

Abstract
Blood sampling is a common practice to monitor health, but it entails a series of drawbacks for patients including pain and discomfort. Thus, there is a demand for more convenient ways to obtain samples. Modern analytical techniques enable monitoring of multiple bioanalytes in smaller samples, opening possibilities for new matrices, and microsampling technologies to be adopted. Interstitial fluid (ISF) is an attractive alternative matrix that shows good correlation with plasma concentration dynamics for several analytes and can be sampled in a minimally invasive and painless manner from the skin at the point‐of‐care. However, there is currently a lack of sampling devices compatible with clinical translation. Here, to tackle state‐of‐the‐art limitations, a cost‐effective and compact single‐microneedle‐based device designed to painlessly collect precisely 1.1 µL of dermal ISF within minutes is presented. The fluid is volume‐metered, dried, and stably stored into analytical‐grade paper within the microfluidic device. The obtained sample can be mailed to a laboratory, quantitatively analyzed, and provide molecular insights comparable to blood testing. In a human study, the possibility to monitor various classes of molecular analytes is demonstrated in ISF microsamples, including caffeine, hundreds of proteins, and SARS‐CoV‐2 antibodies, some being detected in ISF for the first time.
Keywords: COVID‐19, health monitoring medical devices, interstitial fluids, microneedles, painless microsampling
Modern analytical technologies enable biomarker monitoring in microsamples, and thus exploration of less invasive matrices than blood. In this article, a microneedle‐based, cost‐effective, painless device is proposed to collect interstitial fluid from the skin. This solution challenges the use of invasive techniques for applications where microsampling could reveal the pursued information, while minimizing patients’ discomfort and logistic requirements.
1. Introduction
Sampling of biological fluids is essential for patient monitoring, diagnostics, and screening. Today, venous blood sampling followed by plasma analysis is the gold standard for these applications. However, such procedure entails the use of invasive and painful methods, such as venipuncture, which needs to be performed by professional phlebotomists typically at a healthcare facility. In addition to the inconvenience for patients and the cost related to personnel and infrastructures, liquid blood needs to be kept refrigerated and handled as a biohazard, adding up logistic requirements.
The COVID‐19 pandemic has promoted and accelerated the demand for more convenient and patient‐centric approaches. For example, sampling procedures had to be moved closer to the point‐of‐care (PoC), such as homes or decentralized centers, where patients could self‐perform a procedure to collect the desired specimen.[ 1 ] Among different sampling techniques, finger pricking can be self‐performed to obtain smaller amounts of capillary blood at the PoC. However, samples must be stored in a format that is stable over time, and that can readily be analyzed using standard equipment thereafter. Currently, the storage of capillary blood in analytical‐grade paper in the form of dried blood spots (DBS) is gaining interest for use in analytical practice in different fields.[ 2 , 3 , 4 , 5 ] The sample, once dried in paper, is stable, not considered biohazardous, and can be shipped at room temperature without special handling.[ 6 ]
However, also capillary blood is still associated with pain and risk of infection. Instead, dermal interstitial fluid (ISF) offers a more accessible specimen as it can be sampled from the skin tissue in a minimally invasive manner.[ 7 , 8 , 9 , 10 , 11 , 12 ] ISF is an emerging specimen for monitoring biomarkers and biomolecules also due to its physiological relevance. Studies have shown that ISF provides the possibility to detect a large variety of analytes commonly found in blood plasma.[ 9 , 13 ] Dermal ISF is of particular relevance because of its proximity to blood and the altogether strong correlation with blood plasma dynamics, especially for low molecular weight species such as metabolites, drugs, and unbound hormones, which can rapidly diffuse through the capillary walls into the interstitial space.[ 14 ] Moreover, due to the sensitivity of modern analytical tools, microsamples of 1 µL can reliably be analyzed. This enables the practical use of new alternative matrices such as ISF which, while it cannot be sampled in large volumes from patients, can provide other advantages for the patients, in terms of comfort and reduced risks.[ 15 ] Today, ISF is widely used for continuous glucose monitoring in diabetic patients, where sensors are transdermally inserted and worn over time.[ 16 , 17 ] Even though the use of ISF as a measurement matrix has gained substantial interest in the research and medical communities, there is currently still a lack of commercial solutions and of user‐friendly ISF sampling techniques available in general. This hinders a wider translational adoption of ISF as an analytical specimen in research and healthcare.
To access dermal ISF, microneedles offer a minimally invasive solution to penetrate the epidermis and provide access to the dermis.[ 10 ] Due to their size, microneedles are painless both during insertion and after removal, potentially improving patient acceptance and compliance. Microneedles also reduce the risks of infections by drawing minute amounts of fluid and producing only microscopic wounds that do not cause long term damage to the skin.[ 18 , 19 ] Therefore, microneedles provide an attractive solution for repeated sampling for chronic patients if compared to collecting venous blood or performing finger pricks.[ 20 ] State‐of‐the‐art methods for ISF sampling consist in prototype‐stage devices based on vacuum‐generated suction,[ 10 , 21 , 22 ] mechanically applied overpressure,[ 23 , 24 ] or absorbing porous microneedles.[ 25 , 26 , 27 , 28 , 29 ] In fact, unlike blood, sampling ISF requires an external pressure gradient to be applied to extract it from the skin tissue into an external collecting device. Major issues with previously proposed devices are for example their bulkiness,[ 21 , 30 ] user‐dependence,[ 31 ] and lack of scalable manufacturability and analytical compatibility,[ 26 , 28 , 29 , 32 ] which today makes them hardly implementable as cost‐effective and disposable PoC devices in translational applications.
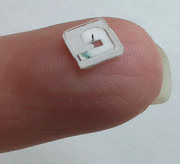
Ultimately, the realization of a complete and simple system that is designed to take care of all steps from the sampling to the generation of reliable and quantitative results is of fundamental importance for the translational adoption of ISF. In this article we present an interstitial fluid sampling device designed to painlessly extract a specific amount of ISF from human skin in ≈5 min. The device is compact and potentially cost‐effective, as it is based on a single microneedle connected to a simple microfluidic chip (Figure 1A,B). ISF is therein stably stored in an analytical‐grade paper matrix in dry format, ready for regular shipping, and subsequent analysis at a laboratory facility (Figure S1A, Supporting Information). The recovered ISF can be analyzed using state‐of‐the‐art equipment (Figure 1C), such as liquid chromatography‐tandem mass spectrometry (LC‐MS/MS), proximity extension assays, or other immunoassays. The use of this characterization equipment is at the forefront of laboratory technology, but it is also gaining traction for clinical adoption, enabling the practical use of ISF microsamples for streamlined monitoring of a broad range of bioanalytes in the near future.
Figure 1.
Overview of the concept and analytical workflow. A) Picture of the disposable part of the system, placed upside‐down on a fingertip. The device is simple, compact, and the microneedle is barely visible due to its size. B) Exploded view of the disposable device components: microneedle, microfluidic laminated plastic and adhesive layers, and analytical‐grade paper matrix. C) Overview of the entire concept, from sample collection to analytical result. The system is used to painlessly sample ISF at the point‐of‐care. The obtained sample is left to dry in the device and shipped to a laboratory using regular mail without refrigeration or special biohazard handling. The paper content can eventually be eluted and characterized using state‐of‐the‐art equipment.
2. Results and Discussion
2.1. Design of the Device
To create a minimally invasive ISF sampling device relevant for practical use, at least six key aspects have to be considered and simultaneously addressed: i) achieve painless access to ISF to limit patient discomfort, ii) preserve simplicity and contain the cost of all disposable parts of the system, iii) perform the sampling procedure within a few minutes, iv) sample ISF without detrimental contamination from other specimen such as blood or sweat, v) ensure that the sampling is compatible with subsequent analytical measurements, and vi) achieve volume metering of the extracted sample to allow analyte quantification.
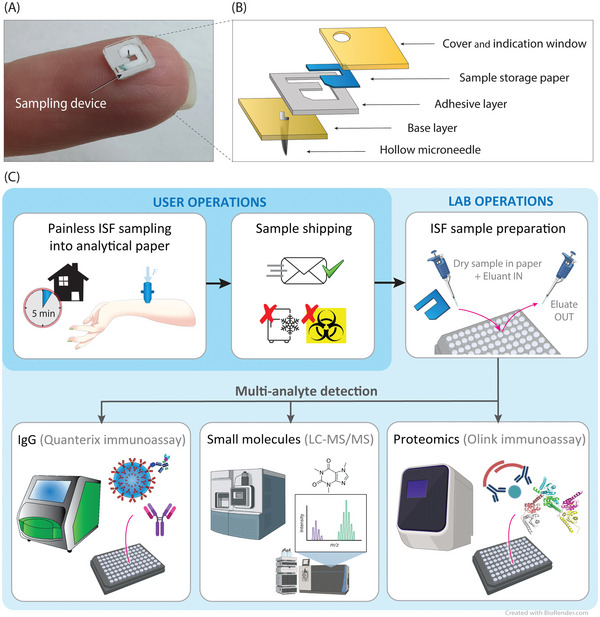
The proposed device design strives to simultaneously combine these key aspects. Figure 2A illustrates the sampling principle of the designed system applied to the skin. The system is composed of a reusable pressuring applicator and a small disposable sampling device. The pressuring applicator is responsible for the correct intradermal insertion of the microneedle and for producing a lateral radial overpressure guiding the ISF into the sampling device. The application of a certain pressure, radially converging toward the needle, is required to overcome the physiological ISF retention exercised by the skin (Figure S1B–D, Supporting Information). To reduce the force required, the applicator should be designed so that the pressure is applied to the skin as close as possible to the microneedle, for the pressure gradient not to be dispersed. Moreover, it is important that the pressure applied is as decoupled as possible from the microneedle itself, to avoid pushing it in depth on compressed skin which would result in greater tissue damage then necessary, potentially also increasing pain sensation and possible bleeding. Therefore, in our design the microneedle is not integral with the pressuring ring as in previous work.[ 30 ] The use of an applicator is also necessary to correctly insert the microneedle in the skin. In fact, the impact velocity plays a fundamental role in the correct application of the device due to the epidermis resistance and the elastic properties of the skin.[ 33 , 34 , 35 ]
Figure 2.
Device operation. A) Illustration of the cross‐section of the system. The applicator applies pressure to the skin, guiding ISF into the microneedle lumen and into the chip where it is then absorbed and stored into a paper matrix. B) Illustration of the top view of the sampling chip. The needle is in the center, where filling of the paper matrix starts to occur. A colored powder dye in the paper allows visualization of the liquid front while it progresses toward the far end of the paper geometry. Sampling is stopped by the operator when the liquid reaches the visualization window area. C–F) Sampling process from the forearm skin overtime after 20, 80, 130, and 190 s, respectively. For visualization purposes, in this device the entire paper matrix is visible through a transparent cover and not only through a visualization window at the end of the filling path. G,H) OCT images showing in vivo forearm skin cross‐sections, while inserted and after device removal, respectively. G) Microneedle, attached to a PMMA base, inserted in the skin to visualize the skin curvature around the insertion location. H) Skin imaged immediately after the device removal, showing the pricking location. Colored lines were added to the images to highlight the different components. I–L) Recovery process of the skin over time after the sampling procedure (same scale applies to all figures). Immediately after a device removal, the signs of the skin prick and the applied ring pressure were visible. Pictures were taken after I) 1 min, J) 25 min, K) 2 h, and L) 5 h, respectively. After 5 h, marks are barely visible.
The disposable sampling device is based on integrating a hydrophilic stainless steel hollow microneedle for intradermal skin penetration and a compact microfluidic chip for sample collection and storage. The use of microneedles obtainable with standard processes from stainless steel hypodermic needles is important to simplify and limit the cost of this disposable component (Figure S1F, Supporting Information), as opposed for example to silicon or polymeric microneedle arrays.[ 36 , 37 , 38 ] In fact, hollow microneedles were obtained by shortening 32 G hypodermic needles made with the standard and inexpensive process used for making these commercially today.[ 39 ] The distal end of the microneedle is assembled into a laminated microfluidic channel where a porous analytical‐grade paper matrix is placed and arranged to be in contact with the open wall of the needle. The assembled sampling device is thus compact and cost effective, due to of the materials and manufacturing technologies involved.
As opposed to previous devices storing ISF in liquid state or in unconventional matrices,[ 26 , 28 , 29 , 30 , 32 ] the presence of analytical‐grade paper and its specific geometry serve multiple critical functions within the device: fluid uptake for enhanced sampling, volume‐metering to enable subsequent quantitative analysis, stable storage upon drying to enable simplified logistics, and easy analyte recovery. The choice of using this particular type of paper is dictated by the proven compatibility of cotton‐linter‐based paper discs with analyte recovery by elution and subsequent analysis. This material has been used in DBS applications in previous internal and external studies, showing recoveries in the range of 86–99.8%, depending on the analyte of interest.[ 3 , 40 , 41 ] In fact, factors affecting dried sample recovery are the paper material, the bioanalyte itself, and the hematocrit,[ 42 ] the latter detrimental factor not playing a role in dried ISF, importantly. The thickness and geometry are instead a trade‐off between wicking speed, volume‐metering resolution, and compatibility with the manufacturing and assembly technologies used. To verify the complete filling of the paper matrix, a color indicator is used. The fluid flow in the paper dissolves a blue powder dye present in the paper matrix itself. When the colored fluid is visible at the dedicated visualization window (Figure 2B), the device can be removed from the skin and stored for subsequent analysis. The designed paper geometry ensures that all the paper matrix volume is filled with ISF before reaching the indicator location. This feature enables volume metering of the sampled ISF, permitting an accurate quantification of the analyte concentration and a comparison across different samples. Moreover, the short distance between the storing paper and the needle tip ensures minimal dead volume (<20 nL) and fluid traveling distance, of particular importance when performing ISF microsampling in the fastest possible manner.
2.2. Evaluation of the Sampling Procedure on Healthy Volunteers
ISF sampling was performed by inserting the device's microneedle into the dermis of the inner forearm of the volunteers and applying a gentle pressure to guide ISF into the device. The applicator was removed when the liquid front reached the indicator location, i.e., the edge of the visualization window illustrated in Figure 2B. The sampling time varied in the range of 1.5–10 min between different tests and individuals (n = 24), with an average of 5 min. Figure 2C–F shows the filling of the paper matrix with ISF over time during sampling. In this case, the complete filling was achieved in 3.2 min. Correct operation on an even larger and diverse population should be tested to prove operator independence and the system's functionality in everyday life conditions.
2.3. Volume‐Metering Reliability
The volume‐metering reliability was assessed by pumping an ISF surrogate solution into the device at known flow rates until it reached the visualization window. The minimum and maximum flow rates used were 0.13 and 0.34 µL min−1, respectively. These values correspond to the fastest and slowest sampling rates observed during in vivo experiments. By imposing a flow rate of 0.13 µL min−1, the indicator location was reached after dispensing 1.06 µL, while with 0.34 µL min−1, 1.13 µL were dispensed. Therefore, the variability of different filling condition was ±3.5% (n = 4), with an average collected volume of 1.1 µL. This shows accuracy and reproducibility of the volume‐metering function of the device.
2.4. Microneedle Insertion and Skin Recovery
The insertion speed and the needle geometry play fundamental roles in the skin penetration process, especially when using microneedles.[ 43 , 44 , 45 , 46 ] Several design parameters and trade‐offs come into play in the optimization of the needle penetration as well as in the minimization of pain and avoidance of unwanted bleeding.[ 47 ] Fundamental factors are application parameters (c.f. Section 4 and the Supporting Information), tip sharpness, needle length and related penetration depth, the number and spacing of needles in the case of arrays, and the uptake area to facilitate fluid sampling. During experiments, the sharpness of the tip was a fundamental factor to avoid bleeding and minimize the pain sensation. In that regard, the usage of commercially made hypodermic needle tips (Figure S1F, Supporting Information) eased penetration and its consistency. In fact, there was a clear difference when using blunter needles, or after repeated insertions of the same needle, in terms of both pain sensation increase and possible blood contamination. Moreover, longer than 1 mm needles also increased the chances of seeing bleeding after removal or traces of blood in the collection paper (Figure S1G, Supporting Information). The use of arrays of the same microneedle type was also investigated to try to accelerate the sampling rate. However, in addition to the know complications of using arrays,[ 35 , 48 ] the pain score interestingly increased significantly, going against one of the main value propositions of the device. This development direction was thus abandoned. Nevertheless, further optimization with arrays of smaller needles might reduce the perceived pain, while allowing for shorter sampling times.
Regarding the penetration depth and needle length, Figure 2G shows an optical coherence tomography (OCT) image of the needle inserted in the skin. The vertical skin bending around the needle formed by its insertion is ≈150 µm. The resulting penetration depth is thus ≈850 µm. The needle bevel opening, starting 250 µm below the device base, is thus completely inserted inside the skin. In fact, to avoid contamination by sweat and impurities on the skin surface, ISF is best sampled directly from within the dermis, by having the entire opening inserted, rather than sampling from the skin surface.[ 31 ] Figure 2H shows the pricking location after device removal. After 1 h, the pricking location could not be visualized using OCT anymore.
The microneedle insertion was experienced as painless and imperceptible by all individuals that participated in the study (n = 24). Figure 2I–L shows pictures of the recovery process of the skin until 5 h after the sampling procedure. Initially, marks from the skin prick and ring pressure were visible at and around the sampling location. The skin then completely recovered within 5 h (Figure 2L), without leaving any permanent mark.
2.5. Measurement of Caffeine Concentration
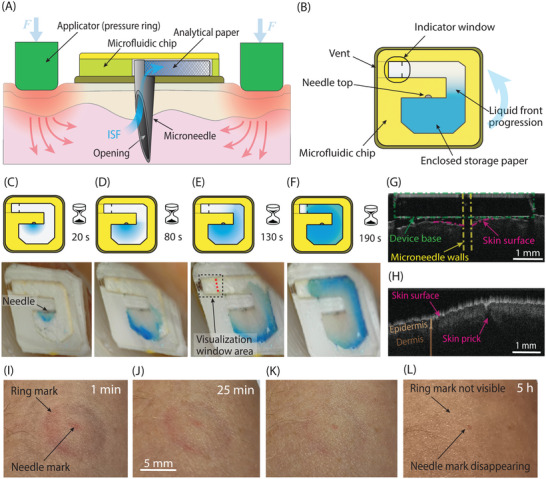
Previously, a good correlation between the concentration and dynamics of therapeutic drugs and narcotics in ISF and blood plasma has been shown.[ 49 ] To evaluate the possibility to measure relevant physiological concentrations of a compound in 1 µL dried ISF samples, we selected caffeine as a model drug. First, the concentration of caffeine in 1 µL spiked ISF surrogate was measured for calibration (inset, Figure 3A), showing the desired linearity. The caffeine concentration was measured using liquid chromatography‐based tandem mass spectrometry (LC‐MS/MS), similarly to what has previously been reported with DBS samples.[ 3 , 50 , 51 , 52 ] As a control for all ISF analytical characterizations, 10 µL of capillary blood were sampled at the same time as ISF sampling via finger pricks and stored dry in the form of DBS, until they were eluted for analysis.
Figure 3.
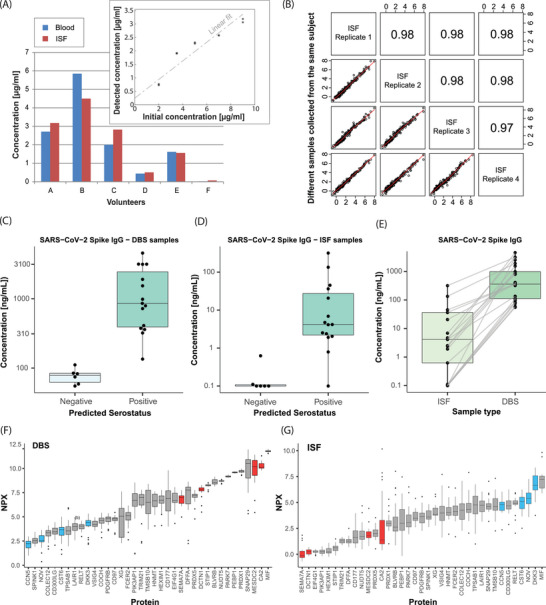
Analytical data. A) Caffeine concentration measured in one ISF and one DBS sample obtained from six different individuals (n = 6), using LC‐MS/MS. Volunteer “F” acts as a negative control. Inset: Calibration curve of spiked 1 µL surrogate ISF samples eluted from paper, in duplicates for each concentration point. B) Spearman correlation between four ISF samples collected from the same individual within minutes using four separate devices, i.e., four replicates (n = 4). C,D) Detection of SARS‐CoV‐2 antibodies (anti spike IgG) in samples collected from the self‐reported positive group (right) and a control group (left), respectively (n = 21). Measured in (C) DBS samples and (D) ISF microsamples, in single replicates. E) Comparison of measured anti spike IgG antibodies in ISF (left) and DBS (right) with lines connecting samples from the same individuals (p = 3.9 × 10−10, Wilcoxon test). F,G) Visualization of relative protein levels measured with Olink in DBS and ISF microsamples, respectively, from 23 individuals in single replicates (n = 23). The plots show the combined top 20 protein targets with the highest signal intensities in each sample type, sorted in ascending order. Highlighted in blue are examples of proteins showing higher signal intensity in ISF in relation to the other targets compared to what they have in blood, whilst the ones in red are vice versa.
Then, the samples collected from six volunteers (n = 6) were analyzed for the caffeine compound, again using LC‐MS/MS. Figure 3A shows the caffeine concentration in ISF and DBS samples, respectively, with volunteer F acting as a negative control. The results show caffeine quantification in ISF collected by using our sampling device, and the results were in concordance with those obtained from DBS samples. This demonstrates that the procedure allows microsampling and measuring relevant physiological caffeine concentrations from eluted dry ISF microsamples.
2.6. Repeatability of the Sampling and Analytical Procedures
The repeatability of the entire procedure was assessed by calculating the coefficient of variation (CV) from four ISF replicates, sampled from the same individual within 30 min using four different devices and then analyzed for protein content, as described in the dedicated section. The obtained average CV was <16.5%. Since this value combines all the possible introduced errors from sampling to storage, transport, elution, and analytical measurement, it shows a good performance of the method overall. The correlation between these four replicates is shown in Figure 3B.
2.7. Detection of SARS‐CoV‐2 Antibodies
The spreading of viral infections in the population and the need for early diagnosis and monitoring of autoimmune diseases are examples of conditions in which less invasive home‐based detection of antibodies could alleviate the burden on the healthcare systems. Previous studies showed the possibility to measure IgG, IgA, IgD, and IgE antibodies in ISF, providing comparable results to blood plasma or serum.[ 53 , 54 ] Here we evaluated the detectability of SARS‐CoV‐2 antibodies (anti‐Spike IgG) in ISF microsamples, for the first time to the best of our knowledge. Determination of the presence of IgG for a specific pathogen is an example of a use case in which a simple positive/negative answer might be sufficient, regardless of the absolute concentrations in blood plasma or any other gold standards. Moreover, the presence of SARS‐CoV‐2 antibodies in dermal ISF is currently unknown in literature.
Volunteers enrolled in the study were individuals who were previously self‐reportedly diagnosed with a COVID‐19 infection, tested positive for SARS‐CoV‐2 antibodies, or as presumably negative controls. From each individual, ISF and DBS samples were analyzed in parallel. Previous studies showed the clinical relevance of measuring COVID‐19 IgG in 10 µL DBS samples, in comparison with venous blood,[ 55 ] as well as the possibility to detect SARS‐CoV‐2 antibodies in 1 µL blood microsamples.[ 56 ] Figure 3C shows the measured anti‐Spike IgG concentration in DBS samples for the groups of presumably seropositive and seronegative volunteers. Figure 3D shows the results obtained from ISF samples. As expected, the measured IgG concentration varied substantially between individuals also within the same group and it was typically higher in subjects who recently recovered from the disease. The anti‐spike IgG concentration in ISF were concordant with but significantly lower than in DBS samples (p < 0.001), as shown in Figure 3E. Values in ISF samples were nonetheless well above the limit of quantification (limit of detection, LOD = 11 pg mL−1; lower limit of quantification, LLOQ = 44 pg mL−1) reported for the used assay (SIMOA, Quanterix), thus providing the correct sought information. Finally, Figure 3E shows clear concordance between the ISF samples and the DBS controls. Therefore, despite the high molecular weight of the antibodies and the low sample volume collected, discrimination of positive and negative subjects could be achieved. Apart from COVID‐19 and in general antiviral antibodies, minimally invasive ISF sampling could prove valuable for testing and monitoring of other antibody types, such as therapeutic antibodies.[ 57 ] These antibodies are engineered for the treatment of several diseases and are gaining traction in clinical use.[ 58 ] However, their pharmacokinetics, dynamics, and specific tissue accessibility must be tested and monitored over time during validation. Repeated ISF microsampling could thus conveniently provide insights and time‐profiling during clinical studies about the perfusion in the interstitial space of this antibody class as well.
2.8. ISF Proteome Profiling
Previous studies showed the possibility to measure a large number of proteins in ISF with variable correlation to blood plasma and some discrepancies in terms of detectable targets.[ 24 , 30 , 59 , 60 ] Another study showed a proteomic analysis of 10 µL DBS samples.[ 61 ] Here, to convey the analytical versatility of the method and the possibility to perform multianalyte detection from such small microsamples (1.1 µL), we evaluated the feasibility of profiling hundreds of proteins from the same ISF microsample eluates used for the analyses discussed in the previous sections.
The proteomic characterization of ISF was performed using a commercial proximity extension assay (Olink). Importantly, this methodology is developed for blood plasma and serum and thus the assay was not optimized for ISF testing specifically. Nevertheless, due to its high sensitivity, it is particularly suited for analysis of microsamples. We used this affinity‐based strategy to target 184 proteins involved in immunological responses, cell motility, and neurogenesis. One‐third of the analyzed proteins have been associated to diseases of various origins from neurodegenerative diseases to inflammatory and cancerous pathologies. In this study we detected in ISF, for example, several low abundant blood proteins in ISF, which are involved in inflammation processes ultimately associated with sepsis, autoimmune disorders, and cancer, such as CCL11, CCL21, IL6, and MILR1.[ 62 , 63 , 64 , 65 ] Out of the 184 targets, 37 proteins (20%) were not detected above the limit of detection of the assay in any of our ISF samples. The remaining 147 proteins were also detectable in DBS (Table S1 and Figure S2B,C, Supporting Information).
Due to the semi‐quantitative nature of the Olink technology, however, we did not directly compare protein concentrations between sample types or between different proteins within the same sample type. Instead, we performed a qualitative evaluation of the protein composition of ISF and DBS, using normalized protein expression (NPX) values as an indication of relative abundance. When protein levels in ISF and DBS from the same individual were compared, the correlation between the two sample types was moderate and protein dependent (0.36 < ρ < 0.7) (Figure S2D, Supporting Information).
Figure 3F,G shows that some proteins are ranked higher in relative abundance within ISF than within DBS. Some examples are DKK3, CCN5, NOV, and CST6 which have shown a potential as disease biomarkers. Markers relatively more abundant within ISF could for example be of interest as specific targets to be detected in ISF samples. DKK3 (dickkopf‐3) and CCN5 (cellular communication network 5) are emerging biomarkers for cardiovascular disease and cardiac fibrosis in hypertensive patients.[ 66 , 67 ] NOV (CNN3 or calponin 3) and CST6 (Cystatin M) are highly expressed respectively in the endothelial cells and in the stratum granulosum of skin.[ 68 , 69 , 70 ] These last two proteins are involved in the progression of different cancer types. In particular, CST6 has a relevant role in cutaneous carcinoma and melanoma and it could therefore be a relevant marker to be monitored in the ISF collected from the skin. Our proteomic investigation confirms ISF as an information‐rich biofluid, with great future potential in minimally invasive testing for screening and biomarker monitoring. Even if the correlation with plasma gold standards is protein‐dependent and molecular‐weight‐dependent,[ 71 ] ISF could still offer similar or potentially complementary insights for a broad range of protein targets.
2.9. Discussion of the Choice of Specimen and Testing Method for Biomarker Analysis
Pure dermal ISF is an interesting alternative and minimally invasive matrix for the detection of various biomarkers and is also appealing for the analysis of samples recovered after collection, drying, and storage. This is due to the absence of interfering cellular components typical of dried whole blood and the compatibility with the workflow of DBS characterization. When sampling ISF, it is important to do so without contamination from sweat, blood, or other sources.[ 72 ] In fact, if the concentration dynamics differ, contamination by other matrices might unpredictably alter the results. Some previously proposed solutions, e.g., based on blotting from the skin surface after pricking, are prone to errors due to contamination by sweat and exogenous impurities on the skin itself.[ 31 , 73 ] Other alternatives, based on blistering, were also shown to provide altered results induced by the sampling action itself, in addition to the substantial tissue damage.[ 74 , 75 , 76 ]
When discussing minimally invasive strategies to sample a fluid at the PoC, there are several applicable possibilities depending on the targeted analytes. For example, noninvasive options may also consist in using other bodily fluids such as saliva, sweat, or urine. However, the information that can be collected in these specimens is often physiologically limited and less reproducible than in blood or ISF. In fact, in many cases both the dynamics and the absolute concentrations of biomarkers and drugs are not strongly correlated to those observed in blood plasma. Moreover, the concentrations are highly dependent on external factors and stimulations, or not yet fully understood.[ 77 , 78 ] Therefore, the most common choice in recent years to circumvent venous blood sampling was the collection of capillary blood, which can be stored in DBS before off‐line analysis. Even though some blood microsampling devices were proposed,[ 79 , 80 ] finger pricks are still perceived as discomfortable and, despite the similarity with venous blood, capillary blood is still not considered to be an equivalent matrix to liquid venous blood. In fact, the dynamics have been shown to slightly differ and especially, once dried in DBS format, the cellular components of blood contaminate the sample by hemolysis, affecting the concentration of several analytes.[ 4 ] As discussed earlier, for a wide range of analytes, and particularly small molecules, metabolites, etc., ISF concentrations are similar to those in blood plasma. However, for other biomarkers, also between plasma and ISF there are discrepancies when comparing ISF concentrations to the values normally obtained from standard venous blood samples. Nevertheless, even when a certain marker is present in ISF at a different absolute concentration or with different dynamics, the sampling of ISF could still prove valuable or complementary. In fact, in many cases such as early screening or binary tests on the presence/absence of a certain biomarker, a qualitative outcome could be sufficient to gather the required information, as in the example of SARS‐CoV‐2 spike IgG shown in our study.
Finally, the proposed solution enables quantitative measurements of multiple analytes from the collected specimen. While the convenience of devices built for a direct readout at the PoC is unquestionable for certain applications,[ 12 , 81 ] such an approach generally entails a series of challenges and limitations. Firstly, this type of PoC devices can typically detect only a fixed and small set of analytes at a time and often with no quantification capability. Secondly, the range of detectable analytes is more limited, and the reliability of the obtained results requires individual optimization and poses challenges in terms of cost‐effectiveness and quality‐control. For applications such as diabetes monitoring, having continuous access to ISF and immediate readouts thanks to a specifically developed chemistry is fundamental to provide the required information in a timely manner. This is more easily implemented in the form of a transdermal sensor, rather than a microsampling device. However, for other applications where discrete datapoints can provide the sought information or when realizing a PoC sensor is problematic, a microsampling solution is better suited. Moreover, the developed device specifically favors repeated applications due to the minimal tissue damage caused, if compared to more invasive sampling methods.
Therefore, for versatile and reliable analysis of small sample volumes, state‐of‐the‐art analytical tools are preferred, since they can reliably provide simultaneous and quantitative information on a broad range of analytes from the same microsample. Nevertheless, the proposed device has also the potential to be compatible with PoC testing for certain analytes, despite the sensitivity challenges. The specimen flow in a paper matrix is in fact similar, for example, to standard lateral flow assays.[ 81 , 82 , 83 ]
3. Conclusion
Painless microsampling of ISF holds a great potential for biomarker monitoring. However, its translational adoption remains largely unexplored and hindered by the lack of technologies that can provide ISF collection in a relatively fast and simple manner, and simultaneously enable straightforward handling and analysis of the obtained microsamples. In this study, we presented a minimally invasive and compact device able to sample dermal ISF from the human forearm. The device is cost‐effective due to the choice of materials and fabrication techniques, the lack of complex actuators, and the reusability of most components. This device collects a controlled amount of ISF (1.1 µL) and allows accurate quantitative multianalyte measurement of caffeine, SARS‐CoV‐2 antibodies, and hundreds of soluble proteins from such microsample. Therefore, the proposed device enables a more convenient PoC and patient‐centric approach in health monitoring. This could be especially valuable for homecare, to monitor chronically ill patients via repeated sampling, for people scared of needles and blood, as well as in pediatric settings. It also has applicability to large‐scale clinical trials and screening studies that are today ethically questionable when using more invasive sampling techniques.
4. Experimental Section
Fabrication
The microneedles were realized by processing standard hypodermic needles to obtain 1 mm protruding microneedles (1.9 mm long overall, considering the portion assembled into the microfluidic chip) with the design illustrated in Figure 1B (and Figure S1F, Supporting Information). The uncoated 32 G stainless steel needles (Novo Nordisk A/S) were produced by a commercial supplier using standard needle‐forming processes,[ 39 ] and then further processed in the laboratory using a femtosecond laser (Spirit, Spectra‐Physics, MKS Instruments) for fast prototyping of the distal‐end cut, to be shortened to the desired length. Importantly, the final shape of the needle could potentially be obtained using purely standard processes, for future large‐scale manufacturing. The microneedle surface was finally cleaned and sonicated in a 10% citric acid solution in deionized (DI) water, resulting in enhanced hydrophilic surfaces, which enhanced the sampling speed and reduced the required pressure required to start extraction.
The microfluidic chip base, in which the microneedle is vertically assembled, was made of polymethyl methacrylate (PMMA). The top part was made of two 170 µm thick layers of a double‐adhesive tape (Tesa SE) and a plastic foil (PP2280, 3 M, Sweden). 340 µm thick analytical‐grade chromatography blotting paper filters (Grade 238, Ahlstrom‐Munksjö) were laser‐processed to obtain a geometry able to contain ≈1 µL of liquid, with the shape illustrated in Figures 1B and 2B. The paper fibers bridge the liquid flow from the needle lumen into the perpendicular microfluidic channel, absorbing the liquid by capillary action. Few µg of dry blue dye powder (Patent Blue VF, Sigma‐Aldrich) were placed on the paper matrix to facilitate the visualization of the front of the ISF flow during sampling. The desired geometries were patterned into the various materials using a CO2 laser (VSL 2.3, Universal Laser Systems Inc.) and were then laminated and assembled. The applicator is designed to be reusable after sterilization. For this study, the applicator's size was not optimized, but it could be further reduced, while preserving a dimension and design compatible with self‐sampling and which can easily and ergonomically be used by patients. The various versions were made of assembled 3D‐printed components (Form 3, Formlabs Inc.) and a metallic spring (Sodemann Industrifjedre A/S).
Importantly, the proposed design and materials involved would allow for scalable and currently in‐use manufacturing methods: the microneedles are already obtained by standard hypodermic needle processing, the compact microfluidic chip is made by lamination technology employing cheap materials (plastic sheets, adhesives, and paper)[ 3 , 84 ] and has a very small footprint (5 × 5 mm2), the needle‐to‐chip integration could be scaled up for example using previously reported techniques,[ 85 ] and the applicator could inexpensively be fabricated by injection molding.
Characterization of the Device Properties
The metering accuracy was evaluated by pumping an artificial ISF solution (a mixture of phosphate buffered saline solution, glucose, bovine serum albumin, and dye) into the microneedle lumen at defined flow rates, which were varied between the different runs (n = 4). At the time in which the liquid crossed the line, corresponding to the visualization window (c.f. Figure 2B), the flow was stopped, and the total amount dispensed by the tool was recorded. For these tests, a syringe pump (NE‐1002X Programmable Microfluidics Syringe Pump, Pump Systems Inc., USA) was used. The experiments were video recorded using a portable microscope camera (43‐AM4113ZTL Dino‐Lite, AnMo Electronics Corporation). To evaluate the penetration depth and the effect of microneedle penetration into the skin, an optical coherence tomography tool (OQ Labscope 2.0, Lumedica Inc.) was used.
Volunteers and Sample Collection
The study was approved by the regional ethical review board in Stockholm (DNR 2020‐03976). All volunteers signed an informed consent form to participate in the study. ISF samples were collected from 24 healthy human volunteers, including male (n = 13) and female subjects (n = 11), different ethnicities, and within an age range between 24 and 65 years old. The chosen sampling location was the inner forearm, because this is easily accessible in most circumstances. Moreover, the thickness of the forearm skin layers allows access to the dermal region, rich of homogeneously distributed ISF with high correlation with concentration dynamics in blood, using 1 mm long microneedles.[ 86 , 87 ] The spring‐loaded applicator and the pressuring ring were used to achieve insertion and create a radial pressure, resulting from an applied force of 10 N, to guide dermal ISF into the microneedle opening. The sampling continued until the filling indicator was visualized by the operator. Nevertheless, the volunteers could decide to interrupt the procedure at any time, if desired.
As a comparative control for analytical characterization, dried blood spots of 10 µL capillary whole blood were collected at the same time of ISF sampling from the volunteers using finger pricking and pipetting the correct amount in a cotton‐linter‐based paper filter disk (Whatman 903 DBS paper, GE Healthcare). Previous studies showed the possibility to correctly detect SARS‐CoV‐2 antibodies and caffeine concentration from this type of samples, with results comparable to venous blood (37).
To guarantee a balanced population for the study of SARS‐CoV‐2 antibodies detection, the volunteers were asked if at any prior stage they had received a positive test result of either virus infection or antibody presence. None of the volunteers at the time of sampling was yet vaccinated against SARS‐CoV‐2 infection. For the caffeine study, the volunteers were also asked if they had an intake of any coffee in order to ensure caffeine presence in the majority of the test subjects, but also to include a negative control. Finally, for proteome characterization, the samples from the same recruited volunteers were used without specific inclusion criteria.
Sample Preparation
Each ISF sample was eluted in 30 µL elution buffer (phosphate buffered saline (PBS) with 0.05% Tween TM 20 and cOmpleteTM Protease Inhibitor Cocktail (Roche, Prod: 04 693 116 001, Lot #41353800)) in a nonskirted 96‐well polymerase chain reaction (PCR) plate (ThermoFisher, Cat number: AB0600) and each DBS sample was eluted in 100 µL elution buffer in a 96‐well tissue culture plate (VWR, Cat number: 10861–562). The samples were eluted for 1 h with gentle shaking at 230 rpm at room temperature. The DBS samples were then spun at 3000 rpm for 3 min, and 70 µg of clear supernatant was transferred into a new PCR plate prior to analysis.
Analysis of Caffeine Concentration
Caffeine was measured using LC‐MS/MS, a method commonly used for characterization of plasma and other biological matrices.[ 50 , 88 , 89 , 90 ] For this purpose, similar LC‐MS/MS protocols were previously developed for analysis in whole blood from DBS paper.[ 6 ] For the calibration measurement in Figure 3A (inset), 1 µL aliquots of dyed artificial ISF with the addition of caffeine at concentrations varying from 0.1 to 9 µg mL−1 were pipetted into different paper pieces, in duplicates. Samples were left to dry into the paper matrix and stored. The dried samples were then eluted and characterized using LC‐MS/MS (at MVZ Labor Dessau GmbH, Germany). For the analysis reported in Figure 3A, 10 µL DBS‐/ISF extracts from the eluates from six individuals were fortified with deuterated internal standard in acetonitrile (625 ng mL−1 extract). After protein precipitation and dilution, 4 µL were injected into a Waters Acquity I‐Class ultra performance liquid chromatography system using a Waters BEH‐Phenyl 1.7 µm, 2.1 × 100 mm2 column with gradient separation connected to an MS‐detector (Waters XEVO TQ‐S, Eschborn). Three transitions were monitored in selective reaction monitoring (SRM) for caffeine and the internal standard (caffeine‐d9).
Quantification of SARS‐CoV‐2 Antibodies
Assays were performed using a Simoa TM SARS‐CoV‐2 Spike IgG kit (Quanterix, Product number: 103762, Lot number: 502822) according to manufacturer's instructions, with adaptation to the analysis of eluates from paper both for ISF and DBS. The recommended plasma dilution for the kit is 1:1000. A dilution of plasma was estimated in DBS eluates of 1:20, and therefore the DBS eluates were then in turn diluted 1:50 in sample diluent. ISF was diluted 1:30 when eluted from the paper and then the eluate was rediluted 1:4 in sample diluent. Kit specific calibrators, controls, and diluted eluates were incubated with SARS‐CoV‐2 Spike antigen coupled beads for 30 min at 30 °C, 800 rpm, followed by a 10 min incubation, 30 °C, 800 rpm, with Detector Reagent. Streptavidin‐ ß‐galactosidase (SßG) enzyme reagent was then diluted 1:2 with SßG Diluent and 100 µL solution was incubated with the beads for 10 min, at 30 °C, 800 rpm. After washing the beads were loaded into the Quanterix SR‐X instrument and mixed with Resorufin ß‐d‐galactopyranoside substrate to generate the signals. The SR‐X instrument allows for capturing images of individual beads for a high sensitivity. Signals are reported as average enzyme per bead, and the instrument implements a four‐parameter logistic curve fit based on the calibrator signals and theoretical concentrations to calculate the concentration of human anti‐SARS‐CoV‐2 Spike antibodies (IgG) in the samples.
Profiling of Protein Composition of ISF and DBS by Affinity Proteomics
The proximity extension assays (Olink Proteomics AB) were performed using the Development panel (Product No. 95352, Lot No. B03102) and the Immune Response panel (Product No. 95320, Lot No. 95320, Lot No. B04304). Affinity‐based assays allow to investigate low abundant proteins.[ 91 ] The assays were performed according to manufacturer's instructions for the analysis of plasma samples, with minor adjustments in sample dilution for the Development panel. In brief, the DBS eluates were run neat on the Immune Response panel and were diluted to 1:5 for the Development panel with a recommended plasma dilution of 1:100, assuming a 1:20 dilution during the elution procedure. The ISF eluates were run neat on both panels. For each panel, the samples were incubated with a mixture of 92 pairs of oligonucleotide‐labeled antibodies overnight. Upon target recognition, the oligonucleotides on the antibody pairs are brought in proximity which allows for hybridization. After subsequent proximity extension, the DNA reporter sequences are amplified by real time PCR and the sequences are quantified using a microfluidic real‐time PCR instrument (Biomark HD, Fluidigm). Data were then processed using NPX manager software (v. 2.2.0.288, Olink Proteomics AB) and protein relative quantification was reported as NPX (values).
Statistical Analysis
Data analysis was performed in R, version 3.6.0,[ 92 ] except for caffeine sample analysis for which Microsoft Excel was used. Data were presented as mean value ± standard deviation, unless stated otherwise. Wilcoxon test was performed using the “stat_compare_means” function from the Ggpubr R package (version 0.4.0) and it was used to analyze statistical significance for ISF and DBS anti‐Sars Cov2 Spike antibody levels. A p‐value <0.05 was considered statistically significant. Correlations between replicated samples were computed using the “corr.test” function of the “psych” R package (version 2.1.9).
Conflict of Interest
F.R., G.S., and N.R. have a pending patent application.
Supporting information
Supporting Information
Acknowledgements
The authors thank all the volunteers who participated in the study. This work was financially supported by the Foundation Olle Engkvist Byggmästare, the Swedish Foundation for Strategic Research (SSF), and the Horizon 2020 Framework Programme (EU‐H2020‐FET Proactive, project WatchPlant, grant agreement ID: 101017899). The authors would also like to thank the Affinity Proteomics Stockholm Unit at SciLifeLab for conducting analytical studies.
Ribet F., Bendes A., Fredolini C., Dobielewski M., Böttcher M., Beck O., Schwenk J. M., Stemme G., Roxhed N., Microneedle Patch for Painless Intradermal Collection of Interstitial Fluid Enabling Multianalyte Measurement of Small Molecules, SARS‐CoV‐2 Antibodies, and Protein Profiling. Adv. Healthcare Mater. 2023, 12, 2202564. 10.1002/adhm.202202564
Contributor Information
Federico Ribet, Email: ribet@kth.se.
Niclas Roxhed, Email: roxhed@kth.se.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. James C. A., Barfield M. D., Maass K. F., Patel S. R., Anderson M. D., Nat. Med. 2020, 26, 1810. [DOI] [PubMed] [Google Scholar]
- 2. Wagner M., Tonoli D., Varesio E., Hopfgartner G., Mass Spectrom. Rev. 2016, 35, 361. [DOI] [PubMed] [Google Scholar]
- 3. Lenk G., Sandkvist S., Pohanka A., Stemme G., Beck O., Roxhed N., Bioanalysis 2015, 7, 2085. [DOI] [PubMed] [Google Scholar]
- 4. Lehmann S., Delaby C., Vialaret J., Ducos J., Hirtz C., Clin. Chem. Lab. Med. 2013, 51, 1897. [DOI] [PubMed] [Google Scholar]
- 5. Mahadeva S. K., Walus K., Stoeber B., ACS Appl. Mater. Interfaces 2015, 7, 8345. [DOI] [PubMed] [Google Scholar]
- 6. Lenk G., Ullah S., Stemme G., Beck O., Roxhed N., Anal. Chem. 2019, 91, 5558. [DOI] [PubMed] [Google Scholar]
- 7. Kiang T. K. L., Ranamukhaarachchi S. A., Ensom M. H. H., Pharmaceutics 2017, 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fogh‐Andersen N., Altura B. M., Altura B. T., Siggaard‐Andersen O., Clin. Chem. 1995, 41, 1522. [PubMed] [Google Scholar]
- 9. Venkatesh B., Morgan T. J., Cohen J., Crit. Care Med. 2010, 38, S630. [DOI] [PubMed] [Google Scholar]
- 10. Samant P. P., Prausnitz M. R., Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim Y., Prausnitz M. R., Nat. Biomed. Eng. 2021, 5, 3. [DOI] [PubMed] [Google Scholar]
- 12. Li C. G., Joung H.‐A., Noh H., Song M.‐B., Kim M.‐G., Jung H., Lab Chip 2015, 15, 3286. [DOI] [PubMed] [Google Scholar]
- 13. Samant P. P., Niedzwiecki M. M., Raviele N., Tran V., Mena‐Lapaix J., Walker D. I., Felner E. I., Jones D. P., Miller G. W., Prausnitz M. R., Sci. Transl. Med. 2020, 12, 10.1126/scitranslmed.aaw0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kiang T. K. L., Schmitt V., Ensom M. H. H., Chua B., Häfeli U. O., J. Pharm. Sci. 2012, 101, 4642. [DOI] [PubMed] [Google Scholar]
- 15. Henion J., Oliveira R. V., Chace D. H., Bioanalysis 2547, 2013, 5. [DOI] [PubMed] [Google Scholar]
- 16. Vaddiraju S., Burgess D. J., Tomazos I., Jain F. C., Papadimitrakopoulos F., J. Diabetes Sci. Technol. 2010, 4, 1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hilgers M. E., Racchini J. R., Hoegh T. B., Diabetes Technol. Ther. 2001, 3, 81. [DOI] [PubMed] [Google Scholar]
- 18. Le Floch J. P., Bauduceau B., Levy M., Mosnier‐Pudar H., Sachon C., Kakou B., Diabetes Care 2008, 31, e73. [DOI] [PubMed] [Google Scholar]
- 19. Lodwig V., Kulzer B., Schnell O., Heinemann L., J. Diabetes Sci. Technol. 2014, 8, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vicente‐Perez E. M., Larrañeta E., McCrudden M. T. C., Kissenpfennig A., Hegarty S., McCarthy H. O., Donnelly R. F., Eur. J. Pharm. Biopharm. 2017, 117, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu H., Li D., Ji Y., Zhang X., Xu K., Key Eng. Mater. 2013, 562, 571. [Google Scholar]
- 22. Wang P. M., Cornwell M., Prausnitz M. R., Diabetes Technol. Ther. 2005, 7, 131. [DOI] [PubMed] [Google Scholar]
- 23. Mukerjee E. V., Collins S. D., Isseroff R. R., Smith R. L., Sens. Actuators, A 2004, 114, 267. [Google Scholar]
- 24. Tran B. Q., Miller P. R., Taylor R. M., Boyd G., Mach P. M., Rosenzweig C. N., Baca J. T., Polsky R., Glaros T., J. Proteome Res. 2018, 17, 479. [DOI] [PubMed] [Google Scholar]
- 25. Chen J., Wang M., Ye Y., Yang Z., Ruan Z., Jin N., Biomed. Microdevices 2019, 21, 63. [DOI] [PubMed] [Google Scholar]
- 26. Cahill E. M., Keaveney S., Stuettgen V., Eberts P., Ramos‐Luna P., Zhang N., Dangol M., O'Cearbhaill E. D., Acta Biomater. 2018, 80, 401. [DOI] [PubMed] [Google Scholar]
- 27. Mandal A., Boopathy A. V., Lam L. K. W., Moynihan K. D., Welch M. E., Bennett N. R., Turvey M. E., Thai N., Van J. H., Love J. C., Hammond P. T., Irvine D. J., Sci. Transl. Med. 2018, 10, 10.1126/scitranslmed.aar2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Romanyuk A. V., Zvezdin V. N., Samant P., Grenader M. I., Zemlyanova M., Prausnitz M. R., Anal. Chem. 2014, 86, 10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang H., Zheng M., Yu X., Than A., Seeni R. Z., Kang R., Tian J., Khanh D. P., Liu L., Chen P., Xu C., Adv. Mater. 2017, 29, 1702243. [DOI] [PubMed] [Google Scholar]
- 30. Miller P. R., Taylor R. M., Tran B. Q., Boyd G., Glaros T., Chavez V. H., Krishnakumar R., Sinha A., Poorey K., Williams K. P., Branda S. S., Baca J. T., Polsky R., Commun. Biol. 2018, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kolluru C., Gupta R., Jiang Q., Williams M., Gholami Derami H., Cao S., Noel R. K., Singamaneni S., Prausnitz M. R., ACS Sens. 2019, 4, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He R., Niu Y., Li Z., Li A., Yang H., Xu F., Li F., Adv. Healthcare Mater. 2020, 9, 1901201. [DOI] [PubMed] [Google Scholar]
- 33. Verbaan F. J., Bal S. M., van den Berg D. J., Dijksman J. A., van Hecke M., Verpoorten H., van den Berg A., Luttge R., Bouwstra J. A., J. Controlled Release 2008, 128, 80. [DOI] [PubMed] [Google Scholar]
- 34. Crichton M. L., Donose B. C., Chen X., Raphael A. P., Huang H., Kendall M. A. F., Biomaterials 2011, 32, 4670. [DOI] [PubMed] [Google Scholar]
- 35. Ranamukhaarachchi S. A., Stoeber B., Biomed. Microdevices 2019, 21, 100. [DOI] [PubMed] [Google Scholar]
- 36. Donnelly R. F., Mooney K., Caffarel‐Salvador E., Torrisi B. M., Eltayib E., McElnay J. C., Ther. Drug Monit. 2013, 36, 10. [DOI] [PubMed] [Google Scholar]
- 37. Smith R. L., Collins S. D., Duy J., Smith R. L., Collins S. D., Duy J., Minogue T. D., SPIE 2019, 10491, 10.1117/12.2299264. [DOI] [Google Scholar]
- 38. Jeong H. R., Lee H. S., Choi I. J., Park J. H., J. Drug Targeting 2017, 25, 29. [DOI] [PubMed] [Google Scholar]
- 39. Moustafa F., Hoverson K., Dover J. S., Arndt K. A., J. Drugs Dermatol. 2021, 20, 44. [DOI] [PubMed] [Google Scholar]
- 40. Bloem K., Schaap T., Boshuizen R., Kneepkens E. L., Wolbink G. J., De Vries A., Rispens T., Bioanalysis 2018, 10, 815. [DOI] [PubMed] [Google Scholar]
- 41. Lee Y., Lee J., Kang H., BioTechniques 2019, 67, 219. [DOI] [PubMed] [Google Scholar]
- 42. De Vries R., Barfield M., Van De Merbel N., Schmid B., Siethoff C., Ortiz J., Verheij E., Van Baar B., Cobb Z., White S., Timmerman P., Bioanalysis 2013, 5, 2147. [DOI] [PubMed] [Google Scholar]
- 43. Römgens A. M., Bader D. L., Bouwstra J. A., Baaijens F. P. T., Oomens C. W. J., J. Mech. Behav. Biomed. Mater. 2014, 40, 397. [DOI] [PubMed] [Google Scholar]
- 44. Coulman S. A., Birchall J. C., Alex A., Pearton M., Hofer B., O'Mahony C., Drexler W., Považay B., Pharm. Res. 2011, 28, 66. [DOI] [PubMed] [Google Scholar]
- 45. Enfield J., O'Connell M.‐L., Lawlor K., Jonathan E., O'Mahony C., Leahy M., J. Biomed. Opt. 2010, 15, 046001. [DOI] [PubMed] [Google Scholar]
- 46. Leone M., van Oorschot B. H., Nejadnik M. R., Bocchino A., Rosato M., Kersten G., O'mahony C., Bouwstra J., Van Der Maaden K., Pharmaceutics 2018, 10, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gill H. S., Denson D. D., Burris B. A., Prausnitz M. R., Clin. J. Pain 2008, 24, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kochhar J. S., Quek T. C., Soon W. J., Choi J., Zou S., Kang L., J. Pharm. Sci. 2013, 102, 4100. [DOI] [PubMed] [Google Scholar]
- 49. Kiang T. K. L., Schmitt V., Ensom M. H. H., Chua B., Häfeli U. O., Int. J. Drug Dev. Res. 2011, 3, 26. [Google Scholar]
- 50. Li W., Tse F. L. S., Biomed. Chromatogr. 2010, 24, 49. [DOI] [PubMed] [Google Scholar]
- 51. Freeman J. D., Rosman L. M., Ratcliff J. D., Strickland P. T., Graham D. R., Silbergeld E. K., Clin. Chem. 2018, 64, 656. [DOI] [PubMed] [Google Scholar]
- 52. Beck O., Kenan Modén N., Seferaj S., Lenk G., Helander A., Clin. Chim. Acta 2018, 479, 38. [DOI] [PubMed] [Google Scholar]
- 53. Kolluru C., Williams M., Yeh J. S., Noel R. K., Knaack J., Prausnitz M. R., Biomed. Microdevices 2019, 21, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arévalo M. T., Rizzo G. M., Polsky R., Glaros T., Mach P. M., J. Proteome Res. 2019, 18, 2381. [DOI] [PubMed] [Google Scholar]
- 55. Roxhed N., Bendes A., Dale M., Mattsson C., Hanke L., Dodig‐Crnković T., Christian M., Meineke B., Elsässer S., Andréll J., Havervall S., Thålin C., Eklund C., Dillner J.., Beck O., Thomas C. E., Mclnerney G.., Hong M.‐G., Murrell B., Fredolini C., Schwenk J. M., Nat. Commun. 2021, 12, 3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Norman M., Gilboa T., Ogata A. F., Maley A. M., Cohen L., Busch E. L., Lazarovits R., Mao C. P., Cai Y., Zhang J., Feldman J. E., Hauser B. M., Caradonna T. M., CHen B., Schmidt A. G., Alter G., Charles R. C., Ryan E. T., Walt D. R., Nat. Biomed. Eng. 2020, 4, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chames P., Van Regenmortel M., Weiss E., Baty D., Br. J. Pharmacol. 2009, 157, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu R. M., Hwang Y. C., Liu I. J., Lee C. C., Tsai H. Z., Li H. J., Wu H. C., J. Biomed. Sci. 2020, 27, 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang Z., Luan J., Seth A., Liu L., You M., Gupta P., Rathi P., Wang Y., Cao S., Jiang Q., Zhang X., Gupta R., Zhou Q., Morrissey J. J., Scheller E. L., Rudra J. S., Singamaneni S., Nat. Biomed. Eng. 2021, 5, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Taylor R. M., Miller P. R., Ebrahimi P., Polsky R., Baca J. T., Lab. Anim. 2018, 52, 526. [DOI] [PubMed] [Google Scholar]
- 61. Fredolini C., Dodig‐Crnković T., Bendes A., Dahl L., Dale M., Mattsson C., Thomas C. E., Beck O., Roxhed N., Schwenk J. M., medRxiv 2021, 10.1101/2021.11.15.21266315. [DOI]
- 62. Polosukhina D., Singh K., Asim M., Barry D. P., Allaman M. M., Hardbower D. M., Piazuelo M. B., Washington M. K., Gobert A. P., Wilson K. T., Coburn L. A., Oncogene 2021, 40, 6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Y., Shao Z., Jiang E., Zhou X., Wang L., Wang H., Luo X., Chen Q., Liu K., Shang Z., J. Cell. Physiol. 2020, 235, 5995. [DOI] [PubMed] [Google Scholar]
- 64. Odler B., Bikov A., Streizig J., Balogh C., Kiss E., Vincze K., Barta I., Horváth I., Müller V., Lupus 2017, 26, 572. [DOI] [PubMed] [Google Scholar]
- 65. Berbers R. M., Drylewicz J., Ellerbroek P. M., van Montfrans J. M., Dalm V. A. S. H., van Hagen P. M., Keller B., Warnatz K., van de Ven A., van Laar J. M., Nierkens S., Leavis H. L., J. Clin. Immunol. 2021, 41, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Piek A., Smit L., Suthahar N., Bakker S. J. L., de Boer R. A., Silljé H. H. W., Sci. Rep. 2021, 11, 8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huang A., Li H., Zeng C., Chen W., Wei L., Liu Y., Qi X., Front. Pharmacol. 2020, 11, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nair V. A., Al‐khayyal N. A., Sivaperumal S., Abdel‐Rahman W. M., World J. Gastrointest. Oncol. 2019, 11, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jin L., Zhang Y., Li H., Yao L., Fu D., Yao X., Xu L. X., Hu X., Hu G., Cell Res. 2012, 22, 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lalmanach G., Kasabova‐Arjomand M., Lecaille F., Saidi A., Cancers (Basel) 2021, 13, 1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Heikenfeld J., Jajack A., Feldman B., Granger S. W., Gaitonde S., Begtrup G., Katchman B. A., Nat. Biotechnol. 2019, 37, 407. [DOI] [PubMed] [Google Scholar]
- 72. Sakaguchi K., Hirota Y., Hashimoto N., Ogawa W., Hamaguchi T., Matsuo T., Miyagawa J. I., Namba M., Sato T., Okada S., et al., J. Diabetes Sci. Technol. 2013, 7, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kolluru C., Williams M., Chae J., Prausnitz M. R., Adv. Healthcare Mater. 2019, 8, 1801262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kiistala U., J. Invest. Dermatol. 1968, 50, 129. [DOI] [PubMed] [Google Scholar]
- 75. Kool J., Reubsaet L., Wesseldijk F., Maravilha R. T., Pinkse M. W., D'Santos C. S., Van Hilten J. J., Zijlstra F. J., Heck A. J. R., Proteomics 2007, 7, 3638. [DOI] [PubMed] [Google Scholar]
- 76. Müller A. C., Breitwieser F. P., Fischer H., Schuster C., Brandt O., Colinge J., Superti‐Furga G., Stingl G., Elbe‐Bürger A., Bennett K. L., J. Proteome Res. 2012, 11, 3715. [DOI] [PubMed] [Google Scholar]
- 77. Pichini S., Altieri I., Zuccaro P., Pacifici R., Clin. Pharmacokinet. 1996, 30, 211. [DOI] [PubMed] [Google Scholar]
- 78. Capiau S., Alffenaar J. W., Stove C. P., in Clinical Challenges in Therapeutic Drug Monitoring, Elsevier Inc., 2016, p. 279. [Google Scholar]
- 79. Li T., Barnett A., Rogers K. L., Gianchandani Y. B., Lab Chip 2009, 9, 3495. [DOI] [PubMed] [Google Scholar]
- 80. Blicharz T. M., Gong P., Bunner B. M., Chu L. L., Leonard K. M., Wakefield J. A., Williams R. E., Dadgar M., Tagliabue C. A., El Khaja R., Marlin S. L., Haghgooie R., Davis S. P., Chickering D. E., Bernstein H., Nat. Biomed. Eng. 2018, 2, 151. [DOI] [PubMed] [Google Scholar]
- 81. Jiang X., Lillehoj P. B., Microsyst. Nanoeng. 2020, 6, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chinnasamy T., Segerink L. I., Nystrand M., Gantelius J., Andersson Svahn H., Analyst 2014, 139, 2348. [DOI] [PubMed] [Google Scholar]
- 83. Yetisen A. K., Akram M. S., Lowe C. R., Lab Chip 2013, 13, 2210. [DOI] [PubMed] [Google Scholar]
- 84. Cassano C. L., Fan Z. H., Microfluid. Nanofluid. 2013, 15, 173. [Google Scholar]
- 85. Rajabi M., Roxhed N., Shafagh R. Z., Haraldson T., Fischer A. C., Van Der Wijngaart W., Stemme G., Niklaus F., PLoS One 2016, 11, e0166330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cengiz E., Tamborlane W. V., Diabetes Technol Ther 2009, 11, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Groenendaal W., Schmidt K. A., von Basum G., van Riel N. A. W., Hilbers P. A. J., Diabetes Technol. Ther. 2008, 10, 283. [DOI] [PubMed] [Google Scholar]
- 88. Chen Z., Fu L., Liu X. A., Yang Z., Li W., Li F., Luo Q., Chin. Chem. Lett. 2022, 33, 3101. [Google Scholar]
- 89. Shu C., Li T. F., Li D., Li Z. Q., Xia X. H., Chin. Chem. Lett. 2022, 33, 916. [Google Scholar]
- 90. Terkelsen T., Pernemalm M., Gromov P., Børresen‐Dale A. L., Krogh A., Haakensen V. D., Lethiö J., Papaleo E., Gromova I., Mol. Oncol. 2021, 15, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Petrera A., Von Toerne C., Behler J., Huth C., Thorand B., Hilgendorff A., Hauck S. M., J. Proteome Res. 2021, 20, 751. [DOI] [PubMed] [Google Scholar]
- 92. Ihaka R., Gentleman R., J. Comput. Graph Stat. 1996, 5, 299. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.