

Abstract
Efferocytosis, mediated by the macrophage receptor MerTK (myeloid‐epithelial‐reproductive tyrosine kinase), is a significant contributor to cardiac repair after myocardial ischemia‐reperfusion (MI/R) injury. However, the death of resident cardiac macrophages (main effector cells), inactivation of MerTK (main effector receptor), and overexpression of “do not eat me” signals (brake signals, such as CD47), collectively lead to the impediment of efferocytosis in the post‐MI/R heart. To date, therapeutic strategies targeting individual above obstacles are relatively lacking, let alone their effectiveness being limited due to constraints from the other concurrent two. Herein, inspired by the application research of chimeric antigen receptor macrophages (CAR‐Ms) in solid tumors, a genetically modified macrophage‐based synergistic drug delivery strategy that effectively challenging the three major barriers in an integrated manner is developed. This strategy involves the overexpression of exogenous macrophages with CCR2 (C‐C chemokine receptor type 2) and cleavage‐resistant MerTK, as well as surface clicking with liposomal PEP‐20 (a CD47 antagonist). In MI/R mice model, this synergistic strategy can effectively restore cardiac efferocytosis after intravenous injection, thereby alleviating the inflammatory response, ultimately preserving cardiac function. This therapy focuses on inhibiting the initiation and promoting active resolution of inflammation, providing new insights for immune‐regulatory therapy.
Keywords: cell therapies, click chemistry, efferocytosis, immune checkpoints, macrophages, myocardial ischemia‐reperfusion injury, reactive oxygen species responsive
A gene‐modified macrophage therapy with the capability of synergistic drug delivery (MACCCR2+MerTK CR‐LipoPEP‐20) is developed. It synergistically challenges all three underlying causes of efferocytosis impairment, thereby enabling precise improvements in cardiac efferocytosis post myocardial ischemia‐reperfusion injury, ultimately leading to improved cardiac function through alleviation of inflammation. This strategy simultaneously demonstrates excellent therapeutic efficacy and biocompatibility.
![]()
1. Introduction
Myocardial infarction, a leading cause of morbidity and mortality worldwide, is characterized by the interruption of coronary artery blood flow, resulting in cell death and intense sterile inflammation.[ 1 ] Timely diagnosis and reperfusion effectively limit cardiac infarct size, but reperfusion itself can cause further cardiomyocyte death and exacerbate the inflammatory response.[ 2 , 3 , 4 , 5 ] Efficient clearance of damaged cells and suppression of inflammatory responses are therefore considered crucial for maximizing the benefits of reperfusion therapy.[ 6 , 7 ] Recent reports from independent groups have indicated that efferocytosis (clearance of apoptotic cells) mediated by the macrophage receptor MerTK (myeloid‐epithelial‐reproductive tyrosine kinase) served as a significant contributor to cardiac repair after myocardial ischemia‐reperfusion (MI/R) injury.[ 6 , 8 ] Efferocytosis efficiently eliminates apoptotic cells, preventing their secondary necrosis and further amplification of inflammation.[ 9 ] Moreover, efferocytosis can actively initiate the resolution of inflammation by activating downstream negative regulatory pathways or by promoting the production of specialized pro‐resolving mediators (SPMs).[ 9 , 10 ]
Apoptosis is one of the major mechanisms of cell death in cardiomyocytes and interstitial cells during MI/R injury.[ 5 , 11 ] Furthermore, neutrophils, as the most abundant inflammatory cells to reach the injury area during the early stage, also rely on apoptosis to be cleared by phagocytes after completing their mission.[ 12 ] However, the natural cardiac efferocytosis function post‐MI/R is impaired due to the following three reasons. First, resident macrophages are the primary executors of efferocytosis, but their absolute numbers are reduced by more than half due to MI/R‐induced cell death.[ 13 ] In the early stage of inflammation, recruited monocytes and neutrophils exhibit low expression of MerTK, while the MerTKhi reparative macrophages peak in the late stage, which collectively impairs the effective sharing of efferocytosis.[ 14 , 15 ] Second, the main effector receptor of efferocytosis, MerTK, is cleaved and inactivated by ADAM17 (a disintegrin and metalloproteinase 17) in the injured area after MI/R due to factors such as reactive oxygen species (ROS) stimulation.[ 15 , 16 , 17 ] Last, MI/R‐induced upregulation of “do not eat me” signal, CD47, on apoptotic cells inhibits efferocytosis, and studies have reported that acute CD47 blockade during MI/R enhances phagocytosis‐associated cardiac repair.[ 18 , 19 ]
For executive cells, effective therapies for cyto‐protection of resident macrophages against MI/R injury are still lacking to date, and the development of drugs specifically stimulating the proliferation of surviving resident macrophages has also stagnated.[ 20 ] For executive receptor, ADAM17 is widely distributed and catalyzes multiple substrates, making it difficult to achieve precise inhibition of MerTK cleavage by ADAM17 inhibition.[ 21 ] In addition, CD47 is widely expressed in normal cells, especially erythrocyte, which poses significant safety concerns for strategies targeting CD47 inhibition to restore efferocytosis. It is noteworthy that strategies aimed at restoring efferocytosis by targeting one single aspect often yield limited effectiveness due to constraints from the other concurrent two. Therefore, the development of efferocytosis reconstruction strategies that organically integrate the three obstacles is challenging but urgently needed.
Herein, inspired by the application research of chimeric antigen receptor macrophages (CAR‐Ms) in solid tumors, we recognized the potential of exogenous macrophages as a direct supplement of efferocytotic executors in the injured heart.[ 22 ] From idea to implementation, exogenous macrophages also face three challenges: how to reach the site of cardiac injury; how to ensure that their own MerTK in the injured cardiac region is not cleaved and inactivated like resident macrophages; and how to overcome the brake effect of CD47 on efferocytosis. Based on this, we genetically modified exogenous macrophages and designed a synergistic drug delivery strategy based on click chemistry (named MACCCR2+MerTK CR‐LipoPEP‐20) (Figure 1 ). By overexpressing CCR2 (C‐C chemokine receptor type 2) and cleavage‐resistant MerTK (MerTKCR), bone marrow‐derived macrophages (BMDMs) are endowed with the ability to chemotaxis to the injured area in pathophysiologic inflammation and ADAM17‐insensitive efferocytosis receptors. By coupling liposomes loaded with PEP‐20 (a CD47 antagonist) on the surface, exogenous macrophages acquire the weapon to mask CD47, thereby antagonizing its inhibition on efferocytosis.[ 23 ] The anchoring of liposomes on the surface of macrophages is achieved through the inverse electron demand Diels‐Alder (iEDDA) reaction, and the liposomes have been subjected to ROS‐sensitive modification to enable their explosive release of PEP‐20 upon reaching the high ROS environment in the injured heart.[ 24 , 25 , 26 ] After intravenous injection, this carrier system can achieve precise guidance toward the injured heart, reaching a considerable number locally while preserving the efferocytosis function intact. This synergistic strategy can effectively restore cardiac efferocytosis following MI/R, thereby inhibiting the initiation and promoting the active resolution of inflammation to preserve cardiac function.
Figure 1.
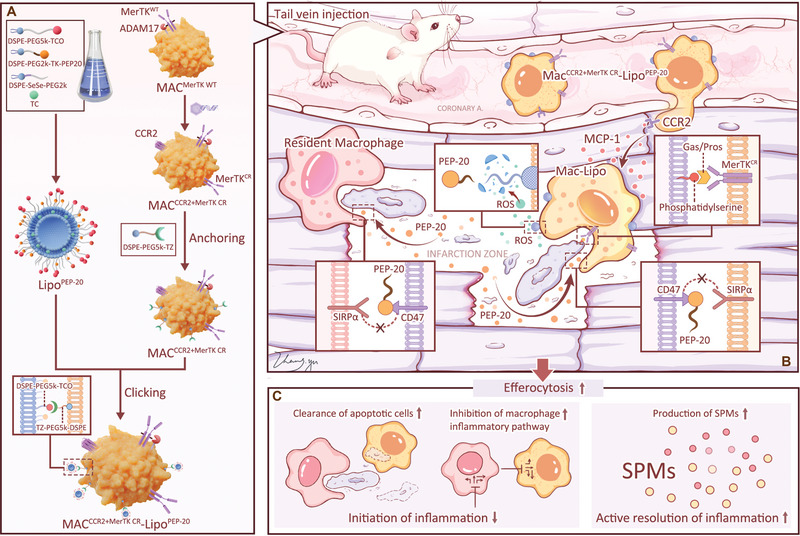
Schematic illustration of MACCCR2+MerTK CR‐LipoPEP‐20 as a potential candidate to reconstruct efferocytosis post‐MI/R injury. A) Fabrication of MACCCR2+MerTK CR‐LipoPEP‐20. B) After intravenous injection, MACCCR2+MerTK CR‐LipoPEP‐20 selectively migrates to the site of cardiac injury through the overexpressed CCR2. The overexpressed MerTKCR on MACCCR2+MerTK CR‐LipoPEP‐20 remains intact and recognizes phosphatidylserine through Gas6 (growth arrest specific 6) or ProS (protein S) bridging, thereby executing efferocytosis function. The anchored PEP‐20 on MACCCR2+MerTK CR‐LipoPEP‐20 is explosively released in response to ROS stimulation, which further enhances the efferocytosis capacity of adoptive macrophages and resident macrophages by antagonizing CD47 on apoptotic cells. C) The cardiac benefits of improved efferocytosis are achieved through inhibiting initiation and promoting active resolution of inflammation.
2. Results
2.1. Genetically Engineering of BMDMs
CCR2 primarily mediates the chemotaxis of circulating monocytes to the site of injury in the inflammatory dynamics following MI/R, and its expression in BMDMs is nearly undetectable, while wild‐type MerTK (MerTKWT) is commonly expressed as a recognized marker of macrophage.[ 27 , 28 ] Previous studies have identified proline‐485 as the MerTK cleavage site and have confirmed that cells expressing MerTK lacking residues 483–488 (MerTKΔ483‐488, to wit MerTKCR) maintained MerTK‐dependent efferocytosis under cleavage‐inducing conditions.[ 10 , 15 , 17 ] We hypothesized that macrophages co‐overexpressing CCR2 and MerTKCR (MACCCR2+MerTK CR) are endowed with the ability to undergo physiological chemotaxis and maintain their efferocytosis functionality intact in cleavage‐inducing environments.
Gene transfer into primary macrophages has been a longstanding challenge. We utilized an adenoviral vector (AD5f35) to achieve synergistic genetic engineering, as macrophages express the docking protein CD46 for this vector.[ 29 ] Primary macrophages were co‐transfected with ADVCCR2 + ADVMerTK, while ADVCCR2 NC + ADVMerTK NC, ADVCCR + ADVMerTK NC and ADVCCR2 NC + ADVMerTK were set as controls. As expected, western blotting (Figure 2A–C), and flow cytometry analyses (Figure 2D–F) indicated that CCR2 and MerTK expression were significantly upregulated following transduction, suggesting that the adenoviral vector can simultaneously upregulate these two genes in primary macrophages. To validate the functional optimization, we conducted transwell cell migration experiments and an efferocytosis assay. In the transwell cell migration system, the CCR2 overexpressed BMDMs exhibited increased responsiveness to the chemokine MCP‐1 (monocyte chemoattractant protein 1), while the co‐overexpression of MerTK did not hinder this improvement (Figure 2G,H). Similarly, in vitro efferocytosis experiments demonstrated that the overexpression of MerTK significantly enhanced the phagocytosis of apoptotic cardiomyocytes by macrophages, while the co‐overexpression of CCR2 did not affect this optimization (Figure 2I,J). The results were also confirmed by flow cytometry (Figure S1A, Supporting Information).
Figure 2.
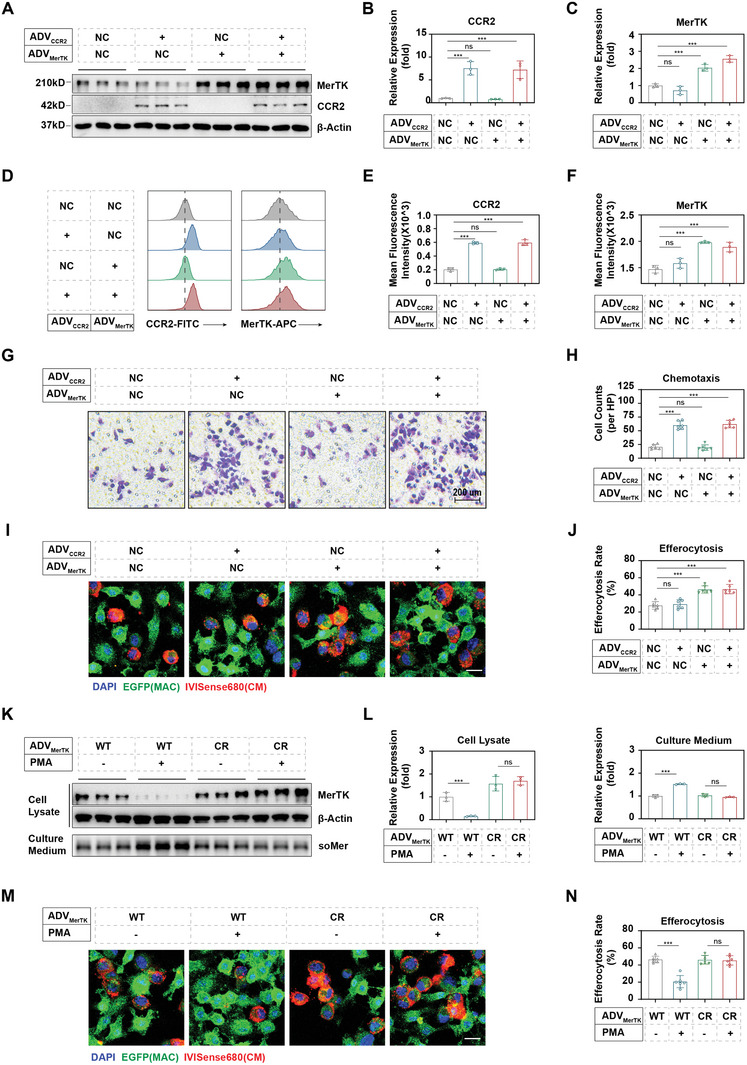
Genetical engineering of macrophages. A) Western blotting of CCR2 and MerTK in macrophages following different combinations of genetically overexpression. “+” refers to macrophages transfected with ADV‐EF1‐CCR2‐6xHis or ADV‐EF1‐MerTK(del483‐486)−3xFLAG, respectively. “NC” refers to macrophages transfected with vector control (ADV‐EF1‐MCS‐6xHis or ADV‐EF1‐MCS‐3xFLAG, respectively). Protein levels of B) CCR2 and C) MerTK were quantified by ImageJ according to the images in (A) (n = 3). D) Flow cytometry assay of CCR2 and MerTK in macrophages following different combinations of genetically overexpression, and quantified in E) CCR2 and F) MerTK (n = 3). G) Transwell assay of macrophages toward 20 ng mL−1 MCP‐1 following different combinations of genetically overexpression (n = 3). H) Quantitative analysis of chemotaxis was performed on the images in (G) by ImageJ (n = 3). I) Confocal imaging of EGFP+ macrophages, engulfing IVISense680‐labeled apoptotic cardiomyocytes, following different combinations of genetically overexpression. J) The percentage of cardiomyocyte‐associated macrophages was calculated as the efferocytosis rate according to images in (I) (n = 3). K) Under PMA stimulation, the cleavage levels of MerTK on the surface of engineered macrophages, with or without the MerTKCR (MerTK cleavage resistance) mutation, were detected by western blotting and quantified in (L) by ImageJ (n = 3). M) Confocal imaging of the engulfing of IVISense680‐labeled apoptotic cardiomyocytes by engineered EGFP+ macrophages, with or without MerTKCR mutation, and efferocytosis rate were calculated in (N) (n = 3). Results are presented as mean ± SD, ns p > 0.05, *p < 0.05, *p < 0.01, *p < 0.001.
We subsequently validated the further improvement of macrophage performance through the overexpression of MerTKCR, instead of MerTKWT in the above experiments. Compared to the MerTKWT group, the MerTKCR macrophages exhibited comparable levels of MerTK overexpression (Figure 2K,L). However, upon the addition of 50 nm PMA (phorbol myristate acetate), a cleavage‐inducing factor, the membrane protein of MerTK in the MerTKWT group significantly decreased, while the MerTKCR group was preserved (Figure 2K,L). Correspondingly, due to PMA stimulation, the soluble MerTK (soMer) in the culture medium of MerTKWT macrophages significantly increased, while the MerTKCR group showed no significant change (Figure 2K,L). Reasonably, the cleavage of MerTKWT induced by PMA significantly impairs macrophage efferocytosis, while MerTKCR macrophages remain unaffected (Figure 2M,N). The results were also confirmed by flow cytometry (Figure S1B, Supporting Information).
All the above suggests that CCR2 and MerTK can be co‐overexpressed in primary macrophages and exert their normal functions without mutual interference. In addition, deletion of the coding sequence corresponding to the cleavage site in the MerTK overexpression vector can endow macrophages with a cleavage‐resistant MerTK receptor and then enable macrophages to retain intact efferocytosis function in the cleavage‐induced environment.
2.2. Fabrication of ROS‐Responsive Liposomes and Their Coupling on Engineered Macrophage Surface
The drug delivery strategy synergizing with engineered macrophages involves three steps (Figure 3A): i) fabrication of trans‐cyslooctene (TCO) modified and PEP‐20‐loading ROS‐responsive liposomes (TCO‐LipoPEP‐20); ii) inserting the anchor of tetrazine (TZ)‐bearing, two‐tailed lipids into the lipid bilayers of engineered macrophage membrane via hydrophobic interactions (go to MACCCR2+MerTK CR‐TZ); and iii) connection of liposomes and macrophages through biorthogonal click reaction between TZ and TCO (go to MACCCR2+MerTK CR‐TZ/TCO‐LipoPEP‐20). To this end, ROS‐responsive bipolar lipids [1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐SeSe‐(poly‐ethylene‐glycol)2000, (DSPE‐SeSe‐PEG2k)]; ROS‐sensitive, hydrophobic tail‐bearing PEP‐20 [1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(poly‐ethylene‐glycol)2000‐TK‐AWSATWSNYWRH, (DSPE‐PEG2k‐TK‐PEP‐20)]; TZ‐bearing lipids [1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(poly‐ethylene‐glycol) 5000‐tetrazine, (DSPE‐PEG5k‐TZ)] and TCO‐bearing lipids [1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(poly‐ethylene‐glycol)5000‐trans‐cyslooctene, (DSPE‐PEG5k‐TCO)] were employed, and their structures are shown in Figure 3A. The rapid, catalyst‐free click reaction at room temperature between DSPE‐PEG5k‐TZ and DSPE‐PEG5k‐TCO was verified by ultraviolet spectroscopy (Figure S2A, Supporting Information).
Figure 3.
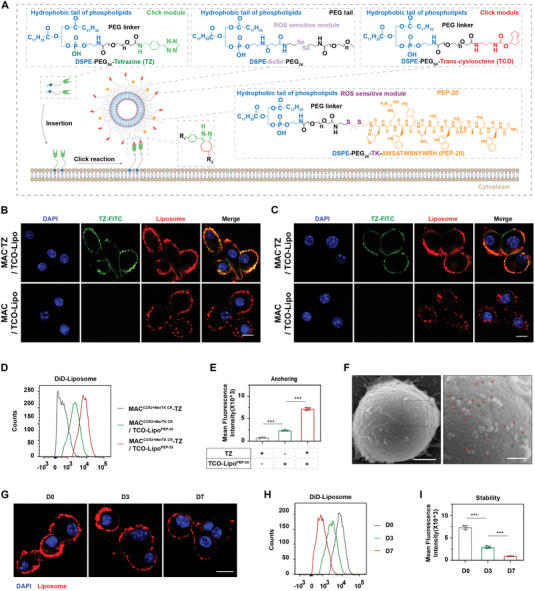
Fabrication of ROS‐responsive liposomes and their anchoring on engineered macrophage surfaces. A) Schematic diagram of liposome structure and anchoring‐click reaction. B) Confocal imaging of MACCCR2+MerTK CR/TCO‐LipoPEP‐20 and MACCCR2+MerTK CR‐TZ/TCO‐LipoPEP‐20 immediately after liposome conjugation. DAPI as a nuclear indicator. Scale bar, 10 µm. C) Confocal imaging of MACCCR2+MerTK CR/TCO‐LipoPEP‐20 and MACCCR2+MerTK CR‐TZ/TCO‐LipoPEP‐20 at 3 h after liposome conjugation. D) The coupling amounts of liposomes on engineered macrophages, with or without TZ anchoring, were evaluated by flow cytometry and quantified in (E) (n = 3). Macrophages without liposome coupling were set as controls. F) SEM images of MACCCR2+MerTK CR/TCO‐LipoPEP‐20. Red arrows indicate the coupled liposomes. Scale bar, 2 µm (left) and 500 nm (right). G) Confocal imaging of MACCCR2+MerTK CR‐TZ/TCO‐LipoPEP‐20 at D0, D3 and D7 after liposome conjugation. Scale bar: 10 µm. H) The retention of liposomes on engineered macrophages was analyzed by flow cytometry at D0, D3, and D7 post conjugation, and quantified in (I) (n = 3). Results are presented as mean ± SD, ns p > 0.05, *p < 0.05, *p < 0.01, *p < 0.001.
TCO‐LipoPEP‐20 was prepared through the thin‐film hydration method, according to our previous report.[ 30 ] The obtained TCO‐LipoPEP‐20 displayed a uniform spherical structure under transmission electron microscopy (TEM) (Figure S2B, Supporting Information). The size and zeta potential of TCO‐LipoPEP‐20 were 68 ± 2.37 nm and −38 ± 0.11 mV, respectively (Figure S2C,D, Supporting Information), measured by dynamic laser scattering (DLS). Moreover, TCO‐LipoPEP‐20 exhibits excellent dispersibility (Figure S2D, Supporting Information) and remains stable for up to 48 h under physiological conditions without detectable aggregation (Figure S2E, Supporting Information).
Cell cytotoxicity testing is of paramount importance, and no impact on macrophage viability was observed for various concentrations of DSPE‐PEG5k‐TZ and TCO‐LipoROS‐PEP‐20 when co‐cultured for 24 h (Figure S3A,B, Supporting Information). To obtain effective and biocompatible parameters for the live‐cell click reaction, we conducted a gradient analysis of substrate concentrations and reaction times for anchoring and clicking. Under various combinations of anchor and click parameters, engineered macrophages were fed with 1,19‐dioctadecyl‐3,3,39,39‐tetra‐methylindo‐dicarbo‐cyanine perchlorate (DiD)‐labeled liposomes. After rinsing away the unbound TCO‐LipoPEP‐20, the semi‐quantification of conjugated liposomes was based on the fluorescence intensity of DiD on the macrophages. We observed a positive correlation between the conjugation of liposomes and the anchoring concentration, anchoring time, clicking concentration, or clicking time, before reaching their respective plateau of 80 µg mL−1, 10 min, 4 mg mL−1 and 20 min (Figure S3C–F, Supporting Information). Henceforth, unless specifically stated, both anchor and click conditions were set according to the plateau parameters. With this strategy, the number of TCO‐LipoPEP‐20 coupled on the surface of MACCCR2+MerTK CR was about 375 liposomes per macrophage, goes to nearly 200 µg of PEP‐20 per million macrophages.
To characterize the resulting MACCCR2+MerTK CR‐TZ/TCO‐LipoPEP‐20, DSPE‐PEG5k‐TZ and TCO‐LipoPEP‐20 were pre‐labeled with fluorescein isothiocyanate (FITC) and DiD, respectively. As shown in Figure 3B, the anchoring of TZ resulted in a significantly higher amount of liposomes coupled on the surface of macrophages compared to natural adsorption. In addition, there was evident co‐localization between TZ and liposomes, further confirming the successful click reaction (Figure 3B). It is noteworthy that after 3 h of contact, naturally adsorbed liposomes were internalized by macrophages, while the anchor‐click‐modified liposomes remained stably distributed on their outer surface (Figure 3C). The enhancement effect of TZ anchoring on the coupling amounts of liposomes to the surface of macrophages was further confirmed by flow cytometry (Figure 3D,E). To visualize the morphology of MACCCR2+MerTK CR‐TZ/TCO‐LipoPEP‐20, we conducted scanning electron microscopy (SEM) imaging. As shown in Figure 3F, a considerable number of liposomes were observed on the surface of macrophages (red arrows). The adherence of DiD‐labeled liposomes on the surface of macrophages was found to persist for up to 7 days, with retention rates of 38.68% and 11.50% on the third and seventh days, respectively, compared to the initial amount (Figure 3G–I). The gradual decrease in the retention of liposomes may be attributed to physiological activities of macrophages, which are accompanied by the generation of trace amounts of reactive oxygen species. Surprisingly, the anchoring quantity of liposomes on macrophages was comparable before and after 1 week of −80 °C cryopreservation (Figure S4, Supporting Information).
Together, the above data demonstrated the successful fabrication of PEP‐20‐loaded liposomes and verified the efficiency and stability of the anchoring‐click strategy for TCO‐LipoPEP‐20 coupling on the engineered macrophage surface.
2.3. The Functional Synergy of Engineered Macrophage and Coupled Liposomes
After intravenous administration, there is a safety concern due to the possibility that PEP‐20, inserted on the surface of macrophage‐coupled liposomes, may directly recognize CD47 on circulating healthy cells. Inspired by a hierarchical targeting strategy, we employed PEG (polyethylene glycol) chains with different lengths to conjugate click module TCO (with long arm PEG5k) and PEP‐20 (with short arm PEG2k) on the surface of liposomes, aiming to mask PEP‐20 through the steric hindrance effect.[ 31 ] As expected, the dual short‐arm strategy (both TCO and PEP‐20 linked with PEG2k) demonstrated significant binding affinity of MACCCR2+MerTK CR‐TZ/TCO‐LipoPEP‐20 (hereafter referred to as MACCCR2+MerTK CR‐LipoPEP‐20) to CD47, while the long‐short‐arm design effectively reduced the binding (Figure 4A). Given that the expression levels of SIRPα (signal regulatory protein α), the specific CD47 receptor, on engineered macrophages from different groups are comparable (Figure 4B), we believe that the long‐short‐arm strategy can effectively prevent the binding of MACCCR2+MerTK CR‐LipoPEP‐20 to CD47 on healthy cells before reaching the target site. It is worth noting that the long‐short‐arm design may also facilitate the click reaction between liposomes and anchoring modules on macrophages by reducing the steric hindrance of PEP‐20 toward TCO.
Figure 4.
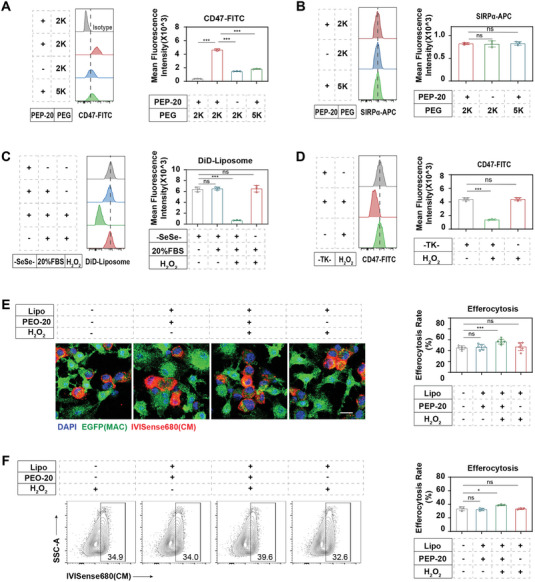
The functional synergy of engineered macrophage and coupled liposomes. A) The CD47‐FITC binding capacity of MACCCR2+MerTK CR‐LipoPEP‐20 and various control groups under the long‐short arm designs was validated by flow cytometry (n = 3). B) The SIRPα expression level of MACCCR2+MerTK CR‐LipoPEP‐20 and various control groups under the long‐short arm designs was detected by flow cytometry (n = 3). C) Flow cytometry analysis of the ROS‐responsive release of liposomes anchored on engineered macrophages (n = 3). D) Flow cytometry analysis of the ROS‐responsive release of PEP‐20 loaded on MACCCR2+MerTK CR‐LipoPEP‐20 (n = 3). E) Confocal imaging and F) flow cytometry analysis of the optimization of synergically loaded PEP‐20 on engineered macrophage's efferocytosis (n = 3). Results are presented as mean ± SD, ns p > 0.05, *p < 0.05, *p < 0.01, *p < 0.001.
The responsive release of PEP‐20 in the injured heart is a prerequisite for achieving synergistic effects with engineered macrophages in efferocytosis. Based on the knowledge of substantial ROS production in the local myocardium during MI/R,[ 32 ] we designed two ROS‐sensitive units for the liposomes: the diselenide bonds (‐SeSe‐) of the skeleton lipid (DSPE‐SeSe‐PEG2k) and the thioketal linkage (‐TK‐) between PEP‐20 and its hydrophobic tail (DSPE‐PEG2k‐TK‐PEP‐20). Through flow cytometry, we verified that the intact liposomal formula can stably attach to macrophages in physiological fluids (PBS [phosphate‐buffered saline] or 20% FBS [fetal bovine serum]) (Figure 4C). The addition of H2O2 at a pathological concentration (100 µm) resulted in a significant reduction in the quantity of liposomes on macrophages (Figure 4C). However, the control group without ‐SeSe‐ in skeletal lipids (DSPE‐SeSe‐PEG2k) showed no sensitivity to H2O2 (Figure 4C). To further validate the ROS‐responsive cleavage of PEP‐20 from its tail, we utilized DSPE‐PEG2k to ensure the stability of liposomes in H2O2, while the dual short‐arm strategy was employed so that FITC‐CD47 could detect PEP‐20. The results demonstrated that H2O2 stimulated the explosive release of PEP‐20, while the absence of ‐TK‐ deprived this responsiveness (Figure 4D). Together, the obtained evidence confirms that the dual ROS‐sensitive units can synergically achieve the responsive release of PEP‐20 from MACCCR2+MerTK CR‐LipoPEP‐20.
We further directly tested the synergistic effect of coupled liposomes on engineered macrophage efferocytosis in a high‐ROS environment. The results from immunofluorescence and flow cytometry both indicated that the responsive release of PEP‐20 from liposomes can further enhance the efferocytotic capacity of engineered macrophages (Figure 4E,F). In addition, we did not observe any influence on macrophage viability due to liposome anchoring for up to 5 days (Figure S5A, Supporting Information), and the coupling of liposomes did not affect the migration ability and polarization state of engineered macrophages (Figure S5B–D, Supporting Information).
2.4. Biodistribution and Targeting Ability of MACCCR2+MerTK CR‐LipoPEP‐20 In Vivo
To evaluate the in vivo performance of our carrier macrophages, the blood circulation retention of EGFP+ C57 BL/6J mice derived MACWT, MACCCR2, MACMerTK CR, MACCCR2+MerTK CR, and MACCCR2+MerTK CR‐LipoPEP‐20 was assessed by flow cytometry after intravenous injection into MI/R mice. The retention rates of macrophages in the circulation were comparable among the groups at three time points after injection (5 min, 1 day, and 3 days) (Figure 5A–C; Figure S6A,B, Supporting Information). Within 5 min after injection, all groups reached around 2.5% of all non‐erythrocyte, equivalent to an absolute number of approximately 7 × 104 mL−1 blood (Figure 5A–C). The retention of macrophages decreased to 35% of the initial amount 1 day after injection, and only 7% of the initial amount remained 3 days later (Figure S6A,B, Supporting Information).
Figure 5.
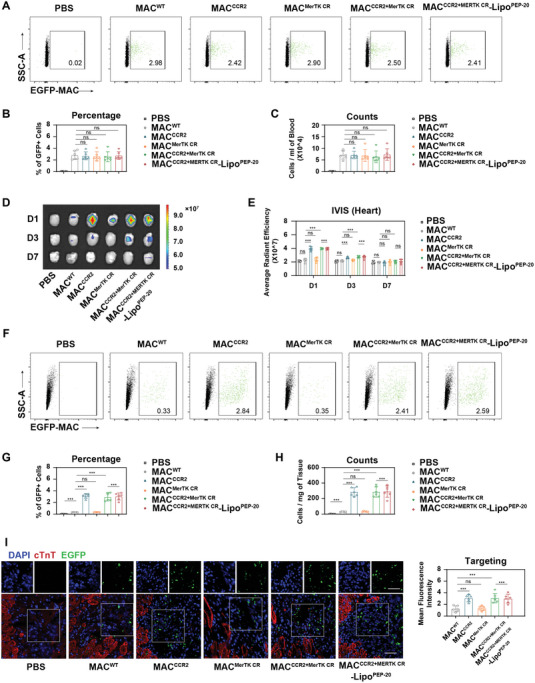
Biodistribution and targeting ability of MACCCR2+MerTK CR‐LipoPEP‐20 in vivo. A) The retention of MACCCR2+MerTK CR‐LipoPEP‐20 and various control MACs in the mice bloodstream at 5 min after intravenous injection was detected by flow cytometry and was quantified in (B,C) (n = 6). D,E) IVIS imaging of the accumulation of IVISense 680 labeled MACCCR2+MerTK CR‐LipoPEP‐20 or various control MACs in MI/R‐induced mice hearts at D1, D3, or D7 post‐i.v. injection (n = 6). F) The accumulation of EGFP+ MACCCR2+MerTK CR‐LipoPEP‐20 or various control MACs in MI/R‐induced mice hearts at D1 post‐i.v. injection was detected by flow cytometry, and were further quantified in (G,H) (n = 6). I) Confocal imaging of the accumulation of EGFP+ MACCCR2+MerTK CR‐LipoPEP‐20 or various control MACs in MI/R‐induced mice hearts after 1 day i.v. injection (n = 6). DAPI (blue) was used as a nuclear indicator. Cardiac troponin T (cTnT) stained the viable myocardium (eed). EGFP (green) is the self‐contained fluorescence of adoptive macrophages. Scale bar, 50 µm. Results are presented as mean ± SD, ns p > 0.05, *p < 0.05, *p < 0.01, *p < 0.001.
We next sought to evaluate the biodistribution of macrophages from each group in major organs by using in vivo spectrum imaging system (IVIS). The results indicated that CCR2 overexpression significantly enhanced the accumulation of macrophages in the injured heart, while the coupling of liposomes does not affect the chemotactic ability of macrophages in vivo (Figure 5D,E). There were no significant differences in the distribution of each group in other healthy organs (liver, spleen, lung, kidney, and brain) (Figure S6C–H, Supporting Information).
We quantitatively analyzed the accumulation of each group of macrophages in the injured heart using flow cytometry (Figure 5F). We found that overexpression of CCR2 led to recruitment of macrophages to the damaged heart at a level of ≈3% of all interstitial cells (Figure 5G), reaching an absolute number of 300 cells mg−1 tissue (Figure 5H), while macrophages that did not overexpress CCR2 were almost undetectable in the injured heart. The anchoring of liposomes does not affect the chemotactic ability of engineered macrophages (Figure 5F–H). The above results were further validated by confocal imaging (Figure 5I). In summary, these pieces of evidence suggest that precise guidance of macrophages toward the injured heart can be achieved by overexpressing CCR2. Notably, adoptive MACCCR2+MerTK CR‐LipoPEP‐20 was nearly 50% retained in the heart on day 3 after injection, 10% on day 7, and virtually undetectable on day 14 (Figure S7, Supporting Information).
2.5. MACCCR2+MerTK CR‐LipoPEP‐20 Improves Cardiac Efferocytosis Post‐MI/R In Vivo
After being recruited to the injured heart, the cleavage resistance of overexpressing MerTKCR in MACCCR2+MerTK CR‐LipoPEP‐20 was evaluated by flow cytometry. Indeed, overexpressed MerTKWT was significantly cleaved locally, while MerTKCR was preserved (Figure 6A,B). It is noteworthy that the cleavage resistance of MerTKCR is independent of liposome anchoring (Figure 6A,B). To evaluate efferocytosis capacity of adoptive or cardiac resident macrophages, α‐MHCCre: Rosa26‐tdTomato mice were were used to model MI/R, whose cardiomyocytes were specifically labeled with red fluorescence (tdTomato). Macrophages that engulf tdTomato+ cardiomyocytes will also be tdTomato positive. Consequently, compared to non‐overexpressed MerTK and overexpressed MerTKWT, overexpressed MerTKCR significantly preserved the efferocytosis capacity of adoptive macrophages (Figure 6C,D). Furthermore, the synergistic co‐delivery of PEP‐20 further enhanced the efferocytosis rate of adoptive macrophages, which may be attributed to its mask on CD47 expressed on apoptotic cells (Figure 6C,D). For resident macrophages, the arrival of their adoptive counterparts from different groups without PEP‐20 packages did not affect their efferocytosis rate (Figure 6E,G). As expected, the synergistic co‐delivery of PEP‐20 also promoted efferocytosis in cardiac resident macrophages (Figure 6E,G). However, the backpacking of MACMerTK CR‐LipoPEP‐20 was unable to affect resident macrophage due to the lack of CCR2 guidance (Figure 6E,G).
Figure 6.
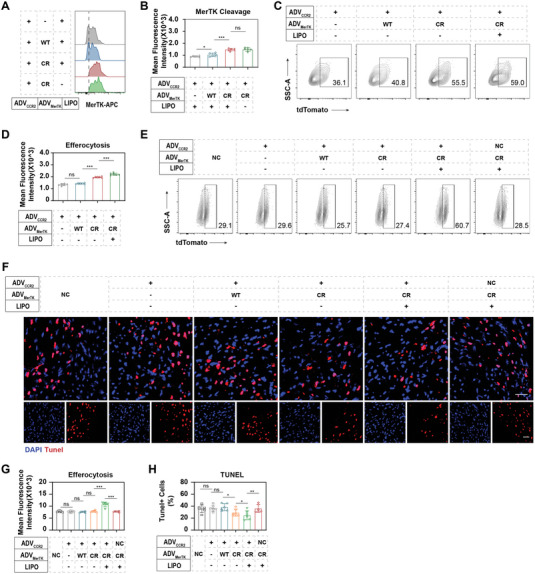
MACCCR2+MerTK CR‐LipoPEP‐20 improves cardiac efferocytosis post‐MI/R in vivo. A,B) Flow cytometry analysis of MerTK cleavage on adoptive MACCCR2+MerTK CR‐LipoPEP‐20 or various control MACs in the local MI/R injured heart (n = 6). C) The efferocytosis of MI/R‐induced cardiomyocytes by adoptive MACCCR2+MerTK CR‐LipoPEP‐20 or various control MACs was detected by flow cytometry, and quantified in (D) (n = 6). “tdTomato” staining represents the phagocytosis of cardiomyocytes by macrophages. E) The impact of PBS or each group of MACs injection on the efferocytosis function of cardiac resident macrophages was also analyzed by flow cytometry, and the results were quantified in (G) (n = 6). F) TUNEL staining of MI/R‐induced heart after PBS, MACCCR2+MerTK CR‐LipoPEP‐20 or various control MACs treatment. DAPI was used as a nucleus indicator. Scale bar: 50 µm. H) The percentage of apoptotic cells (TUNEL+) was calculated by Image J based on the images in (F) (n = 6). Results are presented as mean ± SD, ns p > 0.05, *p < 0.05, *p < 0.01, *p < 0.001.
We therefore quantified the clearance of TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) positive apoptotic cells in the border zone, the main region where apoptosis occurs, of MI/R‐induced heart by confocal imaging (Figure 6F). The results indicated that the MACCCR2+MerTK CR‐LipoPEP‐20 group had the fewest remaining apoptotic cells, followed by the MACCCR2+MerTK CR group, while the other groups did not show statistical significance compared to the PBS group (Figure 6H). Together, the above findings confirmed that MACCCR2+MerTK CR‐LipoPEP‐20 can effectively improve cardiac efferocytosis function in MI/R mice.
2.6. Cardiac Protection of MACCCR2+MerTK CR‐LipoPEP‐20
To verify the cardiac protection of MACCCR2+MerTK CR‐LipoPEP‐20, cardiac function of MI/R‐induced mice was evaluated by echocardiography at 1 and 4 weeks after PBS, MACCCR2, MACCCR2+MerTK WT, MACCCR2+MerTK CR, MACCCR2+MerTK CR‐LipoPEP‐20, and MACMerTK CR‐LipoPEP‐20 administered, respectively. Compared with other groups, the left ventricular ejection fraction (LVEF) of the MACCCR2+MerTK CR‐LipoPEP‐20 group was preserved to the greatest extent, followed by the MACCCR2+MerTK CR group, while the other groups did not show statistical significance compared to the PBS group (Figure 7A). The left ventricular fractional shortening (LVFS) analysis has confirmed that MACCCR2+MerTK CR‐LipoPEP‐20 can effectively preserve the contraction efficiency of cardiomyocytes, while the MACCCR2+MerTK CR group also showed statistically significant benefits (Figure 7B). Statistical analysis of left ventricular end‐systolic volume (LVESV) and left ventricular end‐diastolic volume (LVEDV) suggested that MACCCR2+MerTK CR‐LipoPEP‐20 can effectively prevent the deterioration of mouse cardiac function post‐MI/R (Figure 7C,D). To assess cardiac remodeling, heart paraffin sections were utilized to quantify the preserved left ventricular anterior wall (LVAW) thickness and fibrosis remodeling through Masson's staining (Figure 7E). As expected, the MACCCR2+MerTK CR‐LipoPEP‐20 group has preserved the most myocardium (Figure 7F) and the subsequent fibrosis is the slightest (Figure 7G), compared with other groups. Group MACCCR2+MerTK CR also achieved statistically significant gains, while the other groups did not (Figure 7F,G). These results indicated that MACCCR2+MerTK CR‐LipoPEP‐20 can effectively improve MI/R‐induced cardiac function in mice.
Figure 7.
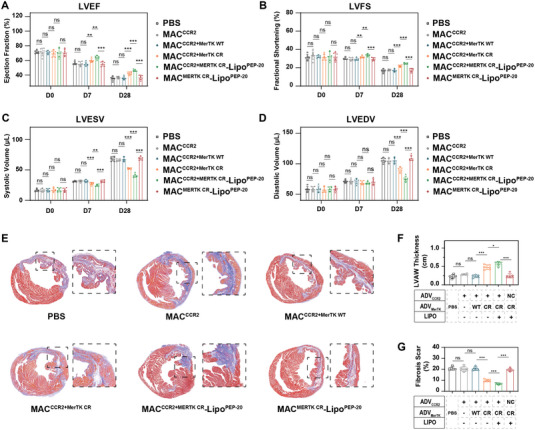
Cardiac protection of MACCCR2+MerTK CR‐LipoPEP‐20. Cardiac function was assessed by echocardiography at 1 or 4 weeks after PBS, MACCCR2+MerTK CR‐LipoPEP‐20 or various control MACs treatment. Statistical evaluation was performed on A) the left ventricular ejection fraction (LVEF), B) left ventricular fractional shortening (LVFS), C) left ventricular end‐systolic volume (LVESV), and D) left ventricular end‐systolic volume (LVESV) (n = 6). E) Masson staining of MI/R heart paraffin sections at 4 weeks after various treatments. F) LVAW thickness and G) fibrosis remodeling in (E) were quantified by using ImageJ software (n = 6). Results are presented as mean ± SD, ns p > 0.05, *p < 0.05, *p < 0.01, *p < 0.001.
2.7. MACCCR2+MerTK CR‐LipoPEP‐20 Dampens the Inflammation of MI/R Heart
To elucidate the underlying mechanisms of MACCCR2+MerTK CR‐LipoPEP‐20 mediated cardio‐protection, we directly focused on the inflammatory response. Previous studies utilizing genetically edited MerTKΔ483‐488 mice have demonstrated that enhancement of efferocytosis can exert cardioprotective effects through the inhibition of the inflammatory response. As expected, MACCCR2+MerTK CR and MACCCR2+MerTK CR‐LipoPEP‐20 produced fewer pro‐inflammatory factors and more anti‐inflammatory factors against MACCCR when fed with apoptotic cardiomyocytes in vitro (Figure S8A, Supporting Information). In vivo, both MACCCR2+MerTK CR and MACCCR2+MerTK CR‐LipoPEP‐20 can significantly reduce the number of infiltrating neutrophils and inflammatory Ly6Chi monocytes in the local area of the injured heart, with the latter showing better efficacy (Figure 8A–D). This may be attributed to the timely clearance of apoptotic cells, which reduces the production of damage‐associated molecular patterns (DAMPs) and other substances that can further amplify inflammation.[ 5 ] In view of this, we examined the concentrations of two typical chemokines in the serum of MI/R mice after PBS, MACCCR2+MerTK CR or MACCCR2+MerTK CR‐LipoPEP‐20 treatment, interleukin‐8(IL‐8) for neutrophils and MCP‐1 for monocytes. MACCCR2+MerTK CR did reduce the levels of these two chemokines, and the co‐loaded PEP‐20 amplified this effect (Figure S8B, Supporting Information). It is worth noting that the reduction in the number of neutrophils may also be partially attributed to an increase in their clearance by MACCCR2+MerTK CR‐LipoPEP‐20 after undergoing apoptosis.[ 10 ] In addition, consistent results were obtained through ELISA (enzyme‐linked immunosorbent assay) analysis of inflammatory (IL‐1β, TNF‐α) and anti‐inflammatory factors (IL‐10, TGF‐β) in the homogenized cardiac tissue (Figure 8E,F).
Figure 8.
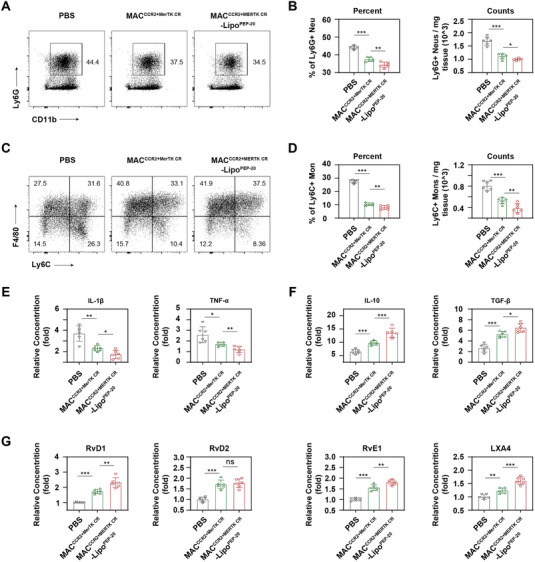
MACCCR2+MerTK CR‐LipoPEP‐20 dampens the inflammation of MI/R heart. A) Flow cytometric analysis of the neutrophils recruitment in the local MI/R‐induced heart following treatment with PBS, MACCCR2+MerTK CR, and MACCCR2+MerTK CR‐LipoPEP‐20, respectively. B) The percentage and absolute number of neutrophils within the CD45+ leukocyte population were quantitatively analyzed (n = 6). C) Flow cytometric analysis of the Ly6Chi monocytes recruitment in the local MI/R‐induced heart following treatment with PBS, MACCCR2+MerTK CR or MACCCR2+MerTK CR‐LipoPEP‐20, respectively. D) The percentage and absolute number of Ly6Chi monocytes within the CD45+CD11b+Ly6G‐ population were quantitatively analyzed (n = 6). E,F) ELISA analysis of inflammatory cytokines (IL‐1β, TNF‐α) and anti‐inflammatory cytokines (TGF‐β, IL‐10) concentrations in heart homogenate of MI/R‐induced mice after treated by PBS, MACCCR2+MerTK CR or MACCCR2+MerTK CR‐LipoPEP‐20 (n = 6). G) ELISA analysis of SPMs (RvD1, RvD2, RvE1, and LXA4) concentrations in heart homogenate of MI/R‐induced mice after treated by PBS, MACCCR2+MerTK CR or MACCCR2+MerTK CR‐LipoPEP‐20 (n = 6). Results are presented as mean ± SD, ns p > 0.05, *p < 0.05, *p < 0.01, *p < 0.001.
Previous studies have also reported that efferocytosis can serve as an initiating factor for the active resolution of acute inflammation by promoting the production of SPMs.[ 8 ] Unsurprisingly, both MACCCR2+MerTK CR and MACCCR2+MerTK CR‐LipoPEP‐20 can significantly enhance the production of SPMs both in vitro and in vivo, with MACCCR2+MerTK CR‐LipoPEP‐20 exhibiting a more profound effect (Figure 8G; Figure S8C, Supporting Information). Taken together, the cardioprotective effects of MACCCR2+MerTK CR‐LipoPEP‐20 after MI/R may be achieved through the regulation of inflammation.
2.8. Biosafety assessment of MACCCR2+MerTK CR‐LipoPEP‐20
Following intravenous injection of PBS and MACCCR2+MerTK CR‐LipoPEP‐20 into healthy mice, a series of biosafety‐related assessments were conducted. Considering the potential impact of co‐delivered PEP‐20 on healthy cells in the circulatory system, we performed a routine blood test. The MACCCR2+MerTK CR‐LipoPEP‐20 treatment had no effect on red blood cells (RBC) and white blood cells (WBC) (Figure S9A, Supporting Information). Regarding immune response, we employed ELISA to determine the levels of IL‐1β and TNF‐α in mouse serum on the third day after MACCCR2+MerTK CR‐LipoPEP‐20 injection, and no positive findings were observed compared to the control group (Figure S9B, Supporting Information). The serum levels of general antibodies, immunoglobulin G (IgG) and IgM, were also quantified by ELISA assay, and no statistical difference was observed between group MACCCR2+MerTK CR‐LipoPEP‐20 and control, indicating that there was no potential immune response after MACCCR2+MerTK CR‐LipoPEP‐20 administration (Figure S9C, Supporting Information). For organ toxicity assessment, we evaluated the liver and renal function in mice following MACCCR2+MerTK CR‐LipoPEP‐20 injection, with PBS injection serving as the control. No statistically significant differences were observed in the measured parameters between the two groups (Figure S9D,E, Supporting Information). Meanwhile, compared with control, no histopathological changes were observed in major organs of healthy mice treated with MACCCR2+MerTK CR‐LipoPEP‐20 (Figure S9F, Supporting Information). The aforementioned evidence collectively indicates that the biological safety of MACCCR2+MerTK CR‐LipoPEP‐20 was up to standard, which is a prerequisite for its clinical translation.
3. Discussion
The publication of a series of clinical trials, especially CANTOS (canakinumab anti‐inflammatory thrombosis outcomes study) and COLCOT (colchicine cardiovascular outcomes trial after myocardial infarction), has made immune modulation a promising strategy to promote cardiac repair.[ 33 , 34 ] However, most studies primarily focus on the negative inhibition of systemic inflammation, which increases susceptibility to infection, while also neglecting the intrinsic demands of local inflammatory responses and the clearance of damaged cells and tissues. Our strategy aims to precisely target and reconstruct local efferocytosis in the heart post‐MI/R, thereby reducing the intrinsic demands that initiate inflammation. Indeed, our results verified that MACCCR2+MerTK CR‐LipoPEP‐20 could effectively improve efferocytosis in the MI/R‐induced heart, alleviate inflammatory response, and consequently preserve better cardiac function. In addition, the biosafety experiments also confirmed that MACCCR2+MerTK CR‐LipoPEP‐20 treatment did not suppress systemic immune markers parameters in mice.
Adoptive cell therapy has revolutionized the clinical approach to cancer treatment.[ 35 ] However, the optimal efficacy of weapon cells at the target site often requires the support of accompanying drugs, such as ensuring the survival of adoptive T cells within tumors or maintaining the polarization state of adoptive macrophages locally.[ 36 , 37 ] Considering the significant differences in biodistribution and pharmacokinetic profiles between drugs and therapeutic cells, the synergistic delivery of drugs in both temporal and spatial dimensions with adoptive cells undoubtedly represents the optimal solution. In this study, we employed click chemistry techniques to anchor PEP‐20‐loaded liposomes onto the surface of engineered macrophages, achieving spatially synergistic delivery without compromising the physiological functions of the adoptive macrophages. Furthermore, we performed ROS‐responsive modification on the liposomes to further optimize the temporal compatibility between PEP‐20 and adoptive macrophages by achieving responsive release of PEP‐20 in the injured area. It is noteworthy that our strategy, like traditional chimeric antigen receptor immune‐cell therapies, is an in vitro cellular agent whose cumbersome and costly preparation process limits its clinical translational potential. Therefore, we will also seek the in vivo construction of this strategy.
Apart from MI/R injury, dysfunction of macrophage efferocytosis has been reported to be closely associated with various diseases, such as atherosclerosis, autoimmune diseases, and Alzheimer's disease, among others.[ 7 ] This provides our formula with more possibilities for clinical application. It is noteworthy that studies have indicated the significant role of tumor‐associated macrophage MerTK in promoting tumor metastasis.[ 38 ] Therefore, the application of MACCCR2+MerTK CR‐LipoPEP‐20 in patients with concomitant tumors needs to be approached with caution, and its safety profile requires further investigation.
4. Conclusion
In summary, our findings verified that MACCCR2+MerTK CR‐LipoPEP‐20 synergistically integrates PEP‐20 and genetically engineered macrophages, thereby enabling precise improvements in cardiac efferocytosis post‐MI/R, ultimately leading to improved cardiac function through the alleviation of inflammation. This strategy simultaneously targets all three underlying causes of efferocytosis impairment, demonstrating excellent therapeutic efficacy and biocompatibility. This therapy focuses on inhibiting the initiation and promoting active resolution of inflammation, providing new insights for immune‐regulatory therapy.
5. Experimental Section
Cell Line and Mice
The mice atrial cardiomyocyte cell line HL‐1 was purchased from the American Type Culture Collection (ATCC, USA). HL‐1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and 1% penicillin‐streptomycin. Male C57 BL/6J mice (21–25 g) aged 6–8 weeks and EGFP+ mice were all purchased from SLRC Laboratory Animal (Shanghai, China). Fluorescent protein expression in cardiomyocytes was induced by crossing α‐MHCCre mice (SLRC Laboratory Animal, Shanghai, China) with Rosa26‐tdTomato mice (SLRC Laboratory Animal, Shanghai, China). Mice were maintained in a 12 h/12 h light/dark cycle environment with a 22 °C constant temperature and free access to standard laboratory chow and tap water. All animal experiments were approved by the Ethics Committee of Zhongshan Hospital, Fudan University, Shanghai, China, and were in compliance with the guidance for the Care and Use of Laboratory Animals published by the National Research Council (U.S.) Institute for Laboratory Animal Research.
Isolation and Genetic Modification of BMDMs
BMDMs were extracted from 6–8 weeks C57BL/6 mice according to the previously reported protocol.[ 30 ] Briefly, after mice were euthanized by rapid cervical dislocation, their femurs and tibias were quickly separated, and then the bone marrow cavity was flushed with sterile PBS through a 70 µm cell strainer to obtain a single cell suspension. The isolated bone marrow cells were centrifuged and resuspended in DMEM containing 10% FBS, 1% penicillin/streptomycin (P/S), and 20 ng mL−1 recombination murine macrophage colony stimulating factor (M‐CSF, PeproTech, USA) for 7 days to obtain mature macrophages. The successful extraction of BMDMs was confirmed by flow cytometry.
CCR2 overexpression (pc‐ADV‐EF1‐CCR2‐6xHis) and vector adenovirus (pc‐ADV‐EF1‐MCS‐6xHis), as well as MerTK overexpression (pc‐ADV‐CMV‐EF1‐MerTK‐3xFLAG or pc‐ADV‐EF1‐MerTK(del483‐486)−3xFLAG) and its vector control (pc‐ADV‐CMV‐EF1‐MCS‐3xFLAG), were purchased from OBIO Technology (Shanghai, China). BMDMs were transduced with these adenoviral particles according to the manufacturer's instructions.
Western Blotting
The overexpression of CCR2 and MerTK, and the cleavage resistance of MerTKCR were validated by Western blotting. Briefly, cell lysates were prepared by lysis on ice using radioimmunoprecipitation assay (RIPA) buffer (Millipore, USA) and centrifuged at 12 000 rpm for 20 min at 4 °C. The proteins were separated by SDS‐polyacrylamide gel electrophoresis (SDS‐PAG), transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA), and probed with anti‐Mer (AF591, R&D System, USA) and anti‐CCR2 (MA5‐41175, ThermoFisher Scientific, USA). Antigen‐antibody complexes were detected using a chemiluminescence kit (Millipore, USA). In the cleavage detection experiments, PMA (50 nm) was used as a cleavage‐inducing agent. The culture medium of BMDMs was subjected to concentration using a centrifuged 100 kD filter device (Millipore, USA) before conducting Western blotting.
Transwell Assay
The chemotaxis of macrophages from each group was investigated using a 24‐well transwell migration assay (Corning, USA). Briefly, 1 × 105 macrophages were added to the upper chamber of the transwell, MCP‐1 (20 ng mL−1) was added into the lower chamber to induce the migration. Incubated for 12 h and stained with 0.1% trypan blue, the cells that migrated to the underside of the transwell membrane were counted under a microscope.
Efferocytosis Assay In Vitro
Fluorescent tracing was employed to evaluate the efferocytosis of apoptotic cardiomyocyte by macrophages from each group in vitro. After extraction from EGFP+ mice, BMDMs were randomly assigned to different groups for genetic engineering. HL‐1 cardiomyocytes were induced to undergo apoptosis with 5 µm staurosporine (HY‐15141, Med Chem Express) after labeling with IVISense 680 Fluorescent Cell Labeling Dyes (NEV12000, Perkinelmer). It was then incubated with BMDMs for 45 min. After washing away the unbound apoptotic cardiomyocytes, co‐localization was observed under a confocal microscope to assess the efferocytosis capacity of macrophages in each group. The percentage of cardiomyocyte‐associated macrophages was calculated as the efferocytosis rate. DAPI (Beyotime, China) was used as a nuclei indicator. The impact of MerTK cleavage resistance on efferocytosis was also validated through immunofluorescence. Macrophages overexpressing MerTKWT or MerTKCR, respectively, underwent PMA‐induced MerTK cleavage prior to the efferocytosis assay.
Flow Cytometry
The cultured cells were digested with 0.25% trypsin (Giboco, USA) and centrifuged at 1000 rpm for 5 min, thereby, the cell pellet was resuspended in 100 µL per test (106 cells) of staining buffer to obtain a single‐cell suspension. For tissue samples, a Multi‐Tissue Dissociation Kit 2 (Miltenyi Biotec, USA) was used to prepare the single‐cell suspension according to the instructions. Briefly, after being cut into small pieces (1–2 mm3), harvested heart tissue was digested in Enzyme Mix at 37 °C for 15 min, followed by mechanical agitation through the Program Multi_G of the gentle MACS Dissociator (Miltenyi Biotec, USA), and then repeated once. After being filtered through the MACS SmartStrainer (70 µm), the resulting sample was centrifuged at 600 g for 5 min to separate single cells. The cell pellet was also resuspended in 100 µL per test (106 cells) staining buffer. The obtained single‐cell suspension was incubated with the primary antibody at room temperature for 40 min. Unbound antibodies were removed by centrifugation, and all samples were tested by BD FACS Aria III (BD Bioscience, USA) within 1 h.
Chemical Materials and Methods
DSPE‐SeSe‐PEG2k (R‐DZP014), DSPE‐PEG5k‐TZ (R‐DP‐040), DSPE‐PEG5k‐TZ‐FITC (R‐DP‐040‐FI), and DSPE‐PEG5k‐TCO (R‐88015) were purchased from Xi'an Ruixi Biological Tecnology Co., Ltd. (China). To successfully achieve the surface coupling of liposomes on macrophages by click reaction, DSPE‐PEG5k‐TZ was inserted into the cell membrane as an anchoring module, while DSPE‐PEG5k‐TCO was incorporated into liposomes as a click moiety. Fluorescently labeled tetrazine derivative (DSPE‐PEG5k‐TZ‐FITC) were utilized for fluorescence imaging.
To achieve responsive release of PEP‐20, two designs were implemented: first, DSPE‐SeSe‐PEG2k was used as the skeleton lipid of liposomes, which possesses ROS‐sensitive diselenide bonds; second, thiol‐ketone (‐TK‐), another ROS‐sensitive covalent bond, was employed to connect water‐soluble PEP‐20 to the phospholipid tail (DSPE‐PEG2k‐TK‐PEP‐20), enabling its insertion onto the surface of the liposomes for PEP‐20 loading. ROS‐insensitive phospholipid analogs [DSPE‐PEG2k (HY‐142979, Med Chem Express, USA) and DSPE‐PEG2k‐PEP‐20] were set as a control.
To synthesize DSPE‐PEG2k‐TK‐PEP‐20, 100 mg of DSPE‐PEG2000‐TK‐NH2 (R‐TK‐PN02, Xi'an Ruixi Biological Tecnology Co., Ltd., China) was dissolved in 3 mL of DMF (N, N‐dimethylformamide, HY‐Y0345, Med Chem Express, USA). PEP‐20 (AWSATWSNYWRH‐COOH peptide, R‐TD‐01, Xi'an Ruixi Biological Tecnology Co., Ltd., China) (1.1 equiv.), PyBop (1H‐benzotriazol‐1‐yloxytripyrrolidinophosphonium hexafluorophosphate, P109336, Aladdin, China) (2.0 equiv.) and triethylamine (3.0 equiv.) were added to the solution and dissolved completely. The reaction was carried out at room temperature for 12 h. The reaction mixture was removed to a dialysis bag (1500 Da) and dialyzed in pure water for 24 h. The dialysate was collected and freeze‐dried to obtain DSPE‐PEG2k‐TK‐PEP‐20. The entire synthesis process and identification results are shown in Figure S7, Supporting Information. For the synthesis of control DSPE‐PEG2k‐PEP‐20, DSPE‐PEG2000‐TK‐NH2 was replaced by DSPE‐PEG2000‐NH2(R‐0038, Xi'an Ruixi Biological Tecnology Co., Ltd., China) in the above procedure.
To evaluate the efficiency of the click‐reaction, the DSPE‐PEG5k‐TZ (1 mm) was dissolved in CHCl3, and a UV spectrophotometer was used for wavelength scanning. After adding the DSPE‐BCN solution (with a final concentration of 1 mm in CHCl3), which was fully mixed and reacted at room temperature for 10 min, wavelength scanning was performed using an ultraviolet spectrophotometer to obtain the absorption curve.
Fabrication and Characterization of MACCCR2+MerTK CR‐LipoPEP‐20
PEP‐20‐loaded ROS‐responsive liposomes were prepared by the thin‐film hydration method. Briefly, DSPE‐SeSe‐PEG2k, DSPE‐PEG2k‐TK‐PEP‐20, DSPE‐PEG5k‐TCO, and cholesterol were dissolved in trihalomethane at a molar ratio of 3:3:3:1. The lipid film was prepared by evaporating the organic solvent in a 37 °C water bath under vacuum. DiD‐labeled lipid films were obtained by adding DiD into the initial solution at a v/v ratio of 1:1000. The evaporated dried film was hydrated with PBS (pH 7.4) and incubated at 37 °C for 30 min, mixed vigorously, and further sonicated to obtain a clear suspension of lipid. The resulting suspension was successively extruded 20 times through 0.4, 0.2, and 0.1 µm polycarbonate membranes (Nuclepore Track‐Etched Membranes, Whatman, UK), using a LiposoFast extruder apparatus (Avestin, Canada), to obtain liposomes with uniform particle size. The morphology and size of the liposome were observed under a TEM (Tecnai G2 Spirits Twin, FEI, USA) after negative staining with 1% phosphotungstic acid. The size distribution and zeta potential were measured by a DLS detector (Zetasizer‐ZS90, Malvern Instruments, UK).
MACCCR2+MerTK CR‐LipoPEP‐20 were obtained by incubating MACCCR2+MerTK CR successively with DSPE‐PEG5k‐TZ (80 µg mL−1, 37 °C, 10 min) and TCO‐LipoPEP‐20 (4 mg mL−1, 37 °C, 20 min). The resulting MACCCR2+MerTK CR‐LipoPEP‐20 were collected by centrifugation at 1000 rpm for 5 min, washed with PBS, and resuspended in medium for in vitro or in vivo studies.
To visualize and quantify the anchoring of liposomes on the surface of macrophage, DSPE‐PEG5k‐TZ‐FITC was substituted for DSPE‐PEG5k‐TZ, and the liposomes were also labeled with DiD. The control group was identical to the experimental group, except for the replacement of DSPE‐PEG5k‐TZ by DSPE‐PEG5k in the insertion into the cell membrane. DAPI was used as a nuclear indicator under confocal microscopy, and the amount of liposome anchoring was evaluated by flow cytometry. To investigate the anchoring stability of liposomes on the surface of macrophages, the aforementioned experiments were extended to three additional time points (immediately, 3 and 7 days post‐anchoring). Further direct observation of the resulting MACCCR2+MerTK CR‐LipoPEP‐20 was conducted using scanning electron microscopy (SEM). Considering that liposomes are prone to breakage during the dehydration process in SEM sample preparation, 50 nm diameter Au cores were incorporated into the liposome preparation to provide support. MACCCR2+MerTK CR‐LipoAu were fixed with a 2.5% glutaraldehyde aqueous solution overnight. After washing with PBS three times, samples were dehydrated with gradient ethanol, coated with a layer of gold, and imaged by SEM (Quanta 250, FEI, USA).
To calculate the number of liposomes anchored on the surface of macrophages, 1 × 106 cells were first incubated with DSPE‐PEG5k‐ TZ or DSPE‐PEG5k (80 µg mL−1) at 37 °C for 10 min. After washing with PBS three times, MACCCR2+MerTK CR were then incubated with the TCO‐LipoPEP‐20 solution in 1 mL (4 mg mL−1, 37 °C, 20 min). The concentration of liposomes in the solution before and after incubation was assessed by nanoparticle tracking analyzer (Malvern NanoSight NS300). The liposome number anchored on the surface of macrophages was calculated as follows: (n DSPE‐PEG5k‐TZ before − n DSPE‐PEG5k‐TZ after) − (n DSPE‐PEG5k before − n DSPE‐PEG5k after) / 106, where n DSPE‐PEG5k‐TZ before and n DSPE‐PEG5k before were the counted numbers of liposomes in the solution before the incubation, and n DSPE‐PEG5k‐TZ after and n DSPE‐PEG5k after were the counted numbers of liposomes in the supernatant after the incubation of liposomes with engineered macrophages. This algorithm eliminates the non‐specific adsorption of liposomes on the surface of macrophages.
Synergistic Effect Assessment of MACCCR2+MerTK CR with TCO‐LipoPEP‐20
The impact of liposome anchoring on the physiological functions of engineered macrophages, including viability, chemotaxis, and polarization state, were first evaluated. On D1, D3, and D5 post‐conjunction, the viability of MACCCR2+MerTK CR and MACCCR2+MerTK CR‐LipoPEP‐20 was assessed by using a CCK‐8 Kit (Beyotime, China) according to the manufacturer's protocol, and the absorbances were measured at 450 nm by an EPOCH 2 microplate reader (BioTek, USA). After 12 h of anchoring, the chemotaxis of MACCCR2+MerTK CR and MACCCR2+MerTK CR‐LipoPEP‐20 to MCP‐1 was compared by the previously described transwell assay, while the impact of liposome anchoring on macrophage polarization status was also assessed by flow cytometry.
The short‐long arm strategy (DSPE‐PEG2k‐TK‐PEP‐20 vs DSPE‐PEG5k‐TCO) for masking PEP‐20 was evaluated by flow cytometry. Compared to intact liposomes anchored on macrophages, the substitution of DSPE‐PEG5k‐TCO with DSPE‐PEG2k‐TCO, or the substitution of DSPE‐PEG2k‐PEP‐20 with DSPE‐PEG2k, were used as single variable controls. FITC‐labeled CD47 protein (CD7‐HF3H3, ACRO Biosystems, China) or isotype control (BSA‐FITC, WH0092607, Weihua Bio, China) was used for the detection of PEP‐20. The expression levels of SIRP‐α (APC‐anti‐SIRP‐α, MA5‐46864, eBioscience, USA) on the surface of macrophages in each group were also detected by flow cytometry.
The ROS‐responsive disintegration of macrophage anchored DiD‐liposomes and the ROS‐responsive cleavage of PEP‐20 from liposomes were also assessed by flow cytometry. For the detection of liposome disintegration, the PBS solution of intact MACCCR2+MerTK CR‐LipoPEP‐20 was the initial formulation. First, 20% FBS was added to the solution, followed by the addition of 100 µm H2O2, and finally replace DSPE‐SeSe‐PEG2k in the liposomes with DSPE‐PEG2k. After each step, the fluorescence intensity of DiD on the surface of macrophages using flow cytometry was measured. For the PEP‐20 cleavage detection, DSPE‐PEG5k‐TCO in MACCCR2+MerTK CR‐LipoPEP‐20 was replaced by DSPE‐PEG2k‐TCO in the initial PBS solution. The intervention conditions involved adding 100 µm H2O2 and replacing DSPE‐PEG2k‐TK‐PEP‐20 with DSPE‐PEG2k‐PEP‐20. After each step, the binding capacity of CD47‐FITC to MACCCR2+MerTK CR‐LipoPEP‐20 was accessed using flow cytometry.
Last, the further optimization effect of PEP‐20′s synergistic co‐delivery on engineered macrophage efferocytosis was also confirmed through the efferocytosis assay. MACCCR2+MerTK CR in PBS solution with no liposome anchored was set as the control. The effects of anchoring intact liposomes without H2O2 addition, anchoring intact liposomes with H2O2 addition, or anchoring empty liposomes (without PEP‐20) with H2O2 addition, respectively, on engineered macrophage efferocytosis were assessed by confocal imaging and flow cytometry. Different from the aforementioned efferocytosis assay, the group requiring H2O2 addition should have 100 µm of H2O2 added to the solution containing apoptotic cells that were being fed to the macrophages.
Myocardial Ischemia‐Reperfusion Injury Animal Model
Mice myocardial ischemia‐reperfusion injury model was constructed by left anterior‐descending coronary artery ligation for 60 min, followed by reperfusion. The injury of mice heart was confirmed by ST‐segment characterized electrocardiogram and left ventricle color alteration.
Biodistribution and Targeting of MACCCR2+MerTK CR‐LipoPEP‐20 In Vivo
For the evaluation of blood pharmacokinetic, 106 EGFP+ mice derived MACWT, MACCCR2, MACMerTK CR, MACCCR2+MerTK CR and MACCCR2+MerTK CR‐LipoPEP‐20 were respectively administered to MI/R‐induced C57 BL/6J mice via tail vein injection (n = 6), and the blood samples were collected at different time points (5 min, 1 day, and 3 days) post injection. Mice injected with PBS were designated as the control group. The collected blood samples were treated with RBC Lysis Buffer (Invitrogen, USA) for 5 min at RT. After centrifugation at 1000 rpm for 5 min, the cell pellets were resuspended in staining buffer, and fluorescence signals were detected by BD FACS Aria III.
To explore the distribution of macrophages from each group in major organs, including heart, liver, spleen, lung, kidney, and brain, MI/R‐induced mice were divided into six groups randomly (n = 6). Each group received injections of PBS, IVISense 680 labeled MACWT, MACCCR2, MACMerTK CR, MACCCR2+MerTK CR, and MACCCR2+MerTK CR‐LipoPEP‐20 at a number of 106, respectively, 3 h after reperfusion. At three time points (1 day, 3 days, and 7 days) post‐administration, major organs were harvested and then ex vivo imaged by the IVIS (PerkinElmer, Inc., Waltham, MA).
To directly observe the accumulation of MACCCR2+MerTK CR‐LipoPEP‐20 in the injured heart, EGFP+ mice‐derived BMDMs were used again as the source of adoptively transferred macrophages. The distribution of adoptive macrophage in the injured heart was evaluated by immunofluorescence staining at 1 day after the administration. Briefly, the injured heart was harvested and embedded in an optimal cutting temperature compound (Sakura Finetek, Japan), and then frozen in liquid nitrogen before being cut into 8‐µm cryosections. Tissue sections were either immediately processed for subsequent staining or frozen at −20 °C for further use. For immunofluorescence staining, the obtained tissue sections were first fixed in acetone for 10 min, and then washed three times with PBS, each time for 5 min. The washed tissue sections were blocked with 3% BSA for 1 h, and then incubated with a specific primary antibody, Rabbit‐anti‐cardiac troponin T (15513‐1‐AP, ProteinTech, USA), overnight at 4 °C. After washing three times with PBS, each time for 5 min, it was stained with fluorescently labeled secondary antibody for 1 h, followed by DAPI to indicate the nucleus. Alexa Fluor 568‐anti‐Rabbit IgG (A‐11011) secondary antibody was purchased from Invitrogen (USA). 1 day after the administration, the distribution of adoptive macrophage in the injured heart was also evaluated by flow cytometry, while detailed methods were the same as described above.
In Vivo Assessment of Cardiac Efferocytosis
EGFP+ mice‐derived MACCCR2‐LipoPEP‐20, MACCCR2+MerTK WT‐LipoPEP‐20, MACCCR2+MerTK CR‐LipoPEP‐20, and MACCCR2+MerTK CR were respectively injected into MI/R‐induced mice (n = 6). After 3 h of injection, the cardiac tissue was collected and prepared into a single‐cell suspension. The surface expression of MerTK on EGFP+ adoptive cells in the suspension was detected by flow cytometry after staining with anti‐MerTK‐APC (175751, eBioscience, USA).
To evaluate the efferocytosis capacity of adoptive or cardiac resident macrophages, α‐MHCCre: Rosa26‐tdTomato mice were used, whose cardiomyocytes were specifically labeled with red fluorescence (tdTomato). When cardiomyocytes were engulfed by macrophages, tdTomato signals could be detected in macrophages. The tdTomato+ signal of adoptive macrophages was detected by flow cytometry. To evaluate efferocytosis capacity of adoptive or cardiac resident macrophages, the four experimental groups were adjusted to EGFP+ mice‐derived MACCCR2, MACCCR2+MerTK WT, MACCCR2+MerTK CR, and MACCCR2+MerTK CR‐LipoPEP‐20. To assess the impact of adoptive MACCCR2+MerTK CR‐LipoPEP‐20 on cardiac resident macrophage efferocytosis, PBS and MACMerTK CR‐LipoPEP‐20 groups were newly added to the previous experiment. The tdTomato+ signal of cardiac resident macrophages was detected by flow cytometry after staining with anti‐CD45‐APC‐Cy7 (557659, eBioscience, USA), anti‐CD11b‐PerCP‐Cy5.5 (45‐0112, eBioscience, USA), anti‐Ly6G‐BV421 (562737, eBioscience, USA), anti‐F4/80‐APC (MF48005, eBioscience, USA).
The efficiency of apoptotic cell clearance in MI/R hearts was also evaluated by TUNEL staining at 1 day after receiving PBS, MACCCR2, MACCCR2+MerTK WT, MACCCR2+MerTK CR, MACCCR2+MerTK CR‐LipoPEP‐20, and MACMerTK CR‐LipoPEP‐20 treatments, respectively. TUNEL staining was performed according to the manufacturer's instructions, with DAPI used as a nuclear indicator.
Cardiac Protection of MACCCR2+MerTK CR‐LipoPEP‐20
MI/R‐induced C57 BL/6J mice were blindly divided into six groups (n = 6) and the groups were injected PBS, MACCCR2, MACCCR2+MerTK WT, MACCCR2+MerTK CR, MACCCR2+MerTK CR‐LipoPEP‐20, and MACMerTK CR‐LipoPEP‐20 treatments, respectively, through the tail vein 3 h after reperfusion. Cardiac geometry structure and function were assessed using the 2D guided M‐mode echocardiography (Visual Sonics Vevo 770, Canada) 1 day before the procedure, 1 and 4 weeks after the treatments. After depilation, mice were anesthetized with controlled isoflurane flow to maintain the heart rate around 450 beats min−1. The parasternal long‐axis view was obtained and LVEF, LVFS, LVESV, and LVEDV were calculated and analyzed. The echocardiographer was blind to the treatment protocol of mice and each measurement index was averaged by six consecutive cardiac cycles. After that, the mouse hearts were harvested to make a 5‐µm‐thick paraffin section. Masson staining of paraffin sections was used to determine the infarct size and the degree of remodeling fibrosis.
The Impact of MACCCR2+MerTK CR‐LipoPEP‐20 on MI/R‐Induced Cardiac Inflammation
The cardiac inflammatory cell dynamics of MI/R mice were evaluated by flow cytometry after treatment with PBS, MACCCR2+MerTK CR or MACCCR2+MerTK CR‐LipoPEP‐20, respectively. Anti‐CD45‐FITC (553083, ebioscience, USA), anti‐CD11b‐PerCP‐Cy5.5 (45‐0112, ebioscience, USA), anti‐Ly6G‐BV421 (562737, ebioscience, USA), anti‐F4/80‐PE (565410, ebioscience, USA), and anti‐Ly6C‐PE‐Cy7 (560593, ebioscience, USA) were used.
The levels of inflammation associated cytokines (IL‐1β, TNF‐α, IL‐10, and TGF‐β) and SPMs (RvD1, RvD2, RvE1, and lipoxin B4) in cardiac tissue homogenate were detected by ELISA assay after treatment with PBS, MACCCR2+MerTK CR or MACCCR2+MerTK CR‐LipoPEP‐20, respectively. ELISA Kit for IL‐1β, TNF‐α, IL‐10, and TGF‐β were all purchased from Biolegend (USA). Resolvin D1 ELISA Kit (500380, Cayman), Resolvin D2 ELISA Kit (501120, Cayman), Resolvin E1 ELISA Kit (TW10239, TW‐Reagent) and Lipoxin A4 ELISA Kit (E‐EL‐0053c, Elabscience) were used according to instruction. Before the detection, the BCA assay was used to quantify the protein concentration of the tissue homogenate, and the subsequent detection results were also corrected according to the corresponding protein concentration.
Biosafety Assay of MACCCR2+MerTK CR‐LipoPEP‐20
Considering the potential threat of PEP‐20 co‐delivery to the viability of circulating healthy cells, complete blood count analysis was performed on mice after treatment with PBS and MACCCR2+MerTK CR‐LipoPEP‐20. For the immuno‐safety, C57 BL/6J mice were randomized and treated with MACCCR2+MerTK CR‐LipoPEP‐20 or PBS (n = 6). The serum level of inflammatory cytokines, TNF‐α and IL‐1β, and general antibodies, IgG and IgM, were all quantified by using ELISA kits (Biolegend, USA) according to the manufacturer's protocols. For organ toxicity, biochemical tests of liver (aspartate aminotransferase, AST; alanine aminotransferase, ALT), and renal function (creatinine, CREA; urea nitrogen, UREA) were performed on the serum of mice after being treated with PBS or MACCCR2+MerTK CR‐LipoPEP‐20. Histopathological changes in major organs were also evaluated by HE staining at 4 weeks post PBS or MACCCR2+MerTK CR‐LipoPEP‐20 treatment.
Statistical Analysis
All quantitative data were presented as means ± standard deviation (SD) of triplicate measurements in vitro and six measurements in vivo. Two‐tailed Student's t‐test was used for comparison between two groups. One‐way analysis of variance (ANOVA) followed by Bonferroni test was used for statistics between multiple groups. The difference between groups was considered none statistically significant for ns, statistically significant for *p < 0.05, very significant for **p < 0.01, and the most significant for ***p < 0.001. Statistical and graph analysis were performed using SPSS Statistics 26.0 (IBM, USA) and GraphPad Prism 9.0 (GraphPad Software, USA), respectively.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
H.T., W.L., Z.P., and X.W. contributed equally to this work. This work was supported by the National Natural Science Foundation of China (82170254 and 82370257 to Y.S., 82070281 to Z.H.), the State Key Clinical Specialty Construction Project (YW2021‐002), Shanghai Rising‐Star Program (23QA1401300), and Shanghai Clinical Research Center for Interventional Medicine (19MC1910300).
Tan H., Li W., Pang Z., Weng X., Gao J., Chen J., Wang Q., Li Q., Yang H., Dong Z., Wang Z., Zhu G., Tan Y., Fu Y., Han C., Cai S., Qian J., Huang Z., Song Y., Ge J., Genetically Engineered Macrophages Co‐Loaded with CD47 Inhibitors Synergistically Reconstruct Efferocytosis and Improve Cardiac Remodeling Post Myocardial Ischemia Reperfusion Injury. Adv. Healthcare Mater. 2024, 13, 2303267. 10.1002/adhm.202303267
Contributor Information
Zheyong Huang, Email: huang.zheyong@zs-hospital.sh.cn.
Yanan Song, Email: song.yanan@zs-hospital.sh.cn.
Junbo Ge, Email: ge.junbo@zs-hospital.sh.cn.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Virani S. S., Alonso A., Benjamin E. J., Bittencourt M. S., Callaway C. W., Carson A. P., Chamberlain A. M., Chang A. R., Cheng S., Delling F. N., Djousse L., Elkind M. S. V., Ferguson J. F., Fornage M., Khan S. S., Kissela B. M., Knutson K. L., Kwan T. W., Lackland D. T., Lewis T. T., Lichtman J. H., Longenecker C. T., Loop M. S., Lutsey P. L., Martin S. S., Matsushita K., Moran A. E., Mussolino M. E., Perak A. M., Rosamond W. D., et al., Circulation 2020, 141, e139. [DOI] [PubMed] [Google Scholar]
- 2. Yu Z. C., Gong H. X., Xue F. Q., Zeng Y. Y., Liu X. L., Tang D. P., Anal Chem 2022, 94, 13233. [DOI] [PubMed] [Google Scholar]
- 3. Yu Z. C., Gong H. X., Gao Y., Li L., Xue F. Q., Zeng Y. Y., Li M. J., Liu X. L., Tang D. P., Small 2022, 18, e2202564. [DOI] [PubMed] [Google Scholar]
- 4. Jennings R. B., Circ. Res. 2013,113,428. [DOI] [PubMed] [Google Scholar]
- 5. Heusch G., Nat. Rev. Cardiol. 2020, 17, 773. [DOI] [PubMed] [Google Scholar]
- 6. Wan E., Yeap X. Y., Dehn S., Terry R., Novak M., Zhang S., Iwata S., Han X., Homma S., Drosatos K., Lomasney J., Engman D. M., Miller S. D., Vaughan D. E., Morrow J. P., Kishore R., Thorp E. B., Circ. Res. 2013, 113, 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rurik J. G., Aghajanian H., Epstein J. A., Circ. Res. 2021, 128, 1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Howangyin K. Y., Zlatanova I., Pinto C., Ngkelo A., Cochain C., Rouanet M., Vilar J., Lemitre M., Stockmann C., Fleischmann B. K., Mallat Z., Silvestre J. S., Circulation. 2016, 133, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doran A. C., Yurdagul A., Tabas I., Nat. Rev. Immunol. 2020, 20, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cai B., Thorp E. B., Doran A. C., Subramanian M., Sansbury B. E., Lin C.‐S., Spite M., Fredman G., Tabas I., Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Konstantinidis K., Whelan R. S., Kitsis R. N., Arterioscler., Thromb., Vasc. Biol. 2012, 32, 1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horckmans M., Ring L., Duchene J., Santovito D., Schloss M. J., Drechsler M., Weber C., Soehnlein O., Steffens S., Eur. Heart J. 2017, 38, 187. [DOI] [PubMed] [Google Scholar]
- 13. Dick S. A., Macklin J. A., Nejat S., Momen A., Clemente‐Casares X., Althagafi M. G., Chen J., Kantores C., Hosseinzadeh S., Aronoff L., Wong A., Zaman R., Barbu I., Besla R., Lavine K. J., Razani B., Ginhoux F., Husain M., Cybulsky M. I., Robbins C. S., Epelman S., Nat. Immunol. 2019, 20, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swirski F. K., Nahrendorf M., Nat. Rev. Immunol. 2018, 18, 733. [DOI] [PubMed] [Google Scholar]
- 15. Deberge M., Yeap X. Y., Dehn S., Zhang S., Grigoryeva L., Misener S., Procissi D., Zhou X., Lee D. C., Muller W. A., Luo X., Rothlin C., Tabas I., Thorp E. B., Circ. Res. 2017, 121, 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arandjelovic S., Ravichandran K. S., Circ. Res. 2013, 113, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thorp E., Vaisar T., Subramanian M., Mautner L., Blobel C., Tabas I., J. Biol. Chem. 2011, 286, 33335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izmirly P. M., Saxena A., Kim M. Y., Wang D., Sahl S. K., Llanos C., Friedman D., Buyon J. P., Circulation 2011, 124, 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang S., Yeap X. Y., DeBerge M., Naresh N. K., Wang K., Jiang Z., Wilcox J. E., White S. M., Morrow J. P., Burridge P. W., Procissi D., Scott E. A., Frazier W., Thorp E. B., JACC Basic Transl. Sci. 2017, 2, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen S., Yu Z., Wang Y., Tang J., Zeng Y., Liu X., Tang D., Anal. Chem. 2023, 95, 14494. [DOI] [PubMed] [Google Scholar]
- 21. Lambrecht B. N., Vanderkerken M., Hammad H., Nat. Rev. Immunol. 2018, 18, 745. [DOI] [PubMed] [Google Scholar]
- 22. Klichinsky M., Ruella M., Shestova O., Lu X. M., Best A., Zeeman M., Schmierer M., Gabrusiewicz K., Anderson N. R., Petty N. E., Cummins K. D., Shen F., Shan X., Veliz K., Blouch K., Yashiro‐Ohtani Y., Kenderian S. S., Kim M. Y., O'connor R. S., Wallace S. R., Kozlowski M. S., Marchione D. M., Shestov M., Garcia B. A., June C. H., Gill S., Nat. Biotechnol. 2020, 38, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu W.‐B., Ye Z.‐H., Chen X., Shi J.‐J., Lu J.‐J., Drug Discovery Today 2021, 26, 561. [DOI] [PubMed] [Google Scholar]
- 24. Yoon H. Y., Lee D., Lim D.‐K., Koo H., Kim K., Adv. Mater. 2022, 34, 2107192. [DOI] [PubMed] [Google Scholar]
- 25. Tang D., Yu Y., Zhang J., Dong X., Liu C., Xiao H., Adv. Mater. 2022, 34, 2203820. [DOI] [PubMed] [Google Scholar]
- 26. Weng X., Tan H., Huang Z., Chen J., Zhang N., Wang Q., Li Q., Gao J., Sun D., Yakufu W., Wang Z., Li W., Zhu G., Pang Z., Song Y., Qian J., Ge J., J. Nanobiotechnol. 2022, 20, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dewald O., Zymek P., Winkelmann K., Koerting A., Ren G., Abou‐Khamis T., Michael L. H., Rollins B. J., Entman M. L., Frangogiannis N. G., Circ. Res. 2005, 96, 881. [DOI] [PubMed] [Google Scholar]
- 28. Mcshane L., Tabas I., Lemke G., Kurowska‐Stolarska M., Maffia P., Cardiovasc. Res. 2019, 115, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaggar A., Shayakhmetov D. M., Lieber A., Nat. Med. 2003, 9, 1408. [DOI] [PubMed] [Google Scholar]
- 30. Tan H., Song Y., Chen J., Zhang N., Wang Q., Li Q., Gao J., Yang H., Dong Z., Weng X., Wang Z., Sun D., Yakufu W., Pang Z., Huang Z., Ge J., Adv. Sci. 2021, e2100787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yin N., Zhao Y., Liu C., Yang Y., Wang Z.‐H., Yu W., Zhang K., Zhang Z., Liu J., Zhang Y., Shi J., Adv. Mater. 2022, 34, 2201322. [DOI] [PubMed] [Google Scholar]
- 32. Pell V. R., Chouchani E. T., Murphy M. P., Brookes P. S., Krieg T., Circ. Res. 2016, 118, 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ridker P. M., Everett B. M., Thuren T., Macfadyen J. G., Chang W. H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S. D., Kastelein J. J. P., Cornel J. H., Pais P., Pella D., Genest J., Cifkova R., Lorenzatti A., Forster T., Kobalava Z., Vida‐Simiti L., Flather M., Shimokawa H., Ogawa H., Dellborg M., Rossi P. R. F., Troquay R. P. T., Libby P., Glynn R. J., N. Engl. J. Med. 2017, 377, 1119.28845751 [Google Scholar]
- 34. Tardif J.‐C., Kouz S., Waters D. D., Bertrand O. F., Diaz R., Maggioni A. P., Pinto F. J., Ibrahim R., Gamra H., Kiwan G. S., Berry C., López‐Sendón J., Ostadal P., Koenig W., Angoulvant D., Grégoire J. C., Lavoie M.‐A., Dubé M.‐P., Rhainds D., Provencher M., Blondeau L., Orfanos A., L'allier P. L., Guertin M.‐C., Roubille F., N. Engl. J. Med. 2019, 381, 2497. [DOI] [PubMed] [Google Scholar]
- 35. Finck A. V., Blanchard T., Roselle C. P., Golinelli G., June C. H., Nat. Med. 2022, 28, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang L., Zheng Y., Melo M. B., Mabardi L., Castaño A. P., Xie Y.‐Q., Li N., Kudchodkar S. B., Wong H. C., Jeng E. K., Maus M. V., Irvine D. J., Nat. Biotechnol. 2018, 36, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shields C. W., Evans M. A., Wang L. L.‐W., Baugh N., Iyer S., Wu D., Zhao Z., Pusuluri A., Ukidve A., Pan D. C., Mitragotri S., Sci. Adv. 2020, 6, eaaz6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maimon A., Levi‐Yahid V., Ben‐Meir K., Halpern A., Talmi Z., Priya S., Mizraji G., Mistriel‐Zerbib S., Berger M., Baniyash M., Loges S., Burstyn‐Cohen T., J. Clin. Invest. 2021, 131, e126089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.