

Abstract
Molecular spintronics seeks to use single or few molecules as functional building blocks for spintronic applications, directly relying on molecular properties or properties of interfaces between molecules and inorganic electrodes. Spin‐crossover molecules (SCMs) are one of the most promising classes of candidates for molecular spintronics due to their bistability deriving from the existence of two spin states that can be reversibly switched by temperature, light, electric fields, etc. Building devices based on single or few molecules would entail connecting the molecule(s) with solid surfaces and understanding the fundamental behavior of the resulting assemblies. Herein, the investigations of SCMs on solid surfaces, ranging from isolated single molecules (submonolayers) to ultrathin films (mainly in the sub‐10 nm range) are summarized. The achievements, challenges and prospects in this field are highlighted.
Keywords: molecular spintronics, spin‐crossover molecules, spinterfaces
Spin‐crossover molecules (SCMs) are one of the most promising candidates for molecular spintronics applications due to their ability to exist in two spin states that can be reversibly switched by light and voltage, among others. Investigations of SCMs on surfaces are summarized, highlighting the achievements and challenges, as well as laying out the future prospects.
1. Introduction
Harnessing the spin of electrons, in addition to their charge—a field known as spintronics—offers the prospect of ultrafast and low‐power devices for data storage and magnetic/information sensing.[ 1 ] Among the novel materials and device concepts that are explored for the realization of spintronic concepts, molecular spintronics has emerged as one of the most exciting approaches that can reproduce existing inorganic spintronic devices such as spin valves and magnetic tunnel junctions, as well as produce radically new device concepts.[ 2 , 3 , 4 ]
The goal of molecular spintronics is to use single or few molecules as functional building blocks for spintronic applications, directly relying on molecular properties or properties of interfaces between molecules and inorganic electrodes. These goals are all the more relevant because of the continued drive toward miniaturization of devices and the impending end of Moore's law for the existing silicon‐based electronics. Although the interest in molecules as spin transport media stems initially from the long spin‐lifetime due to their intrinsically weak spin‐relaxation mechanisms,[ 5 ] it was soon realized that molecules might offer additional options not provided by conventional spintronics. This is due to the fact that the structural, chemical, and electronic properties of molecules can be tuned with atomic precision in an almost infinite number of ways, unlike the materials used in inorganic spintronics. When molecules are brought in contact with inorganic electrodes—which is a prerequisite for realizing single‐ or few molecule‐based devices—their interface interactions can give rise to functionalities otherwise inaccessible in standard inorganic interfaces.[ 3 , 4 ]
Engineering quantum phenomena at the interface between molecules and inorganic electrodes—commonly referred to as “spinterface”—can drastically modify the magnetic properties of both the molecule and the substrate. For instance, zinc methyl phenalenyl deposited on a Co substrate gives rise to a hybrid magnetic layer characterized by an enhanced magnetocrystalline anisotropy with a coercive field of 600 Oe, which is significantly higher than that of the underlying Co substrate (40 Oe).[ 6 ] The hybrid layer can act as a second magnetic electrode that can be addressed independently, enabling the fabrication of spin‐valve devices based on a single magnetic material.[ 6 ] Or, for hydrogen phthalocyanine deposited on two monolayers of Fe on W(110), the spin polarization of Fe is inverted at the molecular site due to interface effects.[ 7 ] While spinterfaces investigated so far have only played a passive role as spin filters or modifiers of spin polarization, the prospect of engineering active spinterfaces such that the interface states can be controlled with external inputs is even more intriguing. This has been recognized in the spinterface community as a way to realize radically new device concepts based on molecules.[ 4 ] The way to achieve active spinterfaces is to incorporate “responsive” molecules like spin‐crossover molecules (SCMs) that change their structural, magnetic, and electronic properties upon exposure to external stimuli like light or electric fields.
SCMs are a class of transition‐metal‐based coordination compounds with a near‐octahedral geometry around the metal and an electronic configuration of 3d4–3d7. The octahedral ligand field splits the d‐orbitals into eg and t2g levels (Figure 1a), and the molecule can assume two spin states—so‐called high spin (HS) and low spin (LS) state—depending upon the ligand‐field strength. In cases where the mean spin‐pairing energy lies between the ligand‐field splitting energy of the HS and the LS state and the enthalpy of the HS state is slightly higher than that of the LS state, the molecule can be switched from one state to the other (LS at low T and HS at high T) by changing the temperature. This process is termed “thermal spin‐crossover (SCO)”. Of the molecules exhibiting thermal SCO, about 90% are based on FeII (3d6), with HS and LS corresponding to paramagnetic and diamagnetic states, respectively. In the bulk, the nature of SCO is governed by cooperative effects; depending upon the strength of cooperativity, the spin switching is gradual, abrupt, or even exhibits hysteresis (Figure 1b). Cooperativity is generally accepted as originating from elastic interactions resulting from the change in molecular size that accompanies the spin transition—SCMs have a larger volume in the HS state than in the LS state.[ 8 ] It should also be mentioned that recent reports indicate significant contributions to cooperative effects deriving from electrostatic and magnetic interactions.[ 9 ] The spin states of SCMs can also be switched from LS to HS by light, a phenomenon commonly referred to as light‐induced excited spin‐state trapping (LIESST). Moreover, HS‐to‐LS switching (reverse‐LIESST) can be affected with near infra‐red light; both phenomena are generally observed at low temperatures. [ 10 , 11 ] In most cases, LIESST is induced by irradiation into metal‐to‐ligand charge‐transfer states, which can be schematically represented in a potential energy diagram as shown in Figure 1c. Apart from light and temperature, the spin states of SCMs can also be switched by other stimuli like pressure, magnetic or electric fields. For a comprehensive understanding of SCMs, the reader may refer to reviews on SCMs in general,[ 12 , 13 ] in nanomaterials,[ 14 ] and in thin films. [ 15 , 16 ]
Figure 1.
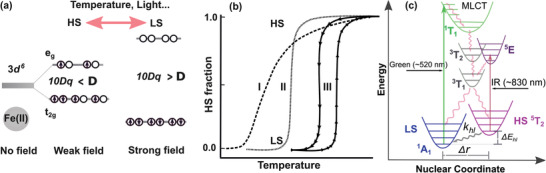
a) Schematic sketch of octahedral ligand‐field splitting of the 3d orbitals into eg and t2g in an Fe(II)‐based SCM and the eventual interplay between the ligand‐field strength (10Dq) and the spin‐pairing energy (Δ) determining the spin states. b) The different curves showing cooperative effects in the thermally induced SCO—dashed curve I for anti‐cooperativity or no to weak cooperativity, dotted curve II for strong cooperativity, and solid curve III for strong cooperativity with hysteresis. The arrows of curve III indicate the direction of temperature variation. c) Metal‐to‐ligand‐charge‐transfer (MLCT) states accounting for the light‐induced LS–HS or HS–LS transitions at low temperatures. b,c) Adapted with permission.[ 12a ] Copyright 1994, Wiley‐VCH.
While SCMs have been known and investigated for a long time—the phenomenon was discovered in the early 1930s[ 17 ]—their investigations on surfaces are relatively recent. The first investigation in this regard was made about a decade ago on [FeII(L)2](BF4)2 (L = bis(pyrazolyl)pyridine) deposited from solution on highly oriented pyrolytic graphite (HOPG).[ 18 ] The field saw an appreciable surge in activity only after reports of vacuum‐evaporable SCM complexes like [FeII(phen)2(NCS)2] (phen = 1,10‐phenanthroline; 1),[ 19 ] [Fe(H2B(pz)2)2)phen] (pz = pyrazolyl, phen = 1,10‐phenanthroline; 2) and [FeII(H2B(pz)2)2)bipy] (bipy = 2,2′‐bipyridine; 3).[ 20 ] More recently, [FeII((3,5‐(CH3)2pz)3BH)2] 4,[ 21 ] [FeIII(pap)2]+ (pap = N‐2‐pyridylmethylidene‐2‐hydroxyphenylaminato) 5,[ 22 , 23 ] and a few other Fe(II) complexes were deposited on surfaces by vacuum evaporation.[ 24 , 25 ] Thin films of SCOs have also been prepared with the Langmuir–Blodgett technique,[ 26 ] by spin‐coating,[ 27 ] drop casting,[ 28 ] and nanopatterning using lithography.[ 29 ] However, vacuum evaporation is particularly suited for investigating the fundamental behavior of SCMs on surfaces at the single‐molecule level or in ultrathin films because it allows precise control of the thickness of the molecular layer and the sample is free from impurities.
Herein, the recent progress on the surface‐based investigations of SCMs is sumamarized—specifically the vacuum‐evaporable ones—ranging from isolated single molecules to ultrathin films. (In the context of this article, ultrathin films would mean thicknesses in the sub‐10 nm range, except in certain cases where higher thicknesses will also be considered for completeness if necessary.) We have excluded the transport measurements in single or assemblies of few molecules as this subject has been reviewed recently.[ 16 , 30 ] An attempt is made to indicate the prospect for this class of molecules to be incorporated into spinterface science. It should be pointed out that this topic is also addressed by a recent article that focused more on the technical aspects of surface‐based studies of SCMs.[ 31 ]
2. Spin‐Crossover Molecules on Surfaces
What happens if a molecule gets in contact with a surface? An exhaustive answer to this question would be outside the scope of the article, but a brief overview is presented, putting it into the context of SCMs. If molecules are brought in close proximity to a surface, bonding interactions may occur, ranging from the weak physisorption to the strong chemisorption. The magnitude of the interaction is measured by the hybridization strength (ΔE), which is the energy difference between, on the one hand, the sum of the energies of the individual molecules and the substrate and, on the other, that of the hybrid system; ΔE > 1 eV is considered the chemisorption regime while physisorption is present for ΔE < 1 eV.[ 32 ] Chemisorption can in turn range from weak to strong. In weak chemisorption, the discrete molecular orbitals of the gas phase are broadened due to interaction with the substrate's valence/conduction band. This may lead to a partial charge transfer, as observed in the case of perylenetetracarboxylic dianhydride (PTCDA) on Ag.[ 33 ] In the strong chemisorption regime—usually observed for reactive surfaces like 3d ferromagnets—hybridization of the molecular orbitals and that of the metals occurs, resulting in a strong modification of both the electronic and the geometric structure of the adsorbed molecule.[ 4 , 32 ] On the other hand, physisorption—originating from weak van der Waals forces and usually observed on inert surfaces such as gold—can also be of different strength. In some cases, the orbitals of the physisorbed molecules are virtually undisturbed, and the molecules retain their gas‐phase behavior.[ 33 , 34 ] Yet in other cases, the molecular orbitals are weakly broadened into the substrate (metal) and may polarize the interface. Such physical effects may cause a subtle realignment of the HOMO and LUMO levels with respect to the gas phase.[ 35 ] Strong chemisorption is still a no‐go zone for SCMs due to the stringent demand placed on the spin switching to happen, such as maintaining the delicate interplay between the ligand‐field strength and the mean pairing energy, and the preservation of octahedral geometrical structure of the molecule. Even physisorption of SCMs on a gold surface results in the loss of SCO either due to molecular fragmentation, as observed for 2 (see below),[ 36 ] or due to the presence of a chemically active element like S on the molecule, as observed in the case of 1 (see below).[ 37 , 38 ]
STM (scanning tunneling microscopy), STS (scanning tunneling spectroscopy), and XAS (X‐ray absorption spectroscopy) are the main techniques used to investigate SCMs on surfaces. While an exhaustive account of the utility of STM and STS in investigating SCMs has been published recently,[ 39 ] a brief survey over the techniques employed in this field, from identifying the spin states to inducing SCO, is given in the following. STM‐ and STS‐based investigations of SCMs rely on the fact that SCMs generally have a higher conductance in the HS than in the LS state,[ 40 ] which is reflected by differential conductivities or tunneling currents in STS and different apparent heights in the STM image. For instance, SCM 1 deposited on Cu and Au surfaces, [ 37 , 41 ] the STS recorded at 4.6 K of HS molecules exhibits a zero‐bias Kondo resonance due to the presence of unpaired electrons. By contrast, the Kondo peak is absent for a weakly coupled molecule‐substrate system such as SCM 4 on Au(111)[ 42 ] or the cationic FeIII complex [Fe(pap)2]+ (pap = N‐2‐pyridylmethylidene‐2‐hydroxyphenylaminato; 5) on Cu2N/Cu(100), both recorded in the region of 4–5 K.[ 23 ] Nevertheless, HS and LS states are distinguishable from the tunneling spectra. The spin switching investigated with STM is usually carried out by placing the STM tip above the center of the molecule and varying the sample voltage (or current). A gas‐phase DFT‐calculated molecular structure, when overlaid on the STM image, can provide a fairly good hint on the molecular orientation on a surface. For example, Figure 2a shows the molecular orientation of SCM 1 on Cu, Au, and on Cu2N/Cu(100), derived from the assumption that the double‐lobed structures correspond to the two phenanthroline ligands. The lobes are farther apart in the HS state than in the LS state, reflecting the volume expansion in the HS state (Figure 2b). However, no pronounced lobe‐spacing difference between the two spin states is observable for the molecules on Cu2N/Cu(100) (Figure 2c). Nevertheless, the two spin‐states can be clearly differentiated based on their apparent height differences and the presence and absence of zero‐bias Kondo resonance in the HS and LS state, respectively.[ 37 ] XAS, on the other hand, is a global technique but, due to its high spin‐state sensitivity and chemical selectivity, is a powerful tool to probe spin‐crossover molecules on surfaces. In particular, the XAS recorded at the Fe L2,3 edge exhibits distinctive patterns depending upon the spin state of the molecule (refer to Section 2.2.1.1).
Figure 2.
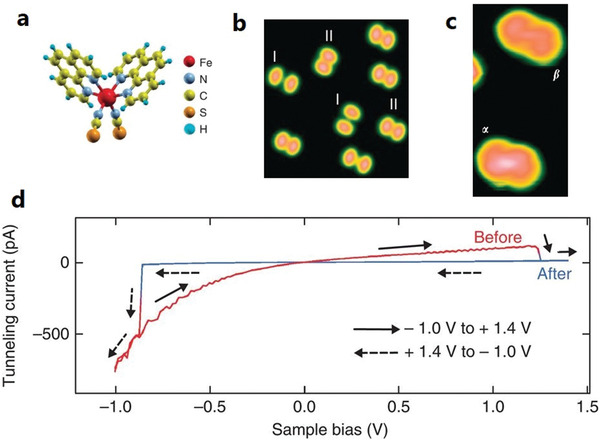
SCM 1 a) Orientation—standing on the two NCS legs—on metal substrates Cu(100), Au(111), and Cu2N‐passivated Cu(100) surface. b,c) STM images on Cu(100) (b) and Cu2N/Cu(100) (c). d) I(V) curves of an isolated SCM 1 on Cu2N/Cu(100) showing a hysteretic switching in going from HS to LS (red), and back (blue). The measurement was carried out at 4.6 K. a–d) Reproduced with permission.[ 37 ] Copyright 2012, Springer Nature.
2.1. Metal Surfaces
2.1.1. [FeII(phen)2(NCS)2] (1)
A complete loss of SCO behavior concomitant with a coexistence of HS and LS at 4.6 K has been reported for the SCM 1 in direct contact with Cu(100),[ 37 , 43 ] Cu(111), and Au(111).[ 41 ] The observed loss of SCO has been attributed to strong coupling between the molecule and the metallic substrates, mediated by the S‐atoms of the thiocyanate ligands. This is supported by ab initio calculations, which show that the adsorption energy difference between the two spin states of the molecule is more than twice the energy difference between the two spin states in the gas phase, accounting for the spin‐state trapping of the adsorbed molecules.[ 41 , 44 ] The spin‐state coexistence at all temperatures is due to the site‐dependent adsorption of the two spin states, as evidenced by the spin switching obtained by a lateral transfer of the molecule with the STM tip. Interestingly, on introducing an ultrathin insulating layer of Cu2N between the SCM 1 and the Cu(100) substrate, the molecules retain their SCO property. This is due to the reduced molecule–substrate interaction, evidenced by the lowering of the Kondo temperature by about 107 K upon going from Cu(100) to Cu2N/Cu(100). Figure 2d shows the voltage‐induced spin switching of the SCM 1 on Cu2N/Cu(100), recorded at 4.6 K. The molecule in the HS state is switched to the LS state on increasing the sample voltage to 1.2 V, indicated by the abrupt drop in the tunneling current to zero. On reversing the sample voltage, i.e., at −0.8 V, the tunneling current suddenly increases (negative direction), indicative of the LS‐to‐HS transition. The system therefore shows memristance behavior. The voltage‐induced spin switching processes were explored in detail by measuring the switching rate at a constant voltage for different values of the tunneling current. It was found that HS‐to‐LS switching is governed by a single electron–molecule interaction, while the reverse switching (LS‐to‐HS) is governed by higher‐order processes involving eight electrons.[ 37 ]
2.1.2. [FeII(H2B(pz)2)2)phen] (2) and [FeII(H2B(pz)2)2)bipy] (3)
In an STM‐based study of a bilayer of the SCM 2 on Au(111) at 5 K, it was reported that by injecting an electron to a given molecule via the STM tip, spin switching in both directions can be carried out by applying different sample voltages: LS‐to‐HS at 3 V, HS‐to‐LS at 1.8 V (Figure 3a–c). The authors proposed the electron‐induced spin switching (HS‐to‐LS or vice versa) to occur via individual electron‐molecule interactions, analogous to LIESST, and termed it electron‐induced excited spin‐state trapping (ELIESST).[ 45 ] However, there are basic differences between the spin switching in the opposite directions, chiefly that HS‐to‐LS switching is achieved by a strong injection of current (0.5 nA, an order of magnitude higher than switching in the reverse direction) and only occurs in the direct vicinity of the STM tip, while LS‐to‐HS switching occurs remotely (up to a nanometer distance from the STM tip position). It should be mentioned that charge‐induced spin‐crossover has been reported previously; i.e., by injecting two electrons into the bpp ligands of [FeII(bpp)2]2+ (bpp = 2,6‐bis(pyrazol‐1‐yl)pyridine) the compound is switched from the LS to the HS state.[ 46 ] The authors attributed the spin transition to an antiferromagnetic coupling between the two extra‐injected electrons, which presumably reside at the ligands, and the electrons of the FeII ion. In a follow‐up investigation on compound 2, using a combination of XAS and STM, it was observed that the molecules in direct contact with the Au(111) surface undergo fragmentation into HS [FeII(H2B(pz)2)2] and the phenanthroline ligand.[ 36 ] It is thus conceivable that the molecules in the second layer, which were found to be switchable by the ELIESST effect (see above), are separated from the gold surface by a monolayer of the dissociated molecules. Notably, in a 7 nm thick film (5–6 ML) of 2 on Au(111), investigated by ultraviolet photoelectron spectroscopy (UPS), the molecules seem to preserve their integrity, as the photoelectron spectra can be reproduced by DFT calculations of the complex in the gas phase. Although a residual LS species is observed at room temperature, full conversion from LS to HS can be affected at 28 K by virtue of the LIESST effect.[ 47 ]
Figure 3.
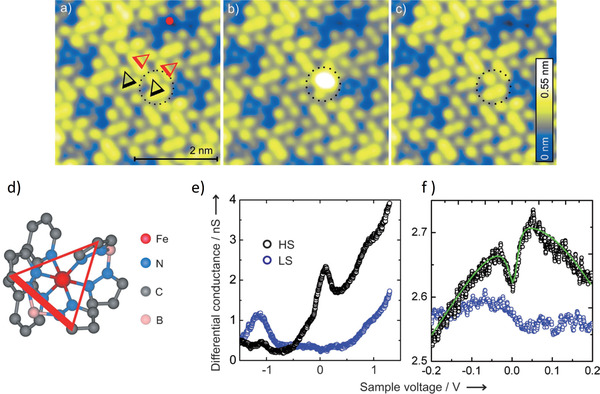
a–c) Constant‐current (50 pA) STM image of a bilayer of the SCM 2 on Au(111). The dotted circles in (a–c) indicate a molecule which is switched from LS (a) to HS (b) and back to LS (c). The LS‐to‐HS switching is achieved by applying a 3 V pulse with the STM tip positioned above the molecule. d) View of the SCM 2 in its bilayer orientation on Au(111). The thicker side of the overlaid triangle indicates the upward‐pointing H2B(pz)2 group and the opposite to it indicates the end of the other H2B(pz)2 group. e) The differential conductance measured as a function of sample voltage of HS (black) and LS (blue) molecules, and f) the zoom‐in of (e) around zero bias voltage. The STM is operated at 5 K. a–f) Adapted with permission.[ 45 ] Copyright 2012, Wiley‐VCH.
Different groups have investigated the behavior of SCM 3 on Au. Although there is a general consensus that the interactions with the Au surface inhibit the SCO property, there is an apparent disagreement on the fraction of molecules undergoing spin transition at lower coverages (submonolayer and bilayer). Based on STM images and local tunneling spectra, and backed by DFT calculations, a bilayer of the SCM 3 on Au(111) was reported to coexist in the two spin states with no evidence of spin switching on lowering the temperature from 300 to 131 K. The authors attributed the spin‐state coexistence and the loss of SCO in the bilayer at all the investigated temperatures to the substrate‐induced packing/molecular assembly.[ 48 ] X‐ray photoemission (XPS)‐based investigations of the same system of higher coverages of up to 50 nm indicated that the SCO behavior is largely retained once the molecules are decoupled from the surface.[ 49 ]
In a subsequent investigation of the bilayer of SCM 3 on Au(111) using a combination of STM, XAS, and XPS, it was reported that as much as 20(6)% of the molecules retain their SCO property.[ 50 ] Yet, a similar percentage of molecules preserving their SCO property was reported in a submonolayer of the SCM 3 on Au(111) using XAS.[ 51 ] The apparent discrepancy could be due to an error in the estimation of the molecular coverage, as has been pointed out elsewhere.[ 52 ]
2.1.3. [FeII((3,5‐(CH3)2pz)3BH)2] (4)
Among the SCM‐on‐Au(111) studies, SCM 4 stands out in that the SCO property is observed for molecules in direct contact with a metallic surface. In the STM‐based investigation carried out at 4.6 K, different superstructures are observed depending on the sample voltage, unlike the previous reports of voltage‐induced spin switching.[ 42 ] In particular, at 0.3 V, one in three molecules appears bright (Figure 4a); a complementary image is obtained at −0.7 V (Figure 4b), and yet the STM image recorded at −1.5 V reveals a well‐ordered monolayer (inset of Figure 4a). A later study using grazing‐incidence X‐ray diffraction established that the SCM 4 molecular layer has an epitaxial relationship with the underlying Au(111) lattice (Figure 4c), the epitaxy being strong enough to give rise to the experimentally observed high‐quality 2D arrangement. However, not all of the molecular lattices are commensurate with the Au(111) lattice. Only in a second‐order commensurate lattice every second molecule is found to be in registry with the underlying Au mesh (Figure 4c). The theoretical DFT calculations show that nearest‐neighbor mixed‐spin pairings (LS–LS–HS or HS–LS–HS) have the lowest energy. Using a mechanoelastic model, a strong case is made that the existence of the HS molecules at low temperatures leads to a partial reduction of the elastic strain energy arising from lattice misfits of the molecular and Au lattices as the system undergoes a transition from the HS to the LS state, leading to the existence of residual HS molecules at low temperatures.[ 53 ] SCM 4 has also been reported to retain its molecular integrity even on the more chemically reactive surfaces Cu and Ag.[ 54 ] The spin‐state coexistence is observed at low temperatures; partial spin‐state switching from HS‐to‐LS is observed on illumination with blue light for the molecule on Cu (and Au). This is an interesting twist to the phenomenon of light‐induced switching in SCMs as blue light—or visible light in general—is known to only affect LS‐to‐HS switching in bulk materials and thin films, as already mentioned in the introduction. The authors attributed the blue light‐induced HS‐to‐LS state switching to low energy photoelectrons from the metal surface interacting with the molecular layer.[ 54 ]
Figure 4.
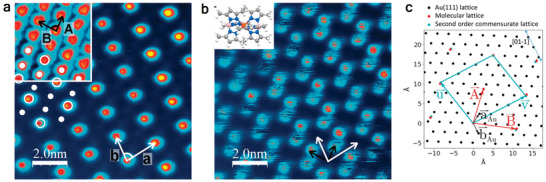
STM image of a monolayer of the SCM 4 on Au(111) recorded at the sample voltage of a) 0.3 V and b) −0.7 V. c) Molecular lattice vectors and and the underlying Au mesh ( and ). The turquoise‐colored dots are the second order commensurate molecular lattices + and . Images in (a,b) were recorded at 4.6 K. a,b) Adapted under the terms of the CC‐BY Creative Commons Attribution 4.0 International license (http://creativecommons.org/licenses/by/4.0/).[ 42 ] Copyright 2016, The Authors, published by Springer Nature. c) Reproduced with permission.[ 53 ] Copyright 2019, American Chemical Society.
2.1.4. [FeIII(pap)2]+ (5)
SCM 5, which is a cationic Fe(III) complex, paramagnetic both in the LS (S = 1/2) and HS (S = 5/2) states, has recently been studied on Au(111) and Cu2N/Cu(100) surfaces, using STM operated at ≈4 K (Figure 5a). While in the bulk it is associated with a negative counter ion , the molecule was deposited on Au(111) with and without the counter ion by sublimation and electrospray deposition, respectively. The STM images in both cases have the same characteristic features; moreover, a very low‐yield voltage‐induced conformational switching—although with no influence on the spin states—was reported.[ 22 ] However, when SCM 5 was sublimed on a Cu2N/Cu(100) surface, its SCO behavior was retained, similar to that of SCM 1 on the same surface, but with the difference that the SCM 5 exhibits three spin states. Further, no zero‐bias Kondo resonance is observed, unlike in the case of SCM 3, which is attributed to weak molecule–substrate interactions. The three spin states are ascribed to one HS state and two LS states, each having different conductance and conformation. Figure 5b shows the tunneling currents corresponding to the three different conformations or spin states, recorded by placing the STM tip on top of the molecule at a sample voltage of 2.5 V, and Figure 5c–e shows the three conformations A (HS), B (LS), and C (LS), respectively. There are also subtle differences in the differential conductance spectra recorded for the three conformations. Further support for assigning the HS state to conformation A is also obtained from the gas‐phase DFT calculations of the molecular geometry.[ 23 ]
Figure 5.
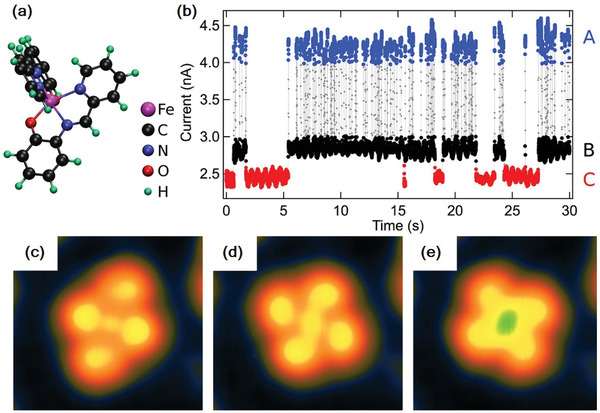
a) Structure of [Fe(pap)2]. b) Tunneling current variation with time of [Fe(pap)2] recorded at a sample voltage of 2.5 V, the three different values reflecting the three spin states: the highest (blue) has the structure (c) and is identified as the HS state; the two lower values (black and red) have the structures (d) and (e), respectively, and are identified as the two LS states. The STM is operated at ≈4 K. Reproduced with permission.[ 23 ] Copyright 2017, American Chemical Society.
2.2. Semimetals
2.2.1. [FeII(H2B(pz)2)2)phen] (2) and [FeII(H2B(pz)2)2)bipy] (3) and Their Derivatives
Highly Oriented Pyrolytic Graphite (HOPG)
Both SCMs 2 and 3 undergo complete spin switching with temperature and light on a HOPG surface. Using these complexes as a testbench, spin switching processes have been investigated in detail to shed light on the distinctive features with respect to the bulk materials, such as the evolution of cooperative effects with thickness, or the effect of ligand modification on the spin switching at submonolayer coverages. Figure 6a shows the chemical structures of SCM 2 and 3 and their methyl‐derivatives all of which have been investigated on the said surface. The spin state has been probed by the distinctive XAS spectral patterns observed for the Fe L2,3‐ (or just the Fe L3‐) edge for the HS and LS states. As an example, Figure 6b shows the Fe L2,3‐edge XA spectral patterns of about 0.4 ML of SCM 2 on HOPG, i.e., the HS spectrum at 300 K (red) and the LS spectrum at 6 K (blue), which is converted to the HS spectrum upon illumination with green light (green). Spin‐state compositions at high‐spin fractions between 1 and 0 can be derived from fitting with a linear combination of the reference HS and LS spectra. The temperature‐induced SCO of 0.4 ML of SCM 2 on HOPG is found to be rather gradual—unlike the bulk material, which exhibits abrupt spin switching with hysteresis. The spin transition without cooperativity can be described by a simple thermodynamical model (van't Hoff's model):
| (1) |
where γHS is the HS fraction; ΔH and ΔS are the enthalpy and entropy differences between the HS and the LS states, respectively, and R is the universal gas constant. The model yields (Figure 7c, black curve) ΔH = 7.2(5) kJ mol−1 and ΔS = 45(3) J K−1 mol−1, similar to the values of the bulk material, which is to be expected as these are single‐molecule properties. Upon illumination with green light of 520 nm wavelength and a flux density of φ = 4.2(8) × 1014 photons mm−2 at 6 K, the molecules are switched from the LS to the HS state by LIESST, while back relaxation to the LS ground state takes place on raising the temperature to 65 K (Figure 6c, green curve). The spin relaxation occurs through quantum tunneling mediated by thermally activated vibrational levels (non‐adiabatic multiphonon relaxation).[ 11 ] The effective energy barrier between the LS and the HS states is estimated to be lower than that of the bulk material by a factor of 2.6, indicating a destabilization of the metastable HS state on the HOPG surface. The light‐induced LS‐to‐HS switching is found to be highly efficient with a time constant of τ = 20.4(7) s.[ 55 ]
Figure 6.
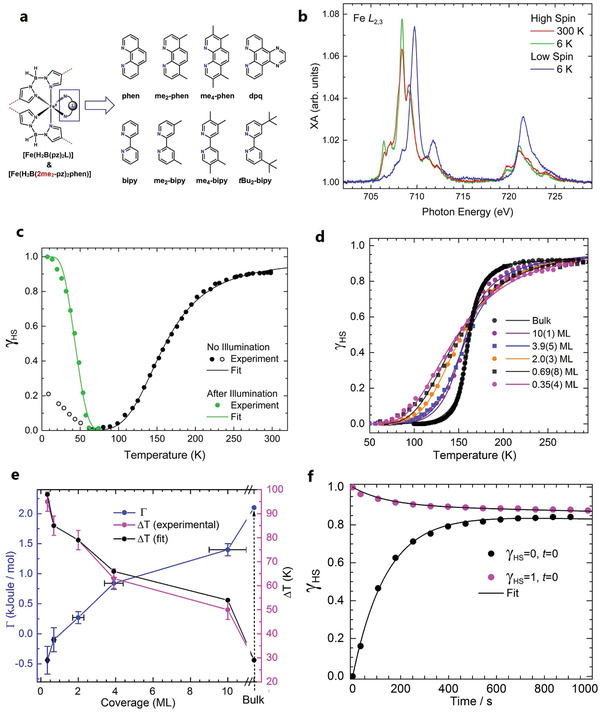
a) Sketch of the SCMs 2 (phen) and 3 (bipy) and derivatives investigated on HOPG. b) Fe L2,3‐edge spectra of 0.4 ML of the SCM 2 on HOPG recorded at 300 K (red), at 6 K before and after illumination with a green light (blue and green, respectively). c) Temperature‐dependent SCO of (b) (black curve), and the relaxation of the light‐induced HS state at 6 K to the ground LS state at low temperatures (green curve). d) Thickness‐dependent SCO of the SCM 3 on HOPG ranging from 0.35(4)‐ML to 10(1)‐ML (and the bulk), the steepness of the curves indicating the magnitude of cooperative effects. e) Variations of Γ (blue) and ΔT (red) as a function of thickness obtained from fitting (d) with the mean‐field Slichter–Drickamer model. f) X‐ray‐induced spin switching (SOXIESST) of 0.8 ML of the SCM 3 on HOPG, recorded at 5 K. a) Reproduced under the terms of the CC‐BY Creative Commons Attribution 3.0 Unported license (https://creativecommons.org/licenses/by/3.0/).[ 25 ] Copyright 2019, IOP Publishing Ltd. b,c) Reproduced with permission.[ 55 ] Copyright 2015, American Chemical Society. Further permission related to the material excerpted is directed to the ACS. d,e) Reproduced under the terms of CC‐BY Creative Commons Attribution 4.0 International license (https://creativecommons.org/licenses/by/4.0/).[ 57 ] Copyright 2018, The Authors, published by Springer Nature. f) Reproduced with permission.[ 62 ] Copyright 2017, IOP Publishing.
Figure 7.
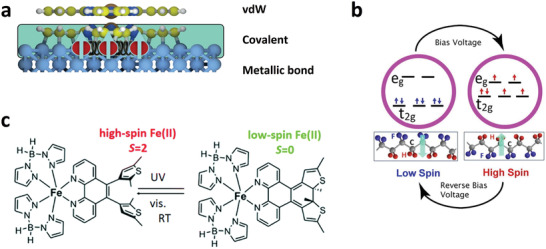
a) The different modes of interactions across a spinterface, with a strong molecule–substrate interaction for molecules at the first layer (shaded area) and weak coupling for molecules at the second layer. b) Molecular magneto‐electric effects of an SCM deposited on a ferroelectric substrate, with the spin state controlled by voltage. c) Combining a photochromic molecule with an SCM, where room‐temperature spin‐state switching is achieved. a) Adapted with permission.[ 4 ] Copyright 2017, Springer Nature. b) Adapted with permission.[ 67 ] Copyright 2019, American Institute of Physics. c) Adapted with permission.[ 78 ] Copyright 2018, The Royal Society of Chemistry.
In the case of SCM 3 on HOPG, a clear build‐up of cooperativity is observed upon increasing the coverage from 0.35 to 10 ML, which is evident from the increasing steepness of the spin‐transition curve (Figure 6d). The evolution of cooperativity with thickness can be described using the Slichter–Drickamer (SD) model:[ 56 ]
| (2) |
where Γ is the interaction parameter, describing the interaction strength between neighboring molecules. ΔS and ΔH values vary minimally across the coverage and are similar to the bulk values, meaning the transition temperature T 1/2 also hovers around the bulk value of 159.5 K for all coverages. What varies widely is the interaction parameter Γ: for the 0.35 ML sample, Γ = −0.44 kJ mol−1; at 0.7 ML, Γ = −0.1 kJ mol−1; and upon further increase of the thickness to 10 ML, Γ monotonically increases to 1.4 kJ mol−1, which is about 67% of the bulk value (Figure 7e). The negative value of Γ at the submonolayers may be interpreted as resulting from anticooperativity between nearest neighbors, favoring “unlike‐spin” pairings (LS–HS). This might be due to a substrate‐induced distribution in energy barriers between the two spin states or to the nature of the molecular distribution on the surface (clustering). On the other hand, the positive values of Γ observed for the higher coverages—starting from the bilayer sample—are an indication that the system favors “like‐spin” pairings of the nearest neighbors (LS–LS or HS–HS) during the spin‐transition process. It is worth noting that Γ and the transition width ΔT—defined as temperature difference at which 80% of the molecules are in the HS and LS states, respectively—have an inverse one‐to‐one correspondence (Figure 6e), meaning that either of the two can be used as a yardstick to measure the degree of cooperativity. The light‐induced LS‐to‐HS switching at 5 K of SCM 3 yields efficiency and effective cross‐section similar to that of SCM 2 and (not surprisingly) is independent of the thickness, as LIESST is a single‐molecule phenomenon. On the other hand, the light‐induced HS state is comparatively unstable at low temperatures (10 to 40 K), and exhibits a stretched‐exponential relaxation behavior—a difference to the bulk behavior, where any appreciable relaxation occurs above 40 K and is sigmoidal.[ 57 ]
In the bulk material, ligand modification can profoundly affect the nature of SCO.[ 58 ] To investigate the effect at mono‐ or submonolayer coverage on HOPG, SCM 2 and 3 have been functionalized with methyl groups. The new compounds shown in Figure 6a—[Fe(H2B(pz)2)2(me2‐phen)], [Fe(H2B(pz)2)(me4‐phen)], [Fe(H2B(4‐me2‐pz)2(phen)], [Fe(H2B(pz)2)2(me2‐bipy)], [Fe(H2B(pz)2)2(me4‐bipy)], [Fe(bpz)2(tbu2‐bipy)], [Fe(bpz)2(dpq)]—are all vacuum evaporable. For the thin films deposited on quartz or glass substrates, all of them exhibit a complete HS‐to‐LS conversion upon going from 300 to about 40 K, with LIESST effects also observed at 5 K upon illumination with green light (519 nm).[ 25 , 58 , 59 ] For submonolayer coverages on HOPG (≤ 0.6 ML) the compounds can be grouped based on two contrasting SCO behaviors. For the samples of [Fe(H2B(pz)2)2(me2‐phen)], [Fe(H2B(4‐me2‐pz)2(phen)], [Fe(H2B(pz)2)2(me2‐bipy)], and [Fe(bpz)2(tbu2‐bipy)], only about 30–40% of the molecules respond to light or temperature, with the rest being locked in the HS state. Samples of [Fe(H2B(pz)2)(me4‐phen)], [Fe(H2B(pz)2)2(me4‐bipy)], and [Fe(bpz)2(dpq)], on the other hand, showed no SCO behavior—the compounds are all trapped in the HS state at temperatures ranging from 300 to 10 K.
Nitrogen K‐edge spectra showed that both the parent molecules 2 and 3 and the HS‐locked [Fe(H2B(pz)2)2(me4‐phen)] are preferentially oriented on the surface in a manner similar to the orientation of a double layer of 2 on Au(111).[ 36 ] The partially switched [Fe(H2B(pz)2)2(me2‐phen)], on the other hand, showed a random orientation on the surface. The role of the surface in inhibiting the spin switching is confirmed by the fact that for [Fe(H2B(pz)2)2(me2‐bipy)] molecules lying at the second layer, 70% exhibit SCO whereas this applies to only 40% of the molecules in contact with the HOPG surface.[ 25 ] The inhibition of SCO may be attributed to CH–π interactions between the methyl groups and HOPG.[ 60 ]
One of the challenges in using X‐rays to investigate SCMs is the LS‐to‐HS spin switching induced by them at low temperatures, the so‐called soft‐X‐ray‐induced excited spin‐state trapping (SOXIESST).[ 61 ] This was investigated on 0.8 ML of the SCM 3 on HOPG at 5 K. At a photon flux of I = 1 × 1011 s−1 mm−2, LS‐to‐HS switching is saturated on reaching 60% HS state, implying that X‐rays switch the spin state both ways (SOXIESST: LS‐to‐HS; reverse‐SOXIESST: HS‐to‐LS). In fact, HS‐to‐LS switching is more dominant when starting from the light‐induced HS state, shown in Figure 6f. Additionally, about 5% of the molecules undergo soft X‐ray‐induced photochemistry, whereby the molecules—as a result of direct interaction with X‐rays—are degraded to a trapped LS state. The rate of the X‐ray‐induced LS‐to‐HS switching is highly dependent on the intensity of the X‐ray beam: by reducing the photon flux by about an order of magnitude, the rate of LS‐to‐HS switching is also reduced roughly by an order of magnitude, while that of HS‐to‐LS switching exhibits no significant change. SOXIESST is rationalized as being due to X‐ray‐induced secondary electrons.[ 62 ]
2.2.2. Bismuth
SCM 2 and its derivative [Fe(H2B(pz)2)(me4‐phen)] were investigated on a Bi(111) surface.[ 59 ] This study was motivated by the fact that bismuth has a low density of states at the Fermi level and is known to exert low van der Waals forces on the deposited aromatic compounds.[ 63 ] Yet, both molecules exhibit only a partial spin transition—with about 50% responding to light or temperature, at 0.3 ML coverage in both cases—and the rest being locked in the HS state. The N K‐edge XA spectra of the two molecules showed a similar pattern with three distinct peaks, analogous to those observed for both molecules on HOPG,[ 25 ] indicating the integrity of the molecules on the surface.[ 59 ]
2.2.3. Others
[FeII(NCS)2L] (L: 1‐N,N‐dimethylmethanamine) was the first SCO molecule ever retaining its spin switching behavior in direct contract with a solid surface. For a submonolayer coverage of this molecule deposited on HOPG, a complete thermal spin transition—in the temperature range from 300 to 75 K—is observed, in a manner similar to that of SCMs 2 and 3 on the same surface.[ 64 ] Recently, a new vacuum‐evaporable SCM [Fe(pypyr(CF3)2)2(phen)] (pypyr = 2‐(2′‐pyridyl)pyrrolide, phen = 1,10‐phenanthroline) was also investigated on HOPG. For 0.4 ML coverage, the molecule exhibits complete light‐induced LS‐to‐HS switching at 10 K, which then relaxes back to the LS state when heating the sample to 100 K.[ 65 ] The SCM [Fe(pypyr(CF3)2)2(phen)] was also investigated on the semimetal surfaces WSe2 and HfS2. For a 0.4 ML coverage on both surfaces, about 80% of the molecules exhibit light‐induced LS‐to‐HS switching at 10 K, which then relaxes back to the ground LS state on raising the sample temperature to about 100 K. [ 65 ]
2.3. Ferroelectric and Dielectric Surfaces
Integrating SCMs into dielectrics and ferroelectrics could yield magneto‐electric effects, with the possibility of voltage control of the magnetic state that could find application in conventional spintronic devices. To our knowledge, studies towards this goal have only been reported for thin films of SCM 3 deposited on some organic ferroelectrics and dielectric substrates. In a 25 ML thick film of the SCM 3 deposited on organic ferroelectric copolymer of 70% vinylidene difluoride and 30% trifluoroethylene (PVDF‐TrFE), the magnetic state can be controlled by varying the polarization direction of the underlying ferroelectric substrate: at 100 K, with the interface dipoles directed towards the SCM, the molecule is pinned in the (paramagnetic) HS state, while on reversing the dipoles’ direction, the molecule is pinned in the (diamagnetic) LS state.[ 66 ] Similar isothermal change in the spin state induced by electric dipoles is also observed at room temperature for thin films of the same molecule deposited on the organic ferroelectric croconic acid (C5H2O5) and poly(vinylidene fluoride) hexafluoropropylene (PVDF‐HFP).[ 67 ] The authors attribute this to magnetoelectric coupling between the molecular layer and the underlying ferroelectric substrate.
Dielectric substrates are also reported to pin the SCM 3 to the LS state. In a study of 5 nm and 20 nm thin films on SiO2 and Al2O3, respectively, the complex is pinned to the LS state at up to 345 K. [ 68 ] Interestingly, LS‐to‐HS switching is observed at room temperature on exposure to X‐rays—stable for up to an hour—reminiscent of the SOXIESST phenomenon observed at low temperatures of SCMs in general. The spin state can be reversed to the LS state upon raising the temperature by about 10 K. The X‐ray‐induced LS‐to‐HS switching rate is highly dependent on temperature—complete HS transition obtained within 13 min exposure at 200 K, 4–5 min at 300 K and 3 min at 345 K. In trying to elucidate the X‐ray‐induced spin transition, the authors identify charge rearrangement and conformational changes to be the likely mechanisms.[ 68 ] It is worth mentioning that the SOXIESST phenomenon is caused by X‐ray‐induced secondary electrons,[ 62 , 69 ] and the cross‐section—investigated in thin films of the SCM 3 deposited on a ferroelectric substrate—can be increased by more than an order of magnitude if the interface is poled positive (interface dipoles pointing toward the molecules), as compared to if the interface is poled in the reverse, underlying the role of interface charge states.[ 36 , 70 ]
2.4. Magnetic Surfaces
Investigating SCMs on 3d ferromagnets is an area of huge interest because of its potential to engineer “active” spinterfaces (see above). However, such systems have rarely been investigated because of the fragility of SCMs and the high reactivity of ferromagnets. There exists only one such report—the investigation of SCM 1 on a bilayer of Co islands on Cu (111). Most of the molecules are reported to undergo fragmentation on Co/Cu. Of the few intact molecules in the HS state, the spin‐polarized STM measurement showed the presence of spin‐polarized density of states close to the Fermi level.[ 71 ] Calculations revealed the spin‐polarization at the Fermi level to be −84%, which is much higher than that of the substrate. Theoretical calculations also showed that, irrespective of the spin state, the molecule is ferromagnetically coupled to Co atoms via indirect superexchange interactions. The ferromagnetic coupling is calculated to persist even when introducing several Cu spacer‐layers (Cu n /Co/Cu).[ 72 ]
3. Conclusions and Future Prospects
The investigation of SCMs on surfaces is a young field, and as such, little is understood on the behavior of SCMs on surfaces (gold and HOPG being notable exceptions). It is therefore important to create a portfolio of SCMs and inorganic/organic substrates to explore the fundamental nature of interface interactions. This can also lead to the discovery of hitherto unknown functionalities, which has proven to be the case in the field of molecular spintronics research. On the molecular side, the lack of vacuum evaporable SCMs and their tendency to lose the spin switching behavior upon contact with surfaces are the main stumbling blocks for progress in the field. Moreover, it is important to look at radically new concepts like how metallic surface properties such as electric and magnetic dipoles can be used to engineer SCO. Recent theoretical work based on the experimental investigation of an FeII‐based organometallic compound on gold has shown that SCO can indeed be engineered on gold by using suitable ligands with the electric polarization of the surface driving the spin switching process.[ 73 ] It is based on the premise that electrostatic interactions play an important role in governing SCO phenomena and may give rise to cooperative effects—a role mainly attributed to elastic forces in bulk materials. The success of SCM 4 in retaining SCO and LIESST even on metallic surfaces like Ag and Cu underlines the importance of engineering new, chemically robust compounds. This can be achieved, for example, by replacing bidentate ligands like dihydrobis(pyrazolyl)borate (bpz) or simple diimines like phen and bipy with tridentate ligands like hydrotris(pyrazolyl)borate (tpz). Finally, it is highly desirable from memory applications point of view to engineer SCMs on surfaces that exhibit large cooperative effects. The general trend so far has been a reduced cooperativity for thin films with no hysteresis. There have been attempts to enhance molecular cooperativity in thin films by modifying the ligands, such as by adding long alkyl chains (C12) to 3. The modified molecule, remarkably, self‐organized as lamellar‐bilayer structures both in the bulk and sublimed films, but showed no noticeable enhancement in the cooperativity as compared to the parent molecule.[ 74 ] There are certain pointers to achieving cooperativity in thin films through a suitable choice of substrates: films with thickness of hundreds of nm of the SCM [Fe(H2B(pz)2)2bipy] deposited on the dielectric Al2O3 is reported to exhibit thermal hysteresis, although no such effect is observed in the bulk material.[ 75 ]
The other challenge is to couple SCMs with ferromagnetic surfaces in order to engineer active spinterfaces, as already mentioned in the introduction. The radical transformations in magnetic properties observed in spinterfaces such as spin inversion and spin filtering are attributed to strong hybridization between 3d ferromagnets and π‐conjugated molecules forming hybrid interface states (HISs).[ 3 , 4 ] By incorporating SCMs into the mix, the interface states can be controlled by external inputs via changing the spin states of the SCM. The idea holds great potential for radically new device concepts. While optical control promises dissipationless and ultrafast coherent manipulation of HISs, voltage control could find direct application in conventional spintronic devices like spin valves and tunnel junctions.[ 4 ] On the other hand, SCMs and the SCO property are unlikely to survive any strong interaction with 3d ferromagnets due to the stringent demands placed for the spin switching to occur, such as in maintaining the octahedral symmetry of the molecule. There are ways to combine SCMs with 3d ferromagnets, such as by introducing a decoupling layer between them (depicted in Figure 7a). The decoupling layer can be a 2D material like graphene or any other well investigated paramagnetic or π‐conjugated organic molecules that already exhibit the properties associated with a spinterface. Another interesting platform to investigate SCMs is with layered 2D ferromagnetic, ferroelectric, and multiferroic materials.[ 76 ] While van der Waals forces of the 2D ferromagnetic or ferroelectric materials are unlikely to affect the SCO property, magnetic or electric proximity effects may result in HISs that can be controlled by electrical or optical means via varying the spin states of the SCMs. In this respect, a recent comprehensive experimental investigation is worth noting that shows that the generation of spin‐polarized HISs is a very general phenomenon that does not require the presence of π‐electrons in the isolated molecules or a fully occupied majority d band in the 3d ferromagnet (strong ferromagnet). This would imply that engineering spinterfaces does not require strong interactions.[ 77 ]
While light‐ and voltage‐induced reversible spin switching in SCMs are generally observed at low temperatures, room temperature (RT) spin switching is necessary for applications. Recent reports have shown this as achievable by two approaches—by pairing a photochromic molecule with an SCM, or by using an organic ferroelectric substrate. In a 5 nm (6–8 ML) film of [Fe(H2B(pz)2)2phen*] (phen* = diarylethene‐functionalized phenanthroline ligand) (Figure 7c) deposited on Au(111), light (UV)‐induced HS‐to‐LS spin switching at RT was reported—the photoswitching at the molecular level driven by the photocyclization of the diarylethene ligand, which triggers SCO of the FeII‐ion remotely. The spin switching, however, is observed in only about 5(1)% of the molecules, but nevertheless, it is a demonstration of photochromic‐ligand driven RT SCO.[ 78 ] The other approach at obtaining RT SCO is reported in the case of thin films of 3 deposited on organic ferroelectrics croconic acid and PVDF‐HFP, where a nonvolatile voltage‐induced reversible spin switching is observed (Figure 7b). The voltage‐induced spin switching is enabled by magneto‐electric coupling of the SCM and the ferroelectric substrate.[ 79 ] In summary, investigation of SCMs on surfaces is an exciting field in terms of engineering multifunctional devices, but a lot more work is necessary to achieve a fundamental understanding of the involved interactions and deduce principles to design molecular spintronic devices.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
Financial support from DFG through Sfb 658 (project B3), Sfb 677, Sfb/TRR 227 and the BMBF with projects 05K16KE3 and 05K19KEA is gratefully acknowledged. L.K. acknowledges funding from the EU Horizon 2020 research and innovation program under grant agreement no. 766726. The TOC graphic is provided by Holger Naggert.
Note: Equation (1) was corrected on June 17, 2021, after initial publication online.
Open access funding enabled and organized by Projekt DEAL.
Biographies
Lalminthang Kipgen studied physics and materials science at the Indian Institute of Technology Delhi. In 2019, he obtained his Ph.D. under Wolfgang Kuch at Freie Universität Berlin on the topic of exploring the behavior of molecular switches in contact with surfaces, using X‐ray absorption spectroscopy. Currently, he is working as a postdoc at Université de Paris (MPQ Laboratory). His research interest includes molecular switches, molecular magnets, surface physics, and layered ferromagnetic and multiferroic materials.

Matthias Bernien studied physics at the Freie Universität Berlin. He received his Ph.D. in 2009 in the group of Wolfgang Kuch working with X‐ray absorption spectroscopy on adsorbed Fe complexes. As a postdoc he continued his work in the same group focusing on spin‐state switching and conformal switching as well as the magnetic coupling of molecules on surfaces. After receiving his habilitation at Freie Universität Berlin in 2017, he switched topics and now works in the field of vacuum metrology at the Physikalisch‐Technische Bundesanstalt.

Felix Tuczek studied chemistry at Johannes‐Gutenberg University Mainz and received his Ph.D. in 1989 in the group of Philipp Gütlich for work in Mössbauer emission spectroscopy. After 2 years as a postdoc at Stanford University in the group of Prof. Ed Solomon he returned to Mainz to work on his habilitation (1997). In 1999 he received a call to Christian Albrechts University Kiel and since then has held the chair of Molecular Inorganic Chemistry in Kiel. His scientific interests include molecular spin switches, spin‐crossover, surface chemistry, and spectroscopy as well as small‐molecule activation (nitrogen fixation, oxygen activation by copper proteins).

Wolfgang Kuch has been a full professor at Freie Universität Berlin (Germany) since 2004. He received his diploma degree in 1989 from the Goethe‐Universität Frankfurt (Germany) and his Ph.D. degree from Universität Stuttgart (Germany) in 1993. Before joining Freie Universität Berlin, he was a staff scientist at the Max‐Planck‐Institut for Microstructure Physics in Halle (Germany). His research interest is on magnetic films, surfaces, and adsorbed molecules, including the dynamic behavior on ultrashort time scales.

Kipgen L., Bernien M., Tuczek F., Kuch W., Spin‐Crossover Molecules on Surfaces: From Isolated Molecules to Ultrathin Films. Adv. Mater. 2021, 33, 2008141. 10.1002/adma.202008141
Contributor Information
Lalminthang Kipgen, Email: kipgen@physik.fu-berlin.de.
Felix Tuczek, Email: ftuczek@ac.uni-kiel.de.
Wolfgang Kuch, Email: kuch@physik.fu-berlin.de.
References
- 1.a) Žutić I., Fabian J., Sarma S. D., Rev. Mod. Phys. 2004, 76, 323; [Google Scholar]; b) Bader S. D., Parkin S. S. P., Annu. Rev. Condens. Matter Phys. 2010, 1, 71; [Google Scholar]; c) Dieny B., Prejbeanu I. L., Garello K., Gambardella P., Freitas P., Lehndorff R., Raberg W., Ebels U., Demokritov S. O., Akerman J., Deac A., Pirro P., Adelmann C., Anane A., Chumak A. V., Hirohata A., Mangin S., Valenzuela S. O., Onbasli M. C., D'Aquino M., Prenat G., Finocchio G., Lopez‐Diaz L., Chantrell R., Chubykalo‐Fesenko O., Bortolotti P., Nat. Electron. 2020, 3, 446. [Google Scholar]
- 2.a) Rocha A. R., Garcia‐Suarez V. M., Bailey S. W., Lambert C. J., Ferrer J., Sanvito S., Nat. Mater. 2005, 4, 335; [DOI] [PubMed] [Google Scholar]; b) Sanvito S., Chem. Soc. Rev. 2011, 40, 3336; [DOI] [PubMed] [Google Scholar]; c) Coronado E., Nat. Rev. Mater. 2020, 5, 87. [Google Scholar]
- 3. Sanvito S., Nat. Phys. 2010, 6, 562. [Google Scholar]
- 4. Cinchetti M., Dediu V. A., Hueso L. E., Nat. Mater. 2017, 16, 507. [DOI] [PubMed] [Google Scholar]
- 5. Dediu V. A., Hueso L. E., Bergenti I., Taliani C., Nat. Mater. 2009, 8, 707. [DOI] [PubMed] [Google Scholar]
- 6. Raman K. V., Kamerbeek A. M., Mukherjee A., Atodiresei N., Sen T. K., Lazic P., Caciuc V., Michel R., Stalke D., Mandal S. K., Blugel S., Munzenberg M., Moodera J. S., Nature 2013, 493, 509. [DOI] [PubMed] [Google Scholar]
- 7. Atodiresei N., Brede J., Lazic P., Caciuc V., Hoffmann G., Wiesendanger R., Blugel S., Phys. Rev. Lett. 2010, 105, 066601. [DOI] [PubMed] [Google Scholar]
- 8.a) Willenbacher N., Spiering H., J. Phys. C: Solid State Phys. 1988, 21, 1423; [Google Scholar]; b) Spiering K. B. H., Linares J., Varret F., Phys. Rev. B 2004, 70, 184106. [Google Scholar]
- 9.a) Kepenekian M., Guennic B. L.e, Robert V., J. Am. Chem. Soc. 2009, 131, 11498; [DOI] [PubMed] [Google Scholar]; b) Kepenekian M., Guennic B. L.e, Robert V., Phys. Rev. B 2009, 79, 094428; [Google Scholar]; c) Banerjee H., Chakraborty S., Saha‐Dasgupta T., Inorganics 2017, 5, 47. [Google Scholar]
- 10. Decurtins S., Gütlich P., Köhler C. P., Spiering H., Hauser A., Chem. Phys. Lett. 1984, 105, 1. [Google Scholar]
- 11. Hauser A., in Spin Crossover in Transition Metal Compounds II (Eds: Gütlich P., Goodwin H. A.), Springer, Berlin, Germany: 2004, p. 155. [Google Scholar]
- 12.a) Gütlich P., Hauser A., Spiering H., Angew. Chem., Int. Ed. Engl. 1994, 33, 2024; [Google Scholar]; b) Gütlich P., Garcia Y., Goodwin H. A., Chem. Soc. Rev. 2000, 29, 419; [Google Scholar]; c) Real J. A., Gaspar A. B., Niel V., Muñoz M. C., Coord. Chem. Rev. 2003, 236, 121. [Google Scholar]
- 13.a) Gütlich P., Goodwin H. A., Spin Crossover in Transition Metal Compounds I–III, Springer, Berlin, Germany: 2004; [Google Scholar]; b) Bousseksou A., Molnar G., Salmon L., Nicolazzi W., Chem. Soc. Rev. 2011, 40, 3313; [DOI] [PubMed] [Google Scholar]; c) Halcrow M. A., Chem. Soc. Rev. 2011, 40, 4119; [DOI] [PubMed] [Google Scholar]; d) Gütlich P., Eur. J. Inorg. Chem. 2013, 2013, 581; [Google Scholar]; e) Murray K. S., Oshio H., Real J. A., Eur. J. Inorg. Chem. 2013, 2013, 577. [Google Scholar]
- 14.a) Molnar G., Rat S., Salmon L., Nicolazzi W., Bousseksou A., Adv. Mater. 2018, 30, 1703862; [DOI] [PubMed] [Google Scholar]; b) Shepherd H. J., Molnár G., Nicolazzi W., Salmon L., Bousseksou A., Eur. J. Inorg. Chem. 2013, 2013, 653. [Google Scholar]
- 15.a) Cavallini M., Phys. Chem. Chem. Phys. 2012, 14, 11867; [DOI] [PubMed] [Google Scholar]; b) Kuch W., Bernien M., J. Phys.: Condens. Matter 2016, 29, 023001. [DOI] [PubMed] [Google Scholar]
- 16. Ruben M., Kumar K. S., Angew. Chem., Int. Ed. 2021, 60, 7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cambi L., Szegö L., Ber. Dtsch. Chem. Ges. 1931, 64, 2591. [Google Scholar]
- 18. Alam M. S., Stocker M., Gieb K., Muller P., Haryono M., Student K., Grohmann A., Angew. Chem., Int. Ed. 2010, 49, 1159. [DOI] [PubMed] [Google Scholar]
- 19. Shi S., Schmerber G., Arabski J., Beaufrand J. B., Kim D. J., Boukari S., Bowen M., Kemp N. T., Viart N., Rogez G., Beaurepaire E., Aubriet H., Petersen J., Becker C., Ruch D., Appl. Phys. Lett. 2009, 95, 043303. [Google Scholar]
- 20.a) Palamarciuc T., Oberg J. C., El Hallak F., Hirjibehedin C. F., Serri M., Heutz S., Létard J. F., Rosa P., J. Mater. Chem. 2012, 22, 9690; [Google Scholar]; b) Naggert H., Bannwarth A., Chemnitz S., von Hofe T., Quandt E., Tuczek F., Dalton Trans. 2011, 40, 6364. [DOI] [PubMed] [Google Scholar]
- 21. Davesne V., Gruber M., Studniarek M., Doh W. H., Zafeiratos S., Joly L., Sirotti F., Silly M. G., Gaspar A. B., Real J. A., Schmerber G., Bowen M., Weber W., Boukari S., Costa V. D., Arabski J., Wulfhekel W., Beaurepaire E., J. Chem. Phys. 2015, 142, 194702. [DOI] [PubMed] [Google Scholar]
- 22. Jasper‐Tonnies T., Gruber M., Karan S., Jacob H., Tuczek F., Berndt R., J. Phys. Chem. Lett. 2017, 8, 1569. [DOI] [PubMed] [Google Scholar]
- 23. Jasper‐Toennies T., Gruber M., Karan S., Jacob H., Tuczek F., Berndt R., Nano Lett. 2017, 17, 6613. [DOI] [PubMed] [Google Scholar]
- 24.a) Shalabaeva V., Rat S., Manrique‐Juarez M. D., Bas A.‐C., Vendier L., Salmon L., Molnár G., Bousseksou A., J. Mater. Chem. C 2017, 5, 4419; [Google Scholar]; b) Rohlf S., Gruber M., Floser B. M., Grunwald J., Jarausch S., Diekmann F., Kallane M., Jasper‐Toennies T., Buchholz A., Plass W., Berndt R., Tuczek F., Rossnagel K., J. Phys. Chem. Lett. 2018, 9, 1491; [DOI] [PubMed] [Google Scholar]; c) Atzori M., Poggini L., Squillantini L., Cortigiani B., Gonidec M., Bencok P., Sessoli R., Mannini M., J. Mater. Chem. C 2018, 6, 8885; [Google Scholar]; d) Mahfoud T., Molnár G., Cobo S., Salmon L., Thibault C., Vieu C., Demont P., Bousseksou A., Appl. Phys. Lett. 2011, 99, 053307. [Google Scholar]
- 25. Ossinger S., Kipgen L., Naggert H., Bernien M., Britton A. J., Nickel F., Arruda L. M., Kumberg I., Engesser T. A., Golias E., Nather C., Tuczek F., Kuch W., J. Phys. Condens. Matter 2020, 32, 114003. [DOI] [PubMed] [Google Scholar]
- 26. Soyer H., Mingotaud C., Boillot M. L., Delhaes P., Langmuir 1998, 14, 5890. [Google Scholar]
- 27.a) Matsuda M., Tajima H., Chem. Lett. 2007, 36, 700; [Google Scholar]; b) Tissot A., Bardeau J.‐F., Rivière E., Brisset F., Boillot M.‐L., Dalton Trans. 2010, 39, 7806; [DOI] [PubMed] [Google Scholar]; c) Félix G., Abdul‐Kader K., Mahfoud T., Gural'skiy I. A., Nicolazzi W., Salmon L., Molnár G., Bousseksou A., J. Am. Chem. Soc. 2011, 133, 15342; [DOI] [PubMed] [Google Scholar]; d) Quintero C. M., Gural'skiy I. A., Salmon L., Molnár G., Bergaud C., Bousseksou A., J. Mater. Chem. 2012, 22, 3745. [Google Scholar]
- 28.a) Galyametdinov Y., Ksenofontov V., Prosvirin A., Ovchinnikov I., Ivanova G., Gütlich P., Haase W., Angew. Chem., Int. Ed. 2001, 40, 4269; [DOI] [PubMed] [Google Scholar]; b) Hayami S., Gu Z., Yoshiki H., Fujishima A., Sato O., J. Am. Chem. Soc. 2001, 123, 11644; [DOI] [PubMed] [Google Scholar]; c) Kuroiwa K., Shibata T., Sasaki S., Ohba M., Takahara A., Kunitake T., Kimizuka N., J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5192; [Google Scholar]; d) Seredyuk M., Gaspar A. B., Ksenofontov V., Reiman S., Galyametdinov Y., Haase W., Rentschler E., Gütlich P., Chem. Mater. 2006, 18, 2513; [Google Scholar]; e) Cavallini M., Bergenti I., Milita S., Kengne J. C., Gentili D., Ruani G., Salitros I., Meded V., Ruben M., Langmuir 2011, 27, 4076. [DOI] [PubMed] [Google Scholar]
- 29. Molnár G., Cobo S., Real J. A., Carcenac F., Daran E., Vieu C., Bousseksou A., Adv. Mater. 2007, 19, 2163. [Google Scholar]
- 30. Lefter C., Davesne V., Salmon L., Molnár G., Demont P., Rotaru A., Bousseksou A., Magnetochemistry 2016, 2, 18. [Google Scholar]
- 31. Gruber M., Berndt R., Magnetochemistry 2020, 6, 35. [Google Scholar]
- 32. Muscat J. P., Newns D. M., Prog. Surf. Sci. 1978, 9, 1. [Google Scholar]
- 33. Tautz F. S., Prog. Surf. Sci. 2007, 82, 479. [Google Scholar]
- 34. Duhm S., Gerlach A., Salzmann I., Bröker B., Johnson R. L., Schreiber F., Koch N., Org. Electron. 2008, 9, 111. [Google Scholar]
- 35.a) Baldo M. A., Forrest S. R., Phys. Rev. B 2001, 64, 085201; [Google Scholar]; b) Hoffmann R., Rev. Mod. Phys. 1988, 60, 601; [Google Scholar]; c) Neaton J. B., Hybertsen M. S., Louie S. G., Phys. Rev. Lett. 2006, 97, 216405. [DOI] [PubMed] [Google Scholar]
- 36. Gopakumar T. G., Bernien M., Naggert H., Matino F., Hermanns C. F., Bannwarth A., Mühlenberend S., Krüger A., Krüger D., Nickel F., Walter W., Berndt R., Kuch W., Tuczek F., Chem. – Eur. J. 2013, 19, 15702. [DOI] [PubMed] [Google Scholar]
- 37. Miyamachi T., Gruber M., Davesne V., Bowen M., Boukari S., Joly L., Scheurer F., Rogez G., Yamada T. K., Ohresser P., Nat. Commun. 2012, 3, 938. [DOI] [PubMed] [Google Scholar]
- 38. Gruber M., Davesne V., Bowen M., Boukari S., Beaurepaire E., Wulfhekel W., Miyamachi T., Phys. Rev. B 2014, 89. [DOI] [PubMed] [Google Scholar]
- 39. Bellec A., Lagoute J., Repain V., C. R. Chim. 2018, 21, 1287. [Google Scholar]
- 40. Aravena D., Ruiz E., J. Am. Chem. Soc. 2012, 134, 777. [DOI] [PubMed] [Google Scholar]
- 41. Gruber M., Miyamachi T., Davesne V., Bowen M., Boukari S., Wulfhekel W., Alouani M., Beaurepaire E., J. Chem. Phys. 2017, 146, 092312. [DOI] [PubMed] [Google Scholar]
- 42. Bairagi K., Iasco O., Bellec A., Kartsev A., Li D., Lagoute J., Chacon C., Girard Y., Rousset S., Miserque F., Dappe Y. J., Smogunov A., Barreteau C., Boillot M. L., Mallah T., Repain V., Nat. Commun. 2016, 7, 12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gruber M., Davesne V., Bowen M., Boukari S., Beaurepaire E., Wulfhekel W., Miyamachi T., Phys. Rev. B 2014, 89, 195415. [DOI] [PubMed] [Google Scholar]
- 44. Gueddida S., Alouani M., Phys. Rev. B 2013, 87, 144413. [Google Scholar]
- 45. Gopakumar T. G., Matino F., Naggert H., Bannwarth A., Tuczek F., Berndt R., Angew. Chem., Int. Ed. 2012, 51, 6262. [DOI] [PubMed] [Google Scholar]
- 46. Meded V., Bagrets A., Fink K., Chandrasekar R., Ruben M., Evers F., Bernand‐Mantel A., Seldenthuis J. S., Beukman A., van der Zant H. S. J., Phys. Rev. B 2011, 83, 245415. [Google Scholar]
- 47. Ludwig E., Naggert H., Kalläne M., Rohlf S., Kröger E., Bannwarth A., Quer A., Rossnagel K., Kipp L., Tuczek F., Angew. Chem., Int. Ed. 2014, 53, 3019. [DOI] [PubMed] [Google Scholar]
- 48. Pronschinske A., Chen Y., Lewis G. F., Shultz D. A., Calzolari A., Buongiorno Nardelli M., Dougherty D. B., Nano Lett. 2013, 13, 1429. [DOI] [PubMed] [Google Scholar]
- 49. Pronschinske A., Bruce R. C., Lewis G., Chen Y., Calzolari A., Buongiorno‐Nardelli M., Shultz D. A., You W., Dougherty D. B., Chem. Commun. 2013, 49, 10446. [DOI] [PubMed] [Google Scholar]
- 50. Beniwal S., Zhang X., Mu S., Naim A., Rosa P., Chastanet G., Létard J. F., Liu J., Sterbinsky G. E., Arena D. A., Dowben P. A., Enders A., J. Phys.: Condens. Matter 2016, 28, 206002. [DOI] [PubMed] [Google Scholar]
- 51. Warner B., Oberg J. C., Gill T. G., el Hallak F., Hirjibehedin C. F., Serri M., Heutz S., Arrio M. A., Sainctavit P., Mannini M., J. Phys. Chem. Lett. 2013, 4, 1546. [DOI] [PubMed] [Google Scholar]
- 52. Kuch W., Bernien M., J. Phys. Condens. Matter 2017, 29, 023001. [DOI] [PubMed] [Google Scholar]
- 53. Fourmental C., Mondal S., Banerjee R., Bellec A., Garreau Y., Coati A., Chacon C., Girard Y., Lagoute J., Rousset S., Boillot M. L., Mallah T., Enachescu C., Barreteau C., Dappe Y. J., Smogunov A., Narasimhan S., Repain V., J. Phys. Chem. Lett. 2019, 10, 4103. [DOI] [PubMed] [Google Scholar]
- 54. Zhang L., Tong Y., Kelai M., Bellec A., Lagoute J., Chacon C., Girard Y., Rousset S., Boillot M. L., Riviere E., Mallah T., Otero E., Arrio M. A., Sainctavit P., Repain V., Angew. Chem., Int. Ed. 2020, 59, 13341. [DOI] [PubMed] [Google Scholar]
- 55. Bernien M., Naggert H., Arruda L. M., Kipgen L., Nickel F., Miguel J., Hermanns C. F., Kruger A., Kruger D., Schierle E., Weschke E., Tuczek F., Kuch W., ACS Nano 2015, 9, 8960. [DOI] [PubMed] [Google Scholar]
- 56. Slichter C. P., Drickamer H. G., J. Chem. Phys. 1972, 56, 2142. [Google Scholar]
- 57. Kipgen L., Bernien M., Ossinger S., Nickel F., Britton A. J., Arruda L. M., Naggert H., Luo C., Lotze C., Ryll H., Radu F., Schierle E., Weschke E., Tuczek F., Kuch W., Nat. Commun. 2018, 9, 2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Naggert H., Rudnik J., Kipgen L., Bernien M., Nickel F., Arruda L. M., Kuch W., Näther C., Tuczek F., J. Mater. Chem. C 2015, 3, 7870. [Google Scholar]
- 59. Ossinger S., Naggert H., Kipgen L., Jasper‐Toennies T., Rai A., Rudnik J., Nickel F., Arruda L. M., Bernien M., Kuch W., Berndt R., Tuczek F., J. Phys. Chem. C 2017, 121, 1210. [Google Scholar]
- 60. Karthikeyan S., Ramanathan V., Mishra B. K., J. Phys. Chem. A 2013, 117, 6687. [DOI] [PubMed] [Google Scholar]
- 61. Collison D., Garner C. D., McGrath C. M., Mosselmans J. F. W., Roper M. D., Seddon J. M. W., Sinn E., Young N. A., J. Chem. Soc., Dalton Trans. 1997, 22, 4371. [Google Scholar]
- 62. Kipgen L., Bernien M., Nickel F., Naggert H., Britton A. J., Arruda L. M., Schierle E., Weschke E., Tuczek F., Kuch W., J. Phys. Condens. Matter 2017, 29, 394003. [DOI] [PubMed] [Google Scholar]
- 63. Schulze G., Franke K. J., Pascual J. I., Phys. Rev. Lett. 2012, 109, 026102. [DOI] [PubMed] [Google Scholar]
- 64. Bernien M., Wiedemann D., Hermanns C. F., Krüger A., Rolf D., Kroener W., Müller P., Grohmann A., Kuch W., J. Phys. Chem. Lett. 2012, 3, 3431. [DOI] [PubMed] [Google Scholar]
- 65. Rohlf S., Grunwald J., Jasper‐Toennies T., Johannsen S., Diekmann F., Studniarek M., Berndt R., Tuczek F., Rossnagel K., Gruber M., J. Phys. Chem. C 2019, 123, 17774. [Google Scholar]
- 66. Zhang X., Palamarciuc T., Letard J.‐F., Rosa P., Lozada E. V., Torres F., Rosa L. G., Doudin B., Dowben P. A., Chem. Commun. 2014, 50, 2255. [DOI] [PubMed] [Google Scholar]
- 67. Hao G., Mosey A., Jiang X., Yost A. J., Sapkota K. R., Wang G. T., Zhang X., Zhang J., N'Diaye A. T., Cheng R., Xu X., Dowben P. A., Appl. Phys. Lett. 2019, 114, 032901. [Google Scholar]
- 68. Zhang X., Costa P. S., Hooper J., Miller D. P., N'Diaye A. T., Beniwal S., Jiang X., Yin Y., Rosa P., Routaboul L., Gonidec M., Poggini L., Braunstein P., Doudin B., Xu X., Enders A., Zurek E., Dowben P. A., Adv. Mater. 2017, 29, 1702257. [DOI] [PubMed] [Google Scholar]
- 69. Vanko G., Renz F., Molnar G., Neisius T., Karpati S., Angew. Chem., Int. Ed. 2007, 46, 5306. [DOI] [PubMed] [Google Scholar]
- 70. Wäckerlin C., Donati F., Singha A., Baltic R., Decurtins S., Liu S.‐X., Rusponi S., Dreiser J., J. Phys. Chem. C 2018, 122, 8202. [Google Scholar]
- 71. Gueddida S., Gruber M., Miyamachi T., Beaurepaire E., Wulfhekel W., Alouani M., J. Phys. Chem. Lett. 2016, 7, 900. [DOI] [PubMed] [Google Scholar]
- 72. Gueddida S., Alouani M., Phys. Rev. B 2016, 93, 184433. [Google Scholar]
- 73. Groizard T., Papior N., Guennic B. L.e, Robert V., Kepenekian M., J. Phys. Chem. Lett. 2017, 8, 3415. [DOI] [PubMed] [Google Scholar]
- 74. Kumar K. S., Studniarek M., Heinrich B., Arabski J., Schmerber G., Bowen M., Boukari S., Beaurepaire E., Dreiser J., Ruben M., Adv. Mater. 2018, 30, 1705416. [DOI] [PubMed] [Google Scholar]
- 75. Jiang X., Hao G., Wang X., Mosey A., Zhang X., Yu L., Yost A. J., Zhang X., DiChiara A. D., N'Diaye A. T., Cheng X., Zhang J., Cheng R., Xu X., Dowben P. A., J. Phys. Condens. Matter. 2019, 31, 315401. [DOI] [PubMed] [Google Scholar]
- 76.a) Gibertini M., Koperski M., Morpurgo A. F., Novoselov K. S., Nat. Nanotechnol. 2019, 14, 408; [DOI] [PubMed] [Google Scholar]; b) Mak K. F., Shan J., Ralph D. C., Nat. Rev. Phys. 2019, 1, 646; [Google Scholar]; c) Cui C., Xue F., Hu W.‐J., Li L.‐J., npj 2D Mater. Appl. 2018, 2, 18; [Google Scholar]; d) Hu T., Kan E. J., WIREs Comput. Mol. Sci. 2019, 9, e1409. [Google Scholar]
- 77. Djeghloul F., Gruber M., Urbain E., Xenioti D., Joly L., Boukari S., Arabski J., Bulou H., Scheurer F., Bertran F., Fevre P. L.e, Taleb‐Ibrahimi A., Wulfhekel W., Garreau G., Hajjar‐Garreau S., Wetzel P., Alouani M., Beaurepaire E., Bowen M., Weber W., J. Phys. Chem. Lett. 2016, 7, 2310. [DOI] [PubMed] [Google Scholar]
- 78. Poggini L., Milek M., Londi G., Naim A., Poneti G., Squillantini L., Magnani A., Totti F., Rosa P., Khusniyarov M. M., Mannini M., Mater. Horiz. 2018, 5, 506. [Google Scholar]
- 79. Poggini L., Londi G., Milek M., Naim A., Lanzilotto V., Cortigiani B., Bondino F., Magnano E., Otero E., Sainctavit P., Arrio M. A., Juhin A., Marchivie M., Khusniyarov M. M., Totti F., Rosa P., Mannini M., Nanoscale 2019, 11, 20006. [DOI] [PubMed] [Google Scholar]