

Abstract
Hydrogels play an important role in the field of biomedical research and diagnostic medicine. They are emerging as a powerful tool in the context of bioanalytical assays and biosensing. In this context, this review gives an overview of different hydrogels and the role they adopt in a range of applications. Not only are hydrogels beneficial for the immobilization and embedding of biomolecules, but they are also used as responsive material, as wearable devices, or as functional material. In particular, the scientific and technical progress during the last decade is discussed. The newest hydrogel types, their synthesis, and many applications are presented. Advantages and performance improvements are described, along with their limitations.
Keywords: bioassays, biosensing, hybridization assays, hydrogels, immunoassays, wearables
To achieve a good biosensing performance it is of great importance how the bioprobes are immobilized on the substrate and how well they are accessible. In that regard, hydrogels can provide several additional features to the performance of a biosensor. Beneficial characteristics include their solution‐like behavior and their ability to function as a visual indicator of target detection.
1. Introduction
The global COVID‐19 (Corona Virus disease 2019) pandemic has demonstrated the critical importance of quickly detecting contagious diseases to prevent their spread. When viable treatments or vaccines are not available, rapid detection methods are crucial for the timely rollout of measures to prevent transmission within societies.[ 1 ] This need exemplifies the importance of fast, efficient, and reliable testing and demonstrates the value of biosensors and bioanalytical tests.[ 2 , 3 ] Even before the COVID‐19 outbreak, however, biosensors and bioanalytical detection methods played a major role in the fields of clinical diagnostics, point‐of‐care testing, personalized medicine, and pharmaceutical research, with a key objective of understanding interaction processes at the molecular level.[ 4 , 5 ] In this area, highly sensitive detection platforms with excellent specificity are required for their ability to provide useful insights into individual's health.[ 6 ] In heterogeneous assays, the contact between the surface and the analyte determines the performance of the sensor, and so the quest for new and improved materials is intensifying. Among the problems encountered are insufficient loading capacity of 2D surfaces, poor accessibility of immobilized bioprobes, and the incorrect orientation of these probes. There are many tools to improve and optimize biosensors, among them the use of hydrogels. Hydrogels are viscoelastic, solid‐like, but deformable materials that consist of a considerably diluted network in an aqueous medium.[ 7 ] The network consists of a chemically or physically crosslinked hydrophilic polymer that entails the structural stability and prevents the dissolution of the polymer into the aqueous media. See Figure 1 for a schematic of a hydrogel at different scales. Maintaining their shape while absorbing a large amount of water or aqueous media makes hydrogels the ideal candidate for the encapsulation of drugs, proteins, and other biomolecules.[ 8 , 9 , 10 ] Furthermore, the 3D matrix displays a high permeability for small molecules, metabolites, oxygen, and other water‐soluble components. These traits and their visco‐elastic properties make hydrogels an excellent candidate for mimicking the extracellular matrix or mucus, and as a carrier material for biomolecules.[ 11 , 12 , 13 ]
Figure 1.
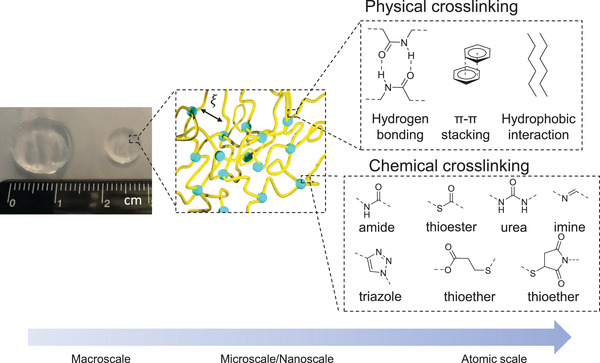
Representative hydrogel scheme from macro‐ to atomic scale. At macroscale we see a prepared gel pad in a swollen (left) and freshly prepared (right) state. On a micro/nanoscale we see the 3D crosslinked polymer structure with a certain pore size (micro) or mesh size ξ (nano). On atomic scale a few typical examples of either chemical or physical crosslinking are represented.
The first hydrophilic gel (hydrogel) was synthesized by Wichterle and Lim in 1960, with the ambitious goal of creating a “plastic” that is in permanent contact with human tissue. They defined the demands for a polymeric network to be placed in a human body and stated the need for a hydrophilic, 3D network.[ 14 ] Ever since, the variety, demand, and range of applications have grown immensely. Hydrogels are used in various fields of biotechnology, such as drug delivery, tissue engineering, wound healing, and molecular diagnostics.[ 15 , 16 , 17 , 18 , 19 , 20 ]
Hydrogels have been generated from a variety of synthetic polymers, including polyethylene glycol (PEG), poly(N‐isopropylacrylamide) (PNIPAAm), poly(2‐hydroxyethyl methacrylate) (PHEMA), as well as natural polymers, such as chitosan, alginate, dextran, and hyaluronic acids. Generally, these materials are resistant to protein and cell adhesion due to their hydration properties.[ 21 , 22 ] Specific properties of hydrogels originate from the molecular units of the polymer chains, and these units allow for further chemical modifications to provide networks with additional or adjustable functional properties. Typical groups, which either possess a function or are used to introduce further functionalities, are carboxylic acids, amino, and hydroxyl moieties. Functional groups influence the hydrogel properties, such as the stiffness, swelling ratio, and processability. Moreover, either inherently or by means of chemical modification, hydrogels can be thermo‐responsive, pH‐responsive, degradable, charged, swell upon stimuli, be self‐healing, or magnetic. In heterogenous bioassays the interface between solution and surface is critical. Through immobilization of these water‐swollen, 3D polymeric networks, we can extend the limited coverage of 2D surfaces and unlock enhanced immobilization capacity and accessibility of biomolecules. The improved signal intensity leads to a lower limit of detection and reduces the background signal for better signal‐to‐noise ratios.[ 23 ] The solution‐like environment created by hydrogel matrices also stabilizes and protects the structure of delicate biomolecules that would otherwise denature on surfaces.[ 24 ] Hydrogels have been reviewed in regard to sensor applications before, for example by Pinelli, Tavakoli, and Spychalska.[ 25 , 26 , 27 ] Our review, however, focuses on the role that hydrogels can adopt in bioanalytical assays and biosensors. After a short introduction to the general concept of biosensing and immobilization methods, we categorized the functions of hydrogels into hydrogels used as the immobilization matrix, as the responsive unit, as a wearable device, and as functional hydrogels using recent, notable examples in each section. A selection of the presented studies is listed in Table 1 .
Table 1.
Selection of the discussed hydrogel materials in the context of biosensing categorized according to the hydrogel's role
| Hydrogel material | Target | Detection method | Probe Immobilization method | Ref. |
|---|---|---|---|---|
| Hydrogels as Immobilization Matrix | ||||
| Dextran | Oligonucleotide | Fluorescence | Covalent, aldehyde amine | [ 23 ] |
| (PEG methacrylate) | Dye, protein, Oligonucleotide | Fluorescence | Covalent, Diels‐Alder cycloaddition | [ 50 , 51 ] |
| Chitosan | Enzymes | Chromogenic (indigo) | Covalent, amide bond | [ 54 ] |
| Chitosan, dextran | Glucose | Electrochemical | Electrostatic | [ 76 ] |
| Responsive Hydrogels | ||||
| Acrylic acid and dimethylaminoethyl methacrylate | Urea | Piezoresistive pressure sensor | Encapsulation | [ 90 ] |
| Shape‐memory DNA film | pH, Ag+/cysteine | Photonic crystal | Hybridization | [ 93 ] |
| Poly(methyl methacrylate‐co‐methacrylic acid) | pH | Naked eye | Dual‐layered hydrogel | [ 96 ] |
| Polyacrylamide | Glucose | Nanoparticle as visual indicator | Hybridization | [ 104 ] |
| Wearable Devices | ||||
| Hydrolyzed keratin and polyacrylamide | Human motion | Electrical | Adhesion | [ 125 ] |
| Agarose | L‐lactate | Potentiometric | Encapsulation | [ 140 ] |
| Functional Hydrogels | ||||
| PEG, hPG | Oligonucleotide, Fab, Ab | Fluorescence | Covalent, amide bond | [ 148 ] |
| PANI/PNIPAAm | BSA | IR | Molecular imprinting | [ 150 ] |
1.1. Biosensing
Generally, a biosensing element (bioprobe), which is usually immobilized on a solid support, is chosen to specifically interact with and sequester a target analyte from solution. The term “biosensor” was coined by Carl Kamman for the first time in 1977 but was only defined by IUPAC in 1997.[ 28 ] A biosensor has to be “a self‐contained integrated device, which is capable of providing specific quantitative or semiquantitative analytical information using a biological recognition element[…], which is retained in direct spatial contact with a transduction element.” Figure 2 shows a schematic representation of a biosensor. A sample can be obtained from a variety of biological sources; either it is used directly as the analyte, or the target is extracted and potentially purified before it is applied to the biosensing device. The biological counterpart of the target, the probe molecules (antibodies, peptides, nucleic acids, enzymes, cells, saccharide, etc.) must be immobilized on a substrate (solid surface, e.g., glass, metal, microbeads, hydrogels, etc.).[ 29 , 30 , 31 , 32 , 33 ] The interaction of the target and the probe are registered physico‐chemically and then transduced into a detectable signal.[ 34 ] Depending on the detection method, the signal can be amplified, but finally it is processed and displayed. The readout is typically accomplished optically with or without labeling, electrochemically, or by determining the mass change.[ 7 , 35 , 36 ] Examples of commonly used biosensors are glucose monitoring devices for diabetic patients and pregnancy tests, which detect human chorionic gonadotropin in urine. Besides these, biosensors are not only applied in health care but play an important role in crime detection, environmental field monitoring, and food analysis.[ 1 ]
Figure 2.
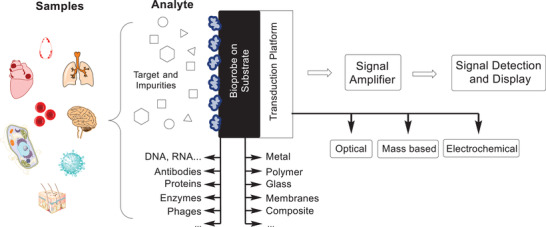
Schematic representation of a biosensor. A sample is collected, from which the analyte is extracted containing the target molecule and potentially impurities. The target will interact with the bioprobe immobilized on a substrate. For the final signal detection, the affinity interaction needs to be transduced into a measurable signal, which can be optical, mass based, or electrochemical.[ 34 ]
Compared to 2D and polymer brush functionalized planar surfaces, the 3D nature of hydrogels confers several advantages in the field of biosensing and bioanalytical systems. The hydrogel allows for the accommodation of more recognition elements and provides a highly aqueous and biocompatible microenvironment. This more natural setting increases the stability of biomolecules and preserves their functionality, which are crucial properties for feasibility, specificity, and sensitivity of a biosensor.[ 34 , 37 ] The hydrogel itself can also serve a purpose like separation of the target from other molecules in a sample.[ 38 ] Key parameters in biosensing are sensitivity, specificity, response time, selectivity toward a specific target, prevention of nonspecific binding, and reproducibility, all of which are dependent on the chemical structure and overall properties of the polymers used during hydrogel formation. This review aims to investigate, how hydrogels contribute to improving these parameters.
1.2. Immobilization Methods
The probe molecule in almost all bioanalytical assays must be immobilized onto a substrate for the purpose of easy separation, incubation, washing, and finally detection. There are several methods to immobilize the probe molecule onto various base materials, called substrates. However, not all substrates work with all detection techniques, and not all immobilization methods work on all substrates. Fortunately, there are many methods and techniques to choose from. Substrates can come in multiple forms and shapes. Commonly used materials are multiwell plates, glass slides, microbeads (fluorescent, magnetic, or both), rods, chips, fibers, and cups. The initial choice for the right substrate dependents mainly on the mode of detection. Once the right platform is determined, substrates usually come in a variety of base materials (glass, metal, polymer, or composites). They can be used unfunctionalized if the material itself interacts with the bioprobe (c.f. Figure 3a). However, for some applications special functionalities are needed, and there are several methods to introduce them. One straightforward approach is the use of plasma treatment, which eliminates surface contamination and inserts chemically reactive functional groups, and can also be used to change wettability, adhesion characteristics, and biocompatibility of the base material.[ 39 ] Alternatively, substrates can be functionalized with a monolayer of functional groups. In this case, linkers are used that carry a substrate binding functionality on one side and a bioprobe reactive group on the other side (c.f. Figure 3b). If a larger distance between substrate and linker is desired, longer linear chains can be implemented. This is intended to make the bioreactive group more easily accessible to the biomolecule of interest. Prominent examples are self‐assembled monolayers (SAMs) on gold and the silanization of glass surfaces, since silanes with diverse functional groups are readily available. However, the probe coverage in this introduced monolayer is often not sufficient to create a detectable signal. Often secondary interactions such as the (strept)avidin‐biotin interaction are utilized (c.f. Figure 3c). This interaction is one of the strongest noncovalent interactions known, which is represented in a high dissociation constant. This results in a good stability at high temperatures and allows for its use in nucleic acid detection under polymerase chain reaction (PCR) conditions.[ 40 ] There are several methods to couple biotin specifically to biomolecules. The biotinylated probe is then captured onto a streptavidin coated surface at high density in a directed orientation. To further increase the number of functional groups available on a substrate, polymer brushes or multivalent linkers like dendrimers, hyperbranched polymers, or star‐shaped polymers are introduced as exemplified in Figure 3d.[ 41 , 42 ] In this case, a polymer is either grafted to or grafted from the surface. With every monomer a new functional group is introduced for the subsequent immobilization of a biomolecule. Although the introduction of multifunctional polymers greatly enhances the number of functional groups, not all these groups are accessible, especially if the biomolecule of interest is rather large.[ 34 ] This is attributed to the dense packing of the functional groups. It is reported for poly(methyl methacrylate) beads grafted with bottle brushes of poly(acrylic acid) that this method can create a high density of up to 50 nmol cm−2 of accessible COOH groups, whereas monolayers of carboxylic groups only create around 10 nmol cm−2.[ 43 , 44 ] Additionally, for coupling reactions at the 2D interface, the number of effective collisions is reduced due to restrictions in translational and rotational degrees of freedom. Therefore, coupling reactions to surfaces show lower reaction rate constants and have lower conversions compared to the respective reaction in solution. It was found that surface DNA hybridization is suppressed 20‐ to 40‐fold in comparison with bulk hybridization, probably owing to interactions between analyte and substrate.[ 45 ] This is sought to be prevented or mitigated by the implementation of a hydrogel matrix for the immobilization of the bioprobe as schemed in Figure 3e. In the solution‐like environment, the probes stay immobilized, but the affinity interactions are less restricted. The distance between the surface and the bioprobe is increased, as well as the surface area for the immobilization of probes.[ 46 ] Different possibilities to immobilize hydrogels on surfaces for bioanalytical purposes are described for example by Prucker et al.[ 47 ] Instead of using the hydrogel simply as an immobilizing matrix, it can be used as the substrate, where its flexibility and low background intensity can be beneficial. Hydrogels can also function as substrate coating, or even function as the sensor or transducer platform in the case of target‐responsive hydrogels. In all these cases hydrogels are very promising, due to their exceptional mechanical, electrical, physicochemical, and optical properties. The hydrogel formation typically proceeds under mild synthetic conditions, with a huge chemical and structural diversity to choose from. Thus, we can not only optimize the chemical and physical properties of hydrogels but also manipulate their nanostructure (e.g., mesh size) and microstructure (e.g., shape) to achieve an optimal sensing platform.
Figure 3.
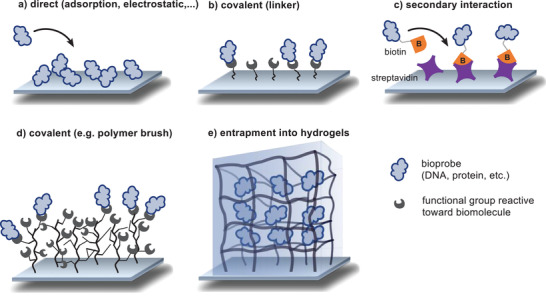
Representation of possible immobilization methods of a bioprobe onto a substrate.
2. Hydrogels as Immobilization Matrix
Compared to 2D types of biointerfaces, the 3D, porous structure of hydrogels increases the surface area of the material, allowing the immobilization of orders of magnitude larger amounts of recognition elements, ranging from small molecules to proteins and even cells. The highly hydrated structure resembles biological tissue; this makes hydrogels optimal for biological interactions and provides an ideal environment for probe molecules to be retained on the substrate. If the probes are entrapped inside the matrix as well as on top of the matrix, they are kept comfortably in the soft and flexible network, are less affected by surface interactions, and can maintain their biological activity.[ 24 , 34 ] Another outstanding feature of hydrogels as an immobilization matrix is the reduction or even prevention of nonspecific binding. This is attributed to the antifouling properties of the polymers and the solution‐like environment created by the hydrogel. As a result, biomolecules that are not chemically addressed will not bind to the surface.
In the following sections, hydrogel features will be discussed in the context of biomolecule immobilization. Here, hydrogels can be beneficial as 1) a coating material, to 2) completely replace the substrate, or as 3) the encapsulation matrix, where biomolecules will be entrapped inside the network.
2.1. Substrate Coating
The flexible nature of hydrogels can be used to increase the accessibility of probes. The rough yet elastic surface leads to a higher surface binding capacity as well as improved exposure of the recognition site. Due to the hydrophilicity of polymers used in hydrogels, nonspecific binding is significantly decreased.[ 48 ] As early as 1990, it was found that a dextran coating operated well in SPR‐based techniques. A gold chip was coated with the dextran hydrogel, and the more flexibly bound antibodies showed performance superior to antibodies directly coupled to a solid support.[ 49 ] More recently, a 3D dextran matrix was compared to a 2D surface in the context of DNA microarrays. Both surfaces provided aldehydes for covalent attachment of the DNA probe strand via imine formation. It was found that the signal intensity of the fluorescence‐based hybridization assay was higher for the hydrogel‐based assay, which was attributed to the higher surface loading paired with improved accessibility. The studied microarray platform also worked well with samples in serum, thus showing great potential for clinical diagnosis. Furthermore, by using the signal enhancing detection method hybridization chain reaction, a limit of detection as low as 10 × 10−15 m was achieved.[ 23 ] In the context of microarray fabrication, another hydrogel coating approach was chosen, where instead of coating the entire substrate, the hydrogel coating was placed only at the position of the array spots themselves. In this case, a synthetic hydrogel was used whereby the maleimide group was exploited in a twofold manner; for crosslinking and bioconjugation. As polymeric components a mixture of hydrophilic PEG‐based methacrylate and maleimide methacrylate were block‐copolymerized with a hydrophilic linear PEG macroinitiator by ATRP polymerization. For hydrogel formation the precursor solution was dropped onto the substrate and covered with a photomask. Crosslinking between the maleimide groups proceeded only at the exposed positions, which led to a micropatterned hydrogel coating in an array format. Maleimide groups, which were not crosslinked, remained readily available for the immobilization of thiol‐containing probe molecules by the Michael‐type addition reaction. Alternatively, a furan‐containing dye was immobilized via the Diels–Alder cycloaddition.[ 50 ] By adding methyl methacrylate as a fiber‐forming substance into the polymer mixture, the same group produced nanofibers via electrospinning and arranged them in a micropatterned array. They immobilized dyes, proteins, and oligonucleotides. The microporous structure of the nanofibers provided a high surface area with good accessibility, making them an ideal coating for biosensing applications.[ 51 ] Using hydrogels as the coating material also has the benefit of providing additional functionality within the network, such as directly immobilizing a chromogen, which only becomes visible for readout upon presence of the target. Nam et al. used hydrogel superimposed glass fiber membrane strips for the colorimetric detection of nitrite ions. An NHS (N‐hydroxy‐succinimid) ester was integrated into a PEG‐diacrylate‐based hydrogel, making the hydrogel susceptible to amines. The hydrogel solution was simply dropcast onto the glass fiber strip and subsequently functionalized with N‐(1‐naphthyl)ethylenediamine dihydrochloride, which formed an azo dye with sulfanilamide in the presence of nitrite ions (Griess reaction).[ 52 ] For the detection of nitrite ions, this strip was administered to a cocktail of sulfanilamide and analyte, containing various amounts of nitrite ions. The intensity of the color produced was correlated to the nitrite concentration in the sample, and the detection range was between 10 and 500 × 10−6 m. The sensor was optimized with respect to the UV time during hydrogel formation, concentration of both the immobilized probe and visibility agent, and the pH.[ 53 ] The use of a hydrogel in this sensor setup combined several benefits. It ensured the immobilization of a large amount of the colorimetric molecule, in contrast to only a monolayer of reactive groups available in a 2D format. Additionally, the solution‐like environment facilitated subsequent modification steps and at the same time guaranteed a good distribution of the analyte solution, which can be especially tricky for charged targets. The concept of integrating the color‐producing compound into the hydrogel to make a sensor suitable for naked‐eye detection was also used for the discrimination between a pathogenic (EHEC) and a nonpathogenic (E. coli. K12) strain. Here, Jia et al. produced shape‐encoded, chitosan‐based substrates, which contained enzyme‐specific probes. The probes simultaneously functioned as transducer, since the initially pale‐yellow hydrogel turned blue upon target detection (c.f. Figure 4 , top). More specifically, one chromogenic substrate based on 5‐bromo‐4‐chloro‐3‐indoyl sugar was covalently immobilized in one of two shapes, which were manufactured with a polydimethylsiloxane (PDMS) mask. The visible indigo dye is only produced upon the presence of the enzyme, which is generated by the respective E. coli. strain. See Figure 4, bottom, for the chemical details. Therefore, the appearance of a blue color corresponded to the existence of a specific E. coli. strain, which could either be pathogenic or nonpathogenic.[ 54 ] In a similar approach, the same group immobilized three different colorimetric substances inside a chitosan hydrogel to detect β‐glucuronidase. The three independent color codes reduced false positive test results by introducing redundancy.[ 55 ] This nicely demonstrates the versatility of hydrogel functionalization and its potential application in multiplex detection approaches. These systems nicely exemplify the utilization of hydrogel assets. The high amount of colorimetric substance in combination with the test strip format make the hydrogels ideal candidates for point‐of‐care testing since no additional instrumentation is needed. Additionally, preparing the hydrogel film by dropcasting simplified the manufacturing process. The flexible nature of the prepared hydrogel films would also allow application of such substrates to fabric or on soft thermoplastics for food packaging.
Figure 4.
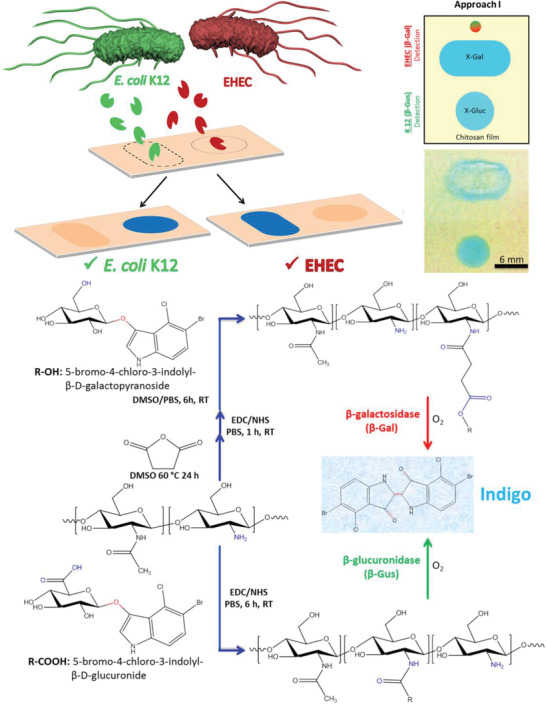
(Top) Pathogenic (EHEC) and nonpathogenic E. coli K12 were detected by shape encoded color change of a chitosan hydrogel substrate. The blue color appears only upon the presence of the respective enzyme secreted from the bacteria. (Bottom) Chemical details of the chitosan modification with the chromogen 5‐bromo‐4‐chloro‐3‐indonyl and its reaction to visible indigo dye after presence of the bacteria specific enzymes. Reproduced with permission.[ 54 ] Copyright 2018, American Chemical Society.
2.2. Substrate Replacement
Hydrogels can also completely replace the substrate instead of being used merely as a substrate coating. For example, mRNA was entrapped in a hydrogel microparticle based on PEG diacrylate. This approach spatially separated different mRNA strands inside one single microparticle (shape‐encoded “intraplex”), as can be seen in Figure 5 . By using PEG600 as a porogen, Choi et al. could tune the porosity to enhance mass transport and achieve a universal platform to detect large mRNA next to small miRNA in a single approach. This method outperformed the indirect method of reverse transcription PCR, which is highly sensitive but includes several steps and requires amplification of complementary DNA strands.[ 56 ]
Figure 5.
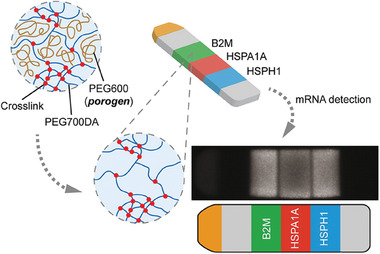
Schematic illustration of hydrogel‐based mRNA detection by a shape‐encoding intraplex hydrogel microparticle. The particle in stripe shape contained a reference fluorophore as positive control (yellow, channel 1) and blanks as negative controls (gray, channel 2 and 6). Channel 3–5 were used for the simultaneous detection of three different targets. Inert PEG600 was added as a porogen to enhance mass transport, whereas PEG diacrylate was used for the hydrogel formation. Reproduced with permission.[ 56 ] Copyright 2012, American Chemical Society
Using a natural hydrogel, King et al. detected DNA sequences with a hydrogel based on a self‐assembling peptide. The peptide sequence (Val‐Lys‐Val‐Lys‐Val‐Glu‐Val‐Lys) is known to form antiparallel β‐sheets, which stack along their long axis to form flexible fibers. The hydrogel‐based fibers were used as 3D substrate onto which oligonucleotides could be immobilized. The biological origin of the peptide‐based hydrogel helped with bioprobe immobilization. The material could be dried, which made the probe oligonucleotides less prone to degradation by nucleases. The rationale behind using a self‐assembling base peptide was that probe oligonucleotides could be introduced on a molecular level by mixing base peptides with modified peptides. Consequently, the probe sequences were exposed on the surface of the β‐sheets. Upon addition of a beacon containing the target sequence, a fluorescence dye, and a quencher as Förster resonance energy transfer (FRET) system, fluorescence intensity was implemented as the chosen detection method.[ 57 ] A hydrogel film based on acrylamide was prepared via redox initiated radical polymerization initiated by potassium persulfate, whereby a rhodamine derivative is copolymerized via its secondary amines. The amine–persulfate system effectively polymerizes vinyl monomers in aqueous conditions. The resulting free‐standing, clear, and stretchable hydrogel film contained the rhodamine dye in its spirolactam form, which was colorless and nonfluorescent. The addition of aluminum ions altered the structure of the dye and transferred it into its ring‐opened form, which showed color and fluorescence as measurable parameters. Consequently, the added rhodamine derivate acted as redox promoter for successful hydrogel formation and as the detection probe for Al3+. Interestingly, the fluorescence could be switched off, thereby enabling reversible on‐off fluorescence switching. This was done by adding ethylenediaminetetraacetic acid, which pulled out the Al3+.[ 58 ] Aluminum ions are known to inhibit the ingestion of other essential metal ions and could be related to diseases, such as Parkinson's disease, anemia, and Alzheimer's disease.[ 59 ] In a study by Shibata et al. hydrogel microbeads were fabricated in a microfluidic setup, whereby a glucose‐responsive fluorescent dye monomer was included. This dye contained a diboronic acid moiety which quenched the fluorescence of anthracene. Upon the presence of glucose, the boronic acid would preferably bind to the hydroxyl groups of glucose and thereby switch on the fluorescence intensity. They injected the microbeads into mice and correlated the fluorescence intensity to the relevant range of glucose concentration in blood and hence, the hydrogel microbeads showed potential to be used as an in vivo continuous glucose monitor.[ 60 ] In vivo biosensors have many advantages since they are designed for continuous monitoring of target analytes in actual biological systems. However, the readout, clearance after measurement, response time, and biocompatibility remain a challenge. More examples on in vivo biosensing targeting a variety of analytes are reviewed elsewhere.[ 61 ]
2.3. Encapsulation Matrix
Preservation of the native structure of biomolecules is a crucial requirement for feasibility, specificity, and sensitivity in biosensing applications. One of the first systems that used biomolecule entrapment was devised in 1967 by Updike and Hicks.[ 62 ] They entrapped the enzyme glucose‐oxidase in a gel of polyacrylamide, thus increasing the operational stability of the enzyme and simplifying the sensor preparation.[ 63 ] There are different approaches for the encapsulation of biomolecules, ranging from size‐selective encapsulation to covalent linkage to entrapment of an object which carries the bioprobe. The typical advantages of this approach over direct immobilization onto substrates are higher loading, better accessibility, and a more biological environment provided by the flexible nature of hydrogels. There are several reports about the encapsulation of biomolecules, particularly enzymes, into hydrogels. It was found to preserve the native protein conformation and thereby improve enzyme activity.[ 64 , 65 ] For example, super‐porous agarose gel was used in a flow‐through electrochemical sensor to immobilize the signal‐producing enzymes in larger amounts. This increased the signal intensity by a factor of more than five while retaining good flow properties.[ 66 ] As an example for a synthetic hydrogel, polyacrylamide‐based gel pads were used in immunoassays for their high‐capacity support, low nonspecific binding, and low background fluorescence. The amount of immobilized antibody was thereby increased by a factor of 20.[ 67 , 68 ]
Our group worked on the covalent and noncovalent entrapment of biomolecules into a biocompatible hydrogel based on PEG and hyperbranched polyglycerol. This setup was used to immobilize the enzyme periplasmatic aldehyde oxidoreductase for electrochemical detection of benzaldehyde. The use of a hydrogel kept the enzyme stable up to 4 days.[ 69 ] Based on the same two macromonomers, a hydrogel was synthesized from linear PEG functionalized with a strained alkyne and crosslinked by the hyperbranched polyglycerol functionalized with azide groups. Gelation proceeded via the strain‐promoted azide‐alkyne cycloaddition (SPAAC) between the two macromolecules. Due to the bioorthogonal nature of the crosslinking reaction, nonspecific binding was minimized, and size‐selective immobilization of large biomolecules (streptavidin and antibody) could be achieved in situ and without additional modification steps of the bioprobe. Several methods of substrate preparation were shown, such as spotting, spin‐coating, and dropcasting of the probe‐hydrogel mixture. It was shown that the streptavidin was immobilized inside the network without leaching, while remaining accessible for ≈3.7 biotin per streptavidin molecule (compared to 4 binding sites in solution). Additional functional groups of the hyperbranched polyglycerol could selectively be used for the covalent immobilization of small molecules. This made the matrix a versatile tool for the immobilization of a range of biomolecules and provided an inert, biocompatible environment in all cases (c.f. Figure 6 ). All the fluorescence‐based assays showed higher loading capacity, better sensitivity, and a lower limit of detection compared to 2D substrates.[ 70 , 71 ]
Figure 6.
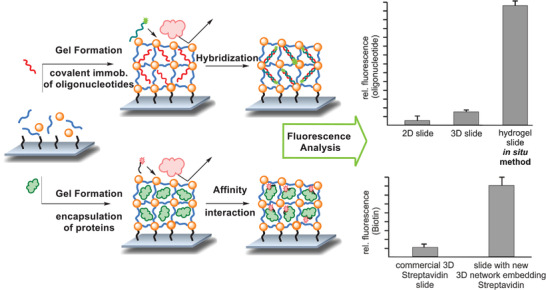
In situ immobilization of probe molecules onto a substrate in a bioorthogonal hydrogel matrix. Immobilization was done either covalently for small molecules, such as a 20mer oligonucleotide sequence or by size for larger proteins such as an antibody and streptavidin. After hybridization or affinity interaction with the respective fluorescence labeled targets, the detection was achieved by measuring the fluorescence intensity. Higher loading compared to reference substrates was found in all cases. Values are presented as mean value (n = 3) and error bars reflect the standard deviation. Adapted with permission.[ 71 ] Copyright 2018, American Chemical Society
Another antifouling hydrogel for the covalent and noncovalent immobilization of proteins and oligonucleotides was used by Díaz‐Betancor et al. The gel was based on 2‐Methacryloyloxyethyl phosphorylcholine and polymerized by UV irradiation. The zwitterionic phosphocholine is the polar, hydrophilic head group of some phospholipids and has good antifouling properties while acting as specific recognition element for C‐reactive protein. Other probe molecules, such as oligonucleotides and an antibody, could also be immobilized via thiol‐acrylate click chemistry. The group demonstrated the in situ hydrogel and probe immobilization onto two different surfaces and studied the signal transduction using labeled and label‐free detection formats. Since the hydrogel formation was initiated by UV radiation, this method has proven excellent for microarray printing by a robotic dispensing system. Additionally, the phosphorylcholine containing hydrogel structure was biocompatible, antifouling, and easy to manufacture.[ 72 ] In the context of automatic dispensing, hydrogels can also be used for 3D printing. Those properties were studied by Schmieg et al., who worked on PEG‐diacrylate‐based hydrogels for the entrapment of an enzyme. For 3D‐printing the viscosity and hardening properties of the hydrogel ink are determining the eligibility of the material. They studied the rheological properties, the printability, and the enzyme activity inside the printed structure. They used viscosity enhancing additives to tune flow properties and to enable degradation.[ 73 ] The hydrogels has proven to be beneficial since both, the biological relevant species and the viscosity enhancing material could be mixed into the ink and the final material had the desired properties regarding biocompatibility and printability. Another instance of probe molecule encapsulation was performed with a composite hydrogel based on gum tragacanth nanoparticles and acrylic acid (AA). It was reacted into a superabsorbent hydrogel by crosslinking with by N,N′‐methylenebisacrylamide (MBA). As a second crosslinker, fluorescein diacrylate (Fl‐DA) was added, which yielded a polymeric network with the fluorescent dye incorporated within the structure. See Figure 7 for a scheme of the chemical structure and crosslinking strategy. By encapsulating quantum dots (CdTe) the fluorescence intensity was enhanced due to the synergistic effect between fluorescein and CdTe quantum dots. As the biorecognition element, the enzyme glucose oxidase was immobilized within the gel, which catalyzed the oxidation of β‐D‐glucopyranose. Thereby hydrogen peroxide was produced and sensed on the surface of the quantum dots, where it quenched their fluorescence intensity. The superabsorbent feature of the hydrogel was intended to enhance mass transport of substrates and products in the highly swollen structure, and the enzyme affinity for glucose was found to be superior inside the hydrogel compared to other gel‐like matrices. The enzyme was stably immobilized inside the gel for 120 h.[ 74 ] This is an excellent example of the multiple purposes a hydrogel can adopt. It combines the biorecognition element, the transducing feature, and the fluorescence‐enhancing component. Through the combination of a natural hydrogel with synthetic moieties, the platform enabled many options for optimization.
Figure 7.
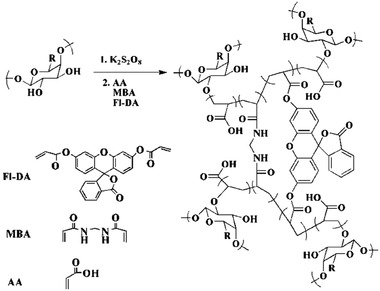
Reaction scheme of the acrylic acid (AA) and gum tragacanth composite hydrogel, crosslinked by N,N′‐methylenebisacrylamide (MBA) and fluorescein diacrylate (Fl‐DA). Adapted with permission.[ 74 ] Copyright 2020, The Royal Society of Chemistry.
To overcome limitations of glucose sensors based on 2D substrates, Liang et al. used a setup based on a natural hydrogel mixture of quaternized chitosan and oxidized dextran. By the formation of reversible Schiff base bonds, the resulting hydrogel had self‐healing properties. Since covalent attachment of the enzyme within the hydrogel can impede with the enzyme's bioactivity,[ 75 ] the researchers encapsulated CeO2/MnO2 hollow nanospheres and modified them with carboxylic groups. Thus, the nanospheres could be covalently immobilized into the hydrogel by EDC/NHS coupling (1‐Ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide/N‐Hydroxysuccinimide). The covalent attachment prevented leaching of the spheres from the hydrogel, which was important for sustaining the sensing performance. See Figure 8 for a schematic overview. The negative charge of the carboxylic groups simultaneously allowed for electrostatic interaction with the enzyme for probe immobilization. The nanospheres used are biocompatible, rather inexpensive, and beneficial for glucose oxidase immobilization. In this setup, the hydrogel should enrich glucose which enhanced the glucose oxidase contact to glucose, thereby broadening the range of detection and improving sensitivity and the response rate. Finally, a flexible polyethylene terephthalate film coated with an indium tin oxide (ITO) conductive coating as well as a gold‐based three‐electrode screen‐printing chip were coated with the nanosphere‐encapsulated hydrogel, and glucose was tested successfully in the physiologically relevant range. The excellent performance found in this study is attributed to the large specific surface area of the hollow nanospheres, which is further increased by embedding them in large numbers in the hydrogel. Furthermore, the hydrophilic environment of the polymeric network facilitated the diffusion and concentration of glucose. The flexible nature with additional self‐healing properties yielded an excellent system for potential application as a continuous glucose monitoring system.[ 76 ]
Figure 8.
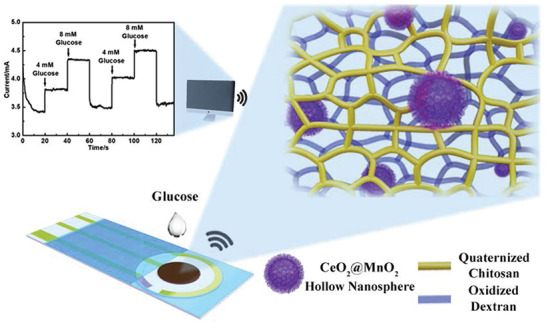
Overview of the glucose sensor based on CeO2/MnO2 hollow nanospheres, which were encapsulated in a natural hydrogel produced from quaternized chitosan and oxidized dextran. The concentration dependent, electrical readout was shown in a repetitive manner. Reproduced with permission.[ 76 ] Copyright 2020, Elsevier B.V.
The embedding of larger objects in a hydrogel was also performed with silver nanoclusters and a DNA hydrogel for the detection of hydroxyl radicals. Silver nanoclusters were synthesized in situ and stabilized inside a DNA hydrogel, whereby cysteine‐rich sequences interacted with silver ions which were then reduced to fluorescent nanoclusters. Silver nanoclusters show excellent performance in bioimaging and biosensing; however, they are rather unstable. Embedded in the hydrogel, the silver nanoclusters performed with improved photo‐ and thermostability next to showing lower toxicity compared with conditions in ambient environment. Generally, composite materials of hydrogels and nanoparticles have attracted much research interest due to the lower toxicity displayed when the nanoparticles are incorporated into a hydrogel. At the same time, the combination increases structural diversity and property enhancement.[ 77 ] In the case of embedded silver nanoclusters, the detection of reactive oxygen species such as hydroxyl radicals in viable cells was done with improved performance. As a consequence of the superior photostability only strong oxidation agents were able to quench the fluorescence and that yielded high selectivity. Furthermore, the silver nanoclusters were protected by the long DNA strands in the hydrogel.[ 78 ] Reactive oxygen species were also detected in the form of hydrogen peroxide (H2O2) by a hydrogel‐based sensor, which was developed based on a peroxidase‐mimicking hemin (DNAzyme) complex. This complex exhibited high peroxidase activity, showed high stability, and its side chain could be tailored for further functionalization. Hydrogen peroxide is typically detected in a colorimetric format by the enzyme‐catalyzed (e.g., horseradish peroxidase) oxidation of 3,3′,5,5′‐tetramethylbenzidine (TMB) or toluidine blue, respectively. This reaction yields a blue product, which can be analyzed and quantified.[ 79 , 80 ] In diluted solution, however, this is not feasible for naked‐eye detection. By incorporating the peroxidase‐mimicking enzyme (DNAzyme) into a hydrogel, the hydrogen peroxide was accumulated locally and produced the visible oxidized TMB in high local concentration. Therefore, a quadruplex with hemin was formed from a DNA strand that contained a hemin‐aptamer sequence in the middle and a poly(thymine) sequence on both ends, whereby the poly(thymine) sequences protruded on all sides (c.f. Figure 9 , top line). To encapsulate the DNAzyme into the hydrogel, an acrydite‐DNA strand is copolymerized with acrylamide, whereby acrydite was used to introduce a methacryl group at the 5’ end. This led to a linear polymeric chain with poly(adenine) sequences protruding (c.f. Figure 9, second line). The hydrogel was formed by hybridization between the poly(thymine) and poly(adenine) chain and thereby encapsulated the DNAzyme complex as schemed in Figure 9. The signal accumulation inside the hydrogel, detected by naked eye, reached a limit of detection comparable to electrochemical H2O2 sensors.[ 81 ] This is an excellent example of a case where the hydrogel enabled a device‐free detection method due to its higher amount of immobilized probe, and where its accumulation capability is utilized. In a different study, a peroxidase‐mimicking hemin‐based quadruplex was stacked by a cationic template to form a nanofibrous hydrogel. The researchers integrated the nanowires into a conductive matrix by in situ deposition of polyaniline. The printing capability of this system provided great potential for designing hierarchical architectures or for manufacturing flexible electronics.[ 20 ]
Figure 9.
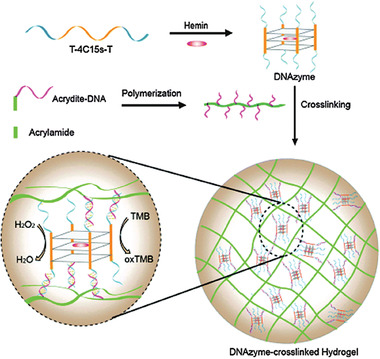
(Top) Illustration of the DNA strand containing a hemin‐aptamer sequence with poly(thymine) sequences on both ends which formed a quadruplex with hemin (DNAzyme). The DNAzyme was crosslinked into an acrylamide‐based hydrogel via its protruding thymine strands utilizing hybridization with a poly(adenine) sequence. (Bottom) The encapsulated quadruplex was used for naked eye detection of peroxide via visible TMB oxidation. Reproduced with permission.[ 81 ] Copyright 2016, The Royal Society of Chemistry.
3. Responsive Hydrogels
Hydrogels that respond to physical or chemical changes in their environment can be used for the detection thereof. A typical sensing approach is to use the network's swelling or its degradation. When the hydrophilic polymers of a hydrogel are immersed in aqueous solution, the osmotic pressure causes the water to diffuse into the material's body. This proceeds until the polymer is completely dissolved, as far as the physical or chemical crosslinks allow it.[ 82 ] This leads to the expansion of the whole network. Opposed to the stretching of the polymer chains stands the retractive force imposed by entropy. The swelling continues until those two forces are balanced; this dynamic is described extensively by the Flory–Rehner theory.[ 83 , 84 ] The swelling behavior and degree of swelling can thus be influenced by modifying the hydrogel's properties. The introduction of reversible bonds can further increase swelling or even lead to complete dissolution of the network. This is because noncovalent or reversible linkages will break in response to certain stimuli, such as mechanical stress or physical changes. Those stimuli include alteration in pH, ionic strength, temperature, or biochemical indications.[ 85 ] Alternatively, a ligand may be present whose affinity toward the crosslinker is stronger than the hydrogel component itself. Consequently, the binding between polymer and crosslinker inside the network will break up, resulting in property changes. In this way hydrogels are beneficial to detect the linear increase (or decrease) of a targeted property over a wide range. This allows for concentration‐dependent detection. Hydrogels as a material are often stable over a broad range of external stimuli but undergo a conformational change once a critical point is reached. The transformation happens due to highly cooperative interactions from the repeating units in the polymeric material.[ 86 ] This can be used for transducing a certain critical target concentration into a visible signal.
3.1. Stimuli‐Responsive
In this chapter, external physical stimuli that have an impact on certain hydrogel structures will be discussed. pH‐responsive hydrogels are a prominent example. One way to obtain pH‐responsive hydrogels is to introduce carboxylic groups or use polymers that intrinsically contain many carboxylic groups (e.g., poly(methacrylic acid)).[ 87 , 88 ] By increasing the pH, the carboxylic groups become charged and the charge repulsion leads to higher swelling.[ 89 ] This increase in swelling can be detected in a sensing setup or it can be used for the release of a previously loaded cargo. For the detection of urea, which is used in fertilizer and is also an important biomedical indicator for diseases, the enzyme urease was encapsulated into a pH‐responsive hydrogel based on acrylic acid and dimethylaminoethyl methacrylate (c.f. Figure 10 , top). Upon the presence of urea, basic by‐products were produced that increased the pH and resulted in swelling of the hydrogel. The swelling was transduced into a measurable signal by a piezoresistive pressure sensor, which is schemed in Figure 10, bottom. The selectivity for urea over similar species was investigated and found to be extremely high. The swelling process was also reversible, allowing for repeated urea detections with the same sensor. The enzyme was physically entrapped by size; however, it did not leach out of the network. It also remained active over the course of at least 8 weeks, yielding a reusable and stable biosensor for the concentration‐dependent detection of urea.[ 90 ] The study was performed at neutral pH, which is where the maximum rate of enzyme activity is known to be reached (pH 7.4).[ 91 ] For the application of such a sensor with real samples with a different pH, further studies should investigate the performance at a broader pH range. The authors discuss other hydrogel‐based urea sensors and find their size‐based entrapment was superior to systems where the urease is covalently immobilized or other hydrogels were used.
Figure 10.
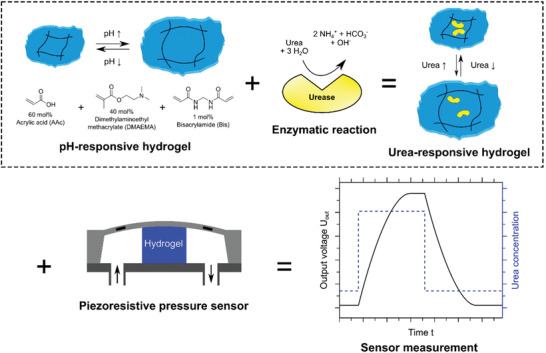
(Top) A pH‐responsive hydrogel was produced from acrylic acid (AA), dimethylaminoethyl methacrylate (DMAEMA), and crosslinked with bisacrylamide (Bis). The enzyme urease was encapsulated noncovalently. The enzymatic hydrolysis of urea at neutral pH led to an increase in pH which deprotonated the acrylic acid groups. As a result to the charge repulsion, the hydrogel swelled and deswelled once the pH dropped. (Bottom) Depicted is the measuring principle, whereby the swelling pressure is transformed into an output voltage via a piezoresistive transducer. Reproduced with permission.[ 90 ] Copyright 2019, MDPI.
Another approach for urea detection was developed by Li et al., who used a hydrogel based on acrylamide and acrylic acid and covalently immobilized urease by EDC/NHS coupling into the hydrogel. The hydrogel was decorated with polystyrene colloidal particles in a 2D crystalline colloidal array. The volumetric change of the hydrogel, which was induced by the reaction products of urea degradation, altered the lattice spacing of the colloidal crystal. This led to a shift in the diffraction wavelength and thus, realized a simple and label‐free detection of urea that was present in the analyte through observing the color change of the material, whereby detection was even possible by naked‐eye. In this case, the shrinkage of the hydrogel relied on the release of the charged ions NH4 + and HCO3 −. These ions shielded the present charges, thus reducing repulsion and decreasing osmotic pressure which finally led to the shrinkage of the gel. This resulted in a decrease of spacing between the polystyrene spheres which was visible as a change in color.[ 92 ] Wang et al. also used polystyrene nanoparticles as a 2D photonic crystal for visualization transduction, but they embedded them in a shape‐memory DNA hydrogel film. The DNA‐based crosslinker served as recognition element for external stimuli (pH and Ag+/cysteine) and as actuating unit, which regulated the spacing between the polystyrene nanoparticles by swelling and shrinking.[ 93 ] Another photonic visualization was studied by Noh et al.; they used an interpenetrating network of polyacrylic acid with a cholesteric liquid crystal for urea detection, whereby the pH‐dependent swelling of the gel altered the photonic structure of the liquid crystal. This change in color could be detected by naked eye.[ 94 ] Urease containing hydrogel sensors were the focus of a theoretical model (MECurea model) that investigated the responsive behavior toward urea stimulus, including the distribution patterns of the pH and of the electrical potential. The model coupled biochemical, electrochemical, and mechanical effects together, and validation with a PNIPAAm hydrogel showed good agreement. The researchers proposed to use urea‐sensitive hydrogels as material for biofuel cells as it exhibits an electrical potential and pH gradient upon the addition of a urea rich solution. This would allow the generation of electricity from urine. This theoretical model provides insight into the fundamental mechanism of the hydrogel in response to the urea cue.[ 95 ] A novel and application for a pH‐sensitive hydrogel was reported for the point‐of‐care diagnosis of a bacterial urine infection, whereby the readout relied on the release of a fluorescent dye (5(6)‐carboxyfluorescein) into the urine bag. For this application, the urine catheter was coated with a dual‐layered hydrogel. The bottom layer, based on poly(vinyl alcohol), functioned as a hydrogel reservoir for the fluorescent dye. It was covered by a pH‐responsive “trigger layer” (poly(methyl methacrylate‐co‐methacrylic acid)) that dissolved upon pH values above 7 and consequently exposed the bottom layer, which released the dye to provide a clear visual detection (c.f. Figure 11 ). The rise in pH correlated with an early indication of urinary catheter blockage as a result of bacterial infection, which could thus be treated early on.[ 96 ]
Figure 11.
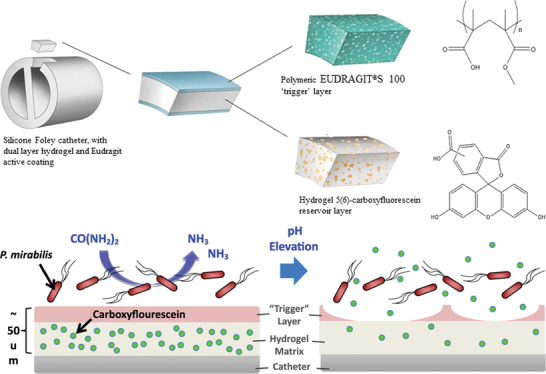
(Top) Schematic illustration of the dual‐layered polymeric architecture for the pH‐triggered release of a visual indicator. Depicted is a silicone foley catheter, which was composed of a hydrogel covered by a polymeric, pH‐responsive “trigger” layer (EUDRAGIT). The hydrogel contained 5(6)‐carboxyfluorescein as visual indicator. (Bottom) The presence of infectious bacteria (P. mirabilis) led to a pH elevation through the production of basic ammonia which dissolved the “trigger” layer and thereby exposed the hydrogel reservoir which enabled the release of fluorescence dye. Reproduced with permission.[ 96 ] Copyright 2016, Elsevier.
If a hydrogel is built up by a mixture of hydrophilic and hydrophobic units, the resulting hydrogel becomes thermo‐responsive, whereby the ratio of the hydrophobic/‐philic units determines the transition temperature.[ 97 ] In solution, thermo‐responsive polymers precipitate above the lower critical solution temperature. As a crosslinked structure, the hydrogel shrinks upon heating and reswells upon cooling below the polymer's volume phase transition temperature. The most commonly known thermo‐responsive hydrogel is poly(N‐isopropylacrylamide) (PNIPAAm) with a volume phase transition temperature of ≈33–35 °C, where the temperature can be adjusted by tuning the composition of the materials.[ 98 ] Fei et al. used a PNIPAAm‐based thermo‐responsive hydrogel as a self‐cleaning coating of a subcutaneously implanted glucose sensor. The problem with implanted devices is that biofilm formation inhibits the glucose diffusion and therefore, limits the lifetime and sensitivity of such transdermal and subcutaneous biosensors. In this study, the introduction of the thermo‐responsive gel still allowed for glucose diffusion during glucose monitoring. In the presence of accumulated biofilm, the thermo‐responsive gel was heated, leading to a deswollen state of the hydrogel coating. The accumulated cells would detach, thereby enabling the self‐cleaning of the glucose monitor. After cooling down to the initial temperature, the hydrogel would reswell and the device would be reset to its initial state. See Figure 12 for a scheme of one self‐cleaning process. The authors could perform the self‐cleaning process for up to 6 cycles.[ 99 ]
Figure 12.
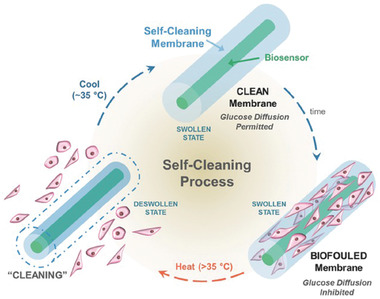
Scheme of a self‐cleaning, thermo‐responsive hydrogel that periodically rids itself of attached cells to maintain glucose diffusion. The thermo‐responsive hydrogel was implemented as self‐cleaning membrane on a glucose biosensor. Over time, cells would attach in vivo which inhibited the glucose diffusion. Heating above 35 °C led to shrinkage of the thermo‐responsive membrane and the cells were cleansed from the sensor. Cooling back to 35 °C reinstated the system, allowing for a new cycle to begin. Reproduced with permission.[ 99 ] Copyright 2016, Wiley‐VCH GmbH.
Droplets created by microfluidic devices already serve as individual microreactors, where cell signals are accumulated in a rather small volume and therefore become detectable.[ 100 ] This concept was combined with the thermo‐responsive behavior of a hydrogel in the transduction segment of the biosensor to further increase signal intensity. Therefore, in a microfluidic setup cells were co‐incubated with thermo‐responsive micrometer‐sized hydrogels (microgels) serving as immunosensor. In the resulting droplets, the cells secreted proteins which diffused into the hydrogels and interacted with the respective, immobilized antibody. After demulsification, the microgels were incubated with the fluorescence‐labeled secondary antibody and finally, heating of the microgel particles led to a shrinkage that induced a fivefold signal enhancement. The thermo‐responsive hydrogel was based on PNIPAAm, copolymerized with acrylic acid. This yielded hydrogels that contained carboxylic groups for antibody immobilization via amide coupling. The addition of inert polyethylene glycol as a porogen during the fabrication process generated an increased mesh size for facilitated diffusion of the antibodies. Using hydrogel microparticles within the microfluidic droplets fulfilled two important functions. First, it created a heterogeneous immunoassay, which allowed for subsequent washing and incubation steps. These are important measures for low background signals and increased specificity. Second, the temperature‐induced shrinkage of the hydrogel yielded an increased signal intensity to reduce the limit of detection. Additionally, differently labeled antibodies were used to enable multiplex detection of three different antibodies in a single run.[ 101 ]
Sun et al. made use of the electrical stimulus that DNA‐doped graphene releases for the label‐free, impedimetric detection of mitochondrial DNA. A bionic hydrogel was developed based on graphene oxide crosslinked by fish sperm DNA. The gelation proceeded by mixing graphene oxide sheets with fish sperm DNA and heating it. Upon noncovalent interaction of the single‐stranded DNA strands with the graphene oxide sheets a stable, self‐healing hydrogel was formed with high water content.[ 102 ] The gel was prepared in a cone bottom‐centrifuge tube, and a copper wire was inserted as the working electrode. Since the hydrogel was negatively charged due to the fish sperm DNA, the immobilization of probe DNA was hampered because of charge repulsion. To circumvent this issue, positively charged polyethyleneimine was added to enable electrostatic immobilization of the probe strand with good efficiency, which proved to yield high sensitivity and selectivity. The detection relied on the change in impedance, which significantly decreased after hybridization between probe and target because the conductivity of graphene oxide improves when it is doped with double‐stranded DNA instead of single strands. Using biological structures, such as the fish sperm DNA as hydrogel material outperformed the use of hydrogel electrodes crosslinked with synthetic polymers. The researchers ascribed the excellent performance to the high‐water content in a natural environment, which drove the target DNA toward hybridization with the probe strand.[ 103 ]
3.2. Target‐Responsive
If the receptors or sensing units are incorporated into the gel‐forming structure, biochemical cues or the presence of the target moiety can be detected by physical changes of the polymeric network, either by forming from solution or dissolving into solution.[ 33 ] To make simpler detection techniques viable, a cargo can be released upon target presence. This makes the use of hydrogels advantageous over lab‐based technologies, such as gas chromatography, immunoassays, or PCR‐based techniques, since it often works by naked‐eye detection.
For the in vitro detection of glucose, a hydrogel was used to entrap gold nanoparticles as visual indicators. In a classical assay where only the target molecules are labeled, the concentration is usually not high enough for naked‐eye detection. By entrapping a visual indicator, such as colored nanoparticles, the presence of low concentrations in the analyte solution can be transduced into a clearly visible signal. Therefore, a hydrogel was crosslinked with an aptamer that was designated to bind a complex between glucose and Shinkai's receptor (c.f. Figure 13A). In the presence of this complex the aptamer was released from the network and led to the dissolution of the gel. The hydrogel backbone was based on linear polyacrylamide, and two short DNA strands were grafted to the chain (PS‐A and PS‐B, c.f. Figure 13B). They were designed to partially hybridize with the glucose‐Shinkai's receptor‐aptamer linker, and thus, crosslinking proceeded via hybridization between the single DNA strands of the polymeric chain and the aptamer. The hydrogel formed featured encapsulated gold nanoparticles, which produced a clearly visible, red‐colored network. The nanoparticles stayed encapsulated as long as the hydrogel network was intact, and the supernatant remained colorless. Upon the introduction of glucose with Shinkai's receptor the aptamer linker preferred to bind with the complex. Therefore, the network was disrupted the previously encapsulated gold nanoparticles were released into solution. The resulting red color in the supernatant could be detected by the naked eye and would become more intense with higher amounts of glucose.[ 104 ]
Figure 13.
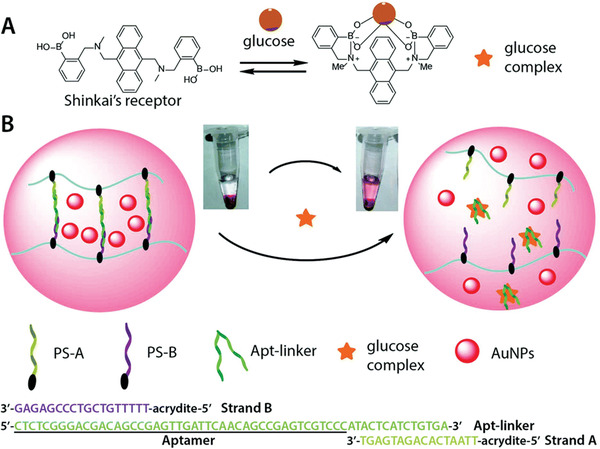
A) Scheme of the glucose complex formation between glucose and Shinkai's receptor. B) Working principle of the DNA hydrogel containing gold nanoparticles (AuNPs) as visual indicators encapsulated inside the hydrogel. The hydrogel, based on acrylamide, contained polymer sidechain A and B (PS‐A and PS‐B). They were partially hybridized with the aptamer linker. Upon the presence of the glucose complex the aptamer would bind more strongly with the complex and thereby led to the dissolution of the hydrogel network. The released AuNPs produced a red‐colored solution which could be detected by naked eye. Reproduced with permission.[ 104 ] Copyright 2018, The Royal Society of Chemistry.
An aptamer‐linked hydrogel setup was also used for the detection of microRNA, which is important for early cancer diagnosis. Here, a hybrid DNA‐polyacrylamide gel was immobilized on a 3‐(trimethoxysilyl)propyl methacrylate‐treated ITO/PET electrode (indium tin oxide/polyethylene terephthalate). In this work, two different DNA strands were incorporated into the bottle brush polymer chains by copolymerizing acrylamide with acrydite‐DNA strands. Subsequently, the polymers were crosslinked into a network by a partially complementary DNA strand. The crosslinker sequence was doped with ferrocene, which allowed for electrochemical detection. In the presence of the target microRNA strand, the crosslinker strand fully hybridized with the target and thereby the hydrogel crosslinks were broken up. Consequently, the network dissolved, producing a loss of ferrocene labeled strands which resulted in reduction of current. This was measured very sensitively by CV and differential pulse voltammetry.[ 105 ]
Another method for visual detection of low concentrations of target was used for the hydrogel‐based detection of sulfide (S2−). The hydrogel was crosslinked and thereby shrunk by copper ions (Cu2+) through an abundance of carboxyl and amine groups, which bind to the copper. It reswelled because the sulfide has a strong affinity toward copper and pulled it out of the hydrogel. This resulted in a less physically crosslinked network, which was visible as swelling. H2S is an important endogenous signaling molecule that is important for homeostasis. The change in volume could be detected by reading the gradations on a measuring container by the naked eye.[ 106 ] Swelling as a result of changes in crosslinking was also used for the detection of lead ions, which are an important indicator of water pollution. For this, short DNA strands were grafted to a polyacrylamide backbone. A crosslinking sequence contained the complementary sequence and also had in its middle part a sequence for interacting with the Pb2+‐dependent GR‐5 DNAzyme. In the presence of Pb2+, the DNAzyme was activated and cleaved the crosslinking sequence, leading to disintegration of the network. This shows that the lead‐ion‐dependent DNAzyme played the roles of response unit and crosslinker in the hydrogel. Once the hydrogel was dissolving, a solution was allowed to flow into a capillary to indicate the Pb2+ ions. The distance and velocity with which the solution rose in the capillary was related to the number of lead ions present. Since the marks on the capillary can be read out in the field by the naked eye, this system is an ideal point‐of‐care device.[ 107 ] The same hydrogel setup for the detection of lead was also implemented by Mao et al.; however, a different visualization strategy was pursued. Instead of using a capillary for indicating the disintegrating hydrogel, they encapsulated compounds which led to etching of gold nanorods in a cascade of reactions. Upon cleavage of the crosslinking sequence in the presence of Pb2+, the encapsulated enzyme glucoamylase was released and catalyzed the reaction from amylose to glucose in the supernatant. Glucose was oxidized by glucose oxidase to generate H2O2, which created hydroxy radicals in the presence of iron ions. The radicals caused etching of the nanorods, which led to different aspect ratios of gold nanorods displaying as different colors.[ 108 ] This nicely shows how versatile and multifunctional the application of hydrogels is in the context of biosensing. Additional biosensing applications of DNA‐based hydrogels are reviewed elsewhere.[ 109 , 110 ] Instead of classical oligonucleotides, Langford et al. integrated morpholino oligonucleotides into a covalently crosslinked polyacrylamide hydrogel for a swelling based detection technique. Morpholino oligonucleotides are synthetic DNA analogues with an uncharged backbone. This increases rigidity and reduces salt dependencies whereby the water solubility remains high. The researchers prepared hydrogel spots in which the probe sequence was partially hybridized with a crosslinker strand for additional crosslinking leading to a shrunken network. Both strands were covalently copolymerized into the hydrogel. Upon full hybridization with the target sequence, the additional crosslinks broke, and the hydrogel swoll (c.f. Figure 14 , left). Analysis was done with a camera and an appropriate image analysis software. Interestingly, a proof‐of‐concept experiment was performed with a clip‐on cell phone magnifying lens attachment. Such a setup could be advantageous for point‐of‐care diagnostic when there is no access to lab equipment.[ 111 ] Similar to the prioritized hybridization of fully compatible oligonucleotide sequences over partial hybridization, a commonly used feature in immunoassays is the weaker binding affinity of immobilized or functionalized biomolecules as compared to their free derivatives.[ 112 ] This competitive binding was used in an immunoassay for the detection of hepatitis B virus core antigen. Usually, the detection of hepatitis B is done with lab based diagnostic tools, such as ELISAs. However, these are laborious, expensive, and time‐consuming, providing a strong motivation to facilitate diagnosis with a hydrogel‐based point‐of‐care system. Therefore, Lim et al. used an activated acrylate (N‐succinimidyl acrylate) to couple the antibody and the antigen via their respective amine group. The resulting biofunctionalized acrylamides were then copolymerized with acrylamide into a polyacrylamide hydrogel. The affinity interaction between antibody and antigen formed additional crosslinks which led to a shrunken hydrogel network. The free antigen exhibited a higher affinity towards the antibody and therefore, its presence inside the network disrupted the interaction between the immobilized biomolecules. This led to swelling of the hydrogel, which was detectable by an increase in volume and weight. See Figure 14, right, for a schematic representation of the hydrogel formation and the detection procedure.[ 113 ] Another swelling based detection was done by Tan et al., who used hydrogel microparticle droplets based on polyacrylic acid and detected Ca2+ and Mg2+ ions in saliva. Here, the carboxylic groups from the hydrogel backbone coordinated with the ions, which made the hydrogel droplets shrink. Measured was not the absolute size but the speed of size reduction, which could be related to the ion concentration.[ 114 ] In another hydrogel droplet‐based study the blood glucose concentration was detected in a dual‐mode approach. The enzyme glucose oxidase was encapsulated together with fluorescent carbon dots. The produced gluconic acid and the hydroxyl radicals led to a size reduction of the hydrogel droplet and quenched the fluorescence intensity of the carbon dots. The hydrogel was based on polyacrylic acid would get protonated at decreasing pH, leading to a decreased charge repulsion.[ 115 ]
Figure 14.
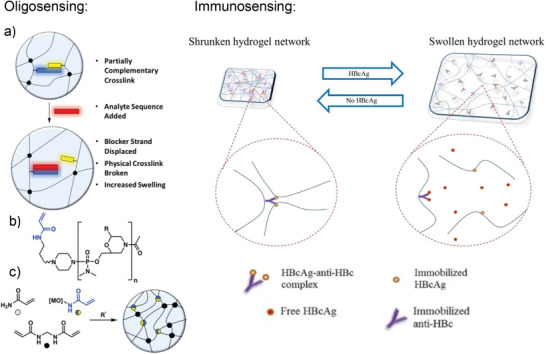
(Left, a) Morpholino oligonucleotide crosslinked hydrogel system showing displacement of the crosslinker strand (yellow) from the probe strand (blue) by the target sequence (red). b) Chemical structure of morpholino oligo with acrylamide, where R is any of the nucleobases. c) UV initiated radical polymerization for hydrogel formation. Adapted with permission.[ 111 ] Copyright 2018, American Chemical Society. (Right) The affinity interaction between immobilized antigen (HBcAg) and antibody (anti‐HBc) provide additional crosslinks and yielded a shrunken hydrogel network. The presence of free antigen led to partial disintegration of the network which was detected as swelling. This process is reversible, and swelling is related to the amount of free antigen present. Adapted with permission.[ 113 ] Copyright 2019, Elsevier B.V.
4. Wearable Devices
Health monitoring is of particular importance for many diseases. If the monitoring can be done continuously and at home, this is a huge relief on the health care system and increases the convenience for the patient. Personalized continuous health monitoring not only has clinical advantages, but has also become more popular and accepted by the public. This holds true for simple diagnostic devices that measure temperature, blood pressure, or pH, but is of even greater importance for glucose monitoring, hormones, lactate, and alcohol measurements.[ 116 ] Contrary to in vivo sensing, where substrate and probe are internalized into the system of interest, wearable devices are usually attached at a specified position from where accurate, real‐time measurements of a specific target are delivered. The target can either be a biomarker or a physiological parameter. The wearable devices can be attached externally (e.g., on skin) as well as internally (e.g., on soft tissue). The applications of wearable devices range from the detection of human movements to force sensing to detection of small molecules.[ 117 , 118 , 119 , 120 ] For example, a wearable device on skin, a flexible silk fibroin patch with encapsulated enzyme served as substrate for a conducting polymer, which was photocrosslinked on top. This flexible and biodegradable skin like patch served as a free‐standing electronic device. If implanted, it could even serve as a platform for an “implant and forget” sensor, due to its biodegradability.[ 34 , 121 , 122 ] The goal of such devices is to satisfy the demands of end users for self‐ or ambulatory testing in relatively simple diagnostic scenarios.[ 123 ] Hydrogels have emerged as excellent candidates for such applications due to their mechanical flexibility, biocompatibility, similarity to biological tissue, and powerful sensory capacity.[ 124 ] While hydrogels have a wide array of applications in wearable devices, the Sections 4.1 and 4.2 will focus on their use in the area of human motion detection and in sweat analysis.
4.1. Human Motion Detection
The combination of suitable skin adhesion and long‐term stability is still a challenge with human‐sensor interfaces. Conductive hydrogels in wearable devices fulfill basic requirements for mechanical flexibility in order to produce hydrogel‐based transducers and to support intelligent sensor technology. If the mechanical stability of a hydrogel alone is not sufficient for application on skin, composite materials can be produced. For example, Gao et al. presented a hydrogel with high sensitivity for use as a transducer to monitor human movements. This hydrogel was produced from a combination of lithium chloride, hydrolyzed keratin, and crosslinked polyacrylamide. The hydrolyzed keratin endowed the material with good mechanical strength and robust adhesion for dynamic and curved surfaces (like skin). The lithium chloride helped the hydrogel to resist against extreme temperatures (−20 to 50 °C) and prevented the loss of moisture. This material showed excellent adhesion to the skin as well as excellent stretchability. In addition, the conductive and transparent hydrogel was extremely sensitive to pressure and stretching over a wide range. Consequently, the obtained transducer was not only able to detect breathing modes, vocal cord vibration, and human movements but also pinpointed handwriting and facial expressions.[ 125 ] In addition to these physiological parameters even slight pulse changes were monitored by another composite hydrogel developed by Liu et al. They fabricated a soft hydrogel network via covalent crosslinking between poly(vinyl alcohol) (PVA) and poly(vinylpyrrolidone) (PVP), which was interconnected with a hard and dynamically crosslinked network of Fe3+ and cellulose nanocrystals. The resulting network was self‐healing, elastic, conductive, and strain‐sensitive.[ 124 ] Another interesting composite material was studied by Sun et al.; the hydrogel patches consisted of hydroxypropyl guar gum, doped with dopamine‐coated reduced graphene oxide. The produced hydrogel patches were highly flexible and due to the graphene oxide also conductive. They tolerated a wide temperature range owing to the addition of glycerol and the reversible bonds were responsible for the self‐healing property. Crosslinking was achieved by adding borax and metal ions (Fe3+), which formed reversible boron‐oxygen as well as metal–ligand interactions. A wide range of human motions detection with fast response time was achieved, ranging from sound vibrations to a full knee flex.[ 126 ] However, there are challenges with conductive hydrogels used as motion detectors on skin. Drawbacks which limit their application at the biointerfaces include mechanical mismatch with skin and a lack of antibacterial properties. This can potentially impede skin adhesion and lead to infections or biofilm formation. This problem was addressed by Zhao et al., who presented an animal‐skin‐inspired, conductive hydrogel consisting of a supramolecular structure of silver nanoparticles (Ag NPs) decorated with polydopamine (PDA), polyaniline (polyAN), 3‐aminophenylboronic acid (ABA), and polyvinyl alcohol (PVA) (c.f. Figure 15 ). Aniline and 3‐aminophenylboronic acid were polymerized to become a conductive aromatic system whereby the boron coordinated both with the dopamine and PVA and thereby functioned as the crosslinker. In addition to the epidermal sensing properties of the conductive hydrogel, this setup also exhibited broad antibacterial activity against Gram‐negative and Gram‐positive bacteria caused by the silver nanoparticles. The antibacterial properties had a therapeutic effect during diabetic wound healing.[ 127 ]
Figure 15.
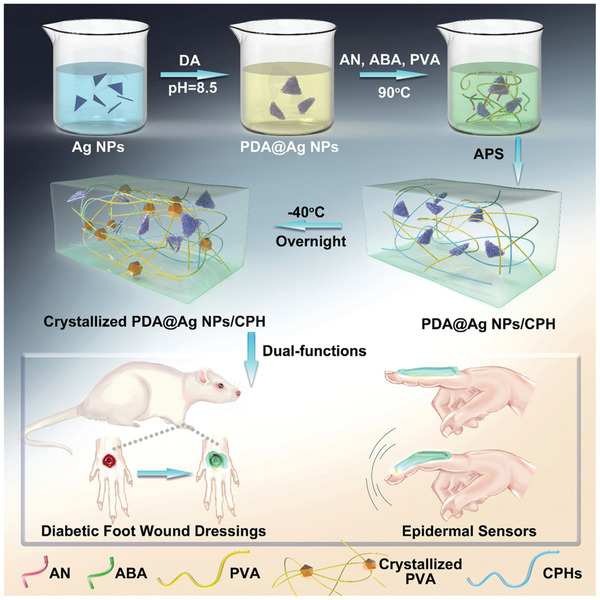
The synthetic procedures of polydopamine decorated silver nanoparticles (PDA@Ag NPs) is shown and their applications in the epidermal sensor and diabetic foot wound dressing. AN is aniline, ABA is 3‐aminophenylboronic acid, and PVA is polyvinyl alcohol. Together they yielded conductive polymeric hydrogels (CPHs). Reproduced with permission.[ 127 ] Copyright 2019, Wiley‐VCH GmbH.
To create conductivity different basic approaches are available: adsorption, percolation, and blending conductive filler materials into the hydrogel, such as graphene, carbon nanotubes, carbon black, metallic nanowires, and nanoparticles.[ 128 , 129 , 130 , 131 ] Problems, which come with the use of hydrogels as a conductive strain sensor, are low conductivity at freezing temperatures and insufficient toughness. This was addressed by Chen et al., who manufactured an ultrastretchable, tough, antifreezing, and conductive cellulose‐based hydrogel. Therefore, a composite hydrogel was produced from hydroxypropyl methylcellulose dissolved in a solution of zinc chloride, onto which acrylonitrile/acrylamide copolymers were grafted. The excellent properties arose from the dipol–dipol interactions, hydrogen bonds, and covalent linkages of the network paired with the metal coordination of cellulose to zinc. Additionally, the zinc provided the material with conductivity and antifreezing properties.[ 132 ] Aiming for similar properties Shao et al. used a combination of poly(vinyl alcohol) as hydrogel matrix and crosslinked it with both phytic acid and amino‐polyhedral oligomeric silsesquioxane, whereby the phytic acid was also responsible for good conductivity. Preparation of the hydrogel was done by three freeze‐thaw cycles which led to crystallization of the poly(vinyl alcohol). A comprehensive characterization of the hydrogel and its application in human motion detection was shown.[ 133 ]
A study which included additional features into their strain‐sensing patches was done by Yi et al., who integrated a supercapacitor for electrochemical energy storage into a strain sensor. It was produced from a composite material consisting of lithium chloride added to a polyacrylamide network with carbon nanotube fibers embedded into the material. The carbon nanotubes functioned as electrodes and the hydrogel material served as electrolyte and as separator simultaneously. The use of a hydrogel as semisolid material as opposed to a liquid electrolyte prevented the risk of leakage and its flexibility and stretchability was advantageous over classical solid electrode materials.[ 134 ] Also aiming for multiple properties in a single wearable device, Chen et al. reported on multifunctional hydrogels which were able to detect human motion next to measuring ambient and skin temperature. Instead of a skin‐adhesive patch they produced fibers for potential use to be woven into fabrics by coaxial wet‐spinning. These core–shell segmental fibers had segments for strain sensing (shell) and a segment for temperature measurement (core). The fiber production method should be up scalable and thus, be suitable for industry and mass production.
They studied a series of four hydrogel materials and characterized their conductivity and stretchability, whereby poly(2‐acrylamido‐2‐methyl‐1‐propanesulfonic acid‐co‐acrylamide) doped with reduced‐graphene‐oxide was identified as the best candidate for the strain sensor material. The temperature sensing segment was based on silicon rubber which was infused with microcapsules altering their color upon temperature changes. Immediate curing after preparation prevented water loss and maintained the shape of the fiber, while it also inhibited short circuit of the conductive hydrogel.[ 135 ]
4.2. Sweat Analysis
Various diseases and other health conditions leave relevant biomarkers in human sweat, including psychological stress, fatigue, and dehydration. The presence of these biomarkers, along with its ease of collection, makes sweat an especially attractive diagnostic tool.[ 136 , 137 , 138 ] On the one hand, the secreted amount of sweat can be analyzed and on the other hand, the composition of the sweat can be determined.[ 139 ] Wearable sensors involving hydrogels, e.g., in the form of tattoos or patches to monitor electrolytes or biomarkers in sweat can provide information about clinically relevant health conditions. Key targets are metabolites such as lactate,[ 140 ] electrolytes,[ 141 ] e.g., chloride, sodium, potassium, pH,[ 118 ] glucose,[ 142 , 143 ] drugs,[ 144 ] and other small molecules. Often the sensors are used together with gel‐like materials that are called gel patches. In many cases, the hydrogels serve as the skin contact material and are used for sample extraction from sweat, as a reservoir, or as an electrolyte for the transducing system. An important step in sweat analysis is sampling, which affects the accuracy of the results. In a publication from Liu et al. commonly used methods for sweat sampling were presented. The focus was on sweat sampling in wearable sensors, including absorbent materials, such as hydrogels or superhydrophobic/superhydrophilic surfaces. The basic procedure, advantages and disadvantages of each sampling method were introduced in detail.[ 145 ] Emaminejad et al. used electrochemically enhanced iontophoresis to periodically generate and to in situ analyze defined amounts of sweat. The electrodes that stimulated sweat secretion were connected to the skin with a thin layer of agonist hydrogel between the electrodes and the skin. The hydrogels were loaded with substances that stimulate cholinergic sweat gland secretion (e.g., pilocarpine). As a proof of function and medical relevance, levels of chloride ions, sodium ions, and glucose were determined, with the first mentioned ions being an important marker for cystic fibrosis.[ 146 ] Nagamine et al. published a study in which both working and reference electrode were covered by an agarose hydrogel immersed with Dulbecco's phosphate buffered saline. Upon contact of the hydrogel with skin, sweat was extracted into the buffer and sweat components could be analyzed. In this proof‐of‐concept study lactate was chosen as analyte due to its prevalence in sweat. Unfortunately, the potentiometrically detected signal was not only dependent on the L‐lactate concentration in sweat but also on the amount of sweat extracted into the agarose gel. The extracted amount of L‐lactate was influenced by the L‐lactate concentration in sweat as well as by the transpiration rate. In addition, skin surface temperature had an effect on the response to the electrochemical sensor. The bioanalytical properties such as lifetime, detection range, and detection limit, have not yet been quantitatively evaluated. This will certainly be the subject of further research.[ 140 ] A similar approach was followed by Lin et al., who presented a thin hydrogel micropatch (THMP) which combined sweat extraction and the electrochemical sensing of sweat analytes. Here, not only lactate but also caffeine was tested as model targets (c.f. Figure 16 ). They demonstrated that the sampling method was suitable to track metabolic patterns and to provide biomarker data for personal monitoring. Also, in this study it was recognized that it is necessary to correlate the biomarker readings with the sweat rate to account for the interfering effects of varying levels of sweat production. For this, concentrations would need to be measured directly in sweat and possibly in blood. In addition, standardized procedures for skin cleaning need to be developed and investigated to minimize skin contamination and to achieve autonomous operation.[ 144 ] Sempionatto and colleagues reported on an enzyme biosensor for noninvasive electrochemical detection of vitamin C in sweat. For this experiment, the enzyme ascorbate oxidase (AAOx) was immobilized on printable tattoo electrodes. The analytical evaluated signal was generated by changing the reduction current of the oxygen cosubstrate. The corresponding substances and enzymes were embedded in a hydrogel‐like matrix consisting of agarose. The flexible patch was fabricated on a polyurethane substrate. Additionally, an iontophoretic sweat stimulation was realized.[ 147 ]
Figure 16.
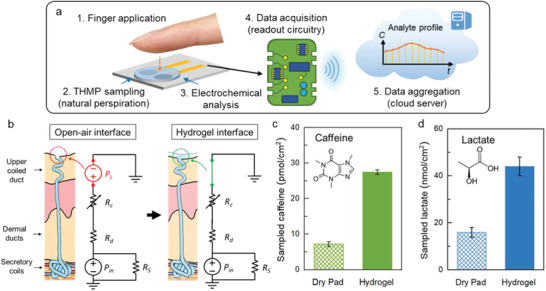
a) Schematic of the thin hydrogel micropatch (THMP) and the naturally perspired analyte sampling and analysis. b) Natural perspiration sampling at the open‐air interface and at the hydrogel interface. c,d) Amount of caffeine and lactate sampled by a dry pad and by the hydrogel, sampled from four different fingers of the same subject; error bars indicate standard error. Reproduced with permission.[ 144 ] Copyright 2019, American Chemical Society.
5. Functional Hydrogels
Hydrogel properties such as swelling, degradation or the release of a cargo can be used as part of the biosensing part itself. However, in this last section we want to focus on examples where the hydrogel served a secondary function, which either facilitates the detection, enables it, or is used to recover valuable parts of the sensor.
For example, in one study fluorescent microbeads were entrapped by a bioorthogonal, PEG‐, and hyperbranched polyglycerol‐based hydrogel for the multiplex detection of differently sized biomolecules. The microbeads entrapped by the hydrogel fully retained their capability to allow hybridization on their surface, which was detected with a fluorescence microscope. The micrometer‐sized beads functioned as the substrate for probe immobilization. Once they were covered by the hydrogel, they would not pass through the nanometer‐sized meshes of the hydrogel. The pores of the crosslinked structure were used to size‐selectively allow penetration of small molecules, whereas larger antibodies were excluded (c.f. Figure 17 ). By separating the detection levels with the hydrogel, different molecules were detected in each level. Thereby a new method of multiplex analysis was established. The hydrogel was able to keep the microbeads in their respective layer even after drying and reswelling. This is especially advantageous in the preparation of a multiplex assay since it enables storage under dry conditions.[ 148 ] The combination of these macromonomers crosslinked by SPAAC into a hydrogel was also used for molecular imprinting. The method of molecular imprinting is a more elaborate version of encapsulation because it shows shape selectivity toward the imprinted biomolecule and therefore, does not need a probe. It was used for the visible detection of influenza A viruses. Gold nanoparticles functionalized with sialic acids were included in the hydrogel network, both as recognition element for the virus and as visible indicator. The hydrogel was bioimprinted with a virus template, which meant the gold nanoparticle‐doped hydrogel formed around the virus shell, which was subsequently removed by incubation in acidic buffer. The bioimprinting increased specificity by shape recognition and introduced a volumetric change since the recognition elements were immobilized within the hydrogel. In the presence of the virus the gold nanoparticles with the sialic acid accumulated around the virus, leading to shrinkage and thus a shift in color originating from the plasmonic feature of gold nanoparticles. If the virus was absent, the hydrogel would reswell and the color disappear.[ 149 ]
Figure 17.
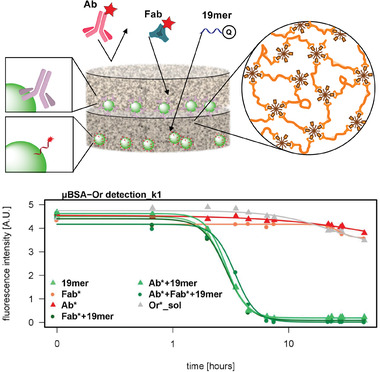
Spatial separation of fluorescent microbeads into two detection levels by a hydrogel. The mesh size of the used hydrogel selected which molecules could penetrate by size exclusion. If a mixture of labeled antibody (Ab), fragment antigen binding (Fab), and oligonucleotide (19mer) was added, each molecule was detected in a different level and the antibody was excluded completely. The graph depicts the detection of the quencher labeled oligonucleotide, which proceeded in the bottom layer, and was achieved successfully in all cases were the 19mer was added (green lines). All negative controls retained their fluorescence intensity (red and grey lines). Adapted with permission.[ 148 ] Copyright 2019, American Chemical Society.
In a different study, the combination of polyaniline (PANI) and graphene oxide sheets were crosslinked into a thermo‐responsive poly(N‐isopropylacrylamide) (PNIPAAm) hydrogel, which was imprinted with bovine serum albumin (BSA). The immobilization onto a glassy carbon electrode resulted in a molecular imprinted, conductive, and mechanically stable hydrogel electrode. The graphene oxide/PANI nanocomposites converted near infrared light into thermal energy which would shrink the thermo‐responsive PNIPAAm. This led to the removal of BSA templates from the hydrogel electrode and sensing nanocavities were obtained with high affinity and specificity toward BSA. Thereby, an infrared‐based detection and cleaning toward the protein BSA was obtained and it exhibited an adsorption/desorption property via alternately turning on and off the laser. Such a system could be useful for cleaning BSA from serum or cleaning a surface without the need of an eluant solution.[ 150 ]
Further examples for molecular imprinted photonic hydrogels used in biosensing are the optical detection of L‐histidine or L‐kyneurenine in human serum.[ 151 , 152 ] In a study which utilized the reversible gel formation of agarose upon heating, the hydrogel was used to embed conductive polypyrrole nanotubes and to immobilize them on a gold electrode. To achieve recovery of the precious sensing material, the natural, nontoxic agarose fulfilled the purpose of recovering the nanotubes after the detection. If agarose was boiled in hot water, it dissolved the hydrogel and recovery of the polypyrrole material was possible. This approach made this sensor reusable and recyclable, which is beneficial with respect to cost and environmental impact. In this study, dopamine was sensed by an aptamer immobilized on the nanotubes. The nanotubes embedded in the hydrogel were compared to direct‐contact nanotubes and nanotubes in solution.[ 153 ] Although this study addresses an important feature, namely the recovery of precious material, the recycling was done only twice and the linearity of signal to concentration was not yet achieved. This leaves room for further improvement.
A final example, in which the hydrogel did not participate in the sensing but was crucial for the collection of a gut sample, demonstrates well the broad spectrum of hydrogel applications. Waimin et al. used a hydrogel to seal a small, noninvasive sampling device which had been passively collecting microbiome samples throughout the entirety of the gastrointestinal (GI) tract. The sampling device used was a 3D‐printed capsule which could be swallowed by a patient. The capsule featured a biodegradable enteric coating that would delay the sample collecting until the capsule reached the GI tract. Once the coating degraded, gut fluids containing microorganisms entered the capsule. Consequently, the hydrogel material inside the capsule became hydrated and the swelling pushed a PDMS membrane onto the sampling aperture. This process sealed the capsule and thereby terminated the sample collection. The hydrogel used provided an ideal living environment for the sampled bacteria to survive until retrieval of the capsule. The absorption speed and compressive force (for pushing the PDMS cap) were optimized by adjusting the monomer ratio. The sealing efficiency was tested to ensure that no fluid would be exchanged after sample collection in the GI tract. Finally, the sealed capsule containing hydrogel‐entrapped bacteria was exposed to hostile solutions (bleach and antibiotics). The bacteria showed excellent viability within the sealed capsule while encapsulated by the hydrogel.[ 154 ]
6. Conclusion and Outlook
Hydrogels in biosensing applications have been the subject of considerable research interest in recent decades. Our review has aimed to reflect the various directions this interest has taken. Undeniably, there is not one particular hydrogel which can resolve all analytical issues. Therefore, we showed a variety of approaches where hydrogels were developed, synthesized, and adapted to refine and possibly enhance the performance of biosensors. Hydrogel‐based systems have proved to offer several advantages, including i) increasing the loading capacity of probe and analyte, ii) preventing nonspecific biomolecule adsorption, iii) decreasing background signal, iv) allowing modulation of the biosensing molecules, v) enabling the combination of sensing with transducing moieties, vi) providing a highly aqueous environment that is biocompatible, and vii) serving as sample collection and storage matrix.[ 23 , 71 , 144 ] The applications of hydrogels discussed in this review ranged from serving as the immobilization matrix for biomolecules to actively participating in the sensing process. We demonstrated that there is a broad range as to what the target was, and which hydrogel was used. Certainly, hydrogels did not always outperform existing techniques, but they did add further functionalities, facilitated detection, and integrated several biosensor components into one system.
The hydrogels used were either based on natural polymers, such as dextran, chitosan, and agarose, or they were based on synthetic polymers, such as PEG‐, PNIPAAm‐, polyglycerol‐, and acrylate‐based, or a combination of both (hybrid materials). There are advantages and disadvantages associated with either of those materials. Natural polymers are easily available, nontoxic, and naturally biodegradable. However, there can be issues regarding reproducibility due to variations in the polymer quality and constitution. Furthermore, they do not tolerate harsh reaction conditions and are structurally more complex. Synthetic polymers, on the other hand, are not biodegradable and are typically less biocompatible than natural polymers. Also, the reaction conditions needed for the hydrogel formation (temperature, UV light, addition of catalysts, etc.) must be considered when choosing the right material. The conditions need to be compatible with the biomolecule's tolerance and with the environment the hydrogel is applied to, e.g., for in situ hydrogels applied to skin or tissue.
If the hydrogel material alone is not sufficient to comply with the demands of the application, composite materials are chosen to introduce further functionalities. Here, a synergistic effect of hydrogels with other traditional analytical tools is utilized. For example, electrical conductivity needed in electrochemical sensors can be generated by either adding salts, using conductive polymers like polyaniline, or by adding conductive materials, such as carbon‐based materials, metal nanoparticles, or nanowires to improve conductivity.[ 155 ] Further composite material approaches include the integration of micro‐ or nanoparticles into the hydrogel matrix and the addition of fluorescent or chromogenic components for optical detection.[ 148 , 156 , 157 , 158 ]
Many bioreceptors were immobilized into or onto the matrix for the detection of a specific target. Such bioassays have wide applications as diagnostic tools in healthcare, and they have traditionally been performed by laboratory methods (e.g., PCR or ELISA).[ 159 ] However, they are tedious and require laboratory equipment and trained personnel.[ 160 ] The cases where hydrogels can replace or facilitate those methods are beneficial for both healthcare worker and patient. However, some limitations still exist, which reveal that there is yet a long way to go before they can be used as commercial healthcare products. Those limitations include long‐term stability and shelf life, handling, storage, and compatibility with the transducer format. With up‐scaling and the end consumers in mind, the cost of building blocks next to synthesis complexity plays a major role. The additional functionality has to justify the higher price or lead to a significantly better performance.
We believe that these remaining issues are the main reason why the presented research is still in the proof‐of‐concept stage. The translation from research to a product that is used by a patient or medical staff has not been managed yet. In addition to such proof‐of‐concept studies, extensive investigations on sensitivity, specificity, response time, reproducibility, and durability (stability) have to be performed. These necessary investigations have not been sufficiently performed yet and need to be addressed in further studies. Moreover, in connection to the approval of in vitro, and even more so, of in vivo applications, additional parameters, such as biocompatibility, toxicity, and extractable components should be considered. According to the current state of knowledge, hydrogels are most likely to be used in products that serve to detect movement, because these are medical devices that have only limited approval requirements. In addition to the application of hydrogels in diagnostic devices, the authors see great potential in the combination of hydrogel‐based sensors coupled with drug delivery systems. Here, bioresponsive hydrogels function as sensor and drug delivery system simultaneously.
In conclusion, it must be pointed out that to date there is no commercial sensor based on hydrogels. Especially for point‐of‐care tests, hydrogels could play an important role as they amplify the readout signal and offer the possibility to integrate the visualization agent into the sensing network. The Covid‐19 pandemic clearly demonstrated the importance of point‐of‐care tests, especially those that can be performed by untrained personnel. This trend should be exploited to invest in further research on healthcare devices and to increase the rate of translation from laboratory to industry.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
The authors would like to thank Benjamin Andrew Allen for language polishing the manuscript. The authors acknowledge the BMBF, KMU‐innovative program: Biotechnology – Biochance and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB 1449 – Projektnummer 431232613 for funding.
Open access funding enabled and organized by Projekt DEAL.
Biographies
Anna Herrmann received her B.Sc. and M.Sc. in chemistry from the Freie Universität Berlin, where she completed her Ph.D. in 2020. Her research focused on the development of functional, synthetic hydrogels which she immobilized onto different surfaces. She characterized the hydrogels regarding their physical and chemical properties and studied their performance in different bioanalytical assays. Recently, she started working at the University of British Columbia as a postdoc in the field of cell surface engineering.

Rainer Haag is full professor in macromolecular chemistry at the Freie Universität Berlin. Since 2021 he is the spokesperson of the collaborative research center 1449 “Dynamic Hydrogels at Biointerfaces.” His research interests are dendritic polymers as multifunctional scaffolds, macromolecular nanotransporters for DNA‐ and drug‐delivery and protein resistant material surfaces. His scientific output is documented by >500 peer review publications and >35 patent applications. In 2019, he became an elected member of the German Academy of Technical Sciences (acatech). For more information see the research group homepage: www.polytree.de.

Uwe Schedler is founder and CEO of PolyAn GmbH (www.poly‐an.de) and has more than 25 years of experience in the field of surface modification technologies, polymer science and photochemistry for diagnostics, life science research, medical engineering, and membrane separation. He received his diploma degree and Ph.D. in chemistry from the Humboldt University Berlin and founded PolyAn GmbH in 1996, after two years in an international research project. He is founder of three other companies and scientific advisor in various organizations and companies. Finally, he works as lecturer and supervisor for bachelor, master, and Ph.D. students at the Freie Universität Berlin.

Herrmann A., Haag R., Schedler U., Hydrogels and Their Role in Biosensing Applications. Adv. Healthcare Mater. 2021, 10, 2100062. 10.1002/adhm.202100062
Contributor Information
Rainer Haag, Email: haag@chemie.fu-berlin.de.
Uwe Schedler, Email: u.schedler@poly-an.de.
References
- 1. Jung I. Y., Lee E. H., Suh A. Y., Lee S. J., Lee H., Anal. Bioanal. Chem. 2016, 408, 2383. [DOI] [PubMed] [Google Scholar]
- 2. Barclay R. A., Akhrymuk I., Patnaik A., Callahan V., Lehman C., Andersen P., Barbero R., Barksdale S., Dunlap R., Goldfarb D., Jones‐Roe T., Kelly R., Kim B., Miao S., Munns A., Munns D., Patel S., Porter E., Ramsey R., Sahoo S., Swahn O., Warsh J., Kehn‐Hall K., Lepene B., Sci. Rep. 2020, 10, 22425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jung I. Y., You J. B., Choi B. R., Kim J. S., Lee H. K., Jang B., Jeong H. S., Lee K., Im S. G., Lee H., Adv. Healthcare Mater. 2016, 5, 2168. [DOI] [PubMed] [Google Scholar]
- 4. Koch W. H., Nat. Rev. Drug Discovery 2004, 3, 749. [DOI] [PubMed] [Google Scholar]
- 5. Hügle M., Behrmann O., Raum M., Hufert F. T., Urban G. A., Dame G., Analyst 2020, 145, 2554. [DOI] [PubMed] [Google Scholar]
- 6. Kim J., Campbell A. S., de Ávila B. E.‐F., Wang J., Nat. Biotechnol. 2019, 37, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mateescu A., Wang Y., Dostalek J., Jonas U., Membranes 2012, 2, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dey P., Schneider T., Chiappisi L., Gradzielski M., Schulze‐Tanzil G., Haag R., Macromol. Biosci. 2016, 16, 580. [DOI] [PubMed] [Google Scholar]
- 9. Peppas N. A., Hilt J. Z., Khademhosseini A., Langer R., Adv. Mater. 2006, 18, 1345. [Google Scholar]
- 10. Li J., Mooney D. J., Nat. Rev. Mater. 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grigoryan B., Paulsen S. J., Corbett D. C., Sazer D. W., Fortin C. L., Zaita A. J., Greenfield P. T., Calafat N. J., Gounley J. P., Ta A. H., Johansson F., Randles A., Rosenkrantz J. E., Louis‐Rosenberg J. D., Galie P. A., Stevens K. R., Miller J. S., Science 2019, 364, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y., Yu J., Ren K., Zuo J., Ding J., Chen X., Biomacromolecules 2019, 20, 1478. [DOI] [PubMed] [Google Scholar]
- 13. Authimoolam S. P., Dziubla T. D., Polymers 2016, 8, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wichterle O., LÍM D., Nature 1960, 185, 117. [Google Scholar]
- 15. Hoffman A. S., Adv. Drug Delivery Rev. 2012, 64, 18. [Google Scholar]
- 16. Billiet T., Vandenhaute M., Schelfhout J., Van Vlierberghe S., Dubruel P., Biomaterials 2012, 33, 6020. [DOI] [PubMed] [Google Scholar]
- 17. Sood N., Bhardwaj A., Mehta S., Mehta A., Drug Delivery 2016, 23, 748. [DOI] [PubMed] [Google Scholar]
- 18. Tang Q., Plank T. N., Zhu T., Yu H., Ge Z., Li Q., Li L., Davis J. T., Pei H., ACS Appl. Mater. Interfaces 2019, 11, 19743. [DOI] [PubMed] [Google Scholar]
- 19. Xiao M., Gao L., Chandrasekaran A. R., Zhao J., Tang Q., Qu Z., Wang F., Li L., Yang Y., Zhang X., Wan Y., Pei H., Mater. Sci. Eng., C 2019, 105, 110067. [DOI] [PubMed] [Google Scholar]
- 20. Zhong R., Tang Q., Wang S., Zhang H., Zhang F., Xiao M., Man T., Qu X., Li L., Zhang W., Pei H., Adv. Mater. 2018, 30, 1706887. [DOI] [PubMed] [Google Scholar]
- 21. Vandermeulen G. W. M., Klok H.‐A., Macromol. Biosci. 2004, 4, 383. [DOI] [PubMed] [Google Scholar]
- 22. Bae J., Park J., Kim S., Cho H., Kim H. J., Park S., Shin D.‐S., J. Ind. Eng. Chem. 2020, 89, 1. [Google Scholar]
- 23. Chao J., Li Z., Li J., Peng H., Su S., Li Q., Zhu C., Zuo X., Song S., Wang L., Wang L., Biosens. Bioelectron. 2016, 81, 92. [DOI] [PubMed] [Google Scholar]
- 24. Larsson A., Ekblad T., Andersson O., Liedberg B., Biomacromolecules 2007, 8, 287. [DOI] [PubMed] [Google Scholar]
- 25. Pinelli F., Magagnin L., Rossi F., Mater. Today Chem. 2020, 17, 100317. [Google Scholar]
- 26. Tavakoli J., Tang Y., Polymers 2017, 9, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spychalska K., Zając D., Baluta S., Halicka K., Cabaj J., Polymers 2020, 12, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Renneberg R., Pfeiffer D., Lisdat F., Wilson G., Wollenberger U., Ligler F., Turner A. P., Adv. Biochem. Eng./Biotechnol. 2008, 109, 1. [DOI] [PubMed] [Google Scholar]
- 29. Filik H., Avan A. A., Talanta 2020, 211, 120758. [DOI] [PubMed] [Google Scholar]
- 30. George S. M., Tandon S., Kandasubramanian B., ACS Omega 2020, 5, 2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu S., Sedgwick A. C., Elfeky S. A., Chen W., Jones A. S., Williams G. T., Jenkins A. T. A., Bull S. D., Fossey J. S., James T. D., Front. Chem. Sci. Eng. 2020, 14, 112. [Google Scholar]
- 32. Kassahun G. S., Griveau S., Juillard S., Champavert J., Ringuedé A., Bresson B., Tran Y., Bedioui F., Slim C., Langmuir 2020, 36, 827. [DOI] [PubMed] [Google Scholar]
- 33. Jung I. Y., Kim J. S., Choi B. R., Lee K., Lee H., Adv. Healthcare Mater. 2017, 6, 1601475. [Google Scholar]
- 34. Herrmann A., Ph.D. Thesis, Freie Universität, Berlin: 2020. [Google Scholar]
- 35. Dhanjai A. S., Kalambate P. K., Mugo S. M., Kamau P., Chen J., Jain R., TrAC, Trends Anal. Chem. 2019, 118, 488. [Google Scholar]
- 36. Dou Q., Zhang Z., Wang Y., Wang S., Hu D., Zhao Z., Liu H., Dai Q., ACS Appl. Mater. Interfaces 2020, 12, 34190. [DOI] [PubMed] [Google Scholar]
- 37. Gehrke S. H., Uhden L. H., McBride J. F., J. Controlled Release 1998, 55, 21. [DOI] [PubMed] [Google Scholar]
- 38. Andersson O., Larsson A., Ekblad T., Liedberg B., Biomacromolecules 2009, 10, 142. [DOI] [PubMed] [Google Scholar]
- 39. Yoshida S., Hagiwara K., Hasebe T., Hotta A., Surf. Coat. Technol. 2013, 233, 99. [Google Scholar]
- 40. Schmidt C., Schierack P., Gerber U., Schröder C., Choi Y., Bald I., Lehmann W., Rödiger S., Langmuir 2020, 36, 628. [DOI] [PubMed] [Google Scholar]
- 41. Mark S. S., Sandhyarani N., Zhu C., Campagnolo C., Batt C. A., Langmuir 2004, 20, 6808. [DOI] [PubMed] [Google Scholar]
- 42. Huang C.‐J., Brault N. D., Li Y., Yu Q., Jiang S., Adv. Mater. 2012, 24, 1834. [DOI] [PubMed] [Google Scholar]
- 43. Hennig A., Borcherding H., Jaeger C., Hatami S., Würth C., Hoffmann A., Hoffmann K., Thiele T., Schedler U., Resch‐Genger U., J. Am. Chem. Soc. 2012, 134, 8268. [DOI] [PubMed] [Google Scholar]
- 44. Li P., Xu J., Wang Q., Wu C., Langmuir 2000, 16, 4141. [Google Scholar]
- 45. Gao Y., Wolf L. K., Georgiadis R. M., Nucleic Acids Res. 2006, 34, 3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jung Y., Jeong J. Y., Chung B. H., Analyst 2008, 133, 697. [DOI] [PubMed] [Google Scholar]
- 47. Prucker O., Brandstetter T., Rühe J., Biointerphases 2018, 13, 010801. [DOI] [PubMed] [Google Scholar]
- 48. Siegers C., Biesalski M., Haag R., Chem. – Eur. J. 2004, 10, 2831. [DOI] [PubMed] [Google Scholar]
- 49. Löfas S., Johnsson B., J. Chem. Soc., Chem. Commun. 1990, 1526. [Google Scholar]
- 50. Cengiz N., Gevrek T. N., Sanyal R., Sanyal A., Bioconjugate Chem. 2020, 31, 1382. [DOI] [PubMed] [Google Scholar]
- 51. Kalaoglu‐Altan O. I., Sanyal R., Sanyal A., ACS Appl. Polym. Mater. 2020, 2, 4026. [Google Scholar]
- 52. Sieben V. J., Floquet C. F. A., Ogilvie I. R. G., Mowlem M. C., Morgan H., Anal. Methods 2010, 2, 484. [Google Scholar]
- 53. Nam J., Jung I.‐B., Kim B., Lee S.‐M., Kim S.‐E., Lee K.‐N., Shin D.‐S., Sens. Actuators, B 2018, 270, 112. [Google Scholar]
- 54. Jia Z., Sukker I., Müller M., Schönherr H., ACS Appl. Mater. Interfaces 2018, 10, 5175. [DOI] [PubMed] [Google Scholar]
- 55. Jia Z., Müller M., Schönherr H., Macromol. Symp. 2018, 379, 1600178. [Google Scholar]
- 56. Choi N. W., Kim J., Chapin S. C., Duong T., Donohue E., Pandey P., Broom W., Hill W. A., Doyle P. S., Anal. Chem. 2012, 84, 9370. [DOI] [PubMed] [Google Scholar]
- 57. King P. J. S., Saiani A., Bichenkova E. V., Miller A. F., Chem. Commun. 2016, 52, 6697. [DOI] [PubMed] [Google Scholar]
- 58. Lee K. M., Oh Y., Chang J. Y., Kim H., J. Mater. Chem. B 2018, 6, 1244. [DOI] [PubMed] [Google Scholar]
- 59. McLachlan D. R. C., Environmetrics 1995, 6, 233. [Google Scholar]
- 60. Shibata H., Heo Y. J., Okitsu T., Matsunaga Y., Kawanishi T., Takeuchi S., Proc. Natl. Acad. Sci. USA 2010, 107, 17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rong G., Corrie S. R., Clark H. A., ACS Sens. 2017, 2, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Updike S. J., Hicks G. P., Nature 1967, 214, 986. [DOI] [PubMed] [Google Scholar]
- 63. Scheller F., Schubert F., Biosensors, Vol. 11, Elsevier Science Publishers B.V., Amsterdam: 1992. [Google Scholar]
- 64. Öztop H. N., Akyildiz F., Saraydin D., Microsc. Res. Tech. 2020, 83, 1487. [DOI] [PubMed] [Google Scholar]
- 65. Badalyan A., Neumann‐Schaal M., Leimkühler S., Wollenberger U., Electroanal 2013, 25, 101. [Google Scholar]
- 66. Khayyami M., Pérez Pita M. T., Peña Garcia N., Johansson G., Danielsson B., Larsson P.‐O., Talanta 1998, 45, 557. [DOI] [PubMed] [Google Scholar]
- 67. Charles P. T., Goldman E. R., Rangasammy J. G., Schauer C. L., Chen M.‐S., Taitt C. R., Biosens. Bioelectron. 2004, 20, 753. [DOI] [PubMed] [Google Scholar]
- 68. Arenkov P., Kukhtin A., Gemmell A., Voloshchuk S., Chupeeva V., Mirzabekov A., Anal. Biochem. 2000, 278, 123. [DOI] [PubMed] [Google Scholar]
- 69. Dey P., Adamovski M., Friebe S., Badalyan A., Mutihac R.‐C., Paulus F., Leimkühler S., Wollenberger U., Haag R., ACS Appl. Mater. Interfaces 2014, 6, 8937. [DOI] [PubMed] [Google Scholar]
- 70. Randriantsilefisoa R., Cuellar‐Camacho J. L., Chowdhury M. S., Dey P., Schedler U., Haag R., J. Mater. Chem. B 2019, 7, 3220. [Google Scholar]
- 71. Herrmann A., Kaufmann L., Dey P., Haag R., Schedler U., ACS Appl. Mater. Interfaces 2018, 10, 11382. [DOI] [PubMed] [Google Scholar]
- 72. Díaz‐Betancor Z., Bañuls M.‐J., Sanza F. J., Casquel R., Laguna M. F., Holgado M., Puchades R., Maquieira Á., Microchim. Acta 2019, 186, 570. [DOI] [PubMed] [Google Scholar]
- 73. Schmieg B., Schimek A., Franzreb M., Eng. Life Sci 2018, 18, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Qasemi S., Ghaemy M., J. Mater. Chem. C 2020, 8, 4148. [Google Scholar]
- 75. Garcia‐Galan C., Berenguer‐Murcia Á., Fernandez‐Lafuente R., Rodrigues R. C., Adv. Synth. Catal. 2011, 353, 2885. [Google Scholar]
- 76. Liang Z., Zhang J., Wu C., Hu X., Lu Y., Wang G., Yu F., Zhang X., Wang Y., Biosens. Bioelectron. 2020, 155, 112105. [DOI] [PubMed] [Google Scholar]
- 77. Dannert C., Stokke B. T., Dias R. S., Polymers 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li J., Yu J., Huang Y., Zhao H., Tian L., ACS Appl. Mater. Interfaces 2018, 10, 26075. [DOI] [PubMed] [Google Scholar]
- 79. Thenmozhi K., Narayanan S. S., Mater. Sci. Eng., C 2017, 70, 223. [DOI] [PubMed] [Google Scholar]
- 80. Hayat A., Marty J.‐L., Radi A.‐E., Electroanal 2012, 24, 1446. [Google Scholar]
- 81. Zhao H., Jiang G., Weng J., Ma Q., Zhang H., Ito Y., Liu M., J. Mater. Chem. B 2016, 4, 4648. [DOI] [PubMed] [Google Scholar]
- 82. Tokarev I., Minko S., Soft Matter 2009, 5, 511. [Google Scholar]
- 83. Flory P. J., Rehner J., J. Chem. Phys. 1943, 11, 521. [Google Scholar]
- 84. Peppas N. A., Bures P., Leobandung W., Ichikawa H., Eur. J. Pharm. Biopharm. 2000, 50, 27. [DOI] [PubMed] [Google Scholar]
- 85. Richter A., Paschew G., Klatt S., Lienig J., Arndt K.‐F., Adler H.‐J. P., Sensors 2008, 8, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mano J. F., Adv. Eng. Mater. 2008, 10, 515. [Google Scholar]
- 87. Sorber J., Steiner G., Schulz V., Guenther M., Gerlach G., Salzer R., Arndt K.‐F., Anal. Chem. 2008, 80, 2957. [DOI] [PubMed] [Google Scholar]
- 88. Richter A., Bund A., Keller M., Arndt K.‐F., Sensors Actuators B: Chem 2004, 99, 579. [Google Scholar]
- 89. Zhang Z., He C., Chen X., Mater. Chem. Front. 2018, 2, 1765. [Google Scholar]
- 90. Erfkamp J., Guenther M., Gerlach G., Sensors 2019, 19, 2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bubanja I. N., Bánsági T., Taylor A. F., React. Kinet., Mech. Catal. 2018, 123, 177. [Google Scholar]
- 92. Li G., Xiao F., Liao S., Chen Q., Zhou J., Wu Z., Yu R., Sensors Actuators B: Chem 2018, 277, 591. [Google Scholar]
- 93. Wang C., Li F., Bi Y., Guo W., Adv. Mater. Interfaces 2019, 6, 1900556. [Google Scholar]
- 94. Noh K.‐G., Park S.‐Y., Adv. Funct. Mater. 2018, 28, 1707562. [Google Scholar]
- 95. Goh K. B., Li H., Lam K. Y., Biosens. Bioelectron. 2017, 91, 673. [DOI] [PubMed] [Google Scholar]
- 96. Milo S., Thet N. T., Liu D., Nzakizwanayo J., Jones B. V., Jenkins A. T. A., Biosens. Bioelectron. 2016, 81, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wischerhoff E., Zacher T., Laschewsky A., Rekaï E. D., Angew. Chem., Int. Ed. 2000, 39, 4602. [PubMed] [Google Scholar]
- 98. Hoffman A. S., J. Controlled Release 1987, 6, 297. [Google Scholar]
- 99. Fei R., Means A. K., Abraham A. A., Locke A. K., Coté G. L., Grunlan M. A., Macromol. Mater. Eng. 2016, 301, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lagus T. P., Edd J. F., J. Phys. D: Appl. Phys. 2013, 46, 114005. [Google Scholar]
- 101. Hsu M. N., Wei S.‐C., Guo S., Phan D.‐T., Zhang Y., Chen C.‐H., Small 2018, 14, 1802918. [Google Scholar]
- 102. Xu Y., Wu Q., Sun Y., Bai H., Shi G., ACS Nano 2010, 4, 7358. [DOI] [PubMed] [Google Scholar]
- 103. Sun L., Hu N., Peng J., Chen L., Weng J., Adv. Funct. Mater. 2014, 24, 6905. [Google Scholar]
- 104. Ma Y., Mao Y., An Y., Tian T., Zhang H., Yan J., Zhu Z., Yang C. J., Analyst 2018, 143, 1679. [DOI] [PubMed] [Google Scholar]
- 105. Liu S., Su W., Li Y., Zhang L., Ding X., Biosens. Bioelectron. 2018, 103, 1. [DOI] [PubMed] [Google Scholar]
- 106. Wu R., Zhang S., Zhang Q., Liu C., Tian G., Lv J., Sensors Actuators B: Chem 2019, 282, 750. [Google Scholar]
- 107. Jiang C., Li Y., Wang H., Chen D., Wen Y., Sensors Actuators B: Chem 2020, 307, 127625. [Google Scholar]
- 108. Mao Y., Li J., Yan J., Ma Y., Song Y., Tian T., Liu X., Zhu Z., Zhou L., Yang C., Chem. Commun. 2017, 53, 6375. [DOI] [PubMed] [Google Scholar]
- 109. Gačanin J., Synatschke C. V., Weil T., Adv. Funct. Mater. 2020, 30, 1906253. [Google Scholar]
- 110. Li F., Lyu D., Liu S., Guo W., Adv. Mater. 2020, 32, 1806538. [DOI] [PubMed] [Google Scholar]
- 111. Langford G. J., Raeburn J., Ferrier D. C., Hands P. J. W., Shaver M. P., ACS Sens. 2019, 4, 185. [DOI] [PubMed] [Google Scholar]
- 112. Cocchi J. M., Trabaud M. A., Grange J., Serres P. F., Desgranges C., J. Immunol. Methods 1993, 160, 1. [DOI] [PubMed] [Google Scholar]
- 113. Lim S. L., Ooi C.‐W., Low L. E., Tan W. S., Chan E.‐S., Ho K. L., Tey B. T., Mater. Chem. Phys. 2020, 243, 122578. [Google Scholar]
- 114. Tan H., Park S.‐Y., ACS Sens. 2021. [DOI] [PubMed] [Google Scholar]
- 115. Park H.‐I., Park S.‐Y., ACS Appl. Mater. Interfaces 2018, 10, 30172. [DOI] [PubMed] [Google Scholar]
- 116. Meng L., Turner A. P. F., Mak W. C., Biotechnol. Adv. 2020, 39, 107398. [DOI] [PubMed] [Google Scholar]
- 117. Zhao Y., Zhang B., Yao B., Qiu Y., Peng Z., Zhang Y., Alsaid Y., Frenkel I., Youssef K., Pei Q., He X., Matter 2020, 3, 1196. [Google Scholar]
- 118. Scarpa E., Mastronardi V. M., Guido F., Algieri L., Qualtieri A., Fiammengo R., Rizzi F., De Vittorio M., Sci. Rep. 2020, 10, 10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liao M., Liao H., Ye J., Wan P., Zhang L., ACS Appl. Mater. Interfaces 2019, 11, 47358. [DOI] [PubMed] [Google Scholar]
- 120. Mishra R. K., Hubble L. J., Martín A., Kumar R., Barfidokht A., Kim J., Musameh M. M., Kyratzis I. L., Wang J., ACS Sens. 2017, 2, 553. [DOI] [PubMed] [Google Scholar]
- 121. Pal R. K., Farghaly A. A., Collinson M. M., Kundu S. C., Yadavalli V. K., Adv. Mater. 2016, 28, 1406. [DOI] [PubMed] [Google Scholar]
- 122. Pal R. K., Farghaly A. A., Wang C., Collinson M. M., Kundu S. C., Yadavalli V. K., Biosens. Bioelectron. 2016, 81, 294. [DOI] [PubMed] [Google Scholar]
- 123. Xu M., Obodo D., Yadavalli V. K., Biosens. Bioelectron. 2019, 124–125, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liu Y.‐J., Cao W.‐T., Ma M.‐G., Wan P., ACS Appl. Mater. Interfaces 2017, 9, 25559. [DOI] [PubMed] [Google Scholar]
- 125. Gao Y., Gu S., Jia F., Wang Q., Gao G., Chem. Eng. J. 2020, 125555. [Google Scholar]
- 126. Sun Z., Wang L., Jiang X., Bai L., Wang W., Chen H., Yang L., Yang H., Wei D., Int. J. Biol. Macromol. 2020, 155, 1569. [DOI] [PubMed] [Google Scholar]
- 127. Zhao Y., Li Z., Song S., Yang K., Liu H., Yang Z., Wang J., Yang B., Lin Q., Adv. Funct. Mater. 2019, 29, 1901474. [Google Scholar]
- 128. Lim G.‐H., Lee N.‐E., Lim B., J. Mater. Chem. C 2016, 4, 5642. [Google Scholar]
- 129. Zhou J., Yu H., Xu X., Han F., Lubineau G., ACS Appl. Mater. Interfaces 2017, 9, 4835. [DOI] [PubMed] [Google Scholar]
- 130. Yang Z., Wang D.‐Y., Pang Y., Li Y.‐X., Wang Q., Zhang T.‐Y., Wang J.‐B., Liu X., Yang Y.‐Y., Jian J.‐M., Jian M.‐Q., Zhang Y.‐Y., Yang Y., Ren T.‐L., ACS Appl. Mater. Interfaces 2018, 10, 3948. [DOI] [PubMed] [Google Scholar]
- 131. Zhao J., Wang G., Yang R., Lu X., Cheng M., He C., Xie G., Meng J., Shi D., Zhang G., ACS Nano 2015, 9, 1622. [DOI] [PubMed] [Google Scholar]
- 132. Chen D., Zhao X., Wei X., Zhang J., Wang D., Lu H., Jia P., ACS Appl. Mater. Interfaces 2020, 12, 53247. [DOI] [PubMed] [Google Scholar]
- 133. Shao L., Li Y., Ma Z., Bai Y., Wang J., Zeng P., Gong P., Shi F., Ji Z., Qiao Y., Xu R., Xu J., Zhang G., Wang C., Ma J., ACS Appl. Mater. Interfaces 2020, 12, 26496. [DOI] [PubMed] [Google Scholar]
- 134. Yi F.‐L., Meng F.‐C., Li Y.‐Q., Huang P., Hu N., Liao K., Fu S.‐Y., Composites, Part B 2020, 198, 108246. [Google Scholar]
- 135. Chen J., Wen H., Zhang G., Lei F., Feng Q., Liu Y., Cao X., Dong H., ACS Appl. Mater. Interfaces 2020, 12, 7565. [DOI] [PubMed] [Google Scholar]
- 136. Bandodkar A. J., Jeang W. J., Ghaffari R., Rogers J. A., Annu. Rev. Anal. Chem. 2019, 12, 1. [DOI] [PubMed] [Google Scholar]
- 137. McCaul M., Glennon T., Diamond D., Curr. Opin. Electrochem. 2017, 3, 46. [Google Scholar]
- 138. Qiao L., Benzigar M. R., Subramony J. A., Lovell N. H., Liu G., ACS Appl. Mater. Interfaces 2020, 12, 34337. [DOI] [PubMed] [Google Scholar]
- 139. Relf R., Eichhorn G., Waldock K., Flint M. S., Beale L., Maxwell N., J. Therm. Biol. 2020, 90, 102577. [DOI] [PubMed] [Google Scholar]
- 140. Nagamine K., Mano T., Nomura A., Ichimura Y., Izawa R., Furusawa H., Matsui H., Kumaki D., Tokito S., Sci. Rep. 2019, 9, 10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Rose D. P., Ratterman M. E., Griffin D. K., Hou L., Kelley‐Loughnane N., Naik R. R., Hagen J. A., Papautsky I., Heikenfeld J. C., IEEE Trans. Biomed. Eng. 2015, 62, 1457. [DOI] [PubMed] [Google Scholar]
- 142. Lee H., Hong Y. J., Baik S., Hyeon T., Kim D.‐H., Adv. Healthcare Mater. 2018, 7, 1701150. [DOI] [PubMed] [Google Scholar]
- 143. Kim G. J., Kim K. O., Sci. Rep. 2020, 10, 18858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lin S., Wang B., Zhao Y., Shih R., Cheng X., Yu W., Hojaiji H., Lin H., Hoffman C., Ly D., Tan J., Chen Y., Di Carlo D., Milla C., Emaminejad S., ACS Sens. 2020, 5, 93. [DOI] [PubMed] [Google Scholar]
- 145. Liu C., Xu T., Wang D., Zhang X., Talanta 2020, 212, 120801. [DOI] [PubMed] [Google Scholar]
- 146. Emaminejad S., Gao W., Wu E., Davies Z. A., Yin Yin Nyein H., Challa S., Ryan S. P., Fahad H. M., Chen K., Shahpar Z., Talebi S., Milla C., Javey A., Davis R. W., Proc. Natl. Acad. Sci. USA 2017, 114, 4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sempionatto J. R., Khorshed A. A., Ahmed A., De Loyola e Silva A. N., Barfidokht A., Yin L., Goud K. Y., Mohamed M. A., Bailey E., May J., Aebischer C., Chatelle C., Wang J., ACS Sens. 2020, 5, 1804. [DOI] [PubMed] [Google Scholar]
- 148. Herrmann A., Rödiger S., Schmidt C., Schierack P., Schedler U., Anal. Chem. 2019, 91, 8484. [DOI] [PubMed] [Google Scholar]
- 149. Randriantsilefisoa R., Nie C., Parshad B., Pan Y., Bhatia S., Haag R., Chem. Commun. 2020, 56, 3547. [DOI] [PubMed] [Google Scholar]
- 150. Wei Y., Zeng Q., Wang M., Huang J., Guo X., Wang L., Biosens. Bioelectron. 2019, 131, 156. [DOI] [PubMed] [Google Scholar]
- 151. Chen Q., Shi W., Cheng M., Liao S., Zhou J., Wu Z., Microchim. Acta 2018, 185, 557. [DOI] [PubMed] [Google Scholar]
- 152. Rizvi A. S., Murtaza G., Yan D., Irfan M., Xue M., Meng Z. H., Qu F., Talanta 2020, 208, 120403. [DOI] [PubMed] [Google Scholar]
- 153. Hwang Y., Park J. Y., Kwon O. S., Joo S., Lee C.‐S., Bae J., Appl. Surf. Sci. 2018, 429, 258. [Google Scholar]
- 154. Waimin J. F., Nejati S., Jiang H., Qiu J., Wang J., Verma M. S., Rahimi R., RSC Adv. 2020, 10, 16313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Agarwala S., Int J Bioprint 2020, 6, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chen M., Grazon C., Sensharma P., Nguyen T. T., Feng Y., Chern M., Baer R. C., Varongchayakul N., Cook K., Lecommandoux S., Klapperich C. M., Galagan J. E., Dennis A. M., Grinstaff M. W., ACS Appl. Mater. Interfaces 2020, 12, 43513. [DOI] [PubMed] [Google Scholar]
- 157. Nguyen D., Behrens D. M., Sen S., Najdahmadi A., Pham J. N., Speciale G., Lawrence M. M., Majumdar S., Weiss G. A., Botvinick E. L., Anal. Chem. 2020, 92, 7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Chen Y., Zhang S., Cui Q., Ni J., Wang X., Cheng X., Alem H., Tebon P., Xu C., Guo C., Nasiri R., Moreddu R., Yetisen A. K., Ahadian S., Ashammakhi N., Emaminejad S., Jucaud V., Dokmeci M. R., Khademhosseini A., LChip 2020, 20, 4205. [DOI] [PubMed] [Google Scholar]
- 159. Perestam A. T., Fujisaki K. K., Nava O., Hellberg R. S., Food Control 2017, 71, 346. [Google Scholar]
- 160. Wu L., Wang Y., He R., Zhang Y., He Y., Wang C., Lu Z., Liu Y., Ju H., Anal. Chim. Acta 2019, 1080, 206. [DOI] [PubMed] [Google Scholar]