

Abstract
Photoelectrochemical (PEC) water reduction, converting solar energy into environmentally friendly hydrogen fuel, requires delicate design and synthesis of semiconductors with appropriate bandgaps, suitable energy levels of the frontier orbitals, and high intrinsic charge mobility. In this work, the synthesis of a novel bithiophene‐bridged donor–acceptor‐based 2D sp2‐carbon‐linked conjugated polymer (2D CCP) is demonstrated. The Knoevenagel polymerization between the electron‐accepting building block 2,3,8,9,14,15‐hexa(4‐formylphenyl) diquinoxalino[2,3‐a:2′,3′‐c]phenazine (HATN‐6CHO) and the first electron‐donating linker 2,2′‐([2,2′‐bithiophene]‐5,5′‐diyl)diacetonitrile (ThDAN) provides the 2D CCP‐HATNThDAN (2D CCP‐Th). Compared with the corresponding biphenyl‐bridged 2D CCP‐HATN‐BDAN (2D CCP‐BD), the bithiophene‐based 2D CCP‐Th exhibits a wide light‐harvesting range (up to 674 nm), a optical energy gap (2.04 eV), and highest energy occupied molecular orbital–lowest unoccupied molecular orbital distributions for facilitated charge transfer, which make 2D CCP‐Th a promising candidate for PEC water reduction. As a result, 2D CCP‐Th presents a superb H2‐evolution photocurrent density up to ≈7.9 µA cm−2 at 0 V versus reversible hydrogen electrode, which is superior to the reported 2D covalent organic frameworks and most carbon nitride materials (0.09–6.0 µA cm−2). Density functional theory calculations identify the thiophene units and cyano substituents at the vinylene linkage as active sites for the evolution of H2.
Keywords: 2D materials, conjugate polymers; covalent organic frameworks; donor–acceptor structure; photoelectrochemical water reduction
A bithiophene‐bridged D–A‐based 2D sp2‐carbon‐linked conjugated polymer (2D CCP‐Th) is synthesized via Knoevenagel polymerization. Benefiting from the robust sp2‐carbon‐linked conjugations and donor–acceptor structures, when employed as a photocathode for water reduction, 2D CCP‐Th demonstrates a superb saturated photocurrent density (5.5 µA cm−2 at 0.3 V and 7.9 µA cm−2 at 0 V vs reversible hydrogen electrode).
Photoelectrochemical (PEC) water reduction, which can convert solar energy into clean, storable hydrogen fuel, has attracted considerable attention to address energy and environmental issues.[ 1 , 2 , 3 ] A PEC water splitting cell is an innovative H2‐production device consisting of solar energy collection (semiconductors) and water electrolysis (catalysts) units.[ 4 , 5 , 6 ] Principally, the semiconductors need to comply with several requirements for efficient PEC applications, such as a wide‐range light harvesting ability, efficient charge transfer, corrosion stability, and a higher‐lying lowest unoccupied molecular orbital (LUMO) energy level than the reduction potential of the proton (H+/H2).[ 1 , 7 ] 2D covalent organic frameworks (2D COFs), as crystalline, layer‐stacked, 2D porous polymers, have emerged as a promising class of materials for photocatalysis and PEC applications recently.[ 3 , 8 , 9 , 10 , 11 , 12 ] Their energy bandgaps, positions of the frontier orbitals, and active centers can be facilely tailored by bottom‐up organic synthesis with abundant building blocks, linkages, and topologies.[ 6 , 13 , 14 ] In particular, 2D π‐conjugated COFs, which belong to the class of 2D conjugated polymers, show notable advantages for PEC applications due to the π‐stacked columns and in‐plane π‐conjugation that can facilitate charge transport.[ 8 , 15 ] Thus far, the solution synthesis of 2D conjugated polymers has been mainly limited to imine‐linked 2D COFs benefiting from the highly reversible reaction nature of the Schiff‐base chemistry.[ 16 ] However, imine‐linked 2D COFs generally exhibit moderate electron delocalization and poor chemical stability, which hamper the potential implementation of these materials in semiconducting applications.[ 8 , 17 , 28 ]
Recently, 2D sp2‐carbon‐linked (or vinylene‐linked) conjugated polymers (2D CCPs) have attracted growing attention due to the extended π‐conjugation over the whole 2D polymer framework as well as the high chemical stability against acids and bases, in contrast to imine‐linked 2D COFs[ 9 , 17 , 19 , 20 ] However, most of the reported 2D CCPs are not suitable candidates for PEC applications due to the inefficient light harvesting and consequently large optical energy gaps in the range of 2.2–2.4 eV (Table S5, Supporting Information).[ 9 , 21 , 22 , 23 , 24 , 25 ] One promising strategy to regulate the optoelectronic properties of 2D conjugated polymers is the incorporation of donor and acceptor (D–A) conjugated structures,[ 26 ] which can be beneficial for PEC applications.
In this work, we demonstrate the synthesis of a novel D–A‐based 2D CCP via Knoevenagel polymerization between the electron‐accepting building block 2,3,8,9,14,15‐hexa(4‐formylphenyl) diquinoxalino[2,3‐a:2′,3′‐c]phenazine (HATN‐6CHO) and electron‐donating monomer 2,2′‐([2,2′‐bithiophene]‐5,5′‐diyl)diacetonitrile (ThDAN), providing the crystalline 2D CCP‐HATN‐ThDAN (2D CCP‐Th) with a dual pore structure (Figure 1a).[ 17 ] We also synthesized the biphenylene‐bridged 2D CCP analog and imine‐linked 2D COF for comparison, namely, 2D CCP‐HATN‐BDAN (2D CCP‐BD) and 2D C=N HATN‐BZD (2D C=N COF‐B), respectively (Figure 1a). Benefitting from the D–A structure, the bithiophene‐bridged 2D CCP‐Th shows a much narrower optical energy bandgap (2.04 eV), a higher‐lying LUMO energy level (−3.41 eV), and higher charge separation and transport than those of biphenyl‐bridged 2D CCP‐BD and 2D C=N COF‐B. Therefore, as the cathode material for PEC water reduction, 2D CCP‐Th demonstrates a superb saturated photocurrent density up to 5.5 µA cm−2 at 0.3 V and 7.9 µA cm−2 at 0 V versus reversible hydrogen electrode (RHE), which is superior to the reported 2D COFs[ 8 , 9 , 13 , 27 , 28 , 29 , 30 , 31 , 32 ] and most carbon nitride (g‐C3N4) materials (0.09–6.0 µA cm−2)[ 33 , 34 , 35 , 36 , 37 ] (Table S6, Supporting Information). Furthermore, density functional theory (DFT) simulations further suggest that the sulfur atoms in the thiophene as well as the cyano groups at the vinylene linkage serve as the active sites for water reduction.
Figure 1.
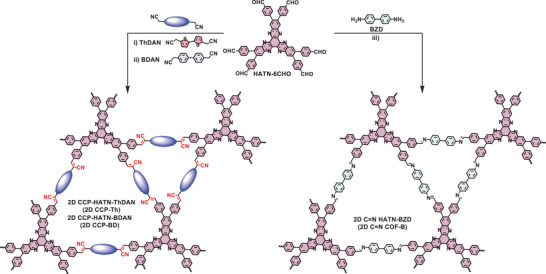
Scheme for the synthesis of 2D CCP‐Th and 2D CCP‐BD (left) and imine‐linked 2D C=N COF‐B (right) from HATN‐6CHO. Reaction conditions: i) for 2D CCP‐Th: NEt4OH (0.1 m), dimethylacetamide (DMAc)/ortho‐dichlorobenzene (o‐DCB) = 3/7, 150 °C, 3 days; ii) for 2D CCP‐BD: Cs2CO3 (0.1 m), DMAc/o‐DCB = 1:1, 120 °C, 3 days; iii) for 2D C=N COF‐B: DMAc/mesitylene (Mes)/acetic acid (HOAc, 6 m) = 5/5/1, 120 °C, 3 days.
After comprehensive screening of the Knoevenagel polymerization conditions (Table S1, Supporting Information), 2D CCP‐Th was synthesized by heating HATN‐6CHO and ThDAN in a mixture of dimethylacetamide (DMAc)/ortho‐dichlorobenzene (o‐DCB)/NEt4OH (0.1 m) = 3/7/1 in a sealed glass ampoule at 150 °C for 3 days. 2D CCP‐BD was facilely achieved by condensation between HATN‐6CHO and 2,2′‐(biphenyl‐4,4′‐diyl)diacetonitrile (BDAN) in a mixture of DMAc/o‐DCB/Cs2CO3 (0.1 m) = 5/5/1 at 120 °C for 4 days. The precipitated powder samples were washed with N,N‐dimethylformamide (DMF), H2O, acetone, and methanol, affording dark‐red powder for 2D CCP‐Th and yellow powder for 2D CCP‐BD with yields of 81% and 78%, respectively. In comparison to the synthesis of 2D CCP‐BD, the formation of 2D CCP‐Th required more harsh reaction conditions by an increased reaction temperature and stronger base as the catalyst due to the electron‐rich feature of ThDAN. Moreover, the imine‐linked 2D C=N COF‐B was synthesized by the Schiff‐base condensation between HATN‐6CHO and benzidine (BZD) (see the Supporting Information for details). After washing the precipitated solid with DMF and acetone, 2D C=N COF‐B was obtained as a yellow powder in 73% yield.
The chemical identities of 2D CCP‐Th, 2D CCP‐BD, and 2D C=N COF‐B were investigated by Fourier transform infrared (FT‐IR) and solid‐state 13C NMR spectroscopy. In comparison to the FT‐IR spectra of the monomers HATN‐6CHO, PDAN, and BDAN, the signals of the —CN groups in the FT‐IR spectra of both 2D CCP‐Th and 2D CCP‐BD (Figures S1 and S2, Supporting Information) shifted from ≈2245 cm−1 (—CH2CN) to ≈2210 cm−1 (—C=C(CN)—). Moreover, the absorptions at ≈2940 cm−1 (—CH2(CN)) and 2830 cm−1 (—CHO) disappeared, indicating an efficient Knoevenagel polymerization. In the solid‐state NMR spectra of 2D CCP‐Th and 2D CCP‐BD (Figure 2a,b), the presence of a chemical shift at ≈118 ppm suggests the successful formation of a [—CH=C(CN)—] linkage. The scanning electron microscopy (SEM) images show a particle morphology with a size of 100–200 nm for 2D CCP‐Th and 2D CCP‐BD (Figures S7 and S8, Supporting Information). High‐resolution transmission electron microscopy (TEM) images manifest the formation of layered structures at the nanoscale for both 2D CCPs (Figures S10 and S11, Supporting Information). The FT‐IR spectrum, solid‐state 13C‐NMR spectrum, and SEM and TEM images of 2D C=N COF‐B are presented in the Supporting Information.
Figure 2.
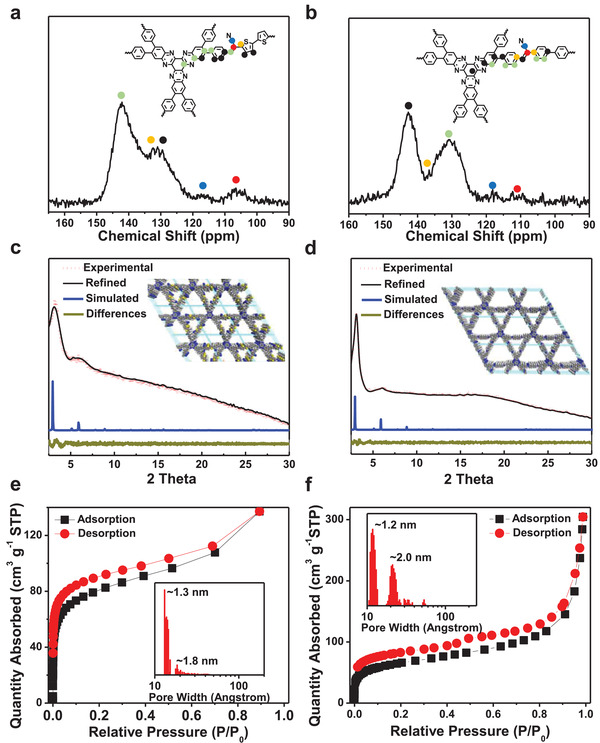
a,b) Solid‐state 13C‐NMR spectra of 2D CCP‐Th (a) and 2D CCP‐BD (b). c,d) Experimental (red dashed line), Pawley‐refined (black line), and simulated (blue line) PXRD patterns and difference (dark green line) plots for 2D CCP‐Th (c) and 2D CCP‐BD (d). e,f) N2 adsorption–desorption isotherms and pore size distributions of 2D CCP‐Th (e) and 2D CCP‐BD (f).
Experimental and simulated powder X‐ray diffraction (PXRD) measurements were conducted to verify the formation of 2D CCPs. As shown in Figure 2c,d, the peaks at ≈3.23° and 6.23° in the experimental PXRD pattern of 2D CCP‐Th as well as the peaks at 3.0° and 6.0° in the PXRDs of 2D CCP‐BD can be attributed to the (100) and (200) planes, respectively, indicating the successful formation of crystalline structures. Afterward, density‐functional tight‐binding method (DFTB+) was conducted to investigate the possible stacking type of 2D CCP‐Th and 2D CCP‐BD with eclipsed (AA), slipped (AA), and staggered (AB) stacking models. The simulations demonstrated that slipped AA stacking type endow the lowest per‐layer stabilization energies of −117.4 and −113.1 kcal mol−1 for 2D CCP‐Th and 2D CCP‐BD, respectively (Tables S2 and S3, Supporting Information). The slipped stacking models were further applied for Pawley refinements (reflex package in Accelrys's Materials Studio 7.0 software). The calculated unit‐cell parameters are a = 33.52 Å, b = 33.30 Å, c = 6.96 Å, α = 90.12°, β = 90.99°, and γ = 59.83°, with R wp = 2.12% and R p = 1.65%, for 2D CCP‐Th and a = 34.81 Å, b = 34.53 Å, c = 6.90 Å, α = 89.97°, β = 90.23°, and γ = 59.50°, with R wp = 1.52% and R p = 1.21%, for 2D CCP‐BD (one unit contains two layers; the PXRD results of 2D C=N COF‐B are presented in the Figure S13 in the Supporting Information). Moreover, the refined diffraction curves are in excellent agreement with the corresponding experimental PXRD data.
N2 adsorption–desorption measurements at 77 K were performed to investigate the porosities of 2D CCP‐Th and 2D CCP‐BD. The sharp rising curves in the low‐pressure range (P/P o = 0–0.01) of all three isotherm plots can be assigned to the type‐I sorption isotherm and suggested the formation of micropores (Figure 2e,f for 2D CCP‐Th and 2D CCP‐BD; Figures S18 and S19 for 2D C=N COF‐B in the Supporting Information). The Brunauer–Emmett–Teller (BET) surface areas were determined to be 290, 441, and 643 m2 g−1 for 2D CCP‐Th, 2D CCP‐BD, and 2D C=N COF‐B, respectively. The pore size distributions of the three 2D conjugated polymers based on nonlocal density functional theory (NLDFT) reveal two main ranges of 1.0–1.3 and 1.8–2.0 nm, which accord with the theoretical values (1.3 and 2.1 nm) and suggest the formation of dual‐pore structures.[ 17 , 38 ]
The optoelectronic properties of 2D CCP‐Th, 2D CCP‐BD, and 2D C=N COF‐B (dispersions in isopropyl alcohol) were further studied by UV–vis absorption and fluorescence spectroscopy. In contrast to the biphenyl‐bridged 2D CCP‐BD and 2D C=N COF‐B with absorption edges at 530 and 555 nm, respectively, the bithiophene‐based 2D CCP‐Th demonstrates a much broader UV–vis absorption with the absorption edge at 674 nm (Figure 3a). Thus, the corresponding Tauc plot analysis (Figure S20, Supporting Information) was calculated from the UV–vis absorption spectrum exhibited an optical energy bandgap (E g opt) for 2D CCP‐Th with 2.05 eV, which is much smaller than the values of 2.41 eV for 2D CCP‐BD and 2.36 eV for 2D C=N COF‐B. The broader absorption and narrower energy bandgap of 2D CCP‐Th can be attributed to the intramolecular charge transfer (ICT) from the donor (bithiophene) to the acceptor (HATN core) moieties.[ 39 ] Notably, 2D CCP‐BD exhibits a strong fluorescence in dispersion with an emission maximum at ≈575 nm (Figure 3b). The absolute photoluminescence quantum yield (PLQY) of solid‐state 2D CCP‐BD was determined to be 8.64%. In contrast, 2D C=N COF‐B and 2D CCP‐Th did not show apparent fluorescence. The fluorescence quenching of 2D C=N COF‐B can be attributed to the thermal dissipation due to the rotationally labile imine bonds,[ 40 , 41 ] while for 2D CCP‐Th, the intimate interaction between the donor and acceptor units can lead to nonradiative recombination processes.[ 42 , 43 ]
Figure 3.
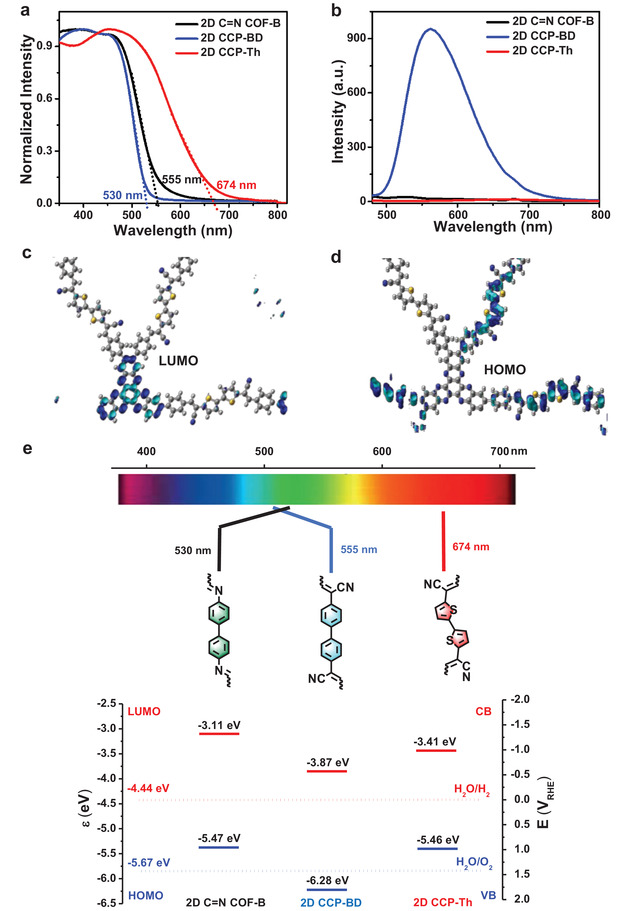
a) UV/vis absorption spectra and b) photoluminescence spectra of 2D CCP‐Th (red line), 2D CCP‐BD (blue line), and 2D C=N COF‐B (black line) from dispersions in isopropyl alcohol (concentration = 0.1 mg mL−1). c,d) Simulated LUMO (c) and HOMO (d) distribution of 2D CCP‐Th. e) Schematic representation of the band positions of BDAN/ThDAN‐linked 2D CCP‐Th, 2D CCP‐BD, and imine‐linked 2D C=N COF‐B along with the H2O/H2 (hydrogen evolution reaction, HER) and O2/H2O (oxygen evolution reaction, OER) redox couples.
Graphical representations of the highest occupied molecular orbital (HOMO) and LUMO distributions of 2D CCP‐Th and 2D CCP‐BD are calculated by DFT. 2D CCP‐Th displays a separated HOMO and LUMO distribution. The LUMO of 2D CCP‐Th is localized at the HATN units (Figure 3c), while the HOMO is fully distributed at the bithiophene units (Figure 3d). In contrast, 2D CCP‐BD shows no separated LUMO and HOMO (Figure S21, Supporting Information): The LUMO of 2D CCP‐BD is localized on the HATN core, while its HOMO is also partially delocalized on the HATN core. The separated HOMO/LUMO distribution of 2D CCP‐Th further confirms the donor–acceptor nature with the HATN core as an acceptor and the bithiophene unit as a donor unit.
To investigate the band positions of the frontier orbitals, cyclic voltammetry (CV) was performed for 2D CCP‐Th and 2D CCP‐BD (Figures S22–S24, Supporting Information). Based on the CV curves, 2D CCP‐Th and 2D CCP‐BD demonstrated HOMO levels of −5.46 and −6.28 eV, respectively. The LUMO energy levels of 2D CCP‐Th and 2D CCP‐BD were calculated as −3.41 and −3.87 eV from the optical energy bandgaps and electrochemically determined HOMO levels (Figure 3e). In comparison to the energy level of the H2O/H2 redox pair (−3.41 eV), the higher‐lying LUMO levels suggest that both 2D CCPs are promising candidates for the transfer of photoexcited electrons to H+ for H2 evolution.
Next, electrochemical impedance analyses were performed to investigate the charge separation and transport of the synthesized 2D conjugated polymers. As shown in Figure 4a, without irradiation, the charge‐transfer resistance (R ct) of 2D CCP‐Th is 8.2 kΩ, which is much lower than the values of 19.8 kΩ for 2D CCP‐BD and 36.7 kΩ for 2D C=N COF‐B. Under light illumination, the three 2D conjugated polymers manifested decreased R ct values of 13.4 kΩ for 2D CCP‐BD and 30.1 kΩ for 2D C=N COF‐B and the lowest R ct value of 6.7 kΩ for 2D CCP‐Th. The decrease in R ct values can be attributed to the generation of photoinduced charge carriers (Figures S27–S29, Supporting Information).[ 6 ] Furthermore, the carrier separation efficiencies of the three 2D conjugated polymers were evaluated according to the equation J ph = J abs × ηsep × ηcat, where J ph is the total photocurrent density, J abs is the photon absorption rate expressed as the current density, ηsep is the carrier separation efficiency, and ηcat is the catalytic efficiency for water reduction. As indicated in Figure 4b, the carrier separation efficiency of 2D CCP‐Th at 0.3 V versus RHE is ≈0.7%, which is much higher than those of 2D CCP‐BD (≈0.2%) and 2D C=N COF‐B (≈0.02%). Clearly, the more efficient charge separation and transport of 2D CCP‐BD than those of 2D C=N COF‐B can be ascribed to the sp2‐carbon‐linked conjugated framework, while the most efficient charge separation and transport for 2D CCP‐Th should further benefit from the donor–acceptor structures.
Figure 4.
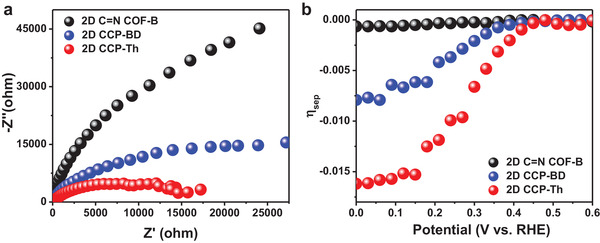
a) Impedance analyses for 2D CCP‐Th (red line), 2D CCP‐BD (blue line), and 2D C=N COF‐B (black line) without light irradiation; b) carrier‐separation efficiency (ηsep) of 2D CCP‐Th (red line), 2D CCP‐BD (blue line), and 2D C=N COF‐B (black line) as a function of the applied potential.
Under AM 1.5 G irradiation and at 0.3 V versus RHE, the three 2D conjugated polymers on fluorine‐doped tin oxide (FTO) electrodes show photocurrents of ≈5.5 µA cm−2 for 2D CCP‐Th, ≈2.8 µA cm−2 for 2D CCP‐BD, and ≈0.06 µA cm−2 for 2D C=N COF‐B (Figure 5a,b). Moreover, 2D CCP‐Th achieves a photocurrent of ≈7.9 µA cm−2 at 0 V versus RHE, which exceeds those of the reported 2D COFs (0.09–6.0 µA cm−2)[ 8 , 9 , 13 , 27 , 28 , 29 , 30 , 31 , 32 ] and most carbon nitride materials (g‐C3N4, 0.3–1.2 µA cm−2) (Table S6, Supporting Information).[ 33 , 34 , 35 , 36 , 37 ] As a key parameter to evaluate the intrinsic PEC performance of photoelectrode materials, the incident‐photon‐to‐current efficiency (IPCE) was measured (Figure S30, Supporting Information). The IPCE of 2D CCP‐Th is up to 6.1%, which is higher than those for 2D CCP‐BD (5.5%), 2D C=N COF‐B (4.1%), the state‐of‐the‐art 2D π‐COF (BDT‐ETTA COF, 0.6%)[ 13 ] and the cocatalyst‐free carbon nitride (g‐C3N4, 1.2%).[ 37 ]
Figure 5.
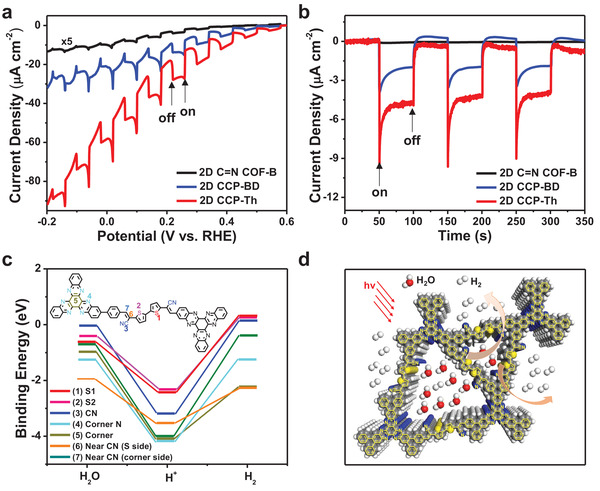
a) Polarization plots of 2D CCP‐Th (red line), 2D CCP‐BD (blue line), and 2D C=N COF‐B (black line) with and without light irradiation. b) Photocurrent–time plots for 2D CCP‐Th (red line), 2D CCP‐BD (blue line), and 2D C=N COF‐B (black line) at 0.3 V versus RHE. On: illumination on; off: illumination off. c) GFN1‐xTB calculated binding energies (eV) of adsorbed species at seven possible active sites of 2D CCP‐Th. d) Schematic illustration of the photoelectrochemical H2 evolution reactions on the reactive CN substitutions on the vinylene linkage and bithiophene units of 2D CCP‐Th under visible light irradiation.
The durability is another important criterion for assessing the PEC hydrogen evolution reaction (HER) performance of a photoelectrode. Therefore, the long‐term PEC HER stability of 2D CCP‐Th was evaluated at 0.4 V versus RHE. After a 6 h PEC reaction, the photocurrent of 2D CCP‐Th revealed no obvious decrease (Figure S31, Supporting Information). The gaseous product generated from 2D CCP‐Th was synchronously analyzed using a gas chromatograph at 0 V versus RHE. The experimentally detected H2 amount revealed a Faradic efficiency of ≈97% for 2D CCP‐Th. After the PEC HER stability test, 2D CCP‐Th was recovered by sonicating the sample from the electrode via sonication treatment. The recovered 2D CCP‐Th was then investigated by SEM and PXRD. The SEM images of 2D CCP‐Th obviously revealed a maintained particle morphology with sizes of 100–200 nm (Figure S32, Supporting Information). The PXRD pattern still displayed two signals at ≈3.23° and 6.23°, indicating retained crystalline structures (Figure S33, Supporting Information). The combination of SEM and PXRD measurements confirmed the excellent electrochemical stability of 2D CCP‐Th.
To reveal the possible active sites for the PEC HER in 2D CCP‐Th, the binding energies of H* and H2 were calculated using GFN‐xTB.[ 44 ] A section of the pore wall consisting of one full linker and two HATN nodes was cut from the global minimum slipped AA structure and further used for calculations. Then, seven possible binding sites on the pore wall were identified (Figure S34, Supporting Information), and the binding energy of each species at each site was calculated (Figure 5c). The two sulfur atoms (sites 1 and 2) can bind H* with energies of −2.40 and −2.36 eV but have positive binding energies for H2 of 0.31 and 0.25 eV, respectively. Similarly, the cyano group (site 3) has binding energies of −3.16 and 0.14 eV for H* and H2, respectively. However, the other four sites (sites 4–7) show binding energies of −4.11, −4.08, −3.52, and −4.01 eV for H* and negative binding energies of −0.75, −2.26, −1.30, and −0.36 eV for H2, respectively (Table S7, Supporting Information). Thus, the simulation results suggest that the sulfur atoms of the bithiophene unit and the CN substituents on the vinylene linkage can serve as the active sites since they can adsorb H* moderately well and allow H2 to desorb (slightly positive binding energies).
In conclusion, we demonstrate the first bithiophene‐bridged D–A‐based 2D CCP (2D CCP‐Th), which can be utilized as a photocathode for superb photoelectrochemical water reduction. Due to the robust sp2‐carbon‐linked conjugations and donor–acceptor structures, 2D CCP‐Th exhibits a broad UV–vis absorption up to 674 nm, a narrow optical energy gap of 2.04 eV, and a high‐lying LUMO level of −3.41 eV. When employed as a photocathode for water reduction, 2D CCP‐Th demonstrates a superb saturated photocurrent density (5.5 µA cm−2 at 0.3 V and 7.9 µA cm−2 at 0 V vs RHE) as a photoelectrode in PEC devices. This work enriches the family of 2D CCPs with thiophene‐containing donor–acceptor structures and provides promising semiconducting organic materials for PEC applications, field‐effect transistors, or photovoltaics.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
S.X. and H.S. contributed equally to this work. The authors thank the financial support from the DFG for the CRC 1415 (No. 417590517), the ERC Consolidator Grant (T2DCP, No. 819698), Coordination Networks: Building Blocks for Functional Systems (SPP 1928, COORNET), EU Graphene Flagship (GrapheneCore3; No. 881603), H2020‐MSCA‐ITN (ULTIMATE, No. 813036), and Center of Advancing Electronics Dresden (cfaed). The authors thank Dr. Petr Formanek (Leibniz Institute for Polymer Research, IPF, Dresden) and S.W.P. for the TEM measurements. The authors thank Dr. Arezoo Dianat for possible calculations, Dr. En Zhang for BET measurements, and Dr. Yungui Li and Dr. Heidi Thomas for PLQY measurements.
Open access funding enabled and organized by Projekt DEAL.
Xu S., Sun H., Addicoat M., Biswal B. P., He F., Park S. W., Paasch S., Zhang T., Sheng W., Brunner E., Hou Y., Richter M., Feng X., Thiophene‐Bridged Donor–Acceptor sp2‐Carbon‐Linked 2D Conjugated Polymers as Photocathodes for Water Reduction. Adv. Mater. 2021, 33, 2006274. 10.1002/adma.202006274
References
- 1. Wang H., Zhang L., Chen Z., Hu J., Li S., Wang Z., Liu J., Wang X., Chem. Soc. Rev. 2014, 43, 5234. [DOI] [PubMed] [Google Scholar]
- 2. Lin L., Yu Z., Wang X., Angew. Chem., Int. Ed. 2019, 58, 6164. [DOI] [PubMed] [Google Scholar]
- 3. Wang X., Maeda K., Thomas A., Takanabe K., Xin G., Carlsson J. M., Domen K., Antonietti M., Nat. Mater. 2009, 8, 76. [DOI] [PubMed] [Google Scholar]
- 4. Ran J., Zhang J., Yu J., Jaroniec M., Qiao S. Z., Chem. Soc. Rev. 2014, 43, 7787. [DOI] [PubMed] [Google Scholar]
- 5. Wang X., Vasileff A., Jiao Y., Zheng Y., Qiao S. Z., Adv. Mater. 2019, 31, 1803625. [DOI] [PubMed] [Google Scholar]
- 6. Sun H., Öner I. H., Wang T., Zhang T., Selyshchev O., Neumann C., Fu Y., Liao Z., Xu S., Hou Y., Turchanin A., Zahn D. R. T., Zschech E., Weidinger I. M., Zhang J., Feng X., Angew. Chem., Int. Ed. 2019, 58, 10368. [DOI] [PubMed] [Google Scholar]
- 7. Zhang T., Hou Y., Dzhagan V., Liao Z., Chai G., Löffler M., Olianas D., Milani A., Xu S., Tommasini M., Zahn D. R. T., Zheng Z., Zschech E., Jordan R., Feng X., Nat. Commun. 2018, 9, 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin E., Lan Z., Jiang Q., Geng K., Li G., Wang X., Jiang D., Chem 2019, 5, 1632. [Google Scholar]
- 9. Bi S., Yang C., Zhang W., Xu J., Liu L., Wu D., Wang X., Han Y., Liang Q., Zhang F., Nat. Commun. 2019, 10, 2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pachfule P., Acharjya A., Roeser J., Langenhahn T., Schwarze M., Schomäcker R., Thomas A., Schmidt J., J. Am. Chem. Soc. 2018, 140, 1423. [DOI] [PubMed] [Google Scholar]
- 11. Wang X., Chen L., Chong S. Y., Little M. A., Wu Y., Zhu W. H., Clowes R., Yan Y., Zwijnenburg M. A., Sprick R. S., Cooper A. I., Nat. Chem. 2018, 10, 1180. [DOI] [PubMed] [Google Scholar]
- 12. Banerjee T., Haase F., Savasci G., Gottschling K., Ochsenfeld C., Lotsch B. V., J. Am. Chem. Soc. 2017, 139, 16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sick T., Hufnagel A. G., Kampmann J., Kondofersky I., Calik M., Rotter J. M., Evans A., Döblinger M., Herbert S., Peters K., Böhm D., Knochel P., Medina D. D., Fattakhova‐Rohlfing D., Bein T., J. Am. Chem. Soc. 2018, 140, 2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun H., Neumann C., Zhang T., Löffler M., Wolf A., Hou Y., Turchanin A., Zhang J., Feng X., Adv. Mater. 2019, 31, 1900961. [DOI] [PubMed] [Google Scholar]
- 15. Chen X., Geng K., Liu R., Tan K. T., Gong Y., Li Z., Tao S., Jiang Q., Jiang D., Angew. Chem., Int. Ed. 2020, 59, 5050. [DOI] [PubMed] [Google Scholar]
- 16. Kandambeth S., Dey K., Banerjee R., J. Am. Chem. Soc. 2019, 141, 1807. [DOI] [PubMed] [Google Scholar]
- 17. Xu S., Wang G., Biswal B. P., Addicoat M., Paasch S., Sheng W., Zhuang X., Brunner E., Heine T., Berger R., Feng X., Angew. Chem., Int. Ed. 2019, 58, 849. [DOI] [PubMed] [Google Scholar]
- 18. Rao M. R., Fang Y., De Feyter S., Perepichka D. F., J. Am. Chem. Soc. 2017, 139, 2421. [DOI] [PubMed] [Google Scholar]
- 19. Zhuang X., Zhao W., Zhang F., Cao Y., Liu F., Bi S., Feng X., Polym. Chem. 2016, 7, 4176. [Google Scholar]
- 20. Jin E., Asada M., Xu Q., Dalapati S., Addicoat M. A., Brady M. A., Xu H., Nakamura T., Heine T., Chen Q., Jiang D., Science 2017, 357, 673. [DOI] [PubMed] [Google Scholar]
- 21. Jadhav T., Fang Y., Patterson W., Liu C.‐H., Hamzehpoor E., Perepichka D. F., Angew. Chem., Int. Ed. 2019, 58, 13753. [DOI] [PubMed] [Google Scholar]
- 22. Becker D., Biswal B. P., Kaleńczuk P., Chandrasekhar N., Giebeler L., Addicoat M., Paasch S., Brunner E., Leo K., Dianat A., Cuniberti G., Berger R., Feng X., Chem. ‐ Eur. J. 2019, 25, 6562. [DOI] [PubMed] [Google Scholar]
- 23. Jin E., Li J., Geng K., Jiang Q., Xu H., Xu Q., Jiang D., Nat. Commun. 2018, 9, 4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen R., Shi J.‐L., Ma Y., Lin G., Lang X., Wang C., Angew. Chem., Int. Ed. 2019, 58, 6430. [DOI] [PubMed] [Google Scholar]
- 25. Zhao Y., Liu H., Wu C., Zhang Z., Pan Q., Hu F., Wang R., Li P., Huang X., Li Z., Angew. Chem., Int. Ed. 2019, 58, 5376. [DOI] [PubMed] [Google Scholar]
- 26. Pachfule P., Acharjya A., Roeser J., Sivasankaran R. P., Ye M. Y., Brückner A., Schmidt J., Thomas A., Chem. Sci. 2019, 10, 8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang F. M., Sheng J.‐L., Yang Z.‐D., Sun X.‐J., Tang H.‐L., Lu M., Dong H., Shen F.‐C., Liu J., Lan Y.‐Q., Angew. Chem., Int. Ed. 2018, 57, 12106. [DOI] [PubMed] [Google Scholar]
- 28. Stegbauer L., Zech S., Savasci G., Banerjee T., Podjaski F., Schwinghammer K., Ochsenfeld C., Lotsch B. V., Adv. Energy Mater. 2018, 8, 1703278. [Google Scholar]
- 29. Cheng Z., Fang W., Zhao T., Fang S., Bi J., Liang S., Li L., Yu Y., Wu L., ACS Appl. Mater. Interfaces 2018, 10, 41415. [DOI] [PubMed] [Google Scholar]
- 30. Jiang Q., Sun L., Bi J., Liang S., Li L., Yu Y., Wu L., ChemSusChem 2018, 11, 1108. [DOI] [PubMed] [Google Scholar]
- 31. Fu Z., Wang X., Gardner A. M., Wang X., Chong S. Y., Neri G., Cowan A. J., Liu L., Li X., Vogel A., Clowes R., Bilton M., Chen L., Sprick R. S., Cooper A. I., Chem. Sci. 2020, 11, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen W., Wang L., Mo D., He F., Wen Z., Wu X., Xu H., Chen L., Angew. Chem., Int. Ed. 2020, 59, 16902. [DOI] [PubMed] [Google Scholar]
- 33. Guo Y., Li J., Yuan Y., Li L., Zhang M., Zhou C., Lin Z., Angew. Chem., Int. Ed. 2016, 55, 14693. [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y., Thomas A., Antonietti M., Wang X., J. Am. Chem. Soc. 2009, 131, 50. [DOI] [PubMed] [Google Scholar]
- 35. Han Q., Wang B., Gao J., Qu L., Angew. Chem., Int. Ed. 2016, 55, 10849. [DOI] [PubMed] [Google Scholar]
- 36. Zhang J., Chen X., Takanabe K., Maeda K., Domen K., Epping J. D., Fu X., Antonieta M., Wang X., Angew. Chem., Int. Ed. 2010, 49, 441. [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y., Schnepp Z., Cao J., Ouyang S., Li Y., Ye J., Liu S., Sci. Rep. 2013, 3, 2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu S.‐Q., Zhan T.‐G., Wen Q., Pang Z.‐F., Zhao X., ACS Macro Lett. 2016, 5, 99. [Google Scholar]
- 39. Lei K., Wang D., Ye L., Kou M., Deng Y., Ma Z., Wang L., Kong Y., ChemSusChem 2020, 13, 1725. [DOI] [PubMed] [Google Scholar]
- 40. Li X., Gao Q., Wang J., Chen Y., Chen Z.‐H., Xu H.‐S., Tang W., Leng K., Ning G.‐H., Wu J., Xu Q.‐H., Quek S. Y., Lu Y., Loh K. P., Nat. Commun. 2018, 9, 2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu S., Li Y., Biswal B. P., Addicoat M. A., Paasch S., Imbrasas P., Park S., Shi H., Brunner E., Richter M., Lenk S., Feng X., Chem. Mater. 2020, 32, 7985. [Google Scholar]
- 42. Körzdörfer T., Tretiak S., Kümmel S., J. Chem. Phys. 2009, 131, 034310. [DOI] [PubMed] [Google Scholar]
- 43. Marras S. A. E., Kramer F. R., Tyagi S., Nucleic Acids Res. 2002, 30, 122e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grimme S., Bannwarth C., Shushkov P., J. Chem. Theory Comput. 2017, 13, 1989. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information