

Abstract
Precision surface engineering is key to advanced biomaterials. A new platform of PEGylated styrene–maleic acid copolymers for adsorptive surface biofunctionalization is reported. Balanced amphiphilicity renders the copolymers water‐soluble but strongly affine for surfaces. Fine‐tuning of their molecular architecture provides control over adsorptive anchorage onto specific materials—which is why they are referred to as “anchor polymers” (APs)—and over structural characteristics of the adsorbed layers. Conjugatable with an array of bioactives—including cytokine‐complexing glycosaminoglycans, cell‐adhesion‐mediating peptides and antimicrobials—APs can be applied to customize materials for demanding biotechnologies in uniquely versatile, simple, and robust ways. Moreover, homo‐ and heterodisplacement of adsorbed APs provide unprecedented means of in situ alteration and renewal of the functionalized surfaces. The related options are exemplified with proof‐of‐concept experiments of controlled bacterial adhesion, human umbilical vein endothelial cell, and induced pluripotent cell growth on AP‐functionalized surfaces.
Keywords: adsorptive surface functionalization, antimicrobial surface properties, cell‐instructive properties, styrene–maleic anhydride copolymers
PEGylated styrene–maleic acid(anhydride) copolymers with systematically varied molecular architecture—anchor polymers—are introduced for the highly versatile adsorptive surface functionalization of materials. The water‐soluble but highly surface affine copolymers can be equipped with a broad range of bioactive ligands introducing cell‐instructive and antimicrobial surface properties.
1. Introduction
Surface functionalized biomaterials and bioresponsive, switchable cell‐instructive hydrogels can effectively direct living systems.[ 1 , 2 , 3 , 4 ] Emerging biotechnologies, including microfluidics and organ‐on‐a‐chip models increasingly rely on such approaches.[ 5 , 6 ] Recent insights into the exogenous regulation of cellular plasticity further highlight the potential of functionalized materials in controlling living systems,[ 7 ] together with and beyond the reconstitution and the templating of extracellular matrix (ECM) cues. However, the related capabilities of engineered materials surfaces will only be fully exploited once a broad range of biofunctional features and molecular signals can be freely combined and controlled in situ by robust and easily applicable techniques.[ 8 , 9 , 10 , 11 , 12 , 13 ]
Addressing this challenge, a range of sophisticated surface functionalization approaches have been developed. Despite being very powerful and multifaceted, most of the developed methods rely on the reactive/covalent immobilization of functional components, which entails considerable effort in the technical implementation,[ 14 , 15 , 16 ] interferes with customization, and limits translation into scalable applications.[ 17 ] Non‐covalent surface functionalization principles are therefore attractive, particularly if applicable in aqueous media (since organic solvents can affect the functionality of bioactive components and cause adverse reactions).[ 18 ] For that purpose, the electrostatically controlled adsorption of polycationic, peptide‐functionalized copolymers from aqueous solutions offers a simple and effective means,[ 19 , 20 ] however, remains restricted to anionically charged surfaces and of limited use in the presence of high salt and protein concentrations. A well‐established alternative for the adsorptive physicochemical modification of hydrophobic surfaces relies on surfactants (e.g., non‐ionic poly(ethylene glycol)‐block‐poly(propylene glycol)‐block‐poly(ethylene glycol) (PEG–PPG–PEG) copolymers).[ 21 ] Several recently published protocols have expanded the underlying principle and utilized hydrophobic interactions in combination with specific biomolecular functionalities: the CRGDS adhesion peptide was tethered to a hydrophobic antimicrobial peptide originating from Bacillus subtilis for non‐covalent immobilization to poly(dimethylsiloxane)‐surfaces, effectively mediating adhesion of fibroblasts and endothelial cells.[ 22 ] Dihydroxyphenylalanine (DOPA)‐containing anchor peptides were utilized to tether heparin‐binding peptides, sulfated hyaluronic acid derivatives, sulfated peptides, and receptor‐derived chemokine binding peptides for stromal‐cell‐derived factor 1 binding to polystyrene tissue culture plates.[ 23 ] A self‐strengthening hydrophobic anchoring unit based on thermal merocyanine‐to‐spiropyran isomerization was combined with adhesion ligand (RGD) peptides to functionalize surfaces, allowing for the fine‐tuned mechanical stimulation of adhering mesenchymal stromal cells.[ 24 ]
Considering the wealth of complementary bioactives to be applied at interfaces, the importance of their physicochemical presentation for control over functionality and the aim of “minimum necessary complexity for a given application”,[ 17 ] we developed a new platform of PEGylated styrene–maleic acid copolymers for the highly versatile, simple, and effective surface functionalization of materials (see Scheme 1 ). The approach relies on a well‐balanced amphiphilicity that renders the polymers water‐soluble while exhibiting a tunable strong affinity to various surfaces. Five copolymers of different molecular weight (MWbb) and maleic acid content (MA%) were converted with PEG chains of different size in different ratios to vary their PEGylation (characterized by the ratio of PEG‐monomer units (—CH2—CH2—O—) to styrene monomer units (—CH2—CH(—C6H6)—) within the respective copolymer: PEG/S‐ratio, see Scheme 1 and Table 1 ), allowing for maximized affinity to specific surfaces, desired structural characteristics as well as for convertibility by homo‐ and heterodisplacement of the adsorbed copolymer layers (see Scheme 1). The synthesis strategy of the respective ‘anchor polymers’ (AP) is versatile but simple: In polymer‐analogous modifications of generally available, variable SMA‐anhydride copolymers in a first step (1) oriented coupling between anhydride groups of the AP‐backbone with primary amines of amine‐containing PEGs (a‐methoxy‐ω‐amino PEG for water solubility, α‐amino‐ω‐tritylthio PEG for biofunctionalization after thiol deprotection during purification step) and amino‐ethylmaleimide (AEM) (for biofunctionalization) is performed in a “one‐pot” reaction (see Scheme 1). In step (2), the PEGylated APs containing thiol or maleimide groups are functionalized with thiol containing cell adhesion ligand peptides (e.g., CWGG‐RGDSP) or maleimide prefunctionalized cytokine‐complexing glycosaminoglycans (e.g., heparin‐Mal) and antimicrobials (e.g., hexetidine‐Mal) in an oriented, superfast (instantly), and nearly quantitative Michael type (click)‐reaction under physiological conditions (see Scheme 1). The APs can be subsequently utilized to produce (bio‐)functional coatings via simple adsorption from aqueous solutions to arbitrary hydrophobic surfaces. Mixing solutions of distinctly functionalized APs allows for an unprecedented and uniquely simple combination of various molecular functions and physicochemical features in adsorbed layers. Targeted in situ alterations of the layer composition (by homo‐ and heterodisplacement using APs of accordingly adjusted characteristics) and lateral patterning (across scales, starting at molecular level spacing) further expand the arsenal of applications.[ 10 ] To illustrate this powerful novel concept, we report first proof‐of‐concept studies with cell‐instructive and antimicrobial AP coatings. Our new, versatile AP technology will facilitate combinatorial high‐throughput surface engineering approaches of previously unrealizable complexity, paving the way for further advanced biomaterials with multimodal features.[ 17 ]
Scheme 1.
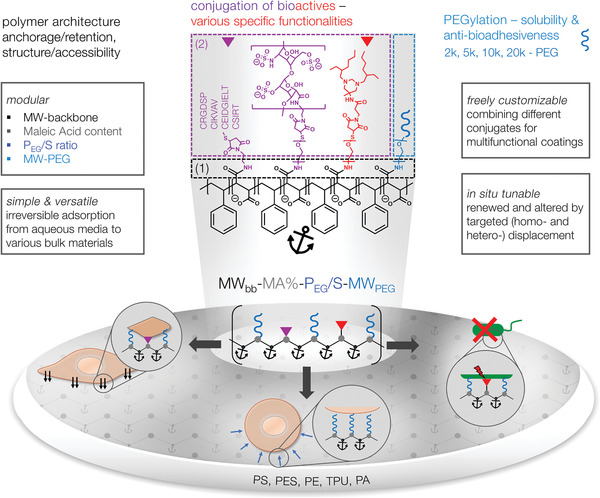
Survey of compositional and functional features of PEGylated styrene (S)–maleic acid (MA) copolymers (“anchor polymers”, APs), conjugated with different bioactives for adsorptive surface engineering. Center: The AP nomenclature: MWbb‐MA%‐PEG/S‐MWPEG, displaying the modularity of the system in terms of the molecular weight of the backbone (MWbb in kDa), the percentage of maleic acid groups (MA in %), the number of PEG monomer units per number of styrene monomer units (PEG/S) and the molecular weight of the PEG chain (MWPEG). The synthesis strategy uses in a first step (1) amine/anhydride reaction between styrene–maleic anhydride copolymers and amine groups of PEG and N‐(2‐aminoethyl)maleimide. In a second step (2), Michael type (click)‐reaction between N‐(2‐aminoethyl)maleimide and thiol containing cell‐adhesion ligands or between thiol groups of PEG and maleimide prefunctionalized glycosaminoglycans (e.g., heparin‐Mal) and antimicrobials (e.g., hexetidine‐Mal) are performed for functionalization. (Table 1 specifies the parameter space actually used.)
Table 1.
Compositional parameter space of the developed AP platform
| Backbone | MWbb a) [kDa] | MA b) [%] | PEG/S c) | MWPEG d) [kDa] | AM e) | CI f) |
|---|---|---|---|---|---|---|
| 1 | 10 | 10 | 3–58 | 2, 5, 10, 20 | — | — |
| 2 | 10 | 30 | 8–165 | 2, 5, 10, 20 | HEX | PEP/HEP |
| 3 | 80 | 30 | 6–32 | 2, 5 | — | — |
| 4 | 120 | 30 | 6–33 | 2, 5 | — | — |
| 5 | 350 | 50 | 22–470 | 2, 5, 10, 20 | HEX | PEP |
Molecular weight of polymer backbone
Maleic acid content of polymer backbone, styrene content (S): 100 – MA
Ratio PEG to styrene units
Molecular weight of the PEG chain
Antimicrobial bioactive: hexetidine (HEX)
Cell‐instructive bioactives: peptides (PEP): CRGDSP, CIKVAV, CEIDGIELT, CSIRT or heparin (HEP); see also Table S3, Supporting Information, for detailed information.
2. Results and Discussion
APs with systematically varied molecular architecture (as displayed by the four‐digit identifier MWbb‐MA%‐PEG/S‐MWPEG) were synthesized; a selected subset was further functionalized with exemplary bioactive units (see Table 1 and Scheme 1). Detailed synthesis protocols and results of basic chemical characterization are provided in the Supporting Information.
As a first selection criterion, we analyzed the water solubility of the PEGylated styrene–maleic acid copolymers using transmission measurements: APs with a PEG/S ratio ≥8 were found to be soluble over the entire tested concentration range c = 0.005–20 g L−1 (Figure 1A). Next, the adsorption kinetics and retention of four representative APs covering the variability of their molecular architectures, that is, PEG/S‐ratios from 16 to 470 (10–30–16–2, 10–30–165–20, 350–50–45–2, and 350–50–470–20), onto polystyrene (PS), polyethersulfone (PES), thermoplastic polyurethane (TPU), polyethylene (PE), and polyamide (PA)—representing biomedically relevant bulk materials—were studied by quartz crystal microbalance with dissipation monitoring (QCM‐D) (for details of the method and results of additional APs see Figure S10, Supporting Information). The APs adsorbed quickly from phosphate buffered saline (PBS) to all surfaces (for kinetics see Figure S11, Supporting Information) and formed hydrated monolayers with thicknesses ranging from 7 to 20 nm (Figure 1B). Furthermore, the adsorption was almost irreversible upon extensive rinsing in PBS with high retention values >77% (Figure 1B) pointing to a good anchoring power of the styrene units independent of the substrate type.
Figure 1.
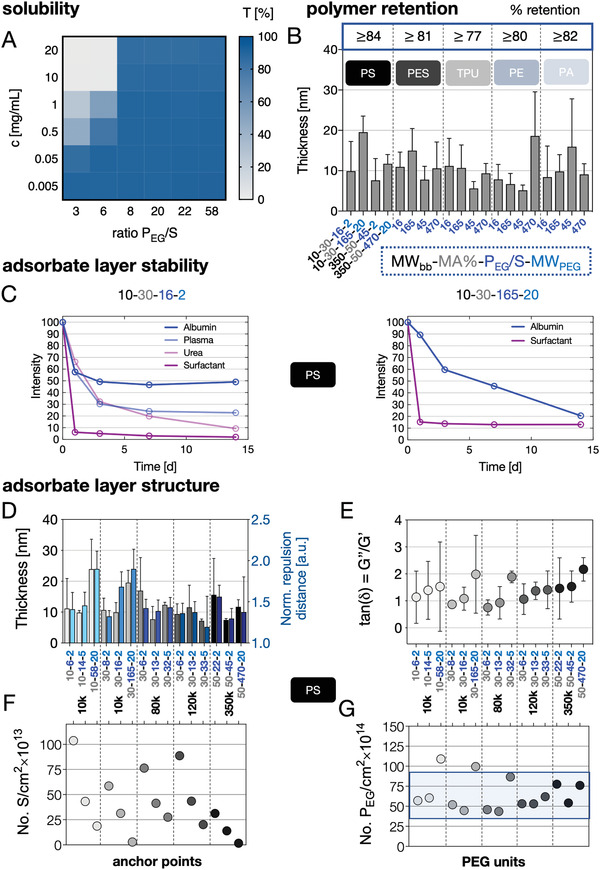
Physicochemical properties of the APs and the resulting adsorbate layers on various substrates. A) Solubility: Transmission recorded at 450 nm of AP solutions (concentration range of c = 0.005–20 g L−1) dissolved in PBS plotted in regard to increasing PEG/S ratios. B) Quartz crystal microbalance with dissipation monitoring (QCM‐D)‐based adsorption measurements from APs (c = 1 g L−1) dissolved in PBS on PS, PES, TPU, PE, and PA substrates. The layer thickness was calculated from frequency and dissipation changes using a viscoelastic model. C) Stability of fluorescently labeled AP (left: 10–30–16–2) and (right: 10–30–165–20) coatings on PS substrates incubated under application relevant conditions (37 °C; plate shaker) for 14 days displayed as a percentage of retained fluorescence intensity. D–G) The impact of the molecular AP architecture on the resulting adsorbate layer structure has been analyzed in detail for PS surfaces using QCM‐D measurements based on a viscoelastic model and with atomic force microscopy (AFM)‐based force spectroscopy measurements. APs with similar backbone molecular weight (MWbb) are grouped and the corresponding thicknesses (QCM‐D) and normalized repulsion distances (AFM) are shown (D). E–G) Ratio of loss to storage moduli as a measure for hydration/viscoelasticity (E), number of anchor points (styrene units) per cm2 (F), and number of PEG units per cm2 are plotted for increasing PEG/S ratios (G).
To further determine the stability of AP layers in presence of competing surface active components at physiological temperature (37 °C), we analyzed the fluorescence of pre‐adsorbed Atto‐647 labeled APs over 14 days upon incubation in: 1) buffered albumin solution (50 × 10−3 m tris(hydroxymethyl)aminoethan (Tris)‐HCL + 4 % bovine serum albumin), 2) citrate‐buffered plasma (0.14 m sodium citrate dihydrate), 3) 6 m urea, and 4) a nonionic surfactant solution (4 × 10−3 m Tween20, Figure 1C, for more AP types on PES, PA, TPU, and PA surfaces the reader is referred to Figure S11, Supporting Information). For adsorbate layers of AP Type 10–30–16–2 on PS, the initial decrease in fluorescence intensity ran into a plateau of the signal after 3 days of exposure to albumin solutions or plasma at physiological temperature, pointing at an excellent long term stability of AP layers nearly invariably containing ≈50% (after exposure to albumin solutions) and ≈25% (after exposure to plasma) of the initially adsorbed amount, respectively. The stability in albumin solutions with ≈50% fits well with the quantification of protein adsorption on the respective layers: for AP Type 10–30–16–2 the adsorbed protein amount was reduced to ≈50% of the value determined for the plain PS surface, that is, pointing at the competitive adsorption of albumin as driving force for polymer replacement (see Figure S12, Supporting Information). We hypothesize that the AP layers contain adsorbed polymers with differing degrees of anchorage, that is, numbers of surface attachment points, of which only the most effectively immobilized ones resist the more challenging biological displacement conditions (plasma at elevated temperature). In contrast, a steady displacement of adsorbed APs in presence of 6 m urea and an almost instant removal of the AP layer by Tween20 were observed (Figure 1C). This effective cleaning by a well‐accepted non‐toxic surfactant can be beneficial in surface regeneration protocols. Similar characteristics of AP Type 10–30–16–2 layers were observed on PES surfaces (with higher plateau retention levels in albumin solutions compared to the layers on PS) and on TPU, PE, and PA surfaces (with lower plateau retention levels in albumin solutions compared to the layers on PS), indicating a superior stability of this AP independent of the surface it is adsorbed to (see Figure S11, Supporting Information). For layers of the AP of the same 10–30 backbone and the highest possible PEGylation with 20 000 Da PEG chains (AP Type 10–30–165–20), a steady decrease in fluorescence intensity was observed in albumin solutions, that is, no long term stability was obtained (Figure 1C). Nevertheless, AP (10–30–165–20) reduced the amount of adsorbed albumin to ≈50% of the value determined for the plain PS surface (see Figure S12, Supporting Information). For the 50% MA–AP Types 350–50–45–2 and 350–50–470–20 on PES surfaces, a stable layer containing about 40% of the initially adsorbed polymer was approached already after 1 day of exposure to albumin solutions, demonstrating stability under application‐relevant conditions (see Figure S11, Supporting Information).
A comparison of the AP layer retention and in‐depth layer analyses can provide insights into the interactions and structural effects governing surface anchorage (beyond the obvious basic principle of hydrophobic interactions).
First, the higher fractions of tightly adsorbed APs on PS and PES surfaces compared to TPU, PE and PA suggest that π–π‐stacking in between aromatic components can contribute to the AP layer stability (see Figure S11, Supporting Information). Along this line, polymer backbone Type 2 (10–30–), that is APs containing 30% MA and 70% styrene, exhibited distinctly higher affinity to PS and PES surfaces pointing at an elevated stabilization due to clustered styrene anchor units (see Figure S11, Supporting Information, stability values and Table 1).
Second, the relevance of the particular molecular AP architecture for the structure of the adsorbed layer was exemplarily analyzed for APs on PS (Figure 1D–G). Applying a viscoelastic model for the analysis of QCM data,[ 25 ] which takes both the adsorbed polymer and bound water into account, layer thickness values in the range of 8–10 nm for most APs were found while APs with the longest PEG‐chains (20 000 Da) as well as the 80–30–6–2 and the 350–50–22–2 variant typically showed values above 10 nm (Figure 1D).[ 25 ] All determined values exceeded the radius of gyration with 1.4–5.7 nm determined for 2000–20 000 Da PEG‐molecules,[ 26 , 27 ] pointing at a brush‐like conformation of the adsorbed APs, resembling comb‐copolymers with an adsorbing backbone and non‐adsorbing side chains.[ 28 ] The QCM‐based layer thickness data correlate very well with force spectroscopy results determining repulsive force distances of the different APs (Figure 1D). The viscoelastic modeling of QCM data further provides the ratio of the loss moduli (i.e., purely viscous behavior of adsorbed layers) to the storage moduli (i.e., purely elastic behavior of adsorbed layers) given as tan(δ) = G″/G′ (Figure 1E). tan(δ) is directly correlated to energy loss/dampening and hydration of the layer. Independent of the polymer backbone molecular weight, we found tan(δ) ≈ 1 for low PEG/S ratios obtained by conjugation of the shortest 2000 Da‐PEG chain, that is, the adsorbed polymer layer resembles a viscoelastic “solid” at the gel point that indicates an ordered brush‐like conformation (were the extension of the PEG‐chains is underpinned by the layer thicknesses of 8–10 nm being not far away from the contour length of a 2000 Da PEG with ≈14 nm [Figure 1D]).[ 29 ] With increasing PEG chain length, we obtained tan(δ) > 1, which indicates that the adsorbate layer resembles more a viscous liquid that correlates with significantly higher water uptake but also a less dense brush regime—a finding that is well‐supported by the bigger differences between the adsorbate layer thickness of 10–15 nm (Figure 1D) and the respective contour length of the 5000–10 000 Da PEG chains with 34–68 nm.[ 29 ] The APs of type 10–30–165–20 and 350–50–470–20 showed the highest hydration with tan(δ) ≈ 2 that manifest in the biggest offset between the layer thickness of 20–22 nm (Figure 1D) and the contour length of the 20 000 Da chain with ≈135 nm pointing to an irregular layer structure with loops and dangling ends going beyond a brush conformation.[ 29 ] Water contact angle data of 10–30–8–2 and 10–30–165–20 AP layers on PS, PES, and TPU showing a strong hysteresis between advancing and receding contact angles pointed to strong swelling phenomena and confirmed the according capability of the APs for effective hydrophilization (see Figure S7, Supporting Information). However, due to the impact of the intrinsic roughness of the substrates and swelling phenomena in contrast to QCM and AFM‐data (compare Figure 1D,E) significant differences between the AP‐architecture could not be resolved utilizing this method.[ 30 ] We further derived the number of anchor points (styrene monomer units) per cm2 and the number of PEG‐monomer units per cm2 from the adsorbed mass data based on QCM measurements (Figure 1F,G, for details see Supporting Information). The number of anchor points per surface area decreased significantly for a given MWbb with increasing PEG/S‐ratio or MWPEG, opposite to the trend of tan(δ). Thus, only very few anchoring units are needed to immobilize strongly hydrophilic APs like 350–50–470–20 to hydrophobic surfaces. The adsorbed APs showed a remarkable similar density of PEG‐monomer units per surface area of about 45–80 × 1014 units per cm2 (with the exception for 10–10–58–20 and 10–30–165–20, which produced a clearly higher PEG surface density > 100 × 1014 units per cm2), indicating rather similar spatial requirements and hydration of the PEG‐monomer units. These results suggest that the structure of the adsorbate layers is determined by a balance of excluding volume expansion forces caused by the hydrated PEG units and spatial retention forces caused by hydrophobic and π–π‐interactions of the anchoring points with the surface or with each other.[ 31 , 32 ] For PLL‐PEG copolymers and mPEG‐DOPA3 adsorbed on anionic surfaces similar values of PEG‐monomer numbers in the range of 5–50 × 1014 units per cm2 have been found (slight differences in absolute values correlate with the lower adsorbed mass in these studies and may be attributed to the sterically more repulsive anchoring units lysin or DOPA compared to the π–π stacking styrene units of our APs).[ 26 , 33 ] For the two identified exceptions, types 10–10–58–20 and 10–30–165–20, both formed out of a small 10 000 Da‐anchoring backbone and long 20 000 Da PEG chains, the higher number of PEG‐monomer units per surface area points at a stronger irregular conformation with loops and defects accompanied by a significantly reduced stability of the adsorbed layers.
The design concept of our AP platform specifically referred to two previously reported properties of styrene–maleic anhydride/acid copolymers (SMAs): 1) the almost linear conformation at neutral pH that favors π‐stacking and hydrophobic interactions of the styrene units and supports self‐association as well as surface affinity (an effect caused by a stabilizing H‐bond between the protonated carboxyl group and the neighboring, deprotonated carboxyl group of the diacid,[ 31 , 32 ]) and 2) the fact that SMAs with a styrene to maleic acid ratio of 2:1 to 3:1 strongly interact with hydrophobic membrane proteins.[ 34 , 35 ] Accordingly, we selected the 10–30, 80–30, and 120–30 backbone types with an optimal styrene to MA ratio of 2.3:1 and PEGylated the SMAs upon reaction of the anhydride with an amine terminated PEG to introduce a stable amide bond.[ 36 ] By selecting a PEG/S‐ratio ≥ 8 we could avoid self‐association (see Figure 1A) while providing a rather pH‐independent solubility (data not shown) and linear conformation stabilized by hydrogen bond formation between the remaining, over a large pH range (pH > 3.5) deprotonated carboxylic acid group, and the NH‐group of the amide bond. Stability tests in hydrogen bond breaking 6 m urea solution on PS surfaces underpin the proposed mechanism by disrupting the hydrogen bonds leading to desorption of the pre‐adsorbed 10–30–16–2 AP (see Figure 1C).
Significantly higher PEGylation of 10–30–165–20 and 10–10–58–20 caused exceptionally high PEG densities per surface area (Figure 1G) and resulted in a slow but nearly complete desorption of the adsorbed APs over 14 days (Figure 1C). We assume that these two APs are bound weakly in a strongly hydrated, irregular loop, and defect rich structure that is not stable over time in polymer free solution. These considerations are further underpinned by a combined mean field and scaling approach to study the adsorption of amphiphilic comb polymers with an adsorbing backbone and hydrophilic side chains to hydrophobic surfaces.[ 37 ] In here, Sartori et al. applied scaling laws (under the assumption of swollen polymer chains) to determine if a polymer of the above‐described architecture with N PEG water soluble repeating monomer units and N B water insoluble backbone monomer units attains mushroom or brush conformation in the adsorbed state or undergoes desorption. If Equation (1) is fulfilled:
| (1) |
the polymer adsorbate layer is in a brush regime. Furthermore, if Equation (2) is fulfilled:
| (2) |
the polymer is beyond the desorption threshold. For the APs included in the datasets displayed in Figure 1, we calculated the respective values (see Table S4, Supporting Information) and found that they are adsorbed in a brush regime with exception of 10–10–58–20 and 10–30–165–20 that are beyond the desorption threshold. These results impressively confirmed the above qualitative considerations and experimental stability data (of note, AP layers of variable but limited stability are of practical interest as “sacrificial layers” for programming the interfacial dynamics to minimize or overcome bioadhesion).
Altogether, our results demonstrate how the modular architecture of APs can afford stable layer formation while simultaneously providing a broad variability of the PEG/S ratio and of architectural features. This allows for the effective interfacial presentation of strongly hydrophobic or hydrophilic bioactives, targeted displacement of the APs for dynamic adjustment of the layer characteristics, and for maximized antibioadhesiveness.
AP layers that were proven to be rather stable against desorption in PBS and displacement by the components of biofluids can nonetheless undergo effective homo‐(self‐)exchange, that is, displacement by similar APs, and heteroexchange, that is, displacement by APs of higher affinity or molecular weight (an effect long known and often referred to for proteins as the “Vroman Effect.”[ 38 , 39 , 40 ] These phenomena can be used for dynamic tuning and/or renewing of materials surface properties produced by adsorbed APs. To elaborate this option, we selected two fluorescence labeled APs with almost similar PEG/S‐ratio but significantly different polymer backbone MWbb (10–30–16–2 and 120–30–13–2) and analyzed the homo‐ and hetero‐ (small vs big and big vs small) displacement of their adsorbed layers on PS, PES, and TPU (Figure 2 ) for a period of 14 days (Figure 2A). The exchange was found to be very effective (≥75%) and independent of the type of AP and surface (Figure 2B).
Figure 2.
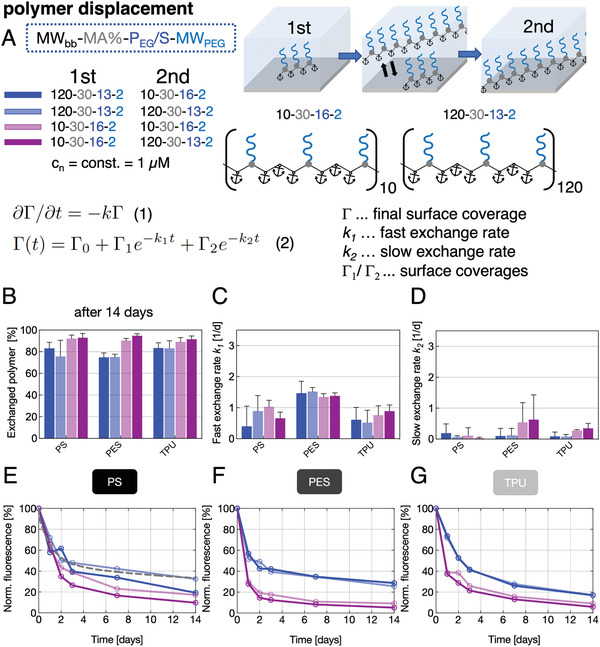
Homo‐ and heterodisplacement of APs adsorbed on PS, PES, or TPU, A) applying APs with identical PEG/S ratio and identical (homodisplacement) or different (heterodisplacement) molecular weights. Differential Equation (1) used to describe the displacement kinetics with Γ being the surface coverage and k the desorption time constant. Equation (2) is fitted to original data (an exemplary fit is indicated by the gray, dashed line in [E]). Four homo‐ and heteroexchange conditions were studied using two APs, each used once as pre‐adsorbed and as displacing component in solution, respectively. B) Exchanged amount of AP after 14 days incubation with displacer solutions, extracted from values of decreasing fluorescence. C,D) One solution of Equation (1) suggests a fast and slow desorption process characterized by two exchange rates of desorbing AP. E–G) Kinetics of AP displacement over a 14‐days period on PS (E), PES (F), and TPU (G) plotted as normalized fluorescence, elucidating the two‐staged desorption process.
To describe the displacement kinetics of the pre‐adsorbed AP, we used a simple differential equation (Figure 2 and Equation (1)) which takes only the pre‐adsorbed AP surface coverage Γ and the fast and slow time constants k into account. However, this model does not consider the displacing AP in solution (of note, a constant molar concentration of 1 × 10−6 m was used for the displacing AP solution in all experiments). We previously utilized this model to describe protein displacement.[ 41 ] Analyzing the data (Figure 2E–G), a fast and slow exchange process are emerging, independent of the type of pre‐adsorbed AP (1) and the AP used for displacement (2) (see Figure 2A). We observed that the fast exchange process on PE and TPU surfaces approaches a plateau value after ≈2 days and on PES surfaces after ≈1 day. Also, the value of the fast exchange rate on PES was at least twice as high compared to the fast exchange rate on PS and TPU (Figure 2C). These results corroborate earlier published data on the exchange of polystyrene in organic solvents with a fraction undergoing fast exchange and a “trapped” fraction hindered from desorption by the newly immobilized molecules (slow exchange).[ 42 ] Furthermore, we observed a clear trend of small pre‐adsorbed APs being much stronger exchanged by small and big APs (Figure 2B,E–G, light red and dark red). On the contrary, big pre‐adsorbed APs are not as efficiently displaced by small or big APs (Figure 2E–G, dark blue and light blue curves). These findings agree with two well‐known features of polymer displacement: 1) identical molecules may exchange each other also in absence of a net driving force due to statistical effects and 2) preferential adsorption of larger molecules due to a net gain in translational entropy resulting from the release of a higher number of smaller molecules.[ 39 ]
Together, the collected insights in correlations of molecular AP architecture and adsorption layer characteristics provide a strong basis for applying the developed polymer platform to precisely adjust biofunctional materials surface properties, an approach that is massively extended and endorsed by the synthesis of AP conjugates carrying various different bioactive molecular units. Subsequently, we illustrate these options with selected proof‐of‐concept data.
As bacterial adhesion precedes the formation of resistant bacterial biofilms and recurrent infections, antimicrobial surface functionalization is considered a key challenge in biomaterials design.[ 43 , 44 , 45 ] We therefore analyzed the antibioadhesive properties of three selected APs, the superior stable adsorbing antiadhesive AP 10–30–16–2, the highly hydrated but slowly “peeling‐off” 10–30–165–20 AP, and one firmly anchored antiadhesive–antimicrobial (10–30–16–2–Hex) AP containing covalently conjugated hexetidine. We tested the initial bacterial adhesion in: i) a solution‐based assay simulating the bacterial surface colonization and in: ii) a direct‐contact assay reflecting the transmission of pathogens between abiotic and biotic surfaces, which is especially prevalent in hospital settings.[ 46 , 47 , 48 , 49 ] To maximize repulsive hydration forces, we selected strongly antiadhesive APs known to effectively adsorb in layers exhibiting a PEG brush architecture, the AP 10–30–16–2, and the slowly “peeling‐off” and highly hydrated AP 10–30–165–20. AP conjugates carrying the antimicrobial component hexetidine were expected to additionally minimize microbial adhesion by killing adherent bacterial cells. We tested the solution adhesion properties in M9 depleted medium for the Gram‐positive species Staphylococcus aureus and the Gram‐negative species Escherichia coli (for details of the method see Supporting Information). The initially adhered amount of bacteria was comparable on all surfaces (Figure 3A,C and Figures S15 and S16, Supporting Information). While for the untreated PS control surfaces for both E. coli and S. aureus a significantly increased total surface‐coverage of 70% and 40% (100% surface coverage corresponds to a completely covered surface) was observed after 24 h, respectively, we observed a 20–100‐fold reduction in the surface coverage of adhered bacteria over the time course of 24 h for PS with all three pre‐adsorbed APs (Figure 3B,D). The kinetic decrease was especially prominent for S. aureus in the first 4 h for the AP with conjugated hexetidine, which we assume to be caused by additional bacterial lysis (Figure 3D). We observed similar trends of reduced adhesion for E. coli, while in here after 24 h, the highly hydrated and slowly “peeling‐off” 10–30–165–20 antiadhesive AP repelled slightly more bacteria than the less hydrated 10–30–16–2‐type.
Figure 3.
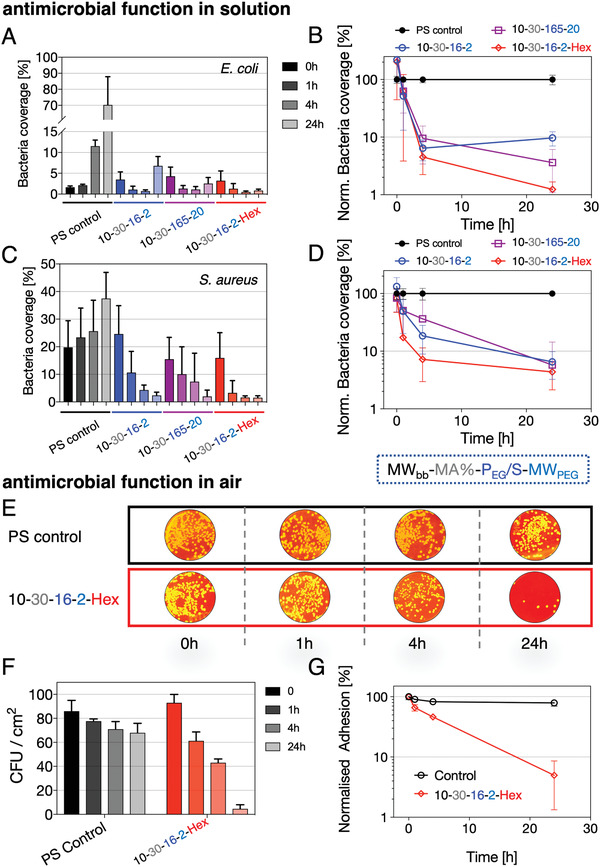
Antibioadhesiveness and antimicrobial functionality of AP layers. Solution‐based assay of E. coli and S. aureus in contact with antiadhesive 10–30–16–2 and 10–30–165 as well as antiadhesive and antimicrobial 10–30–16–2–Hex (hexetidine) AP layers. A,C) Relative bacterial coverage (100% coverage corresponds to completely covered substrate) over 24 h and B,D) normalized (to control surface) bacteria coverage over time for E. coli and S. aureus. E) S. aureus solutions are directly applied to the substrates and the number of colony‐forming units (CFU) is analyzed after pressing the substrates directly on agar plates (false‐colored photographic images of CFUs on agar plates for better visualization) after the indicated incubation times of 0, 1, 4, and 24 h (gray: control surface, green: 10–30–16–2–Hex). F) Analysis of CFU per cm2 for direct‐contact assay. G) Normalized adhesion of CFUs from (F) indicating the relative reduction in adhered colonies over a time period of 24 h compared to the starting condition.
The AP with conjugated hexetidine reduced adhered E. coli colonies by two orders of magnitude (Figure 3B), a significantly stronger drop compared to S. aureus. We further subjected S. aureus to a direct‐contact assay in which bacterial colonies were spread out onto AP‐precoated surfaces in ways to mimic the exposure of widely used plastic surfaces in hospital settings. The samples were then incubated at room temperature in air and at defined time intervals pressed against a solid agar plate to count the surviving colonies (Figure 3E). Control surfaces (PS) showed a 20% reduction in viable colonies compared to the starting condition—indicating a natural decrease in viable colonies on air exposure (Figure 3F), whereas a 20–30‐fold reduction of surviving colonies for the antimicrobial hexetidine‐AP functionalized surface was found (Figure 3F,G).
While all compared AP types afforded antibioadhesive performance, the bifunctional AP 10–30–16–2–Hex (antiadhesive and antimicrobial) provided a reduction in bacterial colony‐forming units by two orders of magnitude compared to the controls in both assays. The result of the contact assay in particular highlights the option to simply spray an aqueous AP‐solution onto polymer surfaces to effectively suppress bacterial contamination, transmission, and infections. This procedure can be extended to a wide range of irregularly shaped objects and devices, with the advantage of easy renewal of functionality while avoiding any visual or haptic surface alterations.
Given the multifactorial exogenous regulation of cell fate control, effectively cell‐instructive engineered materials surfaces require biofunctional features and molecular signals to be freely combined and controlled in situ by robust and easily applicable techniques. To explore the respective options of our newly developed AP technology, we designed AP conjugates carrying adhesion ligand peptides derived from ECM proteins or sulfated glycosaminoglycans capable of complexing cytokines,[ 50 , 51 , 52 ] morphogens, and growth factors, both in combination with a well‐balanced content of PEG‐chains to control hydration and to reduce unspecific protein adsorption. To better cope with the necessity of the dynamic adaptation of cell‐instructive signals, the abovementioned hetero‐ or homodisplacement processes (Figure 2) were employed to alter the composition of adsorbed AP layers applied in cell culture in situ.
A set of exemplary cell adhesion peptides was conjugated to maleimide‐functionalized APs of 10–30–8–2‐type and of 40–50–19–0.5‐type via Michael‐type addition with a universal CWGG‐peptide linker: CWGG‐RGDSP (derived from fibronectin [FN]), CWGG‐IKVAV and CWGG‐SIRT (derived from laminin), and CWGG‐EIDGIELT (derived from tenascin C). The nearly quantitative and biorthogonal conjugation strategy was herein used for preconjugation of peptides or peptide mixtures to APs but also allows for post‐conjugation of maleimide‐functionalized APs after their adsorption to, for example, cell‐culture plates.[ 53 ]
APs functionalized with the FN‐derived αvβ3 and αvβ5 integrin‐binding peptide RGDSP were used to functionalize untreated PS well‐plates and to cultivate human umbilical vein endothelial cells (HUVECs) with an FN coating served as a positive control. HUVECs adhered with similar cell numbers and viability on 10–30–8–2–RGDSP functionalized layers compared to FN in standard endothelial cell media (Figure 4A and Figure S13, Supporting Information), however, exhibiting a slightly more elongated morphology on the 10–30–8–2–RGDSP AP that resembles the tube‐like structures found in 3D‐models more closely.[ 54 ] Antibioadhesive surfaces consisting of adsorbed PEGylated APs without bioactives displayed a slight increase in HUVECs repellency with increasing PEG/S ratio and PEG‐chain length (see results for 10–30–16–2 and 10–30–48–10, Figure 4A,B) that correlates well with the increasing AP layer thickness and hydration (compare Figure 1).
Figure 4.
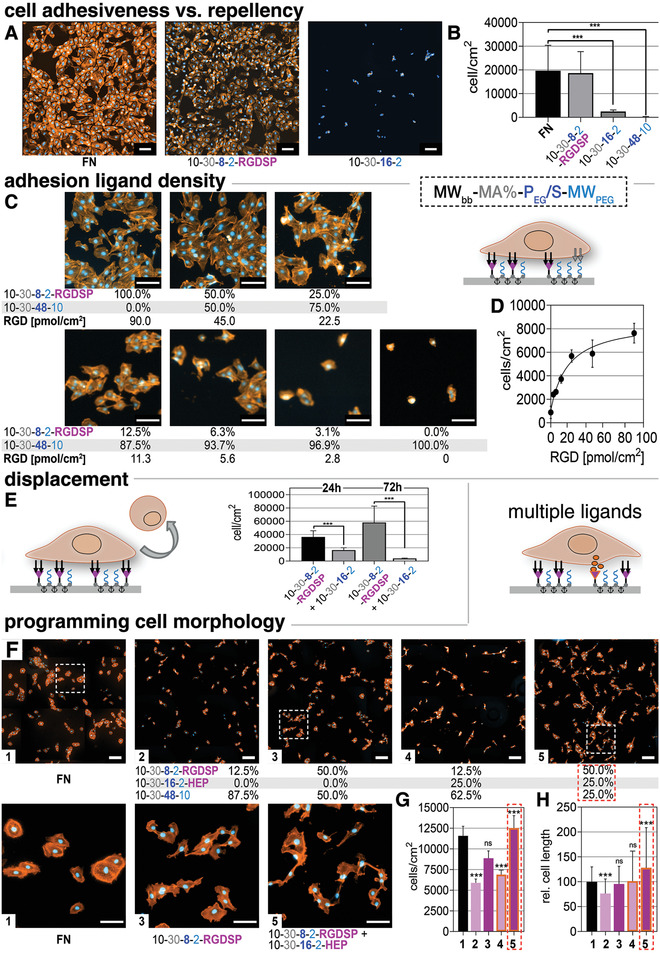
F Cell‐instructive AP layers with distinct functional features for cultivation of HUVECs. A) Adhesion of HUVECs on RGDSP‐functionalized AP (10–30–8–2–RGD) layers, fibronectin (FN), or antiadhesive APs with increasing PEGylation and chain length 10–30–16–2 and 10–30–48–10. B) Quantification of cell numbers. C) Variation of RGDSP ligand density from 0 to 3,1% (2.8 pmol cm−2), 6,1% (5.6 pmol cm−2), 12.5% (11.3 pmol cm−2), 25% (22.6 pmol cm−2), 50% (45 pmol cm−2) as well as 100% RGDSP (90 pmol cm−2). D) Quantification of cell number versus ligand density. E) Switching of surface properties from adhesive to antiadhesive via targeted exchange of AP 10–30–8–2–RGD by AP 10–30–16–2; quantification of adhered cells before and after AP displacement analyzed after 24 or 72 h. F) Programming cell instructive signals by variation of ligand type and concentration by mixtures of 10–30–8–2–RGDSP, 10–30–16–2–HEP, and 10–30–48–10. G,H) Quantification of cell density (G) and of cell elongation (H) after 48 h on the AP‐coated surfaces displaying multiple ligands. Staining (A,C,F): nucleus: Hoechst 33342 (blue), actin: Atto‐555 Phalloidin (orange). Scale bars (A,C,F): 100 µm.
As a particular advantage, our AP technology allows for easily varying the type and density of cell instructive peptide units at interfaces by simply mixing peptide‐conjugated and non‐conjugated APs in solutions used for adsorptive surface functionalization. Since the combined APs exhibit a similar surface affinity and adsorption behavior, the composition of the adsorbed layers can be precisely adjusted in a stochastic but uniform manner. We confirmed the homogeneity of the surface coating in the µm‐range by time‐of‐flight secondary ion mass spectrometry (ToF‐SIMS) measurements and the ligand densities with QCM‐D measurements (see Figure S9, Supporting Information). Mixtures of 10–30–8–2–RGDSP and 10–30–48–10 AP were used to prepare systematically varied RGD‐peptide surface concentrations (0%, 3,1% [2.8 pmol cm−2], 6,1% [5.6 pmol cm−2], 12.5% [11.3 pmol cm−2], 25% [22.5 pmol cm−2], 50% [45.0 pmol cm−2] as well as 100% RGDSP [90 pmol cm−2]) which were shown to cause a gradual increase in the density of adhered cells (Figure 4C,D).[ 55 ]
Our approach thus combines flexibility in the adhesive functionalization with the option of producing a maximal ligand density similar to dedicated protocols using SAMs, gold clusters, or peptide amphiphiles.[ 56 , 57 , 58 , 59 , 60 ] Next, we analyzed the dynamic modulation of the adhesive surface characteristics by targeted AP displacement (Figure 4E). 10–30–8–2–RGDSP was pre‐adsorbed on standard, untreated PS well‐plates and HUVECs were subsequently seeded and allowed to adhere for 2 h. Afterward, the medium was exchanged to contain then antiadhesive AP 10–30–16–2 (directly added to the HUVECs medium) and applied for 24 or 72 h. In result, almost all cells detached from the surfaces after 72 h (Figure 4E). Any negative effects of the antiadhesive APs in solution could be ruled out by the similar viability/metabolic activity and cell morphology that was found when comparing the growth of HUVECs on FN with the antiadhesive AP being optionally present in cell‐culture medium (see Figure S14, Supporting Information). The approach can be considered an option for very gentle cell release, avoiding harsh enzymatic treatment or administration of chelates.
To modulate the presentation of cytokines and growth factors at interfaces we furthermore developed AP conjugates containing sulfated glycosaminoglycans. Eight heparin molecules were linked to the AP 10–30–16–2 via a Michael type addition reaction (see Scheme 1 and Supporting Information). After adsorption of the 10–30–16–2–HEP to PS and excessive washing in PBS, an adsorbate layer of ≈14 nm thickness was obtained displaying a heparin density of ≈30 pmol cm−2 (see Figure S10, Supporting Information). Defined cell culture medium (MV2) containing a reduced serum amount of 0.5% and the heparin‐binding growth factors insulin‐like growth factor 1, epidermal growth factor, fibroblast growth factor 2, and vascular endothelial growth factor were used in experiments using surfaces functionalized by mixtures of 10–30–8–2–RGDSP (adhesive), 10–30–16–2–HEP (growth factor complexing), and 10‐30‐48‐10 (repellent) (see Figure 4F). A low concentration (12.5%; 11.3 pmol cm−2) of RGDSP resulted in significantly reduced cell densities and elongation compared to cells grown on FN (control). Increasing the RGD‐ligand density to 50% RGDSP (45 pmol cm−2), cell density and elongation was higher, pmol cm−2, Figure 4G,H). For the latter condition, the highest cell density was observed and the cells started to fuse (inset, Figure 4F,G).
Finally, different adhesion ligands RGDSP, EIDGIELT, IKVAV, and SIRT (all with 50%) plus 25% HEP were compared (see Figure S13, Supporting Information). Here, the cell elongation and cell area were found to be maximized for 50% EIDGIELT or 50% RGD + 25% HEP respectively, pointing to an additive effect of adhesion ligands and surface‐supported growth factor presentation.
To validate the versatility of the AP technology we also applied it to the even more demanding culture of undifferentiated human induced pluripotent stem cells (hiPSCs). hiPSCs represent a powerful cell resource which holds tremendous promise for cell‐based therapies, drug discovery, disease modeling, and pharmaceutical applications. To date, hiPSCs are mainly cultured on a thin layer of ECM proteins to promote integrin‐mediated cell attachment. The most frequently used substrate is Matrigel, a permissive ECM preparation derived from mouse sarcomas.[ 61 , 62 ] However, its undefined composition and biological origin restricts the use of the cells for therapeutic applications.[ 63 ] To provide a well‐defined, fully synthetic hiPSC growth promoting surface, we conjugated different integrin‐binding RGD containing peptides (cycRGD and PQVTRGDVFTMP) as well as a positively charged peptide sequence (GRKK)4 via a cysteine containing linker unit to a maleimide‐functionalized AP of 40–50–19–0.5‐type via Michael‐type addition (referred to as 40–50–19–0.5–RGD+ coatings subsequently (see Supporting Information)). 40–50–19–0.5–RGD+ coatings were used to modify untreated PS 6‐well plates prior to hiPSC seeding. Matrigel‐coated 6‐well plates, the current gold standard for hiPSC expansion, served as a positive control. For all experiments the hiPSC line CRTD3, derived from the Center of Regenerative Therapies Dresden at the Technische Universität Dresden, was used and cultured routinely on Matrigel‐coated tissue culture plates in mTeSR1 medium (Stem Cell Technologies). After passaging as cell aggregates with the non‐enzymatic reagent ReLeSR (Stem Cell Technologies) we allowed the cells to adapt for one passage to the synthetic surface (for details of cell culture see Supporting Information). After this passage we started to count the passage numbers and characterized the cells regarding their adherence to the 40–50–19–0.5–RGD+‐surface, their proliferation, growth, morphology, and differentiation in each passage.
hiPSCs adhered with similar numbers on the 40–50–19–0.5–RGD+‐ as well as the Matrigel‐coated plates. They grew on both surfaces as densely packed hiPSC colonies composed of round‐shaped cells with clearly visible nucleoli within the nuclei and showed a similar cell morphology in both growth conditions over all passages investigated (Figure 5A). To prove that the cells were still pluripotent, the expression of the pluripotency markers octamer‐binding transcription factor (OCT)3/4 and SRY (sex determining region Y)‐box 2 (SOX2) was analyzed by immunofluorescence (Figure 5B). hiPSCs cultured on 40–50–19–0.5–RGD+‐ or Matrigel‐coated plates displayed a homogeneous expression of both markers (cyan: nuclei, magenta: SOX2; green: OCT3/4, Figure 5B) in almost all cells within the colonies suggesting that the growth on the 40–50–19–0.5–RGD+ surface did not alter the pluripotency of the cells. To quantify the expression of the pluripotency markers after 12 passages we performed fluorescence‐activated cell sorting (FACS) analysis for the nuclear markers OCT3/4 and homeobox protein NANOG (NANOG) as well as the extracellular markers T cell receptor alpha locus (TRA)‐1‐60 and stage‐specific embryonic antigen‐4 (SSEA4) (Figure 5C). A similar percentage of the cells grown on 40–50–19–0.5–RGD+ were double positive for OCT3/4 and NANOG (96.8 ± 0.9% on 40–50–19–0.5–RGD+; 98.1 ± 0.8% on Matrigel) as well as TRA‐1‐60 and SSEA4 (90.7 ± 5.9% on 40–50–19–0.5–RGD+; 88.3 ± 10.4% on Matrigel). This analysis confirmed the results of the immunofluorescence stainings. Last, we immunostained samples for F‐ACTIN (yellow) to determine the nucleus to cytoplasm (N/C) ratio (Figure 5D). A quantification of the N/C ratio revealed comparable values for the cells grown on Matrigel and on the 40–50–19–0.5–RGD+ surface (85.3 ± 2.3% on Matrigel; 85.1 ± 1.8% on 40–50–19–0.5–RGD+). Of note, these values correlate well to the N/C of hESCs.[ 64 ] To conclude, APs functionalized with cell‐instructive peptides can be fine‐tuned to support a variety of applications such as providing an antimicrobial surface or to promote growth of a multitude of cells. The precise adjustment of multiple cell adhesion promoting peptides led to the development of the 40–50–19–0.5–RGD+‐coating which manifested as an alternative culture substrate to animal‐derived Matrigel applicable in regenerative medicine. Our AP approach thus addresses the culture and maintenance of hiPSC by providing an animal‐free, robust coating, allowing experimentation without batch‐to‐batch variety.
Figure 5.
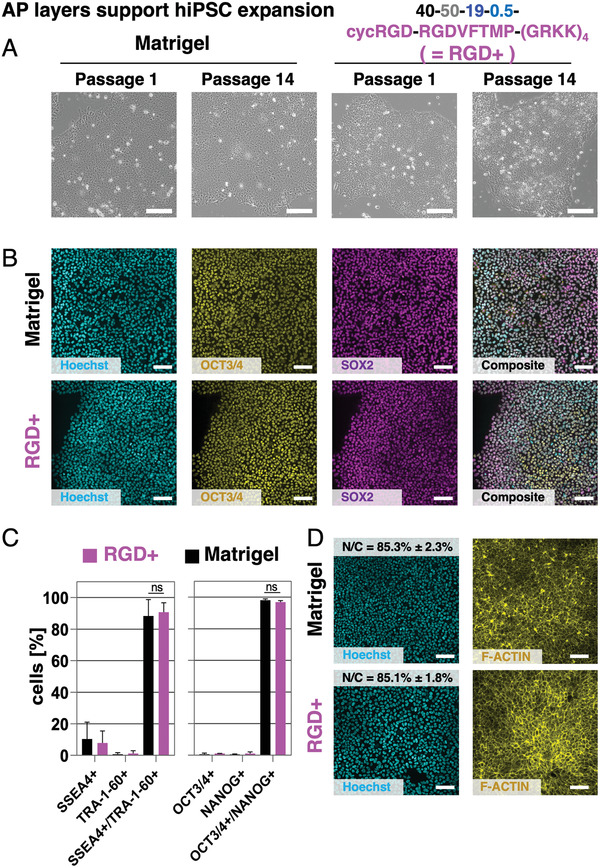
Cell‐instructive AP layers with distinct functionalization promote undifferentiated growth of hiPSCs. A) Growth of hiPSCs on peptide‐functionalized AP layers called 40–50–19–0.5–RGD+ or Matrigel for more than 14 passages visualized by brightfield microscopy. B) Immunofluorescence analyses of cells in passage 12 grown on Matrigel‐coated or AP 40–50–19–0.5–RGD+‐functionalized surfaces revealed that almost all cells co‐express the pluripotency markers OCT3/4 (yellow) and SOX2 (magenta) independent of the surface they were cultured on. C) FACS analysis for the pluripotency markers SSEA4, TRA‐1‐60, OCT3/4, and NANOG in passage 12. The cells grown on the 40–50–19–0.5–RGD+‐functionalized surface express all four pluripotency markers to a similar percentage as to hiPSCs grown on Matrigel. D) Quantification of the nucleus/cytoplasm ratio (N/C) of the cells grown on 40–50–19–0.5–RGD+‐functionalized plates or Matrigel using F‐ACTIN (yellow) and Hoechst (cyan) stained images revealed similar values in both growth conditions. Scale bars (A,B,D): 100 µm.
The examples discussed illustrate how our newly developed AP surface functionalization technology is capable of identifying minimalistic sets of effective cell‐instructive cues. With this unprecedented option, the introduced method can empower combinatorial high‐throughput surface engineering approaches of previously unrealizable complexity, paving the way for further advanced biomaterials with multimodal features.
3. Conclusion
PEGylated styrene–maleic acid(anhydride) copolymers with systematically varied molecular architecture—APs—allow for the highly versatile adsorptive surface functionalization of materials. The simple, well‐controlled, and stable AP layer formation from aqueous solutions on various different bulk material surfaces enables the effective and liberally tunable immobilization of strongly hydrophobic or hydrophilic bioactives. As such, a plethora of different AP‐conjugated bioactives—including cytokine‐complexing glycosaminoglycans, cell adhesion‐mediating peptides, and antimicrobials—can be applied to customize materials for demanding biotechnologies in uniquely versatile, simple, and robust ways. Mixing solutions of distinctly functionalized APs and targeted in situ alterations of the layer composition (by homo‐ and heterodisplacement) allows for an unprecedented and uniquely simple combination of various molecular functions and physicochemical features in adsorbed layers. As illustrated with first proof‐of‐concept data of cell‐instructive and antimicrobial AP coatings to cultivate sensitive human cells like HUVECs and hiPSCs and to prevent bacterial infections, the AP technology will enable combinatorial high‐throughput surface engineering approaches of previously unrealizable complexity, paving the way for further advanced biomaterials with multimodal features.
4. Experimental Section
Synthesis of Anchor Polymers
APs were synthesized utilizing polymer‐analogous modifications by: 1) high‐yield amine/anhydride reaction between styrene–maleic anhydride copolymers and amine groups of homofunctional (a‐methoxy‐ω‐amino) or heterofunctional (a‐amino‐ω‐tritylthio) PEG and AEM in organic solvent at RT over the course of 3–4 days. After purification and optional deprotection of the tritylthio‐group the multifunctional PEGylated APs with reactive maleimide and/or thiol groups were used in a 2) Michael type (click)‐reaction under physiological conditions for functionalization with thiol containing cell adhesion ligand peptides (e.g., CWGG‐RGDSP) or maleimide prefunctionalized cytokine‐complexing glycosaminoglycans (e.g., heparin‐Mal) and antimicrobials (e.g., hexetidine‐Mal). Detailed synthesis protocols and results of basic chemical characterization are provided in the Supporting Information.
Substrate Coating with Anchor Polymers
Coating of arbitrary surfaces, such as PS, PES, TPU, PE, or PA thin films on coverslips/wafers or commercial TCP‐well plates, was achieved via incubating the surfaces of interest in 1 gL−1 aqueous AP solution (PBS or Milli‐Q) for 1 h at RT. After removing the AP‐solution and excessive flushing with Milli‐Q, the surfaces had been directly applied for subsequent investigations or air‐dried and stored at 2–8 °C prior to usage. For further details see Supporting Information.
Conflict of Interest
A related concept for utilizing the AP technology for cell‐instructive coatings is protected by WO2016202329A1 owned by ZetaSCIENCE GmbH, Dresden. ZetaSCIENCE GmbH is a spin‐off enterprise of the Leibniz IPF. U.F., L.D.R., and C.W. hold shares of this company. faCellitate, a venture of Chemovator GmbH, Mannheim, Germany is going to commercialize cell‐instructive coatings for hiPSC expansion.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the Federal Ministry of Education and Research (BMBF), Germany through the Unternehmen Region‐Programm “Zwanzig20 ‐ Partnerschaft für Innovation” project RESPONSE FV11, FV12, and FV17, as well as through “Verbundprojekt Materialien für einen niedrigthrombogenen Blutkreislauf (LTBC), Teilvorhaben: Biofunktionalisierung mittels Ankerpolymeren” 13XP5075C. L.D.R. acknowledges funding by the VolkswagenStiftung. The authors thank Stefanie Hänsel, Nelly Rein, Dagmar Pette, Juliane Drichel, and Judith Horvath for their assistance in performing cell culture experiments, Sofya Rachkevych for her support for testing the antimicrobial/antiadhesive properties of anchor polymer coatings as well as Diana Cozma, Isabell Jeglinski, and Kevin Richter (all IPF Dresden) for their help of anchor polymer synthesis and characterization. Additionally, the authors thank Dr. Mirko Nitschke (IPF Dresden) for performing ToF SIMS measurements.
Open access funding enabled and organized by Projekt DEAL.
Ruland A., Schenker S., Schirmer L., Friedrichs J., Meinhardt A., Schwartz V. B., Kaiser N., Konradi R., MacDonald W., Helmecke T., Sikosana M. K. L. N., Valtin J., Hahn D., Renner L. D., Werner C., Freudenberg U., Amphiphilic Copolymers for Versatile, Facile, and In Situ Tunable Surface Biofunctionalization. Adv. Mater. 2021, 33, 2102489. 10.1002/adma.202102489
Contributor Information
Carsten Werner, Email: werner@ipfdd.de.
Uwe Freudenberg, Email: freudenberg@ipfdd.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Mas‐Moruno C., Su B., Dalby M. J., Adv. Healthcare Mater. 2019, 8, 1801103. [DOI] [PubMed] [Google Scholar]
- 2. Mertgen A. S., Trossmann V. T., Guex A. G., Maniura‐Weber K., Scheibel T., Rottmar M., ACS Appl. Mater. Interfaces 2020, 12, 21342. [DOI] [PubMed] [Google Scholar]
- 3. Ventre M., Netti P. A., ACS Appl. Mater. Interfaces 2016, 8, 14896. [DOI] [PubMed] [Google Scholar]
- 4. Lu Y., Aimetti A. A., Langer R., Gu Z., Nat. Rev. Mater. 2016, 2, 16075. [Google Scholar]
- 5. Hou X., Zhang Y. S., Santiago G. T. D., Alvarez M. M., Ribas J., Jonas S. J., Weiss P. S., Andrews A. M., Aizenberg J., Khademhosseini A., Nat. Rev. Mater. 2017, 2, 17016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang B., Korolj A., Lai B. F. L., Radisic M., Nat. Rev. Mater. 2018, 3, 257. [Google Scholar]
- 7. Nemec S., Kilian K. A., Nat. Rev. Mater. 2021, 6, 69. [Google Scholar]
- 8. Koçer G., Jonkheijm P., Adv. Healthcare Mater. 2018, 7, 1701192. [DOI] [PubMed] [Google Scholar]
- 9. Ruskowitz E. R., Deforest C. A., Nat. Rev. Mater. 2018, 3, 17087. [Google Scholar]
- 10. Maynard S. A., Winter C. W., Cunnane E. M., Stevens M. M., Regener. Eng. Transl. Med. 2020, 10.1007/s40883-020-00180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Satav T., Huskens J., Jonkheijm P., Small 2015, 11, 5184. [DOI] [PubMed] [Google Scholar]
- 12. Nasir A., Thorpe J., Burroughs L., Meurs J., Pijuan‐Galito S., Irvine D. J., Alexander M. R., Denning C., Adv. Healthcare Mater. 2020, 10, 2001448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barata D., Dias P., Wieringa P., Van Blitterswijk C., Habibovic P., Biofabrication 2017, 9, 035004. [DOI] [PubMed] [Google Scholar]
- 14. Park H. J., Yang K., Kim M. J., Jang J., Lee M., Kim D. W., Lee H., Cho S. W., Biomaterials 2015, 50, 127. [DOI] [PubMed] [Google Scholar]
- 15. Tibbitt M. W., Rodell C. B., Burdick J. A., Anseth K. S., Proc. Natl. Acad. Sci. USA 2015, 112, 14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang K., Lee J. S., Kim J., Lee Y. B., Shin H., Um S. H., Kim J. B., Park K. I., Lee H., Cho S. W., Biomaterials 2012, 33, 6952. [DOI] [PubMed] [Google Scholar]
- 17. Darnell M., Mooney D. J., Nat. Mater. 2017, 16, 1178. [DOI] [PubMed] [Google Scholar]
- 18. Krutty J. D., Schmitt S. K., Gopalan P., Murphy W. L., Curr. Opin. Biotechnol. 2016, 40, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. VandeVondele S., Voros J., Hubbell J. A., Biotechnol. Bioeng. 2003, 82, 784. [DOI] [PubMed] [Google Scholar]
- 20. Gribova V., Auzely‐Velty R., Picart C., Chem. Mater. 2012, 24, 854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neff J. A., Tresco P. A., Caldwell K. D., Biomaterials 1999, 20, 2377. [DOI] [PubMed] [Google Scholar]
- 22. Gehlen D. B., De Lencastre Novaes L. C., Long W., Ruff A. J., Jakob F., Haraszti T., Chandorkar Y., Yang L., Van Rijn P., Schwaneberg U., De Laporte L., ACS Appl. Mater. Interfaces 2019, 11, 41091. [DOI] [PubMed] [Google Scholar]
- 23. Clauder F., Möller S., Köhling S., Bellmann‐Sickert K., Rademann J., Schnabelrauch M., Beck‐Sickinger A. G., J. Tissue Eng. Regener. Med. 2020, 14, 1738. [DOI] [PubMed] [Google Scholar]
- 24. Yu L., Hou Y., Xie W., Cuellar‐Camacho J. L., Wei Q., Haag R., Adv. Mater. 2020, 32, 2006986. [DOI] [PubMed] [Google Scholar]
- 25. Reviakine I., Johannsmann D., Richter R. P., Anal. Chem. 2011, 83, 8838. [DOI] [PubMed] [Google Scholar]
- 26. Pasche S., De Paul S. M., Vörös J., Spencer N. D., Textor M., Langmuir 2003, 19, 9216. [Google Scholar]
- 27. Perry S. S., Yan X., Limpoco F. T., Lee S., Muller M., Spencer N. D., ACS Appl. Mater. Interfaces 2009, 1, 1224. [DOI] [PubMed] [Google Scholar]
- 28. van der Linden C. C., Leermakers F. A. M., Fleer G. J., Macromolecules 1996, 29, 1000. [Google Scholar]
- 29. Oesterhelt F., Rief M., Gaub H. E., New J. Phys. 1999, 1, 6. [Google Scholar]
- 30. Meiron T. S., Marmur A., Saguy I. S., J. Colloid Interface Sci. 2004, 274, 637. [DOI] [PubMed] [Google Scholar]
- 31. Garnier G., Duskova‐Smrckova M., Vyhnalkova R., van de Ven T. G. M., Revol J.‐F., Langmuir 2000, 16, 3757. [Google Scholar]
- 32. Malardier‐Jugroot C., van de Ven T., Whitehead M., J. Mol. Struct.: THEOCHEM 2004, 679, 171. [Google Scholar]
- 33. Dalsin J. L., Lin L., Tolsatti S., Vörös J., Textor M., Messersmith P. B., Langmuir 2005, 21, 640. [DOI] [PubMed] [Google Scholar]
- 34. Lazzara T. D., Whitehead M. A., van de Ven T. G. M., J. Phys. Chem. B 2008, 112, 4892. [DOI] [PubMed] [Google Scholar]
- 35. Dörr J. M., Scheidelaar S., Koorengevel M. C., Dominguez J. J., Schäfer M., van Walree C. A., Killian J. A., Eur. Biophys. J. 2016, 45, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buwalda S. J., Dijkstra P. J., Calucci L., Forte C., Feijen J., Biomacromolecules 2010, 11, 224. [DOI] [PubMed] [Google Scholar]
- 37. Sartori A., Johner A., Viovy J.‐L., Joanny J.‐F., Macromolecules 2005, 38, 3432. [Google Scholar]
- 38. Vroman L., Adams A. L., J. Biomed. Mater. Res. 1969, 3, 43. [DOI] [PubMed] [Google Scholar]
- 39. Vroman L., Adams A. L., Surf. Sci. 1969, 16, 438. [Google Scholar]
- 40. Stuart M. A. C., in Biopolymers at Interfaces, 2nd ed. (Ed: Malmsten M.), Dekker, New York: 2003, pp. 1–25. [Google Scholar]
- 41. Renner L., Pompe T., Salchert K., Werner C., Langmuir 2004, 20, 2928. [DOI] [PubMed] [Google Scholar]
- 42. Schneider H. M., Granick S., Macromolecules 1992, 25, 5054. [Google Scholar]
- 43. Renner L. D., Weibel D. B., MRS Bull. 2011, 36, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gristina A. G., Science 1987, 237, 1588. [DOI] [PubMed] [Google Scholar]
- 45. Busscher H. J., Van Der Mei H. C., Subbiahdoss G., Jutte P. C., Van Den Dungen J. J. A. M., Zaat S. A. J., Schultz M. J., Grainger D. W., Sci. Transl. Med. 2012, 4, 153rv10. [DOI] [PubMed] [Google Scholar]
- 46. Sjollema J., Zaat S. A. J., Fontaine V., Ramstedt M., Luginbuehl R., Thevissen K., Li J., van der Mei H. C., Busscher H. J., Acta Biomater. 2018, 70, 12. [DOI] [PubMed] [Google Scholar]
- 47. Russotto V., Cortegiani A., Raineri S. M., Giarratano A., J. Intensive Care 2015, 3, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Otter J. A., Yezli S., French G. L., Infect. Control Hosp. Epidemiol. 2011, 32, 687. [DOI] [PubMed] [Google Scholar]
- 49. Cloutier M., Mantovani D., Rosei F., Trends Biotechnol. 2015, 33, 637. [DOI] [PubMed] [Google Scholar]
- 50. Freeman F. E., Browe D. C., Nulty J., Von Euw S., Grayson W. L., Kelly D. J., Eur. Cells Mater. 2019, 38, 168. [DOI] [PubMed] [Google Scholar]
- 51. Huettner N., Dargaville T. R., Forget A., Trends Biotechnol. 2018, 36, 372. [DOI] [PubMed] [Google Scholar]
- 52. Capila I., Linhardt R. J., Angew. Chem., Int. Ed. 2002, 41, 390. [DOI] [PubMed] [Google Scholar]
- 53. Hoyle C. E., Bowman C. N., Angew. Chem., Int. Ed. 2010, 49, 1540. [DOI] [PubMed] [Google Scholar]
- 54. Chwalek K., Tsurkan M. V., Freudenberg U., Werner C., Sci. Rep. 2014, 4, 4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schuler M., Owen G. Rh., Hamilton D. W., de Wild M., Textor M., Brunette D. M., Tosatti S. G. P., Biomaterials 2006, 27, 4003. [DOI] [PubMed] [Google Scholar]
- 56. Hudalla G. A., Murphy W. L., Soft Matter 2011, 7, 9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hudalla G. A., Murphy W. L., Langmuir 2009, 25, 5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guler M. O., Hsu L., Soukasene S., Harrington D. A., Hulvat J. F., Stupp S. I., Biomacromolecules 2006, 7, 1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spatz J. P., Mössmer S., Hartmann C., Möller M., Herzog T., Krieger M., Boyen H. G., Ziemann P., Kabius B., Langmuir 2000, 16, 407. [Google Scholar]
- 60. Schaufler V., Czichos‐Medda H., Hirschfeld‐Warnecken V., Neubauer S., Rechenmacher F., Medda R., Kessler H., Geiger B., Spatz J. P., Cavalcanti‐Adam E. A., Cell Adhes. Migr. 2016, 10, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Talbot N. C., Caperna T. J., Cytotechnology 2015, 67, 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kleinman H. K., Martin G. R., Semin. Cancer Biol. 2005, 15, 378. [DOI] [PubMed] [Google Scholar]
- 63. Aisenbrey E. A., Murphy W. L., Nat. Rev. Mater. 2020, 5, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wakao H., Kitada M., Kuroda Y., Ogura F., Murakami T., Niwa A., Dezawa M., PLoS One 2012, 7, e48677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.