

Abstract
Cardiac tissue engineering is a promising strategy to prevent heart failure. However, several issues remain unsolved, including efficient electrical coupling and incorporating factors to enhance tissue maturation and vascularization. Herein, a biohybrid hydrogel that enhances beating properties of engineered cardiac tissues and allows drug release concurrently is developed. Gold nanoparticles (AuNPs) with different sizes (18–241 nm) and surface charges (33.9–55.4 mV) are synthesized by reducing gold (III) chloride trihydrate using branched polyethyleneimine (bPEI). These nanoparticles increase gel stiffness from ≈91 to ≈146 kPa, enhance electrical conductivity of collagen hydrogels from ≈40 to 49–68 mS cm−1, and allow slow and steady release of loaded drugs. Engineered cardiac tissues based on bPEI‐AuNP‐collagen hydrogels and either primary or human induced pluripotent stem cell (hiPSC)‐derived cardiomyocytes show enhanced beating properties. hiPSC‐derived cardiomyocytes exhibit more aligned and wider sarcomeres in bPEI‐AuNP‐collagen hydrogels compared to collagen hydrogels. Furthermore, the presence of bPEI‐AuNPs result in advanced electrical coupling evidenced by synchronous and homogenous calcium flux throughout the tissue. RNA‐seq analyses are in agreement with these observations. Collectively, this data demonstrate the potential of bPEI‐AuNP‐collagen hydrogels to improve tissue engineering approaches to prevent heart failure and possibly treat diseases of other electrically sensitive tissues.
Keywords: collagen, gold nanoparticles, heart, tissue engineering
Drug treatment and cardiac tissue engineering are both promising strategies to treat heart disease. Herein, an electroconductive biohybrid hydrogel is developed which allows fabrication of synchronously beating muscle tissue as well as drug release. Human‐induced pluripotent stem cell–derived cardiomyocytes in this biohybrid hydrogel exhibit improved cellular alignment and sarcomere arrangement associated with enhanced beating properties of engineered tissues.
1. Introduction
Cardiovascular diseases (CVD) are the leading cause of death worldwide, responsible for nearly 19 million deaths in 2020[ 1 ] and annually impose an estimated €200 billion on the European countries and $378 billion on the United States.[ 1 , 2 ] Ischemic heart diseases, including myocardial infarction (MI), comprise the major part of CVD, accounting for 39–45% of CVD‐related deaths.[ 2 ] MI results in a cell loss‐dependent impairment of pumping function accompanied by ventricular arrhythmia or tachycardia. Cell therapy has been considered a promising approach to compensate for the loss of cardiomyocytes. However, it suffered from low engraftment and limited survival rate of transplanted cells in the recipient tissue.[ 3 ] Moreover, transplanted human embryonic or induced pluripotent stem cell (hiPSC)‐derived cardiomyocytes resulted in ventricular arrhythmia or tachycardia, which needs to be addressed.[ 4 ] This could be due to inefficient electrical coupling within the graft and between the graft and host tissue.
Over the past 2 decades, engineered biomaterials provided significant advancements in tissue engineering, including enhanced cellular retention, survival, and integration with the host tissue.[ 5 ] Notably, electrically conductive biohybrid systems provide an environment to facilitate the propagation of intercellular electrical signals.[ 6 ] Different electroconductive materials, including nanoparticles, nanorods, nanowires, carbon nanotubes, and polymers have been used in tissue engineering applications.[ 7 ] We have previously shown that introducing electroconductive materials facilitates intercellular communications, enhances cardiomyocyte maturation, and improves the beating properties of engineered cardiac tissues.[ 8 , 9 , 10 , 11 ] We developed electrically conductive electrospun fiber mats based on collagen, hyaluronic acid, and polyaniline to mimic the cardiac extracellular matrix and showed the positive effects of the conductive fabrics on neonatal rat ventricular cardiomyocytes as well as hiPSC‐derived cardiomyocytes.[ 11 ] To investigate the beneficial effects of conductive materials in 3D, we developed an electrically conductive biohybrid hydrogel system based on collagen, alginate, and poly(3,4‐ethylenedioxythiophene) polystyrene sulfonate and showed enhanced maturation and electrical coupling of primary and hiPSC‐derived cardiomyocytes.[ 10 ] Moreover, we have shown that the presence of hydrazide‐functionalized multiwall carbon nanotubes in pericardial matrix‐derived hydrogels enhances contractility, calcium handling, and the response of hiPSC‐derived cardiomyocytes to several pharmaceutical treatments.[ 8 , 9 ]
Recent clinical studies have shown that injecting cell‐free hydrogels into the myocardial wall of infarcted hearts stabilizes the ventricular wall.[ 12 ] Combining this mechanical support with the capability to deliver pharmaceuticals, growth factors, or proangiogenic factors can potentially enhance the function of the host tissue. Moreover, combining pharmaceutical approaches with engineered tissues can improve their beneficial effects. For example, the release of proangiogenic factors will help to facilitate the vascularization of the graft, ensuring survival and optimal function. Other beneficial factors might be pro‐proliferative[ 13 ] or anti‐arrhythmic drugs. Importantly, uncontrolled administration of drugs can have off‐target effects, be prematurely cleared from the body, be difficult to dose, or have a post‐administration burst release. Therefore, it is beneficial to incorporate a drug release system into engineered cardiac tissues. Previously, it has been shown that gold nanoparticles (AuNP)s as drug carriers can prolong the presence of drugs and minimize their burst and off‐target effects.[ 14 ] AuNPs have reportedly been used to deliver drugs and microRNA to the heart to treat heart failure in vivo.[ 15 ] Similarly, branched polyethyleneimine (PEI)‐coated AuNPs have been utilized to release siRNAs[ 16 ] and gapmers.[ 17 ] PEI is a basic, cationic polyamine. The delivery of bioactive factors using PEI is modulated based on the affinity binding of these factors to PEI, which is based on ionic interactions.[ 18 ] Polycationic PEI can condense negatively charged molecules into stable complexes resulting in their retention in the hydrogels. Degradation of PEI or possible pH changes trigger the release of the bioactive factors. Regulating these processes along with a control over the extent of ionic interactions can tailor the release profile.[ 18 ]
Notably, gold is a conductive material whereby AuNPs offer easy fabrication and tailorable geometrical and physicochemical properties. AuNPs have been used for imaging, diagnostics, and tissue engineering application.[ 7 ] Gold is chemically inert[ 19 ] and AuNPs have no effects on the immune response at concentrations below 1% w/v.[ 20 ] Previous studies based on seeding rat cardiomyocytes on scaffolds containing AuNPs collectively showed enhanced alignment of these cells accompanied by increased connexin 43 expression.[ 7 ] Notably, rat cardiomyocytes cultured on a collagen‐AuNP hybrid hydrogel established efficient intercalated discs indicating enhanced intercellular coupling.[ 21 ] However, these studies were limited to post‐fabrication seeding of rat cardiomyocytes on 2D hydrogel films. As an advancement, 3D printing of rat cardiomyocyte‐loaded hydrogels based on gold nanorods and gelatin metacryloyl (Gel‐MA) was reported to enhance connexin 43 expression in these cells.[ 22 ] However, the functionality of the tissue and cellular calcium handling properties were not investigated. Notably, there is a gap in knowledge regarding the effects of AuNPs in a 3D hydrogel system on human cardiomyocytes. Moreover, there are no reports utilizing the dual function of bPEI‐AuNPs for enhancing the conductivity and allowing the release of biologically active materials.
In this study, we have developed an electrically conductive hybrid hydrogel based on collagen type I and bPEI‐coated AuNPs. A positively charged coating of AuNPs allows for incorporating a wide range of negatively charged pharmaceutical drugs and growth factors. We utilized these hydrogel systems and generated engineered cardiac tissues by incorporating neonatal rat ventricular or hiPSC‐derived cardiomyocytes. We have provided an in‐depth analysis of cell survival, subcellular organization, and calcium handling as an effect of bPEI‐AuNPs in a 3D engineered cardiac tissue. Furthermore, we have demonstrated that the here developed hybrid hydrogel system based on bPEI can be used as a carrier platform for drug release and simultaneously increasing electrical conductivity.
2. Results and Discussion
2.1. One‐Step Generation of AuNPs with Different Sizes and Surface Charges
In order to test the suitability of a gold‐based drug releasing system for cardiac tissue engineering, we generated bPEI‐AuNPs by incubating gold(III) chloride trihydrate (HAuCl4·3H2O, from 12.5 to 50 mg mL−1) and bPEI for 1 h at 65 °C (Figure 1a and Table 1 ).
Figure 1.
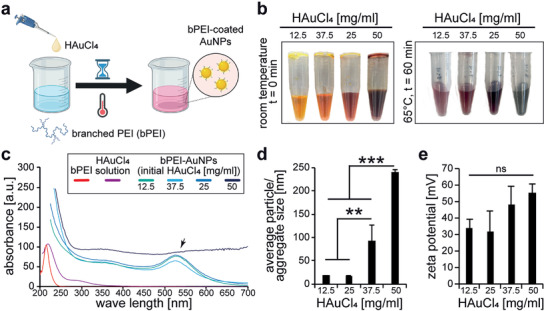
Branched polyethylenimine reduces gold chloride to gold and results in the generation of gold nanoparticles. a) Schematic illustration of the synthesis of gold nanoparticles coated with bPEI. b) Representative photographs of different gold chloride solutions before and after the reaction. c) Representative graphs of UV–vis absorption spectra for the different solutions after the reaction. Absorption graphs of bPEI and HAuCl4·3H2O solution are provided as controls. Black arrow: peak corresponding to the formation of gold nanoparticles. d,e) Quantitative analysis of average particle/aggregate size (diameter, d) and surface charge (e) utilizing dynamic light scattering (DLS) measurements (n = 3). Data are mean ± Standard Deviation (SD) and compared based on a one‐way ANOVA and Bonferroni's multiple comparison test.
Table 1.
Numerical values of particle size and surface charge of bPEI‐AuNPs measured by dynamic light scattering
| Nanoparticles | HAuCl4·3H2O/bPEI | Particle size [nm] | Surface charge [mV] |
|---|---|---|---|
| bPEI‐AuNP‐12.5 | 0.25 | 18.50 ± 0.28 | 33.9 ± 5.6 |
| bPEI‐AuNP‐25 | 0.50 | 18.13 ± 0.62 | 31.6 ± 13 |
| bPEI‐AuNP‐37.5 | 0.75 | 92.75 ± 0.56 | 48.1 ± 11.3 |
| bPEI‐AuNP‐50 | 1.00 | 241.03 ± 0.50 | 55.4 ± 5.48 |
Depending on the initial concentration of HAuCl4·3H2O, an immediate change in the color of the solution was detectable (Figure 1b). Higher concentrations resulted in darker solutions. Analysis of the UV–vis spectra of the different nanoparticle solutions revealed a peak corresponding to the formation of gold nanoparticles for all groups except the solution with the initial concentration of 50 mg mL−1 HAuCl4·3H2O (Figure 1c). In order to eliminate the batch variability effect, we prepared different batches and measured the UV–vis absorption at different dilutions, but all measurements resulted in no peaks for bPEI‐AuNP‐50.
It has been reported that aggregated nanoparticles show several changes in their UV–vis absorption, including changes in the extinction coefficient, maximum wavelength shifts, and formation of new peaks.[ 23 ] It is also reported that alcohol‐induced aggregation of AuNPs results in a time‐dependent decrease of the peak and formation of a new peak at greater wavelengths.[ 24 ] Therefore, we have performed high resolution phase‐contrast microscopic analysis of all different gold nanoparticle solutions (Figure S1a, Supporting Information). This analysis revealed aggregate formation for bPEI‐AuNP‐37.5 as well as bPEI‐AuNP‐50. For bPEI‐AuNP‐12.5 and bPEI‐AuNP‐25, no comparable aggregates were detected. Dynamic light scattering (DLS) analysis, after brief sonication of the samples, were utilized to determine average particle size and surface charge (Figure 1d,e and Table 1).
Our data show that the average particle size and surface charge did not change significantly between bPEI‐AuNP‐12.5 and bPEI‐AuNP‐25 (Figure 1d). However, further increasing the ratio of HAuCl4·3H2O to bPEI resulted in an increase in bPEI‐AuNPs' average particle/aggregate size and surface charge. The initial concentration of 25 mg mL−1 HAuCl4·3H2O resulted in the smallest average particle size of 18.13 ± 0.62 nm (diameter) with a surface charge of 31.6 ± 13 mV compared to the largest average particle/aggregate size of 241.03 ± 0.50 nm and a surface charge of 55.4 ± 5.48 mV observed with an initial concentration of 50 mg mL−1 HAuCl4·3H2O. A closer analysis of the size distribution graphs revealed a narrow distribution of particle size for bPEI‐AuNP‐12.5 and a main peak with a broader distribution for bPEI‐AuNP‐25 (Figure S1b, Supporting Information). In contrast, four similar peaks were detected for bPEI‐AuNP‐37.5. Based on the microscopic analysis (Figure S1a, Supporting Information), the first two peak represents probably single particles (≈25 and ≈105 nm) whereas the other peaks represent two classes of aggregates (≈800 and ≈5 µm). Finally, a very wide peak ≈250 nm and a narrow peak ≈5 µm were detected for bPEI‐AuNP‐50. As the individual particles in Figure S1a, Supporting Information, for bPEI‐AuNP‐50 have a diameter of ≈100 nm, there seems to be no, or at most a few, individual particles detectable by phase contrast microscopy. This might explain why no peak corresponding to the formation of gold nanoparticles was detected in the UV–vis spectra for bPEI‐AuNP‐50.
2.2. AuNPs Enhance the Electrical Conductivity of Collagen Hydrogels
Collagen hydrogels are currently the most advanced hydrogel systems for clinical translation applications. At the beginning of 2021, a clinical trial started with 53 participants with no realistic chance or not being eligible for heart transplantation that will test the performance of engineered cardiac tissues.[ 25 ] Thus, we evaluated the effect of bPEI‐AuNPs on collagen hydrogels. After we obtained bPEI‐AuNPs with different sizes and surface charges, we analyzed whether their incorporation into a pre‐gel solution of collagen I affects gel formation. Nanoparticles were mixed in a pre‐gel solution of collagen I at 1% v/v. The solution was incubated at 37 °C for 1 h to allow thermal‐induced fiber entanglement of collagen I, usually resulting in a gel (Figure 2a).
Figure 2.
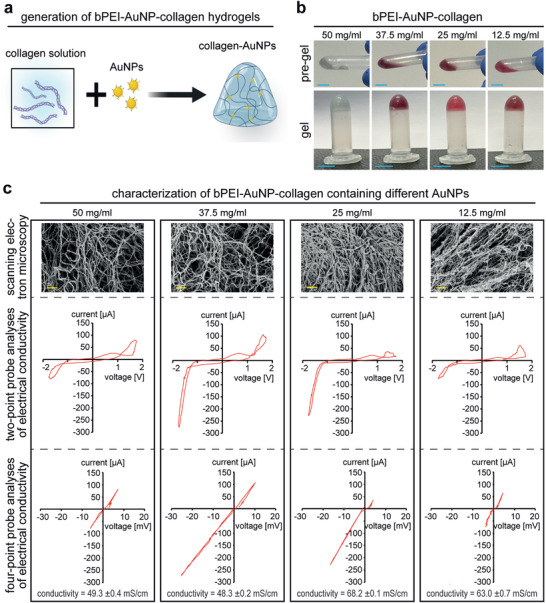
Microstructure and electrical conductivity of bPEI‐AuNP‐collagen hydrogels. a) Schematic illustration of hydrogel formation. b) Representative photographs of different hydrogels before and after gelation. c) Representative scanning electron microscopic images of critically point dried hydrogels containing different types of gold nanoparticles as well as representative graphs of quantitative measurements of electrical conductivity of hydrogels based on two‐point and four‐point analysis of conductivity (n = 3). The concentrations in the figure indicate the initial concentration of HAuCl4·3H2O used for synthesizing bPEI‐AuNPs. Scale bars: 2 µm.
None of the bPEI‐AuNPs prevented gelation (Figure 2b). The generated hydrogels were fixed, dehydrated, critically‐point dried, and sputter‐coated with gold and were analyzed by scanning electron microscopy (SEM). Analysis of their morphology by SEM revealed a fibrous structure in all groups. However, shifting from bPEI‐AuNP‐12.5 toward bPEI‐AuNP‐50 appears to result in less compact fiber networks (Figure 2c).
In order to measure the electrical conductivity of the hydrogels, freshly generated samples were transferred to a custom‐made polytetrafluoroethylene chamber, and their electrical properties were measured based on two‐ and four‐point‐probe analysis of current and voltage (Figure 2c). All different groups of hydrogels showed a typical resistor behavior. Quantitative analyses of electrical conductivity of the hydrogels revealed that smaller AuNPs have a more significant effect on increasing the electrical conductivity of collagen hydrogels. A maximum electrical conductivity of ≈40 mS cm−1 was measured for collagen hydrogels compared to the maximum electrical conductivity of 68.2 ± 0.1 mS cm−1 for collagen hydrogels containing 1% v/v of bPEI‐AuNP derived from HAuCl4·3H2O concentrations of 25 mg mL−1.
To continue our study, we decided against using bPEI‐AuNP‐37.5 and 50 due to their tendency to form aggregates, which could have confounded our experimental results (Figure S1, Supporting Information). However, we found that bPEI‐AuNP‐12.5 and 25 have the same size and surface charge, as confirmed by DLS analysis in Figure 1d,e. Notably, nanoparticles in the size range of 18–20 nm are preferred due to their reduced toxicity in vivo[ 26 ] and lack of damage to DNA in vitro.[ 27 ] In our study, both bPEI‐AuNP‐12.5 and 25 enhanced the electrical conductivity of collagen to similar values of ≈63 and ≈68 mS cm−1. However, as there were no significant differences between these two nanoparticles, we chose bPEI‐AuNP‐25 to continue with our experiments.
2.3. bPEI‐AuNPs Are Cytocompatible
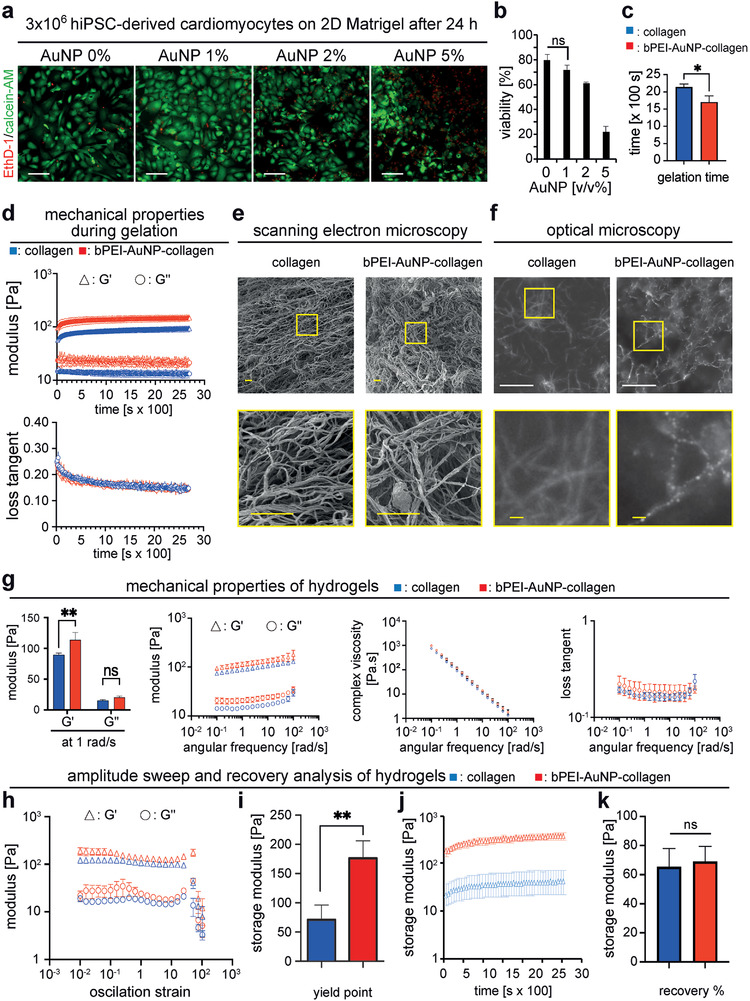
To assess whether the bPEI‐AuNPs are cytocompatible, we tested their toxicity to hiPSC‐derived cardiomyocytes. These cells were seeded at 3 × 105 cells per well of a 24‐well plate and cultured with different concentrations of bPEI‐AuNPs from our bPEI‐AuNP‐25 batch for 24 h (Figure 3a,b). Analysis of live and dead staining images after 24 h showed that the viability of hiPSC‐derived cardiomyocytes treated with 1% v/v was the same as cells cultured without any nanoparticles. However, higher concentrations of bPEI‐AuNPs resulted in a significantly lower viability of cardiomyocytes, indicating cytotoxicity of these nanoparticles at concentrations above 1% v/v. Therefore, we chose 1% v/v bPEI‐AuNPs for the generation of 3D engineered tissues.
Figure 3.
Microstructure and mechanical properties of hydrogels. a) Representative images of hiPSC‐cardiomyocytes cultured on Matrigel for 24 h stained with calcein‐AM (green, living cells) and ethidium homodimer‐1 (EthD‐1, red, dead cells). b) Quantitative analysis of live and dead staining in (a) (n = 3, one‐way ANOVA and Bonferroni's multiple comparison test). c) Quantitative analysis of time to complete gelation of hydrogels performed based on time sweep test (n = 3, two‐tailed t‐test). d) Rheometric analysis of mechanical properties of hydrogels over time during the gelation process (n = 3). e) Representative scanning electron microscopy images of critically‐point dried hydrogels. f) Representative dark‐field optical microscopy images of hydrogels showing collagen fibers and gold nanoparticle–decorated collagen fibers. g) Quantitative post‐gelation rheometric analysis of hydrogels as a function of angular frequency (n = 3, G′ and G′ were compared based on two‐tailed t‐test). h,i) Quantitative rheometric amplitude sweep analysis for 0.01–100% deformation (n = 3) (h) and determination of yield point (i) (n = 3, two‐tailed t‐test). j) Analysis of hydrogel recovery after amplitude sweep test over time (n = 3) and k) quantitative analysis of hydrogel recovery at the end of experiment (n = 3, two‐tailed t‐test). Data are mean ± SD. Scale bars: yellow: 2 µm, white: 20 µm.
2.4. bPEI‐AuNPs Enhance the Mechanical Properties of Collagen Hydrogels
In order to compare bPEI‐AuNP‐collagen hydrogels and collagen hydrogels, we have performed a side‐by‐side analysis of gelation behavior, hydrogel morphology, and mechanical properties of the hydrogels based on rheological analyses. To analyze the gelation kinetics, pre‐gel solutions of the hydrogels were placed on the sample holder of a rheometer at 37 °C, equipped with a solvent trap to minimize dehydration, and their rheological behavior was monitored over time for 45 min. The results of the time sweep test showed a plateau region but no lag or growth phase for storage (G′) and loss modulus (G″) (Figure 3d), which is in agreement with previously reported behavior of collagen I hydrogels studied at 37 °C.[ 28 ] Interestingly, the authors studied the gelation also at lower temperatures. At 32 °C, they detected a growth phase in addition to the plateau region. At 22 or 27 °C, the authors detected three phases; lag, growth, and plateau phase.
In order to study the gelation kinetics more in depth, we set the temperature of the lower plate of the rheometer to 4 °C with a temperature ramp up of 5 °C min−1 to reach 37 °C. These measurements show a lag phase followed by a growth phase ending with a plateau region for both storage modulus (G′) and loss modulus (G″) (Figure S2, Supporting Information). During the growth phase, the G′ values were higher and increased more than G″ resulting in decreased loss tangent values. This indicates a transition toward a more solid‐like material. Notably, no G′/G′' crossover was detected. In order to improve the precision of the measurement, in a second approach the geometry was changed from a 20 to a 40 mm cone plate probe, the sample volume was increased from 200 to 500 µL, and the temperature ramp up was decreased to 1 °C min−1. These measurements resulted in similar profiles whereby the onset of the growth phase was delayed and its duration was slightly increased. Yet, again no G′/G′' crossover was detected. While usually a crossover between G′ and G″ is used as an indication of sol–gel transition, an absence of such a crossover is not necessarily indicating that no sol–gel transition occurs.[ 29 ] We used the G′ as an indicator of gelation considering the storage modulus of complete gelation rather than that of the crossover which indicates initiation of network formation. Time to complete gelation was calculated based on the minimum time required for storage modulus to reach its maximum value. Further analysis of this data showed that the presence of bPEI‐AuNPs resulted in a complete gelation within 28.37 ± 2.46 min which was significantly faster than that for collagen that took 35.73 ± 1.15 min (Figure 3c). After 45 min, storage modulus of collagen was recorded at 91.99 ± 9.46 Pa compared to a significantly greater storage modulus for bPEI‐AuNP‐collagen at 146.22 ± 14.16 Pa (Figure 3d). However, the values of loss tangent, indicating the dampening effect of the hydrogels, were not different. This indicates that incorporation of bPEI‐AuNPs in collagen hydrogels results in a stiffer hydrogel compared to pure collagen. Analysis of SEM images of the critically‐point dried hydrogels showed that collagen and bPEI‐AuNP‐collagen hydrogels exhibit a porous and fibrous structure with no apparent differences (Figure 3e).
To investigate the distribution and adhesion of bPEI‐AuNPs to collagen matrix dark‐field optical micrographs of bPEI‐AuNP‐collagen hydrogel were taken and compared to the images of collagen without any nanoparticles (Figure 3f). Dark‐field imaging mode is here beneficial as it exploits the plasmonic properties of AuNPs making them visible with optical means. Our data show that bPEI‐AuNPs are by large homogeneously distributed across the collagen matrix, along the collagen strands, with typical spacing of the order of a few hundred nanometers. Furthermore, the bPEI‐AuNPs do not appear to coalesce into larger groups. Quantitative analysis of storage (G′) and loss modulus (G″) of hydrogels at 1% shear strain revealed that the presence of bPEI‐AuNPs resulted in a significantly greater storage modulus of 113.71 ± 9.88 Pa at 1 rad s−1 for bPEI‐AuNP‐collagen hydrogels compared to 88.15 ± 1.74 Pa for collagen hydrogels (Figure 3g, Table 2 ). Furthermore, both hydrogels showed a shear thinning behavior evidenced by a decreased viscosity due to increased angular frequency. They showed a similar dampening effect based on the quantitative analysis of the loss tangent (ratio of G″ over G′) over different angular frequencies up to 12 rad s−1.
Table 2.
Summary of mechanical properties of the hydrogels
| end of gelation G′ [Pa] | G′ at 1 rad s−1 [Pa] | |
|---|---|---|
| Collagen | 91.99 ± 9.46 | 88.15 ± 1.74 |
| bPEI‐AuNP‐collagen | 146.22 ± 14.16 | 113.71 ± 9.88 |
We then performed an amplitude sweep test from 0.01% to 100% deformation on the gels (Figure 3h,i). Quantitative analysis of storage and loss modulus of the hydrogels revealed a higher yielding point of 176.12 ± 24.30 Pa at 36.17 ± 1.00% strain for bPEI‐AuNP‐collagen compared to 72.80 ± 24.87 Pa at 24.78 ± 11.92% strain for collagen. However, oscillation displacement at yield was ≈0.013 rad for both bPEI‐AuNP‐collagen and collagen hydrogels. This suggests that while the bPEI‐AuNP‐collagen hydrogel is stronger and can withstand higher stress and strain before yielding, it does not deform significantly more than the collagen hydrogel at yield. Then, we have performed a quantitative analysis of the recovery of the hydrogels after the amplitude sweep test (Figure 3j,k, Figure S3, Supporting Information, and Table 3 ). These data show ≈66% or ≈69% recovery for collagen or bPEI‐AuNP‐collagen hydrogels, respectively. This suggests that the addition of bPEI and AuNP to the collagen hydrogels does not negatively affect the ability to recover. In order to assess the regeneration capacity of the hydrogels, a quantitative analysis of changes in viscosity over time as a function of angular frequency at 0.1 and 100 s−1 was performed (Figure S3, Supporting Information). We noticed great variations in the first measurement phase especially for collagen hydrogels. However, this analysis suggested that bPEI‐AuNPs might slightly enhance the self‐healing potential of collagen hydrogels. These data, summarized in Table 3, indicate that bPEI‐AuNP‐collagen hydrogels compared to collagen alone are similarly shear thinning with the same dampening effects but are significantly stiffer and have greater yield points.
Table 3.
Summary of amplitude sweep and recovery properties of the hydrogels
| Yield point [Pa] | strain at yield [%] | displacement at yield [rad] | recovery [%] | |
|---|---|---|---|---|
| Collagen | 72.80 ± 24.87 | 24.78 ± 11.9 | 0.013 ± 0.004 | 66 ± 12 |
| bPEI‐AuNP‐collagen | 176.12 ± 24.30 | 36.17 ± 1.0 | 0.013 ± 0.000 | 69 ± 8 |
Our data show that the here‐generated hydrogels are shear thinning with a G′ value being in the ranges less than 200 Pa. While being significantly softer than ≈10 kPa reported for the shear storage modulus of human myocardium (end diastolic),[ 30 ] these values are greater than the storage moduli reported in the literature for collagen hydrogels in engineered cardiac tissues that are between 20–30 Pa.[ 31 ] However, very large forces are generated during cardiac cycle that should be tolerated by the material to ensure no drug or cell leakage due to compression cycles. Generating hydrogel‐based engineered cardiac tissues that could bear the mechanical stress of large animals is challenging. However, encapsulated cells secrete their own matrices and extensively remodel their initial microenvironment that usually results in a relatively stiffer and more robust tissue. One possible solution to minimize the risk of tissue rupture post‐transplantation can be the intermittent tissue culture prior to the transplantation. This can give the cells enough time to remodel and strengthen the tissue.
2.5. bPEI‐AuNPs Enhance Primary Cardiomyocyte Contractility in Engineered 3D Tissues
In order to assess the cytocompatibility of these nanoparticles in 3D, neonatal rat ventricular cardiomyocytes were resuspended in bPEI‐AuNP‐collagen pre‐gel solution at 2.5 × 107 cells mL−1 (Figure 4a). Analysis of live and dead staining 7 days after tissue formation revealed that most cells were alive (Figure 4b). These data indicate that the here generated bPEI‐AuNPs at 1% v/v are cytocompatible.
Figure 4.
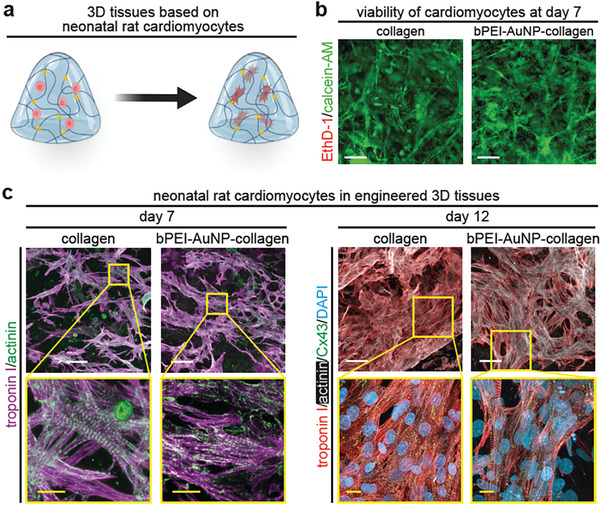
bPEI‐AuNP‐collagen hydrogels support viability and subcellular organization of neonatal rat ventricular cardiomyocytes. a) Schematic illustration of generating engineered cardiac tissues containing neonatal rat ventricular cardiomyocytes. b) Representative images of neonatal rat ventricular cardiomyocytes cultured within hydrogels for 7 days stained with calcein‐AM (green, living cells) and ethidium homodimer‐1 (EthD‐1, red, dead cells). c) Examples of projections of confocal images of neonatal rat ventricular cardiomyocyte‐laden tissue constructs stained for the cardiomyocyte‐specific markers troponin I and sarcomeric‐α‐actinin (day 7 post‐fabrication) and connexin 43 (Cx43, day 12 post‐fabrication). DNA was visualized with DAPI. Scale bars: yellow: 5 µm and white: 25 µm.
In order to evaluate whether the presence of AuNPs affects the contractile apparatus of cardiomyocytes, engineered tissues were fixed and subsequently stained for cardiac troponin I and sarcomeric alpha‐actinin. Cardiomyocytes contained well‐established sarcomeres after 7 and 12 days in both collagen‐ and bPEI‐AuNP‐collagen–based engineered cardiac tissues (Figure 4c). No apparent difference was observed between the two types of engineered cardiac tissues. These data substantiate the conclusion that bPEI‐AuNPs are cytocompatible, exhibiting no toxicity toward neonatal rat ventricular cardiomyocytes.
Finally, we tested whether bPEI‐AuNPs inside collagen‐hydrogels affect the contractile behavior of engineered cardiac tissues containing neonatal rat ventricular cardiomyocytes. For this purpose, we performed quantitative analyses of recorded videos of beating collagen‐ and bPEI‐AuNP‐collagen–based constructs using the MUSCLEMOTION plugin (NIH ImageJ software) (Figure 5 ).[ 32 ]
Figure 5.
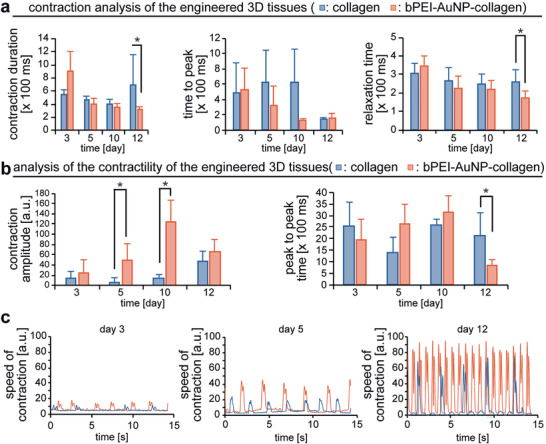
Addition of bPEI‐AuNPs results in enhanced contractility of engineered cardiac tissues containing neonatal rat cardiomyocytes. a,b) MUSCLEMOTION‐based quantitative analysis of contraction (a) and contractility (b). c) Examples of engineered tissues' speed of contraction at different days post‐fabrication. n = 3 in triplicates. Data are mean ± SD and compared based on a two‐way ANOVA and Bonferroni's multiple comparison test. *p < 0.05.
The results revealed that the contraction duration is reduced in the presence of bPEI‐AuNPs due to a reduced relaxation time while the time to peak is not changed (Figure 5a). These changes in contraction behavior were accompanied by a larger contraction amplitude indicating greater force generation, reduced peak‐to‐peak time (equals increased beating frequency), and increased speed of contraction (Figure 5b,c). These data show that engineered cardiac tissues containing bPEI‐AuNPs exhibit enhanced beating properties at 12 days post‐fabrication. Notably, these data are in agreement with the literature showing enhanced contractility of these cells in the presence of AuNPs[ 21 , 22 , 33 ] and with our previous findings that elevated electrical conductivity results in cardiac tissues with enhanced contractile properties.[ 9 , 10 , 11 ]
2.6. bPEI‐AuNPs Enhance Contraction Amplitude of hiPSC‐Derived Cardiomyocytes in Engineered 3D Tissues
While primary cardiomyocytes are a very useful cell source for studying the behavior of cardiomyocytes, they cannot be utilized for therapeutic approaches and exhibit different characteristics than human cardiomyocytes, limiting their usefulness for regenerative therapy and drug testing. Therefore, we analyzed the effect of bPEI‐AuNPs on engineered cardiac tissues containing hiPSC‐derived cardiomyocytes. Engineered cardiac tissues based on collagen I and bPEI‐AuNP‐collagen hydrogels started to beat around 5–7 days after fabrication. Their contractile behavior evolved over time, leading to more regular and synchronous beating. In contrast to neonatal rat cardiomyocyte‐based tissues, quantitative analysis of the contractility of hiPSC‐derived cardiomyocyte‐based tissues using the MUSCLEMOTION plugin of NIH ImageJ revealed no marked changes in contraction behavior or beating frequency (peak‐to‐peak time) in the presence of bPEI‐AuNPs (Figure 6a,b).
Figure 6.
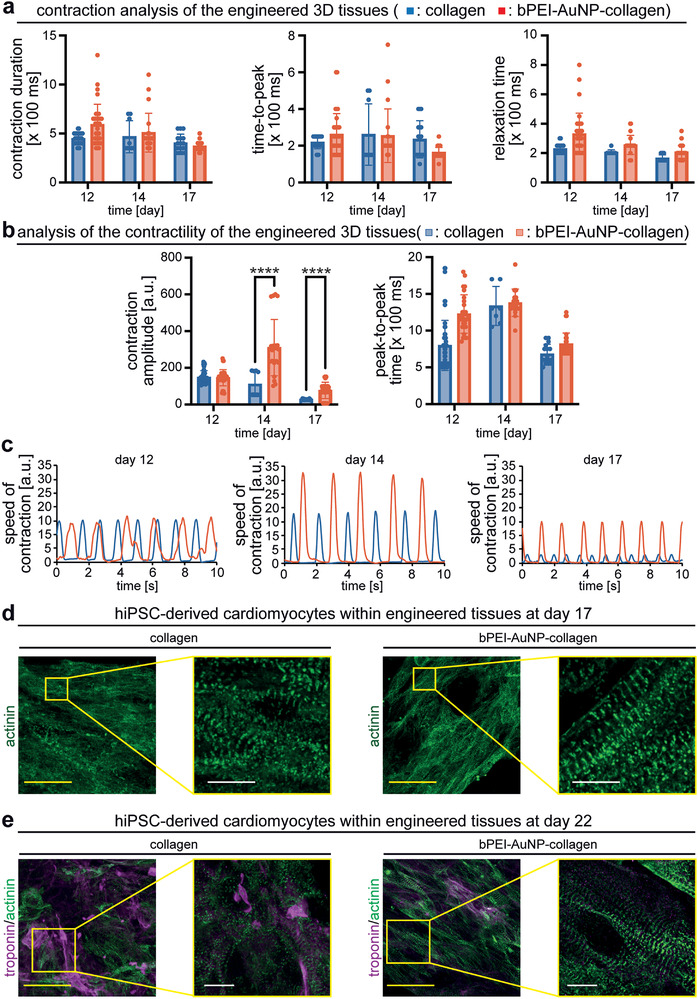
bPEI‐AuNP‐collagen hydrogels support hiPSC‐derived cardiomyocyte function. a,b) MUSCLEMOTION‐based quantitative analysis of contraction (a) and contractility (b) of engineered cardiac tissues. c) Examples of engineered tissues' speed of contraction at different days post‐fabrication. d,e) Examples of projections of confocal images of hiPSC‐derived cardiomyocyte‐laden tissue constructs at 17 days (d) and 22 days (e) post‐fabrication stained for the cardiomyocyte‐specific marker sarcomeric‐α‐actinin and/or troponin I. n = 3. Data are mean ± SD and compared based on a two‐way ANOVA and Bonferroni's multiple comparison test. ****p < 0.0001. Scale bars: yellow: 25 µm and white: 4 µm.
These data are in agreement with our previous observation utilizing electrically conductive polymers.[ 10 , 11 ] The difference between the response of primary and hiPSC‐derived cardiomyocytes could be due to species‐specific properties and/or their different maturation stages. Postnatal rat cardiomyocytes have an intrinsic in vivo beating frequency of 250–500 beats per minute (4.15–8.3 Hz),[ 34 ] while postnatal human heart beats between 80–120 beats per minute (1.3–2 Hz). Importantly, hiPSC‐derived cardiomyocytes primarily represent fetal cardiomyocytes,[ 35 ] while the primary cardiomyocytes are postnatal cells. However, our data show that hiPSC‐derived cardiomyocyte‐based tissues develop a greater contraction amplitude in bPEI‐AuNP‐collagen hydrogels than collagen hydrogels (Figure 6b,c). These data indicate that cardiac tissues engineered with bPEI‐AuNP‐collagen hydrogels exhibit greater forces than collagen hydrogels. This might be due to better‐aligned cardiomyocytes or sarcomeres. Therefore, we have investigated the contractile apparatus of hiPSC‐derived cardiomyocytes within engineered tissues by immunostaining for sarcomeric alpha‐actinin at 17 days post‐fabrication (Figure 6d). Human cardiomyocytes in both collagen and bPEI‐AuNP‐collagen hydrogels contained regular striated sarcomeres. In contrast, sarcomeres were better aligned and wider in cardiomyocytes in bPEI‐AuNP‐collagen hydrogels than collagen hydrogels. In addition, cardiomyocytes appeared more aligned. Analysis of hiPSC‐derived cardiomyocytes for sarcomeric alpha‐actinin and cardiac troponin I at 22 days post‐fabrication (Figure 6e) revealed that the cardiomyocytes in bPEI‐AuNP‐collagen hydrogels remained aligned, and exhibited still more aligned and wider sarcomeres than cardiomyocytes in collagen hydrogels. Furthermore, the here‐generated engineered cardiac tissues beat for 50 days when the experiments were terminated (supplementary movie S4).
2.7. bPEI‐AuNPs Enhance Calcium Handling of hiPSC‐Derived Cardiomyocytes in 3D Tissues
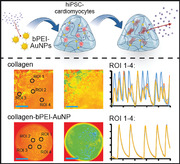
In order to generate maximal force, it is not only crucial that cardiomyocytes contain a well‐organized sarcomeric apparatus and that the cardiomyocytes themselves are aligned but also proper electrical coupling of the cardiomyocytes within these tissues.[ 35 , 36 ] Thus, we investigated the calcium handling of hiPSC‐derived cardiomyocytes within collagen and bPEI‐AuNP‐collagen hydrogels by quantitatively analyzing live videos of Fluo‐4 calcium‐labeled engineered cardiac tissues (Figure 7a,b and Movie S5, Supporting Information). Our data show non‐synchronous calcium flux peaks at different regions of interest in collagen hydrogel‐based tissues. In contrast, we detected synchronous calcium flux peaks when analyzing bPEI‐AuNP‐collagen hydrogel‐based tissues (Figure 7a,b). These data indicate that hiPSC‐derived cardiomyocytes within bPEI‐AuNP‐collagen hydrogels efficiently coupled and exhibited enhanced calcium handling behavior, explaining the increased contraction amplitude (Figure 6b).
Figure 7.
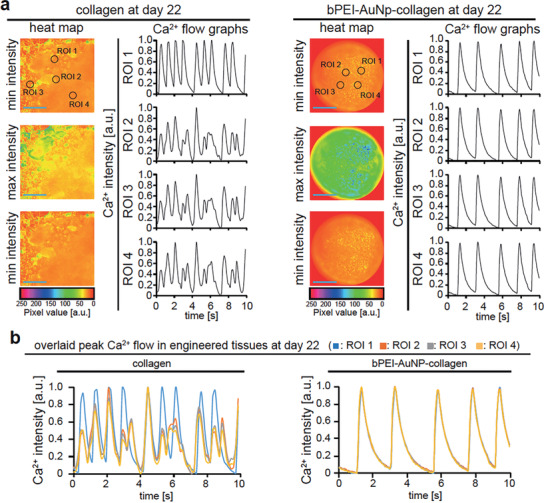
bPEI‐AuNPs enhance calcium handling of hiPSC‐derived cardiomyocytes within hydrogels. a,b) Local calcium minima and maximum concentration based on Fluo‐4 calcium analysis. Analyzed regions of interest (ROI) are indicated by black circles (a). Representative examples of intracellular calcium changes of cardiomyocytes in 3D engineered tissues for different ROIs are provided individually (a) and as overlays (b) to indicate synchronous calcium peaks. Scale bars: blue: 200 µm.
Different models and explanations have been suggested for the mechanism by which electrically conductive materials improve the cell‐to‐cell electrical coupling. The idea is that conductive materials can bridge the electrically resistant obstacles and improve electrical communication between adjacent cells. This was shown when gold nanowires were incorporated within a 3D scaffold of Alginate hydrogel.[ 37 ]
From an activated cell to the neighboring cardiomyocyte, it is suggested that based on the nature of the conductive material, they can either increase the ionic concentration around the recipient cell in the case of ionic conductors or enhance the speed of ion conduction throughout the matrix when electronic conductors are utilized.[ 38 ] AuNPs, as metallic particles, act as electronic conductors. Based on the suggested models, the presence of gold within the matrix increases the signal propagation speed by accelerating the ions that are released from an activated cell. These ions then reach the neighboring cell faster, resulting in an overall enhanced electrical signal propagation.
2.8. bPEI‐AuNPs Improve Calcium Handling, Conduction, and Maturation Based on RNA‐Seq Analyses
Our functional analyses revealed that the presence of bPEI‐AuNPs enhance several important cardiac parameters such as calcium handling, conduction, and sarcomeric apparatus organization. This suggests that the presence of bPEI‐AuNPs has an effect on gene expression in hiPSC‐derived cardiomyocytes. In order to analyze differential expression between the bPEI‐AuNP‐collagen and the collagen groups (each n = 4), RNA‐seq analyses were performed. Figure 8a reveals that there are several differentially expressed genes (Table S1, Supporting Information). The analysis of the expression of a subset of genes previously utilized to demonstrate advanced maturation of human cardiac tissue grown from hiPSC‐derived cardiomyocytes[ 35 ] indicates that the presence of bPEI‐AuNPs also enhances maturation by altering the expression of genes associated to calcium handling, sarcomere maturation, metabolism, and conduction (Figure 8b). Finally, pathway analysis was performed and results were visualized through gene set enrichment networks.[ 39 ] This analysis further confirmed that the presence of bPEI‐AuNPs affects pathways related to “ion transport, muscle contraction, and action potential,” “cell development and growth,” as well as “lipid metabolic process” and “ATP synthesis and metabolics” (Figure 8c and Table S2, Supporting Information). In addition, several unexpected clusters were identified such as “kidney development.” Yet, a closer analysis of the unique genes in each cluster with available RNA consensus tissue gene data showed that on average around 80% of the genes were expressed in the heart (Table S3, Supporting Information). Moreover, on average 75% of the differentially expressed genes in each cluster were expressed in heart.
Figure 8.
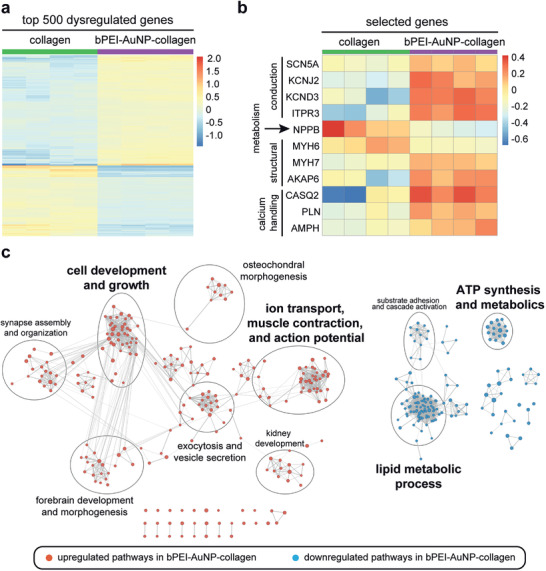
RNA‐seq analyses identify marked expression differences between the collagen and bPEI‐AuNP‐collagen. a) Expression heatmap of the top 500 differentially expressed genes (adjusted p‐value < 0.01) between the bPEI‐AuNP‐collagen (n = 4) and the collagen groups (n = 4). b) Expression heatmap of selected differentially expressed genes (adjusted p‐value < 0.01). Heatmap colors represent expression values (red: up‐regulated; blue: down‐regulated in the bPEI‐AuNP‐collagen). Row/column dendrograms are not shown to increase visibility. c) Gene set enrichment analysis network. Nodes represent significantly dysregulated pathways (FDR < 0.01) and edges (lines connecting nodes) the similarity between two connected pathways based on the proportion of shared genes (measured via the overlap coefficient—OC). Node size is proportional to the number of leading edge genes associated with a given pathway and edge thickness to the OC. Circles identify pathway clusters, and a descriptive label is given for each cluster.
Taken together, the RNA‐seq analyses support our conclusion, based on functional data, that bPEI‐AuNPs enhance maturation of cardiac tissues derived from hiPSC‐cardiomyocytes.
2.9. AuNPs Allow Release of Drugs
To determine whether the here generated bPEI‐AuNPs can be utilized as a drug carrier to release the drug after gelation, we tested our system by loading bPEI‐AuNPs with three different candidate drugs, namely phenylephrine, isoproterenol, and doxorubicin (Figure 9 ). As proof of concept, we initially used doxorubicin, a standard drug utilized in drug delivery studies, to show that our hydrogels allow drug release. However, as doxorubicin is toxic to cardiomyocytes, we utilized in addition phenylephrine and isoproterenol as relevant drugs known to improve heart function. Owing to the cationic bPEI‐dependent positively charged surface of AuNPs, drugs with an overall negative charge can adhere to their surface due to electrostatic forces. It is important to note that different drugs exhibit different surface charges that affect drug loading and subsequent release from bPEI‐AuNPs.[ 40 ] Drugs were loaded on bPEI‐AuNPs by mixing the nanoparticle solution with a stock solutions of phenylephrine (10 mm), isoproterenol (10 mm), or doxorubicin (5 mm) and incubating them for 120 min at room temperature. Then, hydrogels were generated as explained before, yielding a working concentration of phenylephrine (50 µm), isoproterenol (50 µm), or doxorubicin (25 µm) in the hydrogels (Figure 9a).
Figure 9.
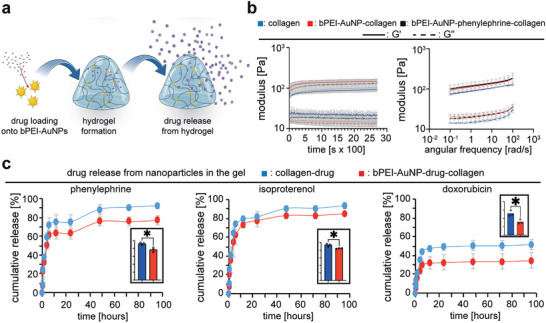
bPEI‐AuNPs function as drug carriers and allow release of drugs from 3D hydrogels. a) Schematic illustration of loading nanoparticles with drugs and subsequent drug release. b) Quantitative rheometric analysis of mechanical properties of hydrogels over time during the gelation process and quantitative post‐gelation rheometric analysis of hydrogels as a function of angular frequency (n = 3, data regarding collagen and bPEI‐AuNP‐collagen is the same as Figure 3 and are provided for comparison purposes). c) Representative graphs of quantitative analyses of drug release from different hydrogels. Measurements were performed based on UV–vis absorption of samples of medium in which hydrogels were incubated (n = 3). Inserted bar graphs show the statistical analysis of the end point release of drugs (n = 3). Data are mean ± SD and compared based on a two‐tailed t‐test.
As a control, collagen hydrogels were generated whereby drugs, without nanoparticles, were added to the pre‐gel solution resulting in the same concentration of drugs. Notably, negatively charged drugs loaded on gold nanoparticles might change the surface charge of these nanoparticles and subsequently affect the mechanical properties of the hydrogels due to a possibly different interaction between nanoparticles and collagen fibers.
To evaluate whether incorporating drug‐loaded nanoparticles result in a different mechanical properties of the hydrogels, we performed rheological analysis on collagen without nanoparticles and hydrogels containing bPEI‐AuNPs or bPEI‐AuNP loaded with phenylephrine (bPEI‐AuNP‐collagen–phenylephrine). The gelation kinetics were monitored based on a time‐sweep test over 45 min followed by a subsequent frequency sweep test to study the frequency dependent behavior of storage and loss modulus in these hydrogels. Further analysis of the data showed no significant differences in mechanical properties of the bPEI‐AuNP‐collagen hydrogels compared to bPEI‐AuNP‐collagen–phenylephrine hydrogels (Figure 9b and Figure S4, Supporting Information).
The quantitative analysis of drug release over 96 h revealed a burst release of ≈60% in the first hours followed by a continuous drug release for all tested hydrogels. The results showed that phenylephrine had a release of 92.32 ± 0.25% when loaded onto bPEI‐AuNPs, while only 77.11 ± 0.45% was released after 96 h. Similarly, isoproterenol had a release of 93.70 ± 0.29% when loaded onto bPEI‐AuNPs, with 84.62 ± 0.08% being released after 96 h. Doxorubicin showed the lowest release rate among the three drugs, with 51.56 ± 0.55% being released when loaded onto bPEI‐AuNPs, and only 34.66 ± 0.83% being released after 96 h. Notably, drug release from hydrogels was less when loaded onto bPEI‐AuNPs compared to when these drugs were mixed in collagen without nanoparticles (Table 4 ). Analysis of the drug release at the end point of the study showed that drug‐loaded bPEI‐AuNPs significantly released lower amounts (retained more) of different drugs (Figure 3c).
Table 4.
Drug release from each hydrogel after 96 h
| Candidate drug loaded on the bPEI‐AuNPs | Drug release after 96 h [%] | |
|---|---|---|
| collagen | bPEI‐AuNP‐collagen | |
| Phenylephrine | 92.32 ± 0.25 | 77.11 ± 0.45 |
| Isoproterenol | 93.70 ± 0.29 | 84.62 ± 0.08 |
| Doxorubicin | 51.56 ± 0.55 | 34.66 ± 0.83 |
Importantly, the net surface charge of a drug affects the loading and subsequent drug release from bPEI‐AuNPs embedded within the hydrogels. Consequently, our data show different release profiles for doxorubicin compared to phenylephrine or isoproterenol (Figure 9c). While the chemical structure of phenylephrine and isoproterenol are relatively similar, doxorubicin contains significantly more aromatic rings resulting in a greater net surface charge. Thus, doxorubicin binds stronger to positively charged bPEI‐AuNPs than phenylephrine or isoproterenol which is reflected by a markedly lower cumulative release (≈35% vs >75%, Figure 9 and Table 4).
Notably, the release trend and the shape of the graphs were similar between collagen and bPEI‐AuNP‐collagen hydrogels. These data show that the here developed system permits release of drugs or potentially growth factors from PEI‐AuNPs inside the hydrogels. However, it is important to note that only negatively charged drugs can be loaded on positively charged bPEI‐AuNPs, and that loading and release of a drug depends on its net surface charge.
2.10. AuNPs Loaded with Phenylephrine Cause an Early Onset of Beating
Our data so far showed that incorporation of bPEI‐AuNPs in collagen promotes contractility and calcium handling of cardiomyocytes embedded within the 3D hydrogel. In order to evaluate the simultaneous effect of drug release and enhanced electrical conductivity by AuNPs, we generated engineered tissues by incorporating bPEI‐AuNPs loaded with phenylephrine and compared them to tissues containing only bPEI‐AuNPs. Phenylephrine binds to α‐1 adrenergic receptors and has inotropic effects on cardiomyocytes.[ 41 ] The presence of phenylephrine‐loaded nanoparticles resulted in an early onset of synchronous contraction of cardiomyocytes in the tissue (Figure 10a). Engineered tissues containing drug‐loaded nanoparticles started to beat as early as day 2 compared to cardiomyocytes in bPEI‐AuNP‐collagen that started their synchronous beating only after 7 days. It is reported that phenylephrine increases the frequency of intracellular calcium flow in stem cell–derived cardiomyocytes through inositol 1,4,5‐triphosphate–dependent enhancement of calcium release from sarcomeric reticulum.[ 42 ] Thus, the early onset of contractions in our tissues might be due to phenylephrine‐dependent enhanced calcium flow in the cells. Furthermore, it is reported that phenylephrine can protect neonatal cardiomyocytes against hypoxia and serum deprivation[ 43 ] thereby resulting in a possible improved contractility in cardiomyocytes.
Figure 10.
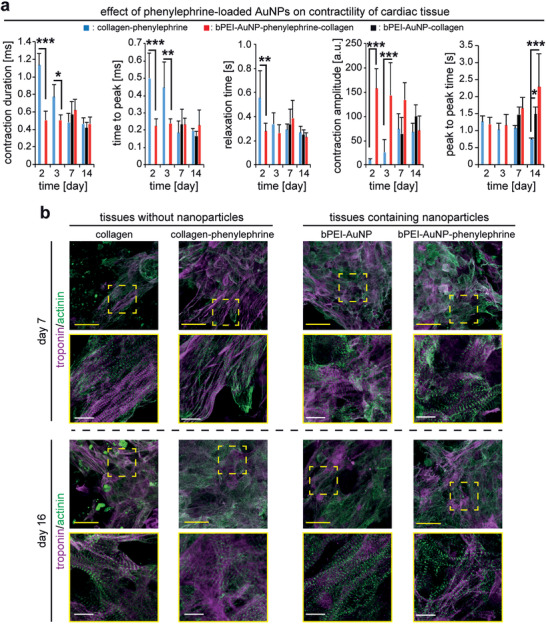
bPEI‐AuNPs function as drug carriers and enhance contractility of cardiomyocytes in 3D engineered tissues. a) MUSCLEMOTION‐based quantitative analysis of contractility of engineered cardiac tissues at different days post‐fabrication (n = 3). b) Examples of projections of confocal images of hiPSC‐derived cardiomyocyte‐laden tissue constructs at 7 or 16 days post‐fabrication stained for the cardiomyocyte‐specific marker sarcomeric‐α‐actinin and troponin I. Data are mean ± SD and compared based on a two‐way ANOVA and Bonferroni's multiple comparison test. Scale bars: yellow: 25 µm and white: 4 µm.
Quantitative analysis of recorded videos of beating tissues revealed that the enhanced contractility of cardiomyocytes was decreased after 7 days, evidenced by a reduced contraction amplitude and increased peak‐to‐peak time. Suggesting that phenylephrine is completely released at this time point which is in agreement with our drug release data showing that more than 90% of phenylephrine is released after 96 h (Figure 9c). Accordingly, the contractility of engineered tissues at day 7 was not different between tissues containing phenylephrine‐loaded nanoparticles and those containing bPEI‐AuNPs. In order to evaluate whether such an effect on the contractility of the tissues was due to the presence of drug‐loaded bPEI‐AuNPs or it could be achieved solely by the presence of phenylephrine, we mixed phenylephrine into the pre‐gel solution of collagen hydrogels and generated engineered cardiac tissues containing 2.5 × 107 cells mL−1 (collagen‐phenylephrine). Our data revealed that the presence of phenylephrine resulted in an early onset of contractility of the tissues from day 2. However, there were signs of arrhythmic beating in collagen‐phenylephrine tissues (Movie S7, Supporting Information). Quantitative analysis of contractility of these tissues revealed a significantly longer time to peak and contraction duration at days 2 and 3 accompanied by a longer relaxation time at day 2 compared to bPEI‐AuNP‐phenylephrine‐collagen tissues (Figure 10a). Moreover, collagen‐phenylephrine tissues showed significantly smaller contraction amplitude than that of bPEI‐AuNP‐phenylephrine‐collagen tissues. However, the contraction amplitude ramped up over time and from day 7 it reached the levels recorded for either bPEI‐AuNP‐phenylephrine‐collagen or bPEI‐AuNP‐collagen tissues (Figure 10a). While peak‐to‐peak time was not different at early time points, cardiomyocytes exhibited shorter peak‐to‐peak time in collagen‐phenylephrine tissues at day 14 (Figure 10a). These data show the beneficial effects of phenylephrine‐loaded bPEI‐AuNPs over administration of freely available drug within the hydrogel, especially at the early time points as hiPSC‐cardiomyocytes at days 2 and 3 beat without arrhythmia with significantly shorter time to peak and contraction duration accompanied by a greater contraction amplitude.
The earlier onset of synchronous contractions can be related to properly formed contractile machinery in cardiomyocytes. Therefore, we immunostained the contractile apparatus of hiPSC‐derived cardiomyocytes for sarcomeric α‐actinin and cardiac troponin I at 7 and 16 days post‐fabrication (Figure 10b). While cardiomyocytes show well‐established contractile machinery in collagen hydrogels based on immunostaining images of sarcomeric alpha actinin and cardiac troponin I images at day 7, presence of phenylephrine in collagen resulted in greater surface coverage of these cells. Moreover, presence of bPEI‐AuNPs or phenylephrine‐loaded bPEI‐AuNPs also resulted in greater surface coverage of the cells as early as day 7 (Figure 10b). Interestingly, regular striated sarcomeres of cardiomyocytes in tissues containing either phenylephrine or drug‐loaded nanoparticles at day 7 were comparable to that of the cells in collagen or tissues containing nanoparticles without the drug at day 16. Moreover, cardiomyocytes maintained their well‐established striations even after 16 days when we terminated the experiment (Figure 10b). These data suggest that incorporation of phenylephrine‐loaded bPEI‐AuNPs in the hydrogels enhances contractility and subcellular organization of contractile machinery of hiPSC‐derived cardiomyocytes embedded within these hydrogels. While these data are exciting, further investigation of cellular maturation in future studies will provide new insight on the efficiency of this approach.
3. Conclusion
Our data show that the incorporation of bPEI‐coated AuNPs in collagen I hydrogels yields an electroconductive hydrogel system that provides the opportunity to simultaneously deliver and release biologically active factors together with cardiomyocytes giving these hydrogels multifunctionality. We found that bPEI‐AuNPs increase the electrical conductivity of collagen hydrogels in the range of 48–68 mS cm−1, depending on their size and surface charge. Smaller nanoparticles had a more significant effect on the electrical conductivity, with ≈20 nm particles resulting in the greatest conductivity. Moreover, incorporating these nanoparticles did not result in a significant difference in the hydrogel's micromorphology but increased its mechanical properties compared to collagen hydrogels. Owing to the positive charge of the bPEI coating of AuNPs, it is possible to load a variety of drugs, peptides, and oligonucleotides, all having an overall negative charge. The novel gel represents a versatile platform, is cytocompatible, and promotes the physiological beating rate and contraction amplitude of neonatal rat cardiomyocytes. Moreover, in electroconductive biohybrid hydrogels, hiPSC‐derived cardiomyocytes exhibited an improved sarcomeric apparatus, elevated contraction amplitude, and advanced calcium handling capacities resulting in a synchronous rhythmic beating. These constructs can be potentially used for pharmaceutical drug screening or as in vitro produced tissues to treat heart disease. Moreover, the here developed system, owing to its enhanced electrical conductivity and possibility of delivering pharmaceutical and growth factors, could be used as a cell‐free system to treat heart diseases. Such a system can be either injected into the myocardial wall using angioplasty approaches or the gel system can be implanted epicardially. A cell‐free approach can avoid the potential risks and ethical concerns associated with cell transplantation, such as immune rejection and tumorigenicity. Additionally, a cell‐free approach can offer greater flexibility and scalability in terms of production and delivery, as well as potentially lower costs.
4. Experimental Section
Synthesis and Characterization of bPEI‐AuNPs
Gold nanoparticles were synthesized as explained previously with slight modifications.[ 44 ] Briefly, HAuCl4·3H2O was dissolved in double distilled water at 100 mg mL−1 and mixed with bPEI (average M w ≈25 000 by LS, average M n ≈10 000 by GPC, branched, Sigma‐Aldrich 408727) 5% v/v (in double distilled water) at different ratios as indicated in Table 5 . The prepared solutions were incubated at 65 °C for 60 min to generate gold nanoparticles.
Table 5.
Composition of reaction solutions
| Solution | Amount of gold (III) Chloride trihydrate [µL] | Amount of bPEI [µL] |
|---|---|---|
| bPEI‐AuNP‐50 | 200 | 200 |
| bPEI‐AuNP‐37.5 | 150 | 200 |
| bPEI‐AuNP‐25 | 100 | 200 |
| bPEI‐AuNP‐12.5 | 50 | 200 |
Solutions at the end of 60 min were diluted 1:20 in double distilled water for the analysis of UV–vis spectrum. Each solution was sonicated for 1 min in a water bath sonication setup, a volume of 100 µL of each solution was transferred into a cuvette and UV–vis absorption was recorded at 200–700 nm using a Bio‐Spectrometer (Eppendorf basic, Germany). Particle size was determined for nanoparticles by the DLS technique. Nanoparticles in double distilled water were sonicated and subsequently, their size was assessed using Zeta Sizer Nano ZS (Malvern Industries Ltd, Malvern, UK). Aliquots of nanoparticles were used to measure particle sizes in a volume of 500 µL. Particle surface charge was determined by the Zeta potential measurements of nanoparticles using a Zetaview PMX 100 (ParticleMetrix, Meerbusch, Germany). Aliquots of freshly prepared diluted bPEI‐AuNPs (200 µL, 1:20 dilution in double distilled water) were used to measure the zeta potential. Each measurement was repeated three times.
Hydrogel Preparation
All solutions were kept on ice in order to minimize thermally‐induced gelation. Using appropriate pipet sizes, per 100 µL of hydrogel, bPEI‐AuNPs (1 µL) were added to 1 µL of Dulbecco's phosphate‐buffered saline (DPBS, Thermofisher Scientific14190). Then, neutralization solution (9.8 µL) and eventually collagen I (88.2 µL, 3.9 mg mL−1, Advanced Biomatrix 5153) was added to this solution resulting in a final concentration of bPEI‐AuNPs at 1% v/v and collagen I at 0.38% w/v. The final mixture was pipetted up and down five times before being dispensed in experimental container. For collagen hydrogels, instead of bPEI‐AuNPs, equivalent volume of DPBS was added. Samples were incubated at 37 °C for a minimum of 30 min to allow induction of the gelation process thermally. Collagen I was generated as a control hydrogel at 0.38% w/v.
Scanning Electron Microscopy
Gels were fixed in a 1:1 mixture of glutaraldehyde and paraformaldehyde (3.7%) for 1 h. They were gradually dehydrated in ethanol series and were immersed in ethanol (100%), critically point dried using an EM CPD300 (Leica, 12 cycles, exchange speed of 5 and 120 s delay), sputter coated with gold (Q150T, Quorum Technologies Ltd., Germany). SEM (FE‐SEM, Auriga, Carl‐Zeiss, Germany) was performed on a cross‐sectional area.
Optical Microscopy
Pre‐gel solutions of collagen and bPEI‐AuNP‐collagen hydrogels were prepared as explained in 4.2. For visualization purposes, we chose bPEI‐AuNP‐37.5. In order to perform optical imaging, 1 µL of the pre‐gel solution was drop‐cast onto silicone (Si) substrates with atomically flat 300 nm thin thermal oxide layer on top. Optical microscopy was carried out with Zeiss AXIO Imager.A2m using 50× magnification in dark field mode.
Electrical Conductivity Measurements
Freshly mixed hydrogels were transferred into a self‐built sealed polytetrafluoroethylene vessel with a capacity of 150 µL and cured for 30 min at 37 °C. The measurement vessel was equipped with four metal probes allowing four‐point electrical measurements in a collinear four‐point probe arrangement.[ 45 ] Two‐ and four‐point current–voltage (I–V) characteristics were taken while sweeping the bias voltage across the outer leads in the range of ±1.75 V. The values of the hydrogel electrical conductivity were estimated based on the four‐point resistance values and accounting for appropriate geometrical corrections associated with the geometry of the measurement vessel.[ 45 ]
During electrical characterization, the samples were kept in a humid environment and at a constant temperature of 37 °C. The measurements for each hydrogel were repeated several times for consistency.
Rheology
Rheological measurements were performed using an DHR‐3 rotational rheometer (TA Instruments, New Castle, DE) with a cone plate probe diameter of 20 mm, as described previously.[ 13 ] The temperature of the rheometer's lower geometry was set at 37 °C. After mixing the components according to hydrogel preparation utilizing either bPEI‐AuNP‐25 alone or loaded with phenylephrine at the final concentration of 1% v/v, 150 µL of the pre‐gel solution was transferred onto the lower measurement geometry using a 1 mL pipet. The gap distance was set to 55.989 µm and a solvent trap filled with deionized water was equipped to prevent edge drying of the samples. For the characterization of gelation kinetics, temperature was set at 37 °C and a time sweep at a deformation of 0.1% and a constant angular frequency of 1 rad s−1 was used. To determine the frequency‐dependent characteristics of the hydrogels, a frequency sweep was performed directly following the prior time sweep. Temperature was kept at 37 °C and the typical frequency range of 0.1–100 rad s−1 at an amplitude of 0.5% was covered for a sufficient analysis of material properties and comparability to existing data sets from the literature. After that, an amplitude sweep test was performed at 0.01 to 100% oscillation strain at 37 °C to study the yield and flow point of the hydrogels. Subsequently, hydrogels were tested at a time sweep at 10 rad s−1 at 37 °C for 5 min to evaluate their recovery after the amplitude sweep test. Self‐healing strain tests were performed to observe the dynamic self‐healing ability of hydrogels when a variable strain was applied cyclically by alternating for three times low (0.1 Hz, recovery phase, 200 s) and high (100 Hz, rupture phase, 200 s) deformations. For time sweep measurements with temperature set to 4 °C, a cone plate probe (20 mm and measurement volume of 200 µL) or cone plate probe (40 mm and measurement volume of 500 µL, gap distance of 65 µm) was used at 5 or 1 °C min−1, respectively, and the final temperature was set to reach 37 °C.
Drug Release from bPEI‐AuNPs in Collagen I Hydrogels
Drugs were loaded on bPEI‐AuNPs by mixing the nanoparticle solution with a stock solutions of phenylephrine (10 mm), isoproterenol (10 mm), or doxorubicin (5 mm) and incubating them for 120 min at room temperature to allow loading of the drug through electrostatic interactions of amine groups from bPEI with negatively charged drugs. After a brief wash in DPBS, bPEI‐AuNPs loaded with either drug were studied for their drug release profile in vitro. bPEI‐AuNPs loaded with the drug were embedded in the collagen matrix as explained in hydrogel preparation yielding a working concentration of phenylephrine (50 µm), isoproterenol (50 µm), or doxorubicin (25 µm) in the hydrogels. Hydrogels containing nanoparticles were incubated in 1 mL DPBS. At specific time points, 200 µL of the supernatant solution was taken out for UV–vis absorption measurements using a BioSpectrometer (Eppendorf, Germany) and replaced with fresh DPBS to maintain the sink volume. At the end of experiment, the hydrogel was digested using collagenase B (2.5 mg mL−1) for 60 min, followed by mechanical agitation, and the remaining amount of the drug was measured.
Primary Cardiomyocyte Isolation
The investigation conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The local Animal Ethics Committee approved the extraction of organs and preparation of primary cell cultures per governmental and international animal experimentation guidelines (protocol TS‐9/2016 Nephropatho). Cardiomyocytes were isolated from 3 days old Sprague Dawley rat's ventricular wall utilizing the gentleMACS dissociation kit (Milteny Biotec) as described.[ 46 ]
hiPSC Culture and Differentiation
F1 hiPSCs[ 47 ] at passage 19 were cultured and differentiated into cardiomyocytes as described previously.[ 10 ] Briefly, mesoderm formation through activating Wnt signaling was induced in hiPSCs more than 85% confluent by introducing CHIR‐99021 (6 µm) for 2 days. After 1 day of incubation in culture medium, Wnt signaling was inhibited by IWR‐1‐endo (5 µm) for 2 days. After another 2 days of incubation in culture medium, cells started to beat. Subsequently, cells were cultured for 2 days in RPMI1640 supplemented with B27 (Thermo Fisher Scientific, 17504044), and on day 9 of differentiation, cells were purified for cardiomyocytes by substituting glucose with lactate for 2 days. The resulting cells were cultured in RPMI1640 supplemented with B‐27.
Formation of 3D Engineered Tissues
The pre‐gel solution of either collagen I (0.38% w/v) or bPEI‐AuNP‐collagen hydrogels (final concentration of bPEI‐AuNPs at 1% v/v and collagen I at 0.38% w/v) were prepared on ice. Subsequently, 2.5 × 107 cells mL−1 were resuspended in the pre‐gel solution. The cell‐laden hydrogels were dispensed in each well of a 24‐well plate and placed in a cell culture incubator at 37 °C and 5% CO2 for 30 min to induce thermal gelation of collagen I. Tissue constructs based on neonatal rat ventricular cardiomyocytes were incubated in Dulbecco's modified Eagle's medium/F12 + Glutamax containing penicillin–streptomycin (100 U mg−1 mL −1), Na pyruvate (3 mm), bovine serum albumin (0.2% w/v), ascorbic acid (0.1 mm), insulin–transferrin–selenium (100×) (0.5% v/v), and 10% fetal bovine serum. Tissue constructs based on hiPSC‐derived cardiomyocytes were incubated in RPMI1640 supplemented with B‐27. For studies related to the effect of phenylephrine on cardiomyocytes in 3D hydrogels, bPEI‐AuNPs loaded with phenylephrine were used at the same protocol explained here.
Live and Dead Assay
The viability of cells in tissue constructs was determined using the Live and dead assay kit (L3224, Invitrogen).[ 10 ] To quantify the viability of hiPSC‐derived cardiomyocytes exposed to AuNPs, cells stained in green or red were manually counted on confocal microscopy images utilizing NIH ImageJ.
Immunostaining
Tissue constructs were immunostained for cardiac‐specific markers as previously described[ 9 ] with the following modifications. Samples were fixed in paraformaldehyde (3.7%) for 30 min and permeabilized in Triton X‐100 (0.25%) for 20 min. The following primary antibodies were used for overnight incubation at 4 °C: mouse anti‐sarcomeric α‐actinin (1:250, Abcam, ab9465), goat anti‐cardiac troponin I (1:250, Abcam, ab56357), and rabbit anti‐connexin 43 (1:250, Abcam, ab11370). The following secondary antibodies were used for 2 h incubation at room temperature in dark: donkey anti‐mouse AlexaFluor 488 (1:500, Abcam A21202), donkey anti‐rabbit AlexaFluor 594 (1:500, Abcam A‐21203), and donkey anti‐goat AlexaFluor 647 (1:500, Abcam A‐21081) DNA was visualized with DAPI (0.5 g mL−1 in 0.1 w/v% NP40, 15 min).
Calcium Handling Properties
Calcium handling properties were analyzed using Fluo‐4 Direct Calcium Assay Kit (ThermoFisher Scientific), as described previously.[ 9 ] Following the manufacturer's instructions, tissue constructs were incubated for 45 min with the labeling solution diluted in culture media (1:3 dilution) and subsequently the fluorescent movies were acquired using a live cell imaging setup (Keyence Microscopy) at 4× objective with a GFP filter at 488 nm.
RNA Sequencing
RNA was isolated from different tissues at day 22 post‐fabrication using TRIzol (Life Technologies). Then, DNA was digested utilizing RNase‐Free DNase set following the manufacturer's instructions (QIAGEN, 79254). Reverse‐stranded mRNA libraries were prepared relying on the Illumina Stranded mRNA Prep kit according to manufacturer's instructions. For each sample, ≈67 and ≈240 ng of total RNA, respectively from collagen and bPEI‐AuNP‐collagen, was used for library preparation. Sequencing of the libraries was performed on the Illumina NovaSeq platform (paired end; 2 × 109 bp). De‐multiplexed raw data (FASTQ files) were pre‐processed with the nf‐core RNA‐seq pipeline v.3.9[ 48 ] using default parameters. Reads were adapter‐ and quality‐trimmed using Trim Galore v.0.6.7 (Babraham, Bioinformatics) and mapped to the Ensembl human genome assembly GRCh38 (release 108)[ 49 ] using STAR v.2.7.10a.[ 50 ] Read counting was performed using Salmon v.1.5.2[ 51 ] relying on the Ensembl gene annotation file release 108.
Further analyses were performed within the R environment v.4.0.3.[ 52 ] Differential expression analysis between the bPEI‐AuNP‐collagen and the collagen group was performed relying on the package DESeq2 v.1.28.1,[ 53 ] starting from the length‐scaled gene‐level counts provided by the nf‐core pipeline. A gene was considered significantly dysregulated between bPEI‐AuNP‐collagen and collagen conditions if its (Benjamini and Hochberg) adjusted p‐value was lower than 0.01. The expression heatmap of the top 500 dysregulated genes (i.e., the 500 genes with the lowest adjusted p‐value) and the heatmap of selected genes were generated using mean‐centered variance‐stabilized transformed counts (DESeq2 function vst, ‘blind’ = FALSE) using the pheatmap package v.1.0.12.
Pathway analysis was performed relying on the Gene Set Enrichment Analysis (GSEA) approach,[ 54 ] as implemented in the universal gene set enrichment analysis function GSEA of the clusterProfiler package v. 3.16.1.[ 55 ] As input list HUGO gene symbols ranked according to decreasing Wald statistic value were given. To this aim, Ensembl Gene Ids were mapped to HUGO gene symbols relying on the biomaRt package v.2.44.4.[ 56 ] Ensembl genes not mapping to any symbols were excluded from the analysis whereas if a gene symbol was associated to more than one Ensembl Gene Id, its Wald statistic was taken equal to the one of the Ensembl Gene Id with the highest absolute value statistic. As query gene sets the Gene Ontology (GO) biological process (BP) collection (release date: 2022‐07‐01) was retrieved from the Molecular Signature Database (MSigDB, https://www.gsea‐msigdb.org/gsea/msigdb/human/collections.jsp – last accessed on 08.12.2022)[ 54 , 57 ] by downloading the associated GMT file with gene symbols (v2022.1.Hs). Pathway analysis results were visualized as gene set enrichment networks relying on the Enrichment Map plug‐in v.3.3.4[ 58 ] of the Cytoscape software v.3.9.1,[ 59 ] as previously described.[ 39 ] Nodes in the enrichment network represent significantly enriched pathways (FDR < 0.01) and edges the similarity between nodes, calculated by means of the overlap coefficient (OC),[ 39 , 58 ] with OC = 0.5 as threshold. Pathway clusters were identified relying on the AutoAnnotate plug‐in v.1.3.5.[ 60 ] The clusters characterized by at least ten pathways were explored in further detail. First, cluster's labels automatically generated by AutoAnnotate were manually revised. Next, the genes belonging to the clusters were further characterized relying on the “RNA consensus tissue gene data” from the Human Protein Atlas[ 61 ] version 22.0 (https://www.proteinatlas.org/ – last accessed on 15.12.2022). This dataset contains consensus transcript expression levels in 54 different tissues, including heart muscle, on the basis of both the human protein atlas (HPA) and the genotype‐tissue expression (GTEx) RNA‐seq data. For our analysis, a gene in a given cluster was considered expressed in heart muscle if the provided normalized transcript expression value (nTPM) was ≥1.
Movie Acquisition
Movies of beating 3D constructs and movies for calcium handling assays were recorded in wells of 24‐well plates for 10 s. All movies were recorded under a Keyence BZ9000 microscope. Analysis of contractility was performed using MUSCLEMOTION plugin for NIH ImageJ.[ 32 ] Calcium movies were analyzed, as explained previously over four different regions of interest per movie.[ 9 ]
Microscopy
Fluorescence images were acquired on an LSM 800 confocal laser scanning microscope (Carl Zeiss). Within individual assays, the same laser power and settings were used for all presented images. The following excitation and emission wavelengths were set for each secondary antibody: AlexaFluor 488 (excitation 493 nm, emission 517 nm, detection wavelength 450–585 nm), AlexaFluor 594 (excitation 280 nm, emission 618 nm, detection wavelength 595–650 nm), AlexaFluor 647 (excitation 653 nm, emission 668 nm, detection wavelength 645–700 nm), DAPI (excitation 353 nm, emission 465 nm, detection wavelength 410–546 nm). The pinhole was set to 1 airy unit (AU).
Statistical Analyses
Data of at least three independent experiments were expressed as mean ± standard deviation (SD). Statistical significance of differences was evaluated by a two‐tailed Student's t‐test (GraphPad Prism) or where appropriate, by one‐way or two‐way analysis of variance (ANOVA) followed by Bonferroni's post hoc test (GraphPad Prism). p < 0.05 was considered statistically significant. For particle size and charge one‐way ANOVA was used. Quantitative analysis of viability of hiPSC‐derived cardiomyocytes exposed to nanoparticles was performed based on a one‐way ANOVA. Two‐tailed Student's t‐test was used to compare storage or loss modulus of collagen to bPEI‐AuNP‐collagen hydrogels for frequency sweep and amplitude sweep experiments. To quantitatively analyze contractility of engineered tissues, two‐way analysis of ANOVA was performed. Quantitative comparison of endpoint drug release from hydrogels was performed based on a two‐tailed student t‐test. Significant differences were indicated by * Symbol where *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Supplemental Movie 3
Supplemental Movie 4
Supplemental Movie 5
Supplemental Movie 6
Supplemental Movie 7
Acknowledgements
The authors would like to thank the Department of Biomaterials of the University of Erlangen‐Nuremberg for providing the access to the scanning electron microscopy. This research was funded by Universitätsklinikum Erlangen ELAN IZKF, grant number P093/Aktenzeichen: 21‐05‐20‐1‐Roshanbinfar, the Deutsche Forschungsgemeinschaft [DFG, German Research Foundation, grant numbers: Projektnummer 326998133—TRR 225 (subproject C01 to F.B.E. and A07 to D.W.S.) and INST 410/91‐1 FUGG, both to F.B.E.), the Manfred Roth Stiftung (to F.B.E.), and the Research Foundation Medicine at the University Clinic Erlangen, Germany (to F.B.E.). Graphical illustrations were generated by BioRender.
Open access funding enabled and organized by Projekt DEAL.
Roshanbinfar K., Kolesnik‐Gray M., Angeloni M., Schruefer S., Fiedler M., Schubert D. W., Ferrazzi F., Krstic V., Engel F. B., Collagen Hydrogel Containing Polyethylenimine‐Gold Nanoparticles for Drug Release and Enhanced Beating Properties of Engineered Cardiac Tissues. Adv. Healthcare Mater. 2023, 12, 2202408. 10.1002/adhm.202202408
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Tsao C. W., Aday A. W., Almarzooq Z. I., Alonso A., Beaton A. Z., Bittencourt M. S., Boehme A. K., Buxton A. E., Carson A. P., Commodore‐Mensah Y., Elkind M. S. V., Evenson K. R., Eze‐Nliam C., Ferguson J. F., Generoso G., Ho J. E., Kalani R., Khan S. S., Kissela B. M., Knutson K. L., Levine D. A., Lewis T. T., Liu J., Loop M. S., Ma J., Mussolino M. E., Navaneethan S. D., Perak A. M., Poudel R., Rezk‐Hanna M., et al., Circulation 2022, 145, e153. [DOI] [PubMed] [Google Scholar]
- 2. Timmis A., Vardas P., Townsend N., Torbica A., Katus H., De Smedt D., Gale C. P., Maggioni A. P., Petersen S. E., Huculeci R., Kazakiewicz D., de Benito Rubio V., Ignatiuk B., Raisi‐Estabragh Z., Pawlak A., Karagiannidis E., Treskes R., Gaita D., Beltrame J. F., McConnachie A., Bardinet I., Graham I., Flather M., Elliott P., Mossialos E. A., Weidinger F., Achenbach S., C. European Society of, G. on behalf of the Atlas Writing , Eur. Heart J. 2022, 43, 716.35016208 [Google Scholar]
- 3. Eschenhagen T., Bolli R., Braun T., Field L. J., Fleischmann B. K., Frisén J., Giacca M., Hare J. M., Houser S., Lee R. T., Marbán E., Martin J. F., Molkentin J. D., Murry C. E., Riley P. R., Ruiz‐Lozano P., Sadek H. A., Sussman M. A., Hill J. A., Circulation 2017, 136, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Liu Y.‐W., Chen B., Yang X., Fugate J. A., Kalucki F. A., Futakuchi‐Tsuchida A., Couture L., Vogel K. W., Astley C. A., Baldessari A., Ogle J., Don C. W., Steinberg Z. L., Seslar S. P., Tuck S. A., Tsuchida H., Naumova A. V., Dupras S. K., Lyu M. S., Lee J., Hailey D. W., Reinecke H., Pabon L., Fryer B. H., MacLellan W. R., Thies R. S., Murry C. E., Nat. Biotechnol. 2018, 36, 597; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chong J. J. H., Yang X., Don C. W., Minami E., Liu Y.‐W., Weyers J. J., Mahoney W. M., Van Biber B., Cook S. M., Palpant N. J., Gantz J. A., Fugate J. A., Muskheli V., Gough G. M., Vogel K. W., Astley C. A., Hotchkiss C. E., Baldessari A., Pabon L., Reinecke H., Gill E. A., Nelson V., Kiem H.‐P., Laflamme M. A., Murry C. E., Nature 2014, 510, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roshanbinfar K., Esser T. U., Engel F. B., Antioxid. Redox Signaling 2021, 35, 143. [DOI] [PubMed] [Google Scholar]
- 6. Ghovvati M., Kharaziha M., Ardehali R., Annabi N., Adv. Healthcare Mater. 2022, 11, 2200055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Esmaeili H., Patino‐Guerrero A., Hasany M., Ansari M. O., Memic A., Dolatshahi‐Pirouz A., Nikkhah M., Acta Biomater. 2022, 139, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roshanbinfar K., Hilborn J., Varghese O. P., Oommen O. P., RSC Adv. 2017, 7, 31980. [Google Scholar]
- 9. Roshanbinfar K., Mohammadi Z., Mesgar A. S.‐M., Mehdi Dehghan M., Oommen O. P., Hilborn J., Engel F. B., Biomater. Sci. 2019, 7, 3906. [DOI] [PubMed] [Google Scholar]
- 10. Roshanbinfar K., Vogt L., Greber B., Diecke S., Boccaccini A. R., Scheibel T., Engel F. B., Adv. Funct. Mater. 2018, 28, 1803951; [Google Scholar]
- 11. Roshanbinfar K., Vogt L., Ruther F., Roether J. A., Boccaccini A. R., Engel F. B., Adv. Funct. Mater. 2020, 30, 1908612. [Google Scholar]
- 12. Traverse J. H., Henry T. D., Dib N., Patel A. N., Pepine C., Schaer G. L., DeQuach J. A., Kinsey A. M., Chamberlin P., Christman K. L., JACC: Basic Transl. Sci. 2019, 4, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hesselbarth R., Esser T. U., Roshanbinfar K., Schrüfer S., Schubert D. W., Engel F. B., Adv. Healthcare Mater. 2021, 10, 2100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J., Ma A., Shang L., Front. Physiol. 2018, 9, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Chen W., Wang Z., Wang L., Chen X., Adv. Mater. 2022, 34, 2106701; [DOI] [PubMed] [Google Scholar]; b) Spivak M. Y., Bubnov R. V., Yemets I. M., Lazarenko L. M., Tymoshok N. O., Ulberg Z. R., EPMA J. 2013, 4, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Shahbazi R., Asik E., Kahraman N., Turk M., Ozpolat B., Ulubayram K., Nanomedicine 2017, 12, 1961; [DOI] [PubMed] [Google Scholar]; b) Shrestha B., Wang L., Zhang H., Hung C. Y., Tang L., Int. J. Nanomed. 2020, 15, 8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. García‐Garrido E., Cordani M., Somoza Á., Pharmaceutics 2021, 13, 2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nguyen M. K., Alsberg E., Prog. Polym. Sci. 2014, 39, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Alkilany A. M., Murphy C. J., J. Nanopart. Res. 2010, 12, 2313; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ray P. C., Yu H., Fu P. P., J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li X., Wang L., Fan Y., Feng Q., Cui F.‐z., J. Nanomater. 2012, 2012, e548389. [Google Scholar]
- 21. Li Y., Shi X., Tian L., Sun H., Wu Y., Li X., Li J., Wei Y., Han X., Zhang J., Jia X., Bai R., Jing L., Ding P., Liu H., Han D., Adv. Mater. 2016, 28, 10230. [DOI] [PubMed] [Google Scholar]
- 22. Zhu K., Shin S. R., van Kempen T., Li Y.‐C., Ponraj V., Nasajpour A., Mandla S., Hu N., Liu X., Leijten J., Lin Y.‐D., Hussain M. A., Zhang Y. S., Tamayol A., Khademhosseini A., Adv. Funct. Mater. 2017, 27, 1605352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crasmareanu E., Plesu N., Muntean S. G., Tara‐Lunga‐Mihali M., Simulescu V., Ilia G., Rev. Chim. 2012, 63, 768. [Google Scholar]
- 24. Li R. M., Cheng C., Wang Z. R., Gu X. F., Zhang C. X., Wang C., Liang X. Y., Hu D. D., Materials 2021, 14, 443.33477518 [Google Scholar]
- 25. Gavenis K., clinicaltrials.gov, 2022, NCT04396899.
- 26. Zhang X. D., Wu D., Shen X., Liu P. X., Yang N., Zhao B., Zhang H., Sun Y. M., Zhang L. A., Fan F. Y., Int. J. Nanomed. 2011, 6, 2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xia Q., Li H., Liu Y., Zhang S., Feng Q., Xiao K., J. Biomed. Mater. Res., Part A 2017, 105, 710. [DOI] [PubMed] [Google Scholar]
- 28. Yang Y. L., Motte S., Kaufman L. J., Biomaterials 2010, 31, 5678. [DOI] [PubMed] [Google Scholar]
- 29. Yang Y. L., Kaufman L. J., Biophys. J. 2009, 96, 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wassenaar P. A., Eleswarpu C. N., Schroeder S. A., Mo X., Raterman B. D., White R. D., Kolipaka A., Magn. Reson. Med. 2016, 75, 1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schlick S. F., Spreckelsen F., Tiburcy M., Iyer L. M., Meyer T., Zelarayan L. C., Luther S., Parlitz U., Zimmermann W. H., Rehfeldt F., Prog. Biophys. Mol. Biol. 2019, 144, 51. [DOI] [PubMed] [Google Scholar]
- 32. Sala L., van Meer B. J., Tertoolen L. G. J., Bakkers J., Bellin M., Davis R. P., Denning C., Dieben M. A. E., Eschenhagen T., Giacomelli E., Grandela C., Hansen A., Holman E. R., Jongbloed M. R. M., Kamel S. M., Koopman C. D., Lachaud Q., Mannhardt I., Mol M. P. H., Mosqueira D., Orlova V. V., Passier R., Ribeiro M. C., Saleem U., Smith G. L., Burton F. L., Mummery C. L., Circ. Res. 2018, 122, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Navaei A., Saini H., Christenson W., Sullivan R. T., Ros R., Nikkhah M., Acta Biomater. 2016, 41, 133. [DOI] [PubMed] [Google Scholar]
- 34. Vornanen M., Acta Physiol. Scand. 1992, 145, 311. [DOI] [PubMed] [Google Scholar]
- 35. Ronaldson‐Bouchard K., Ma S. P., Yeager K., Chen T., Song L., Sirabella D., Morikawa K., Teles D., Yazawa M., Vunjak‐Novakovic G., Nature 2018, 556, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao Y., Rafatian N., Feric N. T., Cox B. J., Aschar‐Sobbi R., Wang E. Y., Aggarwal P., Zhang B., Conant G., Ronaldson‐Bouchard K., Pahnke A., Protze S., Lee J. H., Davenport Huyer L., Jekic D., Wickeler A., Naguib H. E., Keller G. M., Vunjak‐Novakovic G., Broeckel U., Backx P. H., Radisic M., Cell 2019, 176, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dvir T., Timko B. P., Brigham M. D., Naik S. R., Karajanagi S. S., Levy O., Jin H., Parker K. K., Langer R., Kohane D. S., Nat. Nanotechnol. 2011, 6, 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burnstine‐Townley A., Eshel Y., Amdursky N., Adv. Funct. Mater. 2020, 30, 1901369. [Google Scholar]
- 39. Angeloni M., Thievessen I., Engel F. B., Magni P., Ferrazzi F., Biol. Chem. 2021, 402, 953. [DOI] [PubMed] [Google Scholar]
- 40. Benjaminsen R. V., Mattebjerg M. A., Henriksen J. R., Moghimi S. M., Andresen T. L., Mol. Ther. 2013, 21, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang G. Y., McCloskey D. T., Turcato S., Swigart P. M., Simpson P. C., Baker A. J., Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2013. [DOI] [PubMed] [Google Scholar]
- 42. Kapur N., Banach K., J. Physiol. 2007, 581, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu H., McElwee‐Witmer S., Perrone M., Clark K. L., Zilberstein A., Cell Death Differ. 2000, 7, 773. [DOI] [PubMed] [Google Scholar]
- 44. Mikami T., Takayasu Y., Hirasawa I., Chem. Eng. Res. Des. 2010, 88, 1248. [Google Scholar]
- 45. Schroder D. K., Semiconductor Material and Device Characterization, John Wiley & Sons, Hoboken, NJ: 2005. [Google Scholar]
- 46. Petzold J., Aigner T. B., Touska F., Zimmermann K., Scheibel T., Engel F. B., Advanced Functional Materials 2017, 27. [Google Scholar]
- 47. Marczenke M., Piccini I., Mengarelli I., Fell J., Röpke A., Seebohm G., Verkerk A. O., Greber B., Front. Physiol. 2017, 8, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ewels P. A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M. U., Di Tommaso P., Nahnsen S., Nat. Biotechnol. 2020, 38, 276. [DOI] [PubMed] [Google Scholar]
- 49. Cunningham F., Allen J. E., Allen J., Alvarez‐Jarreta J., Amode M. R., Armean I. M., Austine‐Orimoloye O., Azov A. G., Barnes I., Bennett R., Nucleic Acids Res. 2022, 50, D988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., Bioinformatics 2013, 29, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Nat. Methods 2017, 14, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. R Foundation for Statistical Computing , https://www.R‐project.org/ (accessed: November 2022).
- 53. Love M. I., Huber W., Anders S., Genome Biol. 2014, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu G., Wang L.‐G., Han Y., He Q.‐Y., OMICS: J. Integr. Biol. 2012, 16, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.a) Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W., Bioinformatics 2005, 21, 3439; [DOI] [PubMed] [Google Scholar]; b) Durinck S., Spellman P. T., Birney E., Huber W., Nat. Protoc. 2009, 4, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J. P., Bioinformatics 2011, 27, 1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Merico D., Isserlin R., Stueker O., Emili A., Bader G. D., PLoS One 2010, 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Genome Res. 2003, 13, 2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kucera M., Isserlin R., Arkhangorodsky A., Bader G. D., F1000Research 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S., Nat. Biotechnol. 2010, 28, 1248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Supplemental Movie 3
Supplemental Movie 4
Supplemental Movie 5
Supplemental Movie 6
Supplemental Movie 7
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.