

Abstract
One of the key catalytic reactions for life on earth, the oxidation of water to molecular oxygen, occurs in the oxygen‐evolving complex of the photosystem II (PSII) mediated by a manganese‐containing cluster. Considerable efforts in this research area embrace the development of efficient artificial manganese‐based catalysts for the oxygen evolution reaction (OER). Using artificial OER catalysts for selective oxygenation of organic substrates to produce value‐added chemicals is a worthwhile objective. However, unsatisfying catalytic performance and poor stability have been a fundamental bottleneck in the field of artificial PSII analogs. Herein, for the first time, a manganese‐based anode material is developed and paired up for combining electrocatalytic water oxidation and selective oxygenations of organics delivering the highest efficiency reported to date. This can be achieved by employing helical manganese borophosphates, representing a new class of materials. The uniquely high catalytic activity and durability (over 5 months) of the latter precursors in alkaline media are attributed to its unexpected surface transformation into an amorphous MnO x phase with a birnessite‐like short‐range order and surface‐stabilized MnIII sites under extended electrical bias, as unequivocally demonstrated by a combination of in situ Raman and quasi in situ X‐ray absorption spectroscopy as well as ex situ methods.
Keywords: active catalytic structures, electrocatalytic water oxidation, helical manganese borophosphates, selective oxygenation
Helical manganese borophosphates are employed to address two highly demanding yet kinetically controlled reactions: durable electrochemical water oxidation to efficiently produce molecular oxygen and the selective oxygenation of organics to deliver prominent value‐added chemicals.

Increasing depletion of fossil fuels and the rapid growth of energy consumption associated with environmental impacts have motivated the search for renewable and clean energy sources.[ 1 ] In this regard, the electrocatalytic generation of hydrogen (H2) and oxygen (O2) through water electrolysis has recently gained significant attention in both fundamental and applied research.[ 2 ] The reduction of water to H2 is a two‐electron process and comparatively straightforward, while the oxidation of water to O2 represents the bottleneck for water‐splitting.[ 3 ] It involves a complex four‐electron–proton coupled reaction with multiple high energy intermediates, and therefore, sluggish kinetics.[ 2 , 4 ] In nature, water oxidation occurs via photosynthesis and is catalyzed by a protein‐bound oxygen‐evolving Mn4CaO5 complex (OEC) in photosystem II.[ 5 ] Inspired by the vital role in OEC, as well as the combination of low cost, low‐toxicity, natural abundance, and switchable redox states, manganese is an obvious and attractive choice as the active site for artificial photosynthetic systems.[ 6 ] Various strategies have been successfully employed to synthesize different polymorphs of manganese oxides (MnO x ) for electrochemical water oxidation; however, most of them have demonstrated unsatisfactory activity (overpotentials >400 mV) and poor stability (a few hours).[ 7 ] Regardless of which manganese catalyst has been applied for water oxidation, the presence and stabilization of MnIII sites against the disproportionation reaction into inactive MnIV and MnII is essential to enhance the catalytic activity.[ 8 ] MnIII in a Jahn–Teller‐distorted high‐spin t2g 3eg 1 state is expected to provide elongated Mn−O bonds with the required flexibility to facilitate O−O bond formation, and consequently, a mixed MnIII/MnIV state within the same structure has been proposed to be favorable for water oxidation.[ 9 ] Therefore, the future of water electrolysis with Mn‐based anodes relies on the design of an active and sustainable catalyst that can replace the current, expensive and rare precious metals used in state‐of‐the‐art proton‐exchange membrane electrolyzers or Ni‐based electrodes in alkaline water electrolyzers. Herein, we address the following research questions: i) Can one synthesize an unconventional Mn‐based (pre)catalysts beyond oxides that is significantly more active with respect to oxygen evolution reaction (OER)?, ii) What role does the anionic partial structure of such Mn (pre)catalysts play in OER?, iii) How does the structure of the (pre)catalyst influence the OER activity?, iv) What factors stabilize the essential surface‐active MnIII sites throughout the OER, and v) Is it possible to provide a highly robust and efficient Mn‐based electrocatalyst for OER that can be durable for months (a step closer to the performance of the biologically active site in OEC)?
Alternatively, the electrosynthesis of higher value‐added products via oxygenation of organics using water as an oxygen atom source is a highly ambitious reaction and has gained enormous interest in the last couple of years.[ 10 ] Recently, the insufficient performance of precious metals led to the development of Ni‐based electrocatalysts for organic transformations; however, the multi‐step electron transfer process, demanding high energy to overcome the kinetic barrier (similar to water oxidation), fundamentally limits the reaction efficiency.[ 10 , 11 ] In this respect and to the best of our knowledge, although manganese can have variable oxidation states and has been a potent chemical oxidant, its potential and applicability as an electrocatalyst for the coupled oxygenation of organic substrates and OER has not been explored to date. Here, we present a solution on how to combine and boost two kinetically limited challenging reactions: water oxidation and selective oxygenation of organic compounds, using a suitable bifunctional Mn‐based electrocatalyst.
Metal borophosphates (BPOs) are well known for their fascinating structural architectures because of their ability to polymerize borate (BO4/BO3) and phosphate (PO4) units through P‐O‐B linkages to form a great variety of connection patterns and variable extension of the anionic partial structures.[ 12 ] Theoretical calculations have suggested that BPOs have high voltage, gravimetric capacity, specific energy, energy density, and volume change during delithiation, thus, making them suitable materials for electrochemical applications.[ 13 ] However, the potential of BPOs in the field of energy conversion and storage has hardly been explored.
Alkali‐metal manganese BPOs, LiMnII(H2O)2[BP2O8]·H2O (LiMnBPO), and NaMnII(H2O)2[BP2O8]·H2O (NaMnBPO) were synthesized by a mild‐hydrothermal approach. Rietveld refinement of the powder X‐ray diffraction pattern (PXRD), optical and scanning electron microscopy (SEM) along with energy dispersive X‐ray (EDX) analyses, elemental mapping, transmission electron microscopy (TEM), high‐resolution TEM (HRTEM), and its selected area electron diffraction (SAED) pattern, inductively coupled plasma atomic emission spectroscopy (ICP‐AES) analyses, Fourier transform infrared (FT‐IR) spectroscopy, X‐ray photoelectron spectroscopy (XPS) of isotypic materials confirmed the formation of a single‐phase product (Figure 1a–g and Figures S1–S13, Table S1, Supporting Information). The presented BPOs belong to the helical family and crystallize in the chiral space groups P6522 (left‐handed) or P6122 (right‐handed).[ 14 ] The structure consists of 1D infinite loop‐branched BPO helical channels ∞ 1[BP2O8]3− (a spiral ribbon of four‐membered BO4/PO4 rings) built of alternate BO4 and PO4 tetrahedra (Figure S4, Supporting Information), which are strikingly related to α‐quartz (SiO2) and double strands of deoxyribonucleic acid (DNA).[ 15 ] The ribbons are linked via MnO4(OH2)2 octahedra and the free threads of helices are occupied with alkali‐metal ions (Li or Na) with an irregular surrounding of oxygen (Figure 1c and Figure S5, Supporting Information).[ 15 ] Notably, the inner walls of the helical channels are filled with crystal water, which has the capability to enhance the diffusion of ions and electrolytes in OER (Figure 1c).[ 14 , 15 ]
Figure 1.

Structural chemistry of helical manganese BPOs. a) Rietveld refinement of high‐resolution X‐ray diffraction of the LiMnBPO, crystallizing in the hexagonal system, P6522 (space group 179) with unit cell parameters a = 9.5892(1) Å, c = 15.8525(3) Å, V = 1262.39(3) Å3, and Z = 6. b) Representative hexagonal pyramidal single crystal of helical LiMnBPO (Figure S3, Supporting Information). c) Crystal structure of LiMnBPO that is comprised of 1D infinite loop‐branched BPO helices ∞ 1[BP2O8]3− built of alternate BO4 (blue) and PO4 (purple) tetrahedra and is further interconnected through MnO4(OH2)2 coordination (green) octahedra. The free threads of helices are occupied by Li cations (yellow spheres), which are in an irregular surrounding of oxygen atoms (Figures S4 and S5, Supporting Information). The crystal water in the structure is shown as red spheres and located within the helical channels. d) HRTEM image depicting the single crystal with sharp reflection displaying its crystallinity in the SAED pattern (inset). e–g) EDX mapping of hexagonal LiMnBPO (Figure S6, Supporting Information) crystals, where a uniform distribution of Mn (orange), P (yellow), and oxygen (red) was achieved confirming the phase‐purity of the product (elements B and Li are beyond the detection limit of the instrument, and therefore, ICP‐AES was conducted).
The catalytic activity of the materials deposited on fluorine‐doped tin oxide (FTO) (see Figures S14–S16, Supporting Information) was measured in 1 m KOH. Figure 2a shows the cyclic voltammogram (CV) of LiMnBPO/FTO, NaMnBPO/FTO, and bare FTO. Upon applying oxidation potentials, the colorless films turned to light brown, indicating a rapid transformation of the surface structure of the (pre)catalysts (Figure S17, Supporting Information). An overpotential of merely 322 ± 2 mV was attained for LiMnBPO/FTO at a current density of 10 mA cm−2, whereas a slightly higher overpotential (338 ± 2 mV), was observed for NaMnBPO/FTO. Both BPOs exhibited distinct reversible redox features between 1.2 and 1.4 V versus reversible hydrogen electrode (RHE), which could be attributed to the oxidation of low‐valent to higher‐valent Mn sites (Figure 2b). The Tafel slope of LiMnBPO/FTO (41 mV dec−1) was found to be lower than that of NaMnBPO/FTO (47 mV dec−1), confirming the slightly better intrinsically favorable catalytic properties of LiMnBPO (Figure 2c).[ 16 ] The electrochemical double‐layer capacitance (C dl) was 0.079 mF cm−2 for LiMnBPO/FTO, slightly higher than 0.063 mF cm−2 for its Na‐analog (Figure S18, Supporting Information).[ 17 ] To summarize the activity parameters, the measured CVs were also normalized by the C dl, Brunauer–Emmett–Teller (BET) surface area, and mass of the catalyst against the geometrical area (Figure S19, Supporting Information). LiMnBPO is the superior catalyst if the currents are normalized by mass or geometrical surface area but otherwise has identical performance as NaMnBPO if surface roughness (or active surface) is taken into account. Turnover frequencies (TOFs) were obtained by i) normalizing the currents to redox‐active Mn sites and from ii) the amount of Mn loaded on the FTO film (Figures S20 and S21, Supporting Information). The calculated TOFs of BPOs by both methods are superior to any previously reported Mn‐based water oxidation catalysts.[ 18 ] Electrochemical impedance spectroscopy (EIS) revealed favorable charge transfer kinetics of LiMnBPO/FTO compared to NaMnBPO/FTO (for a detailed discussion on the EIS, see Figures S22–S25, Supporting Information).[ 17 ] Contrary to the deactivation of known Mn‐based catalysts within a few hours of operational conditions, the presented BPOs showed outstanding long‐term stability of 24 h in chronoamperometric (CA) conditions (Figure S26, Supporting Information). In order to have a fair comparison of overpotentials in identical conditions, we additionally synthesized and applied highly active Mn‐based, Fe‐, Co‐, and Ni‐oxide/hydroxide/oxyhydroxides, and noble metal‐based oxides for water oxidation (see Figure 2d, synthetic part and Figures S27–S34, Table S2, Supporting Information). Notably, the catalytic performance, TOF, and the stability of BPOs at 10 mA cm−2 are still the highest among all the Mn‐only‐based electro(pre)catalysts reported on FTO and carbon‐based supports for water oxidation, as well as comparable to the highly active nonnoble metal electrocatalysts, applicable in alkaline environment reported thus far (Tables S3–S5, Supporting Information).[ 19 ]
Figure 2.
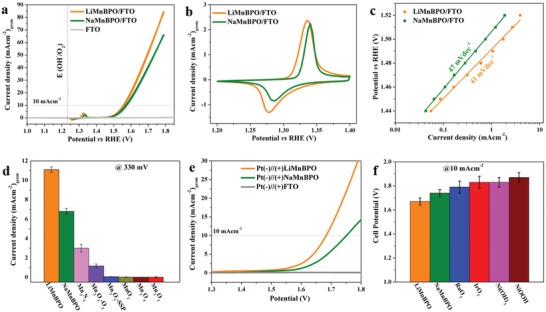
Electrochemical water‐splitting properties of BPOs on FTO electrodes in alkaline electrolyte. a) The geometric area water oxidation activities (CV curves) in aqueous 1 m KOH at a scan rate of 1 mV s−1 with a mass loading of 0.5 mg cm−2. b) Reversible redox couple in the CVs (scan rate 1 mV s−1) indicating oxidation of manganese to higher‐valency, prior to water oxidation. c) The Tafel slopes were obtained by steady‐state measurements applying a stable potential for 600 s between 1.44 and 1.52 V versus RHE. d) The comparison of water oxidation current densities (with error bars) of BPOs with various highly active as‐deposited MnO x catalysts at an overpotential of 330 mV (see Figure S29, Supporting Information). e) Overall water‐splitting performance (linear sweep voltammetry (LSV) curve at a scan rate of 1 mV s−1) of the two‐electrode electrolyzer using BPOs as the anode and Pt/C as the cathode. f) Cell potentials (with error bars) at a current density of 10 mA cm−2–comparison of BPOs, nonnoble‐, and noble‐metal‐based anodes (always Pt/C as the cathode) in the alkaline water electrolyzer.
Various advanced ex situ techniques were employed to identify the origin of the unusual catalytic activity of LiMnBPO/FTO. During cycling, as well as after the 24 h CA experiments, the PXRD pattern did not reveal any significant differences in its crystallinity; however, the ICP‐AES showed substantial loss of Li (70%) along with some amount of B (26%) and P (16%) (Figure S35, Table S6, Supporting Information). SEM and TEM investigations revealed the formation of an amorphous shell on the intact crystalline particles together with a K insertion (Figures S36–S41, Supporting Information).[ 20 ] The FT‐IR spectrum after water oxidation uncovered hydration of the material and is in line with the formation of a birnessite‐type Mn oxide, because of the (obligatory) water‐cation layer between the layered MnO2‐type oxides (Figure S42, Supporting Information). Mn 2p XPS analysis of the LiMnBPO/FTO film after CA revealed a dominant amount of MnIII and MnIV on the surface while the amount of MnII was significantly decreased (Figure S43, Table S7, see Figures S43 and S44 for XPS of Li, P, B, and O, Supporting Information). Overall, the results from ex situ methods after 24 h of CA provided direct evidence for surface‐structural corrosion forming an active amorphous K‐intercalated MnO x phase over crystalline LiMnBPO.
Additionally to the ex situ measurements, we performed ex situ and in situ Raman spectroscopy to gather further insights into the activated structure. The Raman spectra in Figure 3a confirm the surface restructuring of LiMnBPO to a K‐/Li‐birnessite δ‐MnO2 related phase (Figure S45, Note S1, Supporting Information).[ 21 ] The PXRD and SAED data (Figures S35 and S40d, Supporting Information) reveal the absence of a structural periodicity capable to lead to X‐ray or electron diffraction. Therefore, the newly formed phase can best be described as an amorphous phase containing a birnessite‐like short‐range order (called a‐δ‐MnO2 in the following text). The birnessite structure comprises of MnO2 layers accompanied by randomly distributed charge‐neutralizing cations at the interlayer. Due to these cations, some of the Mn sites are reduced from MnIV to MnIII.[ 22 ] It has been shown previously that the transformation of Mn‐based (pre)catalysts to active catalysts can follow various (metastable) intermediate steps.[ 7 , 20 , 23 ] To shed light on the dynamic change in structure, we recorded in situ Raman spectra of LiMnBPO/FTO at a current density of 10 mA cm−2. Between 200 and 700 cm−1, several ν5, ν4, ν3, ν2, and ν1 at 407, 477, 510, 578, and 637 cm−1, respectively (Figure 3b), were observed within a minute, which became more pronounced with increased time intervals (up to 24 h). The ν2 band observed at 578 cm−1 is attributed to the in‐plane Mn—O stretching modes along the octahedral layers in a‐δ‐MnO2, while the bands at 510 (ν3) and 637 cm−1 (ν1) are associated with out‐of‐plane Mn—O vibrations perpendicular to the layers.[ 9a,9b ] Notably, the ν1 band is red‐shifted (653 cm−1) from its positions in MnIV a‐δ‐MnO2 and matched perfectly with K‐birnessite with a mixed MnIII/IV valence state.[ 21 ] The observed ν3 and ν4 bands are only present in intercalated MnIII/IV birnessites. The ν5 band appears as a doublet peak for pure MnIV phases and not as a singlet peak, as observed in here.[ 9 , 21 ] The successive insertion of K was also indicated by the increase in ν2/ν1 ratio, which is in line with the EDX and ICP‐AES results.[ 21 ] The vibration at 948 cm−1 can be assigned to the symmetric stretching of PO4 units, which decreases over time due to the successive loss of P with the growth of the active a‐δ‐MnO2 layer (Figure S46, Supporting Information).[ 24 ] Hence, the Raman spectra show the presence of mixed‐valence MnIII/IV K‐/Li‐intercalated a‐δ‐MnO2 on the surface of LiMnBPO during OER. This behavior stands in contrast to the one of electrochemically synthesized birnessite, where in situ Raman studies revealed the complete oxidation from MnIII/IV at open circuit potential to MnIV under OER conditions.[ 25 ] This demonstrates that the in situ transformation of LiMnBPO is indeed a successful strategy to form a K‐containing a‐δ‐MnO2 phase with abundant MnIII even under highly anodic potentials to efficiently catalyze water oxidation.
Figure 3.
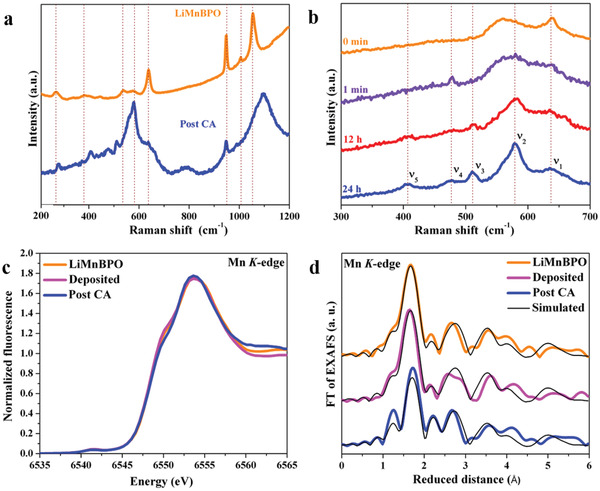
In situ Raman and quasi‐in situ X‐ray absorption spectroscopy of LiMnBPO/FTO under alkaline water oxidation. a) Raman spectra of as‐deposited and post catalytic films suggested complete surface structural transformation under water oxidation potentials. b) Time‐resolved in situ Raman spectra of as‐deposited films at various time intervals evidenced the formation of K‐/Li‐birnessite like MnO x active phase over the surface of the precatalyst. The essential marker bands are represented with ν1, ν2, ν3, ν4, and ν5, respectively. c) XANES and d) Fourier transformed EXAFS measured at the Mn K‐edge of as‐prepared powder, as‐deposited, and the film after water oxidation catalysis further validate the results obtained from Raman spectroscopy. EXAFS simulation parameters and experimental data in k‐space are shown in Tables S9 and S10 and Figure S47 in the Supporting Information.
To investigate the local atomic and electronic structure of LiMnBPO/FTO under water oxidation conditions, quasi‐in situ extended X‐ray absorption fine structure (EXAFS) and near‐edge structure (XANES) measurements were conducted. The XANES spectra in Figure 3c show almost identical edges for both as‐prepared and deposited LiMnBPO, indicating no changes in the starting material during the electrophoretic deposition. The post‐CA film shows only subtle changes in the XANES shape and the average oxidation state of Mn is similar to the precatalytic one (2.1 ± 0.1, Table S8, Supporting Information). The EXAFS spectrum of the as‐prepared LiMnBPO (Figure 3d, the thin black line is a simulation) was successfully simulated based on the crystal structure (Table S9, Supporting Information).
As for the XANES, the EXAFS spectra also indicate only a minimal structural transformation attained for the CA film when compared to the as‐deposited LiMnBPO (Figure 3d). Assuming only surface oxidation of the material and taking into account that X‐ray spectroscopy at the Mn K‐edge is a bulk sensitive method, we simulated the spectrum after 24 h of CA as a linear combination of LiMnBPO powder and short‐range structural δ‐MnO2 (birnessite) motifs. This simulation approach resulted in 0% oxide in the as‐deposited film and 17% after CA (Table S10, Supporting Information). This finding further validates that under water oxidizing potentials, the surface of LiMnBPO was corroded to a‐δ‐MnO2, which may enhance the stabilization of Jahn–Teller‐distorted MnIII sites, preventing the formation of an all‐MnIV state.[ 9b,9g ] The formation of the surface‐induced a‐δ‐MnO2 phase is in agreement with the interpretations deduced from the XPS, HRTEM, SAED, ICP‐AES, EDX mapping, and in situ and ex situ Raman measurements (see the Supporting Information for EXAFS simulation details). As the in situ formed MnO x has an unordered layered structure similar to bulk active CoCat, the electrolyte potentially can diffuse between the layers interacting with more MnIII sites to enhance the activity.[ 26 ] To identify this, we performed a CV after 24 h OER and integrated its MnIII/IV redox peak (Figure S48, Supporting Information). We found that around 50% of the Mn in the newly formed a‐δ‐MnO2 undergoes this redox transition (17% of the LiMnBPO transformed and 8% of the total Mn are redox‐active) exhibiting that is not only the surface Mn sites have electrolyte accessibility.
Motivated by the outstanding performance on FTO, we further deposited and characterized BPOs on 3D porous nickel foam (NF) and evaluated their water oxidation performance in alkaline solution (Figures S49–S51, Supporting Information). The CV curves in Figure 4a displayed the best water oxidation behavior for LiMnBPO/NF with extremely small overpotentials of 228 ± 3 mV at 10 mA cm−2, while the activity of NaMnBPO/NF (262 ± 5 mV) was somewhat lower at the same current density. The bare NF showed limited activity for OER (Figure S52, Supporting Information). Similarly, Tafel slopes, C dl, normalized activities, and EIS were also measured, which indeed substantiated the slightly better intrinsic activity of LiMnBPO/NF over NaMnBPO/NF (Figures S53–S55, Supporting Information). The long‐term stability was carried out for both BPOs at 10 mA cm−2 for 24 h and for LiMnBPO/NF at 20 mA cm−2 over 96 h, showing extraordinary sustainability of the catalysts (Figure S56, Supporting Information). The Mn‐based, Fe‐, Co‐, Ni‐based oxide/hydroxide/oxyhydroxides, and the noble metal‐based electrodes were also prepared on NF and measured in identical conditions to that of the BPOs, and the overpotential of LiMnBPO/NF (228 ± 3 mV) is still the lowest among all measured catalysts, better compared to any Mn‐based catalysts in alkaline, acidic, and neutral conditions and even comparable with the presently most active NiFe‐based (≈200–240 mV) catalysts (for comparison, see Figures S57–S59, Tables S2–S5, Supporting Information).[ 19b ]
Figure 4.
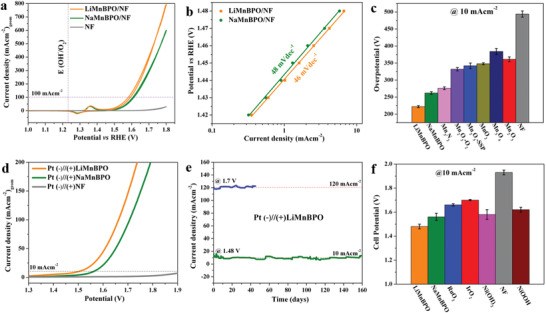
Electrochemical water‐splitting properties of BPOs on NF electrodes in alkaline electrolyte. a) The geometric area water oxidation activities (CV curves) in aqueous 1 m KOH at a scan rate of 1 mV s−1 (mass loading of 1.3 mg cm−2). b) The Tafel slopes obtained by steady‐state measurements applying a stable potential for 600 s between 1.42 and 1.58 V versus RHE. c) The water oxidation overpotentials (with error bars) comparison of a current density of 10 mA cm−2 between BPOs and various highly active as‐deposited MnO x (Figure S54, Supporting Information). d) Overall water‐splitting performance (LSV curve at a scan rate of 1 mV s−1) of the two‐electrode electrolyzer using BPOs as the anode and Pt/C as the cathode. e) Long‐term catalytic stability (CA) test of the alkaline water electrolyzer using LiMnBPO/NF anode and Pt/C cathode at cell potentials 1.48 and 1.7 V. The dashed line is a guide to the eye. f) The cell potentials (with error bars) comparison of a current density of 10 mA cm−2 between BPOs, nonnoble‐, and the noble‐metal‐based anodes (Pt/C as the cathode) in identical conditions of the alkaline water electrolysis.
After the successful demonstration of electrocatalytic water oxidation of BPOs, we constructed a two‐electrode electrolyzer using Pt/C as cathode and BPOs as the anode in the alkaline electrolyte (1 m KOH) using both FTO and NF supports (Figures S60 and S61, Supporting Information). When deposited on FTO, at the current density of 10 mA cm–2, low cell potentials of 1.68 and 1.72 V were attained for LiMnBPO/FTO and NaMnBPO/FTO (Figure 2e), whereas the cell potentials were even dramatically decreased to 1.48 and 1.54 V, respectively, for LiMnBPO/NF and NaMnBPO/NF anodes (Figure 4d). Besides, noble metal‐based and Ni‐based anodes (both on NF and FTO) were also measured in the same conditions but yielded significantly higher cell potentials than the BPOs (Figures 2f and 4f). One of the highlights of the presented work is the durability of BPO electrodes in electrolysis. The LiMnBPO/FTO anode was first subjected to long‐term (CA) electrolysis at 1.68 V, and excellent stability of over 90 h was achieved without deactivation of the catalyst (Figure S60b, Supporting Information). Encouraged by this, we used LiMnBPO/NF anodes for water splitting, where the robust nature of the catalyst was established for 160 days (3850 h) of sustained performance at 10 mA cm−2 and for over 45 days (1100 h) at 120 mA cm−2 (Figure 4d). This long‐term stability is not only the highest reported to date for Mn‐based catalysts but also one of the best reported within the transition metal‐based catalysts in alkaline solution.[ 19b ] Surprisingly, the energetic efficiency of the system even exceeded the 80% level (100% based on the higher heating value) throughout the experiment with a Faradaic efficiency (FE) of ≈100% (Table S11, Supporting Information).
Motivated by the unprecedented stability, we investigated the LiMnBPO/NF anode after 160 days to uncover the active structure of the (pre)catalyst. The SEM, elemental mapping, TEM, HRTEM, SAED, and XPS analysis exhibited a nearly complete transformation of LiMnBPO to a‐δ‐MnO2, which was also supported by EDX and ICP‐AES analysis (Figures S62–S68, Table S12, Supporting Information).[ 9 , 23 ] To show why in situ‐formed catalysts from precatalysts with leaching anions are more active than conventionally synthesized ones, we additionally synthesized a K‐containing amorphous manganese oxide (AMO, K x ‐δ‐MnO2) from a literature reported procedure (Figures S69–S71, Supporting Information), which is structurally similar to the corrosion product that we obtained after the LiMnBPO after 160 days of stability test.[ 8 , 26 ] We studied the electrochemical OER activity of AMO both on FTO and NF in exactly the same conditions as that of LiMnBPO and NaMnBPO (Figure S72, Supporting Information). As anticipated, the attained overpotential comparison showed that the synthetically prepared AMO was almost inactive for water oxidation.
We conclude that during water oxidation a corrosion process is initiated on the surface of LiMnBPO forming a‐δ‐MnO2 structure with the concomitant dissolution of (poly)phosphate and (poly)borate species while the dissolved Li was replaced by K ions from the electrolyte (as predicted by E–pH diagrams).[ 3 , 15 , 27 ] As the CA tests were further prolonged, the borophosphate anions gradually dissolved from the catalyst film. This corrosion process progresses deep inside the core of the particle creating voids that increase the porosity of the films, which leads to the increased density of interfacial Mn atoms in contact with the electrolyte. An important aspect regarding the transformation is that the unit cell of LiMnBPO contains only six Mn atoms in its large volume of 1262.4 Å3 (≈210 Å3 per Mn). Compared to that crystalline birnessite structure has a unit cell volume of ≈51.6 Å3 with one Mn in it. Thus, to form a nonporous densely packed birnessite‐type MnO2 without interlayer cations and water starting from LiMnBPO, the volume of the material would have to shrink by 75%, based on the volume per Mn atom of the unit cells. Due to the rapid transformation and the drastic volume difference, the thermodynamically favored dense crystalline phase is avoided, and instead, a subnanometer porous structure with incorporated electrolyte and a volume closer to the one of LiMnBPO is formed.[ 26 ] Further, a large number of leaching species in (Li/Na)Mn(H2O)2[BP2O8]·H2O compared to remaining Mn lead to the formation of a porous structure. This enables the electrolyte to partly penetrate the catalyst and therefore enables more Mn sites to participate in the catalytic process. Additionally, the large distance in the precatalyst structure between the Mn atoms (6–7 Å) compared to around 2.82 Å in the final structure could be decisive to prevent the formation of ordered layered Mn structures with large domains and could favor the formation of small domains that stack in an unordered way and contain many edge sites. Thus, the finally formed active a‐δ‐MnO2 comprises of structural porosity with an enhanced electrochemical surface area leading to electrolyte permeability and bulk‐activity, fast‐redox switchability, disordered structure, small domains that lead to more edge sites containing the mixed‐valence of Mn (MnIII and MnIV) with a large number of μ‐oxo bridged structures, and abundant water/proton exchange sites to facilitate O2 evolution. This observation is consistent with the proposed OER reaction mechanisms for manganese oxide catalysts (Scheme S1, Supporting Information).[ 28 ]
The anodic chemoselective oxidation of organic substrates for the production of value‐added chemicals (electrosynthesis) is a challenging approach that bears higher economic value than the production of O2.[ 10 , 11 ] In this respect, the oxidation of organic compounds, 5‐hydroxymethylfurfural (HMF), furan‐2‐carboxaldehyde, and acetaldehyde (CH3CHO) to value‐added products through an electrochemical route is environmentally very appealing.[ 11b ] One of the HMF oxidation products, 2,5‐furandicarboxylic acid (FDCA) is an essential precursor for manufacturing polymers, such as polyethylene 2,5‐furandicarboxylate (PEF) and polyethylene terephthalate.[ 11 , 29 ] Further, FDCA can also be used as a surrogate for terephthalic acid for an extensively used constituent in various polyesters.[ 11b ] Similarly, furan‐2‐carboxylic acid (2‐furoic acid), the oxidation product of furan‐2‐carboxaldehyde is widely industrially applied in food products as a preservative acting as bactericide and fungicide as well as a flavoring agent.[ 30 ] In addition to this, it finds applications in biomedical research, optic technology, and nylon preparation.[ 31 ] Finally, acetic acid (CH3COOH), the oxidation product of CH3CHO, has not only been used as a high boiling polar solvent but also in standard organic synthesis.
We first evaluated the catalytic performance for HMF oxidation using a three‐electrode set‐up, where LiMnBPO/NF served as a working electrode in 1 m KOH (Note S2, Supporting Information). A sharp redox peak prior to the water oxidation peak at 1.35 V versus RHE indicated the oxidation of MnIII/MnIV followed by a catalytic wave to further oxidize HMF, and without HMF, only the oxidation of catalysts was observed (Figure 5b). Taking this into account, the electrocatalytic oxidation of organic substrate was performed in a two‐electrode set up (Figure 5a) using LiMnBPO/NF anode at a constant potential in HMF‐added KOH electrolyte. The conversion of organic products was closely monitored by tracing the proton resonance signals from the organic substrates, while the progress of the electrooxidation could be followed by monitoring the gradual change in chemical shift values with time by 1H NMR spectroscopy (Figure 5c). A new peak corresponding to two chemically equivalent ring protons of FDCA appeared at 6.8 ppm after 30 min of bulk electrolysis and complete conversion of HMF to FDCA was observed within 2.5 h when 90 C current was passed through the solution (Figure 5c and Note S2, Scheme S2, Supporting Information). The relative intensity of the proton resonances in 1H NMR allowed us to quantify the FDCA (and HMF) throughout the oxidation reaction and monitor the chemical conversion. Furthermore, the concentration of FDCA formed and HMF consumed during the reaction was measured with time to understand the reaction kinetics, and the time profile with the concentration of FDCA indicated the formation of FDCA followed a pseudo‐first‐order reaction kinetics with an observed rate constant (k obs) value of 9.23 × 10−3 s−1 (Figure 5d). Remarkably, the calculated FE was 98 ± 1%, considering a theoretical six‐electron chemical oxidation of HMF to FDCA, and comparable to a previously reported NiB x /NF electrode.[ 10b ] Moreover, under identical electrochemical conditions, the oxidation of furan‐2‐carboxaldehyde generated furan‐2‐carboxylic acid with high conversion of 86% and FE of 52 ± 1% (Note S2, Scheme S3, Figure S73, Supporting Information). In addition, the oxidation of acetaldehyde produces acetic acid (66%) with high FE (≈84 ± 2%) (Figure S74, Scheme S4, Supporting Information). As no Mn‐based electrode has ever been coupled for highly efficient electrooxidation of organics and OER, this study showcases the opportunity to build robust and cheap Mn‐based electrochemical devices to produce higher commercial value chemicals than conventional fuels.
Figure 5.
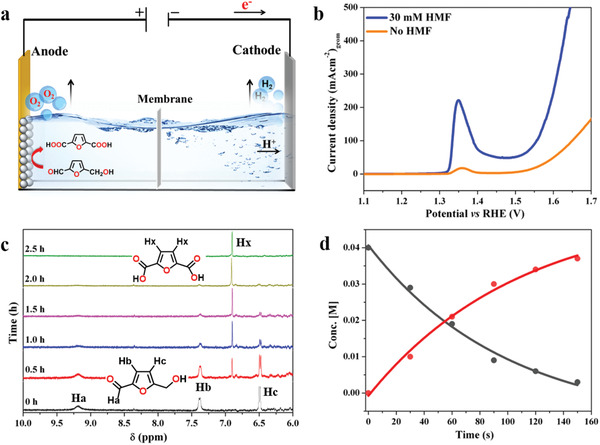
Electrocatalytic oxygenation reaction of HMF to FDCA. a) Fabrication of two‐electrode cell for electrocatalytic oxygenation reaction. b) LSV curves of LiMnBPO/NF in aqueous 1 m KOH solution without HMF (orange) and with 30 × 10−3 m HMF (blue). c) 1H NMR spectra of the reaction solution under electrolysis. The black spectrum (0 h) at the bottom of the panel represents the solution before electrolysis displaying three resonance signals for the HMF while the green spectrum (2.5 h) at the top of the panel consisted of only one proton resonance at 6.8 ppm demonstrating the complete conversion of HMF to FDCA. d) Plot of concentration of HMF and FDCA versus time (in CA conditions). The concentration of the HMF and FDCA was determined from the relative intensities of the proton resonances of HMF and FDCA in 1H NMR. Concentration of HMF (black circles) slowly decreasing, while FDCA (red circles) increasing with time. The fitting of the data points gives rise to a single exponential equation (y = y 0 + A 0 exp(k obs t) and R 2 = 0.99; k obs is the observed rate constant) indicating the electrooxidation, and FDCA formation follows a pseudo‐first reaction kinetics.
In summary, we have successfully addressed the research questions (i)–(v) as mentioned above in the introduction. With respect to question (i), we presented structurally versatile and scalable alkali‐metal manganese BPOs as a new class of anodic precursor materials suitable for alkaline water oxidation, yielding the best catalytic performance compared to any other known Mn‐based catalysts reported to‐date. To answer the questions (ii), (iii), and (iv), we applied a combination of in situ Raman and quasi‐in situ X‐ray absorption (XAS) as well as ex situ methods; and uncovered unequivocally the active catalytic structure, the role of anionic partial structure (BPO) on the performance as well as the electrochemical properties of the evolved Mn‐catalyst. Further, the active Mn‐catalyst exhibited robust and unceasing durability (>5 months), which is unprecedented with respect to any Mn‐based catalysts in alkaline OER, and even comparable to the best active NiFe‐based catalysts reported so far, thereby indisputably addressing the question (v). Besides effective water oxidation, for the first time, we demonstrated that a Mn‐based (pre)catalyst can also be combined with electrochemical selective oxygenation of organic substrates and OER under alkaline conditions.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
C.W., B.C., and J.N.H. contributed equally to this work. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2008/1—390540038. The authors also gratefully acknowledge the financial support of the Bundesministerium für Bildung und Forschung (BMBF cluster project MANGAN; BMBF project Operando‐XAS). The authors thank the Helmholtz‐Zentrum Berlin (HZB) for synchrotron beamtime allocation at KMC‐3/BESSY (Berlin‐Adlershof) and Dr. Ivo Zizak as well as other HZB staff for experimental support. The authors are indebted to Dr. Vitaly Gutkin (HU Jerusalem, Israel) for XPS analysis, Dr. Stefan Hoffmann (TU‐Berlin) for Rietveld measurements and Rodrigo Beltrán‐Suito for elemental mapping.
Open access funding enabled and organized by Projekt DEAL.
Menezes P. W., Walter C., Chakraborty B., Hausmann J. N., Zaharieva I., Frick A., von E. Hauff, Dau H., Driess M., Combination of Highly Efficient Electrocatalytic Water Oxidation with Selective Oxygenation of Organic Substrates using Manganese Borophosphates. Adv. Mater. 2021, 33, 2004098. 10.1002/adma.202004098
Dedicated to Professor Rüdiger Kniep
Contributor Information
Prashanth W. Menezes, Email: prashanth.menezes@mailbox.tu-berlin.de.
Ivelina Zaharieva, Email: ivelina.zaharieva@fu-berlin.de.
Matthias Driess, Email: matthias.driess@tu-berlin.de.
References
- 1.a) Cook T. R., Dogutan D. K., Reece S. Y., Surendranath Y., Teets T. S., Nocera D. G., Chem. Rev. 2010, 110, 6474; [DOI] [PubMed] [Google Scholar]; b) McKone J. R., Lewis N. S., Gray H. B., Chem. Mater. 2014, 26, 407. [Google Scholar]
- 2.a) Boettcher S. W., Nat. Catal. 2018, 1, 814; [Google Scholar]; b) Song F., Bai L., Moysiadou A., Lee S., Hu C., Liardet L., Hu X., J. Am. Chem. Soc. 2018, 140, 7748; [DOI] [PubMed] [Google Scholar]; c) Panda C., Menezes P. W., Driess M., Angew. Chem., Int. Ed. 2018, 57, 11130; [DOI] [PubMed] [Google Scholar]; d) Wei C., Rao R. R., Peng J., Huang B., Stephens I. E. L., Risch M., Xu Z. J., Shao‐Horn Y., Adv. Mater. 2019, 31, 1806296; [DOI] [PubMed] [Google Scholar]; e) Dickens C. F., Kirk C., Norskov J. K., J. Phys. Chem. C 2019, 123, 18960; [Google Scholar]; f) Menezes P. W., Panda C., Loos S., Bunschei‐Bruns F., Walter C., Schwarze M., Deng X. H., Dau H., Driess M., Energy Environ. Sci. 2018, 11, 1287. [Google Scholar]
- 3.a) Hong W. T., Risch M., Stoerzinger K. A., Grimaud A., Suntivich J., Shao‐Horn Y., Energy Environ. Sci. 2015, 8, 1404; [Google Scholar]; b) Menezes P. W., Panda C., Walter C., Schwarze M., Driess M., Adv. Funct. Mater. 2019, 29, 1808632; [Google Scholar]; c) Panda C., Menezes P. W., Yao S. L., Schmidt J., Walter C., Hausmann J. N., Driess M., J. Am. Chem. Soc. 2019, 141, 13306; [DOI] [PubMed] [Google Scholar]; d) Han H., Choi H., Mhin S., Hong Y. R., Kim K. M., Kwon J., Ali G., Chung K. Y., Je M., Umh H. N., Lim D. H., Davey K., Qiao S. Z., Paik U., Song T., Energy Environ. Sci. 2019, 12, 2443. [Google Scholar]
- 4. Dau H., Limberg C., Reier T., Risch M., Roggan S., Strasser P., ChemCatChem 2010, 2, 724. [Google Scholar]
- 5. Umena Y., Kawakami K., Shen J. R., Kamiya N., Nature 2011, 473, 55. [DOI] [PubMed] [Google Scholar]
- 6.a) Wiechen M., Berends H. M., Kurz P., Dalton Trans. 2012, 41, 21; [DOI] [PubMed] [Google Scholar]; b) Walter C., Menezes P. W., Loos S., Dau H., Driess M., ChemSusChem 2018, 11, 2554; [DOI] [PubMed] [Google Scholar]; c) Menezes P. W., Indra A., Littlewood P., Schwarze M., Gobel C., Schomacker R., Driess M., ChemSusChem 2014, 7, 2202; [DOI] [PubMed] [Google Scholar]; d) Melder J., Mebs S., Heizmann P. A., Lang R., Dau H., Kurz P., J. Mater. Chem. A 2019, 7, 25333. [Google Scholar]
- 7.a) Indra A., Menezes P. W., Zaharieva I., Baktash E., Pfrommer J., Schwarze M., Dau H., Driess M., Angew. Chem., Int. Ed. 2013, 52, 13206; [DOI] [PubMed] [Google Scholar]; b) Smith P. F., Deibert B. J., Kaushik S., Gardner G., Hwang S. J., Wang H., Al‐Sharab J. F., Garfunkel E., Fabris L., Li J., Dismukes G. C., ACS Catal. 2016, 6, 2089; [Google Scholar]; c) Ramirez A., Hillebrand P., Stellmach D., May M. M., Bogdanoff P., Fiechter S., J. Phys. Chem. C 2014, 118, 14073; [Google Scholar]; d) Kang Q., Vernisse L., Remsing R. C., Thenuwara A. C., Shumlas S. L., McKendry I. G., Klein M. L., Borguet E., Zdilla M. J., Strongin D. R., J. Am. Chem. Soc. 2017, 139, 1863; [DOI] [PubMed] [Google Scholar]; e) Zhang B., Chen H., Daniel Q., Philippe B., Yu F., Valvo M., Li Y., Ambre R. B., Zhang P., Li F., Rensmo H., Sun L., ACS Catal. 2017, 7, 6311; [Google Scholar]; f) Antoni H., Morales D. M., Fu Q., Chen Y. T., Masa J., Schuhmann W., Muhler M., ACS Omega 2018, 3, 11216; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Park S., Lee Y. H., Choi S., Seo H., Lee M. Y., Balamurugan M., Nam K. T., Energy Environ. Sci. 2020, 13, 2310. [Google Scholar]
- 8.a) Takashima T., Hashimoto K., Nakamura R., J. Am. Chem. Soc. 2012, 134, 1519; [DOI] [PubMed] [Google Scholar]; b) Li A., Ooka H., Bonnet N., Hayashi T., Sun Y., Jiang Q., Li C., Han H., Nakamura R., Angew. Chem., Int. Ed. 2019, 58, 5054; [DOI] [PubMed] [Google Scholar]; c) Guan J., Duan Z., Zhang F., Kelly S. D., Si R., Dupuis M., Huang Q., Chen J. Q., Tang C., Li C., Nat. Catal. 2018, 1, 870; [Google Scholar]; d) Meng Y. T., Song W. Q., Huang H., Ren Z., Chen S. Y., Suib S. L., J. Am. Chem. Soc. 2014, 136, 11452; [DOI] [PubMed] [Google Scholar]; e) Indra A., Menezes P. W., Schuster F., Driess M., J. Photochem. Photobiol., B 2015, 152, 156; [DOI] [PubMed] [Google Scholar]; f) Antoni H., Morales D. M., Bitzer J., Fu Q., Chen Y. T., Masa J., Kleist W., Schuhrnann W., Muhler M., J. Catal. 2019, 374, 335; [Google Scholar]; g) Huynh M., Bediako D. K., Liu Y., Nocera D. G., J. Phys. Chem. C 2014, 118, 17142. [Google Scholar]
- 9.a) Chan Z. M., Kitchaev D. A., Weker J. N., Schnedermann C., Lim K., Ceder G., Tumas W., Toney M. F., Nocera D. G., Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E5261; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Abrashev M. V., Chernev P., Kubella P., Mohammadi M. R., Pasquini C., Dau H., Zaharieva I., J. Mater. Chem. A 2019, 7, 17022; [Google Scholar]; c) Frey C. E., Kwok F., Gonzales‐Flores D., Ohms J., Cooley K. A., Dau H., Zaharieva I., Walter T. N., Simchi H., Mohney S. E., Kurz P., Sustainable Energy Fuels 2017, 1, 1162; [Google Scholar]; d) Zhou L., Shinde A., Montoya J. H., Singh A., Gul S., Yano J., Ye Y., Crumlin E. J., Richter M. H., Cooper J. K., Stein H. S., Haber J. A., Persson K. A., Gregoire J. M., ACS Catal. 2018, 8, 10938; [Google Scholar]; e) Gorlin Y., Lassalle‐Kaiser B., Benck J. D., Gul S., Webb S. M., Yachandra V. K., Yano J., Jaramillo T. F., J. Am. Chem. Soc. 2013, 135, 8525; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Tesch M. F., Bonke S. A., Jones T. E., Shaker M. N., Xiao J., Skorupska K., Mom R., Melder J., Kurz P., Knop‐Gericke A., Schloegl R., Hocking R. K., Simonov A. N., Angew. Chem., Int. Ed. 2019, 58, 3426; [DOI] [PubMed] [Google Scholar]; g) Zaharieva I., Gonzalez‐Flores D., Asfari B., Pasquini C., Mohammadi M. R., Klingan K., Zizak I., Loos S., Chernev P., Dau H., Energy Environ. Sci. 2016, 9, 2433; [Google Scholar]; h) Rabe M., Toparli C., Chen Y. H., Kasian O., Mayrhofer K. J. J., Erbe A., Phys. Chem. Chem. Phys. 2019, 21, 10457; [DOI] [PubMed] [Google Scholar]; i) Speck F. D., Santori P. G., Jaouen F., Cherevko S., J. Phys. Chem. C 2019, 123, 25267; [Google Scholar]; j) Xi L., Schwanke C., Xiao J., Abdi F. F., Zaharieva I., Lange K. M., J. Phys. Chem. C 2017, 121, 12003; [Google Scholar]; k) Risch M., Stoerzinger K. A., Han B. H., Regier T. Z., Peak D., Sayed S. Y., Wei C., Xu Z. C., Shao‐Horn Y., J. Phys. Chem. C 2017, 121, 17682. [Google Scholar]
- 10.a) Sherbo R. S., Delima R. S., Chiykowski V. A., MacLeod B. P., Berlinguette C. P., Nat. Catal. 2018, 1, 501; [Google Scholar]; b) Zhang P. L., Sheng X., Chen X. Y., Fang Z. Y., Jiang J., Wang M., Li F. S., Fan L. Z., Ren Y. S., Zhang B. B., Timmer B. J. J., Ahlquist M. S. G., Sun L. C., Angew. Chem., Int. Ed. 2019, 58, 9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Barwe S., Weidner J., Cychy S., Morales D. M., Dieckhofer S., Hiltrop D., Masa J., Muhler M., Schuhmann W., Angew. Chem., Int. Ed. 2018, 57, 11460; [DOI] [PubMed] [Google Scholar]; b) Liu W. J., Dang L. N., Xu Z. R., Yu H. Q., Jin S., Huber G. W., ACS Catal. 2018, 8, 5533; [Google Scholar]; c) van Putten R.‐J., van der Waal J. C., de Jong E., Rasrendra C. B., Heeres H. J., de Vries J. G., Chem. Rev. 2013, 113, 1499. [DOI] [PubMed] [Google Scholar]
- 12.a) Ewald B., Huang Y. X., Kniep R., Z. Anorg. Allg. Chem. 2007, 633, 1517; [Google Scholar]; b) Ewald B., Prots Y., Menezes P., Natarajan S., Zhang H., Kniep R., Inorg. Chem. 2005, 44, 6431; [DOI] [PubMed] [Google Scholar]; c) Jeazet H. B. T., Menezes P. W., Hoffmann S., Prots Y., Kniep R., Z. Kristallogr. – New Cryst. Struct. 2006, 221, 431. [Google Scholar]
- 13. Hautier G., Jain A., Chen H., Moore C., Ong S. P., Ceder G., J. Mater. Chem. A 2011, 21, 17147. [Google Scholar]
- 14.a) Kniep R., Will H. G., Boy I., Rohr C., Angew. Chem., Int. Ed. 1997, 36, 1013; [Google Scholar]; b) Menezes P. W., Hoffmann S., Prots Y., Kniep R., Z. Kristallogr. – New Cryst. Struct. 2008, 223, 9; [Google Scholar]; c) Menezes P. W., Hoffmann S., Prots Y., Kniep R., Z. Anorg. Allg. Chem. 2009, 635, 614. [Google Scholar]
- 15. Menezes P. W., Indra A., Zaharieva I., Walter C., Loos S., Hoffmann S., Schlögl R., Dau H., Driess M., Energy Environ. Sci. 2019, 12, 988. [Google Scholar]
- 16. Suen N.‐T., Hung S.‐F., Quan Q., Zhang N., Xu Y.‐J., Chen H. M., Chem. Soc. Rev. 2017, 46, 337. [DOI] [PubMed] [Google Scholar]
- 17. McCrory C. C. L., Jung S. H., Peters J. C., Jaramillo T. F., J. Am. Chem. Soc. 2013, 135, 16977. [DOI] [PubMed] [Google Scholar]
- 18. Najafpour M. M., Renger G., Holynska M., Moghaddam A. N., Aro E.‐M., Carpentier R., Nishihara H., Eaton‐Rye J. J., Shen J.‐R., Allakhverdiev S. I., Chem. Rev. 2016, 116, 2886. [DOI] [PubMed] [Google Scholar]
- 19.a) Jung S., McCrory C. C. L., Ferrer I. M., Peters J. C., Jaramillo T. F., J. Mater. Chem. A 2016, 4, 3068; [Google Scholar]; b) Hu C., Zhang L., Gong J., Energy Environ. Sci. 2019, 12, 2620. [Google Scholar]
- 20. Menezes P. W., Walter C., Hausmann J. N., Beltran‐Suito R., Schlesiger C., Praetz S., Verchenko Y., Shevelkov A. V., Driess M., Angew. Chem., Int. Ed. 2019, 58, 16569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen D. C., Ding D., Li X. X., Waller G. H., Xiong X. H., El‐Sayed M. A., Liu M. L., Chem. Mater. 2015, 27, 6608. [Google Scholar]
- 22.a) Peng H., McKendry I. G., Ding R., Thenuwara A. C., Kang Q., Shumlas S. L., Strongin D. R., Zdilla M. J., Perdew J. P., Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9523; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y. F., Liu Z. P., J. Am. Chem. Soc. 2018, 140, 1783. [DOI] [PubMed] [Google Scholar]
- 23. Huynh M., Shi C., Billinge S. J. L., Nocera D. G., J. Am. Chem. Soc. 2015, 137, 14887. [DOI] [PubMed] [Google Scholar]
- 24. Frost R. L., Xi Y., Scholz R., Lopez A., Belotti F. M., Vib. Spectrosc. 2013, 66, 69. [Google Scholar]
- 25. An H. Y., Chen Z., Yang J. X., Feng Z. C., Wang X. L., Fan F. T., Li C., J. Catal. 2018, 367, 53. [Google Scholar]
- 26. Hausmann J. N., Mebs S., Laun K., Zebger I., Dau H., Menezes P. W., Driess M., Energy Environ. Sci. 2020, 13, 3607. [Google Scholar]
- 27.a) Indra A., Menezes P. W., Zaharieva I., Dau H., Driess M., J. Mater. Chem. A 2020, 8, 2637; [Google Scholar]; b) Li W., Xiong D. H., Gao X. F., Liu L. F., Chem. Commun. 2019, 55, 8744. [DOI] [PubMed] [Google Scholar]
- 28.a) Huynh M., Bediako D. K., Nocera D. G., J. Am. Chem. Soc. 2014, 136, 6002; [DOI] [PubMed] [Google Scholar]; b) Tian L., Zhai X. H., Wang X., Li J., Li Z., J. Mater. Chem. A 2020, 8, 14400. [Google Scholar]
- 29. Jiang N., You B., Boonstra R., Rodriguez I. M. T., Sun Y., ACS Energy Lett. 2016, 1, 386. [Google Scholar]
- 30. Varelis P., Hucker B., Food Chem. 2011, 126, 1512. [Google Scholar]
- 31.a) Uma B., Murugesan K. S., Krishnan S., Das S. J., Boaz B. M., Optik 2013, 124, 2754; [Google Scholar]; b) Mariscal R., Maireles‐Torres P., Ojeda M., Sadaba I., Granados M. L., Energy Environ. Sci. 2016, 9, 1144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information