

Abstract
Extracellular vesicles (EV) are an emerging technology as immune therapeutics and drug delivery vehicles. However, EVs are usually stored at −80 °C which limits potential clinical applicability. Freeze‐drying of EVs striving for long‐term stable formulations is therefore studied. The most appropriate formulation parameters are identified in freeze‐thawing studies with two different EV types. After a freeze‐drying feasibility study, four lyophilized EV formulations are tested for storage stability for up to 6 months. Freeze‐thawing studies revealed improved colloidal EV stability in presence of sucrose or potassium phosphate buffer instead of sodium phosphate buffer or phosphate‐buffered saline. Less aggregation and/or vesicle fusion occurred at neutral pH compared to slightly acidic or alkaline pH. EVs colloidal stability can be most effectively preserved by addition of low amounts of poloxamer 188. Polyvinyl pyrrolidone failed to preserve EVs upon freeze‐drying. Particle size and concentration of EVs are retained over 6 months at 40 °C in lyophilizates containing 10 mm K‐ or Na‐phosphate buffer, 0.02% poloxamer 188, and 5% sucrose. The biological activity of associated beta‐glucuronidase is maintained for 1 month, but decreased after 6 months. Here optimized parameters for lyophilization of EVs that contribute to generate long‐term stable EV formulations are presented.
Keywords: extracellular vesicles, freeze‐drying, freeze‐thawing, long‐term stability, outer membrane vesicles, particle preservation, stability testing
Extracellular vesicles (EVs) are lyophilized to improve long‐term stability. Therefore, important formulation principles are evaluated with respect to colloidal stability and biological activity of associated beta‐glucuronidase. In vitro studies confirmed EV stability for one month while particle size and concentration can be preserved for six months.
1. Introduction
Lyophilization is a commonly used method to achieve stable biopharmaceutical products.[ 1 ] Substantial literature is available on freeze‐drying of protein biopharmaceuticals, whereas knowledge about lyophilization of nanoparticulate biopharmaceuticals like vaccines, viruses, or polyplexes is limited.[ 2 ] Due to their different particle properties, nanoparticulate systems require different colloidal and chemical stabilization mechanisms which increases lyophilization complexity.
Extracellular vesicles (EVs) are nanoparticles produced by cells from all branches of the phylogenetic tree. They are surrounded by a lipid‐membrane that contains transmembrane proteins. In their lumen, EVs can contain a plethora of biomolecules, such as proteins, RNA and DNA.[ 3 ] Depending on the producing species, the mechanism of their assembly and the composition of their membrane differs. Mammalian cells produce two main variants of EVs, exosomes derived from multivesicular bodies and microvesicles produced directly by blebbing from the cell‐surface.[ 4 ] EVs derived from gram‐negative bacteria, the so‐called outer membrane vesicles (OMVs), are produced by blebbing from the bacterial outer membrane.[ 5 ] EVs can successfully deliver functional cargo for intercellular communication.[ 6 ] This cargo is encapsulated in EVs and can be composed of proteins and nucleic acids. EVs are therefore intensely investigated as therapeutics,[ 7 , 8 ] drug‐delivery vehicles,[ 9 ] and biomarkers for various diseases.[ 10 ] However, to be viable alternatives to established treatment‐options and to allow for their broad use in clinical settings, many hurdles still need to be overcome.[ 11 ] Besides the reproducibility of their production and purification, storage stability is a big challenge.[ 12 ] EVs may be relatively stable in liquid state for a few weeks at room temperature;[ 13 ] still, clinical use would require extended shelf life. Since physical and biological stability is typically rather limited to a shorter time period, the International Society of EVs recommends storage at −80 °C in phosphate‐buffered saline (PBS).[ 14 ] However, this storage condition is unfavorable in terms of energy consumption, transportation, and most importantly clinical application. In general, freezing‐thawing (FT) is considered to destabilize EVs, for example, by changing EV morphology, function, particle size, and concentration.[ 15 ] EV stability differs by vesicle source and potentially the preparation method.[ 16 ] Pieters et al. demonstrated that milk‐derived EVs are highly stable upon FT while the number of macrophage‐derived vesicles was significantly reduced.[ 17 ]
Freeze‐drying of EVs could accelerate research and offers long‐term stabilization which is an important step towards their application as therapeutics. Moreover, lyophilizates offer new options for administration routes, for example, pulmonary delivery. Nevertheless, lyophilization increases stress during freezing and drying, which can result in EV damage unless appropriate stabilizers are added.[ 18 ] Freezing stress includes mechanical damage due to crystal formation of ice or excipient, exposure to iceliquid interfaces,[ 19 ] pH shifts due to partial buffer salt precipitation,[ 20 , 21 ] and cryoconcentration of the vesicles as well as all solutes, leading to a particle rich phase with increased ionic strength.[ 18 , 19 , 22 ] During drying, the dehydration of the EVs affects their stability. Damage on lyophilized vesicles may also result upon rehydration, for example, with swelling of the amphiphilic molecules forming the vesicle bilayer or osmotic effects.
Frank et al. investigated the stability of different types of EVs during lyophilization.[ 23 ] Particle numbers of lyophilized EVs decreased compared to EVs stored at 4 or −80 °C indicating particle loss or aggregation. They also found a cell type specific freeze‐drying behavior. When comparing different cryoprotective agents, trehalose was found to be superior to mannitol and polyethylene glycol 400. In earlier studies, it was already shown that trehalose is able to protect EVs from freeze‐thawing stress.[ 24 ] Charoenviriyakul et al. also examined the impact of trehalose on aggregation and the biological activity of lyophilized exosomes.[ 25 ] Lyophilization with 50 mm trehalose had no impact on biological activity and polydispersity compared to samples stored at −80 °C. A possible damage already taking place during freezing to −80 °C was not considered. Although lyophilization of EVs seems to be feasible, there is no comprehensive study on their long‐term stability.
Most EV formulations are based on PBS which is known to be critical upon freezing and lyophilization of biopharmaceuticals. During freezing, phosphate buffers cause an acidic pH shift which destabilizes proteins.[ 1 ] This effect might be relevant for surface and/or membrane proteins of EVs. In addition, the pH shift affects the zeta potential and thus the colloidal interactions of EVs.[ 26 ] Furthermore, the high ionic strength in PBS might foster particle aggregation shielding repulsive charge‐based interactions of EVs. To the best of our knowledge, these effects have not been elucidated yet.
The aim of this study was to develop a lyophilized formulation for EVs with long‐term stability of encapsulated cargo up to 6 months at 2–8 °C, including the evaluation of basic formulation components such as buffer agent and cryoprotectant. In order to provide a high number of different formulations, particle characterization was focused on methods using low vesicle concentrations thereby disregarding experiments such as cryo‐EM which would have required increasing vesicle amounts by up to two orders of magnitude.
FT studies were performed to investigate the impact of PBS, various buffers and pH values, and the addition of sucrose and surfactants on EV stability. Here, OMVs derived from SBCy050 myxobacteria and EVs derived from B lymphoblastoid cells (RO cells) were evaluated. RO cells were isolated from the blood of a patient with severe combined immunodeficiency.[ 27 ] As they do not express MHC class II complexes they might be lower in immunogenicity, as this prevents possible MHC‐mismatches.[ 28 ] RO cells can be cultivated under serum‐free conditions which removes the challenges associated with the use of fetal bovine serum.[ 29 ] Thus, RO cell‐derived EVs are a highly interesting basis for EV‐based drug delivery applications.
Based on findings from the FT studies, suitable formulations were selected for freeze‐drying experiments of mammalian RO EVs. The lyophilizates were investigated for their longterm colloidal stability over 6 months at different temperatures. Vesicles were characterized with respect to their hydrodynamic diameter and polydispersity index (PDI) using dynamic light scattering (DLS). Particle number‐based size distribution was examined by tunable resistive pulse sensing (TRPS), while subvisible particle (SVP) numbers were detected by flow cytometry imaging. Lyophilizates were additionally tested for their ability to preserve the biological activity of beta glucuronidase (βGlu) encapsulated into the vesicles as a sensitive model biomacromolecule. Enzyme activity was quantified employing a simple fluorescence‐based assay.
2. Results
2.1. Freeze‐Thawing Studies
2.1.1. Impact of Buffer Type, pH, and Ionic Strength
Figure 1A–D shows cryo‐TEM pictures of EVs from RO cells (mammalian) and SBCy050 OMVs (bacterial) both directly after UC and after an additional step of purification by SEC. Before SEC, the samples still contained non‐vesicular material that was removed after SEC. Purified RO EVs were positive for two typical EV markers, CD9 and CD63 (Figure 1E,F), and negative for the endoplasmic reticulum marker calnexin (Figure S1, Supporting Information). The hydrodynamic particle size and PDI values before formulation preparation, that is, before dialysis, filtration, and excipient addition, were measured by DLS and are summarized in Table S1, Supporting Information. The conducted purification steps (i.e., ultracentrifugation, SEC, and 0.2 µm filtration) help to avoid the presence of foreign particles.
Figure 1.

Characterization of RO EVs and SBCy050 OMVs by cryo‐TEM, both directly after ultracentrifugation (A and C) and after an additional step of SEC‐purification (B and D). Red arrows point to vesicular structures, while white arrows indicate non‐vesicular structures and cell debris. Panels E and F show the analysis of RO EVs by flow cytometry (FACS). RO EVs are positive for both CD9 (E) and CD63 (F).
Different formulation parameters (buffer type, pH, ionic strength) were initially tested in FT studies prior to EV freeze‐drying and extended stability studies. FT was conducted in a freeze‐dryer allowing controlled and thus reproducible ramp freezing (200 µL vial fill volume). Samples were frozen to a minimum of −50 °C since lower temperatures are not relevant for medium and larger‐scale pharmaceutical freeze‐dryers. Buffer type and ionic strength may be critical formulation parameters affecting the stability of colloids upon FT and freeze‐drying.[ 1 ] For this purpose, EVs were prepared in 10 mm Na or K‐phosphate buffer of different pH values, and in PBS. EVs are usually frozen and stored at pH 7.4.[ 14 ] Thus, pH values of 7.4 ± ≈1 pH unit were investigated in this study. As particle size and concentration are important quality criteria, DLS and TRPS measurements were performed before and after FT providing information about colloidal stability. In contrast to DLS, TRPS measurements provide further insight into number‐based particle size distributions. Nanopores with two different size ranges were used to identify FT stable vesicles (NP100) as well as aggregates and/or fused vesicles (NP600). Before FT, SBCy050 OMVs and RO EVs exhibited a mean particle size of 120 and 127 nm (NP100) respectively. DLS revealed particle sizes of 110 nm (SBCy050 OMVs) and 150 nm (RO EVs) with a PDI below 0.4 (Figure S2, Supporting Information).
After three FT cycles, the number of intact RO EVs decreased more drastically (total particle reduction ≈96%) compared to bacterial SBCy050 OMVs (Figure 2 ) indicating a markedly lower FT stability of the RO EVs. For both vesicle types, the total number of larger particles increased after FT; up to 330‐fold for SBCy050 OMVs formulated at pH 8.5, and up to ≈fourfold for RO EVs in PBS (Figure S3, Supporting Information). Interestingly, the number of larger particles became markedly higher for SBCy050 OMVs compared to RO EVs. Thus, SBCy050 OMVs predominantly increased in size (e.g., due to aggregation or vesicle fusion) while RO EVs potentially got disrupted upon freezing. For both vesicle types, flow imaging measurements revealed the highest number of SVPs in PBS‐containing samples (Figure S5, Supporting Information). These results were in line with DLS (Figure S2, Supporting Information).
Figure 2.
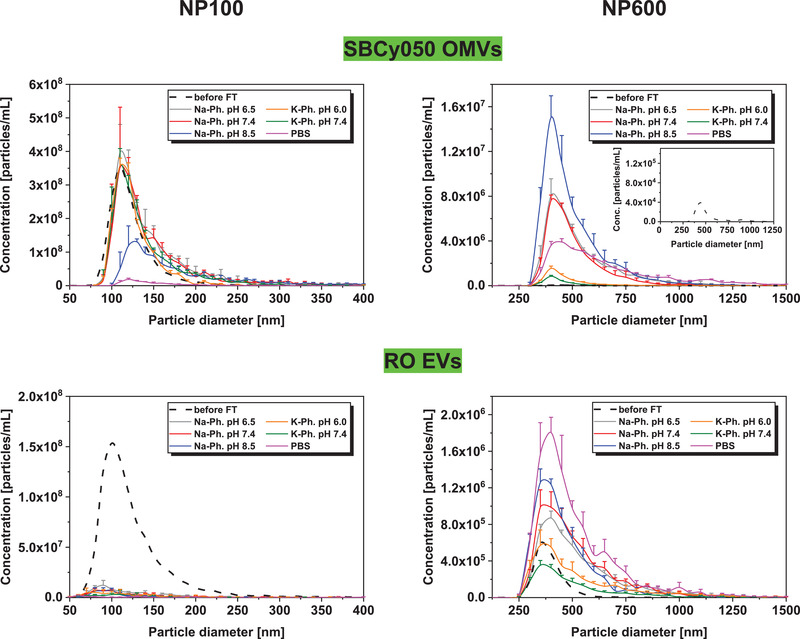
Number‐based particle size distribution of EVs before FT (mean) or three times freeze‐thaw stressed SBCy050 OMVs and RO EVs (TRPS, NP100 and NP600) formulated in 10 mm Na or K‐phosphate buffer at different pH values, and in PBS (mean ± SD; n = 3).
The pH value substantially affected FT stability of EVs. Both vesicle types revealed a lower number of large particles in K‐phosphate buffer at pH 7.4 compared to pH 6.0; total particle reduction ≈54% and ≈45% for SBCy050 OMVs and RO EVs respectively. SBCy050 OMVs exhibited a low number of small particles and simultaneously a high number of large particles at pH 8.5. In case of RO EVs, an increasing pH resulted in a higher quantity of large particles (total number of particles/mL at pH 6.5: ≈5.06E+06; pH 7.4: ≈5.90E+06; pH 8.5: ≈5.99E+06) which was also represented by an increasing particle size in DLS measurements (Figure S2, Supporting Information). In general, K‐phosphate buffers led to a significantly lower number of large particles compared to Na‐phosphate buffers. This effect was more pronounced for SBCy050 OMVs compared to RO EVs.
To further evaluate the impact of buffer pH on colloidal stability, EVs were formulated in K‐phosphate at four different acidic pH values and measured by DLS over 1 h (Figure 3 ). At acidic pH values, larger particle sizes were already measured at T 0 indicating low particle stability, especially for bacterial SBCy050 OMVs. Over time, the particle size of SBCy050 OMVs substantially increased at pH 3, 4, and 5. RO EVs showed less particle growth which corresponded to the behavior upon FT shown before. Thus, a near‐neutral pH proved to be most suitable for particle stability.
Figure 3.
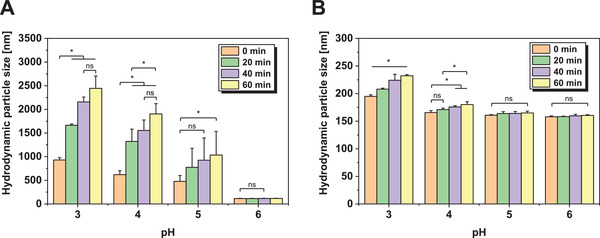
Hydrodynamic particle size (DLS) of A) SBCy050 OMVs and B) RO EVs formulated in K‐phosphate buffer at different pH values over 1 h at 25 °C (n = 3). Each data point represents mean ± SD, n = 3. Two‐way ANOVA, Bonferroni‐Holm post‐hoc test, *p < 0.05, ns = non‐significant.
2.1.2. Impact of Sucrose and Surfactant
Subsequently, different stabilizers were evaluated for their suitability to protect EVs upon freezing. PS20 and P188 were chosen as potential surface‐active stabilizers while sucrose was tested as a cryoprotectant. Vesicles were either formulated in 10 mm Na‐phosphate at pH 7.4 or in PBS, using more critical buffer conditions than K‐phosphate to challenge the stabilizer capacity. K‐phosphate was tested in combination with P188 in the following storage stability study providing a control for the Na‐phosphate formulation. FT‐induced particle growth could be reduced in presence of sucrose (Figure 4 , for DLS results, see Figure S2, Supporting Information). The total number of large particles was reduced by ≈11% and ≈44% for SBCy050 OMVs and by ≈57% and ≈55% for RO EVs in samples formulated in PBS and 10 mm Na‐phosphate respectively (Figure S3, Supporting Information). Still, a pronounced loss of intact vesicles and a high number of larger particles were found in PBS‐containing samples compared to 10 mm Naphosphate. Sucrose better stabilized SBCy050 OMVs compared to RO EVs in 10 mm Na phosphate (measured with NP100). However, particle growth was also more pronounced for SBCy050 OMVs and confirmed for both vesicle types by DLS.
Figure 4.
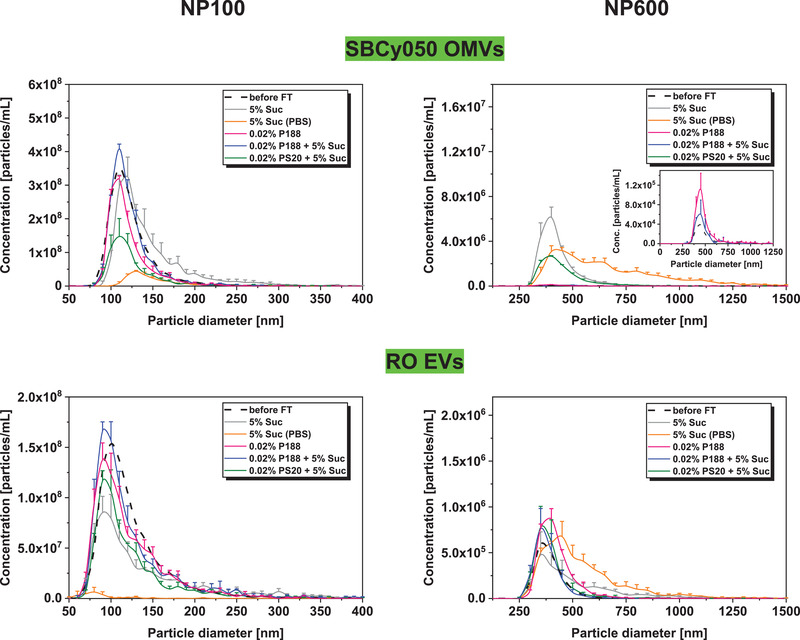
Number‐based particle size distribution of EVs before FT (mean) or three times freeze‐thaw stressed SBCy050 OMVs and RO EVs (TRPS, NP100, and NP600) formulated in 10 mm Na‐phosphate pH 7.4 or PBS with sucrose and/or surfactants (mean ± SD; n = 3).
The addition of P188 led to preserved EV numbers with a slight increase of larger particles (SBCy050 OMVs: ≈1.8‐fold; RO EVs: ≈1.5‐fold; Figure S3, Supporting Information). Moreover, P188 combined with sucrose as cryoprotectant led to the highest number of intact vesicles and the lowest number of aggregated and/or fused particles. The concentration of RO EVs could be preserved in samples containing PS20 and 5% sucrose. In contrast, SBCy050 OMVs showed a loss of approximately 60% of vesicles (6.87E + 08 particles per mL instead of ≈1.70E + 09 particles per mL) already before FT and were thus excluded from the mean particle size distribution (data not shown). Interestingly, in spite of the initial loss, about 90% of SBCy050 OMVs were maintained after FT with an increased number of larger particles compared to P188. SBCy050 OMVs exhibited a mean particle size of 55 nm and a PDI of 1.0 according to the cumulant fit analysis in DLS measurements (Figure S4, Supporting Information). The more suitable regularization fit analysis for non‐monomodal particle size distributions revealed two particle populations: i) a population with a particle size of 130 nm which corresponded to intact vesicles, and ii) a population with a particle size of about 23 nm indicating fragments of disrupted EVs. This effect was not observed for RO EVs which indicates that this population in the 20 nm size range does not reflect PS20 micelles. In DLS, placebos of surfactant‐containing formulations revealed 7 nm sized particles in presence of PS20 representing micelles, while no particles were detected in presence of P188 due to a concentration below the critical micelle concentration[ 30 ] (data not shown). TRPS using NP100 was not suitable to detect micelles in placebo formulations.
Before FT, SBCy050 OMVs and RO EVs revealed a surface charge of ≈−25 and ≈−30 mV respectively, independent of buffer type, pH, sucrose, or surfactant addition (Table S2, Supporting Information). After FT, the surface charge increased by up to 80% in formulations causing a high number of large particles. In contrast, the surface charge changed to 10–30% in surfactant or K‐phosphate‐containing samples.
2.2. Lyophilization of EVs
Consequently, mammalian RO EVs and bacterial SBCy050 OMVs formulated in 10 mm Na‐phosphate buffer in combination with 0.02% P188 and 5% sucrose were tested for their freeze‐drying stability (same EV batches as for FT studies). In addition to the assessment of the colloidal stability, unfiltered RO EVs were lyophilized and analyzed for vesicle morphology by cryo‐TEM and EV markers by FACS. A conservative freeze‐drying cycle was applied (200 µL vial fill volume). The samples were frozen to −50 °C and the product temperature during primary drying was maintained well below the T g′ of sucrose (−32 °C) at 40 mTorr chamber pressure and −20 °C shelf temperature. The lyophilized samples showed a good cake appearance and dissolved instantly (<5 s) upon addition of 190 µL HPW (calculation based on the solid content).
Figure 5A,B shows cryo‐TEM pictures of unfiltered RO EVs after lyophilization and subsequent reconstitution. RO EVs were positive for the two typical EV markers, CD9 and CD63 (Figure 5C,D).
Figure 5.

Characterization of unfiltered RO EVs by cryo‐TEM, directly after lyophilization and reconstitution (A,B). Red arrows point to vesicular structures. RO EVs are positive for both C) CD9 and D) CD63 as shown by flow cytometry (FACS) analysis.
The particle concentration of 0.2 µm filtered was well preserved for both vesicle types with a slight decrease of small particles for RO EVs (Figure 6 ). However, lyophilization led to an increased number of larger particles compared to FT stressed vesicles; this effect was more pronounced for bacterial SBCy050 OMVs (15‐fold increase of the total particle number). In DLS, the mean particle sizes and PDI did not change (Figure S6, Supporting Information).
Figure 6.
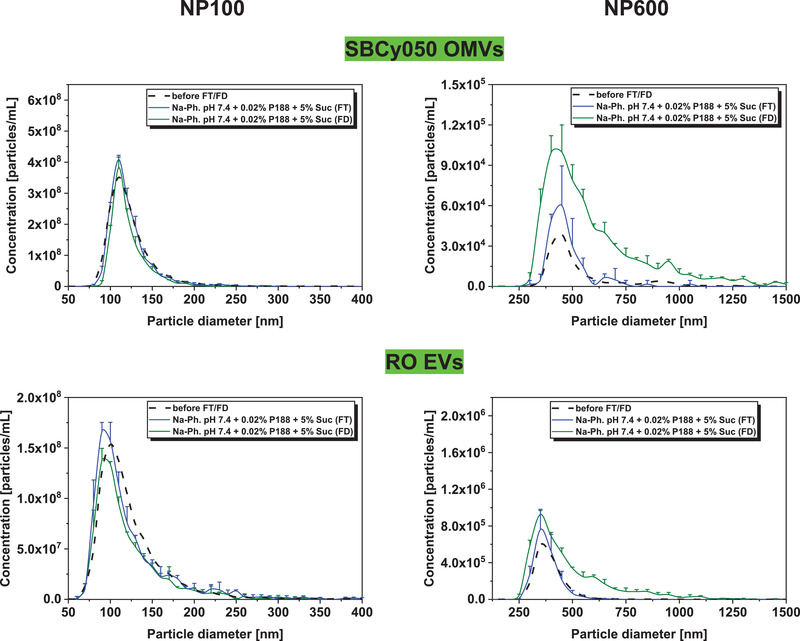
Number‐based particle size distribution of EVs before FT/FD (mean), three times freeze‐thaw stressed (FT), and freeze‐dried (FD) SBCy050 OMVs and RO EVs (TRPS, NP100 and NP600) (mean ± SD; n = 3).
2.3. Long‐Term Stability of Lyophilized EVs
Based on the previous results, four formulations of mammalian RO EVs loaded with ß‐Glu were prepared using saponin‐based encapsulation, with the enzyme acting as an easily quantifiable surrogate for biologically active EV‐cargoes. Samples were investigated regarding long‐term colloidal stability over 6 months at 28, 25, and 40 °C. After reconstitution of the lyophilizates, samples were analyzed for particle size, particle concentration, surface charge, and the biological activity of the associated model enzyme β‐Glu. Furthermore, solid‐state properties of lyophilized placebo formulations were characterized by Karl–Fischer titration and DSC. FT experiments had shown that the addition of P188 drastically improved EV stability. Although colloidal stability issues of EVs in Na‐phosphate buffer can be overcome by the addition of P188, the influence of the buffer salt type on biological activity of the encapsulated enzyme over time was unclear. Therefore, formulations in a Na‐phosphate and K‐phosphate buffer were considered for the long‐term stability study. Furthermore, PVP (MW ≈ 8.000–10.000 Da) was investigated as a surface‐active polymer. It is used in lyophilization due to its cryoprotective properties and the ability to form elegant lyophilizate cakes.[ 31 ] Thus, 0.02% PVP was tested as an alternative to P188, while 5% PVP was evaluated as a sucrose replacement.
2.3.1. Colloidal Stability of Lyophilized EVs
Upon lyophilization and storage, the particle size and concentration of EV samples containing P188 and sucrose remained stable independently of the used phosphate buffer type (Figure 7 ). According to TRPS NP600 measurements, large particles formed during freeze‐drying. Their number slightly further increased with increasing storage temperature and time.
Figure 7.
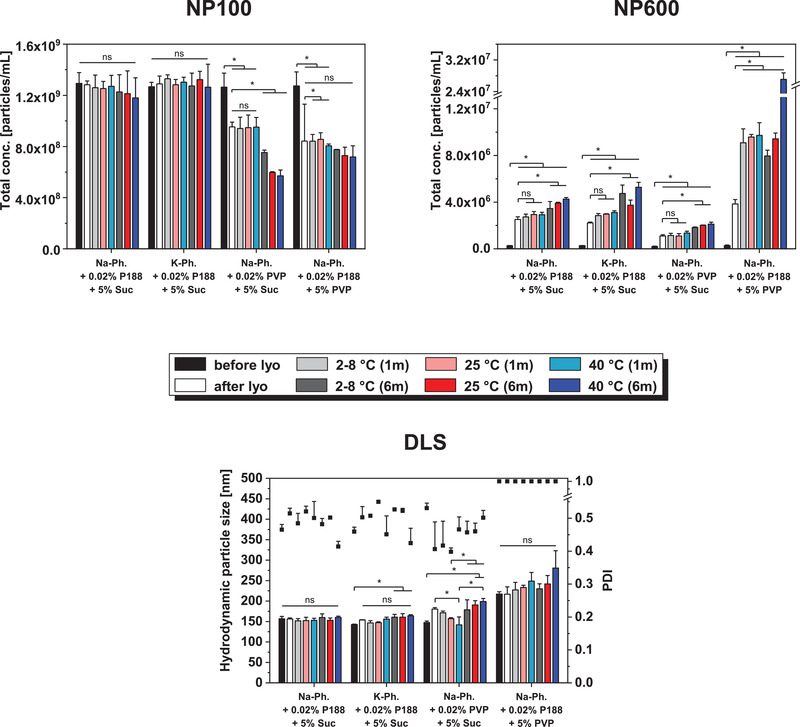
Particle concentration and size of beta glucuronidase encapsulated RO EVs before lyophilization, after lyophilization, and after storage for 1 month and 6 months at 2–8, 25, and 40 °C measured with TRPS using NP100 and NP600, and DLS. Each data point represents mean ± SD, n = 3. One‐way ANOVA, Bonferroni post‐hoc test, *p < 0.05, ns = non‐significant.
Before lyophilization, DLS measurements revealed an EV particle size of about 217 nm in samples containing 5% PVP compared to about 150 nm in the other formulations. This discrepancy was not observed in TRPS measurements. Furthermore, a PDI of 1.0 for these formulations resulted from an additional peak in the low nanometer range in the intensity versus size distribution. This peak was reproducible for EVs and placebo formulations (i.e., 10 mm Na‐phosphate + 0.02% P188 + 5% PVP without EVs) and no additional peaks at bigger particle diameters were detected (data not shown). The particle number measured by TRPS NP100 directly after lyophilization was decreased by 27% and 35% in 0.02% and 5% PVP samples respectively. The number of small particles remained constant over 1 month, but decreased further over 6 months of storage. In parallel, a low number of larger particles was measured for 0.02% PVP samples while a distinct increase was detected in presence of 5% PVP, especially after 6 months at 40 °C. Zeta potential measurements revealed similar surface charge in all formulations (≈−24 mV) which remained mostly unchanged during storage (Table S3, Supporting Information). To ensure that the particles measured were not related to aggregated free enzyme, non‐encapsulated β‐Glu formulated in phosphate buffer containing P188 and sucrose was freeze‐dried. NTA measurements did not reveal enzyme aggregation (Figure S7, Supporting Information).
The residual moisture levels of all lyophilizates increased with increasing storage temperature and longer storage time (Table 1 ). 5% PVP formulations showed higher water contents (0.8–3.4%) compared to 5% Suc formulations (0.7–2.3%). The T g values decreased correspondingly to the increase in moisture. 5% PVP formulations revealed the highest T g values between 111.9 and 86.0 °C.
Table 1.
before lyophilization and T g and RM directly after lyophilization and after storage for 1 month or 6 months at 2–8, 25, and 40 °C (mean ± SD; n = 3)
|
10 mm NaPh. 0.02% P188 + 5% Suc |
10 mm KPh. 0.02% P188 + 5% Suc |
10 mm NaPh. 0.02% PVP + 5% Suc |
10 mm NaPh. 0.02% P188 + 5% PVP |
|||||
|---|---|---|---|---|---|---|---|---|
| T g′/T g [°C] | RM [%] | T g′/T g [°C] | RM [%] | T g′/T g [°C] | RM [%] | T g′/T g [°C] | RM [%] | |
| before lyo | 31.9 ± 0.1 | 32.1 ± 0.1 | 31.6 ± 0.1 | 24.5 ± 0.0 | ||||
| after lyo | 65.3 ± 0.9 | 0.7 ± 0.1 | 65.5 ± 0.5 | 0.7 ± 0.0 | 65.8 ± 1.7 | 0.7 ± 0.1 | 111.9 ± 0.7 | 0.8 ± 0.0 |
| 1 m 28 °C | 65.6 ± 0.4 | 0.7 ± 0.1 | 64.0 ± 0.6 | 0.7 ± 0.0 | 65.6 ± 0.4 | 0.7 ± 0.0 | 111.1 ± 1.1 | 0.9 ± 0.0 |
| 1 m 25 °C | 59.9 ± 0.7 | 1.2 ± 0.1 | 59.4 ± 1.1 | 1.2 ± 0.1 | 60.8 ± 0.5 | 1.3 ± 0.0 | 103.7 ± 2.3 | 1.5 ± 0.0 |
| 1 m 40 °C | 55.6 ± 0.6 | 1.8 ± 0.1 | 54.9 ± 0.4 | 1.9 ± 0.1 | 56.1 ± 1.0 | 1.9 ± 0.1 | 98.3 ± 0.5 | 2.8 ± 0.1 |
| 6 m 28 °C | 54.9 ± 1.5 | 1.4 ± 0.2 | 50.6 ± 2.2 | 1.3 ± 0.0 | 53.9 ± 1.6 | 1.4 ± 0.1 | 95.9 ± 3.5 | 1.6 ± 0.1 |
| 6 m 25 °C | 48.3 ± 2.8 | 2.1 ± 0.0 | 46.0 ± 1.3 | 2.0 ± 0.0 | 47.9 ± 1.2 | 2.1 ± 0.1 | 88.4 ± 5.2 | 3.1 ± 0.1 |
| 6 m 40 °C | 47.1 ± 1.5 | 2.2 ± 0.2 | 44.5 ± 3.2 | 2.2 ± 0.2 | 47.7 ± 2.5 | 2.3 ± 0.1 | 86.0 ± 4.9 | 3.4 ± 0.1 |
2.3.2. Biological Activity of Encapsulated β‐Glu
As the assessment of the colloidal vesicle‐stability already showed that formulations containing the combination of 5% Suc and 0.02% P188 best‐preserved EVs, they were further evaluated regarding the stability of encapsulated βGlu. Figure 8 shows the activity of encapsulated βGlu before and after lyophilization and storage of the formulations containing 5% Suc, 0.02% P188, and either 10 mm Na‐phosphate or 10 mm K‐phosphate. Samples were purified by SEC to remove non‐encapsulated enzyme and the enzyme‐activity in EV‐containing fractions was measured by the conversion of non‐fluorescent fluorescein‐di‐β‐D‐glucuronide to fluorescent fluorescein. EVs from both formulations showed a similar initial fluorescence. After lyophilization, there was a decrease in enzyme activity compared to the initial value, which was more pronounced for K‐phosphate samples. After one month of storage between 66 and 100% enzyme‐activity were recovered for Na‐phosphate and between 52 and 67% for K‐phosphate. After 6 months of storage, the recovered enzyme activity was reduced for all samples to 1525% at 28 and 25 °C. In samples containing Naphosphate, 26% enzyme activity remained at 2–8 °C and 18% at 25 °C, while no activity remained in samples stored at 40 °C. EV samples containing K‐phosphate, stored 6 months at 28 or 40 °C showed a recovery of approx. 20% of the initial enzyme activity, while 15% remained in sample stored at 25≈°C.
Figure 8.
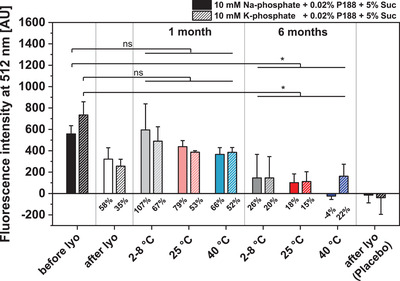
Comparison of the stability of beta glucuronidase encapsulated in RO EVs formulated with 5% Suc, 0.02% P188 and either 10 mm Na‐phosphate or 10 mm K phosphate. Enzyme activity is expressed as the fluorescence‐intensity of fluorescein generated through the enzymatic conversion of fluorescein‐di‐β‐D‐glucuronide. Percent values indicate the recovery rate of active enzyme after lyophilization and after storage for 1 and 6 months compared to samples before lyophilization. Placebo indicates samples containing only the respective buffers and cryoprotectants and no EVs. Each data point represents mean ± SD, n = 3. Two‐way ANOVA, Bonferroni‐Holm post‐hoc test, *p < 0.05, ns = non‐significant.
3. Discussion
We studied the stabilization of EVs derived from RO cells and SBCy050 bacteria by lyophilization. cryo‐TEM showed the typical morphology of EVs and OMVs in the size‐range reported in literature[ 29 , 32 ] and the successful removal of non‐vesicular structures from the samples. The appearance of fewer vesicles was attributed to the dilution of the vesicles due to SEC. The isolated and purified EVs were further processed by dialysis and filtration which may lead to vesicle loss due to adsorption processes[ 33 ] and exclusion of larger vesicles respectively. These steps, however, ensured reliable particle characterization using well‐established techniques (i.e., TRPS, DLS, and SVP measurements). Especially before FT, polydisperse samples do not allow comparison of PDI values (all >0.56) and hamper TRPS measurements due to nanopore clogging using NP100 as seen in preliminary studies (data not shown). TRPS was introduced as a suitable method providing an insight into number‐based particle size distributions. Nonetheless, this method may not be able to discriminate between different types of particles,[ 41 ] such as intact EVs, protein aggregates, or vesicle fragments. Furthermore, particles much smaller than the nanopore may not be detected resulting in an underestimated particle concentration.[ 35 ]
The stability of EVs can be drastically affected by freezing and drying. Both aspects of lyophilization cause several types of stresses, such as cryoconcentration, mechanical damage by ice crystals, interaction at the ice water interfacial area, and loss of a stabilizing hydration shell.[ 1 ] The effect of freezing itself was investigated prior to lyophilization by FT studies and maybe the more aggressive step.[ 36 ] Three FT cycles were conducted to evaluate the stabilizer capacity and the damage promoted by freezing. Recent studies showed a correlation of EV particle and cargo stability upon the freezing and lyophilization process.[ 23 , 37 ] Nevertheless, FT is also reported as a method enabling drug loading into exosomes, however, suffering from low encapsulation efficiency and the formation of large particles most likely due to aggregation.[ 38 ]
Maintenance of biological activity is the most important aspect after lyophilization. A loss of vesicles or the formation of larger particles indicate inappropriate stabilization and have to be avoided, especially as the number of visible particles is restricted in injectable drug products.[ 39 ] Thus, for the sake of simplicity, FT and FD feasibility were performed using unloaded EVs, while EV loading with βGlu was introduced in the subsequent storage stability study. The stabilizer excipients used throughout this study were chosen carefully and are approved for various administration routes, including parenteral use.[ 40 ] Furthermore, side effects due to potassium ions are not expected at the low dosing levels required for EVs.
In general, mammalian RO EVs were more prone to colloidal destabilization upon freezing compared to SBCy050 EVs. The different compositions of the lipid bilayer and surface and/or membrane proteins of EVs originating from mammalian cells and gram‐negative bacteria are assumed to lead to divergent stability profiles. The trend could be observed for different formulation parameters varying in buffer type, pH, the addition of sucrose and a surfactant. Differences in the stability profiles after lyophilization of EVs from different mammalian cell‐lines were also identified by Frank et al.[ 23 ]
The formulation itself drastically affected the propensity of EV's to form larger particles. These larger particles may be formed due to aggregation of intact vesicles, or components thereof, or fusion into larger vesicles as seen for liposomes.[ 41 ] Interestingly, surface charge measurements by TRPS revealed no differences between the formulations before FT/FD; this might be due to, in fact, little surface charge variations or potential charge shielding in presence of 140 mm NaCl. A more detailed investigation of the nature of the particles was beyond the scope of the study. It could be executed by cryo‐TEM which however requires up‐concentration bearing a high risk of artefacts. Without other stabilizers, the number of larger particles was substantially reduced in K‐phosphate compared to Na‐phosphate buffer. Na‐phosphate buffer systems are known to cause an acidic pH shift during freezing due to selective crystallization of buffer components while K‐phosphate buffers are able to maintain the pH.[ 20 ] This pH shift appears to destabilize EVs during freezing. Therefore, the impact of acidic pH was further investigated in the liquid state. DLS measurements over time showed that an acidic pH is unfavorable for colloidal stability leading to the formation of large particles. A detrimental effect of an acidic environment was also shown by Cheng et al. when comparing EV loss upon storage at pH 4 to pH 7 and 10.[ 37 ] A change in pH also affects the EV surface charge and thereby the electrostatic interactions.[ 42 ] In case of SBCy050 EVs, after FT a higher concentration of larger particles was found at pH 8.5 compared to pH 6.5 and pH 7.4. Thus, a pH optimum providing colloidal stability is essential and has to be maintained during the freezing process.
PBS is the most commonly used buffer for the processing of EVs. Therefore, PBS is usually used during storage at −80 °C which is the gold standard for preservation of EVs.[ 14 ] Interestingly, PBS reveals to be unfavorable for both EV types since a high number of large particles was measured after FT. This effect might be driven by the aforementioned pH shift of Na‐phosphate which is part of the buffer system. Furthermore, the salt present in PBS (137 mmol NaCl, 2.7 mmol KCl) can be assumed to trigger particle aggregation. During freezing, the formation of extracellular ice leads to up‐concentration of the extracellular solutes. High salt concentrations are known to negatively impact the stability of colloids by shielding repulsive forces.[ 43 ] Furthermore, surface and/or integral proteins of EVs might be prone to salt denaturation. The cryoconcentration of salts also elevates the extracellular osmotic pressure and rapid water flux through the bilayer can be responsible for physical forces leading to rupture and destabilization.[ 44 ]
Cryo‐ and lyoprotection is an important means to preserve the stability of proteins and colloids during freezing and freeze‐drying. We, therefore, tested the effect of sucrose on the colloidal stability of EVs. Both EV types showed less particle growth in presence of sucrose, but this effect could not be avoided completely. Various mechanisms, such as the preferential exclusion theory, and increased viscosity were discussed in the past in order to explain how cryoprotectants preserve colloids during freezing. The preferential exclusion theory was originally proposed for proteins and later for liposomes.[ 45 ] The theory states that solutes are preferentially excluded from protein surfaces or membranes leading to the formation of a stabilizing solvent layer. Due to cryoconcentration, the solute concentration drastically increases intensifying this mechanism. Furthermore, the increased solution viscosity during freezing is assumed to restrict diffusion and thus colloidal interactions slowing down aggregation and degradation processes.[ 46 ]
Surfactants are known to protect colloids from surface‐induced damage during freezing.[ 1 ] However, the use of surfactants is generally avoided in lipid delivery vehicles due to the fear of disrupting the lipid bilayer. During freezing, this effect is even more critical due to the up‐concentration of the surfactant. PS20 and P188 are approved in parenteral products and were therefore selected in this study.[ 40 ] We could demonstrate here that the type of surfactant has to be chosen carefully. In contrast to RO EVs, our SBCy050 OMVs showed a low number of intact vesicles in presence of PS20 already before FT. The small particles measured by DLS are attributed to EV fragments formed upon lysis. The phenomenon of differential detergent sensitivity was also reported by Osteikoetxea et al. showing surfactant and concentration‐dependent lysis of EVs.[ 47 ] Although surfactants can be detrimental to lipid bilayers, Yu et al. showed that the addition of polysorbate 80 could minimize aggregation and loss of transfection‐activity of lyophilized lipoplexes.[ 48 ]
Interestingly, in presence of P188, size and concentration of EVs were preserved upon FT independently of the vesicle type. Furthermore, the detrimental effect of Na‐phosphate was not observed indicating protective properties of the surfactant in spite of the pH shift during freezing. A stabilizing effect of P188 on lipid membranes was reported by Sharma et al. who observed that P188 decreased the susceptibility of lipid membranes to electroporation.[ 49 ] Further studies revealed that P188 directly inserts into lipid monolayers.[ 50 ] This mechanism was confirmed by later computer simulation studies. It is suggested that hydrophobic chains of P188 get inserted into damaged lipid bilayers, ultimately closing pores.[ 51 ] A stabilizing effect of P188 was also reported for liposomes.[ 52 ] Surfactants protect proteins against surface‐mitigated aggregation which could be the second mechanism for EV stabilization.[ 53 ] The addition of sucrose as a cryoprotectant was not mandatory to preserve the size and concentration of RO EVs in presence of P188. The combination of P188 and 5% Suc only slightly reduced the number of larger particles for SBCy050 EVs. This finding indicates that vitrification may be less critical for EV preservation upon freezing. Freezinginduced damage is rather promoted at the ice–liquid interface or due to hydrophobic interactions which are reduced by surfactants. The differences in molecular weight and hydrophilic/lipophilic balance of P188 (MW ≈ 8400 Da, HLB = 29) and PS20 (MW ≈ 1200 Da, HLB = 16.7) are assumed to lead to different interactions with lipid bilayers and therefore different lysing properties. The gained information is furthermore helpful for current storage practice, that is, storage at −80 °C; our studies confirm that physical stability can be maintained upon repeated FT which may avoid discarding of once thawed but unused sample.
Since sucrose was suitable to provide vesicle stability in presence of P188, it was used as a bulking agent for freeze‐drying to render isotonicity and to obtain an elegant macroscopic cake appearance. Both EV types were evaluated for their lyophilization feasibility using the formulation containing P188 and sucrose in Na‐phosphate buffer at pH 7.4. Particle size and concentration of EVs were well‐preserved after lyophilization showing a negligible increase of larger particles compared to three times FT samples. Furthermore, the vesicle morphology and the typical markers CD9 and CD63 were maintained, as shown for RO EVs. The drying step of lyophilization appears to be less critical compared to freezing and thus FT studies prove to be an important formulation screening tool to evaluate physical stability. Storage stability studies were conducted with vesicles lyophilized in four different formulations:
-
i)
a formulation containing P188 and sucrose in Na phosphate buffer pH 7.4
-
ii)
a formulation containing P188 and sucrose in K‐phosphate buffer pH 7.4 to evaluate the criticality of the pH shift for the freeze‐dried product with a cargo
-
iii)
a formulation with 0.02% PVP as an alternative stabilizer to P188 with similar molecular weight (PVP ≈ 8.000–10.000 Da)
-
iv)
a formulation with 5% PVP as an alternative stabilizer, lyoprotectant, and bulking agent since PVP is commonly used in lyophilization and known for the excellent cake appearance of lyophilizates due to the relatively high T g′ and T g values
RO EVs were chosen for the freeze‐drying storage stability studies due to the greater relevance of EVs derived from mammalian cells in current efforts for clinical translation.[ 7 ] To assess not only the colloidal stability, but also gain insight into the fate of their cargo, βGlu was encapsulated as a surrogate. Saponin‐based encapsulation of hydrophilic cargoes has been used previously without negative impact on EV‐morphology.[ 38 , 54 ] Correspondingly, we did not observe differences in the physicochemical characterization of freshly lyophilized RO EVs with or without encapsulated βGlu.
Before freeze‐drying, the identity of EVs derived from RO cells was confirmed by assessing typical surface‐markers CD9 and CD63 and proving the absence of the endoplasmic reticulum marker calnexin by FACS.[ 55 ] RO EVs exhibited a larger particle size in 5% PVP compared to sucrose formulations. This marked size difference was detected by DLS but not in TRPS measurements. The larger size in DLS is attributed to significantly different osmolality in 5% Suc and 5% PVP solutions. In contrast, TRPS measurements are conducted in presence of 140 mm NaCl which is required to provide sufficient conductivity of the solution. Thus, the osmotic effect of the cryoprotectant may become negligible resulting in vesicles of similar particle size.
After lyophilization and storage, the particle concentration of small particles (NP100) decreased drastically in PVP‐containing formulations while the concentration of large particles (NP600) increased indicating insufficient particle stabilization. These results are in line with findings by El Baradie et al. who observed an EV loss after lyophilization in presence of 100 mm trehalose/5% PVP40.[ 56 ] Thus, the ability of P188 to directly interact with lipid membranes is vital for freeze‐drying success. Furthermore, we conclude that RO EVs are better embedded in sucrose compared to PVP due to enhanced water replacement and interactions between the lyoprotectant and vesicles. Our observation is in accordance with findings by Mensink et al. who showed that molecularly more flexible disaccharides better stabilized proteins during freeze‐drying than molecularly more rigid polysaccharides.[ 57 ] The excess of PVP might furthermore lead to hampered stabilization by P188 due to competitive interactions with the vesicle lipid bilayer.
The solid‐state properties of the lyophilizates showed an increase in residual moisture content over time up to 3.4% which might be due to i) transfer of moisture from stoppers to the formulation, ii) diffusion or transmission of moisture through the stopper, and iii) microleaks in the stopper‐vial seal.[ 1 ] The low fill volume and thus lyophilizate mass contributes to the pronounced increase of water content upon uptake of only little absolute water amounts. The T g of the lyophilizates was decreased by about 27% due to the plasticizing effect of water. Vitrification becomes the limiting factor for storage stability when the storage temperature is closer to or above the T g. In order to avoid residual viscous flow of the lyophilizate, Franks proposed that the T g value should be 20 °C above the storage temperature.[ 58 ] This specification was kept at 28 and 25 °C storage but was exceeded at 40 °C. Still, we did not see any extreme decrease in colloidal stability at 40 °C.
Na and K‐phosphate were both suitable to maintain particle size and concentration of lyophilized and 6 months stored RO EVs. Thus, the pH shift of a Na‐phosphate buffer does not affect particle stability. However, the lyophilization process and further storage led to a decrease of the enzyme activity which is in line with previous findings,[ 59 , 60 ] as freezing and dehydration cause stress to the protein. The increased recovery of βGlu activity after 1 month of storage compared to samples directly after lyophilization speculatively could be an indication for reversible conformational changes of the enzyme inside the EVs.[ 61 ] Storage for 1 month at 2–8 °C revealed no statistically significant changes in encapsulated enzyme activity in both formulations. However, the mean value for K‐phosphate was reduced compared to Na‐containing formulation, which is in contrast with previous findings, where K‐phosphate showed better suitability for the preservation of lyophilized enzymes.[ 62 ] Pikal‐Cleland et al., however, looked at free enzyme, while in our study the EV‐membrane prevents the direct contact between βGlu and buffer. After 6 months of storage, both formulations showed a pronounced reduction in enzyme activity. Storage at 28 °C best‐preserved βGlu revealing an enzyme recovery of 2026%. In general, Na‐phosphate samples exhibited slightly higher recovery rates. Only at 40 °C, the K‐phosphate‐containing samples showed a clear improvement over their Na‐phosphate counterpart. Overall, βGlu activity did not correlate with the high colloidal stability. This points to either enzyme leakage or enzyme degradation within the vesicle as the main causes for the βGlu activity loss. The high enzyme recovery after 1 month suggests that general leakage of the enzyme during freeze‐drying or rehydration was not the reason for the activity‐reduction after 6 months. Thus, we speculate that βGlu encapsulated in the lyophilized EVs degraded over time.[ 59 ] Whether activity‐reduction is related to lyophilization in general or to the specific environment and the conditions inside the EVs should be subject of detailed future studies including freeze‐drying of free enzyme in different formulations with various stabilizers and concentrations.
To the best of our knowledge, there are no publications, concerning the long‐term stability of lyophilizates of liposomes or EVs loaded with enzymes that could allow a closer insight. As hydrophilic molecules do not readily cross the EV‐membrane,[ 55 ] sucrose and P188 will not be present inside the vesicles in sufficient amounts for cryo and lyoprotection of the encapsulated enzyme. Kannan et al. found that during the lyophilization of liposomes, luminal sucrose increased stability, by stabilizing the liposomes from the inside.[ 63 ] A similar effect might be found for EVs if sucrose could be actively encapsulated in sufficient amounts. Nonetheless, compared to previous studies, where after two weeks of storage at 4 °C in a 4% trehalose lyophilizate only 2550% of enzyme‐activity remained,[ 23 ] we showed a substantial improvement in preserving the βGlu activity in this work. βGlu only served as a surrogate to other vesicle cargoes, such as RNA, its sensitive tertiary and quaternary structure making it a good indicator of unfavorable storage‐conditions. Its facile encapsulation into EVs and the straightforward and sensitive evaluation of its stability made it possible to screen manifold EV formulations in parallel. With the knowledge gathered here, the next step would be to characterize the EV integrity and apply our following recommended formulation parameters to other biomedically relevant EVs:
-
■
Buffer: replace PBS with 10 mm Na‐ or K‐phosphate pH 7.4
-
■
Cryo‐ and lyoprotection: 0.02% P188 and 5% sucrose
-
■
Proposed lyophilization conditions:
-
–
Freezing: 1 °C min−1 to −50 °C
-
–
Primary drying: −20 °C, 40 mTorr, manometric endpoint determination (product temperature below T g′)
-
–
Secondary drying: 20 °C, 40 mTorr, 8 h
-
–
Ideal targets could be mesenchymal stem cell‐derived EVs that have shown promising results in many applications,[ 64 ] myxobacterial OMVs with antibacterial activity[ 65 ] or OMVs with protective effects in inflammatory bowel disease,[ 66 ] where lyophilization could provide an ideal basis for the development of solid dosage forms.
4. Conclusion
The growing interest in EVs for various pharmaceutical applications rises the demand for long‐term stable formulations without the need for storage at −80 °C. Lyophilized formulations provide easier shipping and storage, and offer new options for administration, such as pulmonary delivery. We therefore investigated the longterm stability of lyophilized RO‐cell derived EVs regarding colloidal and cargo stabilization for up to 6 months stored at 28, 25, and 40 °C.
Prior to lyophilization, FT studies were performed to select the most effective buffer type and stabilizers. Most freezing induced damage of both SBCy050 OMVs and RO EVs was seen in PBS, which is commonly used in EV isolation and preservation. This damage could be minimized by using 10 mm phosphate buffer without salts. K‐phosphate instead of Na‐phosphate buffer or the addition of sucrose further improved the FT stability. The colloidal stability of both vesicle types could be most effectively preserved by the addition of low amounts of the surfactant P188. PS20 was not suitable for SBCy050 EVs since it reduced the initial particle concentration. The well established cryo/lyoprotectant and bulking agent sucrose was found to be appropriate for successful lyophilization. The lyophilized and stored RO EVs formulated with P188 and sucrose showed comparable particle size and concentrations in Na‐phosphate and K‐phosphate buffer. Colloidal stability was preserved for 6 months while in vitro experiments revealed that the activity of encapsulated βGlu was maintained for at least 1 month. PVP was included in the freeze‐drying study as an alternative stabilizer to either P188 or sucrose, but turned out to be not able to sufficiently preserve mammalian RO EVs after lyophilization and storage.
In conclusion, we could successfully lyophilize mammalian EVs derived from RO cells, maintaining their original particle size and concentration without cargo loss. Storage at 2–8 °C appears suitable for at least 1 month. We further demonstrated that colloidal stability can be provided for at 6 months. Since enzymes are known for their sensitivity during storage, future research might focus on more stable cargos (e.g., RNA, DNA) providing further insight into content retention. In addition, the stabilizing effect of P188 could be elucidated by testing different types of poloxamer.
5. Experimental Section
Materials
RO cells (ACC452) were obtained from DSMZ, Braunschweig, D. Strain SBCy050 of the Cystobacterineae order of myxobacteria was kindly provided by Rolf Müller, Department of Microbial Natural Products, Helmholtz Institute for Pharmaceutical Research, Saarbrücken.
RPMI medium and insulin‐transferrin‐selenium‐ethanolamine were obtained from Thermo Fisher Scientific (Waltham, MA, USA). 2SWC medium was prepared from Bacto Casitone, Bacto Soytone (both Becton, Dickinson, NJ, US), glucose, MgSO4 heptahydrate, CaCl2 dihidrate (all Sigma‐Aldrich, Steinheim, D), maltose monohydrate, cellobiose (both MP Biomedicals SARL, Illkirch–Graffenstaden, FR), HEPES (Carl Roth, Karlsruhe, D) and KOH (Thermo Fisher Scientific, Waltham, MA, US). PBS was prepared from tablets (Thermo Fisher Scientific, Waltham, MA, US). 10 mm Na‐phosphate (Sigma‐Aldrich, Steinheim, D) at pH 7.4 was prepared for size exclusion chromatography. Both buffers were filtered through a 0.2 µm mixed cellulose ester membrane filter (GE Healthcare, Chicago, IL, USA) prior to use. The protein content of EV‐samples was measured using a QuantiPro BCA assay kit (Sigma‐Aldrich, Steinheim, D). Bovine serum albumin (BSA) was obtained from Sigma‐Aldrich (Steinheim, D). To adsorb EVs in FACS experiments, aldehyd/sulfate latex beads with 4 µM diameter (Thermo Fisher Scientific, Waltham, MA, USA) were used.
Stabilizer stock solutions of sucrose (Sigma‐Aldrich, Steinheim, D), poloxamer 188 (Kolliphor P188, BASF, Ludwigshafen am Rhein, D), polysorbate 20 (Tween 20, Merck, Darmstadt, D), and polyvinylpyrrolidone (Kollidon 17 PF, BASF, Ludwigshafen am Rhein, D) were prepared at various concentrations. 20 mm Na‐phosphate and K‐phosphate buffers (VWR International, Ismaning, D) at different pH values were prepared for dialysis. Stabilizer stock solutions were filtered with 0.2 µm polyethersulfone (PES) membrane syringe filters (VWR International, Ismaning, D). For the preparation of buffers and stock solutions, HPW was used. All excipients had analytical or higher grade and were used without further purification.
Carboxylated polystyrene particle standards with a nominal mean diameter of 110 and 950 nm, denoted as CPC100 and CPC1000 were used for TRPS instrument calibration (Izon Science Europe, UK).
2R glass vials (Fiolax clear, Schott, Müllheim, D) with igloo rubber stoppers (B2‐TR coating, West Pharmaceutical Services, Eschweiler, D) were cleaned with HPW and dried for 8 h at 60 °C.
Cell Culture
RO cells (DSMZ, ACC 452) were cultivated as described previously.[ 29 ] Briefly, cells were cultured in RPMI with 1% (v/v) insulin‐transferrin‐selinium‐ethanolamine and a density of 0.75 × 106 cells mL−1 in 45 mL. 25 mL of the total volume were replaced with 50 mL new medium after 3 days. Vesicles were isolated after 7 days from cells cultured until passage 33. RO cell supernatants were centrifuged at 300 × g for 8 min to pellet cells. Subsequently, supernatants were centrifuged at 9500 × g for 15 min and then filtered through a 0.45 µm bottletop filter with a polyvinylidene fluoride membrane (Steritop, Merck, Darmstadt, D). Cell free conditioned medium was subjected to ultracentrifugation (UC) on the same day. UC was performed using a Type 45 Ti rotor (Beckman Coulter, Brea, CA, USA) for 2 h at 100 000 × g with 70 mL polycarbonate tubes (Beckman Coulter, Brea, CA, USA). The resulting EV pellet was resuspended in residual supernatant, left in the tubes after decanting. This resulted in a total volume of approx. 800 µL resuspended EV pellet obtained from one UC run with six tubes.
Bacterial Culture
SBCy050 myxobacteria were cultured in 2SWC medium (0.5% Bacto Casitone, 0.1% Bacto Soytone, 0.2% glucose, 0.1% maltose monohydrate, 0.2% cellobiose, 0.05% CaCl2 dihydrate, 0.1% MgSO4 heptahydrate, 10 mm HEPES, adjusted to pH 7.0 with KOH) for 4 days, starting with an optical density of 0.04±0.01. To obtain conditioned medium the cultures were centrifuged at 9500 × g for 10 min and then filtered through a 0.45 µm pore size bottletop filter (see above). Cell free conditioned medium was subjected to UC on the same day. UC was performed using a SW 32 Ti rotor (Beckman Coulter, Brea, US) for 2 h at 100 000 × g using 32 mL polycarbonate tubes (Beckman Coulter, Brea, US). EV pellets were resuspended as described above.
FACS of RO EVs
EV evaluation by FACS was based on the protocol of Hoppstaedter et al.[ 67 ] The protein content of RO EVs was measured using a bicinchoninic acid assay kit. Then the vesicles were mixed 1:1 with FACS beads (µg mL−1 protein to µL beads), incubated for 15 min at room temperature, diluted to 1 mL with PBS and incubated for 1 h applying mild shaking. Next, 1 mL of a 200 mm glycine stop solution was added to saturate the beads. After 20 min incubation, the beads were centrifuged twice at 2000 × g for 4 min and resuspended in 1% BSA in PBS, in the original volume of FACS beads employed in the initial step. Next, 10 µL of sample were mixed with 10 µL of FITC‐labeled antibody, either anti CD9, anti CD63 or calnexin or the isotype control and incubated on ice for 30 min in the dark (see Table S4, Supporting Information, for further information on antibodies). After dilution to 1 mL using 1% BSA in PBS, samples were centrifuged twice at 2000 × g for 4 min, then measured by FACS (LRS Fortessa, BD Biosciences, NJ, USA) using BD FACSDiva v9.0 software. Data was analyzed using FloJo (version 10.7.0). Negative controls were prepared in the same way as EV‐containing samples, but EVs were replaced with the respective amount of BSA. For the positive calnexin and isotype control with RO cells, cells were centrifuged at 300 × g and resuspended in PBS. One million cells per 100 µL were fixed with 4% paraformaldehyde for 10 min, washed twice with PBS and then incubated with 0.1% saponin and 1 µl calnexin or 2.1 µL of the respective isotype for 30 min. Cells were washed once and resuspended in 400 µL PBS for FACS analysis.
Beta Glucuronidase Encapsulation
βGlu was encapsulated in RO EVs as previously described.[ 68 ] The resuspended pellet was mixed with βGlu and saponin to final concentrations of 1.5 and 0.1 mg mL−1 respectively and incubated for 10 min at ambient temperature.
Size Exclusion Chromatography
To remove impurities carried over from UC and free glucuronidase, EV samples were purified by SEC. SEC was performed using a 1.5 cm diameter glass column (Flex Column, Kimble Chase, Vineland, NJ, USA), filled with 35 mL of sepharose Cl 2b (Cytiva, Marlborough, MA, USA). PBS was used as mobile phase with a flow rate of 1 mL min−1. Up to 750 µL UC pellet were purified per SEC run, purified EVs were collected in 1 mL fractions. The process was automated using an ÄKTA start system equipped with a Frac30 fraction collector (both Cytiva, Marlborough, MA, USA). The vesicle containing fractions were collected and pooled for subsequent characterization and lyophilization. Each SEC run included a 120 mL washing step to ensure the removal of all non‐encapsulated glucuronidase, before the next sample was injected.
Beta Glucuronidase Assay
Glucuronidase activity was measured as described previously.[ 23 ] Free enzyme potentially present from EV leakage during storage was removed by SEC after reconstitution of lyophilized samples. For this purpose, a 1.0 cm diameter column filled with 10 mL of sepharose Cl2b was used with 10 mm Na‐phosphate buffer at pH 7.4 as the mobile phase and a flow rate of 1 mL min−1. Fractions of 0.5 mL were collected. Glucuronidase activity was measured after mixing samples with fluorescein‐di‐β‐D‐glucuronide to a final concentration of 8.3 µg mL−1 in a total volume of 150 µL. Directly after mixing, the fluorescence was measured using a plate reader (Infinite 200Pro, Tecan, Männedorf, CH), with an excitation wavelength of 480 nm and an emission wavelength of 516 nm. After 18 h incubation at 37 °C, the fluorescence was measured again. The difference between t 18 h and t 0 h indicated the enzyme activity found in the respective sample. PBS treated in the same way as EV‐containing samples was measured as a control and subtracted from samples values.
Cryo‐TEM
Cryo‐TEM pictures of EV‐samples both directly after ultracentrifugation and after SEC purification were acquired as previously described.[ 65 ] Briefly, a drop of 3 µL of EV suspension was placed on a holey carbon film (type S147‐4, Plano, Wetzlar, D), blotted for 2 s, then plunged into liquid ethane at T = 108 K with a Gatan cryoplunger model CP3 (Pleasanton, CA, USA). After transferring the frozen samples to a Gatan model 914 cryo‐TEM sample holder, they were imaged in brightfield TEM at T = 100 K with a JEOL JEM‐2100 LaB6 (Tokyo, JP).
Formulation Preparation
After isolation and SEC purification, EVs were dialyzed with 20 mm Na or K‐phosphate buffer in Slide‐A‐Lyzer MINI dialysis devices or cassettes (20K MWCO; Thermo Fisher Scientific, Waltham, MA, USA). After dialysis, EVs were filtered through a 0.2 µm PES membrane syringe filter (VWR International, Ismaning, D) and mixed 1:1 with stabilizer stock solutions to match the respective final buffer and stabilizer concentration (see Table S5, Supporting Information). Unloaded and βGlu loaded RO EVs were investigated from different batches.
Freeze‐Thawing Cycle
Formulations (200 µL in 2R vials) were freeze‐thawed three times on a pilot scale freeze‐drier (FTS LyoStar 3, SP Scientific, Stone Ridge, NY, USA) at −1 °C min−1 to −50 °C followed by a 30 min hold at −50 °C and thawing at 1 °C min−1 to 10 °C followed by a 30 min hold. Before freeze‐thawing, EV concentrations were determined by TRPS and denoted as “before FT”.
Freeze‐Drying Cycle
A lyophilization process stability study was conducted using unloaded RO EVs and SBCy050 OMVs (same batches as for freeze‐thawing studies) formulated in 10 mm Na‐phosphate buffer pH 7.4 in combination with 0.02% P188 and 5% sucrose. Selected formulations of βGlu loaded RO EVs were used for longterm stability studies (Table 2 ), including placebos consisting of identical formulations without EVs. The samples were lyophilized in 2R vials with 200 µL fill volume. Before lyophilization, EV concentrations were determined by TRPS and denoted as “before FD”. Lyophilization was performed on a pilot‐scale freeze‐dryer (LyoStar 3). After an equilibration step at −5 °C for 15 min, the samples were frozen at −1 °C min−1 to −50 °C and held for 120 min. Primary drying was performed at −20 °C and 40 mTorr with manometric end point determination. The product temperature, monitored with thermocouples, was kept below the glass transition temperature of the maximally frozen concentrate (T g′) which was determined by DSC. Secondary drying was performed at 20 °C and 40 mTorr. Samples were stoppered under slight vacuum at 450 Torr nitrogen, and vials were crimped with aluminum seals. The lyophilizates were reconstituted by adding 190 µL of HPW. The reconstitution volume was calculated based on the solid content. The vials were shaken gently to ensure wetting of the complete lyophilizate.
Table 2.
Formulations for freeze drying experiments
| Formulation # | Buffer | Cryoprotectant | Surfactant |
|---|---|---|---|
| 1 | 10 mm Na‐phosphate pH 7.4 | 5% Suc | 0.02% P188 |
| 2 | 10 mm K‐phosphate pH 7.4 | 5% Suc | 0.02% P188 |
| 3 | 10 mm Na‐phosphate pH 7.4 | 5% Suc | 0.02% PVP |
| 4 | 10 mm Na‐phosphate pH 7.4 | 5% PVP | 0.02% P188 |
Long‐Term Stability Testing of Lyophilized Samples
For longterm stability testing, sealed lyophilizates were stored at 28, 25, and 40 °C over a period of 1 and 6 months.
Dynamic Light Scattering
Mean particle sizes and respective polydispersity indices (PDI) were measured using a DLS platereader (DynaPro III, Wyatt Technology, D). 20 µL sample (n = 3) per well of a 384 UV‐well plate (Costar, Corning, Tewksbury, MA, USA) was analyzed at 25 °C using 10 acquisitions with 5 s each. The corresponding preset refractive index parameters were used for all samples. Viscosities of sucrose and PVP formulations required for DLS measurements were determined via an AMVn Automated Micro Viscometer (Anton Paar, Graz, A).
Tunable Resistive Pulse Sensing
Concentration based particle size distributions and zeta potentials were analyzed by TRPS (qNano, Izon Science Europe, UK). Adjustment of nanopore stretch and voltage were optimized according to manufacturer recommendations. In order to provide sufficient conductivity, 27 µL of each sample containing 10 mm phosphate (not PBS) was mixed with 3 µL of a 1.4 M NaCl solution resulting in 140 mm NaCl and 9 mm phosphate. Samples were measured in triplicate with a minimum of 500 particles per analysis. In case of fewer particles a maximum measurement time of 10 min was performed. Calibration of the nanopores NP100 (measurement size range: 50 330 nm) and NP600 (measurement size range: 275–1570 nm) was conducted using carboxylated polystyrene particle standards CPC100 and CPC1000 respectively. Calibration particles were prepared in 10 mm phosphate buffer and mixed with a NaCl solution as described before for sample particles. Zeta potential analysis was conducted according to IZON's instruction “V3.1 Charge Analysis” using NP100 and CPC100 particles. The calibration particles were measured at three applied voltages; particles measured at the highest voltage were measured at two external pressures. Data obtained from measured samples and calibration particles were evaluated using the template “Zeta Template V3.1a” provided by IZON.
Subvisible Particles (SVP)
SVPs were analyzed by flow cytometry imaging (FlowCam 8100, Fluid Imaging Technologies, Inc., Scarborough, ME, USA). A 10× magnification cell was used for the measurements. After 1:10 dilution, 160 µL sample solution was measured with a flow rate of 0.15 mL min−1, an auto image frame rate of 28 frames per s, and a run time of 60 s. After each measurement, the flow cell was flushed with HPW. Particle identification was set with a distance to the nearest neighbor of 3 µm, and a segmentation threshold of 13 and 10 for the dark and light pixels respectively. The software VisualSpreadsheet 4.7.6 was used for measurements and evaluation.
Nanoparticle Tracking Analysis
Nanoparticle tracking analysis was performed using a Nanosight LM‐10 (Malvern Instruments, UK) equipped with a green laser measurement cell. Three videos of 30 s were recorded using a camera level of 14–15 and detection threshold 10 and analyzed using NTA software (NTA 3.1 Software).
Karl–Fischer Titration
The RM of placebo lyophilizates was determined in triplicates by Karl–Fischer titration after lyophilization and after storage. Measurements were performed using an Aqua 40.00 titrator (Analytik Jena AG, Halle, D) equipped with a headspace oven set at 100 °C. Samples of 10 to 20 mg crushed lyophilizates were analyzed in stoppered 2R vials.
Differential Scanning Calorimetry (DSC)
DSC measurements were performed in 40 µL aluminum crucibles using a Mettler Toledo DSC 822e (MettlerToledo GmbH, Giessen, D). In order to determine the glass transition temperature of the maximally freeze‐concentrated solution (T g′), 20 µL of the liquid samples were cooled at −10 °C min−1 from 25 to −60 °C, held at −60 °C for 1 min, and reheated at 10 °C min−1 to 25 °C. For the determination of the glass transition temperature of the lyophilized samples (T g), approximately 10 mg were weighed into the aluminum crucibles. Samples were preheated from 0 to 70 °C, cooled to 0 °C and heated to 150 °C at a scanning rate of 10 °C min−1. T g and T g′ were defined as the inflection point of the glass transition in the heating scan of the DSC experiment. All analyses were performed in triplicate with placebo formulations.
Statistical Analysis
Unless otherwise stated, results are given as mean value ± standard deviation (SD). Statistically significant differences were determined via a two‐tailed student t‐test using Origin 2019b Software (OriginLab Corporation, Northampton, MA, USA). Mean values having p values >0.05 were judged to be not significantly different.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
G.F. acknowledges funding from the Federal Ministry of Research and Education (NanoMatFutur grant, 13XP5029A). M.R. acknowledges funding from Studienstiftung des Deutschen Volkes (German Academic Scholarship Foundation) through a Ph.D. fellowship. The authors thank the Microbial Natural Products group at the Helmholtz Institute for Pharmaceutical Research Saarland for providing myxobacterial strains.
Open access funding enabled and organized by Projekt DEAL.
Trenkenschuh E., Richter M., Heinrich E., Koch M., Fuhrmann G., Friess W., Enhancing the Stabilization Potential of Lyophilization for Extracellular Vesicles. Adv. Healthcare Mater. 2022, 11, 2100538. 10.1002/adhm.202100538
Data Availability Statement
Research data are not shared.
References
- 1. Wang W., Int. J. Pharm. 2000, 203, 1. [DOI] [PubMed] [Google Scholar]
- 2. Kasper J. C., Winter G., Friess W., Eur. J. Pharm. Biopharm. 2013, 85, 162. [DOI] [PubMed] [Google Scholar]
- 3. Woith E., Fuhrmann G., Melzig M. F., Int. J. Mol. Sci. 2019, 20, 5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Niel G., D'Angelo G., Raposo G., Nat. Rev. Mol. Cell Biol. 2018, 19, 213. [DOI] [PubMed] [Google Scholar]
- 5. Schwechheimer C., Kuehn M. J., Nat. Rev. Microbiol. 2015, 13, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yáñez‐Mó M., Siljander P. R.‐M., Andreu Z., Zavec A. B., Borràs F. E., Buzas E. I., Buzas K., Casal E., Cappello F., Carvalho J., Colás E., Cordeiro‐da Silva A., Fais S., Falcon‐Perez J. M., Ghobrial I. M., Giebel B., Gimona M., Graner M., Gursel I., Gursel M., Heegaard N. H. H., Hendrix a., Kierulf P., Kokubun K., Kosanovic M., Kralj‐Iglic V., Krämer‐Albers E.‐M., Laitinen S., Lässer C., Lener T., Ligeti E., Linē A., Lipps G., Llorente A., Lötvall J., Manček‐Keber M., Marcilla A., Mittelbrunn M., Nazarenko I., Nolte‐‚t Hoen E. N. M., Nyman T. A., O‘Driscoll L., Olivan M., Oliveira C., Pállinger É., Del Portillo H. A., Reventós J., Rigau M., Rohde E., Sammar M., Sánchez‐Madrid F., Santarém N., Schallmoser K., Ostenfeld M. S., Stoorvogel W., Stukelj R., van der Grein S. G., Vasconcelos M. H., Wauben M. H. M., de Wever O., J. Extracell. Vesicles 2015, 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wiklander O. P. B., Brennan M. Á., Lötvall J., Breakefield X. O., El Andaloussi S., Sci. Transl. Med. 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuhn T., Koch M., Fuhrmann G., Small 2020, 16, 2003158. [DOI] [PubMed] [Google Scholar]
- 9. Elsharkasy O. M., Nordin J. Z., Hagey D. W., de Jong O. G., Schiffelers R. M., Andaloussi S. E., Vader P., Adv. Drug Delivery Rev. 2020, 159, 332. [DOI] [PubMed] [Google Scholar]
- 10.a) Katsuda T., Kosaka N., Ochiya T., Proteomics 2014, 14, 412; [DOI] [PubMed] [Google Scholar]; b) Vella L. J., Hill A. F., Cheng L., Int. J. Mol. Sci. 2016, 17, 173; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Szabo G., Momen‐Heravi F., Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Lener T., Gimona M., Aigner L., Börger V., Buzas E., Camussi G., Chaput N., Chatterjee D., Court F. A., Del Portillo H. A., O'Driscoll L., Fais S., Falcon‐Perez J. M., Felderhoff‐Mueser U., Fraile L., Gho Y. S., Görgens A., Gupta R. C., Hendrix a., Hermann D. M., Hill A. F., Hochberg F., Horn P. A., de Kleijn D., Kordelas L., Kramer B. W., Krämer‐Albers E.‐M., Laner‐Plamberger S., Laitinen S., Leonardi T., Lorenowicz M. J., Lim S. K., Lötvall J., Maguire C. A., Marcilla A., Nazarenko I., Ochiya T., Patel T., Pedersen S., Pocsfalvi G., Pluchino S., Quesenberry P., Reischl I. G., Rivera F. J., Sanzenbacher R., Schallmoser K., Slaper‐Cortenbach I., Strunk D., Tonn T., Vader P., van Balkom B. W. M., Wauben M., Andaloussi S. E., Théry C., Rohde E., Giebel B., J. Extracell. Vesicles 2015, 4, 30087; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gimona M., Pachler K., Laner‐Plamberger S., Schallmoser K., Rohde E., Int. J. Mol. Sci. 2017, 18, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Ingato D., Lee J. U., Sim S. J., Kwon Y. J., J. Controlled Release 2016, 241, 174; [DOI] [PubMed] [Google Scholar]; b) Kusuma G. D., Barabadi M., Tan J. L., Morton D. A. V., Frith J. E., Lim R., Front. Pharmacol. 2018, 9, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Jeyaram A., Jay S. M., AAPS J. 2017, 20, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Deville S., Berckmans P., van Hoof R., Lambrichts I., Salvati A., Nelissen I., PLoS One 2021, 16, e0245835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Witwer K. W., Buzás E. I., Bemis L. T., Bora A., Lässer C., Lötvall J., Nolte‐‘t Hoen E. N., Piper M. G., Sivaraman S., Skog J., Théry C., Wauben M. H., Hochberg F., J. Extracell. Vesicles 20360, 2013, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Maroto R., Zhao Y., Jamaluddin M., Popov V. L., Wang H., Kalubowilage M., Zhang Y., Luisi J., Sun H., Culbertson C. T., Bossmann S. H., Motamedi M., Brasier A. R., J. Extracell. Vesicles 2017, 6, 1359478; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lőrincz Á. M., Timár C. I., Marosvári K. A., Veres D. S., Otrokocsi L., Kittel Á., Ligeti E., J. Extracell. Vesicles 2014, 3, 25465; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Akers J. C., Ramakrishnan V., Yang I., Hua W., Mao Y., Carter B. S., Chen C. C., Cancer Biomark. 2016, 17, 125; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ayers L., Kohler M., Harrison P., Sargent I., Dragovic R., Schaap M., Nieuwland R., Brooks S. A., Ferry B., Thromb. Res. 2011, 127, 370; [DOI] [PubMed] [Google Scholar]; e) Szatanek R., Baran J., Siedlar M., Baj‐Krzyworzeka M., Int. J. Mol. Med. 2015, 36, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. György B., Hung M. E., Breakefield X. O., Leonard J. N., Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Pieters B. C. H., Arntz O. J., Bennink M. B., Broeren M. G. A., van Caam A. P. M., Koenders M. I., van Lent P. L. E. M., van den Berg W. B., de Vries M., van der Kraan P. M., van de Loo F. A. J., PLoS One 2015, 10, e0121123; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jayachandran M., Miller V. M., Heit J. A., Owen W. G., J. Immunol. Methods 2012, 375, 207; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sokolova V., Ludwig A.‐K., Hornung S., Rotan O., Horn P. A., Epple M., Giebel B., Colloids Surf., B 2011, 87, 146; [DOI] [PubMed] [Google Scholar]; d) Jin Y., Chen K., Wang Z., Wang Y., Liu J., Lin L., Shao Y., Gao L., Yin H., Cui C., Tan Z., Liu L., Zhao C., Zhang G., Jia R., Du L., Chen Y., Liu R., Xu J., Hu X., Wang Y., BMC Cancer 2016, 16, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abdelwahed W., Degobert G., Stainmesse S., Fessi H., Adv. Drug Delivery Rev. 2006, 58, 1688. [DOI] [PubMed] [Google Scholar]
- 19. Tang X., Pikal M. J., Pharm. Res. 2004, 21, 191. [DOI] [PubMed] [Google Scholar]
- 20. Kolhe P., Amend E., Singh S. K., Biotechnol. Prog. 2010, 26, 727. [DOI] [PubMed] [Google Scholar]
- 21.a) Pikal‐Cleland K. A., Rodríguez‐Hornedo N., Amidon G. L., Carpenter J. F., Arch. Biochem. Biophys. 2000, 384, 398; [DOI] [PubMed] [Google Scholar]; b) van den Berg L., Rose D., Arch. Biochem. Biophys. 1959, 81, 319. [DOI] [PubMed] [Google Scholar]
- 22. Franks F., Eur. J. Pharm. Biopharm. 1998, 45, 221. [DOI] [PubMed] [Google Scholar]
- 23. Frank J., Richter M., de Rossi C., Lehr C.‐M., Fuhrmann K., Fuhrmann G., Sci. Rep. 2018, 8, 12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bosch S., de Beaurepaire L., Allard M., Mosser M., Heichette C., Chrétien D., Jegou D., Bach J.‐M., Sci. Rep. 2016, 6, 36162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Charoenviriyakul C., Takahashi Y., Nishikawa M., Takakura Y., Int. J. Pharm. 2018, 553, 1. [DOI] [PubMed] [Google Scholar]
- 26. Midekessa G., Godakumara K., Ord J., Viil J., Lättekivi F., Dissanayake K., Kopanchuk S., Rinken A., Andronowska A., Bhattacharjee S., Rinken T., Fazeli A., ACS Omega 2020, 5, 16701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Préval C., Hadam M. R., Mach B., N. Engl. J. Med. 1988, 318, 1295. [DOI] [PubMed] [Google Scholar]
- 28. Ayala García M. A., González Yebra B., López Flores A. L., Guaní Guerra E., J. Transplant. 2012, 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schulz E., Karagianni A., Koch M., Fuhrmann G., Eur. J. Pharm. Biopharm. 2020, 146, 55. [DOI] [PubMed] [Google Scholar]
- 30. Tian J.‐L., Zhao Y.‐Z., Jin Z., Lu C.‐T., Tang Q.‐Q., Xiang Q., Sun C.‐Z., Zhang L., Xu Y.‐ Y., Gao H.‐S., Zhou Z.‐C., Li X.‐K., Zhang Y., Drug Dev. Ind. Pharm. 2010, 36, 832. [DOI] [PubMed] [Google Scholar]
- 31.a) Kasper J. C., Schaffert D., Ogris M., Wagner E., Friess W., J. Controlled Release 2011, 151, 246; [DOI] [PubMed] [Google Scholar]; b) Schoug A., Mahlin D., Jonson M., Håkansson S., J. Appl. Microbiol. 2010, 108, 1032; [DOI] [PubMed] [Google Scholar]; c) Haeuser C., Goldbach P., Huwyler J., Friess W., Allmendinger A., J. Pharm. Sci. 2020, 109, 807. [DOI] [PubMed] [Google Scholar]
- 32.a) Bitto N. J., Kaparakis‐Liaskos M., Int. J. Mol. Sci. 2017, 18; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cizmar P., Yuana Y., Methods Mol. Biol. 2017, 1660, 221. [DOI] [PubMed] [Google Scholar]
- 33. Ruzicka M., Xiao F., Abujrad H., Al‐Rewashdy Y., Tang V. A., Langlois M.‐A., Sorisky A., Ooi T. C., Burger D., BMC Nephrol. 2019, 20, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vogel R., Coumans F. A. W., Maltesen R. G., Böing A. N., Bonnington K. E., Broekman M. L., Broom M. F., Buzás E. I., Christiansen G., Hajji N., Kristensen S. R., Kuehn M. J., Lund S. M., Maas S. L. N., Nieuwland R., Osteikoetxea X., Schnoor R., Scicluna B. J., Shambrook M., de Vrij J., Mann S. I., Hill A. F., Pedersen S., J. Extracell. Vesicles 2016, 5, 31242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maas S. L. N., de Vrij J., van der Vlist E. J., Geragousian B., van Bloois L., Mastrobattista E., Schiffelers R. M., Wauben M. H. M., Broekman M. L. D., Nolte‐’t Hoen E. N. M., J. Controlled Release 2015, 200, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Date P. V., Samad A., Devarajan P. V., AAPS PharmSciTech 2010, 11, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng Y., Zeng Q., Han Q., Xia W., Protein Cell 2019, 10, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haney M. J., Klyachko N. L., Zhao Y., Gupta R., Plotnikova E. G., He Z., Patel T., Piroyan A., Sokolsky M., Kabanov A. V., Batrakova E. V., J. Controlled Release 2015, 207, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mathonet S., Mahler H.‐C., Esswein S. T., Mazaheri M., Cash P. W., Wuchner K., Kallmeyer G., Das T. K., Finkler C., Lennard A., PDA J. Pharm. Sci. Technol. 2016, 70, 392. [DOI] [PubMed] [Google Scholar]
- 40. Gervasi V., Dall Agnol R., Cullen S., McCoy T., Vucen S., Crean A., Eur. J. Pharm. Biopharm. 2018, 131, 8. [DOI] [PubMed] [Google Scholar]
- 41. Chen C., Han D., Cai C., Tang X., J. Controlled Release 2010, 142, 299. [DOI] [PubMed] [Google Scholar]
- 42. Chi E. Y., Krishnan S., Randolph T. W., Carpenter J. F., Pharm. Res. 2003, 20, 1325. [DOI] [PubMed] [Google Scholar]
- 43.a) Wang W., Int. J. Pharm. 1999, 185, 129; [DOI] [PubMed] [Google Scholar]; b) Fernández M. S., Biochim. Biophys. Acta, Biomembr. 1981, 646, 23; [DOI] [PubMed] [Google Scholar]; c) Ishikawa Y., Katoh Y., Ohshima H., Colloids Surf., B 2005, 42, 53. [DOI] [PubMed] [Google Scholar]
- 44. Muldrew K., McGann L. E., Biophys. J. 1994, 66, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.a) Andersen H. D., Wang C., Arleth L., Peters G. H., Westh P., Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1874; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sydykov B., Oldenhof H., de Oliveira Barros L., Sieme H., Wolkers W. F., Biochim. Biophys. Acta, Biomembr. 2018, 1860, 467. [DOI] [PubMed] [Google Scholar]
- 46. Kasper J. C., Pikal M. J., Friess W., J. Pharm. Sci. 2013, 102, 929. [DOI] [PubMed] [Google Scholar]
- 47. Osteikoetxea X., Sódar B., Németh A., Szabó‐Taylor K., Pálóczi K., Vukman K. V., Tamási V., Balogh A., Kittel Á., Pállinger É., Buzás E. I., Org. Biomol. Chem. 2015, 13, 9775. [DOI] [PubMed] [Google Scholar]
- 48. Yu J., Anchordoquy T. J., J. Pharm. Sci. 2009, 98, 3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sharma V., Stebe K., Murphy J. C., Tung L., Biophys. J. 1996, 71, 3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maskarinec S. A., Hannig J., Lee R. C., Lee K. Y. C., Biophys. J. 2002, 82, 1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adhikari U., Goliaei A., Tsereteli L., Berkowitz M. L., J. Phys. Chem. B 2016, 120, 5823. [DOI] [PubMed] [Google Scholar]
- 52.a) Law S.‐L., Chuang T.‐C., Kao M.‐C., Lin Y.‐S., Huang K.‐J., Drug Deliv. 2006, 13, 61; [DOI] [PubMed] [Google Scholar]; b) Zhang W., Wang G., See E., Shaw J. P., Baguley B. C., Liu J., Amirapu S., Wu Z., J. Controlled Release 2015, 203, 161. [DOI] [PubMed] [Google Scholar]
- 53. Wang W., Roberts C. J., Int. J. Pharm. 2018, 550, 251. [DOI] [PubMed] [Google Scholar]
- 54. Fuhrmann G., Serio A., Mazo M., Nair R., Stevens M. M., J. Controlled Release 2015, 205, 35. [DOI] [PubMed] [Google Scholar]
- 55. Théry C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin‐Smith G. K., Ayre D. C., Bach J.‐M., Bachurski D., Baharvand H., Balaj L., Baldacchino S., Bauer N. N., Baxter A. A., Bebawy M., Beckham C., Bedina Zavec A., Benmoussa A., Berardi A. C., Bergese P., Bielska E., Blenkiron C., Bobis‐Wozowicz S., Boilard E., Boireau W., Bongiovanni A., et al., J. Extracell. Vesicles 2018, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. El Baradie K. B. Y., Nouh M., O'Brien Iii F., Liu Y., Fulzele S., Eroglu A., Hamrick M. W., Front. Cell Dev. Biol. 2020, 8, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mensink M. A., van Bockstal P.‐J., Pieters S., de Meyer L., Frijlink H. W., van der Voort Maarschalk K., Hinrichs W. L. J., de Beer T., Int. J. Pharm. 2015, 496, 792. [DOI] [PubMed] [Google Scholar]
- 58. Franks F., Biotechnology 1994, 12, 253. [DOI] [PubMed] [Google Scholar]
- 59. Montoya A., Castell J., J. Immunol. Methods 1987, 99, 13. [DOI] [PubMed] [Google Scholar]
- 60. Piszkiewicz S., Pielak G. J., Biochemistry 2019, 58, 3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.a) Chang L. L., Pikal M. J., J. Pharm. Sci. 2009, 98, 2886; [DOI] [PubMed] [Google Scholar]; b) Cicerone M. T., Pikal M. J., Qian K. K., Adv. Drug Delivery Rev. 2015, 93, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pikal‐Cleland K. A., Carpenter J. F., J. Pharm. Sci. 2001, 90, 1255. [DOI] [PubMed] [Google Scholar]
- 63. Kannan V., Balabathula P., Thoma L. A., Wood G. C., J. Liposome Res. 2015, 25, 270. [DOI] [PubMed] [Google Scholar]
- 64. Giebel B., Kordelas L., Börger V., Stem Cell Invest. 2017, 4, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schulz E., Goes A., Garcia R., Panter F., Koch M., Müller R., Fuhrmann K., Fuhrmann G., J. Controlled Release 2018, 290, 46. [DOI] [PubMed] [Google Scholar]
- 66. Fábrega M.‐J., Rodríguez‐Nogales A., Garrido‐Mesa J., Algieri F., Badía J., Giménez R., Gálvez J., Baldomà L., Front. Microbiol. 2017, 8, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hoppstädter J., Dembek A., Linnenberger R., Dahlem C., Barghash A., Fecher‐Trost C., Fuhrmann G., Koch M., Kraegeloh A., Huwer H., Kiemer A. K., Front. Immunol. 2019, 10, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Richter M., Fuhrmann K., Fuhrmann G., J. Visualized Exp. 2019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.