

Abstract
Traditional dendritic cell (DC)‐mediated immunotherapy is usually suppressed by weak immunogenicity in tumors and generally leads to unsatisfactory outcomes. Synergistic exogenous/endogenous immunogenic activation can provide an alternative strategy for evoking a robust immune response by promoting DC activation. Herein, Ti3C2 MXene‐based nanoplatforms (termed MXP) are prepared with high‐efficiency near‐infrared photothermal conversion and immunocompetent loading capacity to form endogenous/exogenous nanovaccines. Specifically, the immunogenic cell death of tumor cells induced by the photothermal effects of the MXP can generate endogenous danger signals and antigens release to boost vaccination for DC maturation and antigen cross‐presentation. In addition, MXP can deliver model antigen ovalbumin (OVA) and agonists (CpG‐ODN) as an exogenous nanovaccine (MXP@OC), which further enhances DC activation. Importantly, the synergistic strategy of photothermal therapy and DC‐mediated immunotherapy by MXP significantly eradicates tumors and enhances adaptive immunity. Hence, the present work provides a two‐pronged strategy for improving immunogenicity and killing tumor cells to achieve a favorable outcome in tumor patients.
Keywords: antigen presentation, dendritic cells, endogenous nanovaccines, exogenous nanovaccines, photothermal therapy
A combined strategy by using Ti3C2 MXene‐based nanoplatforms (MXP) to enhance the tumor eradication efficiency via photothermal therapy (PTT) and dendritic cell (DC)‐based immunotherapy. MXP as a photothermal nanoagent and immune vaccine synergistically activating the DC‐based cytotoxic T lymphocyte immune response to significantly eradicate tumors and enhance adaptive immunity.
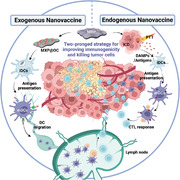
1. Introduction
Dendritic cells (DCs) are the most important antigen‐presenting cells (APCs) that play a central role in tumor immunotherapy.[ 1 ] DCs can process and present tumor antigens and activate antigen‐specific cytotoxic T lymphocyte (CTL) immune responses, which are important for eradicating tumor cells and preventing tumor recurrence.[ 2 ] However, the functions of DCs are generally restricted in tumor patients.[ 3 ] There has been ever‐increasing interest in scientific and clinical research on designing and exploiting various methods to enhance DC activation efficacy for tumor therapy. However, eradicating tumors completely can be difficult owing to the inherent drawbacks of traditional DC vaccines, such as insufficient antigen delivery, inadequate antigen presentation, and high levels of immunosuppression.
Currently, the activation pathway of DCs can be divided into endogenous and exogenous pathways. Promoting tumor cell release of immune components for uptake and processing by DCs is an endogenous pathway,[ 4 ] and synthetic antigens and adjuvants as vaccines injected via to activate systemic DC maturation and antigen presentation are parts of exogenous pathways.[ 5 ] Local tumor tissue can be destroyed, and immunogenic cell death (ICD) can be induced in tumor cells via various treatment methods.[ 6 ] One promising approach for inducing ICD within the tumor and activating the tumor immune microenvironment is near infrared (NIR)‐mediated photothermal therapy (PTT).[ 7 ] Generally, ICD‐released tumor‐associated antigens (TAAs) and damage‐associated molecular patterns (DAMPs), acting as endogenous vaccines in situ, are engulfed by DCs and then presented to T cells.[ 8 ] Meanwhile, endogenous vaccines facilitate the maturation of DCs, activation of CTLs, and secretion of multiple cytokines, thus changing local “cold” immunosuppressive tumors to “hot” immunoresponsive lesions.[ 9 ] However, PTT‐mediated endogenous vaccines alone cannot be effective in ablating distal and metastasizing tumors due to suboptimal systemic immune activation, and high expression of heat shock protein (HSP) increases the heat stress tolerance of tumor cells and limits the thermal effect.[ 10 ] Exogenous immune vaccine processes have the advantages of sufficient antigen delivery and adequate antigen presentation, inhibiting tumor cells through the activation of the body's immune system.[ 11 ] However, the low local immune response rate and high immune‐related adverse effects due to the immunosuppressive tumor microenvironment (TME) limit the curative effect of DC vaccines alone. Recent advances in antitumor therapy have gradually shifted from a focus on monotherapy to combined therapy based on the cooperative enhancement observed with two or more treatments, which may result in powerful superadditive therapeutic effects.[ 12 ] Therefore, more high‐efficiency and advanced multimodal synergistic antitumor therapy modalities are urgently needed. Recently, various MXenes with different structures and compositions have been synthesized.[ 13 ] Benefiting from the excellent absorption of NIR laser and specific surface activity, Ti3C2 MXene has recently been applied in PTT‐mediated antitumor treatment.[ 14 ] However, Ti3C2 only exhibits a single photothermal capability to kill tumor cells with relatively low efficiency; thus, it is necessary to endow Ti3C2 with additional properties to achieve effective tumor eradication.
In this study, we developed a combined strategy by using a 2D Ti3C2 MXene‐based nanoplatform to enhance the tumor eradication efficiency of PTT and DC‐based immunotherapy (Scheme 1 ). To guarantee the desirable physiological stability, polyethyleneimine (PEI), a cationic polymer with extensive biomedical applications, was employed to functionalize the surface of Ti3C2 MXenes. The constructed MXenes@PEI nanosheets (MXP) could be rapidly heated under exposure to an NIR laser (808 nm) and exhibited high photothermal performance for killing tumor cells. Furthermore, MXP can induce ICD of tumors under NIR irradiation. MXP‐mediated PTT provides DCs with “premium” antigens through the destroyed fragments of tumor cells, which form endogenous nanovaccines to promote DC maturation. In addition, MXP with positive charges can deliver antigen ovalbumin (OVA) and CpG as exogenous nanovaccines (MXP@OC), which have the potential to promote antigen internalization, antigen cross‐presentation, and the maturation and migration of DCs. Thus, MXP exerts greater potential as a carrier for delivering tumor antigens and adjuvants. Moreover, the MXP‐mediated PTT and DC‐immunotherapy synergistic strategy showed excellent therapeutic effects in B16‐OVA tumor‐bearing mice. Endogenous/exogenous nanovaccines combine numerous advantages, including improved immunogenicity, increased uptake by DCs and stabilization of the antigens, evoking the CTL immune response, alleviating tumor immunosuppression in the TME, and inducing robust systemic antitumor immune responses. We believe that this synergistic endogenous/exogenous nanovaccine strategy can facilitate DC‐mediated immuno‐oncotherapy with photothermal nanoagents and has potential for future application prospects.
Scheme 1.
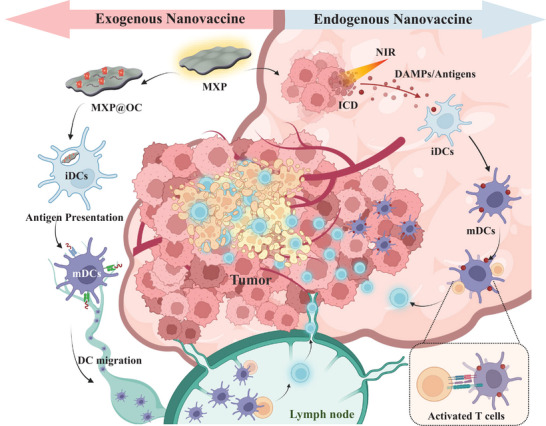
Schematic illustration of the MXP nanoplatform as photothermal nanoagents and immune vaccines synergistically activating the DC‐based antitumor immune cascade to achieve effective tumor destruction.
2. Results and Discussion
2.1. Preparation and Photothermal Properties of MXP
The ideal photothermal agent requires strong NIR light absorption and high photothermal conversion efficiency for the treatment of tumors.[ 13 , 15 ] Among them, carbon‐based Ti3C2 MXenes have been utilized as photothermal theranostic nanoplatforms for tumor therapy due to their unique excellent photothermal conversion rate and low toxicity.[ 16 ] In the present work, single‐layered Ti3C2 MXene flakes (MX) were prepared, and transmission electron microscopy (TEM) images are shown in Figure 1A. Energy dispersive X‐ray (EDX) elemental mapping showed that C and Ti were homogeneously distributed on the whole surface of MX (Figure 1B). MX was subsequently modified by a noncovalent approach with PEI to construct Ti3C2 MXenes@PEI (MXP) (Figure 1C). The zeta potentials of the MX and MXP dispersions were determined to be approximately −28.7 ± 0.5 and 37.6 ± 0.6 mV, respectively (Figure 1D). The MXP having positively charged surfaces indicated that MX was successfully modified by PEI. In addition, fourier transform infrared (FTIR) spectroscopy of MXP at 1650 cm−1 can be assigned to the N—H stretching vibrations from PEI (Figure 1E),[ 17 ] indicating successful modification with PEI.
Figure 1.
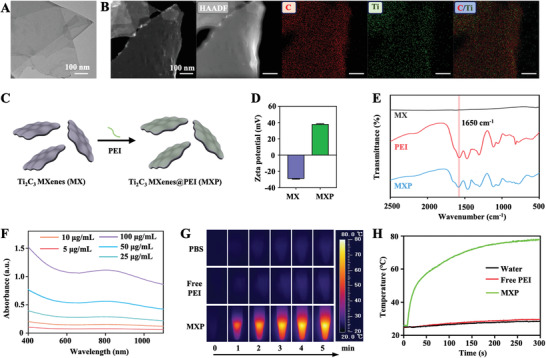
Characterization of MXP. A) TEM images of MX. B) High‐angle angular dark field‐scanning transmission electron microscopy (HAADF‐STEM) and EDX elemental mapping images of MX. Red and green indicate C and Ti elements, respectively. C) Schematic picture of the surface modification of MXP by PEI modification. D) Zeta potentials of MX and MXP. E) Fourier transform infrared (FT‐IR) spectra of MX, free PEI, and MXP dispersions. F) UV–vis spectra of the different concentrations of MXP. G) Thermal images at elevated MXP dispersion under NIR irradiation (808 nm, 1 W cm−2) and H) corresponding photothermal‐heating curves.
As an emerging type of photothermal nanoagent for PTT, MXenes exhibit strong absorption in the NIR region.[ 18 ] The ultraviolet and visible (UV–vis) spectra indicate a stable and extensive optical absorbency from 750 to 850 nm, including both visible and NIR light (Figure 1F). Figure 1G illustrates the temperature elevation of the MXP dispersion treated with 808 nm laser irradiation. The temperature of the MXP dispersion rapidly increased and reached a plateau at approximately 75 °C after 300 s of irradiation at concentrations of 100 µg mL−1 (Figure 1H), which indicated excellent photothermal transformation ability. To further determine the photothermal conversion performance of MXP, different concentrations of MXP were irradiated with an NIR laser (808 nm, 1 W cm−2) for 5 mins. The equilibrium temperature of the MXP dispersion reached approximately 50 and 80 °C at concentrations of 25 and 100 µg mL−1, respectively, after 300 s. This result indicates that the photothermal feature of the MXP dispersion is concentration‐dependent (Figure S1A, Supporting Information). The photothermal conversion efficiency (η) of MXP was calculated to be 35.03% according to the formula (Figure S1B,C, Supporting Information), which is greater than that of the traditional unmodified Ti3C2 MXene (28.17%).[ 19 ] The photothermal stability of MXP was further evaluated by the recycling on‐off irradiation test. The heating/cooling curves show that there is no distinct deterioration of the temperature of the MXP dispersion during multiple cycles (Figure S1D, Supporting Information), indicating the high photothermal stability of MXP. The excellent photothermal performance promises that MXP can be potential photothermal nanoagents for antitumor PTT.
2.2. Photothermal Antitumor Ability of MXP In Vitro
PTT utilizes photothermal materials with high photothermal‐conversion efficiency to convert light energy into thermal energy under NIR illumination, and when the temperature rises to 41 °C, the cell heat shock reaction is initiated, which in turn causes a series of rapid intracellular changes.[ 20 ] The functionalization of photothermal nanoagents under regional NIR irradiation could effectively enhance the efficiency of hyperthermia and decrease the potential side effects.[ 21 ] Based on the above results, MXP possessed great photothermal efficiency. Next, the in vitro PTT properties of MXP for tumor apoptosis was evaluated (Figure 2A). Figure 2B shows that the tumor cell viabilities decreased with increasing MXP concentration under 808 nm NIR irradiation. When the concentration of MXP exceeded 100 µg mL−1 at a power density of 1 W cm−2 for 5 min, nearly 90% of B16‐OVA tumor cells were killed. The viability of tumor cells also decreased with increasing power density at a constant MXP concentration of 100 µg mL−1 (Figure 2C). Dramatic death of B16‐OVA cells was observed when the power density was increased to more than 0.75 W. Therefore, according to the above results, 100 µg mL−1 MXP under 1 W cm−2 for 5 min was selected to conduct PTT on B16‐OVA cells. The visual results of tumor cell death were obtained by calcein‐AM/PI staining. Figure 2D illustrates limited cell cytotoxicity when tumor cells were treated without 808 nm NIR irradiation. The B16‐OVA cells were almost dead in the MXP+NIR group under NIR irradiation for 5 min, while the PBS + NIR group did not show obvious tumor cell death. The penetration and inhibition of MXP for tumor multicellular spheroids were subsequently examined in vitro. MXP was incubated with B16‐OVA multicellular spheroids. Then, the cells were treated with 808 nm laser irradiation for 5 min, and a massive number of dead cells were observed in the center of the multicellular B16‐OVA tumor spheroids (Figure 2E). These data demonstrated that MXP can induce tumor cell death through thermal ablation upon NIR light irradiation.
Figure 2.
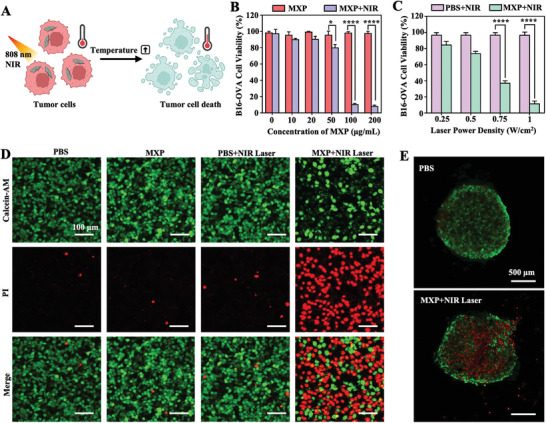
In vitro PTT‐mediated tumor cell death of MXP. A) Schematic illustrating MXP for PTT‐mediated tumor cell death. B) Cell viability of B16‐OVA tumor cells cocultured with MXP nanoparticles at increasing concentrations (0, 10, 20, 50, 100, and 200 µg mL−1) for 2 h with or without irradiation (1 W cm−2, 808 nm, 5 min). C) Relative viability of B16‐OVA tumor cells cocultured with MXP (100 µg mL−1) for 2 h with irradiation by an NIR laser at increasing power densities (0.25, 0.5, 0.75, and 1 W cm−2, 5 min). D) Confocal laser scanning microscopy (CLSM) images of Calcein‐AM/PI‐stained B16‐OVA cells treated with PBS and MXP with or without NIR irradiation. E) CLSM images of B16‐OVA multicellular spheroids treated with PBS and MXP + NIR (100 µg mL−1 MXP under 1 W cm−2 for 5 min).
2.3. Endogenous Nanovaccine Enhanced In Vitro DC Maturation and Activation
Immunogenic cell death (ICD) is triggered by PTT, which can induce tumor cells to release DAMPs and TAAs, activate the antitumor immune response, and regulate the TME.[ 22 ] Therefore, PTT can trigger host immunity to a certain extent.[ 23 ] DAMPs emitted in the course of ICD include calreticulin (CRT) exposed on the cell surface; and non‐histone chromatin‐binding protein high‐mobility group box 1 (HMGB1) and the small metabolite ATP liberated from dying cells into the extracellular space.[ 24 ] DAMPs can be recognized by both the innate and adaptive immune systems, ultimately resulting in the cross‐presentation of tumor antigens to CD8+ CTLs in the context of robust immunostimulation. To evaluate the capacity of MXP to induce ICD in tumor cells, the release of crucial DAMPs, including HMGB1 and ATP, and the exposure of CRT was determined. After the B16‐OVA cells were treated with PBS, NIR, MXP, or MXP+NIR, the release of ATP and HMGB1 from the MXP+NIR group was significantly higher than that from the other groups (Figure S2A,B, Supporting Information). The corresponding Western blot results showed that the intracellular ATP and HMGB1 levels in the MXP+NIR group were significantly lower than those in the other groups (Figure S3C, Supporting Information). Based on the above results, MXP+NIR can induce ICD of tumors and release DAMPs. In addition, treatment of tumor cells with MXP showed remarkable CRT upregulation under NIR irradiation. The CLSM results confirmed the exposure of CRT on the cell surface of the MXP+NIR group (Figure S3, Supporting Information), further verifying the occurrence of ICD.
The effective antigen presentation of DCs is crucial for CTL responses against tumors.[ 25 ] In addition, some carbon‐based nanomaterials have been reported to activate DC maturation after PTT and then trigger subsequent immune responses.[ 7 , 26 ] To determine the PTT‐triggered maturation of DCs, a transwell system was used to determine the ability of MXP to facilitate the maturation of DCs. Promoting the activation and maturation of DCs is an important step in generating effective antigen‐specific T cells. Mature DCs with upregulation of costimulatory molecules, such as CD86, CD80, and CD40, can effectively evoke antitumor immune responses.[ 27 ] The transwell experiment was further applied to determine the immune effect triggered by tumor residues under MXP‐mediated PTT treatment in vitro (Figure 3A). Flow cytometry (FCM) analysis showed that the PBS with laser irradiation and MXP without laser irradiation groups displayed lower CD40 (Figure 3B,C), CD80 (Figure 3D,E), and CD86 (Figure 3F,G) costimulatory molecules than the MXP with laser irradiation group. MXP‐mediated PTT accelerated DC maturation. In addition, DCs can present tumor antigen information by antigen‐MHC I complexes of DCs after the antigen presentation pathway.[ 28 ] To determine whether B16‐OVA tumor cells can release antigen for cross‐presentation of DCs after PTT treatment in vitro, we next determined the efficiency of DC cross‐presentation in different groups. The levels of the SIINFEKL‐H‐2Kb biomarker on DCs in the MXP+NIR groups increased ≈2.5‐fold compared to the levels on DCs within the PBS‐ and MXP‐treated groups (Figure 3H,I). In addition, the secretion of potent Th1‐cytokines (interleukin‐12, IL‐12) and inflammatory cytokines (tumor necrosis factor‐α, TNF‐α) was detected. IL‐12 and TNF‐α were more highly released in the MXP+NIR group than in the PBS and MXP groups (Figure S4A,B, Supporting Information). Therefore, MXP‐based PTT formed an endogenous nanovaccine to inhibit the growth of primary tumors by thermal ablation, and the generated hyperthermia of tumors can also induce ICD and release DAMPs and TAAs under NIR irradiation. The high immunogenicity of residual tumor tissues triggers DC‐mediated immune responses (Figure 3J). These data confirm that MXP is a promising photothermal platform for DC‐mediated tumor immunotherapy.
Figure 3.
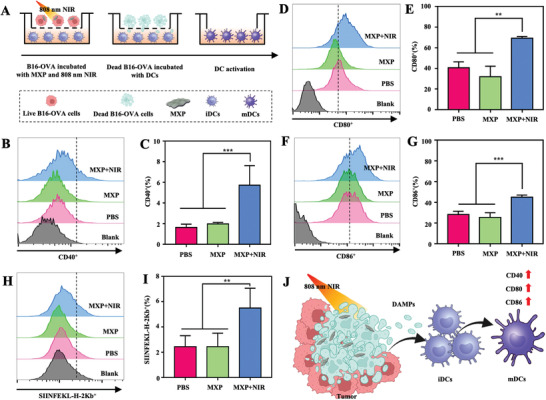
DC maturation triggered by MXP‐mediated PTT in vitro. A) Schematic illustration of the coculture system of B16‐OVA tumor cells and mutuDCs. B16‐OVA cells were cultured in the upper wells, and DCs were cultured in the bottom wells. B16‐OVA cells were treated with MXP and NIR irradiation in the MXP+NIR group. FCM analysis and quantification of B,C) CD40+, D,E) CD80+, and F,G) CD86+ DCs. H) FCM analysis and I) quantification of SIINFEKL‐H‐2Kb+ DCs. J) Schematic illustration of MXP‐mediated PTT through increasing the maturation process of DCs.
2.4. Exogenous MXP@OC Nanovaccine for Promoting Antigen Uptake and Presentation of DCs In Vitro
Enhancing the function of DCs in the circulatory system can be an effective antitumor strategy. Toll‐like receptor (TLR) ligands promote immature DC (iDC) maturation and TLR‐induced cytokine secretion. This process can preferentially guide T cells to differentiate into CTLs[ 29 ] Although immune adjuvants have been widely studied for priming DC‐based immune responses, there are still some substantial limitations in the clinical use of free adjuvants.[ 30 ] For example, CpG‐ODN, a TLR 9 agonist, can initiate humoral and cellular immune responses via immunological cascades. However, the administration of free CpG‐ODN leads to unfavorable biodistribution, especially poor cellular uptake.[ 31 ] Nanocarriers provide an excellent way to achieve carrier functions due to their tunable multicomponent properties, good interaction with cells, and optimal biological distribution.[ 32 ] Current research illustrates that nanocarriers can improve the stability of CpG‐ODN and reduce adverse effects in vivo.[ 33 ]
The advantageous properties of MXenes for application in biomedicine stem from their topology, including their high surface‐to‐volume ratio and mechanical toughness.[ 34 ] In addition, surface modification via conjugation of polymers is aimed at stabilizing MXenes and expanding their biomedical application possibilities to obtain intelligent and versatile therapeutic nanocarriers.[ 35 ] The MXP presented here can also be used as antigen and adjuvant delivery platforms due to the abundant anchors and high specific surface area of MXP. MXP modified with positively charged PEI can promote antigen absorption by DCs, improve antigen escape from lysosomes, and subsequently promote antigen cross‐presentation.[ 36 ] Thus, MXP was used to load OVA antigens and CpG ODNs to construct the MXP@OC nanovaccine, which improved DC‐mediated immune cascade enhancement (Figure 4A). The amount of OVA antigen adsorbed by different concentrations of MXP is shown in Figure S5A, Supporting Information, and MXP also possesses excellent carrying capacity of CpG ODNs (Figure S5B, Supporting Information). The in vitro cell cytotoxicity of the MXP@OC nanovaccine was determined by Annexin V‐PI staining. MXP@OC was practically nontoxic to DCs; even when DCs were treated with 100 µg mL−1 MXP for 24 h, the cell viability was more than 98% (Figure S6A,B, Supporting Information). Moreover, limited cytotoxicity of DCs was shown after treatment with 50 µg mL−1 MXP@OC for 48 h in a Calcein AM‐PI staining experiment (Figure S7, Supporting Information).
Figure 4.
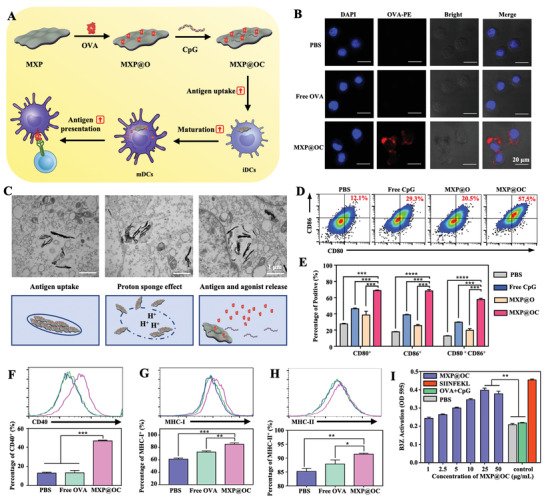
The MXP@OC nanovaccine enhances DC maturation and cross‐presentation. A) Schematic illustration of the preparation of the MXP@OC nanovaccine and promotion of DC maturation and cross‐presentation. B) CLSM results of DCs after treatment with OVA‐PE or MXP@OC‐PE for 6 h. Red, OVA‐PE; blue, nuclei. C) Representative TEM images and schematic illustration showing that treatment with MXP@OC enhances intracellular uptake of antigen, endosomal rupture, and subsequent cytosolic antigen delivery. D) Representative FCM histograms and E) percentages of CD80+ and CD86+ DCs after different treatments. Representative FCM histograms and percentages of F) CD40+, G) MHC I+, and H) MHC II+ DCs after PBS, free OVA, and MXP@OC treatment. I) The relative proportions of activated B3Z cells after coculture with different concentrations of MXP@OC‐treated DCs. Data presented as means ± SD (n = 3). For statistical significance, *P <0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Sufficient antigen internalization is a crucial precondition for the subsequent activation of DCs.[ 37 ] We first determined the antigen cellular uptake of DCs. CLSM results revealed a limited fluorescence signal in DCs treated with free OVA‐PE, while massive amounts of fluorescent PE signal of OVA were observed in DCs treated with MXP@OC‐PE (Figure 4B). The entire process of cellular uptake, endosomal rupture, and cytosolic antigen delivery of MXP@OC in DCs was further determined by TEM. Endosomal membrane destruction and antigen cytosolic exportation were observed in MXP@OC‐treated DCs (Figure 4C). Moreover, free CpG, free CpG with OVA (OC), or MXP‐loaded OVA (MXP@O) were used as controls to determine the adjuvant effect of MXP@OC. The DCs loaded with the MXP@OC nanovaccine showed more upregulation of the CD80 and CD86 costimulatory molecules than PBS‐, free CpG‐, and MXP@O‐loaded DCs (Figure 4D). In addition, CD80+CD86+ DCs are generally considered mature DCs. As shown in Figure 4E, the MXP@OC‐treated DC groups reached a maturity rate of 57.5±0.84%, which was significantly higher than that of the PBS (12.6 ±0.28%), free CpG (29.7 ±0.29%) and MXP@O (19.6 ±1.27%) groups. The CD40 costimulatory molecule was also significantly upregulated ≈3‐fold after MXP@OC treatment of DCs compared with the OC group (Figure 4F).
Additionally, to elicit effective T‐cell immune responses, DCs bind, process, and present antigens to major histocompatibility complex (MHC) molecules on their surface, and then antigens bind to MHC molecules to activate and convert naïve T (Tn) cells into CTLs.[ 38 ] The levels of MHC‐I and MHC‐II were also increased in the MXP@OC group (Figure 4G,H), suggesting the robust immune adjuvant effect of the MXP@OC nanovaccine. We further performed a chlorophenol red‐β‐d‐galactopyranoside (CPRG) assay to assess DC cross‐presentation and B3Z activation.[ 39 ] The quantitative results of B3Z cell activation demonstrated that MXP@OC‐treated DCs induced stronger antigen cross‐presentation of untreated DCs and subsequently increased B3Z‐cell activation similar to the effect of MXP@OC (Figure 4I). Together, these results reveal that MXP can act as antigen carriers in inducing DC‐mediated T‐cell immune responses.
2.5. Exogenous MXP@OC Nanovaccine Promotes DC Migration and Immune Stimulation In Vivo
Lymph nodes (LNs) are important depots of immune cells, and the efficient lymphatic transportation of antigens and adjuvants is crucial for DC‐mediated immune responses.[ 40 ] Studies have shown that activated DCs need to be transported into LNs to initiate T‐cell activation, which is a process necessary for DC‐mediated T‐cell‐dependent immunity.[ 41 ] To verify whether MXP@OC can facilitate the migration of DCs, the vaccines were injected into the footpads of mice. After the subcutaneous injection of PBS, FITC‐labeled OVA with free CpG (OC‐FITC), or MXP@OC‐FITC was injected into the footpads. The proximal popliteal LNs (pLNs) and inguinal LNs (iLNs) of mice were obtained to determine LN draining in vivo (Figure 5A). MXP@OC‐FITC significantly increased the FITC signal in the draining iLNs compared with PBS and OC‐FITC alone.
Figure 5.
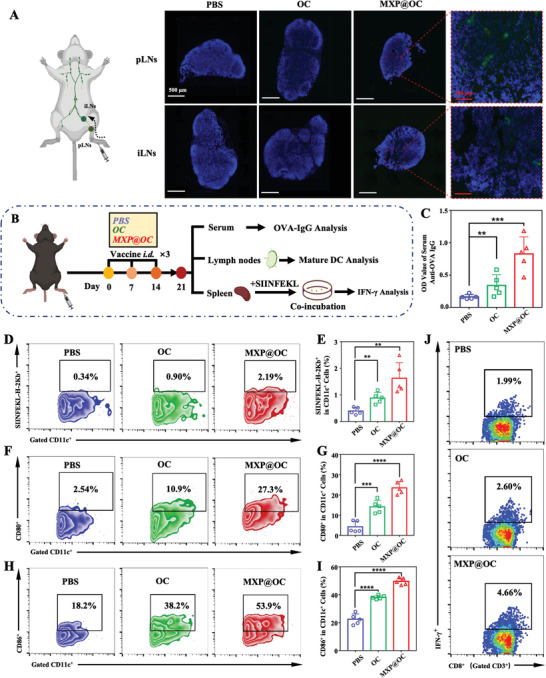
The MXP@OC vaccine enhances lymphatic drainage and DC‐mediated immune responses. A) FITC+ cells in pLNs and iLNs after the administration of PBS, OC, or MXP@OC. Blue, nuclei; green, OVA‐FITC. B) Schematic illustration of the experimental schedule used to evaluate the immune responses to the nanovaccines. C) OVA‐specific immunoglobulin (IgG) production in the harvested serum. D) Ratios of CD11c+SIINFEKL‐H‐2Kb+ cells in LNs as measured by FCM. E) Statistical analysis of CD11c+SIINFEKL‐H‐2Kb+ cells. F,G) Representative FCM plots and statistical analysis of CD11c+ CD80+ cells. H,I) Representative FCM plots and statistical analysis of CD11c+ CD86+ cells. J) Representative FCM images of CD3+CD8+IFN‐γ + T cells in the splenocytes of immunized mice after restimulation with the SIINFEKL peptide. The data are presented as the means ± SD (n = 5/group). For statistical significance, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To determine whether the MXP@OC vaccine can promote DC maturation and activation and subsequently induce CTL immune responses in vivo, fifteen C57BL/6 mice were randomly divided into three groups and then immunized with PBS, OC, or MXP@OC. Immune analysis of harvested serum, iLNs, and splenocytes was performed on day 21 to determine the immunization efficacy (Figure 5B). MXP@OC‐immunized mice had the highest level of OVA‐specific immunoglobulin in harvested serum (Figure 5C), indicating that MXP@OC evoked a greater antigen‐specific response than the other treatments. In addition, to determine whether the MXP@OC nanovaccine can promote DC maturation and activation in iLNs, iLN cells were collected and evaluated by FCM. The ratio of CD11c+SIINFEKL‐H‐2Kb+ DCs in the iLNs of the MXP@OC‐immunized group was greater than that in the PBS or OC groups (Figure 5D,E), which indicated that MXP@OC can activate specific OVA antigen cross‐presentation of DCs. In addition, the number of mature DCs, which highly expressed CD80 and CD86, was significantly increased after MXP@OC immunization (Figure 5F–I), which is consistent with the in vitro transwell results. Greater tumor antigen‐specific DC‐mediated T‐cell immune responses are accompanied by IFN‐γ secretion after a second antigen exposure.[ 42 ] To assess the MXP@OC‐mediated T‐cell response, splenocytes were restimulated with the SIINFEKL peptide, and the intracellular IFN‐γ levels were detected using intracellular cytokine staining (ICS) of FCM. Notably, the ratio of CD3+CD8+IFN‐γ + T cells from the splenocytes of the MXP@OC‐immunized mouse group was ≈2.34‐fold and ≈1.79‐fold higher than those of the PBS‐ and OC‐immunized mouse groups, respectively (Figure 5J, Figure S8A, Supporting Information). Moreover, cell supernatants from restimulated splenocytes were collected and analyzed by ELISA, and it was found that the secretion of IFN‐γ was significantly increased with MXP@OC treatment but not with OC treatment (Figure S8B, Supporting Information). Based on these results, it was concluded that MXP@OC can promote LN transport of DCs and induce stronger antigen‐specific CTL immune responses.
Next, the biocompatibility of the MXP@OC nanovaccine was evaluated before further biomedical application. The hemolysis assay results showed that the MXP@OC nanovaccine had negligible hemolysis effects at 200 µg mL−1 (Figure S9A, Supporting Information). In addition, the in vivo biocompatibility of MXP@OC was investigated by i.v. administration of MXP@OC into C57/BL6 mice. Routine blood and biochemical examinations were completed on the 28th day after i.v. administration of MXP@OC, and the hematology assay indices were within the reference ranges for PBS‐treated mice, indicating that MXP@OC did not cause any physiological disorders (Figure S9B–I, Supporting Information). Moreover, no significant cell death or inflammatory infiltrates were observed in the major organs of the MXP@OC‐treated group (Figure S9J, Supporting Information), further confirming the good biocompatibility of MXP@OC.
2.6. MXP‐Mediated PTT‐Produced Endogenous Nanovaccine Inhibited Tumors In Vivo
Hyperthermia is an effective treatment method to inhibit solid tumors. We further investigated the antitumor efficiency of MXP as photothermal nanoagents under NIR light. Tumor‐bearing mice were randomly divided into four groups for different treatments (PBS, PBS+NIR, MXP, and MXP+NIR) after B16‐OVA tumor model establishment (Figure 6A). The temperature of MXP in the tumor was elevated from 36.6 to 58.0 °C within 5 min, sufficient for tumor apoptosis (Figure 6B,C). When the temperatures reach more than 40 °C, tumor cells can be killed due to denaturation of cell proteins, subsequent disruption of cellular membranes and the cytoskeleton, and damage to DNA. During the 24‐day observation period, the PBS, PBS+NIR, and MXP groups did not show a considerable benefit in tumor suppression; in strong contrast, the MXP+NIR group significantly inhibited tumor growth (Figure 6D, Figure S10, Supporting Information). The different treatment effects of tumor‐bearing mice were shown by survival curves. The survival rate in the MXP+NIR group was significantly improved compared with that in the PBS, PBS+NIR, and MXP groups (Figure 6E). The tumor volumes were evidently decreased in the MXP+NIR group compared to the other groups after 24 days (Figure 6F). Tumors were dissected at day 24, and H&E and TUNEL staining of tumors were performed for histopathological examination (Figure 6G). Significantly broken and small nuclei were observed in the tumors of MXP+NIR‐treated mice. Meanwhile, the TUNEL image showed a significant enhancement of green fluorescence, indicating that MXP‐mediated PTT induces a large amount of apoptosis in tumor cells. In addition, tumors were collected and digested for FCM. The results clearly illustrated that the percentage of mature DCs (CD11c+CD80+ and CD11c+CD86+ cells) from tumor tissue in the MXP+NIR‐treated group was significantly increased (Figure 6H,I). In conclusion, MXP‐basedPTT could effectively inhibit tumor growth, promote intratumoral DC maturation, and extend the survival time of tumor‐bearing mice.
Figure 6.
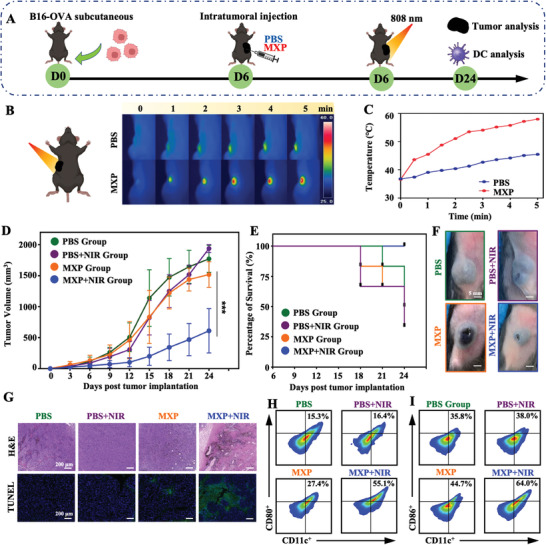
In vivo antitumor experiments of MXP‐mediated PTT. A) Schematic illustration of the establishment of B16‐OVA tumor models and the treatment procedure. B) Temperature elevation in the tumor site after intratumoral injection of PBS or MXP under NIR light irradiation (808 nm, 1 W cm−2). C) Corresponding IR thermal images of the established tumor site. D) Average tumor growth curves of different groups (n = 6 independent mice per group). E) Survival rates after treatment. F) Photographs of B16‐OVA tumor‐bearing mice in different treatment groups. G) Histological microscopy images of tumors after different treatments. H,I) Representative FCM plots of H) CD11c+CD80+ cells and I) CD11c+CD86+ cells in tumor tissues of different groups.
2.7. Combined Endogenous/Exogenous Nanovaccine Efficiently Eradicated Tumors
A combination of photothermal therapy and DC‐mediated immunotherapy may be an ideal synergistic strategy to improve antitumor efficacy. In vitro results inspired us to combine immunoadjuvants with MXP while loading tumor model antigen OVA to enhance DC‐mediated immune stimulation, which can trigger a strong immune response and induce long‐term immunity to antitumor. To determine the effect on eradication of primary tumors, B16‐OVA tumor‐bearing mice were randomly divided into four treatment groups: OC, MXP@OC, MXP+NIR+OC, and MXP+NIR+MXP@OC. The tumor‐bearing mice were sacrificed on day 24, and tumor and immune cells were analyzed (Figure 7A). Tumor growth was relatively rapid in the OC and MXP@OC groups and slow in the MXP+NIR+OC and MXP+NIR+MXP@OC groups, demonstrating synergistic therapeutic efficacy (Figure 7B, Figure S11, Supporting Information). As shown in Figure 7C, the tumor almost disappeared in the MXP+NIR+MXP@OC group, which indicated that synergistic therapy can achieve significant tumor eradication efficiency. More severe damage and volume decrease in tumors were exhibited in the MXP+NIR+MXP@OC group. Next, H&E and TUNEL staining was performed to determine the effect of treatment on the tumor (Figure 7D,E). Compared with the intact and dense tumor tissue in the OC and MXP@OC groups, shrinkage and fragmentation of tumor cells appeared, and ratios of TUNEL+ tumor cells were enhanced in the MXP+NIR+OC and MXP+NIR+MXP@OC groups, especially in the MXP+NIR+MXP@OC group.
Figure 7.
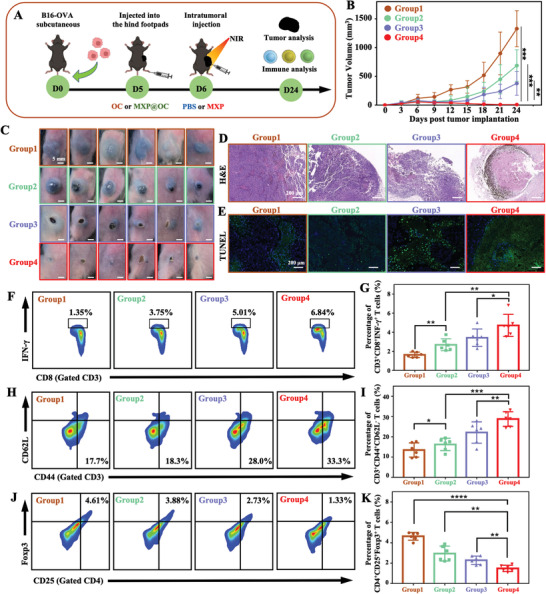
Combining MXP@OC nanovaccine with MXP‐mediated PTT. A) Schematic illustration of the establishment of B16‐OVA tumor models that were then immunized with OC or MXP@OC and treated with MXP combined with NIR light irradiation (808 nm, 1 W cm−2, 5 min, n = 6/group). B) Average tumor growth curves (n = 6 independent mice per group). (Group 1: OC; Group 2: MXP@OC; Group 3: MXP+NIR+OC; Group 4: MXP+NIR+MXP@OC). C) Photographs of B16‐OVA tumor‐bearing mice in the four groups. D) H&E and E) TUNEL staining images of stripped tumors after different treatments. F) Representative FCM plots and G) statistical analysis and the percentages of CD3+CD8+IFN‐γ + T cells. H) Representative FCM plots and I) percentages of CD3+CD44+CD62L− effector memory T cells in tumor tissues. J) Representative FCM plots and K) percentages of CD4+CD25+Foxp3+ Tregs in tumor tissues. For statistical significance, *P<0.05; **P<0.01;
Spleen and tumor tissues were collected to evaluate immune responses and further determine the underlying antitumor mechanism of the synergistic treatment. As a method to assess MXP+NIR+MXP@OC treatment‐induced antigen‐specific T‐cell immunity, splenocytes isolated from mice in different groups were restimulated with the SIINFEKL peptide, and the ICS of FCM was performed to determine the intracellular IFN‐γ levels of CTLs. The percentage of CD3+CD8+IFN‐γ + T cells in splenocytes in the MXP+NIR+MXP@OC group was ≈5.1‐fold, ≈1.8‐fold, and ≈1.3‐fold higher than that in the OC, MXP@OC, and MXP+NIR+OC groups, respectively (Figure 7F,G). Then, we determined the percentages of effector memory (CD3+CD44+CD62L−) T‐cell infiltration in the tumor tissues, which can induce antitumor protection effects against rechallenged tumors.[ 43 ] Mice treated with MXP+NIR+MXP@OC exhibited significantly increased populations of effector memory T cells compared to other groups (Figure 7H,I), indicating that the synergistic strategy effectively induced immunological memory for long‐lasting protection. Moreover, DC vaccines cannot effectively activate the antitumor response in vivo due to immunosuppressive cells from the TME.[ 44 ] These immunosuppressive cells prevent tumor‐infiltrated DCs from evoking CTL responses and then causing tumor progression.[ 45 ] The administration of MXP+NIR+MXP@OC also contributed to a decreased infiltration of immunosuppressive CD4+CD25+Foxp3+ Tregs (1.48±0.13%) in the tumor tissues compared with the OC (4.64±0.16%), MXP@OC (2.94±0.30%), and MXP+NIR+OC (2.29±0.17%) groups (Figure 7J,K). Briefly, synergistic treatment with MXP+NIR+MXP@OC triggered robust CTL immune and immunological memory responses, and decreased the percentage of immunosuppressive Tregs. These results proved that the combination of MXP‐mediated PTT and the MXP@OC nanovaccine was more beneficial for promoting antitumor immunity and eradicating primary tumors.
3. Conclusion
We reported endogenous/exogenous nanovaccines to achieve effective DC‐mediated antitumor therapy. MXP induces ICD of tumor cells under NIR irradiation and thus functions as an endogenous nanovaccine, which subsequently stimulates DC maturation in tumors. Meanwhile, as an exogenous nanovaccine, MXP has excellent biocompatibility and can also deliver antigens and CpG‐ODNs to initiate an efficient DC‐mediated immune response. The photothermal and immune synergistic treatment strategy (MXP+NIR+ MXP@OC) promoted the maturation and antigen processing of DC, evoked the CTL immune responses, alleviated tumor immunosuppression in the TME, and resulted in robust systemic antitumor immune responses in vivo. In summary, this endogenous/exogenous nanovaccine provides a promising synergistic therapeutic strategy for eradicating tumors.
4. Experimental Section
Chemicals and Materials
Concentrated hydrochloric acid (HCl, 37 wt.%) was obtained from Shanghai Jiuyi Chemical Reagent Co., Ltd. (Shanghai, China). Branched PEI (MW, 25 000), dimethyl sulfoxide (DMSO), and OVA were purchased from Sigma–Aldrich (MO, USA). Chlorophenol red‐β‐d‐galactopyranoside (CPRG) was purchased from MREDA (Beijing, China). Dulbecco's modified Eagle's medium (DMEM), Roswell Park Memorial Institute (RPMI) 1640 medium, and phosphate‐buffered saline (PBS) were purchased from HyClone (UT, USA). Fetal bovine serum (FBS) and 2‐mercaptoethanol were purchased from Gibco (CA, USA). The FCM antibodies, including anti‐CD80, anti‐CD86, anti‐CD40, anti‐SIINFEKL‐H2Kb, anti‐CD11c, anti‐MHC I/II, anti‐CD3, anti‐CD4, anti‐CD8, anti‐IFN‐γ, anti‐CD44, anti‐CD62L, anti‐CD25, and anti‐Foxp3, were purchased from eBioscience (CA, USA).
Preparation of MX and MXP
Ti3C2 MXenes (MX) were obtained from Shiyanjia Lab. In brief, MX was fabricated from Ti3AlC2 by chemical exfoliation by first slowly dissolving 0.8 g LiF in 10 mL of HCl and stirring for 5 min. Then, 0.5 g Ti3AlC2 was slowly added, stirred at 45 °C for 24 h, washed with deionized water, and dried overnight. PEI was modified on the surface of MX to endow the nanoparticles (NPs) with the same surface properties. Briefly, the MX suspension (10 mg mL−1) was dispersed in 25 kDa branched PEI solution (10 mg mL−1) and stirred at 4 °C for 12 h. The obtained MXP were collected by centrifugation at 12000 rpm for 30 min with deionized water 3 times, freeze‐dried, and stored at −20 °C.
Assessment of Photothermal Conversion Efficiency
Aqueous suspensions of MXP (100 µg mL−1, 1 mL) were irradiated by an 808 nm NIR laser (1 W cm−2) for 5 min and then cooled naturally. The photothermal conversion efficiency (PTCE) was calculated using the following equation:
| (1) |
(h: heat transfer coefficient, S: exposed surface area of the cuvette, T max: maximum temperature at equilibrium, T amb: minimum temperature at equilibrium, Q 0: heat absorbed by the container, I: incident laser power in W, A: absorbance at 808 nm).
Cell Culture and Mice
The DC2.4 cell line and B16‐expressing OVA (B16‐OVA) tumor cell line were obtained from Procell Life Science & Technology Co., Ltd. (Wuhan, China). The mutuDC cell line and B3Z cell line were gifts from Prof. Hong‐ming Hu (Providence Portland Medical Center; OR, USA). The mutuDCs were cultured in DMEM with 10% FBS (Gibco), 0.05 mm 2‐mercaptoethanol (Gibco), and 2 mm l‐glutamine (Invitrogen, Thermo Fisher Scientific). DC2.4 and B16‐OVA tumor cells were cultured in RPMI 1640 medium with 10% FBS. B3Z cells were cultured in RPMI 1640 medium with 8% FBS, 0.05 mM 2‐mercaptoethanol, and 2 mm L‐glutamine.
Female C57BL/6 mice aged 6–8 weeks were purchased from Shanghai Laboratory Animal Research Center (Shanghai, China). Mouse feeding, care protocols, and experiments strictly followed the ethical standards of animal experiments at the Medical School of Nanjing University (IACUC‐2003034).
OVA Antigen Loading
The loading efficiency of the model antigen OVA in the nanovaccines was determined using the Pierce BCA Protein Assay Kit. Briefly, different MXP suspensions (final concentrations: 5, 100, 200, and 400 µg mL−1) were mixed with OVA solution (1 mg mL−1). After the mixtures were incubated at 4 °C for 12 h, the supernatants were collected after centrifugation at 13 500 rpm for 10 min, and the OVA concentrations in the supernatants were detected using the BCA protein assay kit according to the manufacturer's protocols.
Penetration of MXP in Multicellular Tumor Spheroids
100 µL of warm agarose solution (16 mg mL−1, dissolved in RPMI 1640 medium) was added into 96‐well plates and cooled at room temperature to form an agarose gel. Then, 100 µL of B16‐OVA cells (1 × 105 cells well−1) was seeded on the gel and cultured at 37 °C with 5% CO2 for 7 days to prepare multicellular spheroids. Multicellular spheroids were co‐incubated with MXP (100 µg mL−1) for 4 h and then treated with NIR (1 W cm−2 for 5 min). Multicellular spheroids were transferred to a confocal dish for fluorescence detection. The fluorescence of Calcein‐AM/PI‐stained B16‐OVA multicellular spheroids was observed using a confocal laser scanning microscopy (CLSM).
Immunogenic Cell Death (ICD) Induced by MXP and NIR Irradiation
The intuitive immunofluorescence detection of CRT expressed on the cell surface was carried out. B16‐OVA tumor cells (1 × 105 cells well−1) were seeded into the coverglass bottom dish and cultivated overnight. Next, B16‐OVA cells were treated with PBS, NIR, MXP, or MXP+NIR. In the MXP+NIR group, B16‐OVA cells were incubated with MXP for 4 h, irradiated with an NIR laser (808 nm, 1 W cm−2, 5 min), and cultured for another 12 h. Next, tumor cells in different groups were washed with PBS and fixed using 4% formaldehyde for 15 min. Then, the cells were blocked with 5% BSA solution for 1 h at room temperature, and an anti‐CRT antibody (Invitrogen, dilution 1:100) was added and incubated for 12 h at 4 °C. After rinsing with TBST three times, cells were stained with Alexa Fluor 647‐labeled goat antibody (Abcam, dilution 1:150), nuclei were stained with DAPI, and the cytoskeleton was stained with phalloidin‐FITC (Sigma–Aldrich). Finally, a confocal laser scanning microscope was employed for observation and imaging.
The detection of HMGB1 release was performed using an ELISA kit (Elabscience), and extracellular secretion of ATP was evaluated with a commercially available ATP assay kit (Beyotime). Briefly, B16‐OVA tumor cells (5× 105 cells well−1) were seeded in 6‐well plates and cultivated overnight. Next, the tumor cells were treated with PBS, MXP, or MXP+NIR. After 4 h of incubation, the B16‐OVA cells in the MXP+NIR group were irradiated with an NIR laser (808 nm, 1 W cm−2, 5 min) and cultured for another 12 h. Supernatants were collected and cleared from dying tumor cells by centrifugation (800 g, 5 min) and then immediately analyzed for HMGB1 or ATP abundance. In addition, the detection of intracellular HMGB1 and ATP expression was performed by Western blot. Proteins, harvested from tumor cells in different groups, were separated using SDS‐PAGE gel preparation kit with gel electrophoresis and then transferred to PVDF membranes. After being treated with corresponding antibodies and secondary antibodies, the expression of HMGB1, ATP, and GAPDH was detected.
Transwell Experiment In Vitro
B16‐OVA tumor cells (1 × 105 cells well−1) were seeded into the upper transwell chamber and cultivated overnight. Next, the tumor cells were treated with PBS, MXP, or MXP+NIR. After 12 h of incubation, the B16‐OVA cells in the MXP+NIR group were irradiated with an NIR laser (808 nm, 1 W cm−2, 5 min). Then, mutuDCs (5 × 105 cells well−1) were cultivated in the lower transwell compartment for 12 h. Finally, the DCs were collected and stained with anti‐CD80, anti‐CD86, anti‐CD40, and anti‐SIINFEKL‐H2Kb antibodies and examined by FCM.
Analysis of DC Surface Markers by FCM
Collected mutuDCs were seeded into 12‐well plates at a density of 2 × 105 cells per well and treated with PBS, OVA, MXP, or MXP@OC (50 µg mL−1 for MXP and 50 µg mL−1 for OVA) for 24 h. DCs were harvested and stained with anti‐CD40, anti‐CD80, anti‐CD86, anti‐MHC I, and anti‐MHC II antibodies for FCM analysis.
Chlorophenol Red‐β‐D‐Galactopyranoside (CPRG) Assay
B3Z T cells are a CD8+ T‐cell hybridoma specifically expressing the LacZ gene when the T‐cell receptor recognizes the OVA257–264 epitope (SIINFEKL) in the context of the MHC class I H2Kb molecule. The activation of B3Z T cells could be quantified by the CPRG assay. In brief, mutu DCs (2 × 104 cells mL−1) were treated with different concentrations of MXP@OC for 6 h, and then B3Z T cells (2 × 105 cells mL−1) were added to the wells and cocultured overnight. PBS and OVA alone (100 µg mL−1) were used as negative controls, and the SIINFEKL peptide (1 µg mL−1) was used as a positive control. Then, absorbance at 595 nm was measured to determine the cross‐presentation ability of DCs.
Mouse Immunization
Female C57BL/6 mice were randomly divided into 3 groups (n = 5), and 25 µL of PBS, OC, or MXP@OC (50 µg for MXP and 50 µg for OVA) was subcutaneously injected into the hind footpads on days 0, 7, and 14 as immunizations. Seven days after the last immunization, peripheral blood, iLNs, and spleens were harvested to evaluate the immune response. Peripheral blood was coagulated at 37 °C for 2 h, refrigerated overnight at 4 °C, and then centrifuged at 3000 rpm at 4 °C for 10 min to obtain serum. Serum OVA‐specific IgG levels were measured using ELISA, and the OD at 450 nm represented the antibody concentration. The serum IFN‐γ concentration was detected using a mouse IFN‐γ ELISA kit (MultiSciences) according to the manufacturer's protocol.
The iLNs close to the treated tumors were collected and homogenized to prepare single‐cell suspensions for evaluating DC maturation. After being stained with FITC‐conjugated anti‐CD11C, APC‐conjugated anti‐CD80, and PE‐conjugated anti‐CD86 according to the manufacturer's protocol, these cells were subjected to FCM analysis. The spleens of immunized mice were ground and filtered through 200‐mesh nylon mesh filters to obtain single‐cell suspensions of splenocytes, which were then seeded into 96‐well plates at a density of 1 × 106 cells well−1 and cocultured with RPMI‐1640 medium in the presence of 10% FBS, 1% P/S, 10 µg mL−1 SIINFEKL, and brefeldin A/monensin mixture at 37 °C for 6 h to measure the activation of OVA‐specific cytotoxic T lymphocytes (CTLs). Then, ICS was performed to detect intracellular IFN‐γ levels using FCM. Briefly, SIINFEKL peptide‐restimulated splenocytes were stained with anti‐CD3 and anti‐CD8 antibodies for surface staining, fixed and permeabilized using a BD Cytofix/Cytoperm kit, and intracellularly stained with an anti‐IFN‐γ antibody in BD Perm/Wash buffer.
Thermal Imaging In Vivo
Tumor‐bearing mice were intratumorally injected with PBS (50 µL) and MXP (MXP dose of 100 mg mL−1, 50 µL). At different time intervals (1, 2, 3, 4, and 5 min post injection), the mice were irradiated with an 808 nm laser (1 W cm−2, 5 min), and the highest temperature at the tumor site was recorded.
Photothermal Tumor Therapy In Vivo
When the tumor size exceeded 2000 cm3, the mice were sacrificed according to ethical requirements. Twenty‐four B16‐OVA‐tumor‐bearing mice were randomly divided into four groups (n = 6/group): 1) control group: intratumoral injection of PBS (50 µL,); 2) MXP group: intratumoral injection of MXP dispersion (100 mg mL−1, 50 µL); 3) PBS+NIR group: intratumoral injection of PBS (50 µL) with NIR light irradiation (808 nm, 1 W cm−2, 5 min); and 4) MXP + NIR group: intratumoral injection of MXP dispersion (100 mg mL−1, 50 µL) with NIR light irradiation (808 nm, 1 W cm−2, 5 min). Tumor sizes were measured and photographs of the mice were taken every 3 days. The tumor volumes were measured using a Vernier caliper and calculated using the formula V = 1/2 × a × b2, where a is the largest diameter (length) and b is the smallest diameter (width) of the tumor. The mice were sacrificed on the 24th day. Tumor tissues were also stained with H&E and TUNEL. The primary tumors were collected to analyze CD11c+CD80+ and CD11c+CD86+ DCs by FCM.
Combination Tumor Therapy In Vivo
For in vivo combination treatment, 24 B16‐OVA tumor‐bearing mice were randomly divided into 4 groups and received the corresponding treatments as follows: group 1, OC; group 2, MXP@OC; group 3, MXP+NIR+OC; and group 4, MXP+NIR+MXP@OC. These mice received injection into the hind footpads of OC or MXP@OC and MXP+NIR (808 nm, 1 W cm−2, 5 min). The tumor size of each mouse was recorded until the 24th day. The spleens and tumors were harvested to assess the immune response of tumor‐bearing mice in each group. Intracellular IFN‐γ levels in restimulated splenocytes were detected using ICS as described above. In addition, these treated tumors were also collected and homogenized for the preparation of single‐cell suspensions and stained using corresponding fluorophore‐conjugated antibodies. Then, these stained cells were subjected to FCM analysis to determine the tumor‐infiltrating percentages of CD3+CD44+CD62L− effector memory T cells and CD4+CD25+Fxop3+ Tregs.
Biocompatibility Analysis: Histological Examination Ex Vivo
After treatment, the mice were sacrificed, and the mean organ tissues (heart, liver, spleen, lung, kidney, and tumor) were dissected for histological analysis. After dehydration and staining with hematoxylin and eosin (H&E), all slices were embedded in paraffin cassettes. The H&E‐stained images were collected by microscopy at a magnification of 400×. The blood of the other ten mice was collected for the hematology assay.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software; CA, USA). All data were presented as the means ±SD. Significant differences between two groups were determined using the unpaired two‐tailed Student's t test. One‐way or two‐way ANOVA with multiple comparison tests was performed as indicated. Significant differences were shown as follows: ns, not significant; P >0.05; *P <0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
Y.Z. and Q.L. contributed equally to this work. The authors acknowledge funding from the National Natural Science Foundation of China (81971675), the Key Project of Research and Development Plan of Jiangsu Province (BE2020629), Natural Science Foundation of Jiangsu Province (BK20191382, BK20200710), the Nanjing Medical Science and Technique Development Foundation (YKK19094), and “3456” Cultivation Program for Junior Talents of Nanjing Stomatological Hospital, Medical School of Nanjing University (0222R212). The authors appreciate the assistance of Chaofeng He in the Shiyanjia Lab (www.shiyanjia.com) for the TEM tests and Jiangsu KeyGEN bioTECH for H&E tests. The authors also want to thank BioRender (biorender.com) for providing drawing elements and the professional English language editing service of AJE.
Zhang Y., Li Q., Ding M., Xiu W., Shan J., Yuwen L., Yang D., Song X., Yang G., Su X., Mou Y., Teng Z., Dong H., Endogenous/Exogenous Nanovaccines Synergistically Enhance Dendritic Cell‐Mediated Tumor Immunotherapy. Adv. Healthcare Mater. 2023, 12, 2203028. 10.1002/adhm.202203028
Contributor Information
Yongbin Mou, Email: yongbinmou@nju.edu.cn.
Zhaogang Teng, Email: iamzgteng@njupt.edu.cn.
Heng Dong, Email: dongheng90@smail.nju.edu.cn.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Palucka K., Banchereau J., Immunity 2013, 39, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burbage M., Amigorena S., Nature 2020, 584, 533. [DOI] [PubMed] [Google Scholar]
- 3. Tang M., Diao J., Cattral M. S., Cell. Mol. Life Sci. 2017, 74, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Jiang Y., Huang J., Xu C., Pu K., Nat. Commun. 2021, 12, 742; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu J., Xu L., Wang C., Yang R., Zhuang Q., Han X., Dong Z., Zhu W., Peng R., Liu Z., ACS Nano 2017, 11, 4463; [DOI] [PubMed] [Google Scholar]; c) Tian H., Wang G., Sang W., Xie L., Zhang Z., Li W., Yan J., Tian Y., Li J., Li B., Dai Y., Nano Today 2022, 43, 101405. [Google Scholar]
- 5.a) Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Nat. Mater. 2017, 16, 489; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hassani Najafabadi A., Zhang J., Aikins M. E., Najaf Abadi Z. I., Liao F., Qin Y., Okeke E. B., Scheetz L. M., Nam J., Xu Y., Adams D., Lester P., Hetrick T., Schwendeman A. A.‐O., Wicha M. S., Chang A. E., Li Q., Moon J. A.‐O., Nano Lett. 2020, 20, 7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Min Y., Roche K. C., Tian S., Eblan M. J., McKinnon K. P., Caster J. M., Chai S., Herring L. E., Zhang L., Zhang T., DeSimone J. M., Tepper J. E., Vincent B. G., Serody J. S., Wang A. Z., Nat. Nanotechnol. 2017, 12, 877; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chao Y., Liang C., Tao H., Du Y., Wu D., Dong Z., Jin Q., Chen G., Xu J., Xiao Z., Chen Q., Wang C., Chen J., Liu Z., Sci. Adv. 2020, 6, eaaz4204; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yang Z., Gao D., Guo X., Jin L., Zheng J., Wang Y., Chen S., Zheng X., Zeng L., Guo M., Zhang X., Tian Z., ACS Nano 2020, 14, 17442; [DOI] [PubMed] [Google Scholar]; d) Chen Z., Liu L., Liang R., Luo Z., He H., Wu Z., Tian H., Zheng M., Ma Y., Cai L., ACS Nano 2018, 12, 8633. [DOI] [PubMed] [Google Scholar]
- 7.a) Li X., Lovell J. F., Yoon J., Chen X., Nat. Rev. Clin. Oncol. 2020, 17, 657; [DOI] [PubMed] [Google Scholar]; b) Wang C., Xu L., Liang C., Xiang J., Peng R., Liu Z., Adv. Mater. 2014, 26, 8154; [DOI] [PubMed] [Google Scholar]; c) Huang B., Huang Y., Han H., Ge Q., Yang D., Hu Y., Ding M., Su Y., He Y., Shao J., Chu J., Front. Bioeng. Biotechnol. 2021, 9, 751757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang S., Zhang Q., Luo X. F., Li J., He H., Yang F., Di Y., Jin C., Jiang X. G., Shen S., de Fu L., Biomaterials 2014, 35, 9473. [DOI] [PubMed] [Google Scholar]
- 9. Li Z., Lai X., Fu S., Ren L., Cai H., Zhang H., Gu Z., Ma X., Luo K., Adv. Sci. 2022, 9, 2201734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan W., Yung B., Huang P., Chen X., Chem. Rev. 2017, 117, 13566. [DOI] [PubMed] [Google Scholar]
- 11.a) Chen L., Ma X., Dang M., Dong H., Hu H., Su X., Liu W., Wang Q., Mou Y., Teng Z., Adv. Healthcare Mater. 2019, 8, 1900039; [DOI] [PubMed] [Google Scholar]; b) Li Q., Teng Z., Tao J., Shi W., Yang G., Zhang Y., Su X., Chen L., Xiu W., Yuwen L., Dong H., Mou Y., Small 2022, 18, 2201108. [DOI] [PubMed] [Google Scholar]
- 12. Liu Y., Zhou Z., Hou J., Xiong W., Kim H., Chen J., Zheng C., Jiang X., Yoon J., Shen J., Adv. Mater. 2022, 34, 2206121. [DOI] [PubMed] [Google Scholar]
- 13. VahidMohammadi A., Rosen J., Gogotsi Y., Science 2021, 372, eabf1581. [DOI] [PubMed] [Google Scholar]
- 14. Zhu Y., Wang Z., Zhao R., Zhou Y., Feng L., Gai S., Yang P., ACS Nano 2022, 16, 3105. [DOI] [PubMed] [Google Scholar]
- 15.a) Fernandes N., Rodrigues C. F., Moreira A. F., Correia I. J., Biomater. Sci. 2020, 8, 2990; [DOI] [PubMed] [Google Scholar]; b) Tian Y., Younis M. R., Tang Y., Liao X., He G., Wang S., Teng Z., Huang P., Zhang L., Lu G., J. Nanobiotechnol. 2021, 19, 365; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xuan J., Wang Z., Chen Y., Liang D., Cheng L., Yang X., Liu Z., Ma R., Sasaki T., Geng F., Angew. Chem., Int. Ed. Engl. 2016, 55, 14569; [DOI] [PubMed] [Google Scholar]; d) Song M., Pang S. Y., Guo F., Wong M. C., Hao J., Adv. Sci. 2020, 7, 2001546; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Han X., Huang J., Lin H., Wang Z., Li P., Chen Y., Adv. Healthcare Mater. 2018, 7, 1701394; [DOI] [PubMed] [Google Scholar]; f) Yang D., Chen F., He S., Shen H., Hu Y., Feng N., Wang S., Weng L., Luo Z., Wang L., New J. Chem. 2019, 43, 13256. [Google Scholar]
- 16.a) Liu G., Zou J., Tang Q., Yang X., Zhang Y., Zhang Q., Huang W., Chen P., Shao J., Dong X., ACS Appl. Mater. Interfaces 2017, 9, 40077; [DOI] [PubMed] [Google Scholar]; b) Li Z., Zhang H., Han J., Chen Y., Lin H., Yang T., Adv. Mater. 2018, 30, 1706981; [DOI] [PubMed] [Google Scholar]; c) Liu A., Liu Y., Liu G., Zhang A., Cheng Y., Li Y., Zhang L., Wang L., Zhou H., Liu J., Wang H., Chem. Eng. J. 2022, 448, 137691. [Google Scholar]
- 17.a) Zeng H., Wang L., Zhang D., Yan P., Nie J., Sharma V. K., Wang C., Chem. Eng. J. 2019, 358, 253; [Google Scholar]; b) Deng S., Liu X., Liao J., Lin H., Liu F., Chem. Eng. J. 2019, 375, 122086. [Google Scholar]
- 18. Tao N., Zhang D., Li X., Lou D., Sun X., Wei C., Li J., Yang J., Liu Y. N., Chem. Sci. 2019, 10, 10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu K., Liao Y., Zhou Z., Zhang L., Jiang Y., Lu H., Xu T., Yang D., Gao Q., Li Z., Tan S., Cao W., Chen F., Li G., Biomaterials 2022, 282, 121383. [DOI] [PubMed] [Google Scholar]
- 20.a) Richter K., Haslbeck M., Buchner J., Mol. Cell 2010, 40, 253; [DOI] [PubMed] [Google Scholar]; b) Lin H., Wang X., Yu L., Chen Y., Shi J., Nano Lett. 2017, 17, 384. [DOI] [PubMed] [Google Scholar]
- 21. Yuwen L., Zhou J., Zhang Y., Zhang Q., Shan J., Luo Z., Weng L., Teng Z., Wang L., Nanoscale 2016, 8, 2720. [DOI] [PubMed] [Google Scholar]
- 22.a) Liang R., Liu L., He H., Chen Z., Han Z., Luo Z., Wu Z., Zheng M., Ma Y., Cai L., Biomaterials 2018, 177, 149; [DOI] [PubMed] [Google Scholar]; b) Guo R., Wang S., Zhao L., Zong Q., Li T., Ling G., Zhang P., Biomaterials 2022, 282, 121425. [DOI] [PubMed] [Google Scholar]
- 23. Xu P., Liang F., Int. J. Nanomed. 2020, 15, 9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fucikova J., Kepp O., Kasikova L., Petroni G., Yamazaki T., Liu P., Zhao L., Spisek R., Kroemer G., Galluzzi L., Cell Death Dis. 2020, 11, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong H., Su H., Chen L., Liu K., Hu H. M., Yang W., Mou Y., Cancer Manage. Res. 2018, 10, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang H., Pan X., Wang X., Wang W., Huang Z., Gu K., Liu S., Zhang F., Shen H., Yuan Q., Ma J., Yuan W., Liu H., ACS Nano 2020, 14, 2847. [DOI] [PubMed] [Google Scholar]
- 27.a) Gardner A., Ruffell B., Trends Immunol. 2016, 37, 855; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qin L., Zhang H., Zhou Y., Umeshappa C. S., Gao H., Small 2021, 17, 2006000. [DOI] [PubMed] [Google Scholar]
- 28. Joffre O. P., Segura E., Savina A., Amigorena S., Nat. Rev. Immunol. 2012, 12, 557. [DOI] [PubMed] [Google Scholar]
- 29.a) Haining W. N., Davies J., Kanzler H., Drury L., Brenn T., Evans J., Angelosanto J., Rivoli S., Russell K., George S., Sims P., Neuberg D., Li X., Kutok J., Morgan J., Wen P., Demetri G., Coffman R. L., Nadler L. M., Clin. Cancer Res. 2008, 14, 5626; [DOI] [PubMed] [Google Scholar]; b) Cho H. J., Takabayashi K., Cheng P. M., Nguyen M. D., Corr M., Tuck S., Raz E., Nat. Biotechnol. 2000, 18, 509; [DOI] [PubMed] [Google Scholar]; c) Wang S., Campos J., Gallotta M., Gong M., Crain C., Naik E., Coffman R. L., Guiducci C., Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E7240; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yazdani M., Gholizadeh Z., Nikpoor A. R., Mohamadian Roshan N., Jaafari M. R., Badiee A., Sci. Rep. 2021, 11, 14661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reed S. G., Orr M. T., Fox C. B., Nat. Med. 2013, 19, 1597. [DOI] [PubMed] [Google Scholar]
- 31.a) Zhao Q., Matson S., Herrera C. J., Fisher E., Yu H., Krieg A. M., Antisense Res. Dev. 1993, 3, 53; [DOI] [PubMed] [Google Scholar]; b) Shi F., Hoekstra D., J. Controlled Release 2004, 97, 189; [DOI] [PubMed] [Google Scholar]; c) Shih T. Y., Najibi A. J., Bartlett A. L., Li A. W., Mooney D. J., Biomaterials 2021, 279, 121240; [DOI] [PubMed] [Google Scholar]; d) Krieg A. M., J. Clin. Invest. 2007, 117, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pan J., Wang Y., Zhang C., Wang X., Wang H., Wang J., Yuan Y., Wang X., Zhang X., Yu C., Sun S. K., Yan X. P., Adv. Mater. 2018, 30, 1704408. [Google Scholar]
- 33.a) Wei M., Chen N., Li J., Yin M., Liang L., He Y., Song H., Fan C., Huang Q., Angew. Chem., Int. Ed. Engl. 2012, 51, 1202; [DOI] [PubMed] [Google Scholar]; b) Wilson K. D., de Jong S. D., Tam Y. K., Adv. Drug Delivery Rev. 2009, 61, 233. [DOI] [PubMed] [Google Scholar]
- 34. Huang K., Li Z., Lin J., Han G., Huang P., Chem. Soc. Rev. 2018, 47, 5109. [DOI] [PubMed] [Google Scholar]
- 35. Chen J., Chen K., Tong D., Huang Y., Zhang J., Xue J., Huang Q., Chen T., Chem. Commun. 2015, 51, 314. [DOI] [PubMed] [Google Scholar]
- 36.a) Shen C., Li J., Zhang Y., Li Y., Shen G., Zhu J., Tao J., Int. J. Nanomed. 2017, 12, 5443; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu J., Lv J., Zhuang Q., Yang Z., Cao Z., Xu L., Pei P., Wang C., Wu H., Dong Z., Chao Y., Wang C., Yang K., Peng R., Cheng Y., Liu Z., Nat. Nanotechnol. 2020, 15, 1043; [DOI] [PubMed] [Google Scholar]; c) Gu P., Wusiman A., Zhang Y., Liu Z., Bo R., Hu Y., Liu J., Wang D., Mol. Pharm. 2019, 16, 5000. [DOI] [PubMed] [Google Scholar]
- 37. Cho N. H., Cheong T. C., Min J. H., Wu J. H., Lee S. J., Kim D., Yang J. S., Kim S., Kim Y. K., Seong S. Y., Nat. Nanotechnol. 2011, 6, 675. [DOI] [PubMed] [Google Scholar]
- 38. Joshi M. D., Unger W. J., Storm G., van Kooyk Y., Mastrobattista E., J. Controlled Release 2012, 161, 25. [DOI] [PubMed] [Google Scholar]
- 39. Yang Y. W., Hsu P. Y., Biomaterials 2008, 29, 2516. [DOI] [PubMed] [Google Scholar]
- 40. Trevaskis N. L., Kaminskas L. M., Porter C. J., Nat. Rev. Drug Discovery 2015, 14, 781. [DOI] [PubMed] [Google Scholar]
- 41.a) Sánchez‐Paulete A. R., Cueto F. J., Martínez‐López M., Labiano S., Morales‐Kastresana A., Rodríguez‐Ruiz M. E., Jure‐Kunkel M., Azpilikueta A., Aznar M. A., Quetglas J. I., Sancho D., Melero I., Cancer Discovery 2016, 6, 71; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Harrell M. I., Iritani B. M., Ruddell A., J. Immunol. Methods 2008, 332, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bevan M. J., Nat. Rev. Immunol. 2004, 4, 595. [DOI] [PubMed] [Google Scholar]
- 43. Hu Y., Lin L., Chen J., Maruyama A., Tian H., Chen X., Biomaterials 2020, 252, 120114. [DOI] [PubMed] [Google Scholar]
- 44.a) Giermasz A. S., Urban J. A., Nakamura Y., Watchmaker P., Cumberland R. L., Gooding W., Kalinski P., Cancer Immunol. Immunother. 2009, 58, 1329; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou Z., Liu Y., Song W., Jiang X., Deng Z., Xiong W., Shen J., J. Controlled Release 2022, 352, 793. [DOI] [PubMed] [Google Scholar]
- 45.a) Gabrilovich D., Pisarev V., Curr. Drug Targets 2003, 4, 525; [DOI] [PubMed] [Google Scholar]; b) Hegde P. S., Chen D. S., Immunity 2020, 52, 17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.