

Abstract
The topographic features at the cell–material biointerface are critical for cellular sensing of the extracellular environment (ECM) and have gradually been recognized as key factors that regulate cell adhesion behavior. Herein, a well‐defined nanostructured biointerface is fabricated via a new generation of mussel‐inspired polymer coating to mimic the native ECM structures. Upon the bioinert background presence and biospecific ligands conjugation, the affinity of cancer cells to the resulting biofunctional surfaces, which integrate topographic features and biochemical cues, is greatly strengthened. Both the conjugated bioligand density, filopodia formation, and focal adhesion expression are significantly enhanced by the surficial nano‐features with an optimized size‐scale. Thus, this nanostructured biointerface exhibits high capture efficiency for circulating tumor cells (CTCs) with high sensitivity, high biospecificity, and high purity. Benefiting from the unique bioligands conjugation chemistry herein, the captured cancer cells can be responsively detached from the biointerfaces without damage for downstream analysis. The present biofunctional nanostructured interfaces offer a good solution to address current challenges to efficiently isolate rare CTCs from blood samples for earlier cancer diagnosis.
Keywords: biospecific capture, circulating tumor cells, integrated biointerfaces, reversible release
Multifunctional ECM‐mimic biointerfaces integrated biospecific affinity interaction, antifouling background, reversible biolinker, and well‐defined topographical features are developed to strengthen the cellular interaction for cancer cell isolation. The resulting biointerfaces display an ultra‐high capture efficiency for CTCs with high purity and sensitivity. Benefiting from the competitive binding, the captured cells can be responsively released without damage.
1. Introduction
It is well known that the extracellular matrix is a complex fibrous network with micro‐/nano hierarchical features (ranging from 260 to 410 nm).[ 1 ] Cell growth and functions are tightly associated with the cellular interactions at the cell‐ECM biointerface.[ 2 ] Over the past two decades, a series of nano/microstructured biointerfaces with the abilities to control cell adhesion/detachment and enhance cellular sensing to external signals have been developed.[ 2 , 3 ] These biointerfaces provide possibilities to mimic the natural nanostructure of ECMs and exhibit potential applications in tissue engineering and regenerative medicine. The larger surface area and unique interactions between nanostructured biointerfaces and cells facilitate firm cellular attachment and exhibit great advantages in the isolation and detection of rare cells, for example, the CTCs.[ 4 ] CTCs are cancer cells that detach from primary tumors or metastatic sites and circulate in the peripheral blood as the cellular origin of metastasis.[ 5 ] Clinical researches indicated that the levels of CTCs in metastatic cancer patients are a predictor of survival.[ 6 ] Cytogenetic evidence also matched aneusomic patterns between CTCs and those from the primary tumor.[ 7 ] Enumeration and isolation of CTCs from patients’ blood can provide insight into blood‐borne metastasis, monitor disease progression, and help to guide the therapeutic outcomes of cancer.[ 8 ] Therefore, technologies that can capture and detect CTCs directly from the patient's blood are highly desired from a clinical point of view for early cancer diagnosis. However, the concentration of CTCs in a patient's blood circulatory system is ultra‐low, usually a few to hundreds per milliliter, therefore the isolation of CTCs from a patient's blood with high efficiency and purity is a big challenge.[ 9 ]
In order to detect and isolate these rare CTCs, multiple isolation methods, for example, microfluidic devices, immunomagnetic beads, and microfilter devices, have been developed by utilizing the differences among cellular physical properties or diversity of surface biomarkers of cancer cells.[ 10 , 11 , 12 , 13 ] Among them, the methods based on affinity interaction between the bioligands at the interfaces and the biomarkers on cancer cell membrane yield higher efficiency and good specificity in contrast to mechanical and electrical sorting technique.[ 14 ] But it is still difficult to isolate the CTCs with higher purity and viability.[ 15 ] Recently, increasing evidence indicates that biomimetic micro/nano topographic features at the cell–material interface could greatly improve the CTCs capture efficiency from patient blood.[ 16 , 17 , 18 , 19 , 20 ] Compared to a flatter surface, the nanostructure features at the interface would provide more surface area for the functionalization of antibodies and/or aptamers, suitable sites for cancer cell trapping, as well as further lowering the rolling velocity of cells.[ 15 ] Further results also demonstrate that the nanostructured features may induce the formation of pseudopodia on the nanostructured surface[ 21 ] and improve cell–material surface adhesion force,[ 22 ] which further increases the affinity between cancer cells and nanostructured substrates. Thus, more and more attention has been paid to developing functionalized structured biointerfaces, for example, the integrated NanoVelcro system,[ 18 , 23 , 24 , 25 ] for rare CTCs capture even if it generates another technological challenge to fabricate those nano/micro hierarchical substrates.[ 13 , 26 ]
While higher nonspecific proteins and cells (white blood cell) binding was also consequently observed on the rough nanostructured surfaces in the absence of specific bioligand[ 27 ] and antifouling background.[ 28 ] Nevertheless, the exact relationship between cancer cell capture and the surface roughness or topography of the biointerface has yet to be fully understood. It is noted that different cancer cell lines may show diverse preferred surface features and display dissimilar responsiveness on the same nanostructured surface.[ 29 ] Furthermore, to carry out cell proliferation and downstream analysis, the captured CTCs should be pure enough and able to further detach from the isolation platform with required viability. But the release of the captured cells from the devices possibly causes cell damages and impedes further insights into the biology of CTCs by usual enzymatic and mechanical lift‐off methods.[ 30 ] Thus, responsive linker chemistry between isolation devices and CTCs specific biomarkers is necessary to enable the controllable releasing of the captured cells with minimal damages.
Herein, we presented an advanced biointerface that integrates the biospecific affinity interaction and well‐defined topographical features for cancer cell isolation. A mussel‐inspired coating polymer was developed and used to fabricate the defined nanostructures on the biointerface via a simple dip‐coating method with a series of surface roughness that ranged from the nanometer to the sub‐micrometer scale (Scheme 1 ). Similar to our previous reports about cell adhesion on rough surfaces,[ 31 , 32 ] the adhesion and isolation of cancer cells on this nanostructured biointerface exhibited a unique biphasic manner and an optimized roughness was therefore found. To achieve a high capture specificity, a bioinert hyperbranched polyglycerol (hPG) monolayer coating was introduced onto the defined nanostructured biointerfaces, which served as a strong antifouling background to resist the nonspecific adhesion of proteins and blood cells (red blood cell, white blood cell, platelets, etc.). On the other hand, the chemical property of hPG was then explored to conjugate the epithelial‐cell adhesion molecule antibody (anti‐EpCAM) onto the biointerface via a revisable cis‐diols‐phenylboronic ester binding. Notable evidence revealed that the presented nanostructure features greatly promote the formation of cellular focal adhesion (FA) and filopodia in addition to the increase of surface area for antibody conjugation. Thus, the resulting biointerfaces that combine biospecific affinity interaction and surface nanostructures displayed an ultra‐high capture efficiency for CTCs (>95%) with high purity (>97%) and sensitivity, even for cells with low EpCAM expression, for example, the MDA‐MB‐231 and A549 cells. Moreover, upon exposure to a glycan molecule which has a higher affinity to phenylboronic acid (PBA), that is, sorbitol,[ 33 ] the captured CTCs thus responsively release from the biointerfaces in a short time without damage due to the competitive binding.
Scheme 1.
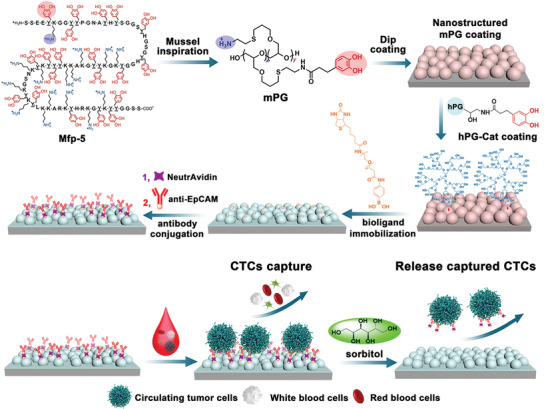
Nanostructured multivalent biointerfaces for specific CTCs adhesion/isolation and responsively release based on mussel‐inspired polymer coating and the unique dynamic cis‐diols‐boronic ester chemistry (Mfp‐5: mussel foot proteins‐5, mPG: mussel‐inspired linear polyglycerol, hPG‐Cat: 5% catecholic hPG).
2. Results and Discussion
2.1. Mussel‐Inspired Polymer Coating with Well‐Defined Surficial Features
To mimic the adhesion properties of mussel foot proteins, we previously developed a mussel‐inspired dendritic polyglycerol (MI‐dPG) coating polymer and versatile coating surfaces have been successively fabricated via this building block.[ 31 , 32 , 34 , 35 ] Due to the multivalent character of the dendritic polyglycerol, most of the functional groups are exposed to the periphery.[ 34 ] The crosslinking of the polymers is greatly accelerated but the resulting roughness and topographic features of the coating are far beyond control. To circumvent these limitations, a new generation of mussel‐inspired polyglycerol‐based coating polymer (mPG) with a linear polymer architecture was thus designed herein. Compared with the dendritic architecture, the linear mPG has a higher hydrodynamic radius and more entanglement conformation. Therefore, the reactivity of the heteromultivalent catechol‐ and amine‐groups on the polymer chain was decelerated and the coating process was more controllable. The average molecular weight of the mPG coating polymer is about 10 kDa, which is similar to the mfp‐5 (≈9 kDa).[ 36 ] While the functionality of the catechol groups in the mPG polymer (70 mol%) is ≈2.5 times higher than that in mpf‐5 (27 mol%[ 37 ]) for rapid deposition. Under basic conditions, the mPG underwent covalent crosslinking and aggregated into nanoparticles, which were partially insoluble in the coating solution (a mixture of methanol and water) and were deposited onto the substrate (Figure 1a and Figure S1, Supporting Information). Furthermore, the deposited particles progressively grew from nanoscale to microscale due to the consecutive Michael addition and/or dopa‐quinone coupling (Figure 1b and Figure S1, Supporting Information). With the increase of incubation time at room temperature, the optical transmittance of the resulting coating surface was progressively decreased (Figure 1c). While the thickness of mPG coating on the substrate continually increased with incubation and reached 340 nm after 4 h and 760 nm after 24 h (Figure 1d), which is quite slow compared with its dendritic analog[ 34 ] but much faster than the crosslinking of dopamine under basic[ 38 ] and even oxidative conditions.[ 39 ] The surface morphology and root mean square roughness (Rq) of the resulting coating with different incubation time were further characterized by atomic force microscopy (AFM) (Figure 1e,f). mPG coating polymers adsorbed and gradually polymerized on the substrate surface and generated well‐defined topographical features ranging from nanometers to micrometers with an increase of coating time (Figure 1e). The surface roughness of the resulting surfaces slowly increased from 56 nm (mPG1: 1‐h incubation) to 571 nm (mPG24: 24‐h incubation) (Figure 1f), which suggests that the catechol‐ and amine‐groups inter/intra‐polymer coupling reaction are greatly affected by the polymer architecture. Thus, the resulting coating process displays a more controllable manner. Except for the difference in the surficial topographic features, the resulting mPG coating surfaces maintain similar surface chemical property (Figure S2, Supporting Information).
Figure 1.
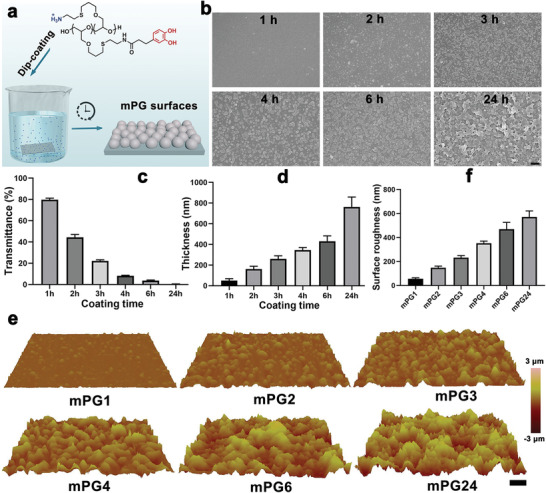
Fabrication of nanostructured surfaces via the mussel‐inspired coating polymer. a) One‐step dip‐coating method is used to prepared surfaces with well‐defined topographic features that result from the self‐polymerization of mPG polymers under the basic condition and the deposition of formed nanoparticles from mixing solution. b) Scanning electron microscope images of the resulting mPG coating surfaces with the variation of dip‐coating time. The scale bar indicates 10 µm. Time‐dependent optical transmittance (c) and thickness (d) of the mPG coating surface. e) The surface morphology and corresponding surface roughness (f) of mPG coating surfaces characterized by AFM. Scale bar indicates 10 µm. The mPG1, mPG2, mPG3, mPG4, mPG6, and mPG24 indicate the resulting mPG coatings with 1, 2, 3, 4, 6, and 24 h dip‐coating time respectively.
2.2. Cellular Response to Nanostructured mPG Surfaces Displayed a Roughness Selective Manner
To investigate cell adhesion on the nanostructured surfaces, human breast cancer cells (MCF7) were seeded and cultured for 3 h. As shown in Figure 2a , the adhesion of MCF7 cells on nanostructured mPG surfaces displayed an interesting biphasic manner. The corresponding cell capture efficiency gradually increased with the increase of surficial roughness and maximized on mPG4 surfaces (Rq = 352 nm, capture efficiency >50%). Once the surface roughness was above 352 nm, the topographic features greatly restricted the cell adhesion, and thus the capture efficiency was drastically reduced. This interesting cellular response to nanostructured surficial features agrees with previous reports on MCF7 cells[ 20 ] and mesenchymal stem cells (MSC)[ 31 , 32 ] but the preferred roughness shifted to a lower region owing to the smaller size of cancer cells.
Figure 2.
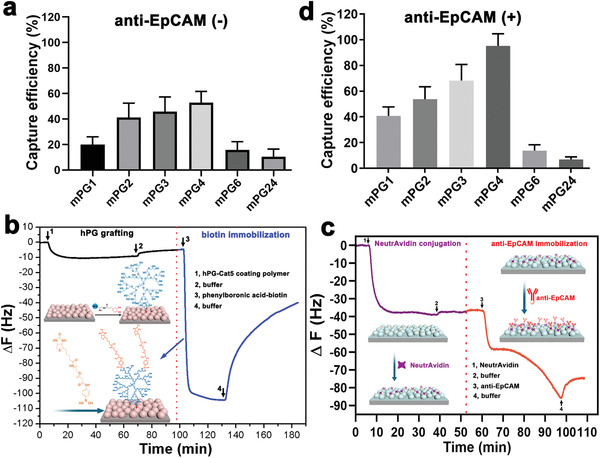
Biospecific adhesion/capture of cancer cells on mPG surfaces with roughness selectivity manner. a) Cell capture on nanostructured mPG surfaces (n = 20–30, 3 technical replicates). b) Real‐time quartz crystal microbalance (QCM) frequency shift of the adsorption of antifouling hPG‐Cat polymers on mPG4 surface and the in situ immobilization of bioligands onto the resulting hPG‐mPG4 surface via the unique diol‐boronic ester bonding between hPG and biotin‐PBA. c) Real‐time QCM frequency shift of the adsorption of NeutrAvidin and anti‐EpCAM onto the biotin functionalized mPG4 surfaces via the bioorthogonal biotin‐avidin interaction, respectively. d) Biospecific CTCs capture efficiency on the antibody (anti‐EpCAM) functionalized mPG coating surfaces (n = 30–35, 3 technical replicates).
Based on this special cell adhesion behavior of nanostructured surfaces, we then asked whether these interfaces could be potentially developed for the capture and isolation of rare CTCs from blood samples. EpCAM (epithelial cell adhesion molecule) is a typical CTCs biomarker, which is overexpressed in most adenocarcinoma CTCs but negative‐expressed in normal blood cells.[ 40 ] To obtain specific CTCs isolation, anti‐EpCAM was then employed as a biospecific ligand and immobilized onto the nanostructured mPG coating surface. As mentioned above, a strong bioinert background is required to resist the interference of nonspecific proteins adsorption and blood cell (red blood cell, white blood cell, and platelets) adhesion during CTCs isolation. Therefore, a monolayer of antifouling hPG was introduced on the defined nanostructured biointerfaces to guarantee the high capture specificity and purity before bioligands conjugation. Catechol functionalized hPG (5% functionality) quickly adsorbed onto the mPG4 coating surface and then coupled with the amino and/or quinone groups on the mPG4 surface to form a stable coating in the basic buffer (Figure 2b). Based on the quantification of QCM measurement, there are 88.4 ng cm−2 of hPG‐Cat polymers chemically anchored onto the mPG4 surface and generate a 1.37 nm monolayer coating on the mPG4h nanostructured surface. No obvious surface roughness change was observed after hPG grafting (Figure S3, Supporting Information), which indicated the mPG4 surface still maintains its nanostructured features. In addition to the excellent antifouling properties, plenty of cis‐diol groups on the hPG polymer also offer an opportunity to immobilize bioligands via the revisable cis‐diol‐phenylboronic ester bonding. Thus, PBA‐modified biotin was grafted onto the hPG‐mPG surfaces. The sharp decrease of frequency in the real‐time QCM curve indicated the rapid biotin‐binding on the hPG‐mPG4 surface through the PBA/cis‐diol esterification (Figure 2b). Subsequently, biotinylated anti‐EpCAM were conjugated onto the coating surfaces via the biorthogonal avidin biolinker, which could form the strongest non‐covalent bonding with biotin.[ 41 ] 651.2 ng cm−2 of NeutrAvidin and 667.1 ng cm−2 of biotinylated anti‐EpCAM were stepwise conjugated onto mPG4 surfaces to generate an anti‐EpCAM functionalized biospecific nanostructured biointerfaces (Figure 2c).
Compared with the nanostructured mPG coating surface, the adhesion of MCF7 cancer cells (EpCAM positive cell line) on anti‐EpCAM functionalized surfaces was significantly enhanced. The corresponding capture efficiency was greatly improved for the surface with lower roughness and maximized on the anti‐EpCAM‐mPG4 surface. More than 95% of spiked cells from the serum‐free medium were captured onto antibody‐functionalized mPG4 surface after 2 h of incubation (Figure 2d). Thereafter, the capture efficiency on higher rough surfaces was drastically reduced. Furthermore, no cells were obviously observed on the hPG‐mPG4 surfaces (Figure S4, Supporting Information), indicating the nanostructured surfaces have an excellent bioinert background. To achieve the highly efficient capture of cancer cells, the nanostructured surfaces still required the functionalization of biospecific ligands to recognize the cells.
2.3. High‐performance Cancer Cell Capture on Biospecific Nanostructured Surfaces
To assess the specifical cancer cell capture on the anti‐EpCAM‐functionalized nanostructured interfaces, EpCAM‐positive cancer cell line (MCF7[ 42 ]) was used as the model cell line, whereas the EpCAM‐negative cell line, that, GFP+‐Hela cells,[ 43 ] and anti‐EpCAM functionalized smooth surfaces were employed as negative control cell line and negative control surface, respectively. For microscopic observation, MCF7 cells were pre‐stained with a fluorescent tracker dye. All experimented cells were spiked in serum‐free RPMI‐1640 culture medium with a concentration of 3000 cells mL−1. Unless otherwise specified, the nanostructured biointerface in the following part was the antibody functionalized mPG4 surfaces with optimized antibody conjugation density (Figure S5, Supporting Information). Benefiting from the nanostructured features, cancer cell capture efficiency on both mPG4 surfaces with and without antibody functionalization was greatly improved compared to the smooth control surfaces (Figure 3a ,b). More than 95% of spiked cells adhered to the anti‐EpCAM‐mPG4 surface whereas only 53% of the spiked cells were captured onto the antibody functionalized smooth surfaces. Under the same condition, less than 5% of EpCAM‐negative HeLa cells were observed on antibody functionalized mPG4 surfaces (Figure 3a,b). The present anti‐EpCAM displayed a good biospecific property and remarkably reduced the nonspecific cell adhesion. Thus, only EpCAM positive MCF7 cells were selectively captured by this biointerface from the MCF7/HeLa mixture (Figure S6, Supporting Information). The hierarchical nanostructured features provide more surface area for cell interaction with the biointerfaces and could further lower the rolling velocity of cells. Therefore, spiked cancer cells tend to interact with the nanostructure features and quickly adhered to the biointerface. The corresponding capture efficiency on nanostructured mPG4 surface reached 90% after 30 min and maximized to 95% after 1‐h incubation, which is about two or three times faster than many reported biointerfaces for cancer cell capture.[ 44 , 45 ] For the smooth control surface, the capture efficiency gradually reached 65% even after incubation for 3 h (Figure 3c). It suggests that the nanostructured features on the surfaces could significantly facilitate the adhesion of cancer cells on the biointerfaces with higher capture efficiency, higher bioselectivity, and shorter detection time, potentially being applied for fast cancer diagnosis. On the other hand, this significant enhancement of biospecificity and bioselectivity may also be ascribed to the excellent bioinert background generated from the hPG layer.
Figure 3.
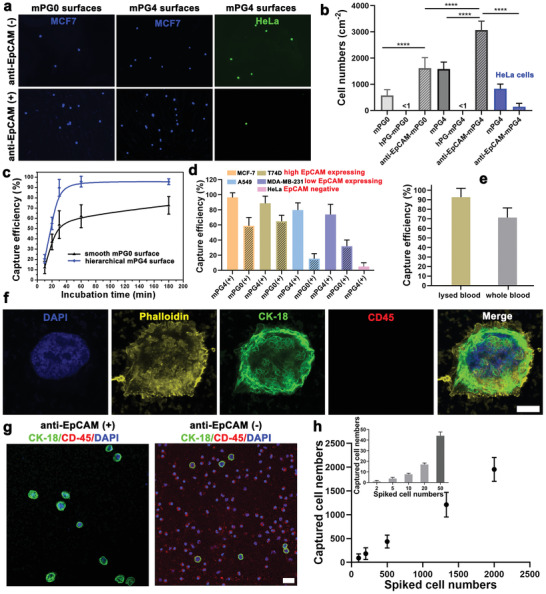
High performance of CTCs captures on antibody functionalized nanostructured mPG4 surfaces. a) Representative images of cells captured on mPG4 surfaces with and without antibody conjugation. MCF7 (EpCAM positive) cells are pre‐stained with CellTrace yellow dye. The scale bar indicates 100 µm. b) Cells captured on smooth mPG0 surface (anti‐EpCAM conjugated hPG monolayer coating on smooth glass slides) and nanostructured mPG4 surfaces (n = 20–25, 3 technical replicates). * indicates a statistically significant comparison with P < 0.05 (one‐way ANOVA). c) Time dependence cell capture on antibody functionalized smooth mPG0 surface and nanostructured mPG4 surfaces. d) Cell capture efficiency on anti‐EpCAM‐mPG4 surfaces for different cell lines with anti‐EpCAM‐mPG0 surfaces as control (n = 15–20, 3 technical replicates). e) Biospecific cancer cell capture on anti‐EpCAM‐mPG4 surfaces from MCF7 spiked RBC lysed blood samples and whole blood samples (n = 20–25, 3 technical replicates). The spiked cell concentration is 2000 cells mL−1. f) Fluorescent micrographs of captured cells on anti‐EpCAM‐mPG4 surfaces from artificial patient blood samples stained for the nucleus (DAPA, blue), actin (phalloidin, yellow), epithelial marker cytokeratin 18 (CK‐18, green), leukocyte marker (CD45, red). The scale bar indicates 5 µm. g) Representative immunofluorescence images of captured cells on the mPG4 surface without anti‐body conjugation and anti‐EpCAM‐mPG4 surfaces from artificial patient blood samples. The scale bar indicates 20 µm. h) Linear correlation between the number of spiked and captured cells on anti‐EpCAM‐mPG4 surfaces from artificial patient blood samples (n = 3 for each condition). The inset indicates the capture performance for ultra‐low concentrations of cancer cells (2, 5, 10, 20, and 50 cells mL−1 of lysed blood, n = 6).
The biomarker expression level of different cancer cell lines greatly determines the cell‐interface binding affinity.[ 46 ] To explore the specificity of these nanostructured biointerfaces for cancer cell capture, three kinds of cell lines, that is, MCF7 and T47D (high EpCAM expression), MDA‐MB‐231 and A549 (low EpCAM expression), GFP+‐HeLa (EpCAM‐negative cells), were spiked into serum‐free medium and incubated with anti‐EpCAM functionalized mPG4 surfaces for 1 h, respectively. Similar to MCF7 cells, high EpCAM expression T47D displayed a higher affinity to the biofunctionalized surfaces and more than 88% of spiked cells were specifically captured onto anti‐EpCAM‐mPG4 surfaces (Figure 3d). Even the MDA‐MB‐231 and A549 cells have a lower EpCAM expression on the cell membrane, there are still ≈75% of MDA‐MB‐231 cells and ≈80% of A549 cells were successively isolated by these biointerfaces. In contrast, the antibody functionalized mPG4 surfaces exhibited a very low capture efficiency for EpCAM‐negative HeLa cells (<5%), which may be nonspecifically trapped on surfaces (Figure 3d). It needs to notice that the capture efficiency for both high‐ and low‐EpCAM expression cancer cells were remarkably enhanced by the surficial nanostructured features, especially for the cell lines with lower EpCAM expression level. More than 5 times and 2 times capture efficiency improvements were found on the anti‐EpCAM functionalized mPG4 surfaces compared to the smooth control surfaces for A549 cancer cells and MDA‐MB‐231 cancer cells, respectively (Figure 3d).
To further verify the potential application of this nanostructured biointerface in cancer diagnosis, we then spiked the cancer cells into human blood and obtained artificial cancer patient's blood samples. As shown in Figure 3e, roughly 90% of MCF7 cancer cells were captured from lysed blood and ≈72% were captured from the whole blood sample by the nanostructured biointerface (anti‐EpCAM‐mPG4) after 1‐h incubation. Immunostaining was used to distinguish the captured cancer cells from blood cells on nanostructured surfaces. Captured cells were identified as CTC cells when they were immunostained positive for anti‐CK18 (a protein marker for epithelial cells) but negative for anti‐CD45 (CD45, a marker for leukocytes) (Figure 3f). Compared with nanostructured surfaces without antifouling background and specific biomarker (mPG4 surfaces), the anti‐EpCAM‐mPG4 nanostructured surfaces displayed a high biospecificity for MCF7 cancer cells (CK18+/CD45‐/DAPI+) and excellent resistance to the adhesion of leukocytes (CK18‐/CD45+/DAPI+) (Figure 3g). The resulting capture purity for anti‐EpCAM‐mPG4 nanostructured surfaces was 93 ± 2.8% (Figure S7, Supporting Information). It is known that the abundance of CTCs in patient blood samples is very rare and ranges from a few to hundreds per milliliter.[ 47 ] Thus, to explore the detection sensitivity of these nanostructured biointerfaces, we further spiked the MCF7 cancer cells into lysed whole blood with a concentration from 2000 to 2 cells mL−1. As shown in Figure 3h, a nice linear correlation between the number of spiked cells and captured cells was observed. These biospecific nanostructured interfaces can still efficiently capture the CTCs from a very dilute suspension, that is, ≈2 cells mL−1 (capture efficiency >75%).
2.4. Nanostructured Topographic Features Enhance Antibody Conjugation and Cancer Cell Adhesion
To explore the underlying mechanism responding for the high cancer cell capture performance on nanostructured surfaces, immunochemistry was first performed to quantify the density of conjugated antibody on topographic surfaces with different surface roughness. As shown in Figure 4a , the antibody conjugation on the coating surface was progressively increasing with the increase of surface roughness, which provides more surface area for biomarkers immobilization. Then the detailed morphology of the captured cells on nanostructured surfaces with different roughness was observed by scanning electron microscope (Figure 4b). This indicates that the cancer cells prefer to spread on surfaces with lower roughness and maintained a round morphology. While the spreading of the cell on nanostructured surfaces slightly decrease but more protruding with the increase of surface roughness. Filopodia are actin‐rich plasma‐membrane protrusions and function as antennae for cells to probe their environment,[ 48 ] which play an important role in cell‐interface interaction during cell adhesion and spreading. The length of the filopodia of the adhered cells on nanostructured surfaces was then quantified. Only a few and short filopodia were observed from cells on the surface with lower roughness (Figure 4b,c). But the filopodia formation on nanostructured surfaces was gradually enhanced with the increase of surface roughness and maximized at mPG4 surfaces. Thereafter, the increased roughness largely restricted the expression of filopodia on nanostructured surfaces. It means the topographical features would strengthen cell‐biointerface interaction if their size were in a suitable range, that is, <350 nm for MCF7 cells. The further increased surface roughness possibly limits cell extension in the horizontal direction and the cells adapt themselves to minimize their contact with those surficial features.[ 3 ] Accordingly, cell membrane mobility and filopodia formation are hindered during cell adhesion.[ 32 ] Thus, this roughness selective manner of cell‐interface interactions explains the roughness‐dependent capture efficiency of cancer cells on nanostructured surfaces. It also demonstrates that the enhancement effect of surficial topographical features on cell capture should be in a preferred range according to the size of cells.[ 19 , 26 ]
Figure 4.
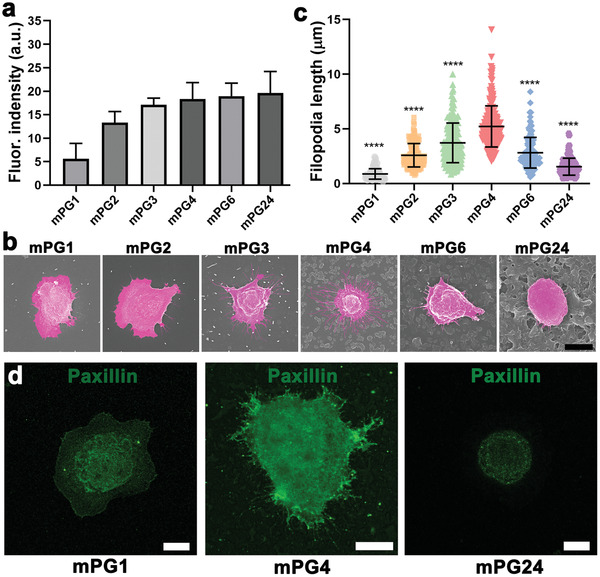
The underlying mechanism for cancer cell capture on nanostructured surfaces with a surface roughness selective manner. a) Quantification of the relative conjugated antibodies on nanostructured surfaces with different surface roughness (n = 4–5, 3 technical replicates). The antibody‐conjugated surfaces are immunofluorescence stained by Cy5‐conjugated goat polyclonal secondary antibody to mouse IgG. b) Representative SEM images of adhered cancer cells on nanostructured surfaces and c) the filopodia length of the adhered cancer cells on nanostructured surfaces (n = 10–15, 3 technical replicates). * indicates a statistically significant comparison with P < 0.05 (one‐way ANOVA). The scale bar indicates 10 µm. d) Representative fluorescence images of paxillin immunostaining cancer cells on mPG0, mPG4, and mPG24 surfaces, respectively. The scale bar indicates 10 µm.
Cell sense and respond to substrate topography through integrin‐mediated mechanotransduction. Paxillin in adhesion complexes was stained to examine the effect of topographical features on FAs formation. As shown in Figure 4d, clear punctate structures and mature FAs in cells on mPG4 surfaces were observed, while no obvious punctate structures and FAs were found on the mPG1 surface with lower roughness (Rq = 56 nm) and the mPG24 surface with higher roughness (Rq = 572 nm). The molecular arrangement of integrin‐mediated FAs formation is sensitive to the surficial nanostructured features but should be in a suitable range, and hence possibly results in distinct adhesion behaviors.
Overall, the nanostructure features on biointerfaces could greatly increase the surface area available for bioligands conjugation and the contact between nanoscale cell‐surface components and the nanofeatures on biointerfaces. Furthermore, the nanostructures that mimic the nanoscale features found in the tissue microenvironment could enhance the formation of filopodia and FA, resulting in highly efficient affinity capture.
2.5. Reversible Release of Captured Cancer Cells Via Dynamic Ligands Bonding
Benefiting from the dynamic cis‐diols/PBA binding chemistry, the competitive dissociation of conjugated bioligands facilitates the responsive detachment of captured cells from the nanostructured surfaces in the presence of higher affinity cis‐diols to PBA, that is, sorbitol[ 25 , 33 ] (Figure 5a ). After adding the glycan into the medium (0.1 M sorbitol), the spread cells on the nanostructured surfaces gradually round‐up (Figure 5b). Both the cell morphology and adhesion status dramatically altered after 10 min incubation at 37 °C. Almost all the captured cells were easily removed by gently rinsing with PBS buffer after 20 min incubation (Figure 5b). This sugar‐responsive cell detachment results from the competitive unbinding of bioligands from basal surfaces. Thus, cells cannot maintain adhesion onto antifouling surfaces anymore in the absence of adhesive ligands and subsequently detach. The sorbitol dose‐dependent and incubation time‐dependent releasing efficiency were also investigated and shown in Figure 5c. Although the detachment of cells under 0.1 M sorbitol solution was comparably quicker than that under 0.05 M sorbitol solution at the beginning, there was no significant difference in their release efficiency after 30 min incubation that 98 ± 1.5% of release efficiency has already been achieved. In addition to the high detachment efficiency, the integrity and viability of the released cells are required and very important for downstream biological analysis. Thus, the released cells were re‐cultured in a fresh medium and the proliferation ability of released cells was quantified via CCK8 assay (Figure 5d,e). Compared with the control group, the released cells still kept high viability, and no significant difference in the proliferation was observed between the control cells and the released cells even using a higher concentration of sorbitol.
Figure 5.
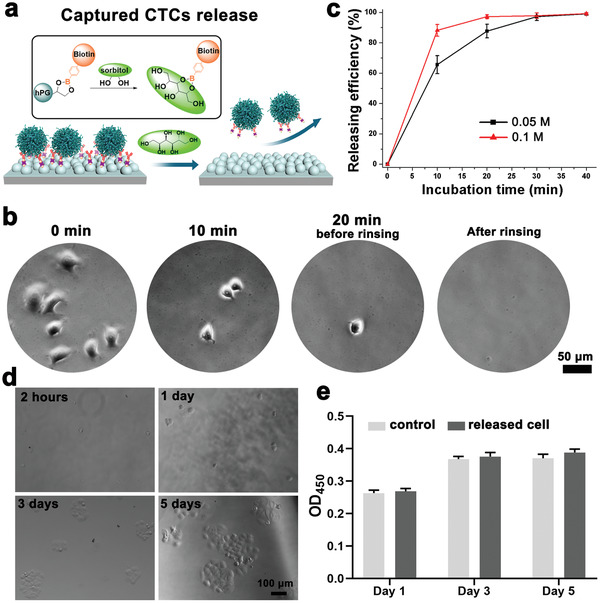
Sorbitol‐induced release of captured cancer cells on nanostructured surfaces. a) Schematic illustration of the mechanism of cancer cell release. The sorbitol has a stronger affinity than the diols from hPG to PBA and results in the release of captured cells from the surface by competitive binding. b) Representative microscopy images of cancer cell responsive release on anti‐EpCAM‐mPG4 surfaces. c) Incubation time and sorbitol concentration‐dependent release efficiency of cancer cells on anti‐EpCAM‐mPG4 surfaces. (n = 15–20, 3 technical replicates). d) The cell viability of released cancer cells from anti‐EpCAM‐mPG4 surfaces by re‐culturing and e) the proliferation analysis of the released cells by CCK‐8 assay. The OD value at 450 nm represents the activity of cancer cells. The scale bar indicates 100 µm.
3. Conclusion
Overall, an advanced biointerface with well‐defined topographical features was developed via a new generation of mussel‐inspired coating polymers. The adhesion of cancer cells on these nanostructured biointerfaces was greatly enhanced compared with smooth surfaces and exhibited an interesting biphasic roughness‐dependent manner. Under the presence of an antifouling coating layer and conjugated biospecific ligands, the generated biofunctional nanostructured surfaces exhibited a high performance for specific cancer cell capture which results from the improvement of bioligand conjugation, increase the cell‐biointerface contact area, and enhancement of filopodia formation and focal adhesion by the topographical features in an optimized size. It should be highlighted that the hPG monolayer on the biointerfaces herein not only provides an excellent bioinert background but also enables directly bioligands conjugation through the PBA‐diols dynamic binding and subsequently detach the bioligands via competitive glycan unbinding. Thus, the resulting surfaces are able to biospecifically capture CTCs with high efficiency (>95%) and purity (>96%) and “on‐demand” release the captured cells quickly without damage. These results provide a new strategy to regulate cancer cell adhesion/capture via the biophysical cues and prospects for designing integrated biointerfaces for advanced cell‐based biomedical studies in the future. Furthermore, the proposed method for in situ conjugating and dissociating bioligands onto the bioinert surface via the responsive diols/PBA binding is not limited to the anti‐EpCAM for cancer cell isolation but also works for versatile homo/hetero‐bioligands immobilization once they have been modified with PBA, which simplifies the multivalent and multifunctional biointerfaces preparation.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors acknowledge the financial support from the Deutsche Forschungsgemeinschaft (DFG). The authors thank the Core Facility BioSupraMol (Freie Universität Berlin, Germany) for the assistance in confocal microscopy measurements support. C.N. and Y.H. acknowledge the finical support from China Scholarship Council (CSC). The authors thank Dr. Pamela Winchester for her careful language polishing of the manuscript.
Open access funding enabled and organized by Projekt DEAL.
Yu L., Tang P., Nie C., Hou Y., Haag R., Well‐Defined Nanostructured Biointerfaces: Strengthened Cellular Interaction for Circulating Tumor Cells Isolation. Adv. Healthcare Mater. 2021, 10, 2002202. 10.1002/adhm.202002202
Contributor Information
Leixiao Yu, Email: leixiaoyu@zedat.fu-berlin.de.
Rainer Haag, Email: haag@chemie.fu-berlin.de.
Data Availability Statement
Research data are not shared.
References
- 1. Abbott A., Nature 2003, 424, 870. [DOI] [PubMed] [Google Scholar]
- 2. Zhu Y., Zhang Q., Shi X., Han D., Adv. Mater. 2019, 31, 1804950. [DOI] [PubMed] [Google Scholar]
- 3. Anselme K., Ploux L., Ponche A., J. Adhes. Sci. Technol. 2010, 24, 831. [Google Scholar]
- 4. Ma Z., Kotaki M., Inai R., Ramakrishna S., Tissue Eng. 2005, 11, 101. [DOI] [PubMed] [Google Scholar]
- 5. Barradas A., Terstappen L. W., Cancers 2013, 5, 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cristofanilli M., Budd G. T., Ellis M. J., Stopeck A., Matera J., Miller M. C., Reuben J. M., Doyle G. V., Allard W. J., Terstappen L. W., N. Engl. J. Med. 2004, 351, 781. [DOI] [PubMed] [Google Scholar]
- 7. Fehm T., Morrison L., Saboorian H., Hynan L., Tucker T., Uhr J., Breast Cancer Res. Treat. 2002, 75, 227. [DOI] [PubMed] [Google Scholar]
- 8. Carter L., Rothwell D. G., Mesquita B., Smowton C., Leong H. S., Fernandez‐Gutierrez F., Li Y., Burt D. J., Antonello J., Morrow C. J., Nat. Med. 2017, 23, 114. [DOI] [PubMed] [Google Scholar]
- 9. Alix‐Panabières C., Pantel K., Nat. Rev. Cancer 2014, 14, 623. [DOI] [PubMed] [Google Scholar]
- 10. Li Y. Q., Chandran B. K., Lim C. T., Chen X., Adv. Sci. 2015, 2, 1500118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Joosse S. A., Gorges T. M., Pantel K., EMBO Mol. Med. 2015, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alix‐Panabières C., Schwarzenbach H., Pantel K., Annu. Rev. Med. 2012, 63, 199. [DOI] [PubMed] [Google Scholar]
- 13. Green B. J., Saberi Safaei T., Mepham A., Labib M., Mohamadi R. M., Kelley S. O., Angew. Chem., Int. Ed. 2016, 55, 1252. [DOI] [PubMed] [Google Scholar]
- 14. Chen J., Yu L., Li Y., Cuellar‐Camacho J. L., Chai Y., Li D., Li Y., Liu H., Ou L., Li W., Haag R., Adv. Funct. Mater. 2019, 29, 1808961. [Google Scholar]
- 15. Wang L., Asghar W., Demirci U., Wan Y., Nano Today 2013, 8, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui H., Wang B., Wang W., Hao Y., Liu C., Song K., Zhang S., Wang S., ACS Appl. Mater. Interfaces 2018, 10, 19545. [DOI] [PubMed] [Google Scholar]
- 17. Meng J., Zhang P., Zhang F., Liu H., Fan J., Liu X., Yang G., Jiang L., Wang S., ACS Nano 2015, 9, 9284. [DOI] [PubMed] [Google Scholar]
- 18. Dong J., Jan Y. J., Cheng J., Zhang R. Y., Meng M., Smalley M., Chen P.‐J., Tang X., Tseng P., Bao L., Sci. Adv. 2019, 5, eaav9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu X., Chen L., Liu H., Yang G., Zhang P., Han D., Wang S., Jiang L., NPG Asia Mater 2013, 5, e63. [Google Scholar]
- 20. Sekine J., Luo S. C., Wang S., Zhu B., Tseng H. R., Yu H. h., Adv. Mater. 2011, 23, 4788. [DOI] [PubMed] [Google Scholar]
- 21. Kim D.‐J., Seol J.‐K., Lee G., Kim G.‐S., Lee S.‐K., Nanotechnology 2012, 23, 395102. [DOI] [PubMed] [Google Scholar]
- 22. Qi S., Yi C., Ji S., Fong C.‐C., Yang M., ACS Appl. Mater. Interfaces 2009, 1, 30. [DOI] [PubMed] [Google Scholar]
- 23. Shen Q., Xu L., Zhao L., Wu D., Fan Y., Zhou Y., OuYang W. H., Xu X., Zhang Z., Song M., Adv. Mater. 2013, 25, 2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ke Z., Lin M., Chen J.‐F., Choi J.‐s., Zhang Y., Fong A., Liang A.‐J., Chen S.‐F., Li Q., Fang W., ACS Nano 2015, 9, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen M. Y., Chen J. F., Luo C. H., Lee S., Li C. H., Yang Y. L., Tsai Y. H., Ho B. C., Bao L. R., Lee T. J., Adv. Healthcare Mater. 2018, 7, 1700701. [Google Scholar]
- 26. Dong J., Chen J. F., Smalley M., Zhao M., Ke Z., Zhu Y., Tseng H. R., Adv. Mater. 2020, 32, 1903663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen W., Weng S., Zhang F., Allen S., Li X., Bao L., Lam R. H., Macoska J. A., Merajver S. D., Fu J., ACS Nano 2013, 7, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang F., Jiang Y., Liu X., Meng J., Zhang P., Liu H., Yang G., Li G., Jiang L., Wan L.‐J., Nano Lett. 2016, 16, 766. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Z., Chen N., Li S., Battig M. R., Wang Y., J. Am. Chem. Soc. 2012, 134, 15716. [DOI] [PubMed] [Google Scholar]
- 30. Canavan H. E., Cheng X., Graham D. J., Ratner B. D., Castner D. G., J. Biomed. Mater. Res., Part A 2005, 75, 1. [DOI] [PubMed] [Google Scholar]
- 31. Hou Y., Yu L., Xie W., Camacho L. C., Zhang M., Chu Z., Wei Q., Haag R., Nano Lett. 2019, 20, 748. [DOI] [PubMed] [Google Scholar]
- 32. Hou Y., Xie W., Yu L., Camacho L. C., Nie C., Zhang M., Haag R., Wei Q., Small 2020, 16, 1905422. [DOI] [PubMed] [Google Scholar]
- 33. Springsteen G., Wang B., Tetrahedron 2002, 58, 5291. [Google Scholar]
- 34. Wei Q., Achazi K., Liebe H., Schulz A., Noeske P. L. M., Grunwald I., Haag R., Angew. Chem., Int. Ed. 2014, 53, 11650. [DOI] [PubMed] [Google Scholar]
- 35. Schlaich C., Camacho L. C., Yu L., Achazi K., Wei Q., Haag R., ACS Appl. Mater. Interfaces 2016, 8, 29117. [DOI] [PubMed] [Google Scholar]
- 36. LaVoie M. J., Ostaszewski B. L., Weihofen A., Schlossmacher M. G., Selkoe D. J., Nat. Med. 2005, 11, 1214. [DOI] [PubMed] [Google Scholar]
- 37. Danner E. W., Kan Y., Hammer M. U., Israelachvili J. N., Waite J. H., Biochemistry 2012, 51, 6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee H., Dellatore S. M., Miller W. M., Messersmith P. B., Science 2007, 318, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wei Q., Zhang F., Li J., Li B., Zhao C., Polym. Chem. 2010, 1, 1430. [Google Scholar]
- 40. Yu M., Bardia A., Wittner B. S., Stott S. L., Smas M. E., Ting D. T., Isakoff S. J., Ciciliano J. C., Wells M. N., Shah A. M., Science 2013, 339, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rusmini F., Zhong Z., Feijen J., Biomacromolecules 2007, 8, 1775. [DOI] [PubMed] [Google Scholar]
- 42. Osta W. A., Chen Y., Mikhitarian K., Mitas M., Salem M., Hannun Y. A., Cole D. J., Gillanders W. E., Cancer Res. 2004, 64, 5818. [DOI] [PubMed] [Google Scholar]
- 43. Jeon S., Moon J. M., Lee E. S., Kim Y. H., Cho Y., Angew. Chem. 2014, 126, 4685. [DOI] [PubMed] [Google Scholar]
- 44. Yang L., Sun H., Jiang W., Xu T., Song B., Peng R., Han L., Jia L., Chem. Mater. 2018, 30, 4372. [Google Scholar]
- 45. Dou X., Li P., Jiang S., Bayat H., Schönherr H., ACS Appl. Mater. Interfaces 2017, 9, 8508. [DOI] [PubMed] [Google Scholar]
- 46. Sollier E., Go D. E., Che J., Gossett D. R., O'Byrne S., Weaver W. M., Kummer N., Rettig M., Goldman J., Nickols N., Lab Chip 2014, 14, 63. [DOI] [PubMed] [Google Scholar]
- 47. van de Stolpe A., Pantel K., Sleijfer S., Terstappen L. W., Den Toonder J. M., Cancer Res. 2011, 71, 5955. [DOI] [PubMed] [Google Scholar]
- 48. Mattila P. K., Lappalainen P., Nat. Rev. Mol. Cell Biol. 2008, 9, 446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.