

Abstract
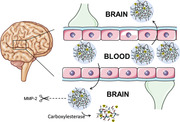
Despite profound advances in treatment approaches, gliomas remain associated with very poor prognoses. The residual cells after incomplete resection often migrate and proliferate giving a seed for highly resistant gliomas. The efficacy of chemotherapeutic drugs is often strongly limited by their poor selectivity and the blood brain barrier (BBB). Therefore, the development of therapeutic carrier systems for efficient transport across the BBB and selective delivery to tumor cells remains one of the most complex problems facing molecular medicine and nano‐biotechnology. To address this challenge, a stimuli sensitive nanogel is synthesized using pre‐polymer approach for the effective delivery of nano‐irradiation. The nanogels are cross‐linked via matrix metalloproteinase (MMP‐2,9) substrate and armed with Auger electron emitting drug 5‐[125I]Iodo‐4”‐thio‐2”‐deoxyuridine ([125I]ITdU) which after release can be incorporated into the DNA of tumor cells. Functionalization with diphtheria toxin receptor ligand allows nanogel transcytosis across the BBB at tumor site. Functionalized nanogels efficiently and increasingly explore transcytosis via BBB co‐cultured with glioblastoma cells. The subsequent nanogel degradation correlates with up‐regulated MMP2/9. Released [125I]ITdU follows the thymidine salvage pathway ending in its incorporation into the DNA of tumor cells. With this concept, a highly efficient strategy for intracellular delivery of radiopharmaceuticals across the challenging BBB is presented.
Keywords: blood−brain barrier, brain tumors, diphtheria toxin receptors, nanogels, radiolabeled thymidine analogue
Matrix metalloprotease (MMP) sensitive nanogels are synthesized for effective delivery of a radiopharmaceutical ([125I]ITdU) across the blood−brain barrier (BBB). The nanogels show efficient transcytosis across the in vitro model of BBB co‐cultured with glioblastoma cells. Once transcytosed, nanogels degrade in an up‐regulated MMP2/9 environment of glioblastoma and release the radiopharmaceutical, which gets incorporated into the DNA of tumor cells by thymidine salvage pathway.
1. Introduction
The nanomedicine drug delivery strategies aim at the improvement of drug therapeutic efficacy. Some of them affect the drug stability and by this, its kinetics, some its specificity. In more than 70% of high‐grade gliomas, the blood−brain barrier (BBB) is disrupted allowing passive targeting by exploiting the enhanced permeability and retention (EPR) effect. However, since high‐grade gliomas rapidly infiltrate the surrounding brain tissue, the drug carriers still need to deal with efficient and tumor‐selective drug delivery across the intact BBB. This barrier acts as a neuroprotective shield that protects the brain by restricting the free passage of molecules from the blood into the central nervous system (CNS) and hence it regulates homeostasis.[ 1 ] While intrathecal‐based drug delivery is considered inefficient, the transvascular route of drug administration, following systemic injection, could virtually treat all neurons in the brain, considering that almost every neuron in the human brain has its capillary.[ 2 ] Another possible strategy is to choose nasal administration, which in combination with a suitable delivery system (e.g., poly(N‐vinyl pyrrolidone)‐based nanogels) exploits retrograde axonal transport.[ 3 ] In the development of drug delivery systems, the most cited strategies to circumvent the BBB refers either to an invasive modulation of the barrier like temporal ultrasound‐mediated disruption via microbubbles[ 4 ] or active transport across it.[ 5 ] Recently, May et al. presented the potential of opening BBB via sonopermeation as a safe and efficient delivery option of nanomedicine formulations to and into the brain.[ 6 ] However, the transvascular route to the brain is virtually impenetrable for the majority of therapeutic substances.[ 1 , 7 ] Therefore, for the treatment of neurodegenerative disorders like Alzheimer's, Parkinson, or brain tumors, the drug or drug delivery system should: i) have the ability for efficient and selective crossing the BBB, ii) be unrecognized by efflux pumps, and iii) be protected from early degradation. With the advances in the field of nanomedicine, tailored drug loaded particles with the size of ten to a few hundred nanometers have been proposed as an intriguing tool to enhance and improve drug transport across the BBB.[ 5 , 8 ] Applying them, the crossing of the BBB is solely dependent on the physicochemical and biomimetic features of the nanoparticle, irrespective of the chemical cargo structure. For functionalized nanoparticles, the most relevant strategies rely either on receptor‐mediated or adsorptive‐mediated transcytosis.[ 5d ] This natively existing cellular mechanism allows transcellular transport of accordingly functionalized nanoparticles upon interaction with the corresponding receptors.[ 5c,e ] To address this, endogenous receptors like insulin‐, transferrin‐, and low‐density lipoprotein receptors have extensively been investigated in preclinical studies. However, the high concentrations of endogenous ligands compete with the delivery system and impair their implementation.[ 5 , 9 ] Recently, the membrane‐bound precursor of heparin‐binding epidermal growth factor‐like growth factor (HB‐EGF), also known as diphtheria toxin receptor (DTR), has been shown to provide a new mechanism to cross the BBB.[ 10 ] Compared to most described receptors, DTR has no endogenous competitors and its expression is strongly up‐regulated on the cerebral blood vessels in gliomas, ischemic stroke, and under hypoxic conditions.[ 11 ] Thus, the functionalization of nanoparticles with a nontoxic mutant of the DTR ligand cross reactive material 197 (CRM‐197), clinically approved as a carrier protein, facilitates a highly specific and efficient delivery system across the BBB at target sites.[ 12 ] However, the delivery of therapeutically relevant doses not only across the BBB but also specifically to the site of the disease still presents a challenging issue. This demands a definition of up‐regulated targetable disease‐associated mechanisms. For glioblastoma, the potential candidates for treatment are mitosis addressing drugs since the proliferating tumor cells are surrounded by quiescent brain cells. Unlike chemotherapeutic drugs, whose effectiveness depends on the concentration achieved at the tumor site; for radiopharmaceuticals, the therapeutic efficiency is primarily related to the energy transferred by the radiation and the range of decay.[ 13 ] The Auger electron emitting nuclides are particularly useful candidates due to their features like the ultra‐short radiation range (1–20 nm) and the high killing potential (one double‐strand DNA break per decay).[ 14 ] Uniquely, when localized in the cell cytoplasm, Auger electron emitters result in mild toxicity; however, when incorporated into DNA, they are highly radiotoxic. In fact, DNA‐incorporated Auger electron emitters can be as radiotoxic as alpha particles.[ 15 ] To achieve the prerequisite DNA proximity, the Auger electron emitters could be linked to nucleoside analogues as vehicles addressing mitotically active tumor cells. As shown in our previous studies, the Auger electron emitter labeled thymidine analogue[125I]ITdU is effectively incorporated into the newly synthesized DNA of malignant cells.[ 16 ] Several in vivo studies visualized its preferential accumulation in tumor tissue and high therapeutic potential resulting in efficient and selective induction of cell death.[ 17 ] However, although Auger electron emitter labeled thymidine analogues were shown to kill glioma cells effectively in vitro, their therapeutic potential in vivo is extremely affected due to the inaccessibility imposed by the BBB. Thus, incorporation of [125I]ITdU into a nano‐formulation and its tumor cells triggered release presents an experimental approach to tackle this limitation.
2. Results and Discussion
In this study, we focused on the development of a transvascular stimuli sensitive drug delivery system for the treatment of glioma, one of the most aggressive and virtually incurable cancers. Nanogels were prepared based on poly(ethylene glycol), which were previously shown to prolong circulation in vivo without nonspecific binding or rapid elimination by the mononuclear phagocyte system.[ 18 ] Six arm star poly(ethylene oxide‐co‐propylene oxide) pre‐polymers with acrylate end groups (Ac‐sPEG) were used to synthesize two types of radiolabeled nanogels. For the quantification of transcytosis experiments, the nanogels were equipped with the chelator NODAGA‐NH2 for 68Ga labeling (NODAGA‐nanogel). For cellular drug delivery experiments, the nanogels were radiolabeled with [125I]ITdU ([125I]ITdU‐nanogel).
2.1. Nanogel Synthesis and Structural Characterization
For NODAGA‐nanogel synthesis, the free amine of the NODAGA was modified to thiol using 2‐iminothiolane (Traut's reagent). 2‐iminothiolane reacted with the primary amine, forming an amidine bond and resulting in a free thiol group. The thiol modified NODAGA was clicked to the Ac‐sPEG by a thiol‐ene Michael addition. The modification of acrylate with NODAGA was kept to a molar ratio of 1:0.3. The successful functionalization of star‐Poly(ethylene glycol‐stat‐ propylene glycol)‐acrylate with NODAGA was confirmed by NMR (Figure S1, Supporting Information). The NODAGA‐modified Ac‐sPEG was further used for the synthesis of nanogels via inverse miniemulsion[ 19 ] using cysteine end‐capped MMP‐2 substrate as a cross‐linker (Ac‐GCRDSGESPAY↓YTADRCG‐NH2) (at a molar ratio of 1:1.5). The kcat value of this specific MMP‐2 cleavable substrate is 2.96 ± 0.72 s−1,[ 20 ] which can be further optimized by tailoring the peptide sequences. The NODAGA‐NG was then post modified with CRM‐197; the peptide was first reacted with Traut´s reagent to obtain thiol group. The remaining free acrylates of the NODAGA‐nanogel were used for the post‐modification of the nanogels with thiolated CRM‐197 (Scheme 1A). Nanogels with NODAGA were radiolabeled with 68Ga as previously described. 18 , 21 ]For microscopic imaging analysis, the nanogels were fluorescently labeled with Alexa‐Fluor 488. To ensure linking of [125I]ITdU to the nanogel via MMP substrate (Scheme 1B), the primary OH‐group of the [125I]ITdU was first reacted with disuccinimidyl carbonate (Scheme 1C) and then added to the MMP‐2 substrate, which was subsequently used for nanogel formation (Scheme 1D). Successful conjugation of the nanogels with the [125I]ITdU was confirmed by radio‐thin layer chromatography. Excess of low molecular weight byproducts and [125I]ITdU was removed with a PD‐10 desalting column. The radiochemical yield (rcy) before purification was 72% ± 8% (Figure S2, Supporting Information). After the desalting column, the product was recovered with a radiochemical purity (rcp) of 90% ± 4% and a specific activity of 2°MBq per mg nanogel.
Scheme 1.
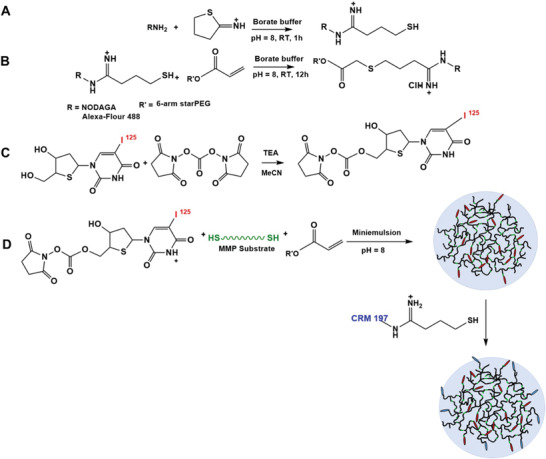
Synthesis of CRM‐197 functionalized MMP‐substrate cross‐linked nanogels. A) The reaction of Traut's reagent with amine‐functionalized fluorophore and/or chelator or CRM‐197. B) The reaction of the thiol‐modified fluorophore and/or chelator resulting in functionalized star‐PEG acrylate. C) The reaction of [125I]ITdU with disuccinimidyl carbonate. D) The disuccinimidyl carbonate modified [125I]ITdU was reacted with cysteine end capped MMP‐substrate during the synthesis of nanogels using acrylate pre‐polymer from step (B); the formed nanogels were post modified with CRM 197.
Successful nanogel formation and size distribution was estimated by dynamic light scattering (DLS) using ALV goniometer (Figure 1A) and TEM micrograph of the nanogels (Figure 1B). In the dry state, the mean hydrodynamic diameter of the microgel is 129°nm ± 31°nm compared to the swollen state where the diameter was 240°nm as measured by DLS. The modification with CRM‐197 resulted in a marginal increase in polydispersity index (PDI = 0.23 versus PDI = 0.26 compared to the non‐functionalized analog. The nanogels showed a zeta potential of −25.6 mV and electrophoretic mobility of −2.005 µm cm Vs−1. The size of the nanogels is in accordance with our previous work, where we show the improved biodistribution of PEG based nanogels with a similar size range. The nanogel cross‐linking by the MMP substrate was investigated by UV–vis spectroscopy. UV spectra of MMP degradable nanogels, the non‐degradable nanogels, and MMP substrate were compared. For the synthesis of non‐degradable nanogels, cross‐linking was performed with 2,2′ ethylenedioxydiethanethiol instead of MMP substrate. The UV–vis spectra of MMP degradable nanogels showed a clear tyrosine peak at 280°nm confirming successful cross‐linking with the MMP substrate when compared to the non‐degradable controls (Figure 1D). Circular dichroism data indicated that CRM‐197 retained its secondary structure after reaction with Traut´s reagent and binding to nanogels (Figure 1E). However, pre‐polymers functionalization with the protein prior to the nanogel synthesis led to the denaturation of the cross‐reactive material (Figure 1E, dashed line). The nondegradable nanogel without CRM‐197 was used as control and blank. he ITdU functionalized nanogels are post‐modified by CRM 197 via a non‐degradable linker. The rationale to use the non‐degradable linkage for CRM‐197 is to facilitate a stable, highly specific, and efficient delivery system across the BBB. Once the nanogels accumulate at the tumor site, the nanogels as well as linked ITdU are designed to be responsive to high concentrations of MMP 2 and 9 present in the tumor microenvironment. However, upon cleavage of the MMP substrate, the ITdU will still carry a fragment of the MMP substrate, which can inhibit its incorporation into the DNA. To overcome this, the MMP substrate is attached to the ITdU with a carboxylesterase cleavable thiocarbonate linkage, which will facilitate the generation of free ITdU for its efficient cell uptake and incorporation into the DNA via thymidine salvage pathway. Thus, the ITdU is linked to the nanogel with two degradable linkages (MMP sensitive and thiocarbonate sensitive).
Figure 1.
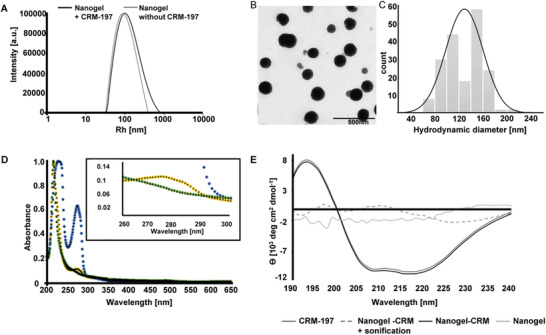
Characterization of nanogels cross‐linked with cysteine end‐capped MMP‐substrate and functionalized with CRM‐197 ligand. A) The size distribution function of nanogels with covalently attached CRM‐197 and without CRM‐197 showed a mean hydrodynamic radius of Rh = 100°nm before and Rh = 120°nm after CRM‐197 functionalization. The modification with CRM‐197 resulted in a slightly higher polydispersity index (PDI = 0.23 versus PDI = 0.26). B,C) TEM micrograph of the nanogels and the histogram showing the distribution of nanogels in the dry state. In the dry state, the mean hydrodynamic diameter of the microgel is 129°nm ± 31°nm compared to the swollen state where the diameter is 240°nm as measured by DLS. D) UV–vis spectra of MMP‐substrates compared to a non‐degradable control and MMP‐cross‐linked nanogels as proof for successful syntheses (tyrosine peak at 28° nm). E) Circular dichroism spectra of CRM‐197 before and after binding to the nanogels in comparison to nanogels without CRM‐197 and to nanogels synthesized after the pre‐polymers were modified with CRM‐197.
2.2. Nanogel Transcytosis via BBB
The suitability of the designed nanogels to cross the BBB was evaluated in an in vitro model consisting of three different brain tissue originating cell types in a trans‐well insert culture system. Cerebral microvascular endothelial cells hCMEC/D3 were seeded apical on the collagen coated membrane, basolateral on the membrane were seeded pericytes, and in the wells were seeded astrocytes (NHA) or glioblastoma cells (human glioblastoma cell lines T3, U‐87, and HT12356) to reproduce physiological or pathological situations, respectively (Figure 2 ). The membrane pore size of 0.4°µm allowed the diffusion and exchange of small molecular substances. Daily performed measurements of transendothelial electrical resistance (TEER), and interim TEM and fluorescence microscopy analysis visualized the integrity and functionality of this BBB model (Figure 2 and Figure S4, Supporting Information). The increased expression of the tight junction (TJ) proteins, like JAM‐1, VE‐Cadherin, and Claudin5, correlated with rising TEER value indicating the tightness and integrity of hCEMC cells when co‐cultured with NHA. In contrast, co‐culture with glioblastoma cells induced a down‐regulation of TJ proteins on the hCMEC cells, leading to a decrease of cell tightness and integrity of the BBB (Figure 2C–F). These results indicate that by secreting soluble factors, glioblastoma cells can degrade TJ actively, and by this, induce BBB disruption. Importantly, all the following experiments were performed in the combined mono‐culture and co‐culture setting starting at day 8 (Figure 2F), to minimize the transport effect caused by the disrupted integrity of BBB.
Figure 2.
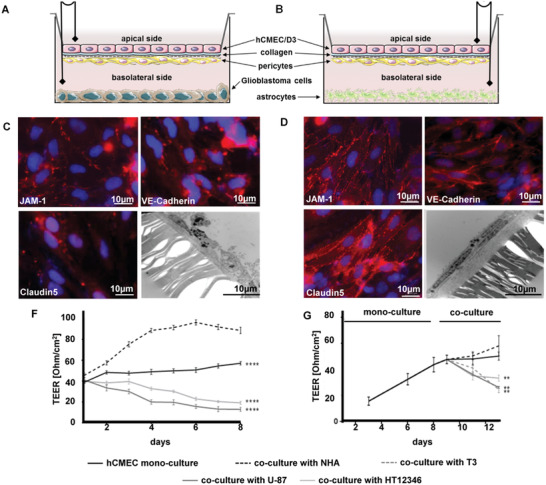
Evaluation and characterization of the in vitro BBB model under physiological and pathological conditions. A) Illustration of in vitro BBB model with human glioblastoma cell lines T3, U‐87, or HT12356. B) Illustration of an in vitro BBB model with normal human astrocytes. C) Fluorescence microscopy analysis for visualization of tight junction proteins expression on hCMEC monolayer in co‐culture with glioblastoma cells for 3 days (red: JAM‐1, VE‐Cadherin, Claudin5, blue: DAPI nucleus staining, 40‐fold magnification) and TEM image of hCMEC cells (3000‐fold magnification). D) Fluorescence microscopy analysis for visualization of tight junction proteins expression on hCMEC monolayer in co‐culture with NHA for 3 days (red: JAM‐1, VE‐Cadherin, Claudin5, blue: DAPI nucleus staining, 40‐fold magnification) and TEM image of hCMEC cells (3000‐fold magnification). E) Transepithelial electrical resistance (TEER) measurements of hCMEC/D3 mono‐culture compared to co‐culture with NHAs or glioblastoma cells. F) TEER measurements were done with mono‐culture followed by co‐culture starting on day 8. (n = 5, **p < 0.01; ****p < 0.001 compared to stable co‐culture; two‐way ANOVA, Dunnett´s multiple comparison test.)
Next to the integrity of the BBB model, the key prerequisites for receptor‐mediated transcytosis (DTR expression on the BBB) and environmental induced nanogel degradation (MMP2 and 9 activities in the media) were analyzed for each co‐culture condition (Figure 3 ). DTR expression was estimated by flow cytometry and fluorescence microscopy after 3 days of co‐culture. The highest endothelial DTR expression was induced in a co‐culture with U‐87 cells (15% ± 3%), while no changes were detected in a co‐culture with NHA (Figure 3A,B). The different effects of investigated glioblastoma cell lines on endothelial DTR expression might at least in part be due to their MMP activities. DTR, which is also expressed in glioblastoma cells, is a precursor of soluble HB‐EGF, which is a mitogenically active growth factor.[ 22 ] The soluble HB‐EGF, which was shown to strongly induce mRNA HB‐EGF expression promotes vascular endothelial growth factor (VEGF‐A) production and by this, the tumor angiogenesis is generated by proHB‐EGF cleavage, a process regulated by MMP.[ 23 ] As visualized by zymography, among the tested conditions, media of co‐cultures with U‐87 and HT12346 cells showed the highest MMP2 and MMP9 activities (Figure 3C,D). MMP2 and MMP9 were shown to be the most abundant and active members of the MMP family in glioblastoma cell lines and glioblastoma brain samples. Importantly, the expression pattern of different MMPs in glioblastoma is highly variable, which is of high impact for the transfer of MMP‐sensitive drug delivery systems to clinical implementation.[ 24 ] However, identification of MMP activity in patient derived biopsy material would allow fabrication of nanogels with corresponding MMP‐sensitive linker for customized drug delivery. Noteworthy, the level of MMP2 activity in U‐87 and HT12346 cells was comparable to that detected in TGFß2‐supplemented media, which supports the known function of TGFß2 as a key factor in glioblastoma pathogenesis (Figure S5, Supporting Information) regulating the MMP2 activity and by this promoting glioma migration.[ 25 ] To estimate the degradation profile under physiological and pathological conditions, radiolabeled CRM‐197‐functionalized nanogels ([68Ga]‐NODAGA‐nanogels) were added apically to the BBB model. After 90 min, the nanogel degradation in basolateral media was investigated using combined non‐reducing SDS‐PAGE/phosphorimager analysis. As visualized by a phosphorimager, the nanogel degradation degree corresponded with MMP2 activities detected by zymography, with the highest in the co‐culture with U‐87 cells producing predominantly low‐molecular weight degradation products of ≈10–20°kDa and intermediate degradation products with MW of 37–50°kDa (Figure 3E). No degradation products were detected on the apical side before transcytosis at 90 min incubation time. The intact nanogel corresponds to bands between 250–100 kDa of the protein standard. Interestingly, compared to the glioblastoma cells, NHA co‐culture induced only partial nanogel degradation. This could be due to the active membrane associated‐MMP2, which is constitutively expressed on astrocytes.[ 26 ]
Figure 3.
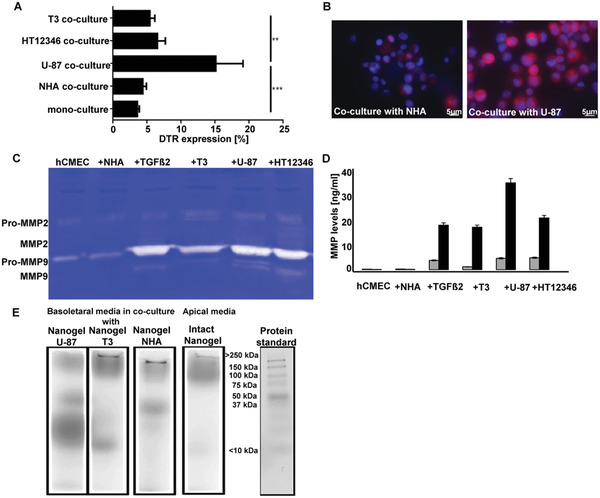
Enzymatic degradation of MMP‐substrate cross‐linked nanogels. A) DTR expression on hCMEC/D3 cells in different co‐cultures after 3 days estimated by flow cytometry (n = 5; one‐way ANOVA; Sidak's multiple comparisons test; **p < 0.01, ***p < 0.001). B) Visualization of membrane associated expression of DTR on hCMEC/D3 cells in co‐culture with NHA or U‐87 using fluorescence microscopy (DAPI: nucleus staining, 40‐fold magnification). C) MMP2 and MMP9 activities after 3 days in the media of mono‐culture, media of co‐culture with NHA, in media supplemented with TGFß2 (25 ng mL−1), in media of co‐culture with glioblastoma cell lines T3, U‐87, or HT12346 were detected by zymography. D) Quantification of MMP 2 (gray bars) and MMP9 (black bars) levels using ELISA (n = 5). E) Native gel electrophoresis /phosphor imager analysis of nanogel from media collected basolateral of the trans‐well, visualizing post transcytosis nanogel degradation products (90 min incubation with 1°MBq per well). The fourth lane visualizes the apical media collected from the co‐culture with U‐87 cells.
To estimate the transcytosis efficiency, nanogels with and without CRM‐197 were added on the apical side of the established BBB model with U‐87 or with NHA. For visualization of transcytosis, all nanogels were additionally labeled with Alexa Fluor 488 (AF‐488). After an incubation time of 90 min, inserts were analyzed via TEM and basolateral seeded cells via fluorescence microscopy (Figure 4A,B). The CRM‐197‐nanogel‐AF488 was visible in hCMEC/D3 cells and in pericytes, which indicated active transcytosis via BBB. As detected by fluorescence microscopy, independent of functionalization, NHA showed lower nanogel uptake than U‐87 cells (Figure 4B). Clearly, the BBB under physiological conditions remained mostly insuperable for the drug delivery systems. Importantly, in pathological situations, conjugation with CRM‐197 increased nanogel availability leading to enhanced intracellular fluorescence signal in U‐87 cells (Figure 4B). This resulted from two successively acting events, the DTR‐CRM‐197 interaction mediated nanogel transcytosis via the BBB and MMP2/9 mediated nanogel degradation at the basolateral side. The last one generated nanogels of lower molecular weight being preferentially taken up by endocytosis.[ 27 ] Thus, an upregulated expression of DTR on glioblastoma cells and their increased MMP2 and endocytic activities are probable mechanisms behind the accumulation of intracellular fluorescence signals in U‐87 cells.[ 28 ]
Figure 4.
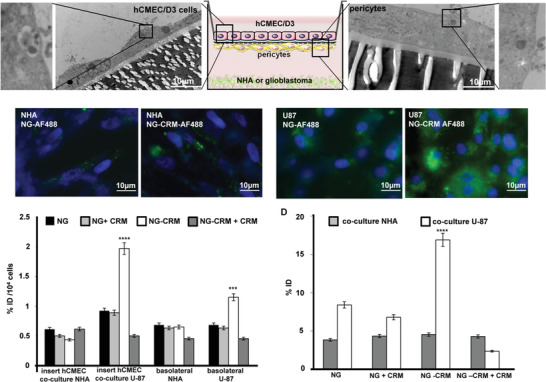
Evaluation of diphtheria toxin mediated transcytosis via BBB. A) TEM images of the trans‐well membrane with hCMEC/D3 cells after 4 h incubation with CRM‐197‐nanogel‐AF488 in co‐culture with U‐87 cells (left, 4600‐fold magnification) and TEM images of the trans‐well membrane with pericytes after 4 h incubation with CRM‐197‐nanogel‐AF488 in co‐culture with U‐87 cells (right, 6000‐fold magnification). B) Fluorescent microscope images of nanogel‐AF488 with CRM‐197 (CRM‐197‐NG‐AF488) and without CRM‐197 (NG‐AF488) in basolateral seeded NHA or U‐87 cells after 4 h incubation with the BBB model (blue: DAPI staining, 66‐fold magnification). C) Gamma‐counter analysis for nanogel transcytosis and uptake quantification. Radiolabeled functionalized and non‐functionalized nanogel accumulation was measured in hCMEC/D3 cells, basolateral media, and seeded NHA after 90 min incubation with 1°MBq per well (n = 5). D) Gamma‐counter analysis for nanogel transcytosis and uptake quantification. Radiolabeled functionalized and non‐functionalized nanogel accumulation was measured in hCMEC/D3 cells, basolateral media, and seeded U‐87 cells after 90 min incubation. For inhibition study, the samples were pre‐treated with the CRM‐197 protein (0.1°mg per well for 60 min) (****p < 0.001; ***p < 0.005, students t‐test).
To quantify the receptor‐mediated transcytosis, the CRM‐197 functionalized and non‐functionalized nanogels were conjugated with the chelator NODAGA and radiolabeled with 68Ga. CRM‐197‐nanogel‐[68Ga]‐NODAGA or nanogel‐[68Ga]‐NODAGA were incubated with the in vitro BBB model. Endothelial cells, basolateral media, and co‐cultured cell lines (U‐87 or NHA) were analyzed for accumulated radioactivity using a gamma‐counter. According to the fluorescence microscopy analysis, under physiological conditions, only low transcellular transport across the BBB regardless of nanogel functionalization was detected (Figure 4C). However, co‐culture with glioblastoma cells promoted DTR‐mediated transcytosis of CRM‐197 functionalized NG (Figure 4D). Importantly, by pre‐treatment with the CRM‐197 protein, the transcytosis was significantly inhibited which indicated the receptor binding specificity and the key function of DTR for the nanogel transendothelial transport. Furthermore, the higher uptake/ transport was a result of the DTR‐mediated transcytosis and not of an unspecific uptake or disruptive BBB.
2.3. Nanogels for Delivery of Radiolabeled Nucleoside Analogue [125I]ITdU
With proof of transcytosis via DTR, we validated the CRM‐197‐functionalized nanogel as a potential and effective system for delivery of the Auger electron emitting nucleoside analogue [125I]ITdU. Although Auger electron emitting nucleoside analogues were shown to have great potential for the induction of cell death at a single cell level in vitro and in vivo,[ 29 ] their application for the treatment of brain cancer is hampered by its inability to cross the BBB upon intravenous injection. As shown for [125I]‐labeled thymidine analogues in experimental and clinical studies, for central nervous system malignancies they need different application routes like intrathecal or intratumoral to generate a therapeutic effect in situ.[ 30 ] However, we can overcome this BBB limitation by the use of nanogels. The second and major challenging aspect is the nucleoside analogue itself. The thymidine salvage pathway is highly specific; thus, any structural modification at the analogue might impact its cellular uptake, phosphorylation by thymidine kinases, and finally the incorporation into the DNA,[ 31 ] last being one of the major limiting factors for this potential radio therapeutics. To avoid this, [125I]ITdU needs to be released after nanogel degradation in its native form. To achieve this, [125I]ITdU was first reacted with disuccinimidyl carbonate and this mixture was added to the cysteine end‐capped MMP substrate and pre‐polymer mixture before inverse miniemulsion (Figure 1B,C). The rationale behind this approach is to use acid degradable carbonate linkage, which in the acidic microenvironment of the glioblastoma cells will degrade, to release the only CO2 without any alternation in the nucleoside analogue structure. To prove this hypothesis, first the [125I]ITdU‐NG was incubated with the BBB model. Enzymatic degradation resulted in a release of [125I]ITdU from the nanogel and polymer structure as indicated by gel electrophoresis and phosphorimager analysis (Figure 5A). Due to glioblastoma‐associated increased activity of MMP2/9, the nanogel degradation was considerably higher in the U‐87 environment with almost no [125I]ITdU release in the presence of normal astrocytes (Figure S6, Supporting Information). Cellular uptake of [125I]ITdU‐CRM‐197‐nanogel (and its released [125I]ITdU) in comparison to net [125I]ITdU was investigated in NHAs and U‐87 cells as mono‐culture. This was compared with the uptake of 68Ga‐labeled nanogels to differentiate between uptake mediated by the vehicle itself and the active transport of the nucleoside analog driven by the human equilibrative nucleoside transporter 1 (hENT1).[ 32 ] In general, all probes showed a considerably lower uptake in NHAs as compared to malignant U‐87 cells (Figure 5B‐i). At 1 h incubation time, the highest uptake was detected for net [125I]ITdU. However, due to the distinct release of the nucleoside analog from the nano‐formulation in the glioblastoma environment, a significantly increased radioactivity uptake in U‐87 cells was detected after incubation with [125I]ITdU‐nanogel. As indicated by native electrophoresis, the nanogels showed a high amount of degradation products already at 1 h post incubation. Interestingly at 4 h post incubation with the [125I]ITdU‐nanogel, the radioactivity was taken up by the cells to an even higher extent than the net nucleoside analogue (Figure 5B‐i). This increased uptake might be explained by two different effects: i) a cumulative uptake effect of the released [125I]ITdU by hENT1 and of the [125I]ITdU still attached to the nanogel via particle endocytosis or DTR‐CRM‐197 mediated uptake as well and ii) the protection of the nucleoside analogue by the nanogel from deiodination.[ 33 ] The effect of treatment with NBMPR on cellular uptake strongly suggests the nucleoside transporter hENT as the main mechanism behind the transport of nanogel released ITdU. Besides the inhibitory effect of NBMPR on the cellular uptake, this is further supported by the drug release as presented in Figure S6, Supporting Information. With more than 30% of U‐87 mediated extracellular cleavage, it provides a tool for efficient and fast cellular uptake of ITdU and its incorporation into the DNA via the salvage pathway. To assess the cellular localization of [125I]ITdU, U‐87 cells were fractionated to obtain the cytosolic and DNA fractions. At 1 h post addition of each compound, 2.7% and 2.0% were detected in the extracted DNA relative to the overall cellular uptake for [125I]ITdU and [125I]ITdU‐nanogel, respectively. After 4 h, the amount of radioactivity incorporated into the extracted DNA was considerably increased for both, the net nucleoside analogue (23.9°%) and the [125I]ITdU‐nanogel (13.0°%) (Figure 5B‐ii). The imaged decrease of the extracellular signal after 4 h (Figure 5A) was not accompanied by a significant increase of cellular localized drug (Figure 5B‐ii), which is probably due to the deiodination of [125I]ITdU after cellular uptake. The significantly lower amount of [125I]ITdU released from the nanogel might be explained by the incomplete degradation of the nanogel structure as indicated by gel electrophoresis and phosphorimaging of the cytosolic fraction (Figure 5C). The presence of higher molecular weight fractions detected in the cytosol also indicated that not only the released [125I]ITdU is taken up by the cells, but also the nanogels‐conjugated [125I]ITdU. This might be due to the receptor mediated endocytosis of [125I]ITdU–conjugated and not completely degraded nanogel fragments, since glioblastoma cells were shown to overexpress DTR, which implements an internalization mechanism after ligand binding.[ 34 ] Importantly, extracellular and intracellular hydrolysis of the generated thiocarbonate by carboxylesterases which were shown to be up‐regulated in tumor cells,[ 35 ] results in the release of the native [125I]ITdU and its efficient incorporation into the DNA of glioblastoma cells. As previously shown, this cellular uptake and DNA incorporation rate via nanogel delivered [125I]ITdU are more than sufficient for a potent induction of apoptosis in glioblastoma cells.[ 29d ]
Figure 5.
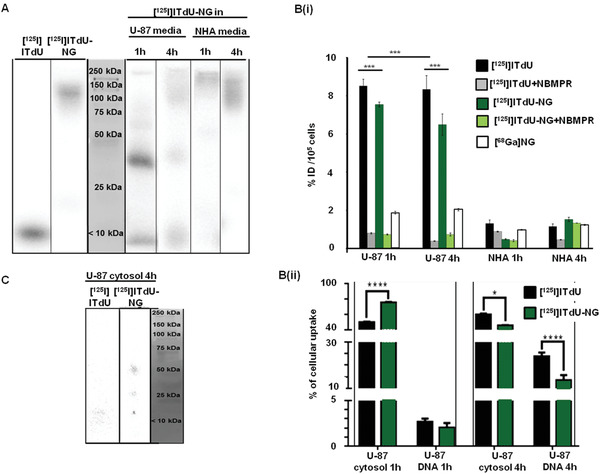
Evaluation of [125I]ITdU‐CRM‐197‐nanogel for delivery, release, and cellular uptake of [125I]ITdU. A) Gel electrophoresis/phosphor imager analysis of intact [125I]ITdU‐conjugated nanogels, and [125I]ITdU as controls, of cell culture media of U‐87 and NHA, after incubation for 1 and 4 h with [125I]ITdU‐CRM‐197‐nanogel. B‐i) Gamma counter analysis for quantification of cellular uptake as % of injected dose [ID = 0.5°MBq] of [125I]ITdU‐CRM‐197‐nanogel in comparison to sole [125I]ITdU, and to 68Ga‐labeled nanogels in NHA and U‐87 cells. For inhibition study, the cells were treated with 0.1 µm L−1 NBMPR for 1 h prior to incubation with [125I]ITdU‐CRM‐197‐nanogel or [125I]ITdU (***p < 0.005, ***p < 0.005, two‐way ANOVA, Bonferroni posttest). B‐ii) Gamma counter analysis for quantification for cytosolic and DNA‐incorporated radioactivity in U‐87 cells (two‐way ANOVA, Sidak's multiple comparisons test). C) Gel electrophoresis/phosphor imager analysis of [125I]ITdU‐nanogels and [125I]ITdU in cytosolic fraction of U‐87 after 4 h.
3. Conclusion
In summary, here we present for the first time an in vitro proof of an innovative concept of MMP‐2/9 degradable CRM‐197‐functionalized‐nanogels for delivery of nano‐irradiation across the BBB and therapy of glioblastoma. In an in vitro model of the BBB, we show i) an efficient and specific receptor‐mediated transcytosis of the nanogels, ii) solely glioblastoma mediated nanogel degradation and efficient release of Auger electron‐emitting [125I]ITdU, and iii) high rate DNA incorporation of the therapeutic [125I]ITdU into glioblastoma cells.This study visualizes the potential of nanogels as a delivery system across the most challenging biological barrier by exploiting DTR as receptor mediated channel which increases the permeability of the BBB. As demonstrated by Wang et al., application of CRM197 in vivo leads to the upregulation of caveolin‐1 protein, an increase of pinocytotic vesicles, and redistribution of tight junction‐associated proteins in the brain microvessels.[ 36 ] Experimental studies using patient‐derived tissue samples presented highly expressed DTR on glioblastoma associated endothelial cells, defining hypoxia as the main factor inducing and regulating DTR expression.[ 37 ] For clinical application, evaluation of an individual DTR expression will be needed to identify patients who will benefit from DTR targeting drug delivery systems. The versatility of nanogels presented here allows a patient expression profile customized design for single or combined targeting of one or more BBB expressed molecules for receptor mediated transport. During pathological processes, at least from the therapeutic point of view, BBB plays a double role as a friend and foe by very tightly controlled transport and highly up‐regulated drug efflux mechanisms. These strongly limit the efficiency of otherwise extremely effective therapeutic agents. The dire need for the achievement of a lethal drug concentration and its prolonged retention at the tumor site despite the BBB is the most critical restrictions in this special scenario. The unique strength of radiopharmaceuticals relies on their lethality which does not require highly efficient drug delivery but depends on the tagged radioisotope. Moreover, the application of radiolabeled nucleoside analogs allows stable and prolonged intratumoral drug retention, provided their DNA incorporation. Additionally, the size of released nucleosides promises a deep and homogeneous penetration of tumor tissue as shown by May et al., for smaller‐sized DDS (10°nm) to accumulate and penetrate brain tumors more efficiently and by this to be more suitable for cellular drug delivery than larger‐sized DDS.[ 6 ] This study together with existing in vivo biodistribution studies with functionalized nanogels and nucleoside analogues suggests a selective and efficient nucleoside uptake by tumor cells and a hepatobiliary clearance of nondegraded nanogels after intravenous application.[ 29 , 38 ] Moreover, due to the restricted accessibility of the deiodination of the payload, the radiolabeled nucleoside is expected to be strongly limited, which would prolong the drug´s in vivo stability and diminishes radioiodine accumulation in clearing organs. Besides clearing organs, a systemic expression of HB‐EGF needs to be considered. HB‐EGF was shown to be expressed on different leukocyte subsets, with monocytes being the most prominent one.[ 39 ] Considering the low mitotic index of PBMC, a serious apoptotic effect in circulating white blood cells is not expectable, since incorporation into the DNA is essential for killing efficiency of auger emitters' labeled drugs. Additionally, evidence derived from a study with [14C]thymidine and [14C]thymidine monophosphate in normal and leukemic cells indicated increased activity of catabolic enzymes thymidine phosphorylase and thymidine phosphatase and low activity of the anabolic enzyme thymidine kinase in normal cells, leading to transient retention of thymidine analogs in normal tissue.[ 40 ] Considering the extremely high and tumor cell specific killing potential of [125I]ITdU[ 16 , 29 ] and its DNA incorporation rate after delivery by nanogels and MMP2/9 induced release presented here, this approach holds a promise to reach potential improvements in therapy outcomes of glioblastoma.
4. Experimental Section
Functionalization of Star Poly(ethylene glycol‐stat‐propylene glycol)‐acrylate with 1,4,7‐Triazacyclononane,1‐glutaric acid‐4,7‐acetic acid (NODAGA)
sP(EG‐stat‐PG)‐acrylate (CHT GmbH, Tübingen, Germany, 100°mg, 5.55×10−6°mol, Mn = 18 000°g mol−1, 1°eq.) was solved in degassed PBS (1.5°mL, 3.70 × 10−3°m) under Ar. The pH was adjusted to 8.4 with Na2CO3 solution (0.1°m, pH = 11.4 ≈0.6°µL). NH2‐NODAGA (CheMatech, Dijon, France; 1.42°mg, 2.77 × 10−6°mol, 0.5°eq) was dissolved in a separate round bottom flask in degassed PBS (100°µL, 2.77 × 10−3°m). The pH was adjusted to 8.5 with 0.1°m Na2CO3 solution (pH = 11.4–2.6°µL). 2‐Iminothiolane • HCl (0.76°mg, 5.55 × 10−6°mol, 1°eq.) was dissolved in degassed PBS (50°µL, 0.11°m) and added to the NH2‐NODAGA solution. The reaction mixture was stirred at room temperature for 90 min before the polymer solution was added dropwise under inert gas. The reaction was allowed to proceed for another 16 h before the raw product was dialyzed (reg. cellulose; MWCO: 3500) against ultrapure water, lyophilized and kept under argon at −20 °C protected from light, prior to further use.
1H°NMR (400 MHz, CDCl3) δ = 6.53–6.34 (m, 0.38 H, ‐CH = CH), 6.20 (dd, J = 17.3, 10.5 Hz, 0.38 H, ‐CH = CH), 5.99 (d, J = 10.5 Hz, 0.39 H, ‐CH = CH), 5.29–4.96 (m, 0.60 H, ‐CH2), 4.33 (s, 0.04 H, ‐CH), 3.98 – 3.42 (m, 110 H, ‐CH2 polymer backbone), 2.83 (d, J = 6.5 Hz, 0.36 H, NODAGA), 2.70 (d, J = 7.1 Hz, 0.38 H, NODAGA), 2.67–2.55 (m, 0.39 H, NODAGA), 2.37 (d, J = 7.5 Hz, 0.07 H, NODAGA), 2.021.84 (m, 0.35 H, NODAGA (15‐16)), 1.35–0.95 (m, 27 H, ‐CH3 (4, 4’)) ppm (Figure S1, Supporting Information).
Radiolabeling of ITdU
Chemicals and solvents were purchased from Sigma‐Aldrich (St. Louis, Missouri, USA) and Merck (Darmstadt, Germany) or otherwise as indicated. No‐carrier‐added (n.c.a.) sodium I‐125‐iodide was obtained from PerkinElmer (Waltham, Massachusetts, USA). No carrier added Na[125I] was used as received. The 5‐trimethylstannyl precursor of ITdU was synthesized according to previously reported methods and provided by PD Dr. rer. nat. Boris Zlatopolskiy (Uniklinik Köln).[ 29c ] ITdU precursor was dissolved in 66% MeOH (in 2°m phosphate buffer pH = 2) and Na[125I] solution in 0.05°mol L−1 NaOH was added before addition of 80°µL Chloroamine T (2°mg mL−1 in 66°% methanol). After 10 min at room temperature, the reaction was stopped by the addition of 50°µL thiosulfate solution (0.1°mol L−1). The crude product was purified via high‐performance liquid chromatography (HPLC) on a C4 column and analytical HPLC was performed on a RP‐18 column for quality control. Total radiochemical yield (rcy) was estimated via thin layer chromatography with >80% and radiochemical purity of >95%. After purification, the radiolabeled product was recovered with a radioactivity concentration of A = 27.7°MBq mL−1 and a specific activity of As = 138°GBq µmol−1 with an overall amount of ITdU of 5.4×10−3°µmol according to HPLC analysis.
Covalent Binding of [125I]ITdU and Nanogel Formation
10–20°MBq [125I]ITdU (in 10% EtOH/90% PBS) was reacted with 0.1°mg (0.39°µmol) disuccinimidyl carbonate. The reaction was allowed to proceed for 10 min before the addition of the MMP substrate (CASLO ApS, Lyngby, Denmark; 1.39°µmol; 0.5°eq.) to yield the radiolabeled peptide. For the preparation of the nanogels, 150°mg surfactants (3:1 weight ratio of SPAN®80 and TWEEN®80) was dissolved in 5°mL n‐hexane and used as an organic phase. The aqueous phase consisting of 25°mg sP(EG‐stat‐PG)‐acr (1.39 × 10−3°mmol, Mn = 18 000°g mol−1) was dissolved in borate buffer (0.5°mL, pH = 8.6). The organic and aqueous phase was pre‐emulsified by magnetic stirring for 5–10 min and additionally ultrasonicated for 60 s using Soniprep 150 plus (10.0 pm amplitude) at 20 °C. Cross‐linking was initiated by the addition of 10°µL cysteine end‐capped MMP substrate (in borate buffer) (0.036°mmol, 15°eq.) and disuccinimidyl carbonate modified [125I]ITdU followed by a further sonication step (60 s). The reaction was quenched by the addition of thiol‐modified CRM‐197 (Sigma Aldrich). The [125I]ITdU‐nanogel was purified using PD‐10 desalting column (Figures S2 and S3, Supporting Information).
Dynamic Light Scattering
Nanogel solutions of about 1°mg mL−1 in double‐distilled water were passed through a 5°µm poly(tetrafluoroethylene) membrane filter. The particle sizes were measured by photon correlation spectroscopy at an angle of 90° with a setup consisting of an ALV‐SP8 goniometer, an ALV‐SIPC photomultiplier, a multiple τ digital real‐time ALV‐7004 correlator, and a solid state laser (Koheras, AZO optics, Birkerod Denmark) with a wavelength of 473°nm as the light source. Sample cuvettes were immersed in a toluene bath and thermostatted within an error of ± 0.1 °C. Autocorrelation functions of intensity fluctuations g2 (q, t) in the self‐beating mode were measured and expressed by the Siegert relation:
| (1) |
where t is the decay time, A is a measured baseline, β is the coherence factor, and g1(q, t) is the normalized first‐order electric field time correlation function related to the measured relaxation rate Γ according to Equation (2):
| (2) |
Deconvolution of the measured intensity autocorrelation was achieved by the DTS software. For pure diffusive relaxation, Γ is related to the translational diffusion coefficient D at q→0 and c→0 according to Equation (3):
| (3) |
The hydrodynamic radius Rh is calculated by the Stokes–Einstein equation as Rh = kBT⁄6πηD with q, kB, T, and η being the scattering vector, the Boltzmann constant, absolute temperature, and solvent viscosity, respectively. A hydrodynamic radius distribution was calculated from the Laplace inversion of g1(t) and was analyzed with CONTIN algorithm, giving the distribution of the relaxation time τ.
Scanning Electron Microscopy
Scanning electron microscopy was performed with a HITACHI S‐ 4800 (Tokia, Japan) instrument in a cryo mode with a secondary electron image resolution of 1.0 nm at 15°kV, 2.0°nm at 1°kV, and 1.4°nm at 1°kV with the beam. The material was fixed on a holder and was rapidly frozen with boiling liquid nitrogen. It was then transferred to the high vacuum cryo‐unit, the Balzers BF type freeze etching chamber. The cryo‐chamber is equipped with a knife, which could be handled from outside using a lever to fracture the sample, for applications in which imaging of the surface of inner structures is aimed. To remove humidity, the samples were cryo‐etched by subliming from 5 to 15 min at −95 °C, after which the sample was further inserted into the observation chamber.
Transmission Electron Microscopy
Transmission electron microscopy (TEM) images were conducted on a Philips CM120 (Amsterdam, The Netherlands) electron microscope at an accelerating voltage of 80°kV. The samples were prepared by dropping nanogel solution onto copper grids coated with a thin film of Formvar and carbon. For all the measurements, aqueous solution of nanogels with a concentration of 1 mg mL−1 was used.
Estimation of Circular Dichroism
Circular dichroism (CD) measurements were carried out with an Olis 17 DSM (Bogart, Georgia, USA); a Cary 17 monochromater was used with a spectral output of 184–260°nm. CD spectra of CMR‐197, CMR‐197 functionalized nanogels with and without sonication and unconjugated nanogels were measured at a concentration of 0.5°mg mL−1 using a cell with 0.090°mm light path.The used wavelength range was between 190 and 260°nm with a bandwidth of 2.00°nm with number of increments of 50 and an integration time of 20 s. CD spectra were expressed in terms of mean residue ellipticity (deg cm2 dmol−1). The data presented are an average of five scans.
Evaluation and Characterization of the In Vitro Model of the BBB
The cell lines used in this study were purchased as follows: U‐87 from DSMZ (Braunschweig, Germany), HT12356 and T3 cells kindly provided by Prof. A. Temme (Section of Experimental Neurosurgery and Tumor Immunology, Department of Neurosurgery, University Hospital Carl Gustav Carus, TU Dresden), hCMEC/D3 from Merck Millipore (Burlington, Massachussets, USA), and NHA from Lonza (Basel, Switzerland). The influence on the tight junctions and potential disruption of the blood brain barrier model were assessed for each cell line used in the co‐culture BBB system (human glioblastoma cell lines U‐87, HT12346, T3) (Figure S4, Supporting Information). The in vitro BBB model consisting of the human brain capillary endothelial cell line hCMEC/D3, normal human pericytes, normal human astrocytes (NHA), or human glioblastoma cell lines was designed as described by Eigenmann et al.[ 41 ] While a co‐culture with NHA resulted in a stable monolayer of the hCMEC/D3 cells, co‐culture with any glioblastoma cell line destroyed the tight junctions within 3–5 days. Co‐culturing with any of the three cell lines initiated on day 1 did not allow the formation of a stable BBB. Thus, for the experimental setup, co‐culturing with different cell lines was performed after the tight junction was established. Immunofluorescence staining for visualization of JAM‐1, VE‐Cadherin, and Claudin 5 (all abcam antibodies) expression were performed according to supplier protocols. Additionally, the BBB integrity was monitored by TEER measurements of hCMEC/D3 mono‐culture compared to co‐culture with NHAs or glioblastoma cells. These ensured that nanogel crossing is not an effect of a leaky BBB model. After each experiment, the membrane was embedded and analyzed via TEM to estimate the model integrity at the time of the experiment. Raster electron microscopy is used to additionally confirm the location and monolayer of the cells on the membrane.
Estimation of Tumor Growth Factor ß2 (TGFß2) Secretion by ELISA
The concentration of TGFß2 in the cell culture supernatant (collected after 3 days culture) was quantified using TGF beta 2 Human ELISA Kit (Abcam, Cambridge, UK) according to suppliers' protocol.
Flow Cytometric Analysis
The DTR expression of DTR on hCMEC/D3 cells was investigated after 3 days in the co‐culture. For evaluation of cell surface expression of DTR by flow cytometry (Cytomics FC 500, Beckman Coulter, Krefeld, Germany), the apical growing endothelial cells (transwell system with 0.4 µm pore size) were harvested by trypsinization. After washing with PBS, the cells were incubated overnight at 4 °C with mouse monoclonal DTR‐specific antibody (Abcam, 5°µg mL−1), followed by incubation with AF555‐conjugated rabbit anti‐mouse antibody (Cell Signaling, Danvers, USA, 1:500 for 1 h at room temperature). The flow cytometry data were analyzed using CXP software (Beckman Coulter). For microscopic analysis (Axio Scope A1), cells were cytospinned on coverslips and co‐stained with DAPI.
Analysis of MMP2/9 Activity
The MMP2/9 activities of investigated co‐culture models were visualized by zymography. As control, the hCMEC/D3 cells were cultivated in media supplemented with TGFß2 (25 ng mL−1). For zymography, basolateral media were collected. The protein concentration was determined using Breadford (Sigma). Of each sample, 15 µg of protein were mixed with non‐reducing Laemmli buffer (4×), and separated by electrophoresis, using a non‐reducing SDS‐PAGE gel embedded with gelatin. The MMP2/9 activities were visualized by staining with Coomassie. MMP levels were quantified using ELISA according to manufactures' protocol (R&D systems, Minneapolis, USA).
Blocking Experiments
Blocking experiments: Transcytosis and uptake of radiolabeled nanogels with covalently bound CRM‐197 (NG‐CRM) and without CRM‐197 modification (NG) with and without competition with CRM‐197 protein (0.1°mg per well for 60 min). Both nanogels (1°MBq per well) are incubated for 90 min under standardized cell culture conditions. After incubation of radiolabeled nanogels, hCMEC/D3 cells were harvested from the trans‐well insert, and uptake was quantified via gamma‐counting. Media from the basolateral side of the BBB model was collected; astrocytes and glioblastoma cells were harvested, counted, and separately analyzed by gamma counting.
Cell Uptake and DNA Incorporation of [125I]ITdU
To investigate the cellular uptake of [125I]ITdU‐CRM‐197‐nanogel, cells were incubated with nanogels (0.5°MBq) for 1 and 4 h. For inhibition study, the cells were treated with 0.1 µm L−1 NBMPR for 1 h prior to incubation with [125I]ITdU‐CRM‐197‐nanogel or [125I]ITdU. For comparison, the cells were incubated with the radiopharmaceutical [125I]ITdU or 68Ga‐labeled nanogels (both 0.5 MBq). After two wash steps with PBS, the U‐87 cells and NHA were harvested and analyzed in terms of nanogel internalization by a gamma counter. The decay corrected cell accumulated activity was calculated relative to the implemented activity. For the isolation of DNA from cultured cells, the DNeasy Blood & Tissue Kit from Qiagen (Hilden, Germany) was used following the exact protocol provided by the company. For phosphorimaging, samples were mixed (1:1 ratio) with a native sample buffer and loaded into a kD TGX gel (Bio‐Rad, Hercules, USA). After run under non‐denaturing conditions, gels were exposed to the phosphorimaging plate overnight and subsequently scanned by phosphorimager (Typhoon FLA 9000, GE, Boston, USA). Detected bands were quantified using Image Quant LAS 4000 software.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
S.S. and N.D. contributed equally to this work. This work was financially supported by the Excellence Initiative of the German federal and state governments in the framework of the I3TM Seed Fund Program (grant no. G:(DE‐82)ZUK2‐SF‐OPSF352), the Faculty of Medicine, RWTH Aachen University, and by Deutsche Forschungsgemeinschaft (DFG) in the framework of the Research Training Group 2375 Tumor‐targeted Drug Delivery (grant no. 331065168). This work was part of the thesis “Evaluation of Radiolabeled Stimuli‐Sensitive Nanogels for Application in Nuclear Medicine” submitted by N.D. in 2019 at tje RWTH Aachen University. The manuscript is an edited version of Chapter 7 “Transcytosis and Delivery of Nano‐irradiation with Biodegradable Nanogels in an In Vitro Blood Brain Barrier Model”.
Open access funding enabled and organized by Projekt DEAL.
Singh S., Drude N., Blank L., Desai P. B., Königs H., Rütten S., Langen K.‐J., Möller M., Mottaghy F. M., Morgenroth A., Protease Responsive Nanogels for Transcytosis across the Blood−Brain Barrier and Intracellular Delivery of Radiopharmaceuticals to Brain Tumor Cells. Adv. Healthcare Mater. 2021, 10, 2100812. 10.1002/adhm.202100812
Contributor Information
Smriti Singh, Email: singh@dwi.rwth-aachen.de.
Agnieszka Morgenroth, Email: amorgenroth@ukaachen.de.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1. Daneman R., Prat A., Cold Spring Harbor Perspect. Biol. 2015, 7, a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zlokovic B. V., Neuron 2008, 57, 178. [DOI] [PubMed] [Google Scholar]
- 3. Picone P., Sabatino M. A., Ditta L. A., Amato A., San Biagio P. L., Mulè F., Giacomazza D., Dispenza C., Di Carlo M., J. Controlled Release 2018, 270, 23. [DOI] [PubMed] [Google Scholar]
- 4.a) Airan R. D., Meyer R. A., Ellens N. P., Rhodes K. R., Farahani K., Pomper M. G., Kadam S. D., Green J. J., Nano Lett. 2017, 17, 652; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mead B. P., Kim N., Miller G. W., Hodges D., Mastorakos P., Klibanov A. L., Mandell J. W., Hirsh J., Suk J. S., Hanes J., Price R. J., Nano Lett. 2017, 17, 3533; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pulicherla K. K., Verma M. K., AAPS PharmSciTech 2015, 16, 223; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Vince O., Peeters S., Johanssen V. A., Gray M., Smart S., Sibson N. R., Stride E., Adv. Healthcare Mater. 2021, 10, 2001343. [DOI] [PubMed] [Google Scholar]
- 5.a) Banks W. A., Nat. Rev. Drug Discovery 2016, 15, 275; [DOI] [PubMed] [Google Scholar]; b) Singh D., Kevadiya B., Nagpal K., Sharma N., Patel S., J. Nanomed. Res. 2017, 5, 00132; [Google Scholar]; c) Kabanov A. V., Batrakova E. V., in Neuroimmune Pharmacology (Eds: Ikezu T., Gendelman H. E.), Springer International Publishing, Cham: 2017, p. 847; [Google Scholar]; d) Tam V. H., Sosa C., Liu R., Yao N., Priestley R. D., Int. J. Pharm. 2016, 515, 331; [DOI] [PubMed] [Google Scholar]; e) Zhou Y., Peng Z., Seven E. S., Leblanc R. M., J. Controlled Release 2017, 270, 290; [DOI] [PubMed] [Google Scholar]; f) Lee S. W. L., Campisi M., Osaki T., Possenti L., Mattu C., Adriani G., Kamm R. D., Chiono V., Adv. Healthcare Mater. 2020, 9, 1901486; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Ayer M., Schuster M., Gruber I., Blatti C., Kaba E., Enzmann G., Burri O., Guiet R., Seitz A., Engelhardt B., Klok H.‐A., Adv. Healthcare Mater. 2021, 10, 2001375; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Zhang T., Lip H., He C., Cai P., Wang Z., Henderson J. T., Rauth A. M., Wu X. Y., Adv. Healthcare Mater. 2019, 8, 1900543. [DOI] [PubMed] [Google Scholar]
- 6. May J. N., Golombek S. K., Baues M., Dasgupta A., Drude N., Rix A., Rommel D., von Stillfried S., Appold L., Pola R., Pechar M., van Bloois L., Storm G., Kuehne A. J. C., Gremse F., Theek B., Kiessling F., Lammers T., Theranostics 2020, 10, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Romermann K., Fedrowitz M., Hampel P., Kaczmarek E., Tollner K., Erker T., Sweet D. H., Loscher W., Neuropharmacology 2017, 117, 182. [DOI] [PubMed] [Google Scholar]
- 8. Arranja A. G., Pathak V., Lammers T., Shi Y., Pharmacol. Res. 2017, 115, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Ivask A., Pilkington E. H., Blin T., Kakinen A., Vija H., Visnapuu M., Quinn J. F., Whittaker M. R., Qiao R., Davis T. P., Ke P. C., Voelcker N. H., Biomater. Sci. 2018, 6, 314; [DOI] [PubMed] [Google Scholar]; b) Paterson J., Webster C. I., Drug Discovery Today: Technol. 2016, 20, 49; [DOI] [PubMed] [Google Scholar]; c) Mager I., Meyer A. H., Li J., Lenter M., Hildebrandt T., Leparc G., Wood M. J. A., Neuropharmacology 2017, 120, 4. [DOI] [PubMed] [Google Scholar]
- 10.a) Wang P., Xue Y., Shang X., Liu Y., Cell. Mol. Neurobiol. 2010, 30, 717; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Robinson M., Lee B. Y., Leonenko Z., Mol. Sci. 2015, 2, 332; [Google Scholar]; c) Fang F., Zou D., Wang W., Yin Y., Yin T., Hao S., Wang B., Wang G., Wang Y., Mater. Sci. Eng. C 2017, 76, 1316. [DOI] [PubMed] [Google Scholar]
- 11. Rip J., Schenk G. J., de Boer A. G., Expert Opin. Drug Delivery 2009, 6, 227. [DOI] [PubMed] [Google Scholar]
- 12. Tosi G., Vilella A., Veratti P., Belletti D., Pederzoli F., Ruozi B., Vandelli M. A., Zoli M., Forni F., Mol. Pharm. 2015, 12, 3672. [DOI] [PubMed] [Google Scholar]
- 13. Carlsson J., Forssell‐Aronsson E., Glimelius B., Mattsson S., Group S. C. S. I., Acta Oncol. 2002, 41, 623. [PubMed] [Google Scholar]
- 14. Kassis A. I., Adelstein S. J., J. Nucl. Med. 2005, 46, 4s. [PubMed] [Google Scholar]
- 15. Howell R. W., Rao D. V., Hou D. Y., Narra V. R., Sastry K. S., Radiat. Res. 1991, 128, 282. [PubMed] [Google Scholar]
- 16.a) Morgenroth A., Vogg A. T. J., Ermert K., Zlatopolskiy B., Mottaghy F. M., Oncotarget 2014, 5, 5483; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morgenroth A., Dinger C., Zlatopolskiy B. D., Al‐Momani E., Glatting G., Mottaghy F. M., Reske S. N., Nucl. Med. Biol. 2011, 38, 1067. [DOI] [PubMed] [Google Scholar]
- 17.a) Morgenroth A., Deisenhofer S., Glatting G., Kunkel F. H., Dinger C., Zlatopolskiy B., Vogg A. T., Kull T., Reske S. N., Clin. Cancer Res. 2008, 14, 7311; [DOI] [PubMed] [Google Scholar]; b) Reske S. N., Deisenhofer S., Glatting G., Zlatopolskiy B. D., Morgenroth A., Vogg A. T., Buck A. K., Friesen C., J. Nucl. Med. 2007, 48, 1000. [DOI] [PubMed] [Google Scholar]
- 18. Drude N., Singh S., Winz O. H., Moller M., Mottaghy F. M., Morgenroth A., Biomacromolecules 2017, 18, 2489. [DOI] [PubMed] [Google Scholar]
- 19. Groll J., Singh S., Albrecht K., Moeller M., J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5543. [Google Scholar]
- 20. Patterson J., Hubbell J. A., Biomaterials 2010, 31, 7836. [DOI] [PubMed] [Google Scholar]
- 21.a) Drude N., Winz O. H., Mottaghy F. M., Roller M., Konigs H., Moller M., Singh S., Morgenroth A., Small 2018, 14, 1704093; [DOI] [PubMed] [Google Scholar]; b) Singh S., Bingöl B., Morgenroth A., Mottaghy F., Möller M., Schmaljohann J., Macromol. Rapid Commun. 2013, 34, 562. [DOI] [PubMed] [Google Scholar]
- 22. Mishima K., Higashiyama S., Asai A., Yamaoka K., Nagashima Y., Taniguchi N., Kitanaka C., Kirino T., Kuchino Y., Acta Neuropathol. 1998, 96, 322. [DOI] [PubMed] [Google Scholar]
- 23. Izumi Y., Hirata M., Hasuwa H., Iwamoto R., Umata T., Miyado K., Tamai Y., Kurisaki T., Sehara‐Fujisawa A., Ohno S., Mekada E., EMBO J. 1998, 17, 7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hagemann C., Anacker J., Ernestus R. I., Vince G. H., World J. Clin. Oncol. 2012, 3, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Wick W., Naumann U., Weller M., Curr. Pharm. Des. 2006, 12, 341; [DOI] [PubMed] [Google Scholar]; b) Baumann F., Leukel P., Doerfelt A., Beier C. P., Dettmer K., Oefner P. J., Kastenberger M., Kreutz M., Nickl‐Jockschat T., Bogdahn U., Bosserhoff A. K., Hau P., Neuro‐Oncol 2009, 11, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ogier C., Bernard A., Chollet A. M., Diguardher T. L. E., Hanessian S., Charton G., Khrestchatisky M., Rivera S., Glia 2006, 54, 272. [DOI] [PubMed] [Google Scholar]
- 27. Zhang S., Li J., Lykotrafitis G., Bao G., Suresh S., Adv. Mater. 2009, 21, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoelzinger D. B., Mariani L., Weis J., Woyke T., Berens T. J., McDonough W. S., Sloan A., Coons S. W., Berens M. E., Neoplasia 2005, 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Morgenroth A., Vogg A. T. J., Zlatopolskiy B. D., Siluschek M., Oedekoven C., Mottaghy F. M., Mol. Cancer Ther. 2014, 13, 144; [DOI] [PubMed] [Google Scholar]; b) Miran T., Vogg A. T. J., Drude N., Mottaghy F. M., Morgenroth A., FASEB. J. 2018, 32, 2803; [DOI] [PubMed] [Google Scholar]; c) Morgenroth A., Deisenhofer S., Glatting G., Kunkel F. H., Dinger C., Zlatopolskiy B. D., Vogg A. T. J., Kull T., Reske S. N., Clin. Cancer Res. 2008, 14, 7311; [DOI] [PubMed] [Google Scholar]; d) Morgenroth A., Vogg A. T. J., Ermert K., Zlatopolskiy B. D., Mottaghy F. M., Oncotarget 2014, 5, 5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Kassis A. I., Wen P. Y., Van den Abbeele A. D., Baranowska‐Kortylewicz J., Makrigiorgos G. M., Metz K. R., Matalka K. Z., Cook C. U., Sahu S. K., Black P. M., Adelstein S. J., J. Nucl. Med. 1998, 39, 1148; [PubMed] [Google Scholar]; b) Kassis A. I., Adelstein S. J., Acta Oncol. 1996, 35, 935; [DOI] [PubMed] [Google Scholar]; c) Kassis A. I., Van den Abbeele A. D., Wen P. Y., Baranowska‐Kortylewicz J., Aaronson R. A., DeSisto W. C., Lampson L. A., Black P. M., Adelstein S. J., Cancer Res. 1990, 50, 5199. [PubMed] [Google Scholar]
- 31.a) Zupanc G. K. H., Horschke I., Comp. Biochem. Physiol. 1996, 114B, 269; [DOI] [PubMed] [Google Scholar]; b) Vernis L., Piskur J., Diffley J. F. X., Nucleic Acids Res. 2003, 31, e120; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McKinley E. T., Ayers G. D., Smith R. A., Saleh S. A., Zhao P., Washington M. K., Coffey R. J., Manning H. C., PLoS One 2013, 8, e58938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pennycooke M., Chaudary N., Shuralyova I., Zhang Y., Coe I. R., Biochem. Biophys. Res. Commun. 2001, 280, 951. [DOI] [PubMed] [Google Scholar]
- 33. Toyohara J., Hayashi A., Sato M., Tanaka H., Haraguchi K., Yoshimura Y., Yonekura Y., Fujibayashi Y., J. Nucl. Med. 2002, 43, 1218. [PubMed] [Google Scholar]
- 34.a) Sery Q., Rabe M., Oliver L., Vallette F. M., Gratas C., Biochem. Biophys. Res. Commun. 2017, 493, 1377; [DOI] [PubMed] [Google Scholar]; b) Höbel S., Appeldoorn C., Gaillard P., Aigner A., Pharmaceuticals 2011, 4, 1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang D., Zou L., Jin Q., Hou J., Ge G., Yang L., Acta Pharm. Sin. B 2018, 8, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang P., Liu Y., Shang X., Xue Y., J. Mol. Neurosci. 2011, 43, 485. [DOI] [PubMed] [Google Scholar]
- 37.a) Chen X., Xie T., Fang J., Xue W., Kang H., Tong H., Guo Y., Zhang B., Wang S., Yang Y., Zhang W., Cancer Biol. Ther. 2018, 19, 416; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ramnarain D. B., Park S., Lee D. Y., Hatanpaa K. J., Scoggin S. O., Otu H., Libermann T. A., Raisanen J. M., Ashfaq R., Wong E. T., Wu J., Elliott R., Habib A. A., Cancer Res. 2006, 66, 867. [DOI] [PubMed] [Google Scholar]
- 38.a) Miran T., Vogg A. T. J., El Moussaoui L., Kaiser H. J., Drude N., von Felbert V., Mottaghy F. M., Morgenroth A., Cancer Med. 2017, 6, 1639; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Drude N., Singh S., Winz O. H., Möller M., Mottaghy F. M., Morgenroth A., Biomacromolecules 2017, 18, 2489; [DOI] [PubMed] [Google Scholar]; c) Drude N., Winz O. H., Mottaghy F. M., Roller M., Königs H., Möller M., Singh S., Morgenroth A., Small 2018, 14, 1704093. [DOI] [PubMed] [Google Scholar]
- 39. Higashiyama S., Abraham J. A., Miller J., Fiddes J. C., Klagsbrun M., Science 1991, 251, 936. [DOI] [PubMed] [Google Scholar]
- 40. Schollenberger S., Taureck D., Wilmanns W., Blut 1972, 25, 318. [DOI] [PubMed] [Google Scholar]
- 41. Eigenmann D. E., Xue G., Kim K. S., Moses A. V., Hamburger M., Oufir M., FluidsBarrires CNS 2013, 10, e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.