

Abstract
Articular cartilage defects caused by traumatic injury rarely heal spontaneously and predispose into post‐traumatic osteoarthritis. In the current autologous cell‐based treatments the regenerative process is often hampered by the poor regenerative capacity of adult cells and the inflammatory state of the injured joint. The lack of ideal treatment options for cartilage injuries motivated the authors to tissue engineer a cartilage tissue which would be more resistant to inflammation. A clustered regularly interspaced short palindromic repeats (CRISPR)‐Cas9 knockout of TGF‐β‐activated kinase 1 (TAK1) gene in polydactyly chondrocytes provides multivalent protection against the signals that activate the pro‐inflammatory and catabolic NF‐κB pathway. The TAK1‐KO chondrocytes encapsulate into a hyaluronan hydrogel deposit copious cartilage extracellular matrix proteins and facilitate integration onto native cartilage, even under proinflammatory conditions. Furthermore, when implanted in vivo, compared to WT fewer pro‐inflammatory M1 macrophages invade the cartilage, likely due to the lower levels of cytokines secreted by the TAK1‐KO polydactyly chondrocytes. The engineered cartilage thus represents a new paradigm‐shift for the creation of more potent and functional tissues for use in regenerative medicine.
Keywords: cartilage tissue engineering, gene editing, inflammation
Here a method to efficiently repair cartilage defects with a novel combination of cells, gene editing, and hydrogel is presented. The highly chondrogenic cells present inflammation‐resistance properties due to gene editing, can be used in off‐the‐shelf and mature into stiff constructs that integrate into mature cartilage when encapsulated in the hyaluronan‐based hydrogel.
![]()
1. Introduction
Articular cartilage has limited potential for self‐repair after injury, and cartilage injuries that result in defects are often accompanied by inflammation.[ 1 ] Although an injury's acute phase can resolve spontaneously, the underlying inflammation often persist for longer periods and frequently results in the onset of arthropathies such as osteoarthritis (OA).[ 2 ] Inflammation in a joint is one of the main factors associated with the progression of cartilage loss, caused by the presence of proinflammatory mediators such as interleukin‐1β (IL‐1β) and tumor necrosis factor α (TNFα) originating from the synovium of post‐traumatic joint injury patients.[ 3 , 4 ] Repair of the cartilage defect by surgical intervention is often necessary in order to restore joint function and mobility.[ 5 ] Cell‐based cartilage regeneration approaches usually rely on chondrogenic cell implantation, as is commonly done in autologous chondrocyte (ACI) or matrix‐assisted autologous chondrocyte implantation (M‐ACT, Codon). However, these techniques often result in the formation of lower‐quality cartilage or fibrocartilage, that predispose to long‐term deterioration.[ 6 , 7 ] The two‐stage surgical procedures requiring good manufacturing practices (GMP) cell processing are very expensive.[ 8 , 9 ] Furthermore, the limited number of cells that can be harvested from the patient necessitates an expansion phase that can result in dedifferentiated chondrocytes with reduced chondrogenic potential.[ 10 ] Using an allogeneic cell source for defect repair is a suitable alternative. It allows a one‐stage surgical repair, reduces cost and above all, may have the potential of improving the quality of the repaired cartilage.
Finger joints removed from young polydactyly patients are a promising, novel, allogeneic source of human chondrocytes.[ 11 ] These cells possess an enhanced ability to produce cartilage extracellular matrix (ECM) compared to adult chondrocytes; they can proliferate faster and retain their chondrogenic potential even after sustained passaging.[ 11 , 12 , 13 ] Furthermore, they lack the major histocompatibility complex (MHC) Class II antigens and B7‐1 and B7‐2 costimulatory molecules and therefore do not stimulate allogeneic T cells in vitro.[ 14 ] These chondrocytes possess immune‐suppressive properties, as demonstrated by the inhibition of interferon γ secretion in activated CD4+ T cells during coculture.[ 11 ] They have already purportedly been used in a clinical trial,[ 15 ] although the origin of those cells ultimately remains unclear.[ 16 ] Polydactyly chondrocytes were therefore chosen for this study, due to their promise as a source for off‐the‐shelf treatments.
One of the main challenges of regenerating cartilage after trauma is overcoming the inflammatory environment of the joint, which promotes cartilage degradation and inhibits the chondrogenic potential of the transplanted cells.[ 5 , 17 ] The acute phase after injury is marked by recruitment and activation of mononuclear cells into the synovial membrane as macrophages. These immune cells react by secreting proinflammatory mediators and chemokines.[ 18 ] The nuclear factor kappa‐B (NF‐κB) pathway plays a central role in regulating the inflammatory signals in chondrocytes. The activation of canonical NF‐κB (p65/p50) signaling in chondrocytes results in the expression of catabolic factors such as matrix metalloproteases (MMPs) and a disintegrin and metalloproteinase with thrombospondin domains (ADAMTSs), in addition to other such proinflammatory cytokines and chemokines as IL‐1β, TNFα, IL‐6, IL‐17 and IL‐8.[ 18 , 19 , 20 ] Chondrocytes can sense both pathogen‐associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) or other bacterial components, and damage‐associated molecular patterns (DAMPs) through their toll‐like receptors (TLRs).[ 21 ] TLR4 is known to be particularly upregulated in OA cartilage,[ 22 ] and it specifically recognizes degraded fibronectin,[ 23 ] fibrinogen[ 23 ] and hyaluronic acid,[ 24 ] among others. There is evidence of bacterial LPS presence in the synovial fluid of OA patients,[ 25 ] a factor that could contribute to the M1 polarization of macrophages and progression toward OA.[ 26 ] TLR4 activation ultimately results in the recruitment of MAP kinases (MAPKs) and activation of NF‐κB.[ 27 ] Activation of the NF‐κB pathway requires a cascade of stimulated MAP kinases that results in the ubiquitination of the NF‐κB inhibitory protein IKKα/β, followed by the translocation of NF‐κB to the nucleus, where it can promote the expression of catabolic genes[ 28 ] (Figure 1A).
Figure 1.
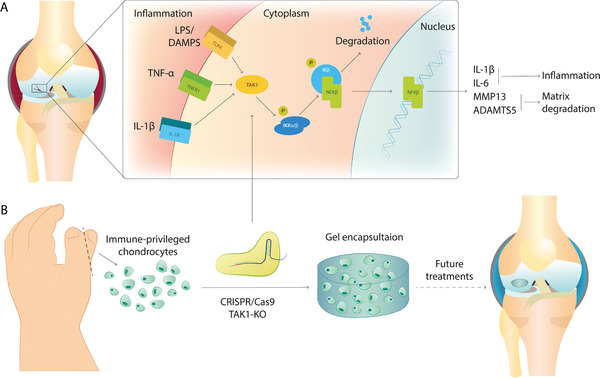
Overview of the inflammation pathways in cartilage injuries and schematic of the methodology used. A) Illustration of the NF‐κB pathway in the injured joint. Activation of the pathway by IL‐1β, TNFα, and LPS or DAMPS leads to phosphorylation of NF‐κB inhibitory protein (IKKα/β) and NF‐κB translocation to the nucleus, that activates the transcription of proinflammatory genes as IL‐1β and IL‐6 and matrix remodeling enzymes as MMP‐13 and ADAMTS‐5. B) Schematic of our methodology. Chondrocytes are collected by corrective surgeries; TAK1 KO is generated by a single‐step electroporation and can be then encapsulated into a hydrogel for future cartilage defect treatments.
The presence of inflammation can be detrimental to successful maturation and integration of tissue‐engineered grafts into native cartilage.[ 29 ] To date, several TE strategies have been developed to overcome this problem. Some hydrogel formulations, such as heparin/heparan sulfate[ 30 ] and sulfated alginate,[ 31 ] can capture IL‐1β, lowering inflammation. The use of anti‐inflammatory cytokine‐loaded hydrogels provides a local delivery of potent signals such as IL‐4 or IL‐10, polarizing macrophages toward the anti‐inflammatory M2 phenotype and lowering inflammation.[ 32 , 33 ] Other approaches include incorporation of anti‐inflammatory drugs, such as dexamethasone or celecoxib, in the hydrogel, to lower inflammation and promote chondrogenesis.[ 34 , 35 , 36 , 37 ] However, the use of biomaterial‐loaded drug depots has a limited temporal window (weeks)[ 38 ] compared to the long process required for cartilage to heal in vivo (months).[ 39 ] Longer‐lasting effects are thus needed and can be achieved with genome engineering. For that reason, many groups have focused on modifying the genes associated with inflammation. Targeting of IL‐1 receptor either by exogenous expression of IL‐1 receptor antagonist (IL‐1Ra)[ 40 , 41 ] or by knockout of IL‐1R[ 42 ] provides protection against the most potent inflammatory cytokine, IL‐1β. However, IL‐1β signaling is only one of the many catabolic pathways that impede cartilage regeneration, and a broader, long‐lasting strategy is needed. Transforming growth factor‐β‐activated kinase 1 (TAK1, alias MAP3K7) has been shown to play a fundamental role in the transmission of the proinflammatory signals and the activation of the NF‐κB pathway[ 43 ] and has been found to be upregulated upon injury in chondrocytes.[ 44 ] Inhibition of TAK1 by the small molecule 5Z‐7 oxozeaenol in a destabilization of the medial meniscus (DMM) OA rat model proved effective in preventing the disease and increasing ECM deposition.[ 45 ] As a critical intersection point of the IL‐1β, TNFα and TLR4 signaling pathways,[ 46 ] TAK1 represents an interesting therapeutic target to abrogate the major molecular mechanisms driving cartilage destruction following trauma and OA onset.
In this study, we combined the promising properties of polydactyly chondrocytes with the inflammation resistance by TAK1 gene editing and a hyaluronan‐based hydrogel to produce a novel off‐the‐shelf treatment for cartilage defects (Figure 1B). Our aim/goal was to obtain robust articular cartilage able to withstand the mechanical compression and inflammatory challenges typical for trauma or OA. To produce the KO, we used the Clustered Regularly Interspaced Short Palindromic Repeats/Crispr associated 9 (CRISPR/Cas9) system delivered as a ribonucleoprotein (RNP) which consisted of Cas9 protein and the single guide RNA (sgRNA) targeting TAK1. The system yielded a high efficiency in production of genome‐edited cells across several donors, which was superior to previous attempts.[ 47 , 48 , 49 ] Next, we characterized the gene expression and matrix deposition of the TAK1‐KO chondrocytes exposed to inflammatory stimuli. Then the ability of TAK1‐KO chondrocytes to produce good quality cartilage in a hydrogel when exposed to an inflamed environment was assessed. Finally, TAK1‐KO chondrocytes encapsulated in hydrogels were subcutaneously implanted in rats to study the in vivo cartilage maturation and the relationship between the construct and the innate immune system. The addition of gene editing to tissue engineering approaches can revolutionize the regenerative medicine field, given the limitations of autologous regeneration.
2. Results
2.1. Efficient TAK1 Editing of Primary Chondrocytes
Because primary chondrocytes undergo dedifferentiation as the number of passages increases,[ 10 ] selecting genome‐edited cells by single clone expansion method would lead to cells that are unable to produce a structurally viable cartilage matrix. In this study, we developed CRISPR‐edited cells with an RNP delivery strategy[ 50 ] to achieve high editing efficiency in a single step. Four sgRNAs were designed to target the first 5 exons of the TAK1 gene, inducing both a disruption of the reading frame and a premature stop codon. All guides were screened to test their ability to produce indels at the cut site (Figure S1A,B, Supporting Information), with the one targeting TAK1 exon 3 giving the highest percentage of edited cells. Sequencing of the cell population's DNA revealed that 94.0 ± 0.8% of sequences were edited (Figure 2A), with the majority of indels in +1 nucleotide insertions and −1 and −2 nucleotide deletions (Figure S1C, Supporting Information). The high editing efficiency with the selected sgRNA was comparable across multiple donors, which demonstrates the method's reproducibility.
Figure 2.
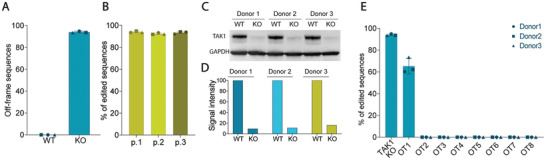
Characterization of the KO by sequencing and protein levels reveals high editing efficiency and low residual levels of TAK1 protein. A) Sanger sequencing of WT and KO samples from 3 different donors, with percentage of off‐frame sequences per total number of alleles (n = 3). B) Sanger sequencing of KO populations for three passages after electroporation from 3 different donors (n = 3). C) Western Blot and D) quantification of TAK1 in WT and KO cells of 3 donors, normalized to GAPDH protein levels. F) Percentages of editing at the 8 most probable off‐target sites by Sanger sequencing (n = 3). Error bars represent SD. OT: off‐target.
2.2. Low Expression of TAK1 Protein Is Maintained over Multiple Passages
Since TAK1 is reported to be involved in cell proliferation,[ 51 ] it was critical to assess whether edited cells can proliferate at the same rate as non‐edited ones. Sequencing of edited cell population over 3 passages showed that the edited percentage of the cell population did not change over time, likely indicating that the KO‐cell pool was homogenous and stable for the several passages of expansion required for cartilage tissue engineering applications[ 52 ] (Figure 2B). While the sequencing reveals the proportion of alleles that were edited in the population, protein expression levels can still be high due to compensation mechanisms in cells that retain one unedited gene copy.[ 53 ] Residual levels of TAK1 protein in edited cells were 12.4 ± 3.3% compared to WT, which is in line with sequencing results (Figure 2C,D).
2.3. TAK1‐KO Does Not Result in Gene‐Associated Off‐Targets
One of the main concerns about the use of CRISPR/Cas9 technology is the possibility of unintended, non‐specific edits in other parts of the genome.[ 54 ] To address this issue, the eight most probable off‐targets were selected for further screening (Table S1, Supporting Information). Sequencing revealed an off‐target cleavage only at the first predicted sequence in 60% of the sequences; however, the cut was not in a coding region of the genome (Figure 2E, and Table S1, Supporting Information). This result suggests a minimal risk for unintended off‐target effects using this specific sgRNA together with the RNP approach.
2.4. TAK1‐KO Reduces Activation of NF‐κB and its Downstream Targets
Activation of the TAK1 pathway by inflammatory stimuli results in the degradation of IKKα/β and nuclear translation of NF‐κB. Cells lacking TAK1 are therefore unable to convey the inflammatory signals from the cell membrane to the nucleus, thus expressing fewer inflammatory cytokines and matrix‐remodeling enzymes. Cells were treated with LPS, TNFα or IL‐1β, factors known to activate the pathway, using the TAK1 inhibitor 5Z‐7 oxozeaenol as a pharmacological control. qPCR of the treated cells shows that NF‐κB mRNA remained at the same level in KO‐chondrocytes in all the conditions, whereas it increased in WT cells, especially in TNFα and IL‐1β‐treated conditions (Figure 3A). Interestingly, LPS treatment did not cause any change in NF‐κB expression in WT cells, even though it is a known activator of the pathway. We therefore investigated whether infant chondrocytes express TLR4 receptor. Expression levels of TLR4 are lower in infant chondrocytes (iCh) when compared to adult ones isolated from a joint injury (jiCh). When treated with LPS, TLR4 levels are overexpressed in jiCh but remain unchanged in iCh in 2D culture (Figure 3B). This result suggests that infant chondrocytes are less sensitive to TLR4‐mediated signaling and represent an even more favorable choice for cartilage cell‐based therapies.
Figure 3.
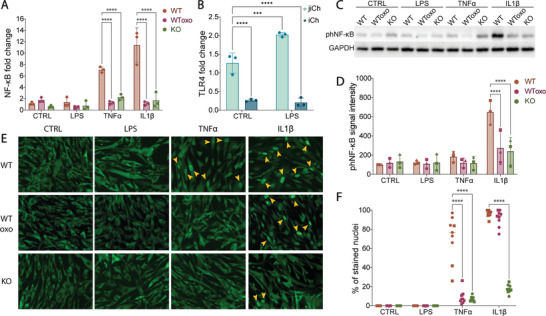
NF‐κB levels and activity are reduced in TAK1‐KO cells. A) qPCR‐based gene expression levels of NF‐κB in WT, oxozeaenol‐treated WT (WToxo), and KO cells treated with inflammatory stimuli for 16 h (n = 9 from 3 donors). B) qPCR‐based gene expression levels of TLR4 in adult chondrocytes derived from joint injury reconstruction (jiCh) and infant chondrocytes (iCh) treated with LPS (n = 3). C) Western Blot and D) quantification of Ser‐536 phosphorylated NF‐κB protein in 30 min‐treatment cells compared to GAPDH levels (n = 3 from 3 donors). E) Immunofluorescence of NF‐κB to detect its translocation to the nuclei after 30 min stimulation with inflammatory signals (yellow arrows indicate the positively stained nuclei) and F) percentage of the nuclei that stained strongly (n = 9 from 3 donors). Error bars represent SD. ***p < 0.001, ****p < 0.0001.
Activation of NF‐κB by Ser‐536 phosphorylation was observed only in IL‐1β‐treated WT cells, but notably did not change with TNFα treatment (Figure 3C,D), which is known to act predominantly on Ser‐903 and Ser‐907.[ 55 ] Nuclear translocation of NF‐κB was observed through immunofluorescence with TNFα (only WT) and IL‐1β (both WT and oxozeaenol‐treated WT) (Figure 3E,F). These findings confirm that the TAK1‐KO can efficiently block NF‐κB activation in the presence of inflammatory stimuli.
2.5. TAK1‐KO Cells Express Fewer Pro‐Inflammatory and Catabolic Factors
Activation of the NF‐κB pathway in chondrocytes is known to cause the upregulation of pro‐inflammatory genes such as IL‐1β, IL‐6 and TNFα and proteases such as MMP‐13 and ADAMTS5, resulting in increased degeneration of the joint cartilage.[ 18 , 19 , 20 ] We investigated the mRNA expression of these genes and observed a statistically significant reduction of their expression in TAK1‐KO cells under almost all conditions when treated with inflammatory stimuli compared to WT cells (Figure 4A). At the protein level, low amounts of IL‐6 and TNFα were measured in TAK1‐KO chondrocytes compared to WT cells. On the other hand, MMP‐13 protein expression was not changed across samples (Figure 4B).
Figure 4.
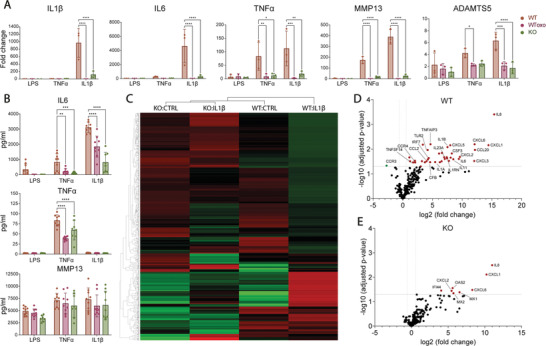
Expression of inflammatory genes is lower in TAK1‐KO cells after induction of inflammation. A) qPCR analysis of indicated genes of WT, oxozeaenol‐treated WT (WToxo), and KO cells (n = 9 from 3 donors). B) Diffused IL‐6, TNFα, and MMP13 protein quantification (n = 9 from 3 donors). C) Hierarchical clustering of inflammation‐related genes of WT and KO cells treated with IL‐β; differential gene expression analysis and volcano plot of D) WT and E) KO cells treated with IL‐1β by nSolver (n = 3). Error bars represent SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
As IL‐1β is a key molecular player driving OA disease pathogenesis,[ 56 ] we decided to further investigate its effects on the TAK1‐KO chondrocytes through gene multiplexing analysis (full data available in Table S2 and Figure S2, Supporting Information). When looking at the broad activation of inflammation‐associated genes, results showed that KO, IL‐1β‐stimulated cells cluster first with KO non‐treated cells, and then with WT non‐treated cells for most genes (Figure 4C). Upon IL‐1β treatment, differential expression analysis of inflammatory markers revealed that 37 genes are upregulated in WT cells (Figure 4D), whereas only 8 are upregulated in KO cells (Figure 4E, and Table S3, Supporting Information). Taken together, these results suggest that edited cells do not transfer the inflammatory signal to the nucleus at the same level as WT cells due to inactive TAK1 protein, resulting in cells that we hypothesize are less prone to inflammation and active degradation of cartilage ECM.
2.6. TAK1‐KO Chondrocytes Deposit and Retain ECM in an Inflamed 3D Pellet Environment
TAK1 has been shown to play a fundamental role in the maturation of cartilage during limb development,[ 57 ] so, it is essential to assess the ability of the edited cells to synthesize cartilage ECM when cultured in pellet form. When WT and KO cells were pelleted and cultured in chondrogenic medium for 21 days they both deposited collagen 2 and low amounts of collagen 1 at a similar rate. KO‐chondrocytes secreted higher amounts of glycosaminoglycans (Figure 5A,B).
Figure 5.
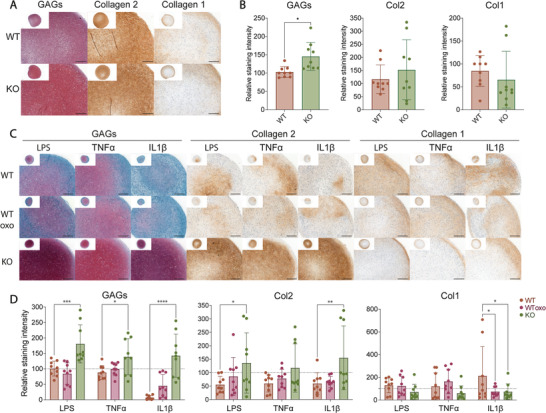
Cartilage ECM deposition is not impeded in TAK1‐KO cells and is enhanced in highly inflamed environment. A) Safranin O (GAGs), collagen 2 and 1 immunostaining of WT and KO pellets and B) quantification of the staining intensity (paired t test, n = 9 from 3 donors). Scale bar, 500 µm. C) Safranin O (GAGs), collagen 2 and 1 immunostaining of WT, oxozeaenol‐treated WT and KO pellets cultured with inflammatory stimuli in the last week of culture and D) quantification of the intensity (two‐way ANOVA, n = 9 from 3 donors). Scale bar, 200 µm. Error bars represent SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Due to the increased expression of catabolic enzymes in response to inflammatory stimuli, we then tested the cells’ ability to preserve the deposited ECM in an inflamed environment. Pellets were stimulated with LPS, TNFα or IL‐1β during the last week of chondrogenesis, with the addition of 5Z‐7 oxozeaenol to WT cells as control (Figure 5C). We chose supraphysiological cytokines’ concentrations to over‐activate the receptors and stress the ability of TAK1‐KO chondrocytes to retain their ECM. Under inflammatory conditions, pellets made from KO cells were able to produce more GAGs and collagen 2, and less collagen 1 compared to WT (Figure 5C,D). Especially in IL‐1β and TNFα stimulated WT pellets, the loss of ECM is visible and consistent with the increased expression of catabolic MMP‐13 and ADAMTS5. In the KO‐chondrocytes pellets, evidence of degradation was not found, as the staining for ECM components was at similar levels to control. Significantly, TAK1‐KO could protect from ECM loss better than oxozeaenol‐treated WT samples.
2.7. KO Chondrocytes Form Mechanically Robust Inflammation‐Resistant Neocartilage that Adheres to Mature Cartilage
The main clinical application we foresee for the genome‐edited neocartilage is the integration into cartilage defects in an inflamed joint. To model this situation, bovine cartilage rings were filled with a cell‐laden hyaluronan‐based hydrogel and cultured in chondrogenic media with or without LPS, TNFα, or IL‐1β for 6 weeks (Figure 6A). To assess ECM deposition quality, the Bern score[ 58 ] was used to grade the repair tissue. This score was specifically developed to assess cartilage quality in tissue engineering, since conventional scores take into consideration also other anatomical features. KO‐chondrocytes retained a normal morphology in the IL‐1β condition, whereas WT cells showed poor ECM deposition and spindle‐shaped morphology in all samples (Figure 6B). TNFα treatment demonstrated a trend in improved cartilage quality score in KO chondrocytes versus WT, albeit the latter was not statistically significant. As expected, LPS treatment did not change the quality of the constructs in both WT and KO conditions, confirming that these cells do not react to it. Histologically, both WT and KO chondrocytes produced cartilage which could efficiently produce ECM throughout the entire defect under control conditions (Figure 6C–H). However, ECM deposition differed greatly between WT and KO cells during IL‐1β stimulation. Based on the staining quantification of GAGs produced by KO chondrocytes, these cells were better at regenerating cartilage tissue (Figure 6C,D). Collagen 2 staining was also more intense in KO samples compared to WT ones (Figure 6E,F), and collagen 1 was more present in IL‐1β treated KO samples compared to WT ones (Figure 6G,H).
Figure 6.
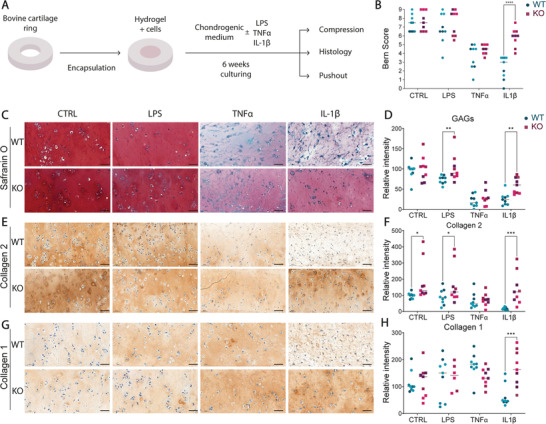
TAK1‐KO cells produce high quality ECM when embedded in a hyaluronan‐based hydrogel in inflamed conditions. A) Schematic of the experiment. Rings were formed from bovine cartilage and filled with hydrogel and cells, then cultured for 6 weeks in the presence of indicated inflammatory stimuli before proceeding with the analysis. B) Bern score to assess the quality of the samples maturation (two‐way ANOVA, n = 9 from 3 donors). C,E,G) Histology of the samples by Safranin O, collagen 2 and 1 immunostaining and D,F,H), Quantification of the staining intensity (two‐way ANOVA, n = 9 from 3 donors). Bovine cartilage ring dimensions: outer diameter, 8 mm, inner diameter 4 mm; hydrogel diameter: 4 mm, 1 mm thick. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The cultured tissue construct achieved a compressive modulus of more than 600 kPa when either WT (636.7 ± 90.0 kPa) or KO (728.5 ± 157.3 kPa) chondrocytes were used (Figure 7A). This stiffening of this cell‐laden hydrogel is in line with what has been reported in the literature[ 59 , 60 , 61 ] and demonstrates the ability of the KO‐chondrocytes to secrete ECM at the level of WT cells. On the other hand, the ability of WT cells to produce functional cartilage was extremely compromised when cultured with inflammatory cytokines, particularly IL‐1β, where a modulus of 48.9 ± 57.7 kPa (87.7% reduction) was measured. Remarkably, when KO samples were treated with IL‐1β, the samples achieved a modulus of 448.3 ± 222.8 kPa, with a 9.2‐fold change increase in stiffness with respect to IL‐1β‐treated WT samples (Figure 7A). TNFα‐treated samples showed a similar trend (317.8 ± 149.8 WT; 535.0 ± 186.5 KO), revealing that KO samples developed better mechanical‐resilient cartilage than WT.
Figure 7.
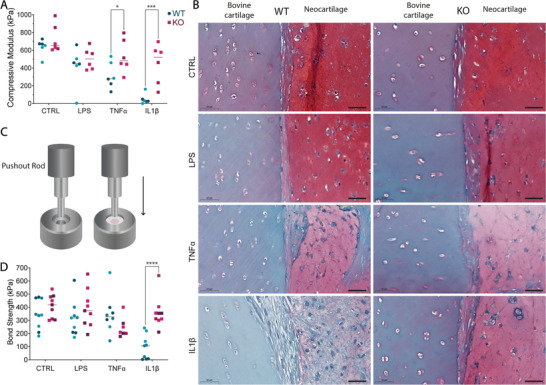
TAK1‐KO cells‐derived neocartilage strongly integrates into mature cartilage and presents high stiffness in inflamed environment. A) Compressive modulus of the formed neocartilage after 6 weeks in culture with or without indicated inflammatory stimuli, normalized by samples’ diameter (two‐way ANOVA, n = 6 from 2 donors). B) Close‐up on the interface between native bovine cartilage (left) and newly formed cartilage (right) from WT or KO cells. Scale bar, 50 µm. C) Representation of the pushout test for integration assessment and D) pushout test representing the bond strength of the samples to bovine cartilage (two‐way ANOVA, n = 9 from 3 donors). Bovine cartilage ring dimensions: outer diameter, 8 mm, inner diameter 4 mm; hydrogel diameter: 4 mm, 1 mm thick. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Histologically, both WT and KO neocartilage samples integrated into native bovine cartilage with a strong matrix deposition at the interface in the control condition (Figure 7B). Based on collagen staining, the interface was predominantly composed of collagen type I and less of collagen type II in all samples, apart from IL‐1β‐treated WT samples (Figure S3A, Supporting Information). Overall, collagen fibers are shown to be aligned along the surface of bovine cartilage based on polarized light microscopy (Figure S3B, Supporting Information). Interestingly, WT samples showed no collagen deposition in IL‐1β‐treated samples, whereas KO samples show collagen presence and alignment at the interface, correlating with push‐out results. TNFα‐treated WT samples show a random orientation of collagen fibers, while KO samples showed consistent collagen content and alignment by polarized light at the interface between the sample and the bovine cartilage. No gaps were observed between the sample and the bovine cartilage in all samples, and some human cells were observed to remodel and infiltrate the bovine cartilage in all conditions (Figure S3C, Supporting Information). WT samples showed fibrotic behavior at the interface when treated with IL‐1β, possibly indicating a worse integration. To quantify the strength of the binding between engineered constructs and native cartilage, we displaced the samples with a push‐out test[ 11 ] (Figure 7C). Under IL‐1β stimulating conditions the differences were striking, where the WT chondrocytes produced cartilage which failed to integrate into the bovine cartilage ring and had low resistance to the applied pushout force (95.6 ± 93.1 kPa WT versus 360.3 ± 118.1 kPa KO) (Figure 7D).
2.8. Inflammation‐Resistant Cartilage Is Stiffer In Vivo and Recruits Few Macrophages
To investigate the performance of TAK1‐KO hydrogels in vivo, WT and KO chondrocytes were encapsulated in the hyaluronan‐based hydrogel and subcutaneously implanted into nude rats after three weeks of preculture to allow neocartilage development (Figure 8A). Six‐weeks post‐implantation, samples were recovered and tested for their mechanical properties. In vivo KO samples developed into significantly stiffer constructs (476.3 ± 277.9) compared to WT samples (203.4 ± 274.1) (Figure 8B), a trend that is observable also in vitro (KO 602.8 ± 300.3; WT 356.6 ± 228.2) (Figure 8C).
Figure 8.
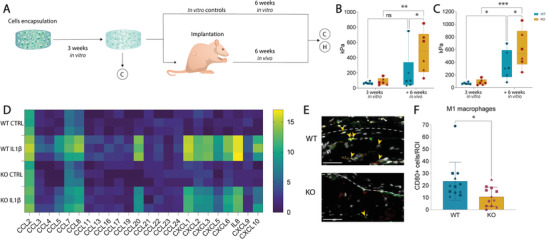
TAK1‐KO cells‐derived neocartilage develops into stiff matrix in vivo and recruits less M1 macrophages than WT‐derived one. A) Schematic of the experiment. WT and KO cells were encapsulated, samples were cultured for 3 weeks before subcutaneous implantation into nude rats and explanted or cultured in vitro after further 6 weeks. Sample diameter and thickness were 4 and 1 mm, respectively. C = compression, H = histology. B,C) Compressive modulus of in vivo (B) and in vitro (C) WT and KO samples at 3 and 9 weeks (two‐way ANOVA, 3 donors, n = 6). D) Gene expression analysis of main chemokines by multiplexing of WT and KO cells ± IL1β (n = 3). E) Immunofluorescence of macrophages infiltration in in vivo samples. External part of the construct above the dotted line, construct below. The pro‐inflammatory M1 macrophages were identified with double positivity for CD68 (pan‐macrophage marker) and CD80 (M1 marker). Red: CD68, green: CD80, grey: Hoechst. Orange arrows pointing to double positive cells for CD68 and CD80. Scale bar, 50 µm. F) CD68–CD80 double positive cells per region of interest (ROI) (Unpaired t test, 3 donors, n = 12). ns: non‐significant. Error bars represent SD. *p < 0.05, **p < 0.01, ***p < 0.001.
Monocytes and macrophages are recruited upon joint injury and can differentiate into a pro‐inflammatory phenotype, making them pivotal mediators of joint destruction and OA progression.[ 62 , 63 ] Based on gene expression analysis, WT chondrocytes treated with IL‐1β express more macrophage‐attracting chemokines (Figure 8D). We then asked if the recruitment of macrophages in vivo would also be affected by the KO: pro‐inflammatory M1 macrophage count confirmed this behavior. M1 macrophages were identified with a double staining for CD68, a pan‐macrophage marker, and CD80, an M1‐specific marker. WT samples showed a higher infiltration of M1 macrophages in the samples compared to KO ones (Figure 8E,F).
3. Conclusion
Current autologous cell‐based strategies for regenerating defective cartilage are still not optimal treatments, because they often result in fibrocartilage deposition[ 6 ] and may lead to post‐traumatic OA.[ 2 ] This effect may be due to the persistence of inflammatory molecules in the synovium.[ 5 , 17 ] Our promising method combines immune‐privileged chondrocytes with gene editing to produce an off‐the‐shelf treatment for cartilage defects that overcomes current limitations.
The two main contributors to cartilage graft failure are the dedifferentiation of adult cells due to their in vitro expansion and the inflammatory signals that promote ECM degradation. Polydactyly‐derived chondrocytes show increased ECM deposition compared to adult cells[ 11 , 12 , 13 ] and are thought to avoid the activation of autoimmunity due to their immune‐evading and ‐suppressive properties.[ 11 , 14 ] In this study, we demonstrated that they are naturally protected from damage‐associated molecular patterns (DAMPs)‐ and pathogen‐associated molecular patterns (PAMPs)‐signaling due to their lower expression levels of TLR4 compared to adult cells. We further engineered this promising cell type to achieve a higher resistance to the inflammatory stimuli that normally lead to matrix degradation and fibrocartilage deposition. By using a single‐step, RNP‐based CRISPR/Cas9 transfection method, we demonstrated that we could obtain an unprecedentedly high gene editing efficiency in a chondrocyte cell population pool. Previous gene editing efforts in primary chondrocytes reported a lower gene editing efficiency, reaching 74% efficiency with MMP13 to reduce ECM degradation,[ 47 ] or 60% for cell cycle inhibitor p21 to increase ECM deposition,[ 49 ] of which only 16% of colonies showed a homozygous deletion. Similar efficiency has only been demonstrated in a recent work targeting miR‐140, where 90–98% of editing was reached;[ 48 ] yet that required two sgRNAs and double transfection, conditions that might enhance post‐transfection cell toxicity and off‐target probability, even if not observed by T7E1 assay. In our editing, we observed a 60% deletion of the first predicted off‐target. Even though the deletion occurred in a region that does not code for any gene and thus is not supposed to cause damage, this is a limitation in our approach that might be avoided with the use of high‐fidelity Cas9 proteins.[ 64 ]
TAK1 is a gene involved in the proliferation of cells.[ 51 ] We analyzed the percentage of edited sequences over three passages after transfection and showed that the proportion of intact TAK1 alleles remained the same, indicating that the edited cells match the proliferation rate of the non‐edited ones. TAK1 also has a crucial role in the development of chondrocytes and cartilage, with TAK1‐deficient mice showing neonatal lethality, decreased limb size, and defects in cartilage.[ 57 ] However, effects of TAK1 deletion vary according to the developmental stage when they are performed. Deletion of TAK1 specifically in limb mesenchyme in Prx1Cre transgenic mice results in joint fusion and delayed maturation toward hypertrophic cartilage, whereas TAK1 deletion in Col2Cre transgenic mice showed delayed chondrocyte maturation and proliferation and prenatal lethality.[ 57 ] It is interesting to note that another study where Col2Cre mice were generated to specifically delete TAK1 in mice showed similar effects but mutants could survive for 2 to 3 weeks postnatally.[ 65 ] These previous studies indicate that TAK1 is crucial during development, with different effects based on the developmental stage. Another study that pharmacologically blocked TAK1 in human OA chondrocytes showed instead an increased expression of aggrecan, collagen type II and Sox9, and no differences in proliferation.[ 45 ] Taken together, these studies suggest that while TAK1 is necessary during development, its deletion does not impair the TGFβ‐associated deposition of ECM or proliferation once cells have reached the differentiation into chondrocytes, as it is also confirmed by our results.
As suggested by Cheng et al., TAK1 inhibition by 5Z‐7 oxozeaenol may prevent the in vivo destruction of cartilage by the inflammatory environment.[ 45 ] TAK1 editing is indeed associated with lower activation of the NF‐κB pathway in inflammatory conditions and with lower expression of inflammation‐associated gene expression. Interestingly, 5Z‐oxozeanol proved to be ineffective at preventing ECM loss after several days of sustained inflammation. A possible explanation could be the dose‐dependent cytotoxicity of 5Z‐7 oxozeaenol and its lack of specificity to TAK1, which could thus negatively affect other cellular pathways and impair ECM deposition.[ 66 , 67 ] Another discrepancy that we observed in our data is MMP‐13 expression in qPCR data and protein secretion after maturation. While being overexpressed after short‐term stimulation in monolayer culture in WT samples, there seems to be little difference between samples after 3D culture maturation. Since histology shows ECM loss in pellets, it is thus probable that most of the MMP‐13 protein is not secreted into the medium but rather entrapped in the pellet, where it is responsible for differences in maturation.
Interestingly, while LPS‐treated 2D cell culture gene expression remained unchanged compared to controls, a significant difference in matrix deposition was observed in LPS‐treated 3D chondrogenic pellets. A likely contributing factor to such an effect is the known differential expression of TLR4 during chondrocyte differentiation.[ 68 ]
The edited cells showed that they can efficiently deposit high quality articular cartilaginous ECM even when exposed to high levels of inflammatory cytokines as IL‐1β and TNFα. Interestingly, edited cells showed higher levels of GAG deposition compared to WT cells in the control condition of pellet culture, where no inflammation was present. Although cytokines are not added to the medium, the in vitro culture conditions are far from being physiological and could stress the differentiating chondrocytes. Normoxic chondrocyte culture is associated with increased MMP expression[ 69 , 70 ] compared to articular hypoxic conditions, and osmolarity commonly present in cell culture conditions is associated with lower ECM deposition compared to physiological values.[ 71 , 72 ] The absence of TAK1 protein in normoxic conditions could therefore be beneficial for ECM deposition and could be associated with increased chondrogenesis by the activation of other pathways unrelated to inflammation. Additionally, TAK1 was associated with the activation of cellular senescence programs,[ 73 ] implying that TAK1‐deficient cells could retain a younger phenotype. Further research is needed to confirm these concepts.
One challenge in cartilage defect repair is the efficient integration of the graft into cartilage.[ 74 ] Here we show that our inflammation‐resistant neocartilage was able to efficiently restore the defect and integrate it into mature cartilage in highly inflamed conditions. Furthermore, we were able to achieve high integration strengths for a tissue‐engineered construct in inflammatory conditions (360 kPa in IL‐1β‐treated KO samples) that are higher than the clinically available fibrin glue (40–50 kPa under ideal conditions)[ 75 ] and other literature values.[ 76 , 77 ] Nevertheless, the values are still an order of magnitude lower than the ones reached when articular cartilage was tested in the same push‐through settings (8.8 ± 0.52 MPa in intact human articular cartilage).[ 78 ] It should be noted that the results are representative of a rather short period of maturation (6 weeks), and longer cultivation periods have been shown to be beneficial for stiffness improvement, therefore potentially resulting in improved integration.[ 79 ] The highly aligned collagen network present at the interface is likely responsible for the strong adhesion of the samples to the native cartilage. The strong polarized light signal is observed in all conditions in the KO samples. In the WT samples, the signal was absent with IL‐1β treatment and less aligned with TNFα treatment, which reflects the results obtained in the pushout test. Additionally, chondrocyte migration from the sample into the mature cartilage suggests a successful integration of the graft. The increased stiffness observed in KO samples reflects the higher ECM content observed in histology. Higher production and retention of GAGs and collagen type II in KO samples under inflamed conditions, although quantified only by staining intensity, is indicative of the reduced susceptibility of KO cells to cytokines. The retention of ECM components in turn results in higher stiffness of the samples found in the mechanical analysis. The remarkable stiffnesses obtained in KO samples after 6 weeks of maturation in control condition (728.5 ± 157.3 kPa) and in IL‐1β‐treated condition (448.3 ± 222.8 kPa) are higher than the ones observed in WT samples (636.7 ± 90.0 kPa control; 48.9 ± 57.7 kPa IL‐1β). Even in inflamed conditions, KO samples could reach a higher stiffness with respect to current tissue engineering approaches performed in control conditions, if polycaprolactone‐reinforced hydrogels are excluded.[ 80 , 81 ] Both in vitro and in vivo, KO samples showed a high stiffness and less infiltration of M1 macrophages than WT samples. This outcome is due to the lower secretion of chemokines by KO cells, and it prevents the worsening of inflammation and matrix degradation by MMPs and ADAMTSs secreted by M1 macrophages.[ 82 ]
In our study, we showed the beneficial effects of TAK1‐KO on chondrocytes in inflammatory conditions and showed their ability to secrete cartilage ECM and integrate into 1 mm thick mature bovine cartilage rings when encapsulated in a hydrogel. Further optimization is required to translate our approach to clinically relevant sizes and thicknesses, as human knee cartilage thickness ranges from 1.65 to 2.98 mm.[ 83 ]
The future application of this technique remains to be explored. While the use of injectable hydrogels is preferred for its ease of use and minimal invasiveness, it poses some limitations. The use of enzymatically‐crosslinked, injectable hydrogels as in our study is preferred for cells and mature tissues with respect to chemical crosslinking methods, being effective at physiological pH and temperature.[ 84 ] However, maturation of the constructs and successful integration achieved by injectable hydrogels rarely match the ones of pre‐cultured samples.[ 85 , 86 ] For this reason, we envision future applications of our technique as a pre‐cultured graft to be press‐fit into the defect area, with the possibility of using the same hydrogel for additional sealing. Additionally, grafts can be prepared with the desired shape in less time with respect to ACI or MACI, due to the use of ready‐to‐use availability of allogeneic polydactyly chondrocytes. Given the high occurrence of polydactyly in population (0.3‐3.6/1000 live births),[ 87 ] the higher cellularity of infant cartilage[ 88 ] and the robust chondrogenesis even at higher passage number of polydactyly chondrocytes,[ 11 ] we are confident that sufficient cell numbers for clinical applications could be made available. In summary, we described a single‐step gene editing method to suppress inflammatory reactions in immune‐privileged chondrocytes. Our findings on their chondrogenic potential in a highly inflamed environment confirm that KO chondrocytes laden in a hyaluronan‐based hydrogel produce superior cartilage quality respect to existing methods. Future large animal studies are required to prove the effectiveness of the method in a cartilage defect model. With the advancing translation of genome‐editing technologies into clinical practice, our method will provide an effective ready‐to‐use, off‐the‐shelf engineered tissue for the treatment of cartilage defects.
4. Experimental Section
Study Design
This study was performed to evaluate a novel approach for cartilage defects treatment that uses a combination of cells, hydrogel and gene editing to avoid detrimental responses to an inflamed environment. To achieve this goal, i) TAK1 gene was knocked out in ii) infant chondrocytes, known to be chondrogenic for more passages than adult cells and do not pose immunological problems for allogeneic treatments. Furthermore, iii) they were encapsulated into a hyaluronan‐based hydrogel developed in the authors’ group to provide them a scaffold to efficiently fill the defect while secreting cartilaginous ECM. All experiments were performed with multiple cells’ donors to take into account donor to donor variability, with biological replicates. The exact n numbers of each experiment are indicated in the respective figure legends.
Primary Infant Chondrocytes
Human chondrocytes were acquired from corrective surgeries in six polydactyly patients aged 8–27 months, as previously described.[ 11 ] Informed consent was obtained from the legal guardians of polydactyly patients, and experiments were approved by the Ethical Committee of Canton Zürich (Kantonale Ethikkommission, Kanton Zürich, license PB_2017‐00510). Briefly, articular cartilage was collected from the joint, washed with PBS containing 1% v/v antibiotic‐antimycotic (Anti‐Anti, Gibco) and then digested in collagenase solution (DMEM [Gibco], 0,1% w/v of collagenase from Clostridium Histolyticum [Sigma Aldrich], 10% v/v fetal bovine serum [FBS, Gibco] and 1% v/v Anti‐Anti) at 37 °C overnight under gentle stirring. The following day, cells were plated at a density of 10 000 cells/cm2 in cell culture flasks with DMEM GlutaMAX (Gibco), 10% FBS, 5 ng mL−1 of FGF2 (Peprotech), and 10 µg mL−1 gentamicin (Invitrogen) at 37 °C, 5% CO2. When cells reached 95% confluence, they were trypsinized and used for transfection. All experiments were performed with n = 3 donors in triplicate up to passage 4.
Adult Joint Injury Chondrocytes
Human chondrocytes were acquired from corrective surgeries on joint traumatic injuries from an adult patient (23 years old). Informed consent was obtained from the patient under approval by the Ethical committee of Canton Zürich (Kantonale Ethikkommission, Kanton Zürich, license 2017‐00510). Cells were isolated and cultured as previously described for infant chondrocytes.
Transfection with RNPs Complexes
Cas9 protein was purchased from Invitrogen (TrueCut Cas9 Protein v2). Guide RNAs were designed by IDT webtool and purchased as a single guide from Synthego. Transfection was performed with Neon Transfection System kit (Invitrogen). For the transfection, the Cas9 protein was mixed with the single guide RNA (sgRNA) at 1:6 ratio, in transfection buffer for 5 µL of total volume and incubated at room temperature for 10 min for the ribonucleoprotein complex (RNP) to be formed. Cells at passage 1 were trypsinized and resuspended in transfection buffer, according to the manufacturer's instructions. The cells and the RNP mix were electroporated in the Neon system, and immediately transferred into a previously prepared flask with fresh medium. Cells electroporated with Cas9 protein without sgRNA were used as a wild‐type control.
Genotyping of CRISPR Editing Analysis
Cells were cultured until confluence was reached and were then trypsinized. 200 000 cells were collected for sequencing analysis, 1 million cells for Western Blot Analysis, and the remaining cells were plated at a 2000 cells/cm2. For DNA isolation, cells were pelleted and resuspended with 30 µL of PCR buffer with 100 µg of Proteinase K from Tritirachium album (Sigma) and incubated for 1 h at 55 °C, to enhance proteinase activity. Proteinase was inactivated for 15 min at 80 °C, and the mixture was directly used for PCR amplification. GoTaq2 polymerase and PCR reagents were all from Promega. The area surrounding the predicted cut was amplified with specific primers: Ex3 Fw: TTCTGGGCAGTCACTTGGTATTT; Ex3 Rv: CACGGTGCTGTATTCTCACTATTT. Amplicons were run on an agarose gel to detect the successful amplification and bands cut from the gel after imaging. PCR product was then purified from the gel with Wizard Gel minicolumn kit (Promega). DNA was then sequenced with the Sanger method (Microsynth AG) using the sequencing primer TCCTTGAACTATTGGGATCACTGCT. Sequences were then analyzed with TIDE web tool (http://shinyapps.datacurators.nl/tide/) to screen for the efficiency of the editing. Predicted off‐targets were identified with IDT CRISPR‐Cas9 guide RNA design checker (https://eu.idtdna.com/site/order/designtool/index/CRISPR_SEQUENCE), and primers flanking the expected cut were designed (Table S1, Supporting Information). Amplicons were sequenced as previously described and analyzed with TIDE. Results were then validated with Western Blot analysis.
Immunofluorescence
For immunofluorescence analysis, 10 000 cells/96 well were plated and cultured in serum deprivation for 24 h. On the day of the experiment, cells were treated with 10 ng mL−1 IL‐1β (Peprotech), 10 ng mL−1 of LPS (LPS from E. coli O111:B4; L4391, Sigma), or 1 ng mL−1 of TNFα (RnD systems). Cells were treated with inflammatory stimuli for 30 min, previously reported as the optimal timepoint to observe NF‐κB nuclear translocation.[ 89 , 90 , 91 , 92 ] Cells were fixed for 20 min with ice cold methanol on ice, washed twice with PBS, and blocked with 2% BSA in PBS (Sigma). Primary antibody anti NF‐κB (Invitrogen, 33–9900) was applied overnight, followed by PBS washes and 2 h RT incubation with secondary antibody Alexa 488 anti‐mouse (Invitrogen, A11001) and Hoechst (Invitrogen). Samples were imaged with Apotome 3 (Zeiss) and positive nuclei counted manually. The experiment was performed with n = 3 donors, in triplicate.
Western Blot
Collected cells were pelleted and resuspended in RIPA buffer, incubated on ice for 15 min and then centrifuged for 10 min at 12 000 g to remove debris and DNA contaminants; then proteins were diluted at 1 mg mL−1. 30 µg of proteins from samples were mixed with 10× reducing agent and 4× loading dye (NuPage) and then denatured for 10 min at 80 °C with 1× proteinase inhibitor (COmplete). Samples were run in a 4–12% Bis‐Tris gel (Thermo Fisher) and then transferred onto a nitrocellulose membrane. The chosen antibodies were rabbit anti‐human TAK1 (Thermo Fisher, 28H25L68, used 1:1000), rabbit anti‐human GAPDH (Cell Signalling, 2118S, used 1:1000), and HRP‐conjugated goat anti‐rabbit IgG (Abcam, 6721, used 1:3000). HRP signal was detected with BioRad DC Protein Assay (BioRad) and Fusion Solo device. Quantification of band intensity was performed by FIJI.
Pellet Culture
To induce pellet formation, 250 000 cells were resuspended in 200 µL of chondrogenic medium (DMEM 31 966 [Gibco] supplemented with 1% ITS [Corning], 40 mg mL−1 L‐proline [Sigma], 1 mL ascorbic acid [Sigma], 1:1000 gentamicin [Gibco], and 10 ng mL−1 TGFβ3 [Peprotech]) and pelleted in a low attachment conical bottom 96 well plate at 250 g for 5 min. Pellets were cultured for 21 days, and medium was changed every second day. Pellets were stimulated with inflammatory cues for the last 7 days of pellet culture. They were treated with chondrogenic medium supplemented with 10 ng mL−1 IL‐1β (Peprotech), 10 ng mL−1 of LPS (LPS from E. coli O111:B4; L4391, Sigma), or 1 ng mL−1 of TNFα (RnD systems). As a control, WT pellets were also treated with 1 mm 5Z‐7‐oxozeaenol (Sigma), an inhibitor of TAK1.
Histological Analysis
After 21 days of chondrogenic stimulation, pellets were collected and fixed with 4% paraformaldehyde solution (Merck). Samples were dehydrated with serial passages in ethanol and processed with LogosJ tissue processor (Milestone Medical SRL), then embedded in paraffin and cut in 5 µm thick slices. Samples were rehydrated with xylene and ethanol cascade before staining. To detect GAGs, samples were stained with Safranin O staining (Thermo Fisher) according to standard protocols. Collagen type I and II immunostainings were performed after antigen retrieval for 30 min at 37 °C with 1200 U/mL hyaluronidase (Sigma Aldrich) and 1 h blocking with 5% BSA in PBS (Sigma). Primary antibody was added overnight at 4 °C (mouse anti collagen I, Abcam, ab6308, 1:1000; mouse anti collagen II, DSHB, II‐II6B3, 1:20) in 1% BSA solution. Secondary HRP‐conjugated goat anti‐mouse (Abcam, ab6789) were diluted 1:1000 in 1% BSA solution and incubated for 1 h at RT. Development of staining was performed with DAB substrate kit (Abcam). Nuclei were counterstained with Weigert's hematoxylin (Sigma); with subsequent 1% acid alcohol solution (5 mL HCL 37% in 500 mL 70% EtOH) and treated with bluing agent (0.5 g sodium bicarbonate in 500 mL dH2O). Detection of M1 macrophages was performed using the broad macrophage marker CD68 and the M1‐specific marker CD80 to specifically detect double positive cells. Antigen retrieval was performed with Proteinase K solution (20 µg mL−1 in TE Buffer, pH 8.0). Primary antibodies (rabbit anti ph‐NFKB, MA5‐15160; rabbit anti rat CD80, BS‐2211R; mouse anti CD68, MA5‐13324; all from Invitrogen) were applied overnight in 1% BSA at 1:200. Secondary antibodies (anti rabbit Alexa 488, A11008; anti mouse Alexa 594, A11005; Invitrogen) were applied 1:1000 with 1:1000 Hoechst for 1 h at RT in the dark. Imaging was performed by the digital slide scanner Pannoramic 250 Flash III by 3DHistech.
Gene Expression Analysis
Cells were plated in triplicate for every condition and for every donor in 24 well‐plates and allowed to proliferate for two days and were then cultivated in serum deprivation for 24 h. Inflammatory stimuli were added at the same concentrations as used for pellet culture, and RNA was collected 16 h afterward with NucleoZol (Macherey Nagel). 1 mg RNA was retrotranscribed with GoScript Reverse Transcriptase kit (Promega) and diluted 1:5 with water. qPCR was performed with GoTaq qPCR Master Mix (Promega). Measurements were from biological triplicates from 3 different donors (n = 9), and measurement performed on two technical duplicates. Statistical analysis was performed with a two‐way ANOVA with GraphPad Prism (Dotmatics).
Cell culture RNA isolates from WT and TAK1‐KO chondrocytes were probed with the NanoString nCounter human inflammation panel v2 (NanoString Technologies, USA), covering 249 genes associated with inflammatory response and six housekeeping genes. Analysis was performed at the Biomedicum Functional Genomics Unit (FuGU) core unit from the University of Helsinki, according to manufacturer's instructions. Gene expression data analysis was normalized to the expression of 15 internal reference genes, and then the WT and TAK1‐KO cells, untreated or treated with IL‐1β for 24 h were compared and cluster analysis was performed using nSolver Analysis Software 4.0 according to the manufacturer (NanoString Technologies). Data from nCounter was normalized and adjusted p value was calculated with the Benjamini–Hochberg method, then plotted in the volcano plot with GraphPad Prism v.9. Cut‐off values were set at 1.3 for p value (−log10(0.05)) and 0.58 for fold change (log2(1.5)) for significance.
Secreted Protein Analysis
Cell culture supernatant was collected from pellets at day 21 and frozen for future analysis. Analysis was performed by Luminex device (Luminex MAGPIX, Luminex Corporation), with IL‐6, MMP‐13 and TNFα ProcartaPlex detection kits (Thermo Fisher) per manufacturer's instructions. Measurements were from biological triplicates from 3 different donors, and measurement performed on two technical duplicates.
Bovine Cartilage Ring Studies
Cartilage from bovine knees was peeled off the bone with a sharp microtome knife at ≈1 mm thickness; then rings of 4 mm of internal and 8 mm of external diameter were created with biopsy punches. The ring hole was filled with a mixture of WT or KO cells suspended at a concentration of 20 mio/mL in a 0.5% hyaluronan, 0.25% alginate‐based hydrogel,[ 59 , 61 ] prepared as previously described.[ 79 ] Constructs were crosslinked for 30 min and then cultured in chondrogenic medium with or without 10 ng mL−1 IL‐1β, 10 ng mL−1 of LPS or 1 ng mL−1 of TNFα for 6 weeks, with biweekly medium change. Samples were then analyzed with histology (biological triplicates from 3 donors) and mechanical testing.
Compression Test
Samples were punched out from the cartilage ring to ensure homogeneous compression. Mature constructs were tested with a TA.XTplus Texture Analyser (Stable Microsystems) with a 500 g load cell and a preload of 0.2 g. Samples were compressed at a loading rate of 0.01 mm s−1, reaching a final strain of 10%. The compressive modulus was calculated as the slope of the stress–strain curve. Measurements were from biological triplicates from 2 different donors (n = 6).
Push‐Out Test
The integration of the engineered cartilage to the bovine cartilage ring was assessed with a displacement push‐out test. The push‐out test was performed using a TA.XTplus Texture Analyser, in which compression via a 3 mm metal rod was applied to the central hydrogel at a speed of 0.5 mm s−1 until breakage. Bond strength was calculated as the maximum force applied before breakage of the sample, divided by the contact area between the hydrogel and inner surface of the ring. Measurements were from biological triplicates from 3 different donors (n = 9).
Animal Subcutaneous Implantation
For the in vivo experiment, 9‐week‐old athymic male rats were used (Rj:ATHYM‐Foxn1nu/nu from JANVIER Labs), in accordance with the ethical license (Application No. ZH147/2019, approved from the Zurich Cantonal Veterinarian Agency). WT and KO cells from three donors were encapsulated in hyaluronan‐based hydrogels as previously described and cast in a polydimethylsiloxane (PDMS) ring of 4 mm of inner diameter. Samples (four samples/condition, two samples/animal) were precultured in chondrogenic medium for 3 weeks and then implanted subcutaneously in the back of the rats. Rats were anesthetized with 5% Isoflurane (Provet AG) and 2 mg kg−1 of Meloxicam (Metacam) were used for analgesia. Anesthesia was maintained with 1.5–3% with Isoflurane during the surgical procedure. Two lateral incisions were performed to create the subcutaneous pockets where samples were inserted. Incisions were closed with surgical staples, removed after one week. After 6 weeks, animals were euthanized with CO2 and samples recovered. Samples were then evaluated histologically with antibodies anti‐CD68 and anti‐CD80 (Thermofisher) and with mechanical testing as described previously.
Statistical Analysis
All data are presented, with individual data points on the graphs, bar plots with error bars representing mean values ± SD. Statistical analysis was performed using the statistical analysis software GraphPad Prism, using a two‐way ANOVA with Fisher's LSD test. For each donor and experimental group technical duplicates or triplicates were performed, as stated in the text. p values < 0.05 were considered significant. For differentially expressed genes analysis, the nCounter dataset p value was adjusted with the Benjamini–Hochberg method, then data were plotted in a volcano plot.
Conflict of Interest
The authors have submitted a patent on the hyaluronan hydrogel.
Supporting information
Supporting Information
Acknowledgements
The authors acknowledge the help of the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich. The monoclonal antibody II‐II6B3 was obtained from the Developmental Studies Hybridoma Bank (DSHB) by Linsenmayer, T.F. (DSHB Hybridoma Product II‐II6B3). G.B. acknowledges the Finnish Cultural Foundation (Grant ID: 00220103).
Open access funding provided by Eidgenossische Technische Hochschule Zurich.
Bonato A., Fisch P., Ponta S., Fercher D., Manninen M., Weber D., Eklund K. K., Barreto G., Zenobi‐Wong M., Engineering Inflammation‐Resistant Cartilage: Bridging Gene Therapy and Tissue Engineering. Adv. Healthcare Mater. 2023, 12, 2202271. 10.1002/adhm.202202271
Contributor Information
Goncalo Barreto, Email: goncalo.barreto@helsinki.fi.
Marcy Zenobi‐Wong, Email: marcy.zenobi@hest.ethz.ch.
Data Availability Statement
The data that support the findings of this study are openly available in ETH Research Collection at doi:10.3929/ethz‐b‐000558251, reference number [93].
References
- 1. Umlauf D., Frank S., Pap T., Bertrand J., Cell. Mol. Life Sci. 2010, 67, 4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Punzi L., Galozzi P., Luisetto R., Favero M., Ramonda R., Oliviero F., Scanu A., RMD Open 2016, 2, e000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee J. H., Ort T., Ma K., Picha K., Carton J., Marsters P. A., Lohmander L. S., Baribaud F., Song X.‐Y. R., Blake S., Osteoarthritis Cartilage 2009, 17, 613. [DOI] [PubMed] [Google Scholar]
- 4. Guilak F., Fermor B., Keefe F. J., Kraus V. B., Olson S. A., Pisetsky D. S., Setton L. A., Weinberg J. B., Clin. Orthop. Relat. Res. 2004, 423, 17. [DOI] [PubMed] [Google Scholar]
- 5. Jungmann P. M., Gersing A. S., Baumann F., Holwein C., Braun S., Neumann J., Zarnowski J., Hofmann F. C., Imhoff A. B., Rummeny E. J., Link T. M., Knee Surg. Sports Traumatol. Arthrosc. 2019, 27, 3001. [DOI] [PubMed] [Google Scholar]
- 6. Clatworthy M., Orthop. J. Sports Med. 2017, 5, 2325967117S00186. [Google Scholar]
- 7. Andriolo L., Merli G., Filardo G., Marcacci M., Kon E., Sports Med. Arthroscopy Rev. 2017, 25, 10. [DOI] [PubMed] [Google Scholar]
- 8. Fuggle N. R., Cooper C., Oreffo R. O. C., Price A. J., Kaux J. F., Maheu E., Cutolo M., Honvo G., Conaghan P. G., Berenbaum F., Branco J., Brandi M. L., Cortet B., Veronese N., Kurth A. A., Matijevic R., Roth R., Pelletier J. P., Martel‐elletier J., Vlaskovska M., Thomas T., Lems W. F., Al‐Daghri N., Bruyère O., Rizzoli R., Kanis J. A., Reginster J. Y., Aging: Clin. Exp. Res. 2020, 32, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mistry H., Connock M., Pink J., Shyangdan D., Clar C., Royle P., Court R., Biant L. C., Metcalfe A., Waugh N., Health Technol. Assess. 2017, 21, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schnabel M., Marlovits S., Eckhoff G., Fichtel I., Gotzen L., Vécsei V., Schlegel J., Osteoarthritis Cartilage 2002, 10, 62. [DOI] [PubMed] [Google Scholar]
- 11. Cavalli E., Levinson C., Hertl M., Broguiere N., Brück O., Mustjoki S., Gerstenberg A., Weber D., Salzmann G., Steinwachs M., Barreto G., Zenobi‐Wong M., Sci. Rep. 2019, 9, 4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adkisson H. D., Martin J. A., Amendola R. L., Milliman C., Mauch K. A., Katwal A. B., Seyedin M., Amendola A., Streeter P. R., Buckwalter J. A., Am. J. Sports Med. 2010, 38, 1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mortazavi F., Shafaei H., Soleimani Rad J., Rushangar L., Montaceri A., Jamshidi M., World J. Plast. Surg. 2017, 6, 183. [PMC free article] [PubMed] [Google Scholar]
- 14. Adkisson H. D., Milliman C., Zhang X., Mauch K., Maziarz R. T., Streeter P. R., Stem Cell Res. 2010, 4, 57. [DOI] [PubMed] [Google Scholar]
- 15. Cherian J. J., Parvizi J., Bramlet D., Lee K. H., Romness D. W., Mont M. A., Osteoarthritis Cartilage 2015, 23, 2109. [DOI] [PubMed] [Google Scholar]
- 16. Evans C. H., Bone Jt Res. 2019, 8, 469. [Google Scholar]
- 17. Fahy N., de Vries‐van Melle M. L., Lehmann J., Wei W., Grotenhuis N., Farrell E., van der Kraan P. M., Murphy J. M., Bastiaansen‐Jenniskens Y. M., van Osch G. J. V. M., Osteoarthritis Cartilage 2014, 22, 1167. [DOI] [PubMed] [Google Scholar]
- 18. Goldring M. B., Otero M., Curr. Opin. Rheumatol. 2011, 23, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen J., Abu‐Amer Y., O'Keefe R. J., McAlinden A., Connect. Tissue Res. 2017, 58, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi M.‐C., Jo J., Park J., Kang H. K., Park Y., Cells 2019, 8, 734.31319599 [Google Scholar]
- 21. Bobacz K., Sunk I. G., Hofstaetter J. G., Amoyo L., Toma C. D., Akira S., Weichhart T., Saemann M., Smolen J. S., Arthritis Rheum. 2007, 56, 1880. [DOI] [PubMed] [Google Scholar]
- 22. Kim H. A., Cho M.‐L., Choi H. Y., Yoon C. S., Jhun J. Y., Oh H. J., Kim H.‐Y., Arthritis Rheum. 2006, 54, 2152. [DOI] [PubMed] [Google Scholar]
- 23. Chen K., Huang J., Gong W., Iribarren P., Dunlop N. M., Wang J. M., Int. Immunopharmacol. 2007, 7, 1271. [DOI] [PubMed] [Google Scholar]
- 24. Liu‐Bryan R., Terkeltaub R., Arthritis Rheum. 2010, 62, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang Z. Y., Stabler T., Pei F. X., Kraus V. B., Osteoarthritis Cartilage 2016, 24, 1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang Z., Kraus V. B., Nat. Rev. Rheumatol. 2016, 12, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawai T., Akira S., Semin. Immunol. 2007, 19, 24. [DOI] [PubMed] [Google Scholar]
- 28. Liu T., Zhang L., Joo D., Sun S.‐C., Signal Transduction Targeted Ther. 2017, 2, 17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Djouad F., Rackwitz L., Song Y., Janjanin S., Tuan R. S., Tissue Eng., Part A 2009, 15, 2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li L., Li Y., Feng D., Xu L., Yin F., Zang H., Liu C., Wang F., Int. J. Mol. Sci. 2016, 17, 1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arlov Ø., Öztürk E., Steinwachs M., Skjåk‐Bræk G., Zenobi‐Wong M., Eur. Cells Mater. 2017, 33, 76. [DOI] [PubMed] [Google Scholar]
- 32. Soranno D. E., Rodell C. B., Altmann C., Duplantis J., Andres‐Hernando A., Burdick J. A., Faubel S., Am. J. Physiol.‐Renal Physiol. 2016, 311, F362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schirmer L., Atallah P., Werner C., Freudenberg U., Adv. Healthcare Mater. 2016, 5, 3157. [DOI] [PubMed] [Google Scholar]
- 34. Choi J. H., Park A., Lee W., Youn J., Rim M. A., Kim W., Kim N., Song J. E., Khang G., J. Controlled Release 2020, 327, 747. [DOI] [PubMed] [Google Scholar]
- 35. Ziadlou R., Rotman S., Teuschl A., Salzer E., Barbero A., Martin I., Alini M., Eglin D., Grad S., Mater. Sci. Eng., C 2021, 120, 111701. [DOI] [PubMed] [Google Scholar]
- 36. Petit A., Redout E. M., van de Lest C. H., de Grauw J. C., Müller B., Meyboom R., van Midwoud P., Vermonden T., Hennink W. E., René van Weeren P., Biomaterials 2015, 53, 426. [DOI] [PubMed] [Google Scholar]
- 37. Koh R. H., Jin Y., Kim J., Hwang N. S., Cells 2020, 9, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Censi R., Dubbini A., Matricardi P., Curr. Pharm. Des. 2015, 21, 1545. [DOI] [PubMed] [Google Scholar]
- 39. Gurer B., Cabuk S., Karakus O., Yilmaz N., Yilmaz C., J. Orthop. Surg. Res. 2018, 13, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nixon A. J., Haupt J. L., Frisbie D. D., Morisset S. S., McIlwraith C. W., Robbins P. D., Evans C. H., Ghivizzani S., Gene Ther. 2005, 12, 177. [DOI] [PubMed] [Google Scholar]
- 41. Frisbie D. D., Ghivizzani S. C., Robbins P. D., Evans H., McIlwraith C. W., Gene Ther. 2002, 9, 12. [DOI] [PubMed] [Google Scholar]
- 42. Brunger J. M., Zutshi A., Willard V. P., Gersbach C. A., Guilak F., Arthritis Rheumatol. 2017, 69, 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takaesu G., Surabhi R. M., Park K.‐J., Ninomiya‐Tsuji J., Matsumoto K., Gaynor R. B., J. Mol. Biol. 2003, 326, 105. [DOI] [PubMed] [Google Scholar]
- 44. Ismail H. M., Didangelos A., Vincent T. L., Saklatvala J., Arthritis Rheumatol. 2017, 69, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cheng J., Hu X., Dai L., Zhang X., Ren B., Shi W., Liu Z., Duan X., Zhang J., Fu X., Chen W., Ao Y., Sci. Rep. 2016, 6, 34497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sato S., Sanjo H., Takeda K., Ninomiya‐Tsuji J., Yamamoto M., Kawai T., Matsumoto K., Takeuchi O., Akira S., Nat. Immunol. 2005, 6, 1087. [DOI] [PubMed] [Google Scholar]
- 47. Seidl C. I., Fulga T. A., Murphy C. L., Osteoarthritis Cartilage 2019, 27, 140. [DOI] [PubMed] [Google Scholar]
- 48. Chaudhry N., Muhammad H., Seidl C., Downes D., Young D. A., Hao Y., Zhu L., Vincent T. L., Osteoarthritis Cartilage 2022, 30, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. D'Costa S., Rich M. J., Diekman B. O., Tissue Eng., Part A 2020, 26, 441. [DOI] [PubMed] [Google Scholar]
- 50. Zhang S., Shen J., Li D., Cheng Y., Theranostics 2021, 11, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao L., Sheu T., Dong Y., Hoak D. M., Zuscik M. J., Schwarz E. M., Hilton M. J., O'Keefe R. J., Jonason J. H., J. Cell Sci. 2013, 126, 5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Francis S. L., Di Bella C., Wallace G. G., Choong P. F. M., Front. Surg. 2018,5, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. El‐Brolosy M. A., Stainier D. Y. R., PLoS Genet. 2017, 13, e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang X.‐H., Tee L. Y., Wang X.‐G., Huang Q.‐S., Yang S.‐H., Mol. Ther.–Nucleic Acids 2015, 4, e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Christian F., Smith E. L., Carmody R. J., Cells 2016, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vincent T. L., F1000Res 2019, 8, 934. [Google Scholar]
- 57. Gunnell L. M., Jonason J. H., Loiselle A. E., Kohn A., Schwarz E. M., Hilton M. J., O'Keefe R. J., J. Bone Miner. Res. 2010, 25, 1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grogan S. P., Barbero A., Winkelmann V., Rieser F., Fitzsimmons J. S., O'Driscoll S., Martin I., Mainil‐Varlet P., Tissue Eng. 2006, 12, 2141. [DOI] [PubMed] [Google Scholar]
- 59. Broguiere N., Cavalli E., Salzmann G. M., Applegate L. A., Zenobi‐Wong M., ACS Biomater. Sci. Eng. 2016, 2, 2176. [DOI] [PubMed] [Google Scholar]
- 60. Levinson C., Lee M., Applegate L. A., Zenobi‐Wong M., Acta Biomater. 2019, 99, 168. [DOI] [PubMed] [Google Scholar]
- 61. Fisch P., Bioprinting Auricular Cartilage for the Treatment of Microtia, ETH Zurich, 2022. [Google Scholar]
- 62. Haubruck P., Pinto M. M., Moradi B., Little C. B., Gentek R., Front. Immunol. 2021, 12, 763702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang H., Cai D., Bai X., Osteoarthritis Cartilage 2020, 28, 555. [DOI] [PubMed] [Google Scholar]
- 64. Kleinstiver B. P., Pattanayak V., Prew M. S., Tsai S. Q., Nguyen N. T., Zheng Z., Joung J. K., Nature 2016, 529, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shim J.‐H., Greenblatt M. B., Xie M., Schneider M. D., Zou W., Zhai B., Gygi S., Glimcher L. H., EMBO J. 2009, 28, 2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li P., Zhao Q.‐L., Jawaid P., Rehman M. U., Ahmed K., Sakurai H., Kondo T., Int. J. Hyperthermia 2017, 33, 411. [DOI] [PubMed] [Google Scholar]
- 67. Fechtner S., Fox D. A., Ahmed S., Rheumatology 2016, kew301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Khodabandehloo F., Aflatoonian R., Zandieh Z., Rajaei F., Sayahpour F., Nassiri‐Asl M., Eslaminejad M. B., J. Cell. Mol. Med. 2021, 25, 5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Markway B. D., Cho H., Johnstone B., Arthritis Res. Ther. 2013, 15, R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ströbel S., Loparic M., Wendt D., Schenk A. D., Candrian C., Lindberg R. L., Moldovan F., Barbero A., Martin I., Arthritis Res. Ther. 2010, 12, R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Erndt‐Marino J., Trinkle E., Hahn M. S., Cartilage 2019, 10, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sieber S., Michaelis M., Gühring H., Lindemann S., Gigout A., BioRes. Open Access 2020, 9, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fujiki T., Miura T., Maura M., Shiraishi H., Nishimura S., Imada Y., Uehara N., Tashiro K., Shirahata S., Katakura Y., Oncogene 2007, 26, 5258. [DOI] [PubMed] [Google Scholar]
- 74. Trengove A., Di Bella C., O'Connor A. J., Tissue Eng., Part B 2022, 28, 114. [DOI] [PubMed] [Google Scholar]
- 75. Arvayo A. L., Wong I. J., Dragoo J. L., Levenston M. E., J. Orthop. Res. 2018, 36, 2406. [DOI] [PubMed] [Google Scholar]
- 76. Sennett M. L., Meloni G. R., Farran A. J. E., Guehring H., Mauck R. L., Dodge G. R., J. Orthop. Res. 2018, 36, 2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Theodoropoulos J. S., De Croos J. N. A., Park S. S., Pilliar R., Kandel R. A., Clin. Orthop. Relat. Res. 2011, 469, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. van de Breevaart Bravenboer J., In der Maur C. D., Bos P. K., Feenstra L., Verhaar J. A., Weinans H., van Osch G. J., Arthritis Res. Ther. 2004, 6, R469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fisch P., Broguiere N., Finkielsztein S., Linder T., Zenobi‐Wong M., Adv. Funct. Mater. 2021, 31, 2008261. [Google Scholar]
- 80. Cipollaro L., Ciardulli M. C., Porta G. D., Peretti G. M., Maffulli N., Br. Med. Bull. 2019, 132, 53. [DOI] [PubMed] [Google Scholar]
- 81. Guo X., Ma Y., Min Y., Sun J., Shi X., Gao G., Sun L., Wang J., Bioact. Mater. 2023, 20, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fernandes T. L., Gomoll A. H., Lattermann C., Hernandez A. J., Bueno D. F., Amano M. T., Front. Immunol. 2020, 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shepherd D. E. T., Seedhom B. B., Ann. Rheum. Dis. 1999, 58, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nezhad‐Mokhtari P., Ghorbani M., Roshangar L., Soleimani Rad J., Int. J. Biol. Macromol. 2019, 139, 760. [DOI] [PubMed] [Google Scholar]
- 85. Galarraga J. H., Locke R. C., Witherel C. E., Stoeckl B. D., Castilho M., Mauck R. L., Malda J., Levato R., Burdick J. A., Biofabrication 2021, 14, 014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Erickson I. E., Kestle S. R., Zellars K. H., Dodge G. R., Burdick J. A., Mauck R. L., Biomed. Mater. 2012, 7, 024110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Umair M., Ahmad F., Bilal M., Ahmad W., Alfadhel M., Front. Genet. 2018, 9, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huch K., Arch. Orthop. Trauma Surg. 2001, 121, 301. [DOI] [PubMed] [Google Scholar]
- 89. Rasmussen M. K., Iversen L., Johansen C., Finnemann J., Olsen L. S., Kragballe K., Gesser B., Inflammation Res. 2008, 57, 329. [DOI] [PubMed] [Google Scholar]
- 90. Bagaev A. V., Garaeva A. Y., Lebedeva E. S., Pichugin A. V., Ataullakhanov R. I., Ataullakhanov F. I., Sci. Rep. 2019, 9, 4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ernst O., Vayttaden S. J., Fraser I. D. C., Methods Mol. Biol. 2018, 1714, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Maguire O., O'Loughlin K., Minderman H., J. Immunol. Methods 2015, 423, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bonato A., Fisch P., Fercher D., Manninen M., Weber D. M., Eklund K. K., Barreto G., Zenobi‐Wong M., 2022, 10.3929/ethz-b-000558251. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are openly available in ETH Research Collection at doi:10.3929/ethz‐b‐000558251, reference number [93].