academy of Inventors (NAI).

Kalairaj M. S., Pradhan R., Saleem W., Smith M. M., Gaharwar A. K., Intra‐Articular Injectable Biomaterials for Cartilage Repair and Regeneration. Adv. Healthcare Mater. 2024, 13, 2303794. 10.1002/adhm.202303794
References
- 1.a) Liede A., Bach B. A., Stryker S., Hernandez R. K., Sobocki P., Bennett B., Wong S. S., J. Bone Jt. Surg., Am. Vol. 1999, 96, 2014. [DOI] [PubMed] [Google Scholar]; b) Yelin E., Weinstein S., King T., Semin. Arthritis Rheum. 2016, 46, 259. [DOI] [PubMed] [Google Scholar]
- 2. Junker S., Frommer K. W., Krumbholz G., Tsiklauri L., Gerstberger R., Rehart S., Steinmeyer J., Rickert M., Wenisch S., Schett G., Matrix Biol. 2017, 62, 75. [DOI] [PubMed] [Google Scholar]
- 3.a) Stockwell R. A., J. Anat. 1967, 101, 753; [PMC free article] [PubMed] [Google Scholar]; b) Naba A., Clauser K. R., Ding H., Whittaker C. A., Carr S. A., Hynes R. O., Matrix Biol. 2016, 49, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tuan R. S., Chen A. F., Klatt B. A., J. Am. Acad. Orthop. Surg. 2013, 21, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) C. Centers for Disease, U. S. D. o. H. S. Human , J. Pain Palliative Care Pharmacother. 2010, 24, 430; [Google Scholar]; b) Zhang W., Ouyang H., Dass C. R., Xu J., Bone Res. 2016, 4, 15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Owen S. G., Francis H. W., Roberts M. S., Br. J. Clin. Pharmacol. 1994, 38, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rai M. F., Pham C. T., Curr. Opin. Pharmacol. 2018, 40, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Zhang Y., Jordan J. M., Clin. Rheum. Dis. 2008, 34, 515; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thysen S., Luyten F. P., Lories R. J., Dis. Models Mech. 2015, 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee J. H., Biomater. Res. 2018, 22, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wojdasiewicz P., Poniatowski L. A., Szukiewicz D., Mediators Inflammation 2014, 2014, 561459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) De Lange‐Brokaar B. J., Ioan‐Facsinay A., Van Osch G. J., Zuurmond A.‐M., Schoones J., Toes R. E., Huizinga T. W., Kloppenburg M., Osteoarthritis Cartilage 2012, 20, 1484; [DOI] [PubMed] [Google Scholar]; b) Goldring M. B., Otero M., Curr. Opin. Rheumatol. 2011, 23, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldring S. R., Goldring M. B., Nat. Rev. Rheumatol. 2016, 12, 632. [DOI] [PubMed] [Google Scholar]
- 13.a) Simkin P. A., J. Rheumatol. 2012, 39, 890; [DOI] [PubMed] [Google Scholar]; b) Lyons T. J., McClure S. F., Stoddart R. W., McClure J., BMC Musculoskeletal Disord. 2006, 7, 52; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Burr D. B., Osteoarthritis Cartilage 2004, 12, 20. [DOI] [PubMed] [Google Scholar]
- 14.a) Akizuki S., Mow V. C., Muller F., Pita J. C., Howell D. S., J. Orthop. Res. 1987, 5, 173; [DOI] [PubMed] [Google Scholar]; b) Roberts S., Weightman B., Urban J., Chappell D., J. Bone Jt. Surg., Am. Vol. 1986, 68, 278; [DOI] [PubMed] [Google Scholar]; c) Calvo E., Palacios I., Delgado E., Sanchez‐Pernaute O., Largo R., Egido J., Herrero‐Beaumont G., Osteoarthritis Cartilage 2004, 12, 878. [DOI] [PubMed] [Google Scholar]
- 15.a) Imhof H., Sulzbacher I., Grampp S., Czerny C., Youssefzadeh S., Kainberger F., Invest. Radiol. 2000, 35, 581; [DOI] [PubMed] [Google Scholar]; b) Lane L. B., Villacin A., Bullough P., J. Bone Jt. Surg., Am. Vol. 1977, 59, 272; [DOI] [PubMed] [Google Scholar]; c) Burr D. B., Schaffler M. B., Microsc. Res. Tech. 1997, 37, 343; [DOI] [PubMed] [Google Scholar]; d) Bullough P. G., Osteoarthritis Cartilage 2004, 12, 2. [Google Scholar]
- 16. Felson D. T., N. Engl. J. Med. 2006, 354, 841. [DOI] [PubMed] [Google Scholar]
- 17.a) Sohn D. H., Sokolove J., Sharpe O., Erhart J. C., Chandra P. E., Lahey L. J., Lindstrom T. M., Hwang I., Boyer K. A., Andriacchi T. P., Robinson W. H., Arthritis Res. Ther. 2012, 14, R7; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Melchiorri C., Meliconi R., Frizziero L., Silvestri T., Pulsatelli L., Mazzetti I., Borzi R. M., Uguccioni M., Facchini A., Arthritis Rheumatol. 1998, 41, 2165. [DOI] [PubMed] [Google Scholar]
- 18. Piccioli P., Rubartelli A., Semin. Immunol. 2013, 25, 425. [DOI] [PubMed] [Google Scholar]
- 19. Martin M. U., Wesche H., Biochim. Biophys. Acta 2002, 1592, 265. [DOI] [PubMed] [Google Scholar]
- 20. Gough N. R., Sci. Signaling 2008, 1, 195. [Google Scholar]
- 21. Roman‐Blas J., Jimenez S., Osteoarthritis Cartilage 2006, 14, 839. [DOI] [PubMed] [Google Scholar]
- 22. Marcu K. B., Otero M., Olivotto E., Maria Borzi R., Goldring M. B., Curr. Drug Targets 2010, 11, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shakibaei M., Schulze‐Tanzil G., John T., Mobasheri A., Ann. Anat. 2005, 187, 487. [DOI] [PubMed] [Google Scholar]
- 24.a) Mengshol J. A., Vincenti M. P., Coon C. I., Barchowsky A., Brinckerhoff C. E., Arthritis Rheumatol. 2000, 43, 801; [DOI] [PubMed] [Google Scholar]; b) Meszaros E., Malemud C. J., Ther. Adv. Chronic Dis. 2012, 3, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Aigner T., McKenna L., Zien A., Fan Z., Gebhard P. M., Zimmer R., Cytokine 2005, 31, 227; [DOI] [PubMed] [Google Scholar]; b) Alaaeddine N., Olee T., Hashimoto S., Creighton‐Achermann L., Lotz M., Arthritis Rheumatol. 2001, 44, 1633. [DOI] [PubMed] [Google Scholar]
- 26.a) El Mansouri F. E., Chabane N., Zayed N., Kapoor M., Benderdour M., Martel‐Pelletier J., Pelletier J. P., Duval N., Fahmi H., Arthritis Rheumatol. 2011, 63, 168; [DOI] [PubMed] [Google Scholar]; b) Hardy M. M., Seibert K., Manning P. T., Currie M. G., Woerner B. M., Edwards D., Koki A., Tripp C. S., Arthritis Rheumatol. 2002, 46, 1789; [DOI] [PubMed] [Google Scholar]; c) Gilman S. C., Chang J., Zeigler P. R., Uhl J., Mochan E., Arthritis Rheumatol. 1988, 31, 126. [DOI] [PubMed] [Google Scholar]
- 27. Afonso V., Champy R., Mitrovic D., Collin P., Lomri A., Jt., Bone, Spine 2007, 74, 324. [DOI] [PubMed] [Google Scholar]
- 28. Farahat M. N., Yanni G., Poston R., Panayi G. S., Ann. Rheum. Dis. 1993, 52, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. MacEwan D. J., Cell Signaling 2002, 14, 477. [DOI] [PubMed] [Google Scholar]
- 30.a) Hsu H., Xiong J., Goeddel D. V., Cell 1995, 81, 495; [DOI] [PubMed] [Google Scholar]; b) Hsu H., Huang J., Shu H. B., Baichwal V., Goeddel D. V., Immunity 1996, 4, 387; [DOI] [PubMed] [Google Scholar]; c) Varfolomeev E., Goncharov T., Fedorova A. V., Dynek J. N., Zobel K., Deshayes K., Fairbrother W. J., Vucic D., J. Biol. Chem. 2008, 283, 24295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Donnell M. A., Legarda‐Addison D., Skountzos P., Yeh W. C., Ting A. T., Curr. Biol. 2007, 17, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rothe M., Pan M. G., Henzel W. J., Ayres T. M., Goeddel D. V., Cell 1995, 83, 1243. [DOI] [PubMed] [Google Scholar]
- 33. Henderson B., Pettipher E., Clin. Exp. Immunol. 1989, 75, 306. [PMC free article] [PubMed] [Google Scholar]
- 34. Saklatvala J., Nature 1986, 322, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Lefebvre V., Peeters‐Joris C., Vaes G., Biochim. Biophys. Acta 1990, 1052, 366; [DOI] [PubMed] [Google Scholar]; b) Verma P., Dalal K., J. Cell. Biochem. 2011, 112, 3507; [DOI] [PubMed] [Google Scholar]; c) Xue J., Wang J., Liu Q., Luo A., Mol. Med. Rep. 2013, 8, 1755. [DOI] [PubMed] [Google Scholar]
- 36.a) Ye Z., Chen Y., Zhang R., Dai H., Zeng C., Zeng H., Feng H., Du G., Fang H., Cai D., Can. J. Physiol. Pharmacol. 2014, 92, 132; [DOI] [PubMed] [Google Scholar]; b) Heraud F., Heraud A., Harmand M. F., Ann. Rheum. Dis. 2000, 59, 959; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lopez‐Armada M. J., Carames B., Lires‐Dean M., Cillero‐Pastor B., Ruiz‐Romero C., Galdo F., Blanco F. J., Osteoarthritis Cartilage 2006, 14, 660. [DOI] [PubMed] [Google Scholar]
- 37. Joos H., Wildner A., Hogrefe C., Reichel H., Brenner R. E., Arthritis Res. Ther. 2013, 15, R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. López‐Armada M. J., Caramés B., Martin M., Cillero‐Pastor B., Lires‐Dean M., Fuentes‐Boquete I., Arenas J., Blanco F., Osteoarthritis Cartilage 2006, 14, 1011. [DOI] [PubMed] [Google Scholar]
- 39.a) Guerne P. A., Carson D. A., Lotz M., J. Immunol. 1990, 144, 499; [PubMed] [Google Scholar]; b) Lotz M., Terkeltaub R., Villiger P. M., J. Immunol. 1992, 148, 466; [PubMed] [Google Scholar]; c) Jones S. W., Brockbank S. M., Clements K. M., Good N. L.e, Campbell D., Read S. J., Needham M. R., Newham P., Osteoarthritis Cartilage 2009, 17, 124. [DOI] [PubMed] [Google Scholar]
- 40. Steel J. C., Waldmann T. A., Morris J. C., Trends Pharmacol. Sci. 2012, 33, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waldmann T., Tagaya Y., Annu. Rev. Immunol. 1999, 17, 19. [DOI] [PubMed] [Google Scholar]
- 42. Scanzello C. R., Umoh E., Pessler F., Diaz‐Torne C., Miles T., Dicarlo E., Potter H. G., Mandl L., Marx R., Rodeo S., Goldring S. R., Crow M. K., Osteoarthritis Cartilage 2009, 17, 1040. [DOI] [PubMed] [Google Scholar]
- 43. Sun J. M., Sun L. Z., Liu J., Su B. H., Shi L., Dis. Markers 2013, 35, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pawlowska J., Mikosik A., Soroczynska‐Cybula M., Jozwik A., Luczkiewicz P., Mazurkiewicz S., Lorczynski A., Witkowski J. M., Bryl E., Folia Histochem. Cytobiol. 2009, 47, 627. [DOI] [PubMed] [Google Scholar]
- 45. Honorati M. C., Neri S., Cattini L., Facchini A., Osteoarthritis Cartilage 2006, 14, 345. [DOI] [PubMed] [Google Scholar]
- 46. Honorati M. C., Bovara M., Cattini L., Piacentini A., Facchini A., Osteoarthritis Cartilage 2002, 10, 799. [DOI] [PubMed] [Google Scholar]
- 47. Chen B., Deng Y., Tan Y., Qin J., Chen L.‐B., J. Int. Med. Res. 2014, 42, 138. [DOI] [PubMed] [Google Scholar]
- 48.a) Okamura H., Tsutsui H., Komatsu T., Yutsudo M., Hakura A., Tanimoto T., Torigoe K., Okura T., Nukada Y., Hattori K., Nature 1995, 378, 88; [DOI] [PubMed] [Google Scholar]; b) Ghayur T., Banerjee S., Hugunin M., Butler D., Herzog L., Carter A., Quintal L., Sekut L., Talanian R., Paskind M., Wong W., Kamen R., Tracey D., Allen H., Nature 1997, 386, 619. [DOI] [PubMed] [Google Scholar]
- 49. Saha N., Moldovan F., Tardif G., Pelletier J. P., Cloutier J. M., Martel‐Pelletier J., Arthritis Rheumtol. 1999, 42, 1577. [DOI] [PubMed] [Google Scholar]
- 50. Peng C. Z., Cao J. M., Xiao T., Peng C., Yang H. B., Chen X., Fang J. Z., Zhongnan Daxue Xuebao, Yixueban 2006, 31, 862. [Google Scholar]
- 51. Corr M., Nat. Clin. Pract. Rheumatol. 2008, 4, 550. [DOI] [PubMed] [Google Scholar]
- 52. Rubinfeld B., Albert I., Porfiri E., Fiol C., Munemitsu S., Polakis P., Science 1996, 272, 1023. [DOI] [PubMed] [Google Scholar]
- 53. Behrens J., Von Kries J. P., Kühl M., Bruhn L., Wedlich D., Grosschedl R., Birchmeier W., Nature 1996, 382, 638. [DOI] [PubMed] [Google Scholar]
- 54. Sassi N., Laadhar L., Driss M., Kallel‐Sellami M., Sellami S., Makni S., Arthritis Res. Ther. 2011, 13, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou B., Lin W., Long Y., Yang Y., Zhang H., Wu K., Chu Q., Signal Transduction Targeted Ther. 2022, 7, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li J., Chen G., Xu X., Abdou P., Jiang Q., Shi D., Gu Z., Regener. Biomater. 2019, 6, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parhi R., Adv. Pharm. Bull. 2017, 7, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu M., Zeng X., Ma C., Yi H., Ali Z., Mou X., Li S., Deng Y., He N., Bone Res. 2017, 5, 17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sobczak M., Int. J. Mol. Sci. 2022, 23, 4421.35457239 [Google Scholar]
- 60.a) Zhang M., Chen X., Yang K., Dong Q., Yang H., Gu S., Xu W., Zhou Y., Carbohydr. Polym. 2023, 301, 120372; [DOI] [PubMed] [Google Scholar]; b) Chu W., Nie M., Ke X., Luo J., Li J., Macromol. Biosci. 2021, 21, 2100109. [DOI] [PubMed] [Google Scholar]
- 61. Hu C., Lu W., Mata A., Nishinari K., Fang Y., Int. J. Biol. Macromol. 2021, 177, 578. [DOI] [PubMed] [Google Scholar]
- 62. Liao J., Wang B., Huang Y., Qu Y., Peng J., Qian Z., ACS Omega 2017, 2, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xu J., Feng Q., Lin S., Yuan W., Li R., Li J., Wei K., Chen X., Zhang K., Yang Y., Wu T., Wang B., Zhu M., Guo R., Li G., Bian L., Biomaterials 2019, 210, 51. [DOI] [PubMed] [Google Scholar]
- 64. Hashemnejad S. M., Badruddoza A. Z. M., Zarket B., Castaneda C. R., Doyle P. S., Nat. Commun. 2019, 10, 2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen W., Li Z., Wang Z., Gao H., Ding J., He Z., J Pain Res 2020, 13, 3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Agrawal P., Pramanik K., Biswas A., Regener. Med. 2018, 13, 545. [DOI] [PubMed] [Google Scholar]
- 67. He T., Zhang C., Vedadghavami A., Mehta S., Clark H. A., Porter R. M., Bajpayee A. G., J. Controlled Release 2020, 318, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Peng Y., Tellier L. E., Temenoff J. S., Biomater. Sci. 2016, 4, 1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Feng Q., Xu J., Zhang K., Yao H., Zheng N., Zheng L., Wang J., Wei K., Xiao X., Qin L., ACS Cent. Sci. 2019, 5, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu T., Chen Y., Liu W., Tong K. L., Suen C. W., Huang S., Hou H., She G., Zhang H., Zheng X., Li J., Zha Z., Mater. Sci. Eng., C 2020, 111, 110757. [DOI] [PubMed] [Google Scholar]
- 71. Gao X., Gao L., Groth T., Liu T., He D., Wang M., Gong F., Chu J., Zhao M., J. Biomed. Mater. Res., Part A 2019, 107, 2076. [DOI] [PubMed] [Google Scholar]
- 72. Almeida H., Eswaramoorthy R., Cunniffe G., Buckley C., O'brien F., Kelly D., Acta Biomater. 2016, 36, 55. [DOI] [PubMed] [Google Scholar]
- 73. Chen W., Li C., Peng M., Xie B., Zhang L., Tang X., Cell Tissue Banking 2018, 19, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cai Z., Li Y., Song W., He Y., Li H., Liu X., ACS Appl. Mater. Interfaces 2021, 13, 59772. [DOI] [PubMed] [Google Scholar]
- 75. Jeong S. H., Kim M., Kim T. Y., Kim H., Ju J. H., Hahn S. K., ACS Appl. Bio Mater. 2020, 3, 5040. [DOI] [PubMed] [Google Scholar]
- 76. Cho H., Kim J., Kim S., Jung Y. C., Wang Y., Kang B. J., Kim K., J. Controlled Release 2020, 327, 284. [DOI] [PubMed] [Google Scholar]
- 77. Zhu N., Chatzistavrou X., Papagerakis P., Ge L., Qin M., Wang Y., ACS Biomater. Sci. Eng. 2019, 5, 4624. [DOI] [PubMed] [Google Scholar]
- 78. Guo C., Cao Z., Peng Y., Wu R., Xu H., Yuan Z., Xiong H., Wang Y., Wu Y., Li W., Kong Q., Wang Y., Wu J., Colloids Surf., B 2022, 218, 112721. [DOI] [PubMed] [Google Scholar]
- 79. Li X., Xiong Y., ACS Omega 2022, 7, 36918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Aisenbrey E. A., Bryant S. J., Biomaterials 2019, 190, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li X., SA, Xu Q., Alshehri F., Zeng M., Zhou D., Li J., Zhou G., Wang W., ACS Appl. Bio Mater. 2020, 3, 4756. [DOI] [PubMed] [Google Scholar]
- 82. Chen J. X., Yuan J., Wu Y. L., Wang P., Zhao P., Lv G. Z., Chen J. H., J. Biomed. Mater. Res., Part A 2018, 106, 192. [DOI] [PubMed] [Google Scholar]
- 83. Damlar I., Esen E., Tatli U., Med. Oral Patol. Oral Cir. Bucal. 2015, 20, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Henrotin Y., Mathy M., Sanchez C., Lambert C., Ther. Adv. Musculoskeletal Dis. 2010, 2, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Iovu M., Dumais G., du Souich P., Osteoarthritis Cartilage 2008, 16, S14. [DOI] [PubMed] [Google Scholar]
- 86. Bai X., Lu S., Cao Z., Ni B., Wang X., Ning P., Ma D., Wei H., Liu M., Carbohydr. Polym. 2017, 166, 123. [DOI] [PubMed] [Google Scholar]
- 87.a) Plamper F. A., Richtering W., Acc. Chem. Res. 2017, 50, 131; [DOI] [PubMed] [Google Scholar]; b) Vinogradov S. V., Curr. Pharm. Des. 2006, 12, 4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.a) Heida T., Otto O., Biedenweg D., Hauck N., Thiele J., Polymers 2020, 12, 1760; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ding S. L., Zhao X. Y., Xiong W., Ji L. F., Jia M. X., Liu Y. Y., Guo H. T., Qu F., Cui W., Gu Q., Adv. Mater. 2023, 35, 2212114; [DOI] [PubMed] [Google Scholar]; c) Chen Z., Zhang F., Zhang H., Cheng L., Chen K., Shen J., Qi J., Deng L., He C., Santos H. A., Adv. Sci. 2021, 8, 2004793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Feng Q., Li Q., Wen H., Chen J., Liang M., Huang H., Lan D., Dong H., Cao X., Adv. Funct. Mater. 2019, 29, 1906690. [Google Scholar]
- 90. Ma T., Gao X., Dong H., He H., Cao X., Appl. Mater. Today 2017, 9, 49. [Google Scholar]
- 91. Feng Q., Li D., Li Q., Li S., Huang H., Li H., Dong H., Cao X., Adv. Healthcare Mater. 2022, 11, 2102395. [DOI] [PubMed] [Google Scholar]
- 92. Lei Y., Wang Y., Shen J., Cai Z., Zeng Y., Zhao P., Liao J., Lian C., Hu N., Luo X., Adv. Funct. Mater. 2021, 31, 2105084. [Google Scholar]
- 93. Teixeira L. S., Feijen J., van Blitterswijk C. A., Dijkstra P. J., Karperien M., Biomaterials 2012, 33, 1281. [DOI] [PubMed] [Google Scholar]
- 94.a) Choi S., Ahn H., Kim S. H., J. Appl. Polym. Sci. 2022, 139, 51887; [Google Scholar]; b) Kim S. H., An Y. H., Kim H. D., Kim K., Lee S. H., Yim H. G., Kim B. G., Hwang N. S., Int. J. Biol. Macromol. 2018, 110, 479. [DOI] [PubMed] [Google Scholar]
- 95. Kim B. S., Kim S. H., Kim K., An Y. H., So K. H., Kim B. G., Hwang N. S., Mater. Today Bio 2020, 8, 100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li G., Liu S., Chen Y., Zhao J., Xu H., Weng J., Yu F., Xiong A., Udduttula A., Wang D., Nat. Commun. 2023, 14, 3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jin Y., Koh R. H., Kim S. H., Kim K. M., Park G. K., Hwang N. S., Mater. Sci. Eng., C 2020, 115, 111096. [DOI] [PubMed] [Google Scholar]
- 98. Noth U., Rackwitz L., Heymer A., Weber M., Baumann B., Steinert A., Schutze N., Jakob F., Eulert J., J. Biomed. Mater. Res., Part A 2007, 83, 626. [DOI] [PubMed] [Google Scholar]
- 99. Sarrigiannidis S. O., Rey J. M., Dobre O., Gonzalez‐Garcia C., Dalby M. J., Salmeron‐Sanchez M., Mater. Today Bio 2021, 10, 100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang Y., Cao Y., Zhao H., Zhang L., Ni T., Liu Y., An Z., Liu M., Pei R., J. Mater. Chem. B 2020, 8, 4237. [DOI] [PubMed] [Google Scholar]
- 101. Park H., Guo X., Temenoff J. S., Tabata Y., Caplan A. I., Kasper F. K., Mikos A. G., Biomacromolecules 2009, 10, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mo C., Xiang L., Chen Y., Macromol. Rapid Commun. 2021, 42, 2100025. [DOI] [PubMed] [Google Scholar]
- 103. Berger J., Reist M., Mayer J. M., Felt O., Peppas N. A., Gurny R., Eur. J. Pharm. Biopharm. 2004, 57, 19. [DOI] [PubMed] [Google Scholar]
- 104. Cao L., Cao B., Lu C., Wang G., Yu L., Ding J., J. Mater. Chem. B 2015, 3, 1268. [DOI] [PubMed] [Google Scholar]
- 105. Li S., Niu D., Shi T., Yun W., Yan S., Xu G., Yin J., ACS Biomater. Sci. Eng. 2023, 9, 2625. [DOI] [PubMed] [Google Scholar]
- 106. Lu S., Gao C., Xu X., Bai X., Duan H., Gao N., Feng C., Xiong Y., Liu M., ACS Appl. Mater. Interfaces 2015, 7, 13029. [DOI] [PubMed] [Google Scholar]
- 107. Naghizadeh Z., Karkhaneh A., Nokhbatolfoghahaei H., Farzad‐Mohajeri S., Rezai‐Rad M., Dehghan M. M., Aminishakib P., Khojasteh A., J. Cell. Physiol. 2021, 236, 2194. [DOI] [PubMed] [Google Scholar]
- 108. Login C. C., Baldea I., Tiperciuc B., Benedec D., Vodnar D. C., Decea N., Suciu S., Oxid. Med. Cell. Longevity 2019, 2019, 1607903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Abou El‐Ezz D., Abdel‐Rahman L. H., Al‐Farhan B. S., Mostafa D. A., Ayad E. G., Basha M. T., Abdelaziz M., Abdalla E. M., Pharmaceuticals 2022, 15, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lu Y., Zhao M., Peng Y., He S., Zhu X., Hu C., Xia G., Zuo T., Zhang X., Yun Y., Zhang W., Shen X., J. Nanobiotechnol. 2022, 20, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Liu S., Deng Z., Chen K., Jian S., Zhou F., Yang Y., Fu Z., Xie H., Xiong J., Zhu W., Mol. Med. Rep. 2022, 25, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Choi S. M., Lee K. M., Ryu S. B., Park Y. J., Hwang Y. G., Baek D., Choi Y., Park K. H., Park K. D., Lee J. W., Cell Death Dis. 2018, 9, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Liu X., Chen Y., Mao A. S., Xuan C., Wang Z., Gao H., An G., Zhu Y., Shi X., Mao C., Biomaterials 2020, 232, 119644. [DOI] [PubMed] [Google Scholar]
- 114. Horisawa E., Hirota T., Kawazoe S., Yamada J., Yamamoto H., Takeuchi H., Kawashima Y., Pharm. Res. 2002, 19, 403. [DOI] [PubMed] [Google Scholar]
- 115. Petit A., Sandker M., Muller B., Meyboom R., van Midwoud P., Bruin P., Redout E. M., Versluijs‐Helder M., van der Lest C. H., Buwalda S. J., de Leede L. G., Vermonden T., Kok R. J., Weinans H., Hennink W. E., Biomaterials 2014, 35, 7919. [DOI] [PubMed] [Google Scholar]
- 116. Gomez‐Gaete C., Retamal M., Chavez C., Bustos P., Godoy R., Torres‐Vergara P., Eur. J. Pharm. Sci. 2017, 96, 390. [DOI] [PubMed] [Google Scholar]
- 117. Li I. C., Moore A. N., Hartgerink J. D., Biomacromolecules 2016, 17, 2087. [DOI] [PubMed] [Google Scholar]
- 118. Dehghan‐Baniani D., Chen Y., Wang D., Bagheri R., Solouk A., Wu H., Colloids Surf., B 2020, 192, 111059. [DOI] [PubMed] [Google Scholar]
- 119. Asgari N., Bagheri F., Eslaminejad M. B., Ghanian M. H., Sayahpour F. A., Ghafari A. M., Stem Cell Res. Ther. 2020, 11, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Teng B., Zhang S., Pan J., Zeng Z., Chen Y., Hei Y., Fu X., Li Q., Ma M., Sui Y., Wei S., Acta Biomater. 2021, 122, 145. [DOI] [PubMed] [Google Scholar]
- 121. Han Y., Yang J., Zhao W., Wang H., Sun Y., Chen Y., Luo J., Deng L., Xu X., Cui W., Zhang H., Bioact. Mater. 2021, 6, 3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tuncay M., Calis S., Kas H., Ercan M., Peksoy I., Hincal A., J. Microencapsulation 2000, 17, 145. [DOI] [PubMed] [Google Scholar]
- 123. Saravanan M., Bhaskar K., Maharajan G., Pillai K. S., J. Drug Targeting 2011, 19, 96. [DOI] [PubMed] [Google Scholar]
- 124. Zhao J., Zhao X., Guo B., Ma P. X., Biomacromolecules 2014, 15, 3246. [DOI] [PubMed] [Google Scholar]
- 125. Bragdon B., Bertone A. L., Hardy J., Simmons E. J., Weisbrode S. E., J. Invest. Surg. 2001, 14, 169. [DOI] [PubMed] [Google Scholar]
- 126. Liang L. S., Jackson J., Min W., Risovic V., Wasan K. M., Burt H. M., J. Pharm. Sci. 2004, 93, 943. [DOI] [PubMed] [Google Scholar]
- 127. Zhang J. X., Yan M. Q., Li X. H., Qiu L. Y., Li X. D., Li X. J., Jin Y., Zhu K. J., Pharm. Res. 2007, 24, 1944. [DOI] [PubMed] [Google Scholar]
- 128. Garcia‐Fernandez L., Olmeda‐Lozano M., Benito‐Garzon L., Perez‐Caballer A., San Roman J., Vazquez‐Lasa B., Mater. Sci. Eng., C 2020, 110, 110702. [DOI] [PubMed] [Google Scholar]
- 129. Li J., Huang Y., Song J., Li X., Zhang X., Zhou Z., Chen D., Ma P. X., Peng W., Wang W., Zhou G., Acta Biomater. 2018, 79, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Li X., Xu Q., Johnson M., Wang X., Lyu J., Li Y., McMahon S., Greiser U., SA, Wang W., Biomater. Sci. 2021, 9, 4139. [DOI] [PubMed] [Google Scholar]
- 131. Yao Y., Wang P., Li X., Xu Y., Lu G., Jiang Q., Sun Y., Fan Y., Zhang X., Acta Biomater. 2020, 111, 197. [DOI] [PubMed] [Google Scholar]
- 132. Han S.‐S., Yoon H. Y., Yhee J. Y., Cho M. O., Shim H.‐E., Jeong J.‐E., Lee D.‐E., Kim K., Guim H., Lee J. H., Polym. Chem. 2018, 9, 20. [Google Scholar]
- 133. Ozturk E., Stauber T., Levinson C., Cavalli E., Arlov O., Zenobi‐Wong M., Biomed. Mater. 2020, 15, 045019. [DOI] [PubMed] [Google Scholar]
- 134. Davachi S. M., Haramshahi S. M. A., Akhavirad S. A., Bahrami N., Hassanzadeh S., Ezzatpour S., Hassanzadeh N., Kebria M. M., Khanmohammadi M., Bagher Z., Mater. Today Commun. 2022, 30, 103230. [Google Scholar]
- 135. García‐Couce J., Vernhes M., Bada N., Agüero L., Valdés O., Alvarez‐Barreto J., Fuentes G., Almirall A., Cruz L. J., Int. J. Mol. Sci. 2021, 22, 5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Agas D., Laus F., Lacava G., Marchegiani A., Deng S., Magnoni F., Silva G. G., Di Martino P., Sabbieti M. G., Censi R., J. Cell. Physiol. 2019, 234, 20013. [DOI] [PubMed] [Google Scholar]
- 137. Kim T., Suh J., Kim W. J., Adv. Mater. 2021, 33, 2008793. [DOI] [PubMed] [Google Scholar]
- 138. Ji X., Shao H., Li X., Ullah M. W., Luo G., Xu Z., Ma L., He X., Lei Z., Li Q., Biomaterials 2022, 285, 121530. [DOI] [PubMed] [Google Scholar]
- 139. Oliveira I. M., Fernandes D. C., Maia F. R., Canadas R. F., Reis R. L., Oliveira J. M., Pharmaceutics 2021, 13, 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Yu C., Gao H., Li Q., Cao X., Polym. Chem. 2020, 11, 3169. [Google Scholar]
- 141. Lei L., Cong R., Ni Y., Cui X., Wang X., Ren H., Wang Z., Liu M., Tu J., Jiang L., Adv. Healthcare Mater. 2024, 13, 2302551. [DOI] [PubMed] [Google Scholar]
- 142. Fan W., Yuan L., Li J., Wang Z., Chen J., Guo C., Mo X., Yan Z., Mater. Sci. Eng., C 2020, 110, 110705. [DOI] [PubMed] [Google Scholar]