

Abstract
Cell–cell interactions regulate intracellular signaling via reciprocal contacts of cell membranes in tissue regeneration and cancer growth, indicating a critical need of membrane‐derived tools in studying these processes. Hence, cell‐membrane‐derived nanoparticles (CMNPs) are produced using tonsil‐derived mesenchymal stem cells (TMSCs) from children owing to their short doubling time. As target cell types, laryngeal cancer cells are compared to bone‐marrow‐derived MSCs (BMSCs) because of their cartilage damaging and chondrogenic characteristics, respectively. Treating spheroids of these cell types with CMNPs exacerbates interspheroid hypoxia with robust maintenance of the cell–cell interaction signature for 7 days. Both cell types prefer a hypoxic environment, as opposed to blood vessel formation that is absent in cartilage but is required for cancer growth. Hence, angiogenesis is inhibited by displaying the Notch‐1 aptamer on CMNPs. Consequently, laryngeal cancer growth is suppressed efficiently in contrast to improved chondroprotection observed in a series of cell and animal experiments using a xenograft mouse model of laryngeal cancer. Altogether, CMNPs execute a two‐edged sword function of inducing hypoxic cell–cell packing, followed by suppressing angiogenesis to promote laryngeal cancer death and chondrogenesis simultaneously. This study presents a previously unexplored therapeutic strategy for anti‐cancer and chondroprotective treatment using CMNPs.
Keywords: cell membranes, cell–cell packing, hypoxia, nanoparticles, Notch‐1 aptamers
Cell‐derived nanoparticles (CMNPs) promote physical tightening of cell–cell interactions and cell packing, thereby inducing hypoxia as a favorable setting for cancer progression and cartilage development. The conjugation of aptamers to CMNPs suppresses Notch‐1 signaling‐mediated angiogenesis with consequent activation of anti‐cancer and chondroprotective effects (“two‐edged sword”) in a xenograft model of human laryngeal cancer.
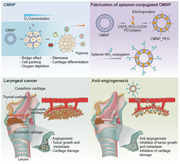
1. Introduction
Cells interact with one another to communicate through the mutual transfer of signaling molecules, thereby establishing their mass characteristics. In addition, cell–cell interactions are implicated in tissue regeneration[ 1 , 2 ] and tumorigenesis[ 2 , 3 ] from development to maturation. Because of the reciprocal interaction of cells with the neighboring cell membranes during tissue regeneration and cancer growth, we employed a cell‐membrane‐derived tool to study these interactions. This strategy involves the use of membrane‐derived mediators such as cell‐membrane‐derived nanoparticles (CMNPs) to promote cell–cell interactions by bridging membrane–membrane contacts. As an unprecedented solution, CMNPs have the following advantages: nanoscale size allowing the tight bridging of two membranes to promote cell–cell packing; membrane characteristics to express cell–cell adhesion molecules (e.g., cadherin) for executing the bridging function at a high resolution; a membrane that could be modified to display a functional molecule, allowing additional functions to be programmed; and a cell source that enables efficient population expansion and maintenance of healthy membrane properties, allowing a sufficient number of CMNPs to be obtained. In this study, tonsil‐derived mesenchymal stem cells (TMSCs) obtained from children were used to produce CMNPs because of their relatively short doubling time.
When cells are packed in a limited space by tightening membrane–membrane contacts, the competition for oxygen consumption among the growing cell population increases despite no change in the oxygen supply, causing oxygen depletion or hypoxia. Hypoxia is commonly accompanied by cartilage development[ 4 ] and cancer progression.[ 5 , 6 ] Culturing of chondrogenic MSCs in a hypoxic mass (e.g., organoid) or under low‐oxygen conditions promotes their differentiation.[ 7 ] Similarly, aggressive cancer cell growth in the form of a mass requires abundant oxygen consumption.[ 8 ] The hypoxic conditions, in turn, drive phenotypic changes in cancer cells and induce stemness[ 9 ] with epigenetic alterations[ 10 ] and increased drug resistance[ 5 , 11 ] these processes contribute to cancer propagation. The present study is based on the concept that a hypoxic environment can set up a trap to activate these causative factors, and consequently induce chondroprotection and suppress cancer propagation by programing the anti‐angiogenic function of CMNPs.
To use the CMNP‐based strategy, we selected laryngeal cancer as the experimental model because it progressively damages the laryngeal cartilage and requires chondroprotection to inhibit cancer growth. Two‐thirds of patients with laryngeal cancer undergo this damage during the early phases, leading to the loss of vocal cords and swallowing ability in the advanced phases of cancer progression. Another function of CMNPs, i.e., suppression of Notch signaling, can prevent this by simultaneously promoting anti‐cancer and chondroprotective functions in a hypoxic environment. The common underlying mechanism of this process is that adult cartilage does not have vasculature; thus, a potent anti‐cancer therapy can inhibit new vascular growth around tumor masses.
To program the two‐edged sword function of CMNPs, the Notch‐1 antagonistic aptamer (apt) was displayed on CMNPs. Notch signaling induces angiogenesis with activation of endothelial cells (ECs) during inflammatory arthritis progression. Inhibition of this signaling leads to chondroprotective effects,[ 12 , 13 ] as supported by the need for an avascular environment for healthy maturation and functional maintenance of articular cartilage.[ 14 ] The apt can inhibit tumor growth and invasion by suppressing Notch‐1 signaling‐mediated tumor angiogenesis by targeting a specific gene or receptor without exerting any side effects.[ 15 ] Therefore, it is hypothesized that displaying the apt on CMNPs can exert dual anti‐cancer and pro‐chondrogenic effects. In this way, a favorable condition (i.e., hypoxia) for both cancer and cartilage cells is established first by promoting cell–cell interaction. Their opposite fates are subsequently induced by inhibiting vessel formation toward an ideal therapeutic scenario of laryngeal cancer to suppress the cancer stemness emerging from the hypoxia trap.
2. Results and Discussion
First, TMSCs were isolated from the palatine tonsils of young human donors (≈10 years old) through tonsillectomy surgeries. Flow cytometry analysis showed that these TMSCs derived from young human donors expressed a series of MSC‐positive markers, including CD73, CD90, and CD105, with nearly no expression of MSC‐negative markers (CD45, CD34, CD11b, CD19, and HLA‐DR) (Figure S1, Supporting Information). These results demonstrate the similar characteristics of TMSCs to those of bone‐marrow‐derived MSCs (BMSCs) and adipose‐derived MSCs (ADSCs). Compared to BMSCs and ADSCs, TMSCs maintained a relatively short doubling time from passages 4 to 9, justifying their selection for producing CMNPs (Figure S2, Supporting Information). CMNPs were then produced from TMSCs through filtering processes in considerably higher yields than exosomes as reported in a previous study,[ 16 ] thereby enabling more cost‐effective production. Filtering the TMSCs via a series of filters with decremented pore sizes resulted in the repetitive destruction and self‐assembly of CMNPs (Figure 1a).[ 17 ] The user‐specified control of filter size enables efficient quality control during production. In addition, filtering of CMNPs resulted in the removal of an MSC marker (CD90) and expression of exosome markers (CD9, CD63, and CD81) as determined via western blotting, indicating the acquisition of an exosome‐like property by CMNPs (Figure 1b). The cell‐membrane‐like characteristics of CMNPs are responsible for membrane contact functions,[ 18 ] thereby improving cell–cell interactions. The expression of major cell–cell interaction mediators (i.e., E‐cadherin and N‐cadherin) was preserved during the multistep production of CMNPs from TMSCs as revealed using western blot (Figure 1c) and transmission electron microscopy (TEM) imaging (Figure S3, Supporting Information) although the series of membrane reassembly processes reduced the expression of N‐cadherin (≈75 kDa).
Figure 1.
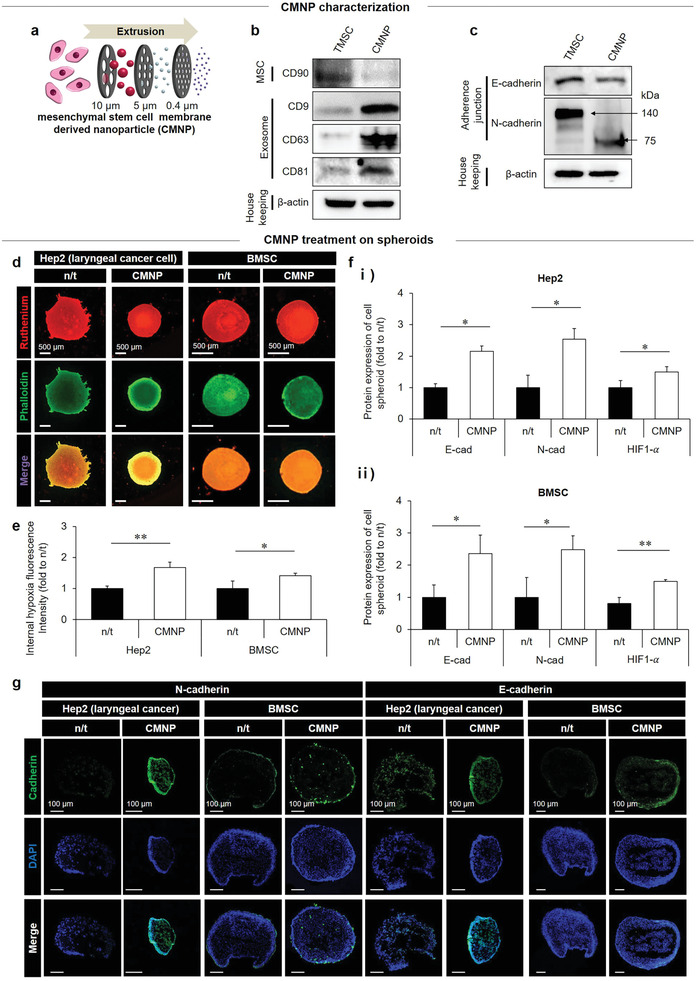
CMNP as a potent promoter of hypoxic cell–cell packing. a) CMNPs were produced by filtering TMSCs using a series of filters with decremental pore sizes. b,c) The expression of marker proteins for MSCs (CD90) and exosomes (CD9, CD63, and CD81) (b) as well as cell–cell adhesion (E‐cadherin and N‐cadherin) in CMNPs (c) was compared to that of TMSCs, indicating the preservation of membrane characteristics in CMNPs post‐production. On day 7 post‐CMNP treatment, d) consequent generation of hypoxia in the spheroids of laryngeal cancer cells (Hep 2) and BMSCs was determined by analyzing incremental ruthenium signals (red) upon hypoxia severance in cell spheroids (green) with e) quantitative analysis of fluorescence intensities. f,g) This result was supported by the expression of HIF1‐α, E‐cadherin, and N‐cadherin according to quantitative analyses of western blots (f) and immunocytochemical images (green: cadherin; blue: cell nucleus) (g). Data are expressed as mean ± standard deviation (SD). *p < 0.05, **p < 0.01, and ***p < 0.001 between lined groups.
Compared with the no‐treatment control (n/t) group, the preservation of CMNP membrane characteristics promoted hypoxia inside spheroids of laryngeal cancer cells (Hep2) and MSCs following treatment with CMNPs (20 µg mL−1) (Figure 1d,e). The generation of inner spheroid hypoxia was determined by ruthenium treatment, in which the red fluorescence intensity increased as the degree of hypoxia was elevated by quenching oxygen.[ 19 ] As a key inducer of hypoxia, CMNP‐mediated cell–cell packing was observed when CMNPs were treated at low confluency (1 × 104 cells per well) in a 2D culture of laryngeal cancer Hep2 cells and BMSCs (Figure S4, Supporting Information). Compared with the n/t group, cell proliferation increased from day 1 to day 3 with more cell–cell interactions and overlapping morphologies on day 3. In addition, cell migration was promoted by reducing the wound area significantly for 3 days post‐CMNP treatment. The quantitative analyses of western blots (Figure 1f; Figure S5, Supporting Information) and immunohistochemistry images (Figure 1g) indicated that expression of HIF1‐α, N‐cadherin, and E‐cadherin types in both Hep2 and BMSC spheroids increased in the CMNP treatment group compared to the n/t group. Moreover, direct evidence of CMNP‐mediated bridging function was shown by CMNP localization in‐between cell membranes of BMSC pellets using TEM and confocal imaging on day 7 and day 1 post‐CMNP treatment, respectively (Figure S6a,b, Supporting Information). In addition, these imaging results were supported by the consequent expression of tight (Zonula occludens‐1; ZO‐1) and adherent (Adherens Junctions Associated Protein 1; AJAP‐1) junction markers in Hep2 and BMSC pellets on day 7 post‐CMNP treatment (Figure S6c, Supporting Information). Last, universal testing machine (UTM) analysis revealed a greater degree of hardness of the BMSC pellet on day 7 post‐CMNP treatment compared with the n/t group, indicating a crucial role of CMNP in packing the pellet (Figure S7, Supporting Information).
Since articular cartilage is exposed to hypoxic conditions, chondrocytes are adapted to a low‐oxygen environment, thereby maintaining their phenotype.[ 20 ] The hypoxia‐induced pro‐chondrogenic effect was determined by injecting CMNPs into the knee joint of rats after a 6‐week surgical induction of osteoarthritis (OA). Cartilage regeneration was observed for the next 6 weeks during the 12‐week experiment (Figure 2a). The CMNP treatment (20 µg mL−1) reinforced cartilage to the level obtained with kartogenin (KGN) treatment, as evidenced by the end‐point magnetic resonance imaging (MRI) analysis (Figure 2b). KGN serves as a control inducer of pro‐chondrogenesis by regulating the CBFβ‐RUNX1 transcriptional program, as reported previously.[ 21 ] Compared with the vehicle control (PBS), the CMNP group exhibited KGN‐like, clearer cartilage regeneration with a higher pixel number in the region of interest (ROI) from the femur to the tibia. The MRI results were supported by Safranin‐O staining (Figure 2c) and immunohistochemistry of aggrecan and type II collagen (Figure 2d). These results indicate that CMNP treatment‐induced hypoxia can create a favorable environment for cartilage maturation and homeostasis. However, the normal group was significantly superior to the other groups, and hypoxia can induce angiogenesis and thereby trigger arthritis progression.[ 22 ] Therefore, angiogenesis should be suppressed by inhibiting Notch‐1 signaling by displaying the apt on CMNPs.
Figure 2.
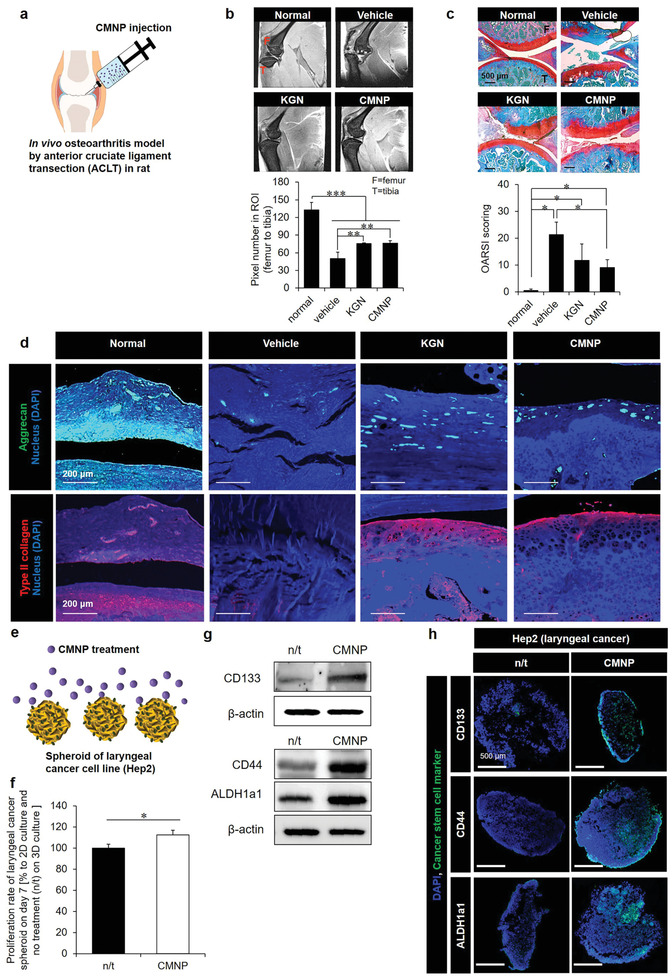
Chondroprotection and cancer survival due to the prohypoxic effect of CMNPs as a promoter of cell–cell interaction. a) After 6 weeks of induction of ACLT osteoarthritis in rats, CMNP treatment promoted cartilage reinforcement to the level obtained with KGN treatment for the next 6 weeks (of the 12 weeks total experimental period). b) This was evidenced by (top) the end‐point MRI images of sagittal knee joints and (bottom) quantitative analysis of pixel numbers in the ROI from the femur to the tibia using ImageJ (F = femur, T = tibia). c) Safranin‐O staining confirmed the 12‐week results: (top) the histology images (red = cartilage, blue = bone, white = synovium) and (bottom) OARSI scores from the analysis of medial tibial plateaus (F = femur, T = tibia), as supported by d) the immunohistochemistry images of aggrecan (green) and type II collagen (red). e) Induction of inner spheroid hypoxia by 7‐day treatment of CMNPs (20 µg mL−1) promoted f) laryngeal cancer cell (Hep2) proliferation with increased protein expression of cancer stem cell markers (CD133, CD44, and ALDH1a1) according to g) western blotting and h) immunocytochemistry analyses. The proliferation rate of Hep2 cells in 3D hypoxic culture was normalized to that of 2D nonhypoxic culture to exclude the effects of unidentified constituents inside CMNPs, thereby capturing only the effect of hypoxia. Data are expressed as mean ± standard deviation (SD). *p < 0.05, **p < 0.01, and ***p < 0.001 between the lined groups.
Because CMNP‐induced hypoxia can promote cancer survival, the promotive effects of hypoxia on cancer survival were examined by administering CMNPs (20 µg mL−1) on normoxic 2D tissue culture polystyrene (TCPS) versus a hypoxic 3D spheroid culture of Hep2 cells (Figure 2e). Compared to that in the 2D TCPS culture, hypoxia increases when cells are cultured in a 3D spheroid because cell aggregation increases the competition for oxygen consumption in each spheroid space, limiting oxygen transport to the inner core of the spheroid. Proaggregation and consequent prohypoxia effects were observed when Hep2 spheroids were treated with CMNPs (Figure 1). In addition, unidentified constituents inside CMNPs can affect hypoxia‐mediated actions. Hence, normalizing the cell survival rate in a 3D hypoxic culture to that of 2D nonhypoxic culture with and without CMNP treatment can exclude other effects to demonstrate only the effects of hypoxia on Hep2 cell survival (Figure 2f). As another evidence of increased Hep 2 cell survival following CMNP treatment, a higher level of Hep2 cell viability (compared with that in the n/t group) was maintained even after 7 days of treatment with CMNPs (Figure S8, Supporting Information). In addition, western blot (Figure 2g) and immunohistochemical (Figure 2h) analyses of the expression of cancer stem cell markers (CD133, CD44, and ALDH1a1) revealed that enhancing hypoxia‐mediated cancer survival by CMNP treatment induced the emergence of cancer stem cells. These results represent a previously unconsidered but significant aspect of this study: the induction of massive cell death can suppress the main causative factor of cancer recurrence by programing an anti‐angiogenic function, such as the inhibition of Notch‐1 signaling.
Notch‐1 signaling drives proangiogenesis to promote tumor growth and invasion,[ 15 ] the latter of which is indicative of apt‐mediated effect on laryngeal cancer cells. Treatment of human melanoma cells (A375P) with 10 × 10−9 m apt to suppress this signaling reduced the cell survival, as evidenced by the significantly decreased cell proliferation (Figure 3a) and the higher TUNEL+ cell number (Figure 3b) than that in the n/t group. In addition, quantitative analysis (Figure 3c) revealed significantly reduced tube formation of human umbilical vein endothelial cells (HUVECs) during culture on Matrigel for 3 days. In addition, apt treatment reduced Notch‐1 expression in melanoma cells (A375P), laryngeal cancer cells (Hep2), HUVECs, and BMSCs, as examined by immunohistochemistry (Figure S9, Supporting Information). On the other hand, the overactivation of Notch‐1 signaling reduced cartilage regeneration,[ 23 ] and sustained Notch‐1 signaling in the developmental stages suppressed chondrogenesis.[ 24 ] Therefore, co‐treatment with apt (10 × 10−9 m) and chondrogenic induction media exerted synergistic pro‐chondrogenic effects on BMSC pellets in a 3D hypoxic environment. These findings are supported by Safranin‐O staining images acquired on day 21 (Figure 3d) and the gene expression of chondrogenic markers (aggrecan, SOX9, and Col12a1) determined on day 7 and day 10 (Figure 3e).
Figure 3.
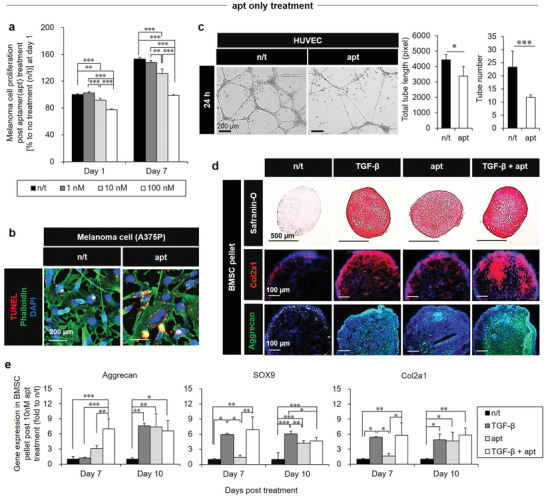
The anti‐cancer, anti‐angiogenic, and pro‐chondrogenic effects of apt. a) Suppression of human melanoma cell (A375P) proliferation on day 1 and day 7 after apt treatment (0 × 10−9, 1 × 10−9, and 10 × 10−9 m), was supported by b) the TUNEL+ (red: apoptosis) cell number, as shown in the representative images on day 3 of A375P culture after treatment with 10 × 10−9 m apt. c) An anti‐angiogenic effect of apt treatment (10× 10−9 m) was evidenced by (left) the disturbed tube formation in a 24 h culture of HUVECs on Matrigel by optical imaging with (right) quantitative analyses of total tube length and number, suggesting the two‐edged sword function using hypoxia as a trap. d,e) Following treatment with chondrogenic induction media, the pro‐chondrogenic effects of apt (treatment with 10 × 10−9 m with media change every 3 days) without or with TGF‐β (positive control) on BMSC pellets were determined by Safranin‐O (red = cartilage), Col2a1 (red), and aggrecan (green) staining on day 21 (d), and by gene expression of chondrogenesis markers (aggrecan, SOX9, and Col2a1) on day 7 and day 10 (e). Data are expressed as mean ± standard deviation (SD). *p < 0.05, **p < 0.01, and ***p < 0.001 between the lined groups.
Because the anti‐angiogenic effects could not be examined in the in vitro models of chondrogenesis and cancer survival (Figure 3), a combined in vivo model (i.e., laryngeal cancer grafting), as discussed later (Figure 6), is required. The pellet form of BMSCs was used to improve chondrogenic differentiation through forced aggregation (Figure 3d,e).[ 25 ] Although transforming growth factor (TGF‐β) regulates several mechanisms, we used it as a positive control for chondrogenic inducers.[ 26 ] Overall, the pro‐chondrogenic effects of apt treatment were evident when compared with the n/t group; however, chondrogenesis of BMSC pellets with apt treatment occurred later compared to that with TGF‐β treatment with or without apt, as shown in the comparison between day 7 and day 10 (Figure 3e). The final production of chondrogenic extracellular matrix (ECM) components by apt treatment on day 10 was not superior to that of other treatment groups. Moreover, apt treatment reduced cancer cell proliferation by less than 50% compared to the no treatment group (Figure 3a). This effect was evident only after 7 days, indicating the need to combine apt with CMNPs and apply the hypoxic effect to establish a favorable environment for activating a phenotype change (e.g., CD 133+ cancer stem cells) as a trap. This combination of apt functions with anti‐angiogenic action can further promote cartilage formation while minimizing cancer recurrence with massive cancer death.
Figure 6.
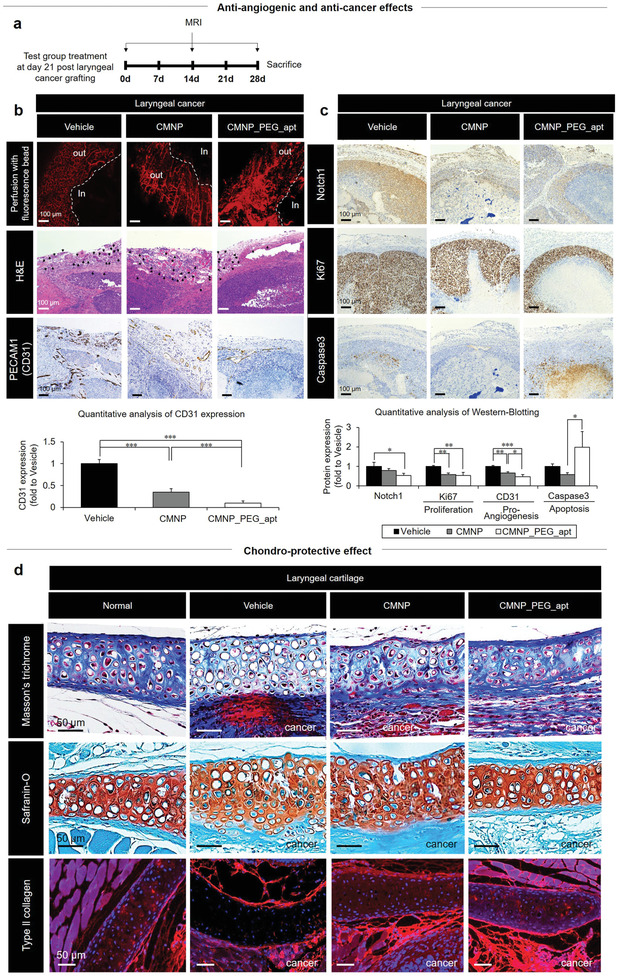
CMNP_PEG_apt exerts anti‐angiogenic effects with the generation of internal hypoxia as a part of the two‐edged sword to promote anti‐cancer growth and chondroprotection post‐orthotopic xenografting of Hep2 cells in nude mice. a) Nude mice were injected with PBS (vehicle) on day 21 post‐orthotopic xenografting of Hep2 cells. Tumor tissues from each group were harvested on day 28 post‐sacrifice. b) To demonstrate the anti‐angiogenic effect of CMNP_PEG_apt, the tissues were examined using (top) fluorescence bead perfusion, H&E staining (black arrow: blood vessel), and (bottom) quantitative analysis of CD31 immunochemistry. c) Protein expression of Notch‐1, proliferation (Ki67), and apoptosis (caspase‐3) markers was determined using (top) immunohistochemistry and (bottom) quantitative analysis of western blots. Data are expressed as mean ± standard deviation (SD; n = 10). *p < 0.05, **p < 0.01, and ***p < 0.001 between the lined groups. d) Larynx tissues of each group were harvested on day 28 post sacrifice to examine the pro‐chondrogenic effect of CMNP_PEG_apt using Masson's trichrome staining, Safranin‐O staining, and Col2a1 (red) immunohistochemistry.
Therefore, CMNP_PEG_apt was produced via PEGylation (PEG) to prevent blood protein adsorption, thereby prolonging the circulation time. Efficient PEGylation post conjugation of DSPE‐PEG‐COOH was evident from the C—H and C=O stretching peaks in Fourier‐transform infrared (FTIR) spectroscopy (Figure S10a, Supporting Information). The electroporation method was selected for PEGylation after comparison with sonication and heating reaction (60 °C) between CMNP and DSPE‐PEG‐COOH because successful outward display of PEG was evidenced by the increased anti‐adsorption property (Figure S10b, Supporting Information). Dynamic light scattering (DLS) analysis (Figure 4a) and TEM images (Figure 4b) validated the conjugation reactions by the decrease in size from CMNPs and CMNP_PEG to CMNP_PEG_apt (average ≈100 × 10−9 m in diameter). Moreover, these results were supported by the average apt loading efficiency (LE) of 7.4% (11.75 × 10−9 m) with the loading content (LC) peak in FTIR (Figure S10c,d, Supporting Information). The input amount of apt (160 × 10−9 m) was set to display 11.75 × 10−9 m of apt based on 7.4% of LE in alignment with the results from screening the apt‐only concentration (10 × 10−9 m; Figure 3a). The reduction in size observed between DLS and TEM results was possibly driven by the neutralization of the negative apt charge through interactions with membrane proteins, in addition to the consequent dense aggregation of CMNP_PEG_apt to separate them from water. Long‐term stability was considered an important criterion to determine the performance of CMNP_PEG_apt. After 6 months of storage at 4 °C, CMNP_PEG_apt showed an intact size distribution around 100 nm in diameter, undisturbed particle morphologies, and unaltered chemical compositions of DSPE‐PEG‐COOH and NH2‐apt as indications of PEGylation and peptide bond formation (Figure S11, Supporting Information). These properties were matched with those of fresh samples immediately after production (Figure 4a,b; Figure S10, Supporting Information).
Figure 4.
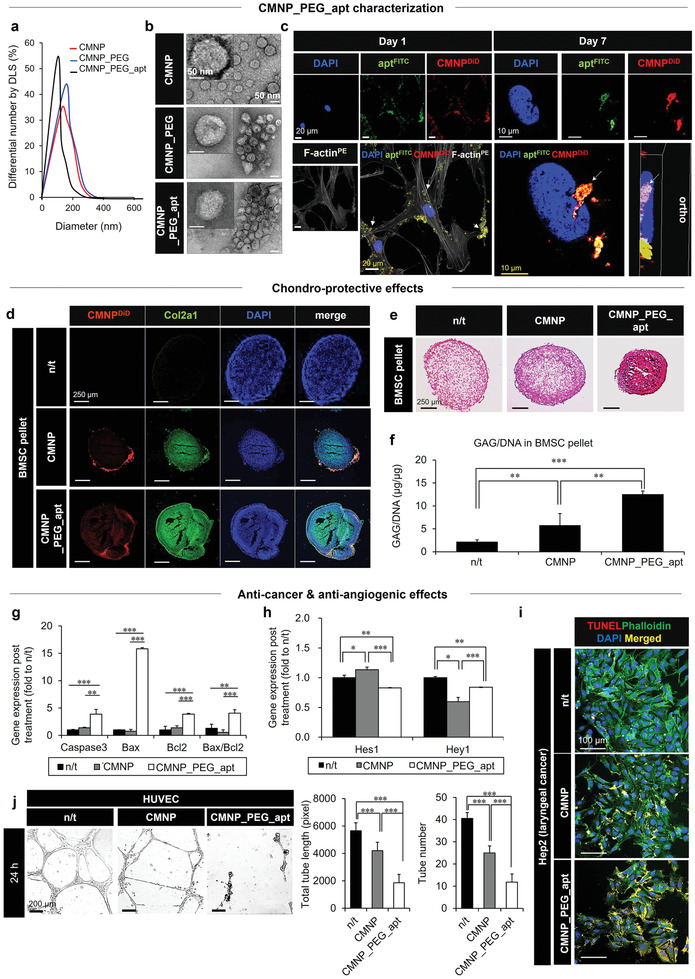
Combination of prohypoxic CMNP (trap) with anti‐angiogenic apt (two‐edged sword) as a form of CMNP_PEG_apt to promote anti‐cancer and chondroprotective effects. a) The size (DLS) and b) morphology (TEM) among the test groups were determined. c) CMNP_PEG_apt dominantly adhered to the cell membrane (white arrows) on day 1 to promote cell–cell interactions, and subsequently to the nuclear membrane on day 7 by penetrating the cell membrane, as visualized by confocal microscopy (blue, nuclei; green, apt; red, CMNP; gray, PE‐loaded F‐actin). d–f) After treatment with chondrogenic induction media, the chondroprotective effect of each CMNP test group (20 µg mL−1 with media change every 3 days) on BMSC pellets was examined on day 21. d–f) The results were determined through immunocytochemistry analysis of Col2a1 (green) production in BMSCs (blue nuclei) following CMNP infiltration (red: DiD loading) (d), Safranin‐O staining of cartilage production (red) (e), and GAG production (µg) by ELISA, normalized to the corresponding BMSC DNA content (µg) (f). g–i) The anti‐cancer effect of CMNP_PEG_apt on Hep2 cells was determined on day 3 post‐treatment by observing gene expression of apoptosis (caspase‐3, bax, and bcl2) (g), Notch‐1 downstream (Hey1 and Hes1) markers (h), and TUNEL+ Hep2 cells (yellow apoptotic cells, green cytoskeleton, and blue nuclei in merge) (i). j) As evidence of the two‐edged sword function, an anti‐angiogenic effect of each test group was shown by (left) disturbed tube formation in a 24 h culture of HUVECs on Matrigel by optical imaging with (right) quantitative analyses of total tube length and number. Data are expressed as mean ± standard deviation (SD). *p < 0.05, **p < 0.01, and ***p < 0.001 between the lined groups.
The initial bridging status of CMNP_PEG_apt among cell membranes was repeatedly visualized by confocal microscopy on day 1 post‐treatment (Figure 4c; Figure S12, Supporting Information). In contrast, the progressive changes in the localization of CMNP_PEG_apt were visualized from the cell membrane into the cytosol on day 3 and subsequently onto the nuclear membrane on day 7 through cell membrane penetration, indicating a critical advantage for efficiently inhibiting Notch‐1 downstream signaling over time. The combination of CMNPs with apt markedly improved the chondrogenesis of BMSC pellets when CMNP_PEG_apt (20 µg mL−1) treatment was administered for 21 days with replacement of the chondrogenic media every 3 days (Figure 4d–f). The results were confirmed through increased Col2a1 production, more efficient adhesion of CMNP_PEG_apt to the BMSC pellet (Figure 4d), and higher cartilage (Figure 4e) and GAG production (Figure 4f) than those in the CMNP and n/t groups. The progressive changes in the diameters of BMSC pellets between the CMNP treatment and n/t groups were not significantly different during the 3‐day culture (Figure S13, Supporting Information). These results indicate that the cell–cell packing effect of CMNPs was accompanied by volumetric maintenance of pellets to the level of n/t group, possibly due to increased cell proliferation (Figure 2f; Figure S4a, Supporting Information). The pellets of both groups stabilized with internal integration for the first 12 h, followed by maintenance of pellet sizes until 72 h post‐culture.
On the other hand, the anti‐cancer effects of CMNP_PEG_ apt were effective on Hep2 cells on day 3 post‐treatment (20 µg mL−1) (Figure 4g–i). Compared to the similar levels between the CMNP and n/t groups, the apoptosis marker genes (caspase‐3, bax, and bcl2) were more markedly expressed in the CMNP_PEG_apt group (Figure 4g). The downstream marker genes of Notch‐1 (Hey1 and Hes1) were significantly less expressed (Figure 4h), with an increase in the number of dead cancer (TUNEL+ Hep2) cells (Figure 4i) in the CMNP_PEG_apt group. As a critical supporting factor of these anti‐cancer activities, HUVEC tubulogenesis was markedly suppressed by treatment with CMNP_PEG_apt during the 3‐day culture on Matrigel than those in the CMNP and n/t groups (Figure 4j). This result was supported by the significant reduction in protein secretion levels of vascular endothelial growth factor (VEGF) and endothelial nitric oxide synthase (eNOS) in media supernatants following CMNP_PEG_apt treatment as compared with that in the n/t group (Figure S14, Supporting Information). The addition of apt to CMNPs significantly reinforced the anti‐angiogenic activities. These results indicate the promising roles of the combined design in exerting dual pro‐chondrogenic and anti‐cancer effects based on the anti‐angiogenic activity as a common regulator, thereby justifying the follow‐up animal experiments.
The in vivo activities of CMNP_PEG_apt were examined after subcutaneous xenografting of a human melanoma cell line (A375P) into nude mice (Figure 5 ). First, to monitor cancer targeting, the test CMNP groups were labeled with a fluorescent dye and injected with PBS (vehicle) into mice on day 10 post‐cancer cell grafting (Figure 5a). Intraperitoneal (IP) injection was selected because i) it has been used to treat cancer patients in clinics; ii) IP spreading of drugs in connection with blood circulation can treat not only target cancer cells but also metastatic cancer cells in unknown parts of the body; and iii) IP injection induces longer lasting drug effects than intravascular (IV) injection.[ 27 ] IVIS imaging (Figure 5b) and quantitative analysis (Figure 5c) of fluorescence intensities at the tumor‐grafted sites showed the superior 10‐day targeting of CMNP_PEG_apt over the other test groups, with the maximum signal on day 1, followed by a decremental pattern over time. In addition, these results were supported by the visualization and analysis of fluorescence signals at tumor sites post‐sacrifice (Figure 5d,e). Overall, the vehicle group exhibited a significantly lower targeting efficiency than the other test groups after 10 days. PEGylation slightly reduced the targeting efficiency as compared with the CMNP group, probably because the prolonged circulation time by PEGylation led to a short stay in the tumor site, resulting in nonspecific targeting over the entire body. These results indicate a key role of apt conjugation in improving cancer targeting, thereby maximizing therapeutic efficacy.
Figure 5.
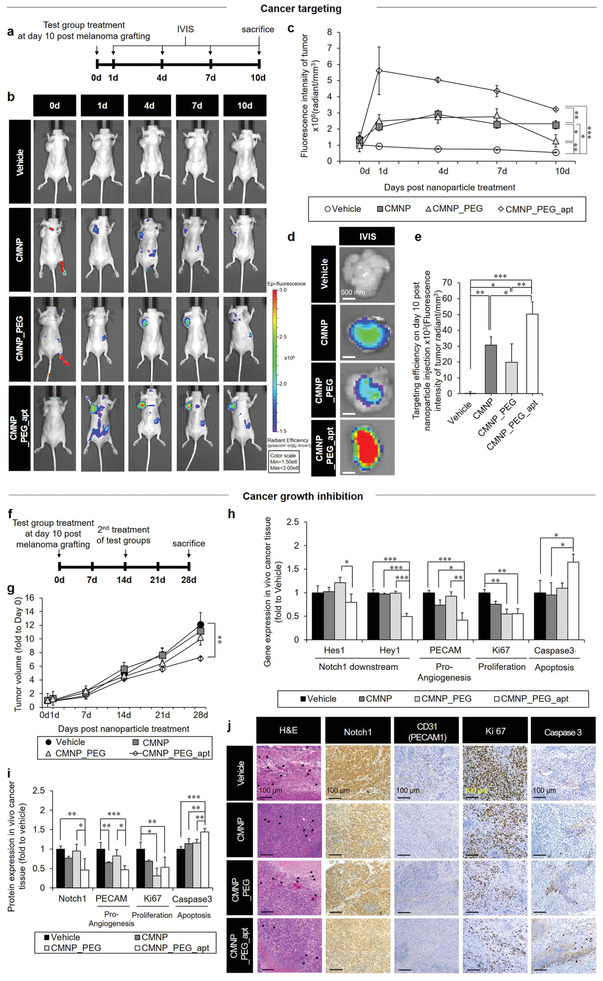
In vivo cancer targeting and growth inhibition by CMNP_PEG_apt post‐subcutaneous xenografting of the human melanoma cell line (A375P) in nude mice. a) To monitor cancer targeting, test groups were labeled with DiD fluorescence dye and injected into mice with PBS (vehicle) on day 10 post‐cancer cell grafting. b–f) The cancer‐targeting efficiency of each test group was visualized by IVIS imaging on day 1, day 4, day 7, and day 10 (sacrifice) post‐injection (b) and determined by analyzing the corresponding fluorescence intensity normalized to the tumor volume (c). Next, tumor tissues were harvested and subjected to IVIS imaging (left) (d) and quantitative image analysis (e). f) To test cancer growth inhibition, test groups were injected into mice with PBS (vehicle) on day 10 post‐cancer cell grafting, followed by g) analysis of tumor volume on day 1, day 7, day 14, day 21, and day 28 with normalization to the 0‐day volume of each group. h) Gene expression of Notch‐1 downstream (Hey1 and Hes1), apoptosis (caspase‐3), proliferation (Ki67), and proangiogenesis (PECAM) markers on day 28. Tumor samples were quantitatively analyzed using real‐time qPCR. The protein expression of Notch‐1, proliferation (Ki67), angiogenesis (CD31), and apoptosis (caspase‐3) markers was measured on day 28. i,j) Tumor tissues were examined through quantitative analyses of western blots (i) and H&E‐staining images (black arrow: blood vessel) and immunohistochemistry (j). Data are expressed as mean ± standard deviation (SD). *p < 0.05, **p < 0.01, and ***p < 0.001 between the lined groups.
Next, we used the hypoxia‐mediated acquisition of CD133‐positive cancer stem cells as a trap and subsequently suppressed the growth of this cell population by anti‐angiogenic and anti‐cancer functions of apt. After preparation in PBS (vehicle), the test groups (vehicle, CMNP, CMNP_PEG, and CMNP_PEG_apt) were injected into mice on day 10 post melanoma cell (A375P) xenografting, followed by second treatment of test groups and sacrifice on day 14 and day 28 post xenografting, respectively (Figure 5f). Immunohistochemical analyses of the harvested tissues revealed that the CD133+ cell number was significantly decreased in the CMNP_PEG_apt group than in the CMNP group without apt, which promoted only internal hypoxia (Figure S15, Supporting Information). The anti‐cancer effect of CMNP_PEG_apt was evidenced by the progressive inhibition of cancer growth because, among the test groups, only CMNP_PEG_apt treatment significantly reduced the tumor volume compared with the vehicle group (Figure 5g; Figure S16, Supporting Information).
The above result was further supported by the significant suppression of Notch‐1 downstream (Hey1 and Hes1), proliferation (Ki67), and proangiogenesis (PECAM) gene expression with the induction of apoptosis (caspase‐3) as compared with the vehicle group (Figure 5h). The protein expression data aligned with these results, as shown via western blotting (Figure 5i; Figure S17, Supporting Information), hematoxylin and eosin (H&E) staining, and immunohistochemistry (Figure 5j). In particular, CMNP_PEG_apt treatment sufficiently reduced the expression of the proangiogenic marker CD31, indicating the potential working efficiency of the two‐edged sword function. Aligning the results between gene and protein expression (Figure 5h,i), the expression of Ki67 in CMNP_PEG was significantly lower than that of vehicle, and the expression of caspase 3 in CMNP was significantly lower than that of CMNP_PEG_apt. Altogether, these results indicate that the unique effects of combined CMNP and apt treatment to reinforce cancer targeting and anti‐cancer actions can be exerted by the complete form of CMNP_PEG_apt. In addition to xenografted tumor sites, test groups were distributed to the heart, liver, spleen, lungs, and kidneys after IVIS examination on day 10 post‐xenografting, resulting in no significant indication of organ toxicity when analyzed histologically after sacrifice (Figure S18, Supporting Information).
Only on day 28 after the first treatment of test groups (on day 14 post‐second treatment), CMNP_PEG_apt reduced the tumor volume significantly more than the other test groups (Figure 5g). Hence, it took 14 days to exert anti‐cancer effects after the second treatment although superior tumor targeting was initiated even on day 1 (Figure 5c). Considering that CMNP_PEG_apt took 7 days to reach the nuclear membrane (Figure 4c; Figure S12, Supporting Information), its anti‐cancer effect was potentiated for 7 days after all the injected CMNP_PEG_apt reached the nuclear membrane. These results indicate a unique advantage of the 7‐day procedure to regulate Notch‐1 activation on the cell membrane and intracellular downstream signaling such as Hes1 and Hey1[ 28 ] (Figures 4h and 5h) sequentially by minimizing systemic side effects (Figure S18b, Supporting Information).
Finally, the two‐edged effects of CMNP_PEG_apt on anti‐cancer growth and cartilage reinforcement were examined in vivo after orthotopic xenograft of Hep2 cells in nude mice (Figure 6 ). Because this laryngeal cancer model represents both cancer growth and cartilage damage simultaneously, the pro‐chondrogenic effect of CMNP was first confirmed in a rat model with anterior cruciate ligament transection (ACLT)‐induced osteoarthritis (Figure 2), followed by validation of anti‐angiogenic and anti‐cancer effects of CMNP_PEG_apt in a mouse model established by xenografting human melanoma cells (A375P) (Figure 5). Thus, the combined effects of CMNP_PEG_apt were analyzed by comparing these with the validated results from each of the two previous models. The nude mice were injected with PBS (vehicle) on day 21 post‐xenografting (Figure 6a), followed by MRI imaging (Figure S19, Supporting Information) and end‐point analyses of the larynx area on day 0 and day 28 (sacrifice) post‐injection. Among the test groups, the anti‐angiogenic effect of CMNP_PEG_apt was evident from the formation of a disturbed vascular network, as observed by the sporadic signals of fluorescence bead perfusion (Figure 6b). This result was supported by the smaller number of blood vessels in the CMNP_PEG_apt group, which had significantly lower CD31 expression compared with that in the other test groups.
Furthermore, CMNP_PEG_apt injection efficiently induced anti‐cancer effects by reducing the expression of Notch‐1 and proangiogenic (CD31) markers, thereby suppressing cancer proliferation and inducing cancer cell apoptosis (Figure 6c). The pro‐chondrogenic effect of CMNP_PEG_apt resulted in cartilage reinforcement in the larynx tissues, as shown by the normal tissue‐like cartilage structure with the visibly richer proteoglycan and collagen contents as compared with other test groups (Figure 6d). These results confirmed the potent synergistic effects of targeting and inhibiting laryngeal cancer, together with cartilage protection by equipping CMNPs with a two‐edged sword function (i.e., apt conjugation). In the in vivo models, CMNP_PEG_apt treatment suppressed blood vessel growth surrounding the tumor, as evidenced by the significant reduction in PECAM/CD31 expression, which most likely suppressed melanoma cell propagation with apoptosis induction. In contrast, the absence of blood vessels in vitro affected CMNP_PEG_apt treatment to promote cell proliferation (Figure 2f; Figure S4a, Supporting Information). No significant organ toxicity was detected when the distributed organs were analyzed histologically after sacrifice (Figure S20, Supporting Information).
Since a previously reported method[ 17 ] was applied to produce CMNPs, the oligonucleotide content including miRNA was determined from the source cells as described in that report.[ 17 ] Identifying the effect of each intra‐CMNP constituent (e.g., miRNA) was impossible, and certain constituents exerted controversial effects (such as pro‐ vs anti‐angiogenic, promotion vs suppression of cancer progression) when each or even grouped miRNA content was analyzed. Hence, the entire set of experiments was designed to focus on i) the physical function of CMNPs to promote cell–cell packing, and ii) the anti‐Notch‐1 function of apt to suppress angiogenesis. Each result was produced by relying on the dominant effects of these two functions. Hypoxia generation in cancer mass resulted from physical cell–cell interactions, and the anti‐angiogenesis effect was potentiated significantly by presenting apt on CMNPs.
3. Conclusion
We have demonstrated a previously unidentified function, i.e., the physical tightening of cell–cell interactions by CMNP treatment. The cell membrane characteristics (e.g., cadherin expression) of CMNPs promoted membrane–membrane contacts. The consequent generation of hypoxia due to increased cell packing established new angles of cancer treatment and cartilage protection based on a trap function, considering the common environment between the two events. In addition, using the two‐edged sword concept, the conjugation of apt to CMNPs appeared to share the underlying mechanism that reinforced the vasculature‐free cartilage tissue and led to anti‐vascularization around the tumor site, thereby inhibiting cancer growth. This function includes inhibition of cancer stem cell proliferation, potentially suppressing its recurrence.[ 29 ] Moreover, the adhesion ability of CMNP_PEG_apt to both plasma (day 1) and nuclear (day 7) membranes maximized the regulatory effects on both cell–cell interaction and intracellular signaling transduction over time. These orchestrated actions represent an unprecedented design concept for achieving a two‐edged sword function. The antagonistic effect of the high‐voltage factor Notch‐1 suppresses cancer growth and invasion by inhibiting angiogenesis[ 15 ] and promoting chondrogenic MSC differentiation.[ 12 ] Therefore, these combined therapeutic factors can exert potent two‐edged effects that have never been reported in the field of advanced nanoscale materials.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
H.‐S.K. and Y.M.S. contributed equally to this work. This study was financially supported by the National Research Foundation of Korea (NRF) (grant nos. 2016M3A9E9941743, 2019R1A2C2010802, 2016R1C1B1010086, 2020R1I1A1A01074423, and 2021R1A6A3A13044778) and the Brain Korea 21 PLUS Project for Medical Science in Yonsei University College of Medicine. Human benign melanoma (A375P) and human laryngeal cancer (Hep2) cell lines were purchased from the Korean Cell Line Bank (KCLB, Seoul, Republic of Korea). HUVECs were purchased from American Type Culture Collection (ATCC, VA, USA). All animal experiments were performed in accordance with the Korean Food and Drug Administration (KFDA) guidelines and protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the Yonsei Laboratory Animal Research Center (YLARC) (Permit Nos. 2017‐0059 and 2019‐0084). The Institutional Review Board (IRB) of Yonsei University College of Medicine (IRB No. 4‐2019‐1260) approved the isolation and use of TMSCs from young donors. The IRB of Dongguk University Ilsan Hospital (IRB No. 22‐34) approved the isolation and use of BMSCs and ADSCs from human donors. Informed consent from the donor or their parent or guardian was obtained.
Kim H.‐S., Shin Y. M., Chung S., Kim D., Park D. B., Baek S., Park J., Kim S. Y., Kim D.‐H., Yi S. W., Lee S., Lee J. B., Ko J.‐Y., Im G.‐I., Kang M.‐L., Sung H.‐J., Cell‐Membrane‐Derived Nanoparticles with Notch‐1 Suppressor Delivery Promote Hypoxic Cell–Cell Packing and Inhibit Angiogenesis Acting as a Two‐Edged Sword. Adv. Mater. 2021, 33, 2101558. 10.1002/adma.202101558
Contributor Information
Mi‐Lan Kang, Email: milan511@tmdlab.co.kr.
Hak‐Joon Sung, Email: hj72sung@yuhs.ac.
Data Availability Statement
Research data are not shared.
References
- 1.a) Grellier M., Bordenave L., Amedee J., Trends Biotechnol. 2009, 27, 562; [DOI] [PubMed] [Google Scholar]; b) Mueller M., Rasoulinejad S., Garg S., Wegner S. V., Nano Lett. 2020, 20, 2257; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xu Y., Peng J., Dong X., Xu Y., Li H., Chang J., Acta Biomater. 2017, 55, 249. [DOI] [PubMed] [Google Scholar]
- 2. Brucher B. L., Jamall I. S., Cell. Physiol. Biochem. 2014, 34, 213. [DOI] [PubMed] [Google Scholar]
- 3.a) Trosko J. E., Ruch R. J., Front. Biosci. 1998, 3, d208; [DOI] [PubMed] [Google Scholar]; b) Hanahan D., Coussens L. M., Cancer Cell 2012, 21, 309. [DOI] [PubMed] [Google Scholar]
- 4.a) Schipani E., Ryan H. E., Didrickson S., Kobayashi T., Knight M., Johnson R. S., Genes Dev. 2001, 15, 2865; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Provot S., Zinyk D., Gunes Y., Kathri R., Le Q., Kronenberg H. M., Johnson R. S., Longaker M. T., Giaccia A. J., Schipani E., J. Cell Biol. 2007, 177, 451; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Makris E. A., Responte D. J., Paschos N. K., Hu J. C., Athanasiou K. A., Proc. Natl. Acad. Sci. USA 2014, 111, E4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harris A. L., Nat. Rev. Cancer 2002, 2, 38. [DOI] [PubMed] [Google Scholar]
- 6.a) Semenza G. L., Nat. Rev. Cancer 2003, 3, 721; [DOI] [PubMed] [Google Scholar]; b) Pennacchietti S., Michieli P., Galluzzo M., Mazzone M., Giordano S., Comoglio P. M., Cancer Cell 2003, 3, 347. [DOI] [PubMed] [Google Scholar]
- 7.a) Robins J. C., Akeno N., Mukherjee A., Dalal R. R., Aronow B. J., Koopman P., Clemens T. L., Bone 2005, 37, 313; [DOI] [PubMed] [Google Scholar]; b) Taheem D. K., Foyt D. A., Loaiza S., Ferreira S. A., Ilic D., Auner H. W., Grigoriadis A. E., Jell G., Gentleman E., Stem Cells 2018, 36, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y., Cairns R., Papandreou I., Koong A., Denko N. C., PLoS One 2009, 4, e7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Heddleston J. M., Li Z., Lathia J. D., Bao S., Hjelmeland A. B., Rich J. N., Br. J. Cancer 2010, 102, 789; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qiu G.‐Z., Jin M.‐Z., Dai J.‐X., Sun W., Feng J.‐H., Jin W.‐L., Trends Pharmacol. Sci. 2017, 38, 669; [DOI] [PubMed] [Google Scholar]; c) Keith B., Simon M. C., Cell 2007, 129, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Jones P. A., Baylin S. B., Cell 2007, 128, 683; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Esteller M., N. Engl. J. Med. 2008, 358, 1148. [DOI] [PubMed] [Google Scholar]
- 11. Carmeliet P., Jain R. K., Nat. Rev. Drug Discovery 2011, 10, 417. [DOI] [PubMed] [Google Scholar]
- 12. Grogan S. P., Olee T., Hiraoka K., Lotz M. K., Arthritis Rheum. 2008, 58, 2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao W., Sweeney C., Walsh C., Rooney P., McCormick J., Veale D. J., Fearon U., Ann. Rheum. Dis. 2013, 72, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hallmann R., Feinberg R. N., Latker C. H., Sasse J., Risau W., Differentiation 1987, 34, 98. [DOI] [PubMed] [Google Scholar]
- 15. Nowell C. S., Radtke F., Nat. Rev. Cancer 2017, 17, 145. [DOI] [PubMed] [Google Scholar]
- 16. Jang S. C., Kim O. Y., Yoon C. M., Choi D. S., Roh T. Y., Park J., Nilsson J., Lötvall J., Kim Y. K., Gho Y. S., ACS Nano 2013, 7, 7698. [DOI] [PubMed] [Google Scholar]
- 17. Yoon J. K., Kim D. H., Kang M. L., Jang H. K., Park H. J., Lee J. B., Yi S. W., Kim H. S., Baek S., Park D. B., You J., Lee S. D., Sei Y., Ahn S. I., Shin Y. M., Kim C. S., Bae S., Kim Y., Sung H. J., Small 2020, 16, 2000012. [Google Scholar]
- 18. Yeo R. W., Lai R. C., Zhang B., Tan S. S., Yin Y., Teh B. J., Lim S. K., Adv. Drug Delivery Rev. 2013, 65, 336. [DOI] [PubMed] [Google Scholar]
- 19. Yadav A., Janaratne T., Krishnan A., Singhal S. S., Yadav S., Dayoub A. S., Hawkins D. L., Awasthi S., MacDonnell F. M., Mol. Cancer Ther. 2013, 12, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lafont J. E., Int. J. Exp. Pathol. 2010, 91, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson K., Zhu S., Tremblay M. S., Payette J. N., Wang J., Bouchez L. C., Meeusen S., Althage A., Cho C. Y., Wu X., Schultz P. G., Science 2012, 336, 717. [DOI] [PubMed] [Google Scholar]
- 22. Konisti S., Kiriakidis S., Paleolog E. M., Nat. Rev. Rheumatol. 2012, 8, 153. [DOI] [PubMed] [Google Scholar]
- 23. Mead T. J., Yutzey K. E., Proc. Natl. Acad. Sci. USA 2009, 106, 14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oldershaw R. A., Tew S. R., Russell A. M., Meade K., Hawkins R., McKay T. R., Brennan K. R., Hardingham T. E., Stem Cells 2008, 26, 666. [DOI] [PubMed] [Google Scholar]
- 25. Johnstone B., Hering T. M., Caplan A. I., Goldberg V. M., Yoo J. U., Exp. Cell Res. 1998, 238, 265. [DOI] [PubMed] [Google Scholar]
- 26. Blobe G. C., Schiemann W. P., Lodish H. F., N. Engl. J. Med. 2000, 342, 1350. [DOI] [PubMed] [Google Scholar]
- 27. Massachusetts General Hospital Cancer Center , Intraperitoneal Chemotherapy, General Hospital Corporation, Boston, MA, USA: 2016, https://www.massgeneral.org/assets/MGH/pdf/cancer-center/gyn/intraperitoneal-chemotherapy.pdf. [Google Scholar]
- 28.a) Roy A., Basak N. P., Banerjee S., Cell Death Dis. 2012, 3, e380; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schroeter E. H., Kisslinger J. A., Kopan R., Nature 1998, 393, 382. [DOI] [PubMed] [Google Scholar]
- 29.a) Hoffman E. J., Cancer and the Search for Selective Biochemical Inhibitors, CRC Press, Boca Raton, FL, USA: 2007; [Google Scholar]; b) Brem H., Folkman J., J. Exp. Med. 1975, 141, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.