

Abstract
Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) share many cellular and molecular features with cancer cells. Taking advantage of these similarities, stem cells are effective vaccines against cancers in animal models. However, the molecular basis is not well understood, which hinders the development of effective cancer vaccines. Here, prophylactic and therapeutic bladder cancer vaccines composed of allogeneic ESCs and CpG with or without granulocyte macrophage colony stimulating factor are tested. The ESC‐based cancer vaccines are able to induce specific antitumor immunity including stimulating cytotoxic CD8+ T cells and memory CD4+ T cells, reducing myeloid‐derived suppressor cells, and preventing bladder cancer growth in mouse models. Furthermore, several genes that are overexpressed in both ESCs and tumors are identified. An epitope‐based vaccine designed with shared overexpressed proteins induces specific antitumor immunity and reduces bladder cancer growth. Functional epitopes underlying the action of stem cell‐based vaccines against bladder cancer are identified and it is confirmed that ESC‐based anticancer vaccines have great potential. A systematic approach is provided here to developing novel effective epitope‐based cancer vaccines in the future.
Keywords: embryonic stem cells, epitopes, tumor immunity, vaccines
Embryonic stem cell (ESC)‐based vaccines stimulate cytotoxic T‐cell immunity and prevent bladder cancer growth. A platform is established to systematically identify the shared tumor‐associated antigens and CD8+ T cell epitopes expressed in ESCs and tumor cells. The antitumor effects of these peptides are validated which may be used for further cancer therapy.
1. Introduction
Bladder cancer is one of the top ten most common cancers worldwide, with ≈550 000 new cases annually.[ 1 ] The most common type of bladder cancer is nonmuscle invasive bladder cancer (NMIBC), accounting for 70% of cases. Almost 25–75% of high‐risk NMIBCs progress to muscle‐invasive cancer and metastatic cancer due to poor treatment.[ 2 ] In the past, the treatment of bladder cancer was limited to surgery and chemotherapy. However, recent extensive analysis of the molecular features in bladder cancer has led to novel therapeutic treatments. Here, we identified several tumor‐associated antigens (TAAs) expressed by embryonic stem cells (ESCs) that can effectively inhibit bladder cancer growth.
Both ESCs and iPSCs share many genetic and molecular features with cancer cells.[ 3 ] A recent study analyzing and comparing RNA sequencing (RNA‐seq) data showed many upregulated genes in both cancer cell lines and iPSC cell lines.[ 4 ] These cancer‐related genes were expressed at high levels in ESCs and iPSCs, but only at low levels in somatic tissues. Mice preimmunized with irradiated iPSCs or ESCs induced tumor antigen‐specific T cells that showed antitumor effects.[ 4 , 5 ] The TAAs expressed by ESCs or iPSCs contain potential major histocompatibility complex (MHC) class I or class II epitopes, which could be used to develop therapeutic cancer vaccines. Vaccines based on oncogenic proteins or epitopes have shown remarkable success in preventing cancers by stimulating the host immune system.[ 6 ] There have been numerous clinical studies on patients immunized against TAAs from pluripotent cells, including oncofetal peptide, cancer‐testis antigen, melanoma‐specific antigen 3, Glypican‐3, and Mucin‐1, which were shown to have no side effects.[ 3 , 7 ] Studies in transgenic mouse models have demonstrated that multiantigen vaccines are significantly more effective than single‐antigen vaccines in inhibiting the progression of cancers.[ 7 ] Therefore, the identification and selection of effective TAAs and the prediction of potential epitopes that can stimulate a strong T‐cell‐associated immune response would be valuable in the treatment of bladder cancer.
We developed a strategy that uses predictive immune‐informatics algorithms to predict potential TAAs and epitopes expressed in ESCs based on their antitumor effects. To identify effective TAAs and epitopes, we focused on two major characteristics of the protein candidates, namely 1) the shared overexpression of proteins in both cancers and ESCs compared to normal tissues and 2) their cellular functions. Aberrant overexpression of a protein in the malignant state is a major mechanism by which that protein becomes immunogenic.[ 8 ] Moreover, the protein's function in the cell, including cell growth, cell cycle, signaling transduction, or cell motility,[ 9 ] is important, as the expression of a protein could promote cancer pathogenesis and negatively influence the prognosis.
In the current study, we demonstrated the antitumor effects of ESCs in bladder cancer and analyzed changes in the immune cell profiles. We identified that the therapeutic effects relied on the induction of T cell‐specific immunity, CD8+ effector, and memory T cell responses and decreased MDSCs. We identified potential TAAs and effective epitopes in ESCs and demonstrated the antigenicity of cancer‐associated proteins (CCNB1, ANLN, ECT2, and MELK). We further validated the T cell epitopes as candidates for therapeutic vaccines by designing vaccines based on these antigens, which consistently inhibited tumor development in a bladder cancer mouse model.
2. Results
2.1. ESC‐Based Vaccine Inhibits Bladder Cancer Growth
We used two ESC (129 and C57) cell lines as the allogeneic whole‐cell vaccines. The ESC (129) cell line shares the same MHC expression type with C57BL/6 mice. For the prophylactic model, the ESC‐based vaccine consisted of irradiated ESCs (129) and an immune adjuvant CpG‐ODN. We primed C57BL/6 mice with the ESC‐based vaccine for 4 weeks. The mice were then injected subcutaneously with 5 × 104 MB49 cancer cells and the subsequent tumor size was measured by calipers every 3 d (Figure 1a). The ESC‐based vaccine effectively inhibited bladder cancer growth (Figure 1b,c). For the therapeutic model, the ESC‐based vaccine contained CpG‐ODN and an immune mediator granulocyte macrophage‐colony stimulating factor (GM‐CSF). The adjuvant GM‐CSF is beneficial in cancer therapies, as it stimulates and activates antigen‐presenting cells (APCs) that can process and present tumor antigens to CD4+ helper T cells and CD8+ cytotoxic T lymphocytes.[ 10 ] Here, we primed the C57BL/6 mice and divided them into three groups: 1) PBS, 2) CPG + GM‐CSF, and 3) ESC (129) + CPG + GM‐CSF. Briefly, 5 × 105 bladder cancer cells were implanted subcutaneously for 1 week. Mice were then immunized with PBS or ESC vaccines for 4 weeks (Figure 1a). Tumor growth was significantly inhibited in the ESC (129) + CPG + GM‐CSF group compared with the CPG + GM‐CSF group (Figure 1d,e). The ESC (129) + CPG + GM‐CSF group also showed more potent and durable protection against tumor growth than with ESC (129) alone, ESC (129) + GM‐CSF or ESC (129) + CPG (Figure S1a,c, Supporting Information). Similarly, ESC (C57) alone also showed significant inhibitory effects on bladder cancer growth compared with E (C57) +CpG (Figure 1f,g and Figure S1b,c, Supporting Information). These results demonstrate the effectiveness and inhibitory activity of ESC‐based vaccines on bladder cancer growth.
Figure 1.
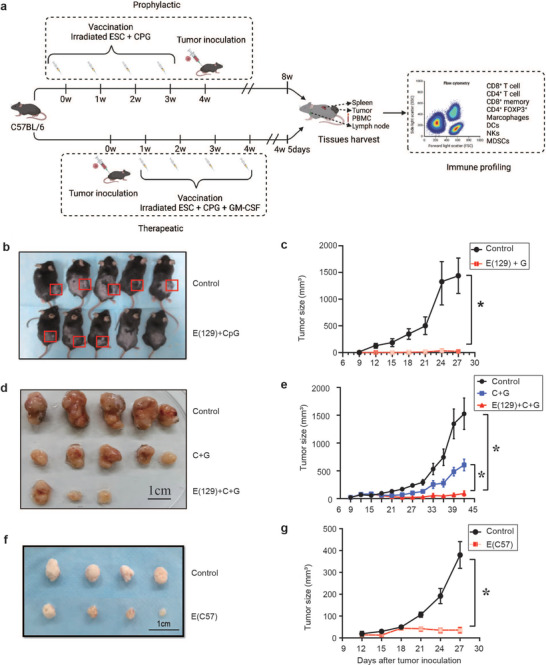
ESC vaccination prevents bladder cancer growth. a) Scheme of immunization. Vaccines were prepared with sorting ESCs, irradiation, resuspension in adjuvant solution, and s.c. injection. b,c) In the prophylactic mouse model, Vaccination of mice with ESC vaccine resulted in a complete rejection of the cancer cells by 4 weeks and overall reductions in MB49 tumor size (n = 5 per group). Representative images showed in (b) and Quantification of the tumor size data presented in (c). Experiments were repeated three times. d,e) In the therapeutic mouse model, Vaccination of C57BL/6 mice with ESC (129) + CpG + GM‐CSF resulted in significant reduction of MB49 tumor sizes (n = 5). Representative images showed in (d) and Quantification of the tumor size data presented in (e). Experiments were repeated three times. f,g) In the therapeutic mouse model, Vaccination of C57BL/6 mice with ESC (C57) resulted in significant reduction of MB49 tumor sizes (n = 4). Representative images shown in (d) and quantification of the tumor size data was presented in (e). Experiments were repeated three times. Data in (c), (e), and (g) represent mean ± SEM (Student's t‐test ANOVA with Tukey's multiple comparison test). *p < 0.05, **p < 0.001, ***p < 0.001, ****p < 0.0001.
2.2. ESC‐Based Vaccines Induce Bladder Cancer Immunity by Upregulating Cytotoxic T Cell Activity
To evaluate the ESC‐based vaccine‐induced immune changes, mice were sacrificed 2 weeks after tumor inoculation in the prophylactic model or after the third vaccine injection in the therapeutic model. We profiled the immune cells from blood and/or draining lymph nodes (dLNs) by flow cytometry. Both ESC (129 or C57) vaccine groups had significantly increased effector/memory cytotoxic T cell and helper T cell populations in the blood and dLNs, whereas the Treg cell population was not affected (Figure S1d, Supporting Information, Figure 2a–c, Figure S6a–e, Supporting Information). The T cells and helper T cells play important roles in tumor growth inhibition and increase the persistence of the antitumor immune response.[ 11 ] The cytotoxic effects of T cells and helper T cells were confirmed by CD8 depletion and CD4 depletion, respectively (Figure 2d). Depletion of CD8+ T and CD4+ T cells abrogated the antitumor effects of both ESC vaccines, which resulted in shorter survival similar to that of untreated mice (Figure 2d). In addition, immunosuppressive MDSCs were significantly inhibited in the blood and dLNs of the ESC (129) vaccine group (Figure 2a,b and Figure S6b,d, Supporting Information). Moreover, NK cells were increased in the ESC (129) + CPG + GM‐CSF group compared to the control group (Figure 2a,b and Figure S6b,d, Supporting Information). There were also increased levels of antigen‐presenting dendritic cells (DCs) in the blood and dLNs of the ESC (129) vaccine group (Figure 2a,b and Figure S6b,d, Supporting Information). The tumor in the ESC (129) or ESC (C57) vaccine group showed infiltration of CD4+ T cells, CD8+ T cells, and CD11c+ cells (Figure S2c–e, Supporting Information), decreased infiltration of Tregs, indicating the effective recruitment of immune cells to the tumor. Both ESC vaccines reduced tumor growth by inducing cytotoxic tumor immunity, which was found to be mediated by both CD8+ T cells and NK cells. We also investigated cytokines changes in the prophylactic and therapeutic models using the ESC (129) vaccine. Although most cytokines were not significantly changed in each model, IFN‐γ was increased and IL‐6 was decreased in the prophylactic model, whereas IL‐17a was decreased in the therapeutic model (Figure S2a,b, Supporting Information). These findings indicate that the ESC vaccines are safe to use and can stimulate antitumor cytokines.
Figure 2.
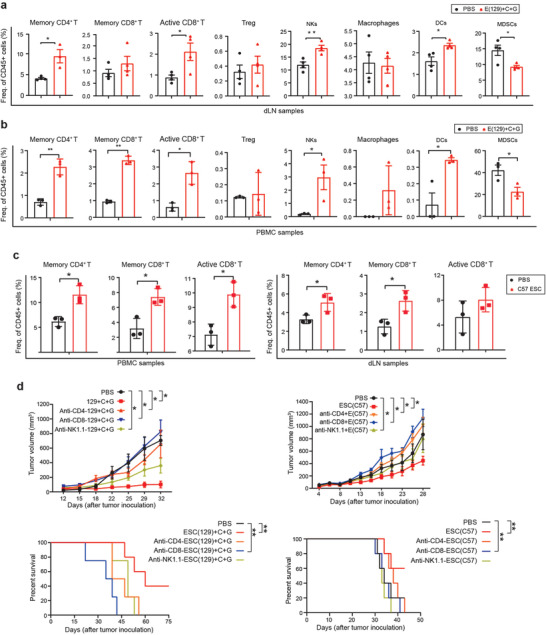
ESC vaccination induces effector and memory CD8+ T cells and antigen‐presenting cells. a,b) 2 weeks after ESCs introduction, ESC (129) + CpG + GM‐CSF‐vaccinated C57BL/6 mice showed a significant increase in cytotoxic T cells (CD8+granzyme‐B+), effector/memory CD8+ T cells (CD8+CD44+), reduction in percentages of MDSCs (CD11c+Gr‐1+), and increase in NK+ cells and upregulation of antigen‐presenting cells (APCs) such as DCs (CD11c+MHC II+CD86+) in the dLN (n = 4) and PBMCs (n = 3). c) 2 weeks after ESC (C57) injection, dLNs and PBMCs of ESC (C57)‐vaccinated C57BL/6 mice revealed an increased frequency of cytotoxic T cells (CD8+granzyme‐B+), effector/memory CD8+T cells (CD8+CD44+), as well as T helper cells (CD4+CD44+) (n = 3). d,e) CD8+ T cells are required for the antitumor effects of ESCs in a bladder cancer model. Male C57BL/6 mice (n = 5) were implanted with 5 x 105 MB49 bladder cancer cells s.c. in the right flank. From day 1, 3, 6, and 9 after tumor inoculation, 10 mg kg−1 of CD8+ T cell depletion antibody, 10 mg kg−1 of CD4+ T cell depletion antibody, 10 mg kg−1 of NK cell depletion antibody were injected intraperitoneally in mice. Tumor volume and survival time of MB49 tumor‐bearing mice were measured. Experiments were repeated three times. Data represent mean ± SEM (n = 5 per group). ANOVA with Tukey's multiple comparison test). *p < 0.05, **p < 0.001, ***p < 0.001, ****p < 0.0001.
2.3. Identification of Tumor‐Associated Genes Expressed in ESCs
We performed RNA‐seq on ESC (129) and ESC (C57) cell lines and compared the expressed genes with cancer cell lines (MB49, ML1, and Hepa1‐6) and healthy tissues. We set up an in silico pipeline to select potential therapeutic TAAs (Figure 3a) based on shared genes expressed in the ESCs but minimally expressed in normal tissues. Based on the expression profiles of the three tumor cell lines, the two ESC cell lines, and 18 normal tissues, we retained 481 genes expressed highly in tumor and ESC from 29 971 genes. To further reduce the number of candidates for experimental testing, we applied a second filter to evaluate the genes in human tumors using RNA‐seq data from the Cancer Genome Atlas (TCGA) Consortium. We retained 60 genes based on two inclusion criteria: 1) expressed in humans and 2) significantly upregulated in different cancer types (Figure S3a, Supporting Information). These 60 genes code proteins mainly involved in nuclear division (GO:0000280, adjusted P value: 4.82e‐28) and mitotic cell cycle regulation (GO:0007346, adjusted P value: 2.26e‐15). By correlating the gene expression level with survival status in TCGA, we selected eight genes for further analysis: Racgap1, Anln, Top2α, Ccnb1, Ect2, Hmmr, Melk, and Igf2bp1, which play important roles in tumor progression (Figure 3b). We compared the expression levels of these genes in other tumors and healthy tissues (Figure S3a, Supporting Information). This analysis revealed that these genes are higher expressed in ESCs and multiple tumor cell lines compared with normal mouse tissues or in tumor tissues compared with adjacent and healthy tissues (Figure 3b and Figure S3b, Supporting Information). Gene ontology analysis indicated that these eight genes are mainly involved in cortical actin cytoskeleton organization and stem cell population maintenance (Figure 3c). These genes have also been widely reported to be associated with multiple cancer types.
Figure 3.
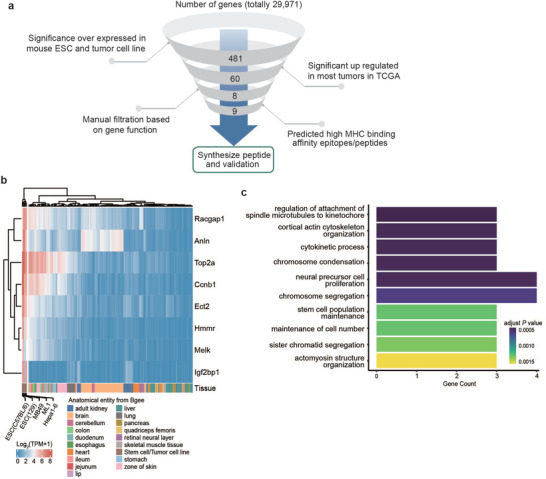
Identification of cancer signature genes in ESCs. a) Illustration of the in silico filtration process. Genes highly expressed in ESC and tumor cell lines were further validated in TCGA expression data, manual filtration based on the gene function, and epitope‐MHC binding affinity prediction was then conducted, with candidate peptides then synthesized. b) Expression profiles of selected eight genes compared between mouse ESC and tumor cell lines to normal healthy tissues. c) Top ten significance enriched gene ontology function of the selected eight genes.
We next assessed the presence of T cell epitopes among these genes, focusing on human lymphocyte antigen A, B, and C, which are common HLA class I alleles, and mouse H‐2 kb and H‐2Db, which are MHC expressed in C57BL/6 mice.[ 12 ] This conservative approach led to the identification of 18 epitope candidates from these genes (Figure S3c, Supporting Information). To ensure peptides work equally in both mice and human, we selected protein sequence with high similarity in human and mice common to screened epitopes which can fulfill: 1) epitopes consistent with MHC alleles expressed in C57BL/6 mice and 2) epitopes containing high population coverage in humans. We successfully selected and synthesized eight epitopes for further biological validation and immunological assays (one CCNB1, one ANLN, one MELK, two TOP2α, and three ECT2). We analyzed the consistent sequence of the epitopes from these genes in humans and mice (Table S2, Supporting Information). We next synthesized long peptides containing the epitopes to evaluate the antigen‐specific T cell immune response.
2.4. The Shared TAA‐Derived Epitopes Induced Strong and Polyfunctional T Cell Responses
The immunogenicity of the eight epitopes (EP‐1, EP‐2, EP‐3, EP‐4, EP‐5, EP‐7, EP‐8, and EP‐13) with the highest in silico predicted immune response was experimentally evaluated in C57BL/6 mice (Figure 4a). To determine the antigen‐specific T cell responses induced by the ESC (129) + C + G and ESC (C57) vaccines, we performed ex vivo IFN‐γ enzyme‐linked ImmunoSpot (ELISPOT) assays. First, mice were immunized with the ESC (129 or C57) vaccines once a week for 2 weeks. The mice were then sacrificed and immune cells were isolated from the spleen. Significant T‐cell responses against seven out of eight epitopes (except the Top 2α‐1 epitope) were detected in C57BL/6 mice vaccinated with ESC (129). However, ANLN‐specific and ECT2–3‐specific T cell responses were only detected in mice vaccinated with ESC (C57) (Figure 4a). The experiment was repeated using a tumor‐bearing mouse model. First, C57BL/6 mice were subcutaneously inoculated with MB49 cancer cells. After 1 week, mice were primed with ESC (129) or ESC (C57) vaccine once a week for 2 weeks. ELISPOT was repeated to detect specific T cell responses against these epitopes. Strong T cell responses against epitopes of ANLN, CCNB1, MELK, ECT2 (3), and Top 2α (2) were detected in C57BL/6 mice vaccinated with ESC (129) or ESC (C57) (Figure 4b). These data confirm that the shared epitopes expressed in ESCs have potential to induce high antitumor immunity.
Figure 4.
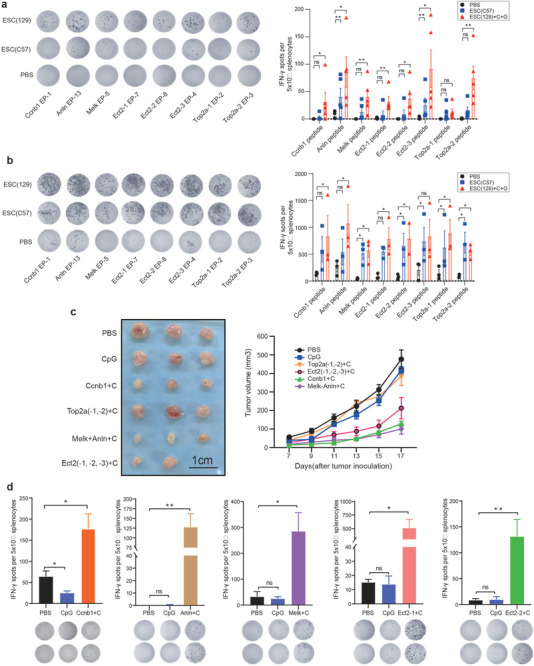
Immunogenicity detection of epitopes derived from ESCs. a) ELISPOT assay for immune cell activation of splenocytes in the ESC (C57) or ESC (129) + CpG + GM‐CSF vaccinated group (n = 5) compared to PBS (n = 5) group upon exposure to selected epitopes. Quantitative analysis of the ELISPOT assay for IFN‐γ secretion. b) C57BL/6 mice were inoculated with bladder cancer cells for 1 week, and injected with PBS, ESC (C57) or ESC (129) + CpG + GM‐CSF once a week for 2 weeks. ELISPOT assay was performed. Significant increase in number of IFN‐γ spots in the ESC (C57) and ESC (129) + CpG + GM‐CSF‐vaccinated C57BL/6 mice (n = 3) compared to PBS group (n = 3) upon exposure to prediction epitopes. Quantitative analysis of the ELISPOT assay for IFN‐γ secretion. c) Antitumor effects of each epitope were detected in bladder cancer‐inoculated mice. C57BL/6 mice (n = 5) were implanted with 3 x 105 MB49 bladder cancer cells s.c. in the right flank, after 1 week, mice were immunized with PBS or different epitope peptides on day 3, day 6, and day 9 and boosted on day 15. Tumor growth was measured every 2 d. d) Five days after the last vaccination, splenocytes (5 × 105) were cocultured with different peptides (10 µg) for the duration of 20 h, cellular immune responses were measured using ELISPOT assay (n = 4). Experiments were repeated three times. Quantitative analysis of the ELISPOT assay for IFN‐γ secretion. Data represent mean ± SEM, *p < 0.05, **p < 0.001, ***p < 0.001, ****p < 0.0001 (Student's t‐test).
Next, we evaluated the capacity of the selected epitope candidates to induce antitumor effects. To assess the therapeutic effects of the eight peptides, we subcutaneously inoculated mice with tumor cells. After 5 d, mice were primed with the peptide vaccines adjuvanted with CpG (Figure 4c). The tumor size was measured by calipers every 2 d. Peptides containing ANLN, MELK, or CCNB 1 epitopes and two combined ECT2‐1 and ECT2‐2 epitopes showed tumor inhibition effects (Figure 4c). However, combined TOP 2α epitopes did not show tumor inhibition effects. Notably, effective epitope peptides also induced specific T cell responses (Figure 4d). In agreement with the bioinformatic prediction, specific T cell induction was observed in immune cells from the spleen. The ELISPOT analysis of CD8+ T cell responses showed ECT2‐1 and ECT2‐2 epitopes, but not ECT2–3 epitope, were the most immunogenic peptides resulting in strong T cell responses and the highest tumor inhibition effects. Notably, ANLN, CCNB 1, and MELK epitopes also induced large CD8+ T cell responses. Although TOP 2α epitopes induced CD8+ T cell responses, they did not show any antitumor effects (Figure S4a, Supporting Information).
We further studied five selected epitopes (two ECT2s, one ANLN, one CCNB 1, and one MELK) by combining them into a multipeptide vaccine, which showed more effective antitumor abilities than any individual peptide (Figure S4c, Supporting Information). CpG was shown as the most effective adjuvant to induce more specific T cells compared with other adjuvants (Figure S4b, Supporting Information). We found that E(129) vaccine induce T cell infiltration into tumors (Figures S2d and S4e, Supporting Information), including specific T cells for our selected peptides (Ccnb1) (Figure S4d, Supporting Information). Flow cytometry analysis showed there were increased memory CD8+ T cells and memory CD4+ T cells, increased proportions of DC cells, and reduced MDSCs in dLNs of peptides‐treated mice, which was consistent with changes in the immune cell profile of mice vaccinated with ESC (129) or ESC (C57) (Figure 5a,b). However, the antitumor effects of the combined epitopes from different genes were less potent than the ESC (129 or C57) vaccines under the same conditions (Figure 5c), indicating the whole‐cell vaccines have greater antitumor potential.
Figure 5.
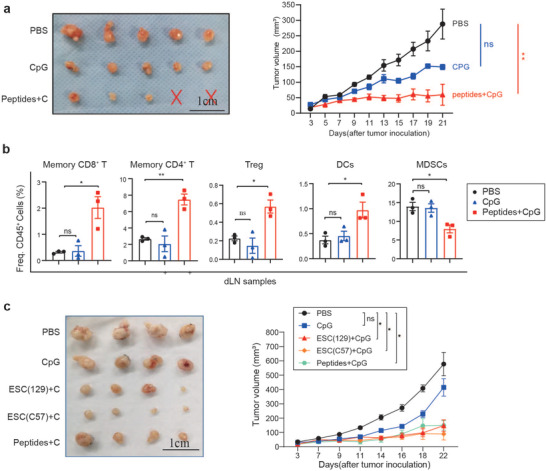
Antitumor effects of mixture epitope‐containing peptides. a) C57BL/6 mice (n = 5) were implanted with 3 x 105 MB49 bladder cancer cells s.c. in the right flank, after 1 week, mice were immunized with PBS or mixed peptides on day 3, day 6, day 9 and boosted on day 15. Tumor volume was detected every 2 d. Experiments were repeated three times. b) Peptide vaccination increased memory CD8+T cells and memory CD4+T cells, as well as DCs, but decreased MDSCs (n = 3). c) ESC‐based vaccines showed more effective tumor growth inhibition effects than peptide vaccines. C57BL/6 mice (n = 5) were implanted with 3 x 105 MB49 bladder cancer cells s.c. in the right flank, after 1 week, mice were immunized with PBS or mixed peptides on day 3, day 6, day 9 and boosted on day 15. tumor volume was detected every 2 d. Quantification of the tumor volume over time, with values being expressed as means ± SEM (*p < 0.05, **p < 0.001, ***p < 0.001, ****p < 0.0001, Tukey's multiple comparison test).
3. Discussion
The development of potent tumor vaccines requires the identification of effective tumor‐specific antigens, which are largely unknown. Effective approaches are needed to identify more tumor antigens. The current study supports the idea that antigenic similarities between stem cells and cancer cells can be used to identify tumor antigens and develop cancer vaccines. We developed a protocol to systematically identify effective antitumor antigens in ESCs. The protocol consists of 1) computational identification of TAAs candidates, 2) molecular identification of shared epitope targets, 3) in silico prediction of epitope‐MHC binding affinity, 4) immunogenicity testing of epitopes, and 5) integrated study of tumor growth inhibition and immune cell changes in mice vaccinated with ESC vaccines and ESC‐derived epitopes.
We used two types of allogeneic murine ESCs as the whole‐cell vaccines, which were reported to be more immunogenic than single antigens.[ 13 ] The ESC vaccines were effective in reducing bladder cancer growth long term in both models by inducing an immune response including upregulating mature APCs in the dLNs, increasing helper T cells and cytotoxic T cells, and increasing NK cells. The impact of the ESC (129 and C57) vaccines on the CD8+ T cell compartment may be direct or indirect. In vivo depletion studies confirmed that the CD8+ T cells, CD4+ T cells, and NK cells were required for the antitumor effects induced by the ESC vaccines. Although the preventive effects of ESC vaccines required CD8+ T cells rather than CD4+ T cells and NK cells, the recruitment of CD4+ T cells during priming can promote the formation and maintenance of the pool of memory CD8+ T cells.[ 14 ] Moreover, they are also critical for the generation and maintenance of long‐term memory and recall responses in vaccinated animals.[ 15 ]
The critical mechanism of the antitumor effects of ESC vaccination is still unclear, but it could involve the increase in cytotoxic T lymphocytes and memory cells in lymph organs as well as the recruitment of immune cells into the tumor. The ESC + CpG + GM‐CSF combined vaccine did not alter Treg ratios in the dLNs and periphery blood. These findings are consistent with a previous study of an ESC‐based vaccine for lung cancer.[ 13 ] We also observed a significant decrease in the percentage of MDSCs in the dLNs and periphery blood of mice treated with ESC vaccines compared to control mice. The infiltration of MDSCs suppresses tumor immunity by inducing immunosuppressive cytokines like IL‐10 and transforming growth factor beta (TGF‐β), and reducing T cell proliferation, and blocking the entry of CD8+ T cells to tumors[ 16 ] These results are consistent with an earlier study that showed an ESC‐based vaccine‐induced potent humoral and cellular immune responses as well as reduced MDSCs in the spleen of a mouse model of transplantable colon carcinoma, leading to strong antitumor effects.[ 17 ]
A recent study reported that the protection against cancers afforded by iPSC/ESC vaccine involves a number of shared antigens.[ 3 , 5 ] The iPSC/ESC‐based vaccines were able to induce strong immune recognition of multiple antigens to limit tumor cells escaping from immune detection and destruction.[ 18 ] The discovery and identification of effective tumor antigens expressed in ESCs/iPSCs as antigenic components of a vaccine could be more effective in reducing bladder cancer growth and would alleviate the limitations of targeting single antigens.
We established a pipeline to identify potential antigens from ESCs with the following characteristics: 1) nmTAAs or nmTSAs are highly expressed in ESCs and cancers, have high immunogenicity, and induce a stronger and longer antitumor immune response and 2) most of them are involved in cell growth and cell function, including tumor growth, cell cycle, and cell signaling transduction. These characteristics are important to reduce the possibility of immune escape variants. We selected eight signature genes that do not have clear antitumor immunity‐related functions. Among these genes, Racgap1, Ect2, Hmmr, and Melk are expressed in the plasma membrane, and Anln, Ccnb1, Top2α, and Igf2bp1 are expressed in the nucleus. Many of these genes were reported to be tumor marker genes and stemness‐related genes.[ 19 ] For example, MELK, CCNB1, and IGF2BP1 maintain cancer stem cell properties involved in tumor growth,[ 20 ] whereas CCNB1, RACGAP1, and TOP2 are stemness‐related genes significantly associated with immune infiltration (negatively), which could benefit the development of clinical treatments.[ 21 ] Mice treated with an ESC‐based vaccine for 20 h, resulted in the degradation of ESCs by host immune cells (Figure S5, Supporting Information). After digestion, the intracellular antigens in ESCs can then be acquired by DCs.
Overall, iPSC‐ and ESC‐based epitopes represent a large source of tumor vaccine candidates. A wide range of vaccines based on oncofetal antigens derived from ESCs are currently being tested in preclinical studies, and some single epitopes are already in stage I/II clinical trials.[ 3 ] Our approach identified several genes expressing several CD8+ T cell epitopes shared between humans and mice. An important finding of our study is that ESC immunization induced strong specific CD8+ T cells. We predicted that the binding affinity of the peptide‐MHC complex was sufficient to elicit a cytolytic T‐cell response. Consequently, our results demonstrated that the injection of epitope‐containing peptides was able to induce antigen‐specific memory T cells in a mouse model of bladder cancer. We also showed our prediction pipelines can successfully identify potential antigens.
The tumor inhibition effects of the selected epitopes were mediated through the induction of functional T cells that secreted IFN‐γ. Generally monovalent cancer vaccines are inefficient and thus targeting one antigen alone is unlikely to generate effective antitumor immune responses for tumor inhibition because of the rapid appearance of escape mutants.[ 22 ] The selected epitopes induced CD8+ T cell clones with high predicted affinity in tumor‐bearing mice. The five epitopes effectively increased the frequency of DCs, activated CD8+ T and memory CD4+ T cells in dLNs, and decreased MDSCs. Moreover, these changes are consistent with the antitumor effects of ESC‐based vaccines. However, we found the antitumor effects of these epitopes were less effective than whole‐cell ESC (129 and C57) vaccines, suggesting there are other more potent epitopes expressed in ESCs.
In summary, our data demonstrated that the ESC‐based vaccine can inhibit tumor growth, induce effective cytotoxic T cell and memory T cell responses, and reduce MDSCs in a bladder cancer mouse model. We also provided evidence that the antitumor effects of ESC‐vaccine are dependent on shared TAAs in both ESCs and tumor cells. We established a platform to systematically identify shared TAAs and CD8+ T cell epitopes and to validate the effectiveness of these epitopes in inducing antitumor immune responses and preventing tumor growth. Tumor and ESCs coexpressed cancer‐signature antigens could be developed into peptide‐based cancer vaccines for bladder cancer.
4. Experimental Section
ESC Culture
Mouse embryonic fibroblast (MEFs) and ESC (129) were gifts from Prof. Pentao Liu of the University of Hong Kong. The MEFs were grown in DMEM Glutamax (ThermoFisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS, ThermoFisher Scientific). The ESC (C57) (C57BL/6, SCSP‐218; American Type Culture Collection (ATCC) number: SCRC‐1002) were purchased from the National Collection of authenticated cell cultures (Shanghai, China). The ESCs were cultured in Knockout DMEM (ThermoFisher Scientific) supplemented with 15% (vol/vol) FBS, 1 × (vol/vol) Minimum Essential Medium (MEM) NEAAs (Stemcell, BC, Canada), 1 × (vol/vol) glutamine–penicillin–streptomycin (ThermoFisher Scientific), 0.1 µm 2‐mercaptoethanol, and 103 U mL−1 human leukemia inhibitory factor (LIF). The ESCs and iPSCs were grown for several passages and transferred to 0.1% gelatin‐coated plates for SSEA‐1 magnetic bead sorting (Miltenyi, Germany).
Cancer Cell Lines and Animal Model
The MB49 bladder cancer cell line (SCC148, Sigma‐Aldrich, St. Louis, MO, USA) is syngeneic to C57BL/6 mice and has low‐grade lymphoid metastatic potential for the lungs. The cancer lines were grown in DMEM with 10% FBS under normal culture conditions. For the mouse models, 5 × 105 or 5 × 104 cancer cells were resuspended in 100 µL PBS and injected subcutaneously in the lower back of the C57BL/6 mice to generate the therapeutic model and prophylactic model, respectively.
Young adult male C57BL/6J mice (6–8 weeks old) were randomly assigned to the different treatment groups. Tumor‐bearing mice were excluded from the experiment if their physical condition required euthanasia before the experimental deadline, such as tumor size exceeding 1.5 cm3, visible distress, pain, or illness. Tumor growth was monitored every 3 d and longitudinal and transverse diameters (in mm) were measured using digital calipers. All mice were handled by the Association for Assessment and Accreditation of Laboratory Animals Care international guidelines. Animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC), Shenzhen Institute of Advanced Technology (SIAT), Chinese Academy of Science (SIAT‐IACUC‐210202‐HCSJ‐ML‐A1542).
Vaccination with ESC‐Based Vaccines
For the ESC‐based vaccine, 2 × 106 SSEA‐1‐sorted syngeneic murine ESCs were irradiated at 6000 rads before injection. Cells were suspended in 100 µL of 5 µm CpG (Invivogen, San Diego, USA) in PBS with or without 20 ng GM‐CSF (Novoprotein, China). Mice were placed in an induction chamber and anesthetized with 2% isoflurane (Isothesia, Butler Schein) in 100% oxygen at a delivery rate of 2 L min−1 until loss of righting reflex, according to the IACUC guidelines at SIAT. Cells were injected subcutaneously (s.c.) into the right flank of male C57BL/6 mice (lacking MHC class II I‐E expression) using a 1/4 cc insulin syringe (Terumo). Body weight and overall appearance were monitored weekly to detect early signs of autoreactivity to the vaccine. The same vaccine dosage and preparation were used for the prophylactic, therapeutic, and adjuvant treatments. The prophylactic and therapeutic vaccination studies were repeated several times. Tumor growth was measured by digital calipers every 3 d.
Isolation of Inflammatory Cells and Serum from Blood, Spleen, and Draining Lymph Nodes (dLNs)
The C57BL/6 experimental mice were sacrificed at 4 and 2 weeks after tumor inoculation, respectively. Tissues were isolated and minced in a digestion buffer containing Roswell park memorial institute (RPMI) medium, FBS, collagenase, DNase, and 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), followed by shaking at 37 °C for 20 min. Samples were filtered through a 100 µm strainer, spun down, and resuspended in ammonium chloride potassium (ACK) lysis buffer to remove any red blood cells. The cell suspension was washed in PBS and used in the subsequent analyses. Dissociated tumors were washed on a percoll gradient (General Electric Healthcare) to remove nonimmune cells and to isolate tumor‐infiltrating leukocytes (TILs). Blood was also collected in two separate tubes for PBMC (ethylenediaminetetraacetic acid (EDTA)‐containing tube) and serum (uncoated tube) isolation.
Staining of Inflammatory Cells for FACS Analysis
Single cells from the spleen or other tissues were resuspended in 100 µL FACS buffer (Dulbecco's phosphate buffered saline (DPBS), 2% FBS) and blocked with an FcR‐blocking Reagent (BioLegend, San Diego, CA, USA) and then divided into two tubes. One tube was stained with a surface marker panel containing CD3, CD4, CD25, CD8α, CD44, and CD45 (BioLegend), and intracellular markers Granzyme‐B and FoxP3 (BioLegend). The second tube was stained for F4/80, MHC‐II, CD86, CD11b, CD11c, NK1.1, Ly6‐G, and CD45 (BioLegend). A rat IgG2b κ isotype control (BioLegend) was used for CD3, CD11b, CD4, CD11c, CD25, Gr‐1, CD44, CD45, Foxp3, and Granzyme B; a rat IgG2a κ isotype (BioLegend) was used for CD86 and F4/80, CD8a and NK1.1; and a rat IgG1 κ isotype (BioLegend) was used for MHC II. Extracellular staining was performed prior to fixing and permeabilizing the samples for staining with intracellular markers. Samples were analyzed on a Beckman Flow Cytometer in the Beckmann FACS facility.
LEGENDplex Multiplex Cytokine Assay
Serum was collected from the vaccinated mice for cytokine profiling in a Flow Cytometer using fluorescence‐encoded beads (LEGENDplex, BioLegend) prepared using a Mouse Th Cytokine Panel (category no. 741044, 12‐plex for IL‐2, IL‐4, IL‐5, IL‐6, IL‐9, IL‐10, IL‐13, IL‐17A, IL‐17F, IL‐22, IFN‐γ, and TNF‐α).
In Vivo T Lymphocyte Subset Depletion
Lymphocyte subset depletion was performed using ascites monoclonal antibodies (mAb) (Bio X Cell, Lebanon, USA). For CD8 depletion, mice were treated with anti‐CD8 (10 mg kg−1, clone 53–6.7, Bio X Cell) or isotype control (10 mg kg−1, clone LTF‐2, Bio X Cell) 1 d before tumor cell inoculation, and then treated every 3 d for a total of four times. For CD4 depletion, anti‐CD4 (10 mg kg−1, clone GK1.5, Bio X Cell) or anti‐NK (10 mg kg−1, clone PK136, Bio X Cell) was administered by intraperitoneal injection on days 1, 3, 6, and 9 after tumor inoculation. Depletion efficiency was checked by flow cytometry analysis of cheek bleeds on day 7 using appropriate antibodies. Tumor size was measured by calipers three times weekly for the duration of the experiment. Mice were euthanized after the tumor size reached 1500 mm3. Survival time of the MB49 tumor‐bearing mice was recorded from the day of tumor cell inoculation. The Kaplan–Meier method was used for survival analysis and the log‐rank test was used to calculate the P‐value.
ELISPOT Assay
Splenocytes (5 × 105 cells) were isolated as described above and cocultured with different peptides (10 µg) for 20 h. The secretion of IFN‐γ was measured by ELISPOT according to the manufacturer's instructions (MabTech, Nacka Strand, Sweden). Adobe Photoshop CS6 software was used to calculate the size and number of IFN‐γ‐positive spots.
Selection of Upregulated Genes Shared by Tumors and Stem Cells
Gene expression data of normal tissue and the ESC (C57) from C57BL/6 mice were downloaded from BgeeDB (version 14.0).[ 23 ] The RNA‐seq data of three tumor cell lines (ML1, MB49, and Hepa1‐6) and ESC (129) were processed accordingly, and two expression measurements (raw count and transcript per million (TPM)) were obtained with the help of kallisto.[ 24 ] Gene expression analysis was performed between the three mouse tumor cell lines and two ESC cell lines and multiple selected normal tissues in DESeq2[ 25 ] using gene raw counts as the input with default parameters. Genes with adjusted P‐value <0.005 and log2 fold change >3 were considered to be significant. The 60 significantly upregulated genes in tumors across multiple cancer types from TCGA were retained. The differential expression results were retrieved from OncoDB[ 26 ] with a cutoff of a more than twofold change and an adjusted P‐value of >0.05 for significantly upregulated genes, resulting in more than 12 cancer types retained. Manual reviews were carried out on the 60 genes based on the following factors: 1) genes highly expressed (by fold change and adjust P‐value) in tumor tissues of multiple tumor types in TCGA data compared to normal tissues using the GEPIA2 online tool[ 27 ] and 2) genes with important biological function in ESCs or during development stages. Genes with less than 50% protein sequence identity between humans and mice were removed. 8‐mer and 15‐mer stepwise MHC I and MHC II epitope prediction were carried out using NetMHCpan with all supported MHC alleles, respectively. Epitopes strongly binding to both C57BL/6 mouse MHC (H‐2‐Db, H‐2‐Kb) and HLAs with populations coverage larger than 10% were kept for downstream synthesis and validation. Moreover, HLAs with strong binding epitopes in any of the eight selected genes were added to the population coverage calculation using the analysis resource from The Immune Epitope Database (IEDB).[ 28 ] All gene ontology analyses were conducted using the clusterProfile package in R.[ 29 ]
Evaluation of CD8+ T Cell Epitope Peptide Immunogenicity in C57BL/6 Mice
Epitope peptides were synthesized by GL Biochem (Shanghai, China). Peptides were dissolved in dimethyl sulfoxide (DMSO) or PBS. Vaccine formulations were prepared by mixing 100 µg of peptides with 1 µm CpG ODN 1826 in phosphate‐buffered saline at a final injection volume of 100 µL per mouse. Control vaccine formulations were prepared using PBS in place of the epitope peptides. Vaccine mixtures were administered to C57BL/6 mice on days 3, 6, and 9, and a booster was given on day 15. All vaccines were administered subcutaneously into the left or right flank of each mouse. After 5 d postvaccination, the cellular immune response in splenocytes was measured by IFN‐γ ELISPOT assay. Splenocytes (5 × 105 cells) were cocultured with different peptides (10 µg) for 20 h, and the level of secreted IFN‐γ was measured according to the manufacturer's instructions (MabTech, Nacka Strand, Sweden). Adobe Photoshop CS6 software was used to calculate the size and number of IFN‐γ positive spots, with CD3+ and CD28+ antibodies used as positive controls.
Quantification and Statistical Analyses
All values in bar graphs and curves are expressed as means ± SEM. Intergroup differences were appropriately assessed by either unpaired two‐tailed Student's t‐test or one‐way analysis of variance (ANOVA) with multiple comparison tests using PRISM GraphPad software. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
M.J. and J.H. contributed equally to this work. M.J. and J.‐D.H. designed the experiment. M.J. and J.H. performed the experiments. L.T. and B.‐Z.Z. participated in the study. M.J. and J.‐D.H. wrote the paper. M.J. and J.‐D.H. analyzed the data. All authors contributed to the article and approved the submitted version.
Supporting information
Supporting Information
Acknowledgements
The research was supported by the National Key Research and Development Program of China (2018YFA0902701). The research was supported by the Natural Science Foundation of China (32101225) to M.J. The research was supported by Guangdong Science and Technology Department (2020B1212030004) to J.H. The authors would like to thank the L&T Charitable Foundation and the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2019BT02Y198) for their support.
Jin M., Hu J., Tong L., Zhang B.‐Z., Huang J.‐D., The Epitope Basis of Embryonic Stem Cell‐Induced Antitumor Immunity against Bladder Cancer. Adv. Healthcare Mater. 2023, 12, 2202691. 10.1002/adhm.202202691
Data Availability Statement
Research data are not shared.
References
- 1. Richters A., Aben K. K. H., Kiemeney L., World J. Urol. 2020, 38, 1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cumberbatch M. G. K., Jubber I., Black P. C., Esperto F., Figueroa J. D., Kamat A. M., Kiemeney L., Lotan Y., Pang K., Silverman D. T., Znaor A., Catto J. W. F., Eur. Urol. 2018, 74, 784. [DOI] [PubMed] [Google Scholar]
- 3. Ouyang X., Telli M. L., Wu J. C., Front. Immunol. 2019, 10, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kooreman N. G., Kim Y., de Almeida P. E., Termglinchan V., Diecke S., Shao N. Y., Wei T. T., Yi H., Dey D., Nelakanti R., Brouwer T. P., Paik D. T., Sagiv‐Barfi I., Han A., Quax P. H. A., Hamming J. F., Levy R., Davis M. M., Wu J. C., Cell Stem Cell 2018, 22, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ouyang X., Liu Y., Zhou Y., Guo J., Wei T. T., Liu C., Lee B., Chen B., Zhang A., Casey K. M., Wang L., Kooreman N. G., Habtezion A., Engleman E. G., Wu J. C., Stem Cell Rep. 2021, 16, 1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stephens A. J., Burgess‐Brown N. A., Jiang S., Front. Immunol. 2021, 12, 696791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corulli L. R., Cecil D. L., Gad E., Koehnlein M., Coveler A. L., Childs J. S., Lubet R. A., Disis M. L., Front. Immunol. 2021, 12, 729809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Chen J., Li Z., Jia X., Song W., Wu H., Zhu H., Xuan Z., Du Y., Zhu X., Song G., Dong H., Bian S., Wang S., Zhao Y., Xie H., Zheng S., Song P., Oncogene 2022, 41, 3118; [DOI] [PubMed] [Google Scholar]; b) Tang Q., Li W., Zheng X., Ren L., Liu J., Li S., Wang J., Du G., Signal Transduction Targeted Ther. 2020, 5, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Xu D., Wang Y., Wu J., Zhang Z., Chen J., Xie M., Tang R., Chen C., Chen L., Lin S., Luo X., Zheng J., Cell Death Dis. 2021, 12, 162; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reinhard K., Rengstl B., Oehm P., Michel K., Billmeier A., Hayduk N., Klein O., Kuna K., Ouchan Y., Woll S., Christ E., Weber D., Suchan M., Bukur T., Birtel M., Jahndel V., Mroz K., Hobohm K., Kranz L., Diken M., Kuhlcke K., Tureci O., Sahin U., Science 2020, 367, 446. [DOI] [PubMed] [Google Scholar]
- 10. Shi Y., Liu C. H., Roberts A. I., Das J., Xu G., Ren G., Zhang Y., Zhang L., Yuan Z. R., Tan H. S., Das G., Devadas S., Cell Res. 2006, 16, 126. [DOI] [PubMed] [Google Scholar]
- 11. Waldman A. D., Fritz J. M., Lenardo M. J., Nat. Rev. Immunol. 2020, 20, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flavell R. A., Allen H., Huber B., Wake C., Widera G., Immunol. Rev. 1985, 84, 29. [DOI] [PubMed] [Google Scholar]
- 13. Yaddanapudi K., Mitchell R. A., Putty K., Willer S., Sharma R. K., Yan J., Bodduluri H., Eaton J. W., PLoS One 2012, 7, e42289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahrends T., Busselaar J., Severson T. M., Babala N., de Vries E., Bovens A., Wessels L., van Leeuwen F., Borst J., Nat. Commun. 2019, 10, 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahrends T., Spanjaard A., Pilzecker B., Babala N., Bovens A., Xiao Y., Jacobs H., Borst J., Immunity 2017, 47, 848. [DOI] [PubMed] [Google Scholar]
- 16. Kumar V., Patel S., Tcyganov E., Gabrilovich D. I., Trends Immunol. 2016, 37, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Zeng H., Xu R. H., Liu B., Li Z., Stem Cells 2009, 27, 3103. [DOI] [PubMed] [Google Scholar]
- 18. Zitvogel L., Perreault C., Finn O. J., Kroemer G., Nat. Rev. Clin. Oncol. 2021, 18, 591. [DOI] [PubMed] [Google Scholar]
- 19.a) Wang C., Hao K., Dong L., Wang J., Zhao L., Xu L., Xia Y., Jiang Q., Qin J., J. Biol. Chem. 2022, 298, 101701; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou Z., Li Y., Hao H., Wang Y., Zhou Z., Wang Z., Chu X., Cell Transplant. 2019, 28, 76S; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pan S., Zhan Y., Chen X., Wu B., Liu B., Front. Oncol. 2019, 9, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen H. M., Lin C. C., Chen W. S., Jiang J. K., Yang S. H., Chang S. C., Ho C. L., Yang C. C., Huang S. C., Chao Y., Liao T. T., Hwang W. L., Teng H. W., Int. J. Mol. Sci. 2021, 22, 6940.34203267 [Google Scholar]
- 21. Zeng H., Ji J., Song X., Huang Y., Li H., Huang J., Ma X., Front. Genet. 2020, 11, 549213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J., Fu M., Wang M., Wan D., Wei Y., Wei X., J. Hematol. Oncol. 2022, 15, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bastian F. B., Roux J., Niknejad A., Comte A., Fonseca Costa S. S., de Farias T. M., Moretti S., Parmentier G., de Laval V. R., Rosikiewicz M., Wollbrett J., Echchiki A., Escoriza A., Gharib W. H., Gonzales‐Porta M., Jarosz Y., Laurenczy B., Moret P., Person E., Roelli P., Sanjeev K., Seppey M., Robinson‐Rechavi M., Nucleic Acids Res. 2021, 49, D831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bray N. L., Pimentel H., Melsted P., Pachter L., Nat. Biotechnol. 2016, 34, 525. [DOI] [PubMed] [Google Scholar]
- 25. Love M. I., Huber W., Anders S., Genome Biol. 2014, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang G., Cho M., Wang X., Nucleic Acids Res. 2022, 50, D1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang Z., Kang B., Li C., Chen T., Zhang Z., Nucleic Acids Res. 2019, 47, W556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bui H. H., Sidney J., Dinh K., Southwood S., Newman M. J., Sette A., BMC Bioinformatics 2006, 7, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., Fu X., Liu S., Bo X., Yu G., Innovation 2021, 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.