

Abstract
No T cell receptor (TCR) T cell therapies have obtained clinical approval. The lack of strategies capable of selecting and recovering potent T cell candidates may be a contributor to this. Existing protocols for selecting TCR T cell clones for cell therapies such as peptide multimer methods have provided effective measurements on TCR affinities. However, these methods lack the ability to measure the collective strength of intercellular interactions (i.e., cellular avidity) and markers of T cell activation such as immunological synapse formation. This study describes a novel microfluidic fluid shear stress‐based approach to identify and recover highly potent T cell clones based on the cellular avidity between living T cells and tumor cells. This approach is capable of probing approximately up to 10 000 T cell–tumor cell interactions per run and can recover potent T cells with up to 100% purity from mixed populations of T cells within 30 min. Markers of cytotoxicity, activation, and avidity persist when recovered high cellular avidity T cells are subsequently exposed to fresh tumor cells. These results demonstrate how microfluidic probing of cellular avidity may fast track the therapeutic T cell selection process and move the authors closer to precision cancer immunotherapy.
Keywords: avidity, cytotoxicity, immunotherapy, microfluidics, shear stress, synapses, T cell receptors
This study describes a novel microfluidic fluid shear stress‐based approach to identify potent T cell clones based on cellular avidity between living T cells and tumor cells. This method is capable of analyzing thousands of T cell–tumor cell interactions and can discriminate and recover highly potent T cells with up to 100% purity from mixed populations of T cells.
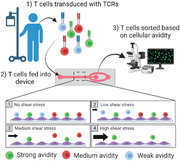
1. Introduction
TCR T cell therapy is emerging as a promising mode of cancer immunotherapy. In this therapy, engineered or native tumor‐specific CD8 T cells are selected, expanded, and injected to target tumor cells that present antigenic peptides presented by the major histocompatibility complexes (pMHC) on cell surfaces.[ 1 ] Clinical trials of TCR T cell therapies have demonstrated efficacious anti‐tumor responses for cancer patients with metastatic melanoma,[ 2 ] synovial sarcoma,[ 2b,c ] and myeloma.[ 3 ]
A major hurdle for TCR therapies is the process of selecting TCRs with potent clinical efficacy. The structural avidity of TCRs to its cognate pMHC is commonly used to identify TCRs for study. Given the diversity of the human TCR repertoire that consists of billions of distinct TCR clonotypes,[ 4 ] it is unsurprising that only a small minority of these clonotypes are reactive against a particular pMHC and an even smaller subset of these are capable of provoking optimal cell‐mediated immune responses.[ 5 ] The limits of existing technologies for detecting these rare T cell clones can exert great time and financial burdens on the development of immunotherapies if nonoptimal candidates are selected.[ 6 ] Surface plasmon resonance is capable of detecting the molecular kinetics and affinity of TCR‐pMHC binding. However, affinity readouts disregard the interactions of multiple TCR‐pMHC complexes and co‐receptors.[ 7 ] This results in poor predictions of T cell functionality.[ 8 ] The development of reversible pMHC multimer technology enabled the probing of avidity readouts using flow cytometry.[ 9 ] This technique provides high‐throughput analysis of multiple TCR‐pMHC interactions as well as their CD8 co‐receptor.[ 7a ] In particular, novel NTA–His tag‐containing multimer technology (NTAmers) generates direct measurements of TCR‐pMHC dissociation rates (k off) that effectively correlate on antitumor responses for a wide range of antigen‐specific T cell clones.[ 6 , 9 ] On the other hand, multimers are not capable of selecting T cell clones independent of the pMHC and occasionally fails to detect functional T cells resulting in false negative readouts of bulk populations.[ 5 , 10 ]
Moreover, all of the above technologies fail to account for the collective strength of other cell–cell interactions such as integrin binding,[ 11 ] gap junction proteins,[ 12 ] and other co‐receptors[ 13 ] that form the IS. The IS has numerous important functions for regulating T cell mediated immune responses including promoting TCR triggering[ 14 ] and integrating numerous co‐stimulatory and co‐inhibitory (checkpoint) signals.[ 15 ] The lack of technologies available to evaluate the overall strength of cellular interactions between activated T cells and tumor cells may be a contributor to the absence of clinically‐approved TCR T cell therapies. Thus, novel strategies are required to measure the sum of these interactions, which we define as “cellular avidity.”
Stronger TCR‐pMHC interactions typically result in more potent anti‐tumor responses.[ 9 , 16 ] This is important for TCR T cell therapy that aims to enhance T cell reactivity against tumors, as the vast majority of reactive endogenous T cells express receptors with weak TCR‐pMHC avidity due to mechanisms of central and peripheral tolerance that eliminate high avidity T cells.[ 1a ] T cells engineered with supraphysiological affinity or avidity however can conversely lead to poor functionality[ 9 , 16 ] and greater risk of cross‐reactivity to self‐antigens resulting in autoimmune diseases.[ 17 ] There is therefore an optimal balance between improved anti‐tumoral activity and autoimmunity[ 1 , 17 ] that needs to be carefully considered to achieve successful clinical outcomes. Therefore, the ability to sort TCR T cells based on their cellular avidities may allow us to select the most efficacious and safe TCRs to advance through clinical trials.
Microfluidics enables the investigation of immunotherapeutic strategies in vitro with numerous advantages[ 18 ] such as its ability to i) analyze individual cell–cell interactions, ii) monitor cell behaviors (e.g., activation and cytokine release) in real‐time, and iii) provide precise control over environmental conditions. Notably, researchers have previously used droplet microfluidics to co‐encapsulate tumor cells and TCR T cells into subnanolitre droplets using a microfluidic droplet generator.[ 19 ] This technique accurately identifies rare TCR T cell clones at the single‐cell level under high‐throughput by analyzing green fluorescent protein (GFP) activation. However, this approach is slow, qualitative, and does not directly assess the strength of T cell‐tumor cell avidity. Furthermore, acoustic microfluidics[ 20 ] has been used by researchers to measure cell–cell interactions for chimeric antigen receptor (CAR) T cells bound to tumor cells, which strongly correlated with in vivo efficacy in mice.[ 21 ] However, this strategy has limited throughput and is incapable of recovering and sorting T cells based on their avidities for further downstream analyses.
Here, we developed a novel fluid shear stress‐based microfluidic method that sorts and recovers primary human and mouse T cell variants based on their cellular avidity to melanoma cells. Using this method, we successfully isolated TCR T cells with high cellular avidity and cytotoxic with up to 100% purity. Each device is easy to operate and capable of probing up to 10 000 TCR‐tumor cell interactions within 30 minutes. This strategy has the potential to fast track the therapeutic process by accelerating the isolation of optimal TCR T cell clones based on functional live‐cell interactions, and may have promising applications in precision cancer medicine.
2. Results
Proof‐of‐concept microfluidic shear devices were designed to discriminate and collect T cells based on their cellular avidity to monolayers of tumor cells. The main component of these devices is a T cell–tumor cell interaction region that consists of a 1.0 × 0.1 × 50 mm (W × H × L) straight microchannel on which T cells interact with adherent tumor cells (Figure 1a). This geometry allows cells interacting within the center portion of the microchannel to experience near identical shear stress (±1%) at a given flow rate to be collected at the product outlet. In contrast, cells located 0.1 mm or less from either sidewall of the channel experienced non‐uniform shear and were diverted to a waste outlet by tailoring the lengths of the outlet channels to control the relative resistances of these paths (Figure 1b). We chose a variety of shear stresses to apply by simply adjusting the input flow rate over the course of an experiment (Tables S1,S2, Supporting Information). In this manner, T cells of varying cellular avidities were subsequently fractionated for downstream processing (Figure 1c,d).
Figure 1.
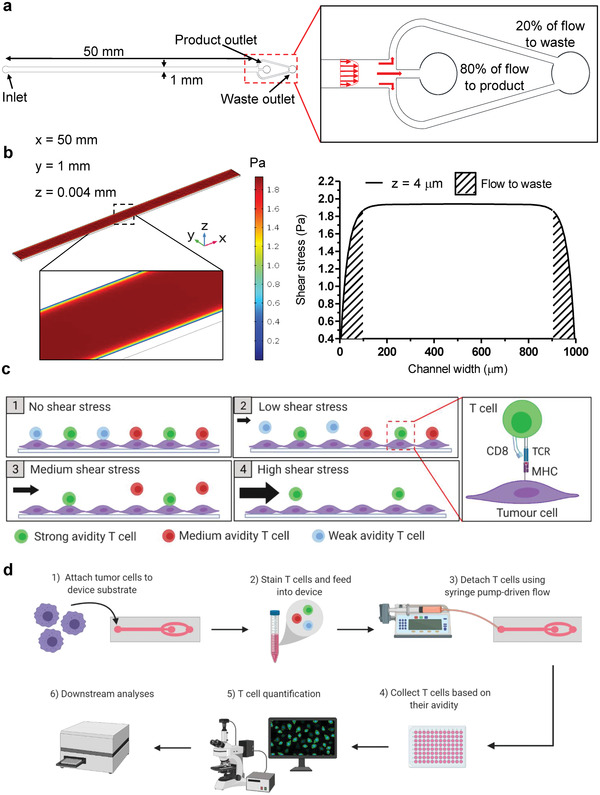
Microfluidic device and operation. a) Schematic of device designed to collect 80% of uniform shear flow (not to scale). b) Fluid dynamic simulation: shear stress contour plot (left) and parabolic shear stress plot (right) at 200 µL min−1 flow rate 4 µm above the surface, hashed area indicates flow sent to waste. c) Operating principles for shear‐induced sorting of T cells based on cellular avidity. d) Experimental workflow.
2.1. Tumor Cell Monolayers Remained Attached under Shear
To assess the adhesion of tumor monolayers under shear, devices seeded with Me290, NA8, and B16F10 melanoma cells were subjected to ramping shear stresses from 0 to 19.2 Pa (Figure S1, Supporting Information). Our data indicate that cells collected under applied shear stresses of 0–1.9 Pa contained poorly adherent cells or those bound non‐specifically (Figure S2, Supporting Information), and thus we considered this initial shear range as a wash step. Greater than 99.9% of all three cell types remained attached under 0–11.5 Pa shear stress. At the highest experimental shear stress of 19.2 Pa, 98.1 ± 1.8%, 99.7 ± 0.4%, and 98.5 ± 0.6% of Me290, NA8, and B16F10 cells remained attached, respectively (Figure 2a,b). Me290 and NA8/B16F10 melanoma cells bound most strongly to device substrates treated with PLL and fibronectin, respectively. In contrast, poor initial cell adhesion was observed on untreated and collagen treated device substrates (Figure S3, Supporting Information).
Figure 2.
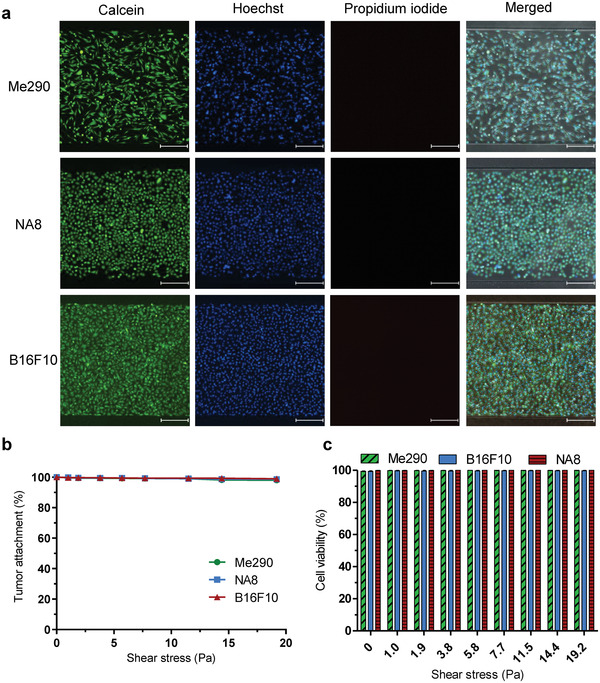
Validation of tumor cell adhesion and viability. a) Representative images of Me290 (NY‐ESO‐1+), NA8 (NY‐ESO‐1−), and B16F10 (gp100+) melanoma cells stained with calcein‐AM (green), Hoechst (blue), and propidium iodide (red) after application of 19.2 Pa shear stresses. Scale bar: 250 µm. b) Quantification of melanoma cell adhesion Me290, NA8, and B16F10 under shear stresses of 0–19 Pa. c) Quantification of cell viability. Data are represented as mean ± SEM error bars, N = 3.
2.2. Tumor Cell Monolayers Remained Viable under Shear
Viability of tumor cells under shear stress were examined using propidium iodide (PI), a nuclear stain for dead cells (Figure 2a). This test was to ensure that tumor cell monolayers remained viable in our device under high shear stress so that cell–cell and cell–substrate analyses are not compromised by poor cell viability. Confluent monolayers of Me290, NA8, and B16F10 melanoma cells were subjected to increasing shear stress ranging from 0 to 19.2 Pa by varying inlet flow rates. Viability data revealed that >99% of tumor cells remained viable even under high shear stress (Figure 2c), with a cell lysis solution used as a control (Figure S4, Supporting Information).
2.3. Activation of Tumor Cell‐Bound SUPT1 T Cells Correlates with Increasing Avidity
To ensure that T cell binding resulted in physiological T cell activation, we probed the concentrations of intracellular calcium, a critical signaling event during early T cell activation, in bound TCR variants using fluorescent dyes. SUPT1 human T cell variants transduced with TCRs of varying avidities to the NY‐ESO‐1 antigen previously reported[ 9a ] were used to validate activation in our device. Unmodified wild‐type (WT, k off = 4.08 × 10−2 s−1) represented a normal physiological range clonotype, Non‐transduced (NT, k off = n/a) represented non‐specific TCRs and double mutant beta (DMβ, k off = 0.78 × 10−2 s−1) represented high avidity TCRs. SUPT1 T cells were stained with a calcium binding dye and then infused into devices. T cells bound to Me290 device monolayers showed activation that corresponded to their relative avidities as determined by analyzing the calcium flux (Figure 3a) and Movie S1, Supporting Information. Automated MATLAB scripts revealed that the percentage of SUPT1 T cell activation increased, as the avidity to the NY‐ESO‐1 antigen increased (Figure 3b). The fraction of DMβ T cells that exhibited activation was 90.3 ± 1.2% on Me290s, whereas WT and NT T cells recorded significantly lower (p < 0.01) activation of 46.7 ± 12.6% and 31.3 ± 1.7%, respectively. Moreover, SUPT1 T cell variants in contact with NA8 cells (NY‐ESO‐1−) exhibited significantly lower levels of activation (Figure 3b). These results validated that initial signaling events involved in T cell activation are influenced by TCR avidity and specific antigen recognition was required for T cell activation.
Figure 3.
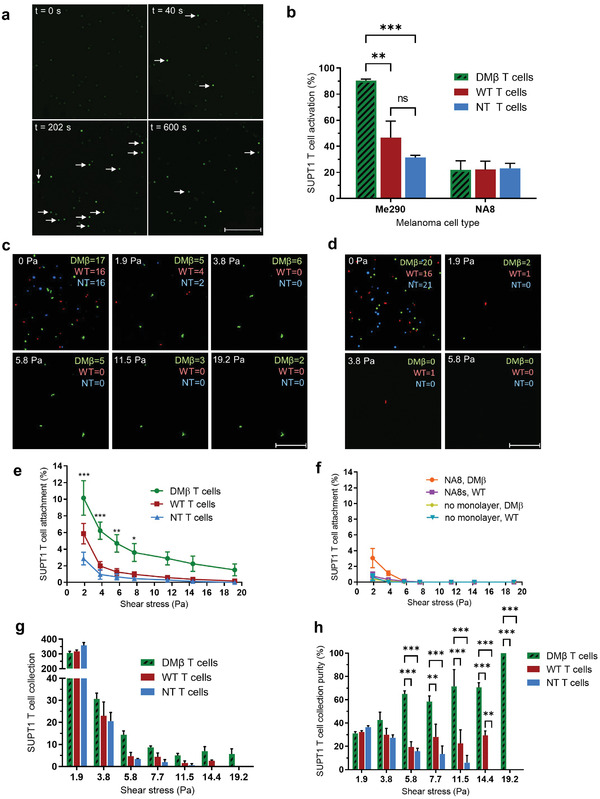
SUPT1 Transduced T cell activation, attachment, and collection. Human SUPT1 T cells transduced with TCRs of varying structural avidity against HLA‐A0201/SLLMWITQC (NY‐ESO‐1 antigen), DMβ with high avidity, wild type (WT) with intermediate avidity, and non‐transduced (NT) with weak avidity. Melanoma cells Me290 (NY‐ESO‐1+) and NA8 (NY‐ESO‐1−). a) Representative calcium flux time‐lapse images of DMβ T cells flowed onto Me290 melanoma monolayer. White arrows indicate activated T cells. b) Quantification of SUPT1 T cell activation on melanoma monolayers. c) Representative images of SUPT1 detachment from Me290. d) Representative images of SUPT1 detachment from NA8. e) SUPT1 that remain attached to Me290 under shear. f) SUPT1 attachment to NA8 monolayers and monolayer‐free devices. g) Collection of SUPT1 T cells from the product outlet under increasing shear stress. h) Percent purity of each SUPT1 T cell variant collected. Scale bar: 250 µm. Data are represented as mean ± SEM error bars, N = 3, p‐values are calculated using two‐way ANOVA with Bonferroni's multiple comparisons test, *p < 0.05,** p < 0.01, and *** p < 0.001.
2.4. SUPT1 T Cells can be Collected Based on Cellular Avidity
SUPT1 T cell variants dyed with calcein variants were infused into devices seeded with monolayers of Me290 (NY‐ESO‐1+) and NA8 (NY‐ESO‐1−) melanoma cells (Figure 3c,d) and subjected to 0–19.2 Pa shear stresses at 37 °C/5% CO2 after 10 min of cell–cell interactions under no flow. Automated image analyses were conducted on up to 10 000 SUPT1 T cells per condition bound in the T cell–tumor cell interaction region and used to determine cellular avidity.
As expected, the adhesion of SUPT1 T cells followed their relative avidities to the NY‐ESO‐1 antigen. DMβ T cells with high avidity remained more strongly adhered compared to WT and NT T cells (Figure 3e), as shear stress was increased from 1.9 to 19.2 Pa. Notably, 1.5 ± 0.7% of DMβ T cells remained adhered at 19.2 Pa; whereas, 0.2 ± 0.1% and 0% of WT and NT T cells remained attached, respectively. In contrast, SUPT1 T cells displayed weaker adhesion to NA8 (NY‐ESO‐1−) melanoma cells and monolayer‐free bare devices (Figure 3f). Under these conditions, all SUPT1 T cell variants were detached by 7.7 Pa and thus, this verified that cellular avidity was dominated by the recognition of the specific NY‐ESO‐1 antigen. Furthermore, avidity bar plots at 1.9 Pa, after the initial wash phase of nonspecific binders, showed differences in avidity between each T cell variant bound to Me290 monolayers (Figure S5a, Supporting Information) and Movies S2–S6, Supporting Information displayed SUPT1 T cell detachment on melanoma monolayers under increasing shear stresses. Interestingly, we observed a large proportion of poorly adherent T cells detaching under low shear flows during the wash phase (0–1.9 Pa). Increasing cell–cell interaction times to 15 min before inducing shear improved initial DMβ T cell adhesion up to 6% at 1 Pa; however, a large proportion of T cells still detached under low shear (Figure S6, Supporting Information).
Effluents of devices under 1.9–19.2 Pa shear stresses were then collected and the numbers of SUPT1 T cell variants quantified using NIS‐Elements Advanced software (Figure 3g,h). There was no statistical difference (p > 0.05) among the T cell purities collected under shear stresses of 1.9–3.8 Pa. However, fractions collected under shear stresses of 5.7–19.2 Pa exhibited a statistically significant higher purity of DMβ T cells in comparison to WT and NT T cells. Importantly, at 19.2 Pa 100% of collected fractions contained only DMβ T cells. These results correlated with the data obtained from T cell attachment experiments described above T cells with stronger cellular avidities were therefore successfully isolated from weaker avidity counterparts.
2.5. Primary T Cells can be Collected Based on Cellular Avidity
To investigate the response of primary mouse T cells, we isolated T cells from splenocytes of Pmel‐1 TCR‐transgenic mice (Pmel) and WT C57BL/6 mice.[ 17b ] Pmel T cells are gp100 antigen‐specific with high avidity for the gp100 antigen presented on the surface of B16F10 cells whereas WT T cells are polyclonal and non‐specific. Activated Pmel and WT T cells were infused into the device coated with adhered B16F10 cells pretreated with 1 μ m of hgp100 peptide 30 min prior to experiments (Figure 4a). Pmel T cells bound more strongly to B16F10 cells compared to WT T cells, as shear stress was increased (Figure 4b). Pmel T cells were differentiated from WT T cells even at low shear stresses of 1.9 Pa, with 19.4 ± 1.7% and 2.4 ± 1.4% of Pmel and WT T cells that remained bound to the tumor monolayer, respectively (Figure 4b; Figure S5b, Supporting Information). These datasets were obtained under infusion ratios of 1:1 (Pmel:WT T cells); however, clear separation was also observed for more stringent ratios of 1:10 (Figure S7, Supporting Information). We found that pretreating B16F10 melanoma cells with hgp100 peptide was an important step in improving separation since early experiments revealed fivefold weaker T cell binding capabilities for T cells adhered to untreated B16F10 cells (Figure S8, Supporting Information). Data gathered here demonstrated that our device can differentiate and select primary activated antigen specific T cells (Pmel) from WT adhered onto tumor cells.
Figure 4.
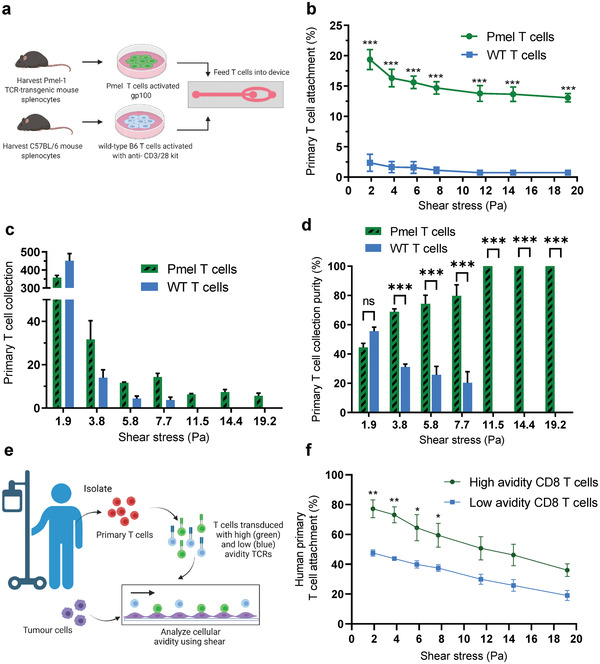
Primary T cell attachment and collection. a) Schematic of mouse T cell isolation, expansion, and seeding. b) Quantification of Pmel (antigen‐specific) and WT T cells (non‐specific) attachment to B16F10 melanoma monolayers (gp100+) under increasing shear stress. c) Quantification of primary mouse T cell collection from the product outlet under increasing shear stress. d) Purity of primary mouse T cell fractions collected. e) Schematic of human primary CD8 T cells extracted from a cancer patient, followed by TCR transduction and microfluidic seeding. f) Quantification of human primary CD8 T cells, high and low avidity clones to Me290 melanoma monolayers under increasing shear stress. Data are represented as mean ± SEM error bars, N = 3, p‐values are calculated using two‐way ANOVA with Bonferroni's multiple comparisons test, *p < 0.05,** p < 0.01, and *** p < 0.001.
Primary mouse T cells were then collected from the product outlet and quantified in order to determine the percentage of Pmel and WT T cells collected after each shear stress challenge (Figure 4c,d; Figure S9, Supporting Information). We observed that Pmel and WT T cells collected were not significantly different (p > 0.05) after 1.9 Pa. In contrast, we found statistically different (p < 0.001) percentages of Pmel and WT T cells collected when we increased shear stress to ≥3.8 Pa. Notably, at applied shear stresses ≥ 11.5 Pa, 100% pure Pmel T cell fractions were successfully isolated from weaker non‐specific WT T cells, indicating that primary mammalian cells can be successfully isolated based on their cellular avidity.
We then assessed the binding of primary human CD8 T cells to Me290 melanoma monolayers (NY‐ESO‐1+). T cells were extracted from a melanoma patient and transduced with high or low avidity TCRs specific to NY‐ESO‐1 antigens and were infused into the device at a 1:1 ratio (Figure 4e). Similar to SUPT1 T cell experiments, human primary T cells transduced with high avidity TCRs bound more strongly to Me290 monolayers compared to T cells with low avidity TCRs, as shear stress was increased from 1.9 to 19.2 Pa (Figure 4f; Figure S5c, Supporting Information). Interestingly, at the lowest shear stress (1.9 Pa) far fewer human primary T cells were detached compared to SUPT1 T cells (77.2 ± 6% and 47.6 ± 2% of high avidity and low avidity T cells remained bound to the tumor monolayer, respectively).
2.6. Primary T Cells with High Cellular Avidity Express Higher Levels of Immunological Synapse Markers and Cytokine Secretion
We next examined if differences in IS formation were observable in cells bound at different shear stresses. Previous studies revealed that the gap junction protein Connexin‐43 (Cx43) accumulates at the IS in an antigen‐dependent manner.[ 12 , 22 ] Representative images of the accumulation of Cx43 gap junction proteins and F‐actin at the IS under high (14.4 Pa), low (1.9 Pa), and no shear stress conditions using immunofluorescence staining with confocal microscopy are presented inFigure 5a. By measuring the mean fluorescence intensity of Cx43 at the synapse we found that antigen‐specific Pmel T cells formed a stronger IS with B16F10 tumor cells compared to WT T cells at 0 Pa (Figure 5b). Moreover, we found that Pmel T cells that remained attached to B16F10 cells after high shear stress expressed significantly higher levels of Cx43 accumulation, with 17% and 24% increases in fluorescent intensity compared to low and no shear conditions, respectively. Representative control images of immunofluorescently stained SUPT1, Pmel, and B16F10 cells can be found in Figure S10, Supporting Information.
Figure 5.
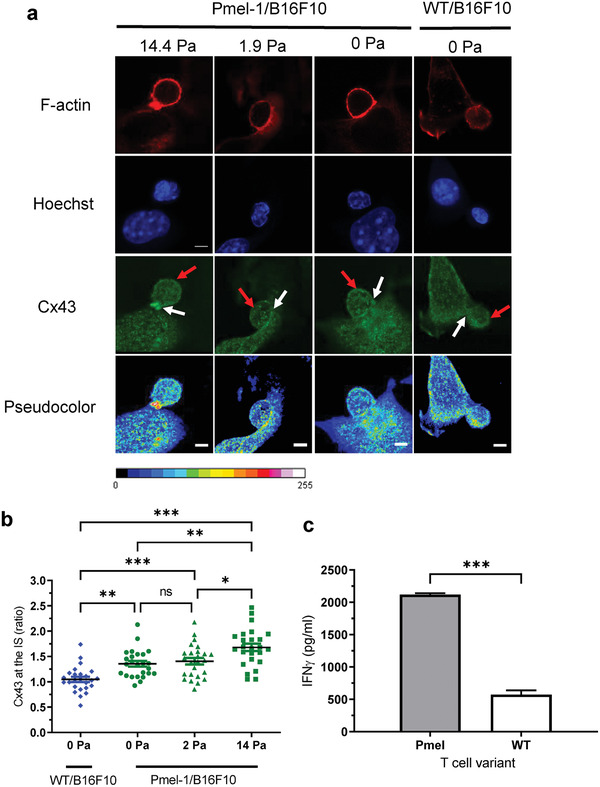
Primary T cells with high cellular avidity express higher levels of IS markers and cytokine secretion. a) Representative immunofluorescence images of F‐actin (red) and Cx43 (green and pseudocolor) at the IS of Pmel T cells (left) and WT T cells (right) remained adhered to the B16F10 melanoma monolayer under high (14.4 Pa), low (1.9 Pa), and no (0 Pa) shear stress conditions. White arrows indicate T cell/tumor cell synapse and red arrows indicate opposite site for Cx43 quantification. b) Quantification of the mean fluorescent intensity ratio of Cx43 accumulation at the IS (white arrows) to the opposite site (red arrows) after high, low, and no shear‐induced flow conditions, with 25 cell conjugates analyzed per condition. c) IFN‐γ secretion of T cell–tumor cell co‐culture media collected at the device outlet. Data are represented as mean ± SEM error bars, N = 3, p‐values are calculated using one‐way ANOVA with Bonferroni's multiple comparisons test (b) or a two‐tailed Student's t test (c), *p < 0.05,** p < 0.01, and *** p < 0.001.
Furthermore, IFN‐γ secretion levels were measured from media collected downstream from the outlet reservoirs of devices co‐cultured overnight with a 2:1 ratio of primary T cells to melanoma cells. Pmel T cells with high avidity to the gp100 antigen presented on B16F10 melanoma cells released significantly higher levels of IFN‐γ at 2120 ± 12 pg mL−1 compared to WT T cells at 573.3 ± 37 pg mL−1 (Figure 5c).
2.7. Recovered Primary Mouse T Cell Fractions with High Cellular Avidity Maintain Improved Cytotoxic, Activation, and Binding Functionalities
We then explored if T cells with superior anti‐tumor capabilities could be isolated with this technology by conducting multiple downstream functional analyses on collected T cell fractions (Figure 6a). Pmel and WT T cells were collected in three fractions based on their cellular avidity to the tumor monolayer (as described above): “strong” binders collected at 11.5–19.2 Pa, “medium” binders at 3.8–7.7 Pa, and “weak” binders at 1.9 Pa. Each fraction was examined for its ability to lyse B16F10 melanoma cells 8 h after collection using cytotoxicity measures. The number of lysed tumor cells correlated with the cellular avidity of T cells to gp100 positive tumor monolayers (Figure 6b). The strongest cellular avidity T cell fraction, collected at11.5–19.2 Pa, induced the most potent anti‐tumor response in vitro, with 31.2 ± 0.8% of tumor cells lysed. In contrast, significantly fewer tumor cells were lysed with medium and weak T cell fractions, which exhibited 14 ± 1.2% and 6.5 ± 1.1% cytotoxicity, respectively.
Figure 6.
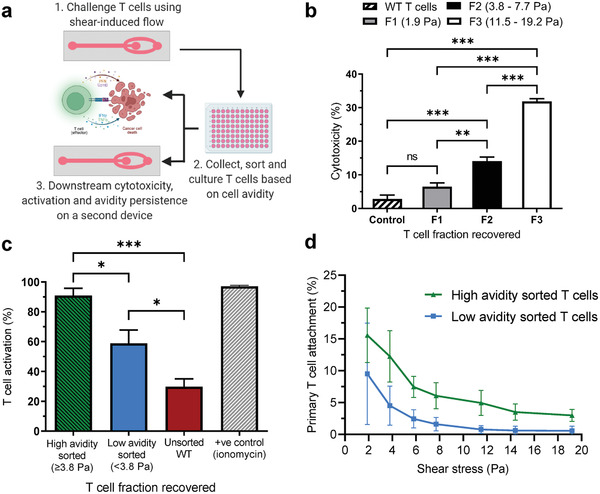
Persistent functionality of T cells after selection by cellular avidity. a) Schematic of T cell recovery and downstream functional analyses. b) Cytotoxicity of primary T cell fractions collected from the device outlet based on their cellular avidity when re‐exposed to B16F10 melanoma cells: F1 (1.9 Pa), F2 (3.8–7.7 Pa), and F3 (11.5–19.2 Pa). c) Quantification of T cell activation by calcium flux indicator from sorted and unsorted T cell fractions on B16F10 melanoma monolayers. d) Quantification of persistence of cellular avidity after second device challenge. Primary T cell binding to B16F10 monolayer devices from high and low avidity T cell fractions recovered under increasing shear stress. Data are represented as mean ± SEM error bars, N = 3, p‐values are calculated using one‐way or two‐way ANOVA with Bonferroni's multiple comparisons corrections: *p < 0.05,** p < 0.01, and *** p < 0.001.
To further explore persistence of the high avidity phenotype, we sorted T cells collected from devices into two fractions: those collected after application of “high” shear (≥3.8 Pa) or “low shear” (<3.8 Pa). Cells were cultured for 24 h in fresh medium at 37 °C/5% CO2 and then stained with a Fluo‐8 calcium flux indicator dye before being transferred onto new devices with fresh B16F10 tumor monolayers. Our results indicate that T cells recovered with high cellular avidities showed significantly greater activation (90.9 ± 4.9%) compared to T cells recovered with low avidities (58.9 ± 8.9%) and an unsorted (29.8 ± 5.3%) WT mixed population (Figure 6c). We then examined if the binding characteristics of primary T cells collected based on cellular avidity persisted when exposed to fresh tumor monolayers. Primary T cell fractions were sorted into high and low shear fractions as described above, cultured for 8 h, then differentially labeled with green and red calcein‐AM dyes, respectively, mixed together and fed into new devices coated with fresh tumor monolayers. High cellular avidity primary T cells from devices maintained significantly stronger avidity to fresh B16F10 tumor monolayers compared to their low cellular avidity counterparts at all tested shear challenges from 1.9 to 19.2 Pa (Figure 6d). These results validated that primary T cells selected based on higher cellular avidities maintained their phenotypic behaviors including cytotoxicity, T cell activation and binding after recovery and culture.
3. Discussion
The discovery of novel TCR T cell therapy relies on our ability to identify TCRs that provoke strong and specific responses against tumor cells. Currently deployed methods[ 9a,c ] for selecting TCR clonotypes based on the affinity of TCR and pMHC interactions often rely on acellular peptide conjugates and are therefore incapable of probing the physiological strength of the collective, non‐covalent intercellular interactions such as integrins and gap junctions proteins involved in the IS. Here we developed a novel microfluidic shear‐based method that is capable of identifying and recovering TCR T cell clones based on the concept of cellular avidity, the overall binding strengths between activated T cells and tumor cells. This technology identifies and recovers highly potent T cells with up to 100% purity from mixed populations of T cells within 30 min by simply varying applied shear stresses.
In comparison to methods that measure avidity using acellular peptides, our method of measuring cellular avidity allows us to assay avidity and physiological signaling events which may provide an opportunity to identify TCR T cell clones with greater chance of success in the clinic. First, the formation of intercellular adhesion complexes such as integrins or gap junction proteins that form as part of the IS contributes to greater levels of cellular avidity between T cells and tumor cells. Probing cellular avidity may therefore allow us to predict successful immune activation more reliably within the context of co‐stimulatory and co‐inhibitory factors that modulate T cell activation. Second, the use of living cells provides the opportunity to assay key moderators of T cell cytotoxic function simultaneous with measures of cellular avidity. One such moderator is IFNɣ, which enhances T cell cytotoxicity and whose secretion is dependent upon IS formation.[ 23 ] Furthermore, the microfluidic device designed in this study was capable of collecting detached T cells under known uniform shear stresses based on their avidity to tumor cells. This is a novel approach since previously reported shear‐induced microfluidic platforms do not probe T cell binding to tumor cell monolayers nor identify high activity T cells based on avidity of interactions.[ 18 , 24 ] Importantly, this feature enables downstream off‐device functional and genomic analyses to be conducted on collected cells such as cytotoxicity, activation, and the TCR sequencing to more rapidly identify optimal clones of interest.
Beyond the ability of this platform to isolate T cell clones based on cellular avidity and other indirect measures of functional activity, the superior optical properties and tailorability of microfluidic devices may permit the direct identification of T cell activation. By mounting our devices on a standard inverted microscope with incubated enclosure, we monitored thousands of live T cell and tumor cell interactions per device in real time. As a proof of concept, we examined if T cells exposed to tumor monolayers could activate a calcium flux response, a key step in initiating potent anti‐tumor responses.[ 25 ] In good agreement with previous studies,[ 9 , 26 ] we found that T cell activation was correlated with higher cellular avidity in an avidity‐dependent manner. The integration of methods such as surface acoustic waves with microfluidics[ 27 ] could be used to induce the local detachment of activated T cells on demand. Interestingly, we recorded background activation from a small proportion of T cells that do not express specific TCRs to antigens presented on melanoma cells, previously reported in several studies.[ 28 ] Finally, our platform could be used for other applications in the future such as validation of selected TCR T cell clones or evaluation of combinatorial immunotherapy with chemotherapies,[ 29 ] drug‐loaded lipid nanoparticles,[ 30 ] nanogels,[ 31 ] or others.
We successfully separated and recovered human SUPT1 and primary mouse T cells into fractions based on their relative cellular avidities. Notably, we found markers of cytotoxicity, T cell activation, and binding characteristics in primary T cells sorted by cellular avidity persisted after recovery and short term culture. Although T cell fractions collected under high shear were very pure, it is important to highlight that in our proof‐of‐concept device, small absolute numbers of T cells were recovered compared to the initial starting population. This was due to a large proportion of poorly adherent T cells detaching during the initial wash phase under low shear. One consequence of this is that it is currently challenging to recover sufficient numbers of T cells for use in the clinic with our current device. For instance, our collected primary T cells did not demonstrate appreciable levels of proliferation during culture for downstream analyses. We believe that this was because primary T cells require high cell densities to proliferate rapidly .[ 32 ]
To improve the number of recovered cells, we explored numerous parameters to improve the adhesion and separation characteristics of general T cell populations including modifications to media composition, calcium/salt concentrations, pre‐shear binding durations (Figure S6, Supporting Information), cell loading concentrations, pH, bovine serum albumin (BSA) to inhibit non‐specific binding (Figure S11a, Supporting Information), E‐cadherin blocking antibody (Figure S11b, Supporting Information), and different cell stains, none of which significantly improved bulk T cell binding (data not shown, unless stated). However, we did find that specific‐peptide pulsing of B16F10 cell monolayers increased the number of primary mouse T cells initially bound compared to non‐pulsed monolayers (Figure 4b; Figure S8, Supporting Information, respectively). This suggests that pMHC density may be an important optimization parameter for better recovery of high cellular avidity T cells since B16F10 melanoma cells exhibit a low density of MHC on its surface[ 33 ] rendering the antigen difficult to recognize by gp100+ CD8 T cells. Therefore, pulsing B16F10 cells with hgp100 peptides likely increased the density of MHC I presentation gp100 on the surface as previously reported.[ 34 ] We attempted peptide pulsing of NY‐ESO‐1+ monolayers for our human TCR experiments, but the peptide presentation compromised tumor cell monolayer adherence to the chip for flow experiments (data not shown). Nonetheless, we have demonstrated modulating antigen presentation may be one route to improving the recovery of high functional TCR T cells and with future optimization and parallelization of our method could dramatically improve the number of rare TCR T cells recovered downstream. This would enable further proliferative and phenotypic studies to be conducted by flow cytometry.
Although human primary T cells were successfully discriminated on‐chip, we did not conduct collection experiments due to the rare and limited supply of primary human T cells available. Interestingly, primary human CD8 T cells bound more strongly than the SUPT1 T cells to the NY‐ESO‐1+ melanoma monolayers. We believe that this behavior is due to the greater ability of primary human T cells to initiate late signaling events involved in immune activation such as IS formation in comparison to SUPT1 T cell lines which have impaired ability for this. On‐chip immunofluorescent stains show undetectable levels of Cx43 IS stain in SUPT1 T cells bound to tumor monolayers appears to support this assertion (Figure S10, Supporting Information). Future work should be conducted to isolate pure fractions of high avidity primary human T cells by applying higher shear stresses or reducing cell–cell interaction times before the initiation of shear challenge, to reduce IS formation resulting in lower overall binding strength between partners. Further refinement of this approach has the potential to isolate rare high avidity T cell clones from a pool of heterogeneous T cells extracted from cancer patients or healthy donors.
While melanoma antigens were used in this study, our platform can be easily adapted to investigate a wide range of tumors and antigens. Recently identified mutation‐derived neoantigens are only found on the surface of tumor cells and thus, may provide a safer route for effective T cell therapy.[ 35 ] Current strategies to find cells with optimal avidity to neoantigens is even more laborious than self‐antigens, as every patient expresses distinct neoantigens.[ 36 ] This platform could therefore potentially accelerate the identification of suitable patient‐specific neoantigen T cell candidates. Moreover, other immune cell types could be sorted based on their cellular avidity, such as CAR T cells that have shown a strong correlation between avidity and functional activity.[ 21a,b ] Thus, cell avidity analysis can provide critical preclinical information for a range of tumor antigen targets and cell therapies.
4. Conclusion
Microfluidic devices have the potential to improve current T cell selection strategies by rapidly identifying rare high avidity T cell clones for adoptive cell therapies in this shear stress‐based selection method. The device developed in this work was able to analyze thousands of cell–cell interactions in real‐time under precisely controlled environments using a cellular avidity approach. This provides a more physiological analysis of T cell—tumor cell interactions that incorporate key immune signaling events such as IS formation. The ability to rapidly identify and recover TCRs of optimal functional activity through cellular avidity may accelerate our expansion of immunotherapeutic strategies and move us closer toward precision cancer medicine.
5. Experimental Section
Reagents were obtained from Thermo Fisher Scientific (UK) unless stated otherwise.
Cell Culture
Me290 (HLA‐A2+/NY‐ESO‐1+) and NA8 (HLA‐A2+/NY‐ESO‐1−) human melanoma cells (RRID:CVCL_S597 and CVCL_S599, respectively) were derived from melanoma patients according to relevant regulatory standards and upon approval by the ethical commissions and regulatory agency of Switzerland (clinical trial NCT00112242). Patient recruitment, study procedures, and blood withdrawal were completed following written informed consent. SUPT1 human T cells (ATCC CRL_1942) were maintained in RPMI 1640 medium. B16F10 (gp100+) mouse melanoma cells were originally acquired from the American Type Culture Collection (ATCC; Manassas, VA, USA) and maintained in DMEM, high glucose, and GlutaMAX medium. All culture media was supplemented with 10% v/v FBS and 1% v/v penicillin/streptomycin. Human primary HLA A*0201pos CD8+ T lymphocytes were obtained following positive enrichment using anti CD8‐coated magnetic microbeads (Miltenyi Biotec), and cultured in RPMI supplemented with 8% human serum (HS) and 150 U mL−1 recombinant human IL‐2. All tumor cell lines were cultured at 37 °C/5% CO2 and passaged every 2–3 days using 0.25% trypsin‐EDTA and used at passage numbers 20 or lower. Cells were validated free of mycoplasma contamination monthly.
TCR Transduction
Full‐length codon‐optimized TCR (AV23.1/BV13.1) chain sequences of a dominant NY‐ESO‐1+specific T cell clone of patient LAU 155 were cloned in the pRRL third‐generation lentiviral vectors as an hPGK‐AV23.1‐IRESBV13.1 construct. Structure‐based amino acid substitutions were introduced into the WT TCR sequence, as previously described.[9c] This generated a panel of TCR‐transduced SUPT1 T cell variants of varying avidity to the NY‐ESO‐1+ antigen: NT (weak avidity), WT (intermediate avidity), and DMβ (high avidity). Primary human T cells were purified from a melanoma patient enrolled in clinical trial NCT00112242. The studies were designed and conducted according to the relevant regulatory standards, upon approval by the ethical commissions and regulatory agency of Switzerland. Patient recruitment, study procedures, and blood withdrawal were done upon written informed consent. Lentiviral production was performed using the standard calcium‐phosphate method and concentrated supernatant of lentiviral‐transfected 293T cells was used to infect TCRα knockout CD8+ SUPT1 T cells or primary bulk CD8 T cells. Levels of transduced TCR expression were measured using flow cytometry with PE‐labeled HLA‐A*0201/NY‐ESO‐1 specific multimers and FITC‐conjugated BV13.1 antibody (Beckman Coulter, USA).
Primary Mouse T Cell Culture and Activation
All the mouse studies were approved by the Swiss authorities (Canton of Vaud, animal protocol ID 3206 and 3533) and performed in accordance with guidelines from the Center of PhenoGenomics (CPG) in EPFL. 6–8 week‐old female Thy1.2+ C57BL/6 (C57BL/6J) mice were purchased from Charles River Laboratories (Lyon, France). TCR‐transgenic Thy1.1+ pmel‐1 (Pmel) mice (B6.Cg‐Thy1a/Cy Tg(TcraTcrb)8Rest/J) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained in the animal facility in the CPG in EPFL. Spleens from Pmel or C57BL/6J mice were mechanically disrupted and grounded through a 70‐µm strainer. Red blood cells were lysed with ACK lysis buffer (2 mL per spleen) for 5 min at 25 °C. The splenocytes were washed twice with phosphate‐buffered saline (PBS) which contained RPMI‐1640, FBS (10% v/v), HEPES (1% v/v), penicillin/streptomycin (1% v/v), and β‐mercaptoethanol (0.1% v/v). Splenocytes were then resuspended at a cell density of 2 × 106 cells mL−1 in complete RPMI medium supplemented with mouse IL‐2 (10 ng mL−1, PeproTech, UK) and IL‐7 (2 ng mL−1, PeproTech), as well as human gp100 (1 µm, GenScript) or an anti‐CD3/28 T cell activation kit (Miltenyi Biotec) for Pmel or C57BL/6J splenocytes, respectively. After 3‐day culture, live cells were enriched by density gradient centrifugation (Ficoll‐Paque PLUS, GE Healthcare, UK), followed by 2‐day culture at a cell density of 0.5–1.0 × 106 cells mL−1 in complete RPMI medium supplemented with mouse IL‐2 (10 ng mL−1) and IL‐7 (10 ng mL−1) to obtain activated CD8 + T cells with purity > 95% validated by flow cytometry (as described above). On day 6–7, activated T cells were frozen down in medium composed of 90% FBS, 10% DMSO at 10 m cells mL−1 and stored at −80 °C for at least 1 day before shipment in dry ice. Upon reception, cells were thawed and cultured in complete RPMI supplemented with mouse IL‐2 (10 ng mL−1) and IL‐7 (10 ng mL−1).
Microfluidic Device Design and Fabrication
Photolithographic masks were printed out on fine grain emulsion film (Micro Lithography Services, UK). Master molds were fabricated using standard soft photolithography techniques in a class 1000 cleanroom (Imperial College London, UK). SU‐8 (GM1070 series, Gersteltec, Switzerland) negative photoresist was spin coated onto 4‐inch diameter plasma treated silicon wafers (Siegert Wafer, Germany) at 950 rpm for 45 s to obtain 100 μm high features. Wafers were baked at 65 and 100 °C for 2 and 10 min, respectively. Resist‐coated wafers were then patterned using UV photolithography with a mask aligner (Karl Süss MJB‐3, Süss MicroTec, Germany) to define masters containing shear devices with a geometry of 1 × 0.1 × 50‐mm3 (width by height by length) coupled with 2‐mm wide fluidic inlets and outlets. The master wafer was completed through a series of post‐bakes at 65 and 100 °C for 2 and 10 min, respectively. This was followed by a 10 min SU‐8 development step using propylene glycol methyl ether acetate (PGMEA, Sigma Aldrich, UK) and a 10 min hard‐bake at 100 °C after washing with iso‐propyl alcohol. The SU‐8 features were verified to be within ± 10% of the target height set using a surface profilometer (AlphaStep 200, KLA‐Tencor, UK). Polydimethylsiloxane (PDMS, Dow Corning, UK) mixed with its cross‐linker at a 10:1 w/w was poured onto silicon master molds, degassed for 1 h using a vacuum desiccator, and cured at 65 °C for 24 h in an oven. Cured PDMS devices were removed from the molds using a scalpel and punched using a 2‐mm biopsy punch (Agar Scientific, UK) to obtain fluidic inlets and outlets. Punched PDMS devices and glass microscope slides were bonded together after plasma treatment (Emitech K1050X, Quorum Technologies Ltd, UK) at 300 mmTor O2 at 50 W for 1 min) and transferred to a hot plate for 10 min at 100 °C.
Melanoma Cell Seeding
Devices were handled using aseptic techniques and sterilized with 70% ethanol for 1 min followed by washing with sterile PBS. Devices were then coated with poly‐L‐lysine (0.01%, Sigma Aldrich) to improve Me290 melanoma attachment or fibronectin (10 µg mL−1, Sigma Aldrich) to improve NA8 and B16F10 cell adhesion. All coatings were left for 1 h at 37 °C/5% CO2 followed by 3× sterile PBS washes. Melanoma cells were detached from culture flasks (as described above) and 10 µL of cell suspensions concentrated to 4 × 106 cells mL−1 were fed into each device. Cells were permitted to adhere to devices for 15 min, followed by feeding with 500 μL of complete RMPI medium for overnight incubation (37 °C/5% CO2). Melanoma cells were cultured in the device for 1–2 days until ≈95% confluence. Devices were fed daily with fresh media. Adhesion experiments were conducted on melanoma cells stained with calcein‐AM (5 μ m, ThermoFisher) and Hoescht (1 μ m, ThermoFisher) for 40 min at 37 °C/5% CO2 followed by 3× PBS washes. Shear stress was induced by adjusting the flow rates of DMEM media infused into the device via TYGON inlet tubing (0.76 mm ID, VWR) using a programmable syringe pump (PHD ULTRA, Harvard Apparatus, UK). Flow rates were ramped up from 0 μL min−1 to the set experimental value for 5 s and kept at a constant flow for 50 s before ramping down to 0 μL min−1. Shear stresses examined were set as the following: 0, 1, 1.9, 3.8, 5.8, 7.7, 11.5, 14.4, and 19.2 Pa. Fluorescent and phase‐contrast images of the 1 × 50‐mm channel at a height of 3 μm were recorded using a 10× objective. All live‐cell imaging was performed on a Nikon Eclipse Ti2 Inverted Microscope (Nikon, Japan) with an incubated humidified chamber (Okolab, Italy) at 37 °C/5% CO2. The microscope was attached with a monochrome Nikon DS‐Qi2 camera and LED illuminator for phase‐contrast and fluorescent imaging. Filters were set as the following: DAPI (excitation 358 nm/emission 461 nm), GFP (excitation 488 nm/emission 509 nm), and RFP (excitation 558 nm/emission 583 nm).
Melanoma Cell Viability
Devices with a confluent monolayer (≈95%) of melanoma cells were stained with PI (1 μ m, ThermoFisher), a red‐fluorescent nuclear and chromosome counterstain used to identify dead cells. Hoescht solution (1 μ m) was used as a nuclear stain to determine total percent viability. Monolayers were transferred to a fluorescent microscope and attached to a syringe‐driven pump via the inlet reservoir. Staining media was loaded into the syringe and shear stresses from 0 to 19.2 Pa were tested by adjusting input flow rates. The monolayer experienced shear‐induced flow for 1 min per flow rate examined and images were acquired after each flow rate tested. Control experiments used a cell lysis solution (10×, Promega Corporation) infused into melanoma coated devices to validate the working mechanism of the dye. Thresholding and size constraint analysis were used to quantify percent cell viability as a function of shear stress.
T Cell Selection by Cellular Avidity
SUPT1 human T cells, patient‐derived transduced T cells and primary mouse T cells were labeled with calcein‐AM (5 μ m, 1 × 106 cells mL−1) for 1 h at 37 °C/5% CO2 followed by 3× PBS washes. SUPT1 DMβ, primary Pmel, and high avidity patient‐derived T cells were stained with calcein green; SUPT1 WT, primary mouse WT T cells, and low avidity human primary T cells were stained with calcein red; SUPT1 NT were stained with calcein blue. The devices containing attached melanoma cells were transferred to the microscope stage and incubated (37 °C/5% CO2) with 1% BSA (Sigma Aldrich) for 1 h to block nonspecific T‐cell adhesion. SUPT1 T cell experiments were initiated by mixing DMβ:WT:NT T cells at 1:1:1 ratios before infusion into Me290 (NY‐ESO‐1+) or NA8 (NY‐ESO‐1−) coated devices using a pipette tip followed by inserting inlet and outlet tubing (0.76 mm ID, VWR) connected to the syringe pump and collection wells, respectively. For primary mouse experiments, B16F10 melanoma cells were treated with 1 µm hgp100 peptide (GenScript, USA) 30 min prior to T cell infusion. Pmel:WT T cells were mixed at 1:1 or 1:10 ratios (Figure S7, Supporting Information) and fed into the device using the same method described previously. T cells were left to attach to melanoma cells for 10 min under static conditions. T cell detachment and collection was examined under shear‐induced flow in the range of 0–19.2 Pa using a programmable syringe pump, as previously described. For recovery experiments, primary mouse T cells were collected from devices into fractions with “high” (≥3.8) and “low” avidities (<3.8 Pa) and their cellular avidity persistence was examined. High and low avidity T cell fractions were recovered from multiple devices under shear flow, cultured for 8 h at 37 °C/5% CO2, and stained with calcein green and red, respectively. These T cells fractions were then mixed and fed into new devices seeded with fresh B16F10 tumor monolayers and cellular avidity was assessed under shear flow, as described above. Fluorescent and phase‐contrast images were recorded after each shear stress tested using the large‐image acquisition feature on NIS‐Elements Advanced software (Nikon). Device images and collection wells were captured with a scan area of 25 × 2 fields and 4 × 4 fields, respectively, and stitched together with 15% overlap. Collection wells were transferred to an incubator where T cells were left to proliferate overnight for further downstream analyses.
T Cell Activation Experiments
SUPT1 human T cells were stained using a Fluo‐8 calcium flux assay kit (5 μ m, Thermo Fisher Scientific) in serum free DMEM media for 60 min at 37 °C/5% CO2 followed by 3× PBS washes. SUPT1 T cells were resuspended in complete DMEM media and 10 µL of cell suspension was fed into the device coated with confluent monolayers of Me290 or NA8 under the microscope (37 °C/5% CO2). For recovery experiments, T cell fractions with high and low avidities were collected from devices, as described previously. Recovered T cell fractions sorted based on cell avidity and an unsorted WT T cell mixed population were then stained using a Fluo‐8 calcium binding dye and fed into new devices with B16F10 tumor monolayers under the microscope. Positive control tests were conducted using ionomycin (1 µm, Sigma Aldrich). Fluorescent and phase‐contrast time‐lapse images were captured at the middle of the channel with a 1 × 1 mm2 field of view and taken every 2 s for 10 min under static conditions.
IFN‐γ Secretion
Fibronectin pre‐treated microfluidic devices were seeded with 12 500 B16F10 melanoma cells and left to attach for 1 h at 37 °C/5%CO2 followed by 1 µm hgp100 peptide simulation 30 min prior to T cell introduction. 25 000 Pmel or WT T cells (2:1, effector:target ratio) were suspended in modified RPMI media supplemented with IL‐2 and IL‐7 and introduced into the device. The co‐cultured devices were incubated overnight and conditioned media was collected after 24 h from the outlet reservoir. The collected media was centrifuged and 50 µL of the cell‐free supernatants were removed for analysis. The IFN‐ γ levels were evaluated by using a highly sensitive mouse IFN‐ γ specific enzyme‐linked immunosorbent assay kit (LEGEND MAX, BioLegened, UK) according to the manufacturer's instructions. Absorbance values for standards and samples were recorded at 450 nm using a microplate reader (Infinite F50, Tecan, Switzerland).
T Cell Cytotoxic Functionality
Primary mouse T cells collected in wells from shear stress experiments were split into 3‐fractions based on cellular avidity to B16F10 melanoma cells: strong collected at 11.5–19.2 Pa, medium at 3.8–7.7 Pa, and weak at 1.9 Pa. Each fraction was tested for its ability to lyse melanoma cells using a non‐radioactive cytotoxicity assay (CytoTox 96 kit, Promega Corporation) by the release of lactate dehydrogenase (LDH) and its conversion into red formazan product. B16F10 tumor cells were incubated at 2000 cells per well in non‐treated 96‐well round bottom plates with T cell fractions collected from the shear device product outlet. Each T cell fraction was diluted to 50 cells per well to match the limited number of T cells collected under the highest shear fraction (11.5–19.2 Pa). The plate was then incubated for 8 h at 37 °C/5% CO2. Spontaneous release of LDH in T cells and tumor cells were measured as well as the culture media background in separate control wells. All samples were run in triplicate and made up to a total volume of 100 μL per well in phenol red‐free RPMI supplemented with 10% FBS, 1% penicillin/streptomycin, and 10 ng mL−1 IL‐2 and IL‐7. The maximum LDH release by tumor cells was measured by adding 10 μL of lysis solution (10×) to yield complete lysis. At the end of the incubation period, the well plate was centrifuged and 50 μL of supernatant from each well was transferred into a fresh flat bottom 96‐well plate and mixed with 50 μL per well of LDH CytoTox 96 reagent. The plate was light protected and incubated at room temperature for additional 30 min, and the reaction was stopped by the addition of the stop solution. Absorbance data at 492 nm was measured using a 96‐well plate reader (Infinite F50, Tecan) alongside T cell‐free and melanoma‐free controls. The percentage of specific lysis was calculated using Equation (1):
| (1) |
Immunofluorescence Staining
Microfluidic devices were plasma bonded to glass cover slips (25 × 60‐mm). Devices were treated with 70% ethanol followed by 3× PBS washes and incubated for 1 h with fibronectin (10 µg mL−1). 12 500 B16F10 melanoma cells were seeded into the device and left to adhere for 1 h at 37 °C/5% CO2 followed by treatment with 1 µm hgp100 peptide 30 min prior to T cell introduction. Primary mouse Pmel or WT T cells were fed into the device at a 2:1 ratio (effector:target cells) and left to bind to the melanoma monolayer for 10 min. High (14.4 Pa), low (1.9 Pa), and no (0 Pa) shear stress conditions were induced using a programmable syringe pump. Cell conjugates were fixed with 4% paraformaldehyde for 20 min, blocked, and permeabilized with 1% BSA and 0.5% saponin (all Sigma Aldrich). Cells were then incubated with primary antibodies (Anti‐Connexin 43/GJA1 antibody, abcam, UK) 1/200 diluted in 1% BSA in PBST (PBS + 0.1% Tween 20) overnight at 4 °C, then washed 3× with PBS. Cells were then treated with secondary antibodies (Goat Anti‐Rabbit IgG, Alexa Fluor 488, abcam) and Phalloidin (Phalloidin‐iFluor 594 Reagent, abcam) 1/200 diluted in 1% BSA for 1 h in the dark. The IS for 20 cell conjugates per condition were captured using a confocal microscope (SP8, Leica). Immunofluorescently stained control samples were observed using DMβ T cells tumor adhered to Me290 and B16F10 monolayers, which showed no Cx43 accumulation at the synapse (Figure S10, Supporting Information)
Image Processing
NIS‐Elements Advanced software (Nikon) was used for image processing and analysis. Large‐image acquisitions of devices were cropped to 25 × 0.8 mm2 (length by width) so that cells located 0.1 mm either side of the channel walls were excluded from the analysis due to experiencing non‐uniform shear stress. The object count tool was used to quantify T cells based on thresholding with applied size and circularity constraints. Fluid dynamic simulation of shear stress in the device was modeled using laminar flow physics on COMSOL 5.3 (COMSOL Inc., Sweden) and the Navier–Stokes equation describing the motion of Newtonian fluids. In each simulation, a no slip boundary condition was used and inlet velocities were inputted based on experimental flow rates tested. For Newtonian fluids, shear stress was proportional to the gradient of velocity in the direction perpendicular to flow, calculated using Equation (2).
| (2) |
where τ is the shear stress in Pa, μ is the dynamic viscosity in Pa s, u is the velocity of the fluid along the boundary in m s−1, and is the shear rate (i.e., velocity gradient).
Shear stress plots were measured at a z‐plane of 4 μm across the channel width (x‐plane) to represent the height of the T cell to tumor cell interaction across the device. Activation was analyzed using a custom MATLAB (MathWorks, USA) script to determine active versus inactive T cells. The criterion for an active T cell was characterized by the standard deviation divided by the mean intensity using a threshold value of 0.2. The accumulation of Cx43 at the synapse was quantified using ImageJ software by measuring the ratio of the mean grey values at the synapse and opposite end of the T cell. BioRender.com (BioRender, Canada) was used to create figures. Prism software (version 9.0, GraphPad Software Inc., USA) was used to present graphical data.
Statistical Analysis
All data were presented as mean ± standard error of the mean (SEM) unless otherwise stated. Experiments were performed independently and repeated three times for each condition. Statistical analyses were performed using GraphPad Prism software. Two‐tailed Student's t‐test, or one‐way or two‐way analysis of variance (ANOVA) was used followed by Bonferroni's multiple comparisons corrections for experiments with multiple groups as indicated in the figure legends. All tests were two‐sided unless otherwise specified. Significance was considered for p < 0.05 and figures were annotated with the following: single asterisk show *p < 0.05, double asterisk show ** p < 0.01, triple asterisk show *** p < 0.001.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
J.F.A. developed devices, conducted experiments, and prepared figures. S.H.A. conceptualized the idea for TCR T cell selection devices and helped plan experiments. N.K.C. conducted primary human T cell and rebinding experiments. J.S., L.T., A.H., and A.K. provided transduced T cells and project support. C.K. assisted with activation experiments and development of MATLAB code. J.F.A. and S.H.A. wrote and all authors contributed to editing the manuscript.
Supporting information
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Supplemental Movie 3
Supplemental Movie 4
Supplemental Movie 5
Supplemental Movie 6
Acknowledgements
The authors are grateful to Nils Kling for assistance with developing MATLAB code for activation analysis, Miguel Hermida Ayala for cell culture support, and Imperial College for research funding.
Ashby J. F., Schmidt J., KC N., Kurum A., Koch C., Harari A., Tang L., Au S. H., Microfluidic T Cell Selection by Cellular Avidity. Adv. Healthcare Mater. 2022, 11, 2200169. 10.1002/adhm.202200169
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.a) Hebeisen M., Allard M., Gannon P. O., Schmidt J., Speiser D. E., Rufer N., Front. Immunol. 2015, 6, 582; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rosenberg S. A., Restifo N. P., Science 2015, 348, 62, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Morgan R. A., Dudley M. E., Wunderlich J. R., Hughes M. S., Yang J. C., Sherry R. M., Royal R. E., Topalian S. L., Kammula U. S., Restifo N. P., Science 2006, 314, 126; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Robbins P. F., Morgan R. A., Feldman S. A., Yang J. C., Sherry R. M., Dudley M. E., Wunderlich J. R., Nahvi A. V., Helman L. J., Mackall C. L., Kammula U. S., Hughes M. S., Restifo N. P., Raffeld M., Lee C. C., Levy C. L., Li Y. F., El‐Gamil M., Schwarz S. L., Laurencot C., Rosenberg S. A., J. Clin. Oncol. 2011, 29, 917; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Robbins P. F., Kassim S. H., Tran T. L., Crystal J. S., Morgan R. A., Feldman S. A., Yang J. C., Dudley M. E., Wunderlich J. R., Sherry R. M., Kammula U. S., Hughes M. S., Restifo N. P., Raffeld M., Lee C. C., Li Y. F., El‐Gamil M., Rosenberg S. A., Clin. Cancer Res. 2015, 21, 1019, ; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Johnson L. A., Morgan R. A., Dudley M. E., Cassard L., Yang J. C., Hughes M. S., Kammula U. S., Royal R. E., Sherry R. M., Wunderlich J. R., Lee C. C., Restifo N. P., Schwarz S. L., Cogdill A. P., Bishop R. J., Kim H., Brewer C. C., Rudy S. F., VanWaes C., Davis J. L., Mathur A., Ripley R. T., Nathan D. A., Laurencot C. M., Rosenberg S. A., Blood 2009, 114, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rapoport A. P., Stadtmauer E. A., Binder‐Scholl G. K., Goloubeva O., Vogl D. T., Lacey S. F., Badros A. Z., Garfall A., Weiss B., Finklestein J., Kulikovskaya I., Sinha S. K., Kronsberg S., Gupta M., Bond S., Melchiori L., Brewer J. E., Bennett A. D., Gerry A. B., Pumphrey N. J., Williams D., Tayton‐Martin H. K., Ribeiro L., Holdich T., Yanovich S., Hardy N., Yared J., Kerr N., Philip S., Westphal S., et al., Nat. Med. 2015, 21, 914, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Laydon D. J., Bangham C. R., Asquith B., Philos. Trans. R. Soc., B 2015, 370, 1675, [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Warren R. L., Freeman J. D., Zeng T., Choe G., Munro S., Moore R., Webb J. R., Holt R. A., Genome Res. 2011, 21, 790, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang S.‐Q., Parker P., Ma K.‐Y., He C., Shi Q., Cui Z., Williams C. M., Wendel B. S., Meriwether A. I., Salazar M. A., Sci. Transl. Med. 2016, 8, 341ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Kaiser A. D., Assenmacher M., Schroder B., Meyer M., Orentas R., Bethke U., Dropulic B., Cancer Gene Ther. 2015, 22, 72; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Allard M., Couturaud B., Carretero‐Iglesia L., Duong M. N., Schmidt J., Monnot G. C., Romero P., Speiser D. E., Hebeisen M., Rufer N., JCI Insight 2017, 2, e92570; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) June C. H., Riddell S. R., Schumacher T. N., Sci. Transl. Med. 2015, 7, 280ps7; [DOI] [PubMed] [Google Scholar]; d) Wouters O. J., McKee M., Luyten J., JAMA, J. Am. Med. Assoc. 2020, 323, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Schmidt J., Dojcinovic D., Guillaume P., Luescher I., Front. Immunol. 2013, 4, 218; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vigano S., Utzschneider D. T., Perreau M., Pantaleo G., Zehn D., Harari A., Clin. Dev. Immunol. 2012, 2012, 153863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sibener L. V., Fernandes R. A., Kolawole E. M., Carbone C. B., Liu F., McAffee D., Birnbaum M. E., Yang X., Su L. F., Yu W., Dong S., Gee M. H., Jude K. M., Davis M. M., Groves J. T., Goddard W. A. 3rd, Heath J. R., Evavold B. D., Vale R. D., Garcia K. C., Cell 2018, 174, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Hebeisen M., Schmidt J., Guillaume P., Baumgaertner P., Speiser D. E., Luescher I., Rufer N., Cancer Res. 2015, 75, 1983; [DOI] [PubMed] [Google Scholar]; b) Gannon P. O., Wieckowski S., Baumgaertner P., Hebeisen M., Allard M., Speiser D. E., Rufer N., J. Immunol. 2015, 195, 356; [DOI] [PubMed] [Google Scholar]; c) Irving M., Zoete V., Hebeisen M., Schmid D., Baumgartner P., Guillaume P., Romero P., Speiser D., Luescher I., Rufer N., Michielin O., J. Biol. Chem. 2012, 287, 23068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dolton G., Lissina A., Skowera A., Ladell K., Tungatt K., Jones E., Kronenberg‐Versteeg D., Akpovwa H., Pentier J. M., Holland C. J., Godkin A. J., Cole D. K., Neller M. A., Miles J. J., Price D. A., Peakman M., Sewell A. K., Clin. Exp. Immunol. 2014, 177, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dimitrov S., Gouttefangeas C., Besedovsky L., Jensen A. T. R., Chandran P. A., Rusch E., Businger R., Schindler M., Lange T., Born J., Rammensee H. G., Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Tittarelli A., Navarrete M., Gleisner M. A., Gebicke‐Haerter P., Salazar‐Onfray F., Int. J. Mol. Sci. 2020, 21, 3736; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hofmann F., Navarrete M., Alvarez J., Guerrero I., Gleisner M. A., Tittarelli A., Salazar‐Onfray F., Int. J. Mol. Sci. 2019, 20, 4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Bashour K. T., Gondarenko A., Chen H., Shen K., Liu X., Huse M., Hone J. C., Kam L. C., Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 2241; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Skanland S. S., Moltu K., Berge T., Aandahl E. M., Tasken K., Biochem. J. 2014, 460, 399. [DOI] [PubMed] [Google Scholar]
- 14. Lee K.‐H., Dinner A. R., Tu C., Campi G., Raychaudhuri S., Varma R., Sims T. N., Burack W. R., Wu H., Wang J., Science 2003, 302, 1218. [DOI] [PubMed] [Google Scholar]
- 15. Dustin M. L., Cancer Immunol. Res. 2014, 2, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Schmid D. A., Irving M. B., Posevitz V., Hebeisen M., Posevitz‐Fejfar A., Sarria J. C., Gomez‐Eerland R., Thome M., Schumacher T. N., Romero P., Speiser D. E., Zoete V., Michielin O., Rufer N., J. Immunol. 2010, 184, 4936; [DOI] [PubMed] [Google Scholar]; b) Dutoit V., Rubio‐Godoy V., Dietrich P.‐Y., Quiqueres A.‐L., Schnuriger V., Rimoldi D., Liénard D., Speiser D., Guillaume P., Batard P., Cancer Res. 2001, 61, 5850. [PubMed] [Google Scholar]
- 17.a) Zhong S., Malecek K., Johnson L. A., Yu Z., Vega‐Saenz de Miera E., Darvishian F., McGary K., Huang K., Boyer J., Corse E., Shao Y., Rosenberg S. A., Restifo N. P., Osman I., Krogsgaard M., Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 6973; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Overwijk W. W., Theoret M. R., Finkelstein S. E., Surman D. R., de Jong L. A., Vyth‐Dreese F. A., Dellemijn T. A., Antony P. A., Spiess P. J., Palmer D. C., Heimann D. M., Klebanoff C. A., Yu Z., Hwang L. N., Feigenbaum L., Kruisbeek A. M., Rosenberg S. A., Restifo N. P., J. Exp. Med. 2003, 198, 569; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yee C., Thompson J. A., Roche P., Byrd D. R., Lee P. P., Piepkorn M., Kenyon K., Davis M. M., Riddell S. R., Greenberg P. D., J. Exp. Med. 2000, 192, 1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Sarkar S., Sabhachandani P., Stroopinsky D., Palmer K., Cohen N., Rosenblatt J., Avigan D., Konry T., Biomicrofluidics 2016, 10, 054115; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sarkar S., Sabhachandani P., Ravi D., Potdar S., Purvey S., Beheshti A., Evens A. M., Konry T., Front. Immunol. 2017, 8, 1736; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guo F., Li P., French J. B., Mao Z., Zhao H., Li S., Nama N., Fick J. R., Benkovic S. J., Huang T. J., Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 43; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pavesi A., Tan A. T., Koh S., Chia A., Colombo M., Antonecchia E., Miccolis C., Ceccarello E., Adriani G., Raimondi M. T., Kamm R. D., Bertoletti A., JCI Insight 2017, 2, e89762; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sarkar S., Motwani V., Sabhachandani P., Cohen N., Konry T., J. Clin. Cell. Immunol. 2015, 6, 334; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ma C., Fan R., Ahmad H., Shi Q., Comin‐Anduix B., Chodon T., Koya R. C., Liu C.‐C., Kwong G. A., Radu C. G., Ribas A., Heath J. R., Nat. Med. 2011, 17, 738; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Stockslager M. A., Bagnall J. S., Hecht V. C., Hu K., Aranda‐Michel E., Payer K., Kimmerling R. J., Manalis S. R., Biomicrofluidics 2017, 11, 064103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segaliny A. I., Li G., Kong L., Ren C., Chen X., Wang J. K., Baltimore D., Wu G., Zhao W., Lab Chip 2018, 18, 3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamsma D., Bochet P., Oswald F., Alblas N., Goyard S., Wuite G. J. L., Peterman E. J. G., Rose T., Cell Rep. 2018, 24, 3008. [DOI] [PubMed] [Google Scholar]
- 21.a) de Larrea C. F., Staehr M., Lopez A. V., Ng K. Y., Chen Y., Godfrey W. D., Purdon T. J., Ponomarev V., Wendel H.‐G., Brentjens R. J., Blood Cancer Discovery 2020, 1, 146; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Katsarou A., Sjöstrand M., Naik J., Mansilla‐Soto J., Kefala D., Kladis G., Nianias A., Ruiter R., Poels R., Sarkar I., Sci. Transl. Med. 2021, 13, eabh1962; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Larson R. C., Kann M. C., Bailey S. R., Haradhvala N. J., Llopis P. M., Bouffard A. A., Scarfó I., Leick M. B., Grauwet K., Berger T. R., Stewart K., Anekal P. V., Jan M., Joung J., Schmidts A., Ouspenskaia T., Law T., Regev A., Getz G., Maus M. V., Nature 2022, 604, 563; [DOI] [PubMed] [Google Scholar]; d) Halim L., Das K., Larcombe‐Young D., Ajina A., Candelli A., Benjamin R., Dillon R., Davies D., Maher J., Front. Immunol. 2022, 13, 836549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Tittarelli A., Mendoza‐Naranjo A., Farías M., Guerrero I., Ihara F., Wennerberg E., Riquelme S., Gleisner A., Kalergis A., Lundqvist A., J. Immunol. 2014, 192, 1313; [DOI] [PubMed] [Google Scholar]; b) Mendoza‐Naranjo A., Bouma G., Pereda C., Ramírez M., Webb K. F., Tittarelli A., López M. N., Kalergis A. M., Thrasher A. J., Becker D. L., J. Immunol. 2011, 187, 3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhat P., Leggatt G., Waterhouse N., Frazer I. H., Cell Death Dis. 2017, 8, e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Kusunose J., Zhang H., Gagnon M. K. J., Pan T., Simon S. I., Ferrara K. W., Ann. Biomed. Eng. 2013, 41, 89; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu H., Koo L. Y., Wang W. M., Lauffenburger D. A., Griffith L. G., Jensen K. F., Anal. Chem. 2004, 76, 5257; [DOI] [PubMed] [Google Scholar]; c) Dura B., Dougan S. K., Barisa M., Hoehl M. M., Lo C. T., Ploegh H. L., Voldman J., Nat. Commun. 2015, 6, 5940. [DOI] [PubMed] [Google Scholar]
- 25. Anikeeva N., Grosso D., Flomenberg N., Sykulev Y., Nat. Commun. 2016, 7, 13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen J. L., Morgan A. J., Stewart‐Jones G., Shepherd D., Bossi G., Wooldridge L., Hutchinson S. L., Sewell A. K., Griffiths G. M., van der Merwe P. A., Jones E. Y., Galione A., Cerundolo V., J. Immunol. 2010, 184, 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bussonnière A., Miron Y., Baudoin M., Matar O. B., Grandbois M., Charette P., Renaudin A., Lab Chip 2014, 14, 3556. [DOI] [PubMed] [Google Scholar]
- 28.a) Ramanathan S., Gagnon J., Ilangumaran S., Arch. Immunol. Ther. Exp. 2008, 56, 311; [DOI] [PubMed] [Google Scholar]; b) Monjazeb A. M., Hsiao H.‐H., Sckisel G. D., Murphy W. J., J. Immunotoxicol. 2012, 9, 248; [DOI] [PubMed] [Google Scholar]; c) Alves N. L., Hooibrink B., Arosa F. A., van Lier R. A., Blood 2003, 102, 2541. [DOI] [PubMed] [Google Scholar]
- 29. Rosenberg S. A., Restifo N. P., Yang J. C., Morgan R. A., Dudley M. E., Nat. Rev. Cancer 2008, 8, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng Y., Tang L., Mabardi L., Kumari S., Irvine D. J., ACS Nano 2017, 11, 3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang L., Zheng Y., Melo M. B., Mabardi L., Castano A. P., Xie Y. Q., Li N., Kudchodkar S. B., Wong H. C., Jeng E. K., Maus M. V., Irvine D. J., Nat. Biotechnol. 2018, 36, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a) Ma Q., Wang Y., Lo A. S.‐Y., Gomes E. M., Junghans R. P., J. Biomed. Biotechnol. 2010, 2010, 386545 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ghaffari S., Torabi‐Rahvar M., Aghayan S., Jabbarpour Z., Moradzadeh K., Omidkhoda A., Ahmadbeigi N., BMC Immunol. 2021, 22, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fujiwara S., Nagai H., Shimoura N., Oniki S., Yoshimoto T., Nishigori C., J. Invest. Dermatol. 2014, 134, 1884. [DOI] [PubMed] [Google Scholar]
- 34. Overwijk W. W., Restifo N. P., Curr. Protoc. Immunol. 2000, 39, 20.1. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Kunert A., Straetemans T., Govers C., Lamers C., Mathijssen R., Sleijfer S., Debets R., Front. Immunol. 2013, 4, 363; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schumacher T. N., Schreiber R. D., Science 2015, 348, 69. [DOI] [PubMed] [Google Scholar]
- 36. Garber K., Nat. Biotechnol. 2018, 36, 215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Supplemental Movie 3
Supplemental Movie 4
Supplemental Movie 5
Supplemental Movie 6
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.