

Abstract
In this work, the covalent attachment of an amine functionalized metal‐organic framework (UiO‐66‐NH2 = Zr6O4(OH)4(bdc‐NH2)6; bdc‐NH2 = 2‐amino‐1,4‐benzenedicarboxylate) (UiO‐Universitetet i Oslo) to the basal‐plane of carboxylate functionalized graphene (graphene acid = GA) via amide bonds is reported. The resultant GA@UiO‐66‐NH2 hybrid displayed a large specific surface area, hierarchical pores and an interconnected conductive network. The electrochemical characterizations demonstrated that the hybrid GA@UiO‐66‐NH2 acts as an effective charge storing material with a capacitance of up to 651 F g−1, significantly higher than traditional graphene‐based materials. The results suggest that the amide linkage plays a key role in the formation of a π‐conjugated structure, which facilitates charge transfer and consequently offers good capacitance and cycling stability. Furthermore, to realize the practical feasibility, an asymmetric supercapacitor using a GA@UiO‐66‐NH2 positive electrode with Ti3C2T X MXene as the opposing electrode has been constructed. The cell is able to deliver a power density of up to 16 kW kg−1 and an energy density of up to 73 Wh kg−1, which are comparable to several commercial devices such as Pb‐acid and Ni/MH batteries. Under an intermediate level of loading, the device retained 88% of its initial capacitance after 10 000 cycles.
Keywords: 2D materials, asymmetric supercapacitors, covalent assemblies, metal‐organic frameworks, MXenes
A facile method for the preparation of graphene/metal–organic framework (MOF) hybrids with an amine‐functionalized MOF and carboxylate‐functionalized graphene via amide bonds for capacitance applications is demonstrated.
Metal‐organic frameworks (MOFs) have emerged as promising materials for various energy and environmental applications because of their unique textural properties with excellent specific surface area and tailored pore size distribution.[ 1 , 2 , 3 , 4 , 5 ] However, the use of MOFs for practical electronic and electrochemical applications is hindered by their limited conductivity and poor mechanical stabilities.[ 6 , 7 , 8 ] On the other hand, graphene, a monoatomic layer of sp2‐bonded carbon exhibits exotic physicochemical properties such as high surface area, excellent electric conductivity, and good mechanical strength.[ 9 , 10 , 11 ] Nevertheless, due to the strong van der Waals force between neighboring layers, graphene sheets are inclined to restack into aggregates a few layers thick, losing their intrinsic characteristics. Hybrid materials based on MOFs and functionalized graphene enable the realization of synergistic properties, allowing the tailored design of materials with properties not possessed by either of the individual components.[ 12 , 13 , 14 ]
Several research groups have devoted their efforts toward exploring various functionalized graphene‐MOF hybrids for different applications based on their structure and property relationships.[ 15 , 16 , 17 , 18 , 19 , 20 ] The use of MOFs to prevent stacking layers of graphene and graphene derivatives such as highly fluorinated graphene oxide (GO) or reduced GO is well established in the literature.[ 21 , 22 , 23 , 24 , 25 , 26 ] Despite the absence of covalent bonds between the MOF and the graphene structure, many of their synergistic properties rely on the stability of the pi–pi, and hydrogen bonds between them, likely to limit the mechanical stability of the materials. Functionalized graphene plays an important role for making graphene‐MOF hybrids, where MOFs are grown selectively at specific functional groups and are covalently attached to graphene nanosheets providing stronger binding and greater stability. Hence, covalent coupling is an important strategy to obtain new and stable graphene‐MOF hybrids with tunable electronic properties, introducing strong bonds between each of the pristine components. Reports on covalently linked functional‐MOF graphene hybrids are still scarce in the literature. The introduction of reactive chemical moieties that are homogeneously distributed over the graphene surface is desirable to enable facile immobilization and strong binding of other molecules and expanded structures on graphene. In particular, carboxyl functionalities are attractive as they readily undergo diverse conjugation reactions (for instance with hydroxyl groups or amines) and increase the material's hydrophilicity.[ 27 , 28 , 29 , 30 ] However, direct, and exclusive attachment of —COOH groups to a graphene surface is very challenging. Typically, GO contains low amounts of carboxyl groups, however they are mostly located at the edges and at defects. Recently, a two‐step process for the synthesis of graphene acid (GA) was reported where fluorographene (FG) is transformed into the fluorine‐free cyanographene (G‐CN, or graphene‐nitrile), which is subsequently hydrolyzed to form carboxylated graphene (G‐COOH) or GA.[ 31 , 32 , 33 , 34 ]
In the present investigation, we report the covalent assembly of GA with the amine functionalized MOF Zr6(OH)4(O)4(bdc‐NH2)6 (denoted as UiO‐66‐NH2, with bdc‐NH2 = 2‐amino‐1,4‐benzenedicarboxylate) (Uio‐Universitetet i Oslo). It shows hierarchical porous behavior. The micro pores originate from octahedral UiO‐66‐NH2 nanocrystals.[ 35 , 36 ] and the mesoporosity stems from the GA‐GA interlayer distances, which are developed through the covalent bonding of GA with the MOF spacers via an amide bridge (Figure 1 ; denoted as GA@UiO‐66‐NH2). Moreover, their covalent assembly is anticipated to impart novel properties, including hierarchical pores for rapid ion transportation, accessible interaction sites, and an interconnected conductive network, that are beneficial for supercapacitor applications. Moreover, the uniformly distributed pores in UiO‐66‐NH2 and the interconnected conductive network of graphene improve the rate capability and long cycling stability. Further, we investigate the material for its capacitive performance, highlighting high performance, and long‐term stability in the positive potential range. Following the electrochemical investigation of the individual material as a positive electrode, we couple it to the 2D Ti3C2T x MXene (where T represents the surface termination, either O, OH, and/or F),[ 37 , 38 , 39 , 40 ]which is an established high performing negative electrode material; the pairing of these two materials could generate a novel kind of high voltage aqueous asymmetric supercapacitor (ASC). The asymmetric combination of electrode materials working in different potential windows is an emerging architecture for continuing the improvement of the energy density of supercapacitors. The subsequent energy density of a device is proportional to the square of its operational voltage. The constructed device in this work is operated at up to 1.7 V and demonstrates a strong performance and stable cyclability of over 10 000 cycles.
Figure 1.
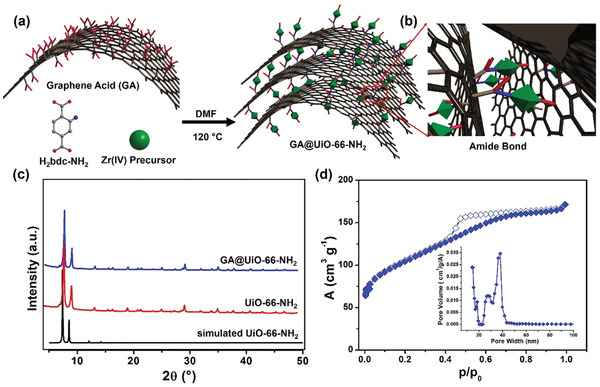
a) Schematic illustration of the covalent assembly of GA and UiO‐66‐NH2 into GA@UiO‐66‐NH2 under solvothermal conditions via b) amide bonds. Carbon, oxygen and nitrogen are represented in grey, red, and blue respectively. Hydrogens have been omitted for clarity. Green octahedrons represent UiO‐66‐NH2 nanocrystals; c) PXRD patterns of UiO‐66‐NH2 (black simulated pattern, red measured pattern) and GA@UiO‐66‐NH2 (blue pattern). d) N2 adsorption isotherm of GA@UiO‐66‐NH2 measured at 77 K, filled symbols represent adsorption, empty symbols desorption. Inset shows pore size distribution determined through NLDFT method.
The Zr‐based MOF UiO‐66‐NH2 was prepared using literature known solvothermal synthesis methods (see Experimental Section Figure S1a, Supporting Information) obtaining highly pure, nanocrystals (Powder X‐ray diffraction (PXRD) in Figure S2, XPS in Figure S3, Supporting Information).The microscopy analysis reveals a morphology featuring octahedral crystals with sizes in the range of 150–200 nm (Figures S4,S5, Supporting Information). Gas adsorption measurements on the material using N2 at 77 K showed a typical type‐I curve. A specific surface area of 780 m2 g−1 was calculated from the isotherm using the Brunauer–Emmett–Teller (BET) method in the p/p0 range of 0.05–0.3 (Figure S1b, Supporting Information). The size of the micropores was calculated using nonlocal density functional theory (NLDFT), giving the micropore size distribution shown (Figure S1 inset, Supporting Information). The GA was prepared under solvothermal conditions using pristine G‐CN derived from FG.[ 30 ] Fourier‐transform infrared spectroscopy confirmed the presence of carboxylic functional groups at high concentration (band at ≈1720 cm−1, Figure S6, Supporting Information), which are distributed homogeneously on the basal plane of graphene rather than on the edges, in contrast to the case of GO. High‐resolution transmission electron microscopy (HRTEM) images of GA reveal transparent nanosheets with lateral dimensions of ≈150–200 nm and elemental mapping from energy‐dispersive X‐ray spectroscopy proves homogeneous distribution of carbon and oxygen throughout the sample (Figure S7a, Supporting Information) and atomic force microscopy (AFM) reveals an average of thickness of the GA nanosheets of around 0.8 nm (Figure S7b, Supporting Information). The nitrogen adsorption isotherm of GA shows a typical type‐II curve which represents surface adsorption and a surface area of around 25 m2 g−1 (Figure S8, Supporting Information).
The GA@UiO‐66‐NH2 hybrid was prepared by combining GA with ZrCl4, and H2bdc‐NH2 using 4‐amino‐benzoic acid as a modulator under solvothermal conditions at 120 °C for 48 h, producing a black gel (see photograph in Figure S9, Supporting Information). The PXRD of the sample possesses all the reflections of pristine Uio‐66‐NH2, confirming that the crystal structure of the MOF is still intact after anchoring to GA (Figure 1c), all index planes shows its phase purity and structural integrity. X‐ray photoelectron spectroscopy (XPS) proves the formation of amide bonds between the carboxylate groups of GA and the amino groups and establishes that only H, C, O, N, and Zr are present in the sample (i.e., no Cl from the ZrCl4 precursor is left in the sample; for the XPS spectra see Figure S10a, Supporting Information). The Zr 3d feature shows one peak corresponding to Zr—O bonds (Figure 2a). The C1s signal was deconvoluted into four peaks assigned to different carbon bonding states, C—C (sp2) (from the graphene basal plane and the MOF linker), C—C(sp3) (from the edges of the GA), C—O (from the carboxylate functionalities of the GA and the linker), and amide/O=C—O (from the covalent attachment of MOF and GA) (Figure 2b). The high resolution N1s spectrum shows three peaks attributed to amine, amide, and partially oxidized nitrogen (Figure 2c). The O1s region of the XPS spectra shows three different bound states for oxygen, O—C, C=O, and amide (Figure 2d), representing the carboxylic acids on the GA, the carboxylates in the MOF structure, the OH termination on the Zr6O4(OH)4 cluster and the covalent amide bridge between the GA and the NH2 functionalized MOF nanocrystals.
Figure 2.
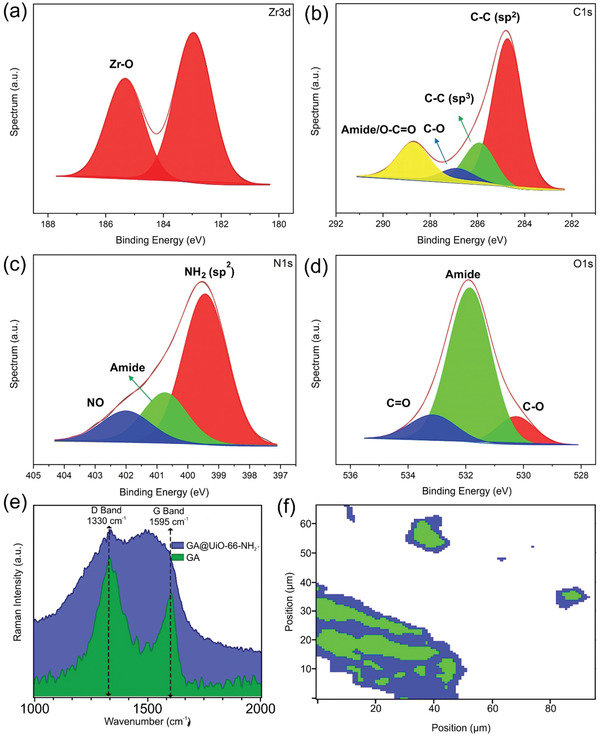
High‐resolution XPS spectra of GA@UiO‐66‐NH2 a) Zr 3d, b) C1s, c) N1s, and d) O1s; e) Raman spectra of pristine GA and GA@UiO‐66‐NH2; f) Raman Map obtained by a hierarchical cluster analysis of the Raman image of GA@UiO‐66‐NH2, blue corresponding to the D band and green corresponding to amide bond.
The Raman spectrum of GA@UiO‐66‐NH2 after covalent attachment, shows two bands present at 1340 (D‐band) and 1593 cm−1 (G‐band) (Figure 2e), but the shape of the D and G bands of the hybrid differ substantially from GA. Additionally, Raman imaging was performed on a representative section of the GA@UiO‐66‐NH2 sample combined with a follow‐up hierarchical cluster analysis via Ward's method (for complete details, please see Supporting Information) (Figure 2f).[ 41 , 42 ] The nitrogen sorption isotherm of the hybrid measured at 77 K exhibits a typical type‐IV isotherm which is characteristic for mesoporous materials. The BET and Langmuir surface areas amount to 600 and 890 m2 g−1 respectively, with a large pore volume of 1.09 cm3 g−1 (Figure 1d). The textural parameters of the hybrid supports the presence of the micropores of pristine UiO‐66‐NH2 (0.1–1.2 nm) and mesopores constructed by the UiO‐66‐NH2 nanocrystals that are covalently binding with oxygen functional groups of GA and acting as a spacer between GA sheets (see micro and meso pore distribution curves in Supporting Information Figure 1d inset). Furthermore, HRTEM analysis of dried gels was used to show morphological evolution of the hybrid. A fibrous micro/mesoporous network structure with a disordered arrangement of interconnected void space of UiO‐66‐NH2 nanoparticles was found, whereas the as‐prepared GA@UiO‐66‐NH2 reveals a complex 3D fibrillary micro/mesoporous network where GA acts as a structure directing agent (Figure 3b–d; Figure S10b, Supporting Information). Additionally, we also observed that the size of UiO‐66‐NH2 nanoparticles decreases 5–10 nm, with GA controlling their morphology. A typical high angle annular dark field (HAADF)‐TEM image of the GA@UiO‐66‐NH2 hybrid shows a uniform distribution of C, N, O, and Zr throughout the sample (Figure 3e–i).Therefore, the collected characterization data substantiates our concept of a covalent‐assembly process of the hybrid, where the amine functionalized UiO‐66‐NH2 nanoparticles are covalently bonded with the carboxylate functional groups of GA through amide bonds. To further underline the covalent bond between GA and UiO‐66‐NH2, we prepared UiO‐66, GA@UiO‐66, a physical mixture of GA and UiO‐66‐NH2 (denoted as GA/UiO‐66‐NH2) for comparison (the complete experimental details are provided in the Supporting Information). In order to gain information on the structural organization, coordination environment, textural, and morphological properties, we measured PXRD, Raman, XPS, FE‐SEM, and N2 physisorption for the comparative materials as well (Figures S12–S22, Supporting Information). These experiments explicitly suggest that the covalent linkage between GA and UiO‐66‐NH2 nanocrystals through amide bond is responsible for the unique textural features of GA@UiO‐66‐NH2.
Figure 3.

Microscopic analysis of GA@UiO‐66‐NH2 a) HRTEM image of GA shows its layered nature b–d) GA@UiO‐66‐NH2 shows fibrous network, where GA nano fibers are interconnected by octahedral UiO‐66‐NH2 nanocrystals. Elemental mapping shows homogenous distribution of all corresponding elements, e) HAADF image, f) carbon, g) zirconium, h) nitrogen, and i) oxygen maps.
The combination of MOF and MOF‐derived materials with graphene and its derivatives showed in previous studies already good improvements on the cycle stability and specific capacitance of supercapacitor materials.[ 43 , 44 , 45 ] This is mostly due to the presence of accessible pores from the MOF/MOF derived material, the conductive nature of the graphene species, more facile access to surface sites and the overall improvement of diffusion throughout the material. The hybrid GA@UiO‐66‐NH2 shows these desired properties like significant surface area, hierarchical pores, and a conductive porous network that is ideally poised for energy storage applications. The material was tested as an active electrode by spray‐depositing it on carbon fabric (CF), without any binder or conductivity enhancing additives (See Supporting Information for details of electrode fabrication). The electrochemical measurements were performed using three‐electrode configuration in 1 m Na2SO4 electrolyte. The GA@UiO‐66‐NH2 was found to be electrochemically stable when swept in the potential range of −0.1 to 1.0 V (versus Ag/AgCl) as seen from Figure 4a. The absence of any narrow redox peaks in the traces at any scan rate and the high degree of symmetry between the charging and discharging sections of each curve, suggest a dominantly capacitive mechanism. Nevertheless, the shapes of cyclic voltammetry (CV) curves are not ideally rectangular and that suggests there is charge contribution from the pseudocapacitive mechanism, which might be originating from the nitrogen based functional amide groups stabilized at the resultant hybrid GA@UiO‐66‐NH2. As described earlier, the surface modification due to the insertion of oxygen, nitrogen or phosphorus functional groups on the surface of GO facilitates the enhancement of total capacitance effects of the electrodes as well as improving the wettability of porous carbon materials with electrolyte.[ 46 , 47 , 48 ] In order to demonstrate the importance of covalent NH2‐bonding, we have performed CV curves for GA and the physical mixture GA/UiO‐66‐NH2 prepared via grinding of GA and UiO‐66‐NH2 samples (see Figure S23, Supporting Information) and compared them with the covalently assembled hybrid GA@UiO‐66‐NH2. The highest current under the CV curves was recorded for GA@UiO‐66‐NH2, suggesting the highest specific capacitance among the tested materials and highlighting the advantage of the covalent bonding.
Figure 4.
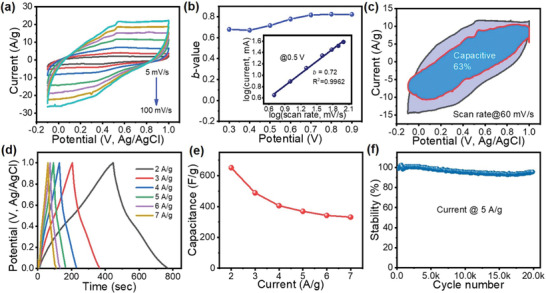
Electrochemical performance of a GA@UiO‐66‐NH2 electrode in a three‐electrode configuration: a) cyclic voltammetry curves recorded at different scan rates in 1 m Na2SO4 electrolyte. b) Variation of b‐values with anodic potential scan. Inset shows power law dependence of peak current density at 0.5 V (versus Ag/AgCl). c) Voltammetry response at a scan rate of 60 mV s−1. The capacitive contribution to the total current is shown by the shaded region (63% of total charge contribution). d) Galvanostatic charge/discharge curves measured at different current densities. e) Variation of specific capacitance as a function of current density. f) Long‐term cycling stability measured at 5 A g−1 over 20 000 cycles.
Contributing factors for the capacitance of the electrode are on the one hand electric double layer capacitance (EDLC) formation and highly surface localized pseudocapacitance (surface redox reactions), but on the other hand also Faradaic redox processes. The intercalation/diffusion based energy storage can be distinguished from the capacitive contributions using the power law.[ 49 ]
| (1) |
In Equation (1), v is a CV scan rate, i is the corresponding current, a is constant, and the b‐value in the equation is a constant constrained to fall in the range of 0.5–1. If b = 1 the system is capacitive. If b = 0.5 the charge storage mechanism is dominated by diffusion controlled processes. The b‐value for GA@UiO‐66‐NH2 is obtained by taking the log of current versus the log of voltage scan rate for each data point. The resulting b‐values for GA@UiO‐66‐NH2 at multiple voltage points lay between 0.7 to 0.8 as shown in Figure 4b. Such a value can be explained by a mechanism that is either a capacitive process or a fast‐redox reaction process that has no diffusion limitation, resulting from the small size of the particles and the highly exposed surface of the electrode materials (nanosize effect). Therefore, these materials are referred to as “extrinsic pseudocapacitors.” The inset of Figure 4b shows an example of plotting the log of Equation (1) at the 0.5 V point of each CV scan.
If we further assume that the avb term can be split into two components as follows,
| (2) |
The values for k 1 and k 2 may be solved for, and these would indicate the relative contribution of the capacitive (EDLC and pseudocapacitance) and the diffusion‐controlled mechanism, respectively. Figure 4c shows the 60 mV s−1 scan with the purple shaded region separating out the capacitive contribution, in this case, 63% of the complete CV scan area, noting that the area of the scan is proportional to the capacitance. It is worth adding that pseudocapacitance being grouped with EDLC as capacitive contributions, constrains the definition of pseudocapacitance in this case to high‐speed surface localized redox reactions that are indistinguishable from EDLC capacitance.[ 50 ]
The capacitance of the material was calculated from galvanostatic charge/discharge (GCD) cycling using the discharge side of the curve. The shapes of the GCD test results are reasonably triangular with a negligible voltage drop, shown in Figure 4d. This is supportive of the claims from CV data that the material is largely capacitive. The specific capacitance values were calculated at numerous current densities and a maximum of 651 F g−1 was recorded at 2A g−1. The material retained just over half of this capacitance when the charge/discharge current was increased threefold to 7 A g−1, where a value of 331 F g−1 was recorded (Figure 4e). The high rate performance is further demonstrated by recording charge/discharge curves at very high current densities (Figure S25, Supporting Information). The high initial capacitance of the material and reasonable rate capability supports our claims that a hierarchical porous structure with a high SSA of covalently attached Uio‐66‐NH2 to GA is facilitating electrolyte ion transport to a large amount of interfaces for the formation of capacitive contributions. By covalently attaching the MOF onto GA, a consecutive charge transfer channel might be generated, resulting in the high performance of the hybrid electrode. In addition, the MOF's ability of adsorbing electrolyte in its pores as well as pseudocapacitance formed due to the amine functional groups also contributed to improve the supercapacitive performance. Even at very high current densities, where slower diffusion reactions are constrained, the electrode material still demonstrates a specific capacitance that is exceptional amongst graphene‐based electrode materials.[ 51 ] For comparison, we have recorded charge/discharge curves for GA, GA/UiO‐66‐NH2 (physical mixture prepared via grinding) and GA@UiO‐66‐NH2 (covalent assembly) as shown in Figure S26, Supporting Information. It was revealed that GA@UiO‐66‐NH2 (covalent) shows excellent performance including Coulombic efficiency 3 A/g using charge/discharge cycling for 100 cycles (Figure S27, Supporting Information). In addition, the GA@UiO‐66‐NH2 electrode showed that it could be cycled at 5 A g−1 for 20 000 cycles and still retain 94% of its initial capacitance, as seen in Figure 4e. This further indicates the covalent bonding approach as a very promising means of anchoring MOF nanoparticles to a conductive graphene framework. Such high retention clearly indicates the mechanical stability of the as‐synthesized hybrid in operando. This was further validated by characterizing the electrodes after charge/discharge cycles. As can be seen from Figure S28, Supporting Information, no significant change in the surface morphology was noticed via SEM measurements. These findings suggest that the capacitive behavior of GA@UiO‐66‐NH2 composite is better than that in conventional graphene based composites (Table S2, Supporting Information), which can be explained as follows: First, the interaction between GA and UiO‐66 in GA@UiO‐66‐NH2 hybrid covalently linked by the amide group is stronger than the van der Waals interaction in conventional composites. The chemical bonding makes the interconnection between GA and UiO‐66‐NH2 more intimate, which reduces the interfacial resistance between GA and UiO‐66‐NH2 and consequently, improves the electrochemical performance.[ 52 ] Second, the electron‐withdrawing carbonyl group (C=O) shown in Figure 1b play a key role as an electron bridge in the formation of the π‐conjugated system containing UiO‐66‐NH2 and GA. Using the π‐conjugated structure, the charges produced by the faradic/pseudocapacitive reaction of UiO‐66‐NH2 can be transferred effectively through the highly conductive GA. The low interfacial resistance and the formation of a π‐conjugated structure can prevent the charges from accumulating, thereby decelerating the change in the structural conformation of UiO‐66‐NH2 with repeated charge/discharge cycles.[ 53 ] This result agrees with that reported in a previous study, wherein low interfacial resistance is highly desirable to improve the electrochemical cycling stability and achieve rapid charge/discharge characteristics at high discharge current densities.[ 52 , 54 ] In addition, the UiO‐66‐NH2 MOFs inserted in between the basal planes of GA layers via amide bonding serve as pillars that prevent the restacking of GA layers and allows full utilization of the materials surface during the electrochemical reactions.
The results showed the improved capacitive performance and electrochemical cycling stability of GA@UiO‐66‐NH2, which can be explained by conjunction of the following factors: i) UiO‐66‐NH2 MOF in between the layers of GA not only acts as a spacer but also provides high surface area and facilitates the rapid transport of the electrolyte ions within the electrode, thereby improving the redox capacitance of UiO‐66‐NH2 and the EDL capacitance of GA. ii) The carbonyl groups in the amide linkage plays a key role in the formation of π‐conjugated structures that effectively transfer the charges, decelerating structural changes of UiO‐66‐NH2 with repeated ion exchange and improving cycling stability. iii) The effective reduction of oxygen‐containing functional groups increases the conductivity of the electrode materials. The result demonstrates that the amide group plays a key role in the formation of the π‐conjugated structure.
After individually testing the GA@UiO‐66‐NH2 material, we used it to assemble an ASC device. The initial electrochemical assessment of the novel hybrid material shown above, promises a strong performance as a positive electrode material. It was decided to pair it with Ti3C2T x MXene as a negative electrode. Like the GA@UiO‐66‐NH2, the MXene electrode was manufactured by spray‐depositing the material onto a CF current collector. Ti3C2T x has been shown to offer stable operation in the negative potential range.[ 55 ] The details of synthesis and characterizations of Ti3C2T x are provided (Figures S29–S33, Supporting Information). Before proceeding to build and test the proposed asymmetric device, we have tested Ti3C2T x /CF using a three‐electrode cell in 1 m Na2SO4 electrolyte. As shown in Figure S31 and S32, Supporting Information, CVs and CD curves of Ti3C2T x /CF clearly show a pair of very broad redox signatures in the chosen potential window (−0.7 to 0.4 V versus Ag/AgCl), which is indicative of pseudocapacitive behavior of Ti3C2T x . The Ti3C2T x /CF electrode delivers a specific capacitance of 406 F g−1 (at a current density of 6.9 A g−1) and long‐term cycling stability over 12 000 cycles. It can be seen that the GA@UiO‐66‐NH2 operates in −0.1 V to 1.0 V versus Ag/AgCl while Ti3C2T x within 0.4 V to −0.7 V versus Ag/AgCl, thereby satisfy the requirements to assemble asymmetric SCs.[ 56 ] From the extrema of these sweeps, the stable potential range for an asymmetric device was determined to be 1.7 V.
Using the two‐electrode configuration, a single cell device was constructed with 1 m Na2SO4 electrolyte. GCD in Figure 5a shows improved symmetry in its triangular appearance compared with the hybrid electrode alone, as expected in a two‐electrode cell, in the same way that the CV is smoothened. The symmetry in appearance is maintained at current densities up to 20 A g−1, where the cell exhibits only a small voltage drop of ≈0.05 V, demonstrating its high‐power handling ability (see inset of Figure 5a). This feature must be attributed to fast charge transport and a large interfacial surface area for EDL formation and surface redox processes. Figure 5b shows the variation of specific capacitance with applied current densities.
Figure 5.
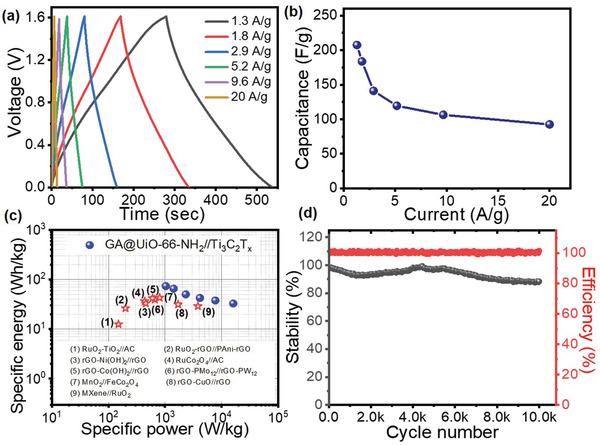
Electrochemical performance of GA@UiO‐66‐NH2//Ti3C2T x asymmetric supercapacitor in 1 m Na2SO4: a) CD curves of device recorded at different current densities. b) Variation of specific capacitance as a function of current density. c) Ragone plot with comparison to other asymmetric devices. d) Cycling stability measured at 5.2 A g−1 over 10 000 cycles with corresponding Coulombic efficiency.
The high‐power cycling ability of the device is supported by its low equivalent series resistance. The collective resistive contributions from current collectors, electrode materials, and electrolyte resistance were found to be 0.6 Ω as obtained from the intercept in the high frequency region of Nyquist plot (see Figure S33, Supporting Information). Power handling and energy density values for the complete cell were calculated from capacitance values obtained at various current densities as seen in Figure 5c. These measurements reveal that the device can produce around 73 Wh kg−1 of energy density at 1 kW kg−1, and even at the much higher power of 16 kW kg−1 the device retained an energy density of 32 Wh kg−1. These values are significantly higher than that for other state of the art asymmetric devices,[ 57 , 58 , 59 , 60 ] and comparable to typical commercial devices such as rechargeable lithium batteries (30–55 Wh kg−1), Pb‐acid batteries (30–45 Wh kg−1), and Ni/MH batteries (60–120 Wh kg−1).[ 61 , 62 ] The drop off in power is much larger than typical EDL‐only SCs. This is related to the limited speed of redox reactions occurring in the device, that is, contributions from near‐surface pseudocapacitance and intercalation reactions. Therefore, the combination of the stable and hierarchically porous chemically‐active GA@UiO‐66‐NH2 hybrid with the pseudocapacitive Ti3C2T x enables a device that behaves as a powerful supercapacitor under high load, but transitions to a slightly battery‐like device under lower power demand.
In the case where storage becomes somewhat battery‐like it may be insightful to consider cell performance using the metric of the C‐rating, where 1 C is equal to discharging the nominal capacity of the cell in 1 h. At the lowest measured discharge current of 1.3 A g−1 the device discharges 100% of its capacity (≈360 Coulombs) in approximately 240 s, equal to 15 C, where it delivers power of 1 kW kg−1. Now consider a constant current discharge of 5.2 A g−1, which is equal to around 60 C. At this rate, the device holds ≈60% of its initial capacity, which equates to an energy density of ≈42 Wh kg−1 at a power of 4.1 kW kg−1. After being cycled at this rate for 10 000 charge/discharges the device retained 88% of its initial storage capacity (Figure 5d). By comparison, state of the art lithium‐ion and all‐solid‐state lithium batteries drop to around 80% capacity at or before a discharge rate of only 5 C, and extrapolation suggests this would be well below 60% at 30 C[ 62 , 63 ] Furthermore, it is known that lithium devices will fail sooner if continuously cycled at high rates and even at modest cycling rates and lower depth of discharge, lithium batteries have an upper life span of around 5000 charge/discharge cycles.[ 64 , 65 ] This comparison highlights the strong potential opportunity that the hybrid material and asymmetric arrangement offers in energy storage development.
In summary, we demonstrated a facile method for the preparation of GA@UiO‐66‐NH2 covalent assembly of GA with amine functionalized Uio‐66‐NH2 via amide bonds. The obtained hybrid provides large surface area, hierarchical pores along with conductive porous network for energy storage applications. The nanocomposite shows significantly high capacitive performance, enhanced rate‐capability along with exceptional cyclic stability. The improved electrochemical performance can be attributed to the fact that the covalent insertion of UiO‐66‐NH2 nanocrystals in between the layers of GA improves the surface area of the materials and facilitates the rapid charge transportation, improving the overall capacitance of the composite. In addition, the amide groups form a π‐conjugated structure that effectively transfer the charges, decelerating structural changes in UiO‐66‐NH2 with repeated ion exchange and improves cycling stability. Thus the result demonstrates that the amide group plays a key role in the formation of the π‐conjugated structure. Furthermore, we demonstrated an asymmetric device arrangement GA@UiO‐66‐NH2 as a positive electrode with a Ti3C2T X MXene as a negative electrode. The device features characteristics of high‐power capacitive devices and high‐energy battery‐like devices, with a power density of up to 16kW kg−1 and an energy density of up to 73 Wh kg−1. Furthermore, the device showed robustness toward repeated charge/discharge cycles with retention of 88% of the initial capacity over 10 000 cycles. It is expected that this kind of stable and hierarchical porous covalent graphene‐MOF hetero 2D nanosheets have promise for application in energy storage, solar cells, and biology applications.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
K.J. and M.H. contributed equally to this work. K.J.R. acknowledges support from Indian Institute of Technology Jammu for providing a seed grant (SGT‐100038) and SERB SRG/2020/000865. D.P.D. acknowledges Queensland University of Technology (start‐up grant: 323000‐0424/07) and Australian Research Council (ARC) for the Future Fellowship (FT180100058). This research was funded by the Operational Programme Research, Development and Education—European Regional Development Fund, Projects (CZ.02.1.01/0.0/0.0/16_019/0000754 and CZ.02.1.01/0.0/0.0/15_003/0000416) of the Ministry of Education, Youth and Sports of the Czech Republic and AB and RZ acknowledge the support from the Czech Science Foundation, project No. 19‐27454X. MO acknowledges the ERC grant (683024) from the EU Horizon 2020 Research and Innovation Programme. HS thankful to IIT Jammu for PhD Fellowship. A.S. gratefully acknowledges the Fonds der Chemischen Industrie for a Liebig Fellowship. Sandia National Laboratories is a multi‐mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration (NNSA) under contract DE‐NA‐0003525. Finally, the work was supported by the German Research Foundation (DFG) within e‐conversion (Fundamentals of Energy Conversion Processes, EXC 2089).
Open access funding enabled and organized by Projekt DEAL.
Jayaramulu K., Horn M., Schneemann A., Saini H., Bakandritsos A., Ranc V., Petr M., Stavila V., Narayana C., Scheibe B., Kment Š., Otyepka M., Motta N., Dubal D., Zbořil R., Fischer R. A., Covalent Graphene‐MOF Hybrids for High‐Performance Asymmetric Supercapacitors. Adv. Mater. 2021, 33, 2004560. 10.1002/adma.202004560
Contributor Information
Kolleboyina Jayaramulu, Email: jayaramulu.kolleboyina@iitjammu.ac.in, Email: jayaram72@gmail.com.
Deepak Dubal, Email: deepak.dubal@qut.edu.au.
Radek Zbořil, Email: radek.zboril@upol.cz.
Roland A. Fischer, Email: roland.fischer@tum.de.
References
- 1. Horike S., Shimomura S., Kitagawa S., Nat. Chem. 2009, 1, 695. [DOI] [PubMed] [Google Scholar]
- 2. Rosi N. L., Eckert J., Eddaoudi M., Vodak D. T., Kim J., O'Keeffe M., Yaghi O. M., Science 2003, 300, 1127. [DOI] [PubMed] [Google Scholar]
- 3. Jiao L., Wang Y., Jiang H. L., Xu Q., Adv. Mater. 2018, 30, 1703663. [DOI] [PubMed] [Google Scholar]
- 4. Shekhah O., Liu J., Fischer R. A., Wöll C., Chem. Soc. Rev. 2011, 40, 1081. [DOI] [PubMed] [Google Scholar]
- 5. Woellner M., Hausdorf S., Klein N., Mueller P., Smith M. W., Kaskel S., Adv. Mater. 2018, 30, 1704679. [DOI] [PubMed] [Google Scholar]
- 6. Stavila V., Talin A. A., Allendorf M. D., Chem. Soc. Rev. 2014, 43, 5994. [DOI] [PubMed] [Google Scholar]
- 7. Burtch N. C., Heinen J., Bennett T. D., Dubbeldam D., Allendorf M. D., Adv. Mater. 2018, 30, 1704124. [DOI] [PubMed] [Google Scholar]
- 8. Xiao X., Zou L., Pang H., Xu Q., Chem. Soc. Rev. 2020, 49, 301. [DOI] [PubMed] [Google Scholar]
- 9. Geim A. K., Novoselov K. S., Nat. Mater. 2007, 6, 183. [DOI] [PubMed] [Google Scholar]
- 10. Cheng C., Li S., Thomas A., Kotov N. A., Haag R., Chem. Rev. 2017, 117, 1826. [DOI] [PubMed] [Google Scholar]
- 11. Li C., Shi G., Adv. Mater. 2014, 26, 3992. [DOI] [PubMed] [Google Scholar]
- 12. Van Ngo T., Moussa M., Tung T. T., Coghlan C., Losic D., Electrochim. Acta 2020, 329, 135104. [Google Scholar]
- 13. Kumar R., Jayaramulu K., Maji T. K., Rao C. N. R., Chem. Commun. 2013, 49, 4947. [DOI] [PubMed] [Google Scholar]
- 14. Du M., Li Q., Zhao Y., Liu C.‐S., Pang H., Coord. Chem. Rev. 2020, 416, 213341. [Google Scholar]
- 15. Zheng Y., Zheng S., Xue H., Pang H., Adv. Funct. Mater. 2018, 28, 1804950. [Google Scholar]
- 16. Liu X.‐W., Sun T.‐J., Hu J.‐L., Wang S.‐D., J. Mater. Chem. A 2016, 4, 3584. [Google Scholar]
- 17. Wang Z., Huang J., Mao J., Guo Q., Chen Z., Lai Y., J. Mater. Chem. A 2020, 8, 2934. [Google Scholar]
- 18. Li S.‐L., Xu Q., Energy Environ. Sci. 2013, 6, 1656. [Google Scholar]
- 19. Zhang K., Kirlikovali K. O., Le Q. V., Jin Z., Varma R. S., Jang H. W., Farha O. K., Shokouhimehr M., ACS Appl. Nano Mater. 2020, 3, 3964. [DOI] [PubMed] [Google Scholar]
- 20. Jahan M., Bao Q., Yang J.‐X., Loh K. P., J. Am. Chem. Soc. 2010, 132, 14487. [DOI] [PubMed] [Google Scholar]
- 21. Huang X., Zheng B., Liu Z., Tan C., Liu J., Chen B., Li H., Chen J., Zhang X., Fan Z., Zhang W., Guo Z., Huo F., Yang Y., Xie L.‐H., Huang W., Zhang H., ACS Nano 2014, 8, 8695. [DOI] [PubMed] [Google Scholar]
- 22. Mao J., Ge M., Huang J., Lai Y., Lin C., Zhang K., Meng K., Tang Y., J. Mater. Chem. A 2017, 5, 11873. [Google Scholar]
- 23. Bu Y., Li F., Zhang Y., Liu R., Luo X., Xu L., RSC Adv. 2016, 6, 40560. [Google Scholar]
- 24. Jayaramulu K., Dubal D. P., Schneemann A., Ranc V., Perez‐Reyes C., Stráská J., Kment Š., Otyepka M., Fischer R. A., Zbořil R., Adv. Funct. Mater. 2019, 29, 1902539. [Google Scholar]
- 25. Jayaramulu K., Geyer F., Petr M., Zboril R., Vollmer D., Fischer R. A., Adv. Mater. 2017, 29, 1605307. [DOI] [PubMed] [Google Scholar]
- 26. Jayaramulu K., Datta K. K., Rosler C., Petr M., Otyepka M., Zboril R., Fischer R. A., Angew. Chem., Int. Ed. 2016, 55, 1178. [DOI] [PubMed] [Google Scholar]
- 27. Georgakilas V., Otyepka M., Bourlinos A. B., Chandra V., Kim N., Kemp K. C., Hobza P., Zboril R., Kim K. S., Chem. Rev. 2012, 112, 6156. [DOI] [PubMed] [Google Scholar]
- 28. Bottari G., Herranz M. Á., Wibmer L., Volland M., Rodríguez‐Pérez L., Guldi D. M., Hirsch A., Martín N., D'Souza F., Torres T., Chem. Soc. Rev. 2017, 46, 4464. [DOI] [PubMed] [Google Scholar]
- 29. Chen D., Feng H., Li J., Chem. Rev. 2012, 112, 6027. [DOI] [PubMed] [Google Scholar]
- 30. Bakandritsos A., Pykal M., Błoński P., Jakubec P., Chronopoulos D. D., Poláková K., Georgakilas V., Čépe K., Tomanec O., Ranc V., Bourlinos A. B., Zbořil R., Otyepka M., ACS Nano 2017, 11, 2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lenarda, Bakandritsos A., Bevilacqua M., Tavagnacco C., Melchionna M., Naldoni A., Steklý T., Otyepka M., Zbořil R., Fornasiero P., ACS Omega 2019, 4, 19944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Papadakis I., Kyrginas D., Stathis A., Couris S., Potsi G., Bourlinos A. B., Tomanec O., Otyepka M., Zboril R., J. Phys. Chem. C 2019, 123, 25856. [DOI] [PubMed] [Google Scholar]
- 33. Bakandritsos A., Kadam R. G., Kumar P., Zoppellaro G., Medved M., Tuček J., Montini T., Tomanec O., Andrýsková P., Drahoš B., Varma R. S., Otyepka M., Gawande M. B., Fornasiero P., Zbořil R., Adv. Mater. 2019, 31, 1900323. [DOI] [PubMed] [Google Scholar]
- 34. Bakandritsos A., Jakubec P., Pykal M., Otyepka M., FlatChem 2019, 13, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cavka J. H., Jakobsen S., Olsbye U., Guillou N., Lamberti C., Bordiga S., Lillerud K. P., J. Am. Chem. Soc. 2008, 130, 13850. [DOI] [PubMed] [Google Scholar]
- 36. Howarth A. J., Liu Y., Li P., Li Z., Wang T. C., Hupp J. T., Farha O. K., Nat. Rev. Mater. 2016, 1, 15018. [Google Scholar]
- 37. Anasori B., Lukatskaya M. R., Gogotsi Y., Nat. Rev. Mater. 2017, 2, 16098. [Google Scholar]
- 38. Wang H., Zhu C., Chao D., Yan Q., Fan H. J., Adv. Mater. 2017, 29, 1702093. [DOI] [PubMed] [Google Scholar]
- 39. Choudhary N., Li C., Moore J., Nagaiah N., Zhai L., Jung Y., Thomas J., Adv. Mater. 2017, 29, 1605336. [DOI] [PubMed] [Google Scholar]
- 40. Xiong D., Li X., Bai Z., Lu S., Small 2018, 14, 1703419. [DOI] [PubMed] [Google Scholar]
- 41. Overman S. A., Thomas G. J., Biochemistry 1995, 34, 5440. [DOI] [PubMed] [Google Scholar]
- 42. Overman S. A., Thomas G. J., Biochemistry 1998, 37, 5654. [DOI] [PubMed] [Google Scholar]
- 43. Chu X., Deng T., Zhang W., Wang D., Liu X., Zhang C., Qin T., Zhang L., Zhang B., Chen C., Zheng W., J. Energy Chem. 2018, 27, 507. [Google Scholar]
- 44. Choi K. M., Jeong H. M., Park J. H., Zhang Y.‐B., Kang J. K., Yaghi O. M., ACS Nano 2014, 8, 7451. [DOI] [PubMed] [Google Scholar]
- 45. Mohammadi Zardkhoshoui A., Hosseiny Davarani S. S., Nanoscale 2020, 12, 1643. [DOI] [PubMed] [Google Scholar]
- 46. Fu D., Zhou H., Zhang X.‐M., Han G., Chang Y., Li H., ChemistrySelect 2016, 1, 285. [Google Scholar]
- 47. Rahmanifar M. S., Hesari H., Noori A., Masoomi M. Y., Morsali A., Mousavi M. F., Electrochim. Acta 2018, 275, 76. [Google Scholar]
- 48. Zhong Y., Cao X., Ying L., Cui L., Barrow C., Yang W., Liu J., J. Colloid Interface Sci. 2020, 561, 265. [DOI] [PubMed] [Google Scholar]
- 49. Wang J., Polleux J., Lim J., Dunn B., J. Phys. Chem. C 2007, 111, 14925. [Google Scholar]
- 50. Horn M., Gupta B., MacLeod J., Liu J., Motta N., Curr. Opin. Green Sustainable Chem. 2019, 17, 42. [Google Scholar]
- 51. Simon P., Gogotsi Y., Dunn B., Science 2014, 343, 1210. [DOI] [PubMed] [Google Scholar]
- 52. Biswas S., Drzal L. T., Chem. Mater. 2010, 22, 5667. [Google Scholar]
- 53. Otero T. F., Padilla J., J. Electroanal. Chem. 2004, 561, 167. [Google Scholar]
- 54. An J., Liu J., Zhou Y., Zhao H., Ma Y., Li M., Yu M., Li S., J. Phys. Chem. C 2012, 116, 19699. [Google Scholar]
- 55. Lukatskaya M. R., Kota S., Lin Z., Zhao M.‐Q., Shpigel N., Levi M. D., Halim J., Taberna P.‐L., Barsoum M. W., Simon P., Gogotsi Y., Nat. Energy 2017, 2, 17105. [Google Scholar]
- 56. Dubal D. P., Ayyad O., Ruiz V., Gómez‐Romero P., Chem. Soc. Rev. 2015, 44, 1777. [DOI] [PubMed] [Google Scholar]
- 57. Dubal D. P., Chodankar N. R., Holze R., Kim D.‐H., Gomez‐Romero P., ChemSusChem 2017, 10, 1771. [DOI] [PubMed] [Google Scholar]
- 58. Dubal D. P., Chodankar N. R., Vinu A., Kim D.‐H., Gomez‐Romero P., ChemSusChem 2017, 10, 2742. [DOI] [PubMed] [Google Scholar]
- 59. Jiang Q., Kurra N., Alhabeb M., Gogotsi Y., Alshareef H. N., Adv. Energy Mater. 2018, 8, 1703043. [Google Scholar]
- 60. Zhang J., Jiang J., Li H., Zhao X. S., Energy Environ. Sci. 2011, 4, 4009. [Google Scholar]
- 61. Burke A., Electrochim. Acta 2007, 53, 1083. [Google Scholar]
- 62. Campbell P. G., Merrill M. D., Wood B. C., Montalvo E., Worsley M. A., Baumann T. F., Biener J., J. Mater. Chem. A 2014, 2, 17764. [Google Scholar]
- 63. Quartarone E., Mustarelli P., J. Electrochem. Soc. 2020, 167, 050508. [Google Scholar]
- 64. Randau S., Weber D. A., Kötz O., Koerver R., Braun P., Weber A., Ivers‐Tiffée E., Adermann T., Kulisch J., Zeier W. G., Richter F. H., Janek J., Nat. Energy 2020, 5, 259. [Google Scholar]
- 65. Horn M., MacLeod J., Liu M., Webb J., Motta N., Econ. Anal. Policy 2019, 61, 93. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information