

Abstract
Volumetric printing (VP) is a light‐mediated technique enabling printing of complex, low‐defect 3D objects within seconds, overcoming major drawbacks of layer‐by‐layer additive manufacturing. An optimized photoresin is presented for VP in the presence of cells (volumetric bioprinting) based on fast thiol–ene step‐growth photoclick crosslinking. Gelatin‐norbornene (Gel‐NB) photoresin shows superior performance, both in physicochemical and biocompatibility aspects, compared to (meth‐)acryloyl resins. The extremely efficient thiol–norbornene reaction produces the fastest VP reported to date (≈10 s), with significantly lower polymer content, degree of substitution (DS), and radical species, making it more suitable for cell encapsulation. This approach enables the generation of cellular free‐form constructs with excellent cell viability (≈100%) and tissue maturation potential, demonstrated by development of contractile myotubes. Varying the DS, polymer content, thiol–ene ratio, and thiolated crosslinker allows fine‐tuning of mechanical properties over a broad stiffness range (≈40 Pa to ≈15 kPa). These properties are achieved through fast and scalable methods for producing Gel‐NB with inexpensive, off‐the‐shelf reagents that can help establish it as the gold standard for light‐mediated biofabrication techniques. With potential applications from high‐throughput bioprinting of tissue models to soft robotics and regenerative medicine, this work paves the way for exploitation of VPs unprecedented capabilities.
Keywords: bioprinting, gelatin, photoclick, thiol–ene, volumetric
Volumetric printing enables the generation of centimeter‐sized free‐form 3D objects within seconds. Optimized photoclick‐based photoresins show ideal biophysical and photochemical properties resulting in fast and biocompatible volumetric bioprinting. Thiol–norbornene photo‐crosslinking outperforms chain‐growth systems permitting significant reduction of radical generation and polymer concentration. This simple and scalable synthesis method opens new avenues for widespread use of photoclick resins in light‐mediated biofabrication.
1. Introduction
the last two decades, photochemical reactions have become increasingly used in the field of tissue engineering and biofabrication.[ 1 ] Light as a remote trigger has enabled fine spatiotemporal control over biophysical[ 2 , 3 ] and biochemical properties[ 4 , 5 , 6 ] of photoactivated materials. Several 3D bioprinting techniques ranging from deposition‐based (i.e., extrusion and droplet) to vat‐polymerization‐based (i.e., Digital Light Processing (DLP) and stereolithography (SL)) have exploited the versatile power of light to develop complex biomimetic cellular constructs within a broad range of size and resolution.[ 1 , 7 , 8 ] Despite the great advances made with these techniques, however, there are still major limitations associated with the lengthy layer‐by‐layer manufacturing. First, the long printing process required to generate centimeter‐scale constructs can impair scalability and clinical translation. The extended biofabrication time potentially compromises cell viability in addition to the intrinsic stress and cell death caused by the printing method itself (i.e., shear‐stress during extrusion printing).[ 9 , 10 , 11 , 12 , 13 ] Also, printing in a layer‐by‐layer fashion is accompanied by structural limitations, such as the difficulties in generating unsupported overhangs and the presence of layer‐layer and line‐line defects.
Recently, there has been growing interest in a novel approach: volumetric printing (VP, also known as tomographic volumetric additive manufacturing or computed axial lithography), which promises to overcome current limitations of printing with cell‐laden materials (bioprinting) and generate low‐defect, free‐form, complex large grafts within seconds.[ 14 , 15 , 16 , 17 ] This powerful tool is based on the projection of dynamically evolving light patterns onto a rotating photosensitive resin (photoresin) container. When the resulting 3D light‐dose accumulation locally exceeds the photo‐crosslinking threshold, the desired solid object is generated. Recent reports have defined some of the key physicochemical features that theoretically define an optimal VP photoresin.[ 14 , 15 ] Viscosity emerged as a critical parameter affecting the printing resolution. Viscosity values >10 Pa s are necessary to effectively counterbalance the sedimentation of the solidified features during the printing process as demonstrated with the use of synthetic acrylate resins (i.e., SR399, polyethylene glycol diacrylate (PEG‐DA)).[ 14 , 15 ] In addition, such high viscosity limits radicals and molecular diffusion‐induced blurring.[ 14 ] It follows that an ideal VP photoresin will benefit from reversible gelation properties. The first few reports on this method have indeed profited from the thermal gelation of photosensitive gelatin.[ 14 , 16 ] In contrast to other vat‐based techniques such as DLP and SLA, photoresin transparency is another essential feature, since light patterns need to pass through the entire depth of the rotating build volume. Because the vast majority of synthetic and naturally derived polymers do not absorb at the wavelength commonly associated with light‐based printing (365–405 nm), photoresin transparency is determined by the photoinitiator (PI) concentration. However, when designing an ideal volumetric bioprinting (VBP) photoresin, physical and optical properties cannot be separated from its photochemical performances. Photochemical reaction kinetics and mechanisms play a pivotal role in determining printing time, resolution, PI and polymer concentration. In addition, for biological applications, the interplay between physical and chemical properties should also guarantee high cell viability and the generation of a cell‐permissive and cell‐interactive matrix. To date, these requirements have been only partially fulfilled with the chain‐growth based crosslinking of highly concentrated (10%) gelatin methacryloyl (Gel‐MA).[ 16 ] More efficient chemical strategies, such as thiol–ene photoclick reactions, have gained interest in recent years,[ 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 ] and represent a viable way to overcome current limitations and satisfy VBP photoresin requirements. Thiol–ene step‐growth crosslinking has a number of advantages compared to Gel‐MA chain‐growth polymerization.[ 1 , 2 , 22 , 27 ] Due to their exceptionally rapid reaction kinetic, thiol–ene photoresins require a significant lower generation of potentially harmful radicals compared to chain‐growth mechanisms (i.e., Gel‐MA) (Figure S1, Supporting Information).[ 26 , 28 ] This benefit is accompanied by the possibility to use a significantly lower polymer content, which results in a more permissive cellular matrix.[ 29 , 30 , 31 ] Furthermore, step‐growth crosslinking efficiency makes it possible to significantly reduce the degree of substitution (DS), therefore largely retaining native biopolymer bioactive and physical properties.[ 32 , 33 ] In addition, oxygen insensitivity and selective reaction between the two complementary groups (thiol and –ene functionalities) pose a superior control over crosslinking reactions and lead to a homogeneous network formation with reduced shrinkage and mechanical stress, thereby overcoming another Gel‐MA drawback that results from the generation of non‐biodegradable kinetic chains and network defects.[ 28 , 33 , 34 ] Finally, Gel‐MA has been the gold‐standard for the past two decades in light‐based biofabrication and its success, besides the desirable biophysical properties of gelatin, can be attributed to its ease of production and cost‐effectiveness.[ 34 ] The straightforward synthesis and material handling made it possible for any lab to synthesize their own Gel‐MA and resulted also in its commercialization. Therefore, for the field to adopt an alternative to Gel‐MA, the photoresin should have the listed properties, but also be simple to produce at a large scale.
In this work we report on a gelatin–norbornene (Gel‐NB)‐based photoresin that fulfills all requirements as an ideal VBP photoresin. Starting from the establishment of a simple and scalable synthesis method, we demonstrate high tunability over rheological properties of Gel‐NB step‐growth based photo‐crosslinkable resin and excellent biocompatibility, to serve as an ideal material for VBP, reporting the fastest centimeter‐scale bioprinting process to date (≈10–11 s).
Among the various synthetic and naturally derived polymers that have been used in light‐mediated biofabrication, we chose gelatin, by far the most widely studied material platform.[ 34 , 35 , 36 ] Obtained via acidic or basic denaturation of collagen, the main component of human extracellular matrix, various gelatin derivatives have been successfully employed as a biocompatible matrix for virtually any cell type, ranging from various primary cells[ 35 , 37 ] to cancer and stem cells.[ 38 , 39 , 40 ] In addition to the retention of desirable collagen properties such as biocompatibility, enzyme‐mediated biodegradability, and integrin binding sites (RGD), gelatin offers another key feature for VBP, represented by its reversible thermal gelation. By printing within a physically gelled bioresin, the printing resolution is maximized and cell sedimentation eliminated. In this study, gelatin has been functionalized with NB groups to obtain a resin crosslinkable via step‐growth mechanism with optimal performance. In contrast to other –ene functionalities (i.e., vinyl sulfones and (meth)acryloyl), NB does not undergo Michael‐type addition with free thiols, therefore limiting undesired side reactions.[ 41 ] Moreover, NB ring‐strain conformation leads to an extremely fast relief upon thiyl radical addition, which outperforms other reported –ene photoreactive moieties and makes it an ideal candidate for our purposes.
2. Results and Discussion
The synthesis of Gel‐NB is performed using carbic anhydride (CA) which, similar to the widely adopted synthesis method of Gel‐MA with methacrylic anhydride,[ 42 ] reacts with free amino groups of the polypeptide chain. Compared to other previously reported Gel‐NB synthesis strategies,[ 28 , 43 , 44 ] the use of CA offers several benefits. Among these, and of major importance for a widespread use of the resulting resin, CA is commercially available, inexpensive, and its reaction can be performed in aqueous solutions, thus avoiding the need for toxic organic solvents and complex multistep synthesis.[ 28 ] The use of an alkaline buffer (carbonate–bicarbonate 0.1 m, pH 9) limits gelatin free amino groups protonation, which inhibits the reaction with CA, and counterbalances the solution acidification that takes place during the formation of NB–dicarboxylic acid upon opening of the cyclic anhydride. Furthermore, the use of sequential pH adjustments and CA loading enables a better control over the reaction conditions and resulted in a 20‐fold reduction of required reagent in addition to a major reduction in reaction time compared to previous reports (from 2–3 days to 1 h) (Figure 1 ).[ 43 , 44 ] In short, to evaluate the most efficient synthesis strategy, we screened three different methods that differed in interval times between sequential CA loading and pH adjustment (Figure 1B). For each method, we also tested different Gel:CA w/w ratios (100:1, 50:1, 10:1) in order to target different DS. We showed that an interval time increase between CA additions, from 30 min (Method 1, M1) to 1 h (Method 2, M2), does not lead to a higher grafting yield (% of NB that has been successfully bounded to gelatin), thus suggesting a fast reagent consumption. In line with this observation, comparable DS and grafting yield can be obtained with an interval time reduced to just 10 min (Method 3, M3) (Figure 1C; Table S1, Supporting Information). To fulfill another desirable requirement, the production of large‐scale batches, we adopted the fast M3 procedure to produce Gel‐NB in synthesis scale up to 50 g. Thus, with a total reaction time of just 1 h, we show that a broad range of DS can be obtained, ranging from ≈0.01 mmol g−1 (≈3%) to ≈0.16 mmol g−1 (≈50%), and in reaction scales up to 50 g (Figure 1D; Table S1, Figure S2, Supporting Information). The high photochemical performances of SH:NB photoclick reactions make it possible to use medium DS (≈50%) for most light‐assisted bioprinting techniques, including high definition two‐photon stereolithography as recently demonstrated by Dobos et al.[ 21 ] Such DS enables the better preservation of gelatin bioactive and mechanical properties by largely leaving RGD motifs unmodified and by limiting the influence that functionalization can have on physical gelation.[ 33 , 45 , 46 , 47 ] The reported inexpensive, accessible to nonexperts and scalable method for synthesizing Gel‐NB with off‐the‐shelf reagents can contribute to its establishment as a gold‐standard in light‐mediated biofabrication techniques and its commercialization. On the other hand, in contrast to Gel‐MA, Gel‐NB based resins require the use of thiolated crosslinkers which can be, for example, represented by thiol‐functionalized biopolymers (i.e., HA‐SH, Gel‐SH) or by the chemically defined and widely adopted bi‐ or multifunctional PEG thiol derivatives. High tunability, well‐established safety profile, and commercial availability in gram scale[ 48 , 49 ] make PEG‐based crosslinkers an attractive solution for standardized and relatively inexpensive Gel‐NB based photoresins. As a rough estimation, by using the example of the VBP process demonstrated later in this work, a Gel‐NB 50 g batch synthesis targeting a DS ≈ 50% allows for about 250 to 500 prints (≈4 mL per print) using a Gel‐NB concentration of 5% or 2.5%, respectively.
Figure 1.
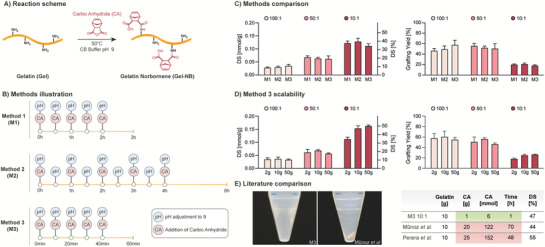
Overview of gelatin‐norbornene (Gel‐NB) synthesis. A) Schematic of the reaction: carbic anhydride (CA) grafting on nonprotonated gelatin‐free amines in pH 9 carbonate–bicarbonate (CB) buffer. B) Illustration of Gel‐NB synthesis methods investigated in this study with varying interval time between sequential addition of CA and pH adjustment. C) Comparison of degree of substitution (DS, left) and norbornene grafting yield (right) obtained with the three different methods in 2 g scale synthesis. The synthesis was performed in 100:1, 50:1, and 10:1 Gel:CA w/w ratio. For DS comparison the right y axis (%) refers only to average values, standard deviations refer to left y axis (mmol g−1). D) Comparison of DS and grafting yield resulting from Gel‐NB synthesis at different reaction scales (2 g, 10 g, 50 g) using the fastest method (M3). The synthesis was performed with different Gel:CA ratio in order to target different DS. For DS comparison the right y axis (%) refers only to average values, standard deviations refer to left y axis (mmol g−1). E) Comparison of Gel‐NB synthesis (≈50% DS) with previous reports using CA.[ 43 , 44 ] Compared to the commonly used protocol developed by Muñoz et al.,[ 43 ] no visible excess of unreacted reagent is obtained upon centrifugation step at pH 7.4 using M3‐based synthesis with 10:1 Gel:CA ratio (left). On the right, main improvements resulting from the use of M3 are highlighted.
We then showed strong control over the reaction's kinetic and mechanical properties obtainable by varying DS, Gel‐NB concentration, SH:NB ratio, and thiolated crosslinker type (Figure 2 ). Photorheology was performed with 0.05% w/v LAP (Figure 2A), which has become the state‐of‐the‐art PI for biofabrication purposes, thanks to its excellent water solubility, molar absorptivity, and better cytocompatibility compared to previous commonly used PIs such as I2959.[ 50 ] Importantly, LAP‐based photoresins are compatible with most light‐mediated bioprinting techniques using 405 nm LED or laser diode light sources. Unless otherwise indicated, photoreology and VP have been performed with a formulation of Gel‐NB and 4‐arm‐PEG‐thiol (PEG4SH) in SH:NB equimolar amount containing 0.05% w/v LAP. As an example, a photoresin with a 1:1 SH:NB molar ratio formulation composed of 2.5% Gel‐NB (DS ≈ 50%) contains around 1.1% PEG4SH, for a total polymer content ≈3.6%. First, Gel‐NB DS showed excellent correlation with hydrogel stiffness (Figure 2B). Absence of hydrogel formation for DS ≈ 3% suggests that a lower DS limit for a successful photo‐crosslinking stands between 3% and 10%. In addition to tuning the mechanical properties based on DS, we tested the influence of Gel‐NB concentration (DS ≈ 50%), showing that a broad range of storage moduli (≈40 Pa to ≈15 kPa) can be attained by changing the Gel‐NB concentration from 1% to 10% (Figure 2C). In addition, the mechanical properties of the homogeneous step‐growth photo‐crosslinked network can be influenced by the SH:NB ratio (Figure 2D). In this respect, while keeping constant the Gel‐NB concentration at 5% (DS ≈ 50%), the use of 5× SH or 5× NB excess led to a less densely crosslinked network and therefore to a final lower storage modulus compared to a resin with 1:1 SH:NB ratio (Figure 2D). Notably, the alteration of the SH:NB ratio resulted in hydrogels with an excess of SH or NB moieties, which are therefore available for further functionalization steps (i.e., via photoclick chemistry or Michael‐type addition). Furthermore, the use of different thiolated crosslinkers can influence the hydrogel properties (Figure 2E). In accordance with what has been recently shown by Van Hoorick et al.,[ 20 ] the use of multifunctional crosslinkers such as PEG4SH resulted in a higher storage modulus due to the grafting of multiple NB moieties in one junction knot. Less densely packed networks are instead formed with bifunctional linkers such as PEG2SH or 3,6‐dioxa‐1,8‐octanedithiol (DODT). Also, differences in the thiolated crosslinker chain length can have an impact on the final hydrogel properties, as has been shown here with the much lower storage modulus of Gel‐NB/DODT formulation compared to Gel‐NB/PEG2SH (Figure 2E). The extremely fast and highly tunable photo‐crosslinking of Gel‐NB represents an attractive advance for several 3D biofabrication techniques. In particular, in this work we report, for the first time, on its application in VP using a high‐performance commercially available stand‐alone tomographic 3D printer (Tomolite, Readily3D SA)[ 51 ] (Figure 3A). To assess the VP performance of the photoresins, we used a built‐in software function called “Dose Test” (see Supplementary Methods, Figures S3–S5, Supporting Information). This function permits the study of photoresin behavior directly with the printer's light source, light path, and settings, therefore overcoming the problem of determining VP printability with measurement systems like photorheology. By projecting light into a nonrotating cuvette filled with resin, a matrix of dots with varying time exposure and light intensity is generated, allowing one to estimate the critical gelation threshold (CGT) for each photoresin formulation. CGT is the critical parameter for VP and can be defined as the minimum required light dose to form a stable gel. Below this threshold, the light absorbed by the photoresin is not sufficient to generate a stable crosslinked network, while above it overexposure occurs. The estimated CGT found with the parametric gelation map of the Dose Test facilitates the subsequent optimization of VP parameters which can slightly differ due to the lensing effect, rotating‐mode, and volumetric absorption. During the printing process, the light‐mediated gelation induces a change in refractive index of the crosslinked object which can be seen with a built‐in camera (Video S1, Supporting Information). This change helps to identify the point in the printing process at which the object is formed and determines, after some reiteration, the optimal light dose. For sake of completeness, the CGTs found in this work are reported as light doses delivered from the printer in areal units (aCGT, mJ cm2−1) and, more accurately for such VP, in terms of volumetric absorbed energy (vCGT, mJ cm3−1) which takes into account build volume diameter and photoresin composition (PI absorption at excitation wavelength and concentration).
Figure 2.
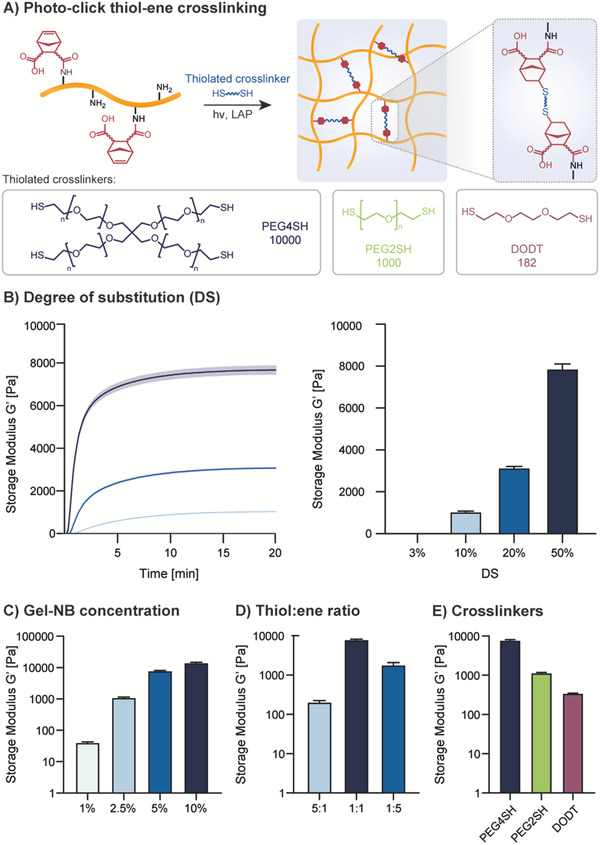
Photorheology characterization of Gel‐NB‐based resin using 0.05% w/v LAP as photoinitiator. Unless otherwise specified, photoresins are composed of 5% Gel‐NB (DS ≈ 50%) and PEG4SH at 1:1 SH:NB molar ratio A) Thiol–ene crosslinking scheme illustration of photoresin composed of Gel‐NB and a thiolated crosslinker. Upon 405 nm excitation of LAP, the generation of radical initiation species leads to step‐growth crosslinking (top). Structures and MW of thiolated crosslinkers used in this study (bottom). B) Investigation of DS influence on final hydrogel mechanical properties. The wide DS range results in tunable hydrogel stiffness. DS of ≈3%, obtained with a Gel:CA ratio of 500:1, is also shown to be not enough to guarantee hydrogel formation. C) Investigation of Gel‐NB concentration influence on final hydrogel mechanical properties. Highest storage modulus is observed for Gel‐NB 10%. A reduction of polymer content is associated with a reduction of the final mechanical properties due to a less densely crosslinked network. D) Influence of SH:NB ratio on final hydrogel mechanical properties. The use of 5× norbornene or 5× thiols results in much weaker gels. E) Influence of different thiolated crosslinker on final hydrogel mechanical properties. The highest storage modulus is observed with PEG4SH, while a drastic reduction is shown with the use of bifunctional crosslinkers. A direct comparison between bifunctional crosslinker with different MW shows that also chain length plays an important role in determining hydrogel stiffness.
Figure 3.
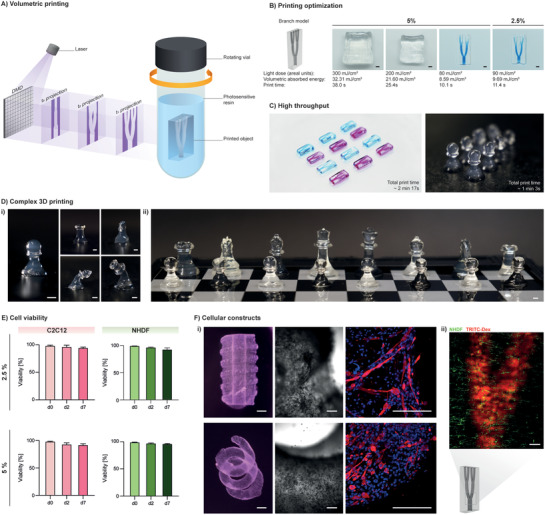
Volumetric printing with Gel‐NB. A) Illustration of volumetric printing principle of operation. A 405 nm laser beam (light purple) is directed toward a digital‐micromirror‐device (DMD), which generates dynamically evolving projection images (dark purple) in synchrony with the rotation of the glass vial containing the photosensitive resin. The desired object is solidified where the local light dose accumulation exceeds the gelation threshold. B) Printing parameter optimization using 5% and 2.5% Gel‐NB/PEG4SH resin (DS ≈ 50%). A branch model perfusable with a high‐MW blue‐dextran solution is obtained with a low light dose, corresponding to extremely fast (≈10–11 s) printing (scale bar: 2 mm). C) Print upscaling with 2.5% Gel‐NB/PEG4SH. The potential of fast printing is shown with the generation of replicas of twelve perfusable branch models (left, alternating perfusion with blue‐dextran and TRITC‐dextran) and eight pawn models (right). D) Printing of various 3D complex objects. i) VP with 2.5% Gel‐NB/PEG4SH. From left to right: pawn, rook (top‐left), knight (top‐right), bishop (bottom‐left), and queen (bottom‐right). Due to the hydrogel softness, tall structures such as the bishop and queen models tend to bend when not submerged in liquid. ii) Printing at higher concentration (5% Gel‐NB/PEG4SH) results in stiffer objects that can easily stand (scale bars: 2 mm). E) High cell viability (>90%) after bioprinting is shown for mouse myoblasts (C2C12) and normal human dermal fibroblasts (NHDF) over 1 week of culture in both 5% and 2.5% Gel‐NB/PEG4SH resin. F) Cellular constructs. i) Bioprinting of C2C12‐laden complex free‐form objects with 2.5% Gel‐NB/PEG4SH resin (left, scale bars: 2 mm). Bright‐field close up on cellular construct after 1 week of culture showing cell spreading and proliferation on the soft matrix (right, scale bars: 200 µm). Immunofluorescence evidence of myotubes differentiation after 3 weeks of culture (Myosin Heavy Chain: red, Nuclei: blue, scale bars: 200 µm). ii) Confocal imaging of branch model bioprinted with NHDF‐laden 2.5% Gel‐NB/PEG4SH photoresin and perfused with TRITC‐dextran (red) after 1 week of culture (scale bar: 500 µm).
We show that, due to optimal physical and photochemical properties, Gel‐NB/PEG4SH photoresins can be used to print perfusable branch models at relatively low light doses (80–90 mJ cm2−1, volumetric absorbed energy: 8.59–9.69 mJ cm3−1), which corresponds to a printing time of only ≈10–11 s, the fastest reported to date, with a writing resolution of ≈200 µm (Figure S6, Supporting Information). Higher light‐doses, and therefore longer printing time, led to overexposure and undesired photo‐crosslinking (Figure 3B). Notably, the excellent NB photochemical properties allowed the developed resin to be printed at much lower Gel‐NB concentrations (2.5%) compared to previous studies operating with 10% Gel‐MA. Generation of low polymer content constructs is of paramount importance for biological applications, in order to enhance nutrient and catabolic waste diffusion, cell potency, proliferation, and migration.[ 29 , 30 , 31 , 52 ] Furthermore, the combination of fast printing and lower polymer concentration significantly improves VP throughput. Taking into account that the printing time for VP is defined by the resin properties and not by the construct size, as with most of the other additive printing strategies, SH‐NB photoclick reaction represents an unprecedented opportunity for upscaling printing of centimeter‐sized complex geometries (Figure 3C). Moreover, in contrast to Gel‐MA,[ 16 ] fast step‐growth crosslinking of Gel‐NB/PEG4SH makes a further postprinting curing step unnecessary, thereby limiting the total photo‐crosslinking time to the few seconds of the printing process. In fact, when printed at the optimal CGT, the SH‐NB based resin reaches ≈35–40% of the fully crosslinked plateau stiffness (Figure S7, Supporting Information) upon a 8–14.5% conversion of NB groups (Figure S8, Supporting Information), which makes the generated object stable enough to be washed from uncrosslinked resin with warm solutions and to be stable under cell culture conditions for at least 3 weeks as shown later in this work. In this way, a wide variety of complex and stable 3D objects can be obtained with 2.5% Gel‐NB/PEG4SH (Figure 3D i; Figure S9, Supporting Information) or 5% Gel‐NB/PEG4SH photoresin (Figure 3D ii).
Low polymer content, low DS, low light exposure, and radical production are among the numerous desirable properties for a cell‐containing photoresin (bioresin). Taking the retention of gelatin's biophysical properties together with its step‐growth processability, Gel‐NB emerges as an ideal material platform for VBP. Interestingly, we also observed that, in contrast to a recent work by Cook et al.,[ 17 ] Gel‐NB/PEG4SH does not require the addition of TEMPO as a radical scavenger, which in turn can have cytotoxic and genotoxic effects.[ 53 , 54 ]
In fact, the addition of a radical scavenger can be adopted to tune the gelation threshold by prolonging the initial induction period (exposure time without gelation). The resulting nonlinear gelation kinetics can help to improve the contrast between crosslinked and uncrosslinked parts during printing, but it does not represent a fundamental requirement for VP which is based on a pure threshold effect. For the generation of cell‐encapsulated constructs, Gel‐NB/PEG4SH photoresin was warmed to 37 °C and mixed with cells. The resulting bioresin was then transferred to the glass vial followed by thermal gelation upon cooling to 4 °C, to ensure high resolution printing and maintenance of homogeneous cell distribution. We showed excellent cell viability (>95%) upon printing and across 7 days of culture (>90%) (Figure 3E; Figure S10, Supporting Information) of encapsulated mouse myoblasts (C2C12) and normal human dermal fibroblasts (NHDF) for both the 2.5% and 5% Gel‐NB conditions tested.
Although it has been observed that, due to the material's softness, some high‐aspect‐ratio designs printed with 2.5% Gel‐NB/PEG4SH are not self‐supporting in air (Figure 3D i, Bishop and Queen models), we show that complex free‐form cell‐laden structures can be bioprinted and easily cultured with good stability in cell culture media (Figure 3F). C2C12 muscle cells proliferated, spread, and differentiated into multinucleated contractile myotubes in the soft crosslinked matrix (Figure 3F i; Video S2, Supporting Information), opening new possibilities for rapid bioprinting of complex models and “living” biohybrid soft robotics.[ 55 ] Also, as a first tissue‐on‐a‐chip proof‐of‐concept, we demonstrated the ability to bioprint a cell‐laden (NHDF) perfusable branch model with mesoscale vasculature lumen size (Figure 3F ii).[ 56 ] Thanks to the high scalability given by the combination of simple Gel‐NB synthesis and fast printing process, VBP holds great promise for high‐throughput generation of complex tissue models such as skeletal muscle.
3. Conclusion
We have developed an optimized material platform (Gel‐NB) for the novel VBP method and a simple and scalable synthesis strategy for its production. We anticipate that these findings will help overcome the limitations of Gel‐MA and open new avenues for a more widespread use of high‐performance Gel‐NB resins in the field of tissue engineering and regenerative medicine. In addition, while pure gelatin‐based resins have been shown to provide a good material platform for VBP, their limited bioactivity can be further tailored by relatively simple biofunctionalization with adhesion sites,[ 57 ] drugs,[ 58 , 59 , 60 ] and growth factors[ 61 ] to target tissue‐specific applications. We foresee that this VBP proof‐of‐concept will stimulate the development of other photoresins based on thermosensitive materials or non‐thermosensitive, but highly viscous polymers modified for thiol–norbornene photoclick chemistry. For example, while maintaining Gel‐NB as starting material, bringing in alternative biodegradable, cell‐interactive thiol donors (i.e., HA‐SH, Gel‐SH, cysteines containing MMP cleavable peptides) will further broaden the VBP resin palette and improve the biological outcome.
4. Experimental Section
All chemicals were purchased from Merck and cell culture reagents from Gibco unless indicated otherwise.
Synthesis of Gel‐NB
Gelatin type A from porcine skin was dissolved at 10% in 0.1 m pH 9 carbonate–bicarbonate buffer at 50 °C. Then, 1/5 of the total cis‐5‐norbornene‐endo‐2,3‐dicarboxylic anhydride (carbic anhydride, CA) necessary to get the desired Gel:CA ratio was added to the solution. Depending on the method used, the reaction was left to proceed for 30 min (Method 1 and 2) or 10 min (Method 3) prior to pH adjustment to 9 with NaOH 0.5 m solution. Under continuous stirring, pH adjustment and sequential addition of CA were repeated five times following the interval time defining each method (see Figure 1B). The solution was then diluted twofold with mQ H2O prewarmed to 40 °C and the pH adjusted to 7.4 with a solution of HCl 0.5 m. Upon centrifugation for 15 min at 3000 rcf, unbound CA was deposited as white precipitate. The supernatant was then dialyzed at 40 °C against mQ H2O with frequent water changes for 3–4 days and then freeze‐dried. Gel‐NB degree of substitution (DS) was determined by 1H‐NMR (Bruker Ultrashield 400 MHz, 1024 scans). In short, Gel‐NB was solubilized at 40 mg mL−1 in a solution of 0.5 mg mL−1 3‐(trimethylsilyl)‐1‐propanesulfonic acid (DSS) in D2O (Apollo Scientific). DSS was used as an internal standard to calculate NB millimoles per gram of gelatin by comparing integrals of the DSS nine methyl protons (≈0.5 to −0.5 ppm) with the two NB–ene protons (≈6.21–6.00 ppm) (n = 3, see Table S1, Figure S2, Supporting Information). DS given as a percentage was calculated based on the lysine + hydroxylysine content of porcine skin gelatin type A (0.325 mmol g−1) estimated by Claaßen et al.[ 62 ]
Synthesis of Gel‐MA
Gel‐MA was synthesized as previously described.[ 63 ] DS was estimated with 1H‐NMR (Bruker Ultrashield 400 MHz, 1024 scans) in D2O (Apollo Scientific). Gel‐MA lysine integration signal (2.95–3.05 ppm) was compared to unmodified gelatin lysine integration signal (2.95–3.05 ppm). Phenylalanine signal (7.2–7.5 ppm) was used as internal reference. DS was found to be ≈55%.
Photoresin Preparation and Photorheology
Gel‐NB photoresins were prepared by first dissolving the freeze‐dried polymer in PBS at 37 °C. Either thiolated crosslinker, 10 kDa PEG4SH (JenKem Technology), 1 kDa PEG2SH or 3,6‐dioxa‐1,8‐octanedithiol (DODT) was then added from a freshly prepared stock solution in PBS to get the desired SH:NB ratio. Similarly, the photoinitiator (PI) lithium phenyl‐2,4,6‐trimethylbenzoylphosphinate (LAP), was diluted in the photoresin mixture at 0.05% w/v from a freshly prepared 2% w/v stock solution in PBS. Gel‐MA was prepared following the same procedure, without the addition of thiolated crosslinker.
Photoreology analyses were carried out on an Anton Paar MCR 301 equipped with a 20 mm parallel plate geometry, 6 mm glass floor, and Omnicure Series1000 lamp (Lumen Dynamics) used at 20% output power with 400–500 nm filter. All tests were performed at 37 °C to avoid physical gelation. A wet tissue paper was used in the chamber to prevent the sample from drying during the measurement. Samples were left to equilibrate for 5 min prior to starting the analysis. Oscillatory measurements were performed in triplicates (n = 3) at 5% shear rate and 2 Hz frequency with 200 µm gap and 10 s measuring point duration.
Volumetric Printing
2.5% Gel‐NB/PEG4SH and 5% Gel‐NB/PEG4SH photoresins were prepared as indicated above, filtered through a 0.45 µm filter to remove potentially scattering particles and 3–4 mL were transferred into the glass vial container. The photoresin was left to physically gel at 4 °C for 10–15 min. Printing was then performed on a commercially available volumetric printer (Tomolite, Readily3D SA)[ 51 ] followed by heating in a warm bath to 37 °C to dissolve the uncrosslinked photoresin. Printed objects were washed in PBS prewarmed to 37 °C, while uncrosslinked photoresin was recovered for further use. For in‐vial pictures, the printed objects were washed with PBS in the glass vial and imaged with printer built‐in camera system. For in‐air pictures, the printed objects were imaged with a Fujifilm XT‐3 camera equipped with a macroextension tube. Branch models were perfused with high MW 2 MDa blue‐dextran or 40 kDa tetramethylrhodamine isothiocyanate‐dextran (TRITC‐dextran).
Volumetric Bioprinting Procedure and Cell Viability
Gel‐NB/PEG4SH photoresins were prepared as indicated above and filtered sterilized through a 0.20 µm filter. NHDFs were isolated from juvenile foreskin skin biopsies. Biopsies were taken under parental informed consent and their use for research purposes was approved by the Ethical Committee of Canton Zurich (BASEC‐Request‐Nr. 2018‐00269). C2C12 and NHDF were added to Gel‐NB/PEG4SH photoresin at 1 million cells mL−1 and the resulting bioresin was transferred to glass vials sterilized via sequential EtOH 70% washing and UV‐sterilization. After printing, the vials containing the printed objects were warmed up to 37 °C in a water bath. The cell‐laden hydrogel was then washed under sterile conditions twice in warm PBS and then submerged in 6 mL cell culture media composed of DMEM + GlutaMAX‐I + 2% horse serum + 1% ITS (Cornig) + 10 µg mL−1 gentamicin for C2C12 and DMEM + GlutaMAX‐I + 10% fetal bovine serum + 10 µg mL−1 gentamicin for NHDF in a 6‐well suspension culture plate (Greiner CELLSTAR). Media were changed every 4 days.
For the cell viability assay, samples after 0, 2, and 7 days of culture were incubated for 45 min in FluoroBrite DMEM supplemented with 1:2000 CalceinAM (Invitrogen), 1:500 Propidium Iodide (PI, Fluka), and 1:1000 Hoechst 33342 (Invitrogen). Imaging was performed on a Leica SP8 microscope (Leica) and Olympus Fluoview 3000 (Olympus) equipped with a 10× objective. Z‐stacks were acquired from the samples surface at 5 µm steps and 300 µm into the sample. The presented pictures resulted from maximum intensity z‐projection. Cell viability after printing was assessed by counting viable (CalceinAM) and dead (PI) cells throughout the entire z‐range with the ImageJ Analyze particle function. Starting from day 2, due to the spread cell morphology, the total number of cells was more reliably calculated based on Hoechst 33342 stained nuclei.
Volumetric Bioprinting of Cellular Constructs
Cellular constructs were bioprinted following the described volumetric bioprinting procedure at 1 million cells mL−1 for C2C12‐laden spiral models and 10 000 cells mL−1 for NHDF‐laden branch models. Bright‐field images of spiral‐shaped cellular constructs and myotubes contraction videos were taken on a Axio Observer.Z1 (Zeiss) with a 5× and 10× objective, respectively. For immunofluorescence, after 3 weeks of culture, C2C12 constructs were washed 3× in PBS and fixed in 4% paraformaldehyde for 2 h at 4 °C and washed again 3× in PBS + 0.02% BSA prior to permeabilization with 1% Triton‐X100 in PBS for 15 min at room temperature. After washing three times in PBS + 0.02% BSA, the constructs were blocked for a further 15 min in a solution of 1% BSA, 1% Tween‐20 in PBS, and then incubated with primary antihuman myosin heavy chain antibody (MF‐20, DSHB, 1:20 in PBS + 0.02% BSA) for 2 h at room temperature. They were then washed three times with PBS, incubated with secondary antibody (Invitrogen, goat anti‐mouse Alexa488, 1:1000 in PBS + 0.02% BSA), and Hoechst 33342 (Invitrogen, 1:1000 in PBS + 0.02% BSA) for 4 h at 4 °C. Samples were washed in PBS and imaged on Leica SP8 microscope (Leica) equipped with a 25× water immersion objective.
NHDF‐laden branch models were stained by incubating them for 45 min in FluoroBrite DMEM supplemented with 1:2000 CalceinAM (Invitrogen). Prior to imaging on Leica SP8 microscope (Leica) equipped with a 10× objective, the samples were transferred to a glass coverslip and perfused with 40 kDa TRITC‐dextran.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supplemental Video 1
Supplemental Video 2
Acknowledgements
D.R. and H.L. contributed equally to this work. M.Z.‐W. acknowledges ETH Grant application ETH‐38 19‐1 for their support. The authors further acknowledge Philipp Fisch for the help with compression testing, Dr. René Verel for the help with magic angle spinning NMR, and the assistance from ETH (ScopeM) imaging facility. Source .stl files for chess pieces were obtained from www.thingiverse.com/thing:585218 (under CC BY‐SA 3.0 license). Source .stl files for glass, Klein bottle and ok‐hand models were obtained from www.thingiverse.com/thing:460 (under GPLv2 license), www.thingiverse.com/thing:5217 (under Public Domain Certification) and www.thingiverse.com/thing:2216106 (under CC BY 4.0 license), respectively. [Correction added on 19 May 2022, after first online publication: CSAL funding statement has been added.]
Open Access Funding provided by Eidgenossische Technische Hochschule Zurich.
Rizzo R., Ruetsche D., Liu H., Zenobi‐Wong M., Optimized Photoclick (Bio)Resins for Fast Volumetric Bioprinting. Adv. Mater. 2021, 33, 2102900. 10.1002/adma.202102900
Data Availability Statement
The data that support the findings of this study are openly available in the ETH Research Collection (https://www.research-collection.ethz.ch/) at https://doi.org/10.3929/ethz-b-000479062.
References
- 1. Lee M., Rizzo R., Surman F., Zenobi‐Wong M., Chem. Rev. 2020, 120, 10950. [DOI] [PubMed] [Google Scholar]
- 2. Brown T. E., Anseth K. S., Chem. Soc. Rev. 2017, 46, 6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tibbitt M., Kloxin A. M., Dyamenahalli K. U., Anseth K. S., Soft Matter 2010, 6, 5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shadish J. A., Benuska G. M., DeForest C. A., Nat. Mater. 2019, 18, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Broguiere N., Lüchtefeld I., Trachsel L., Mazunin D., Rizzo R., Bode J. W., Lutolf M., Zenobi‐Wong M., Adv. Mater. 2020, 32, 1908299. [DOI] [PubMed] [Google Scholar]
- 6. Qin X.‐H., Wang X., Rottmar M., Nelson B. J., Maniura‐Weber K., Adv. Mater. 2018, 30, 1870070. [DOI] [PubMed] [Google Scholar]
- 7. Schwab A., Levato R., D'Este M., Piluso S., Eglin D., Malda J., Chem. Rev. 2020, 120, 11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lim K. S., Galarraga J. H., Cui X., Lindberg G. C. J., Burdick J. A., Woodfield T. B. F., Chem. Rev. 2020, 120, 10662. [DOI] [PubMed] [Google Scholar]
- 9. Solis L. H., Ayala Y., Portillo S., Varela‐Ramirez A., Aguilera R., Boland T., Biofabrication 2019, 11, 045005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campbell A., Mohl J. E., Gutierrez D. A., Varela‐Ramirez A., Boland T., Front. Bioeng. Biotechnol. 2020, 8, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Snyder J., Rin Son A., Hamid Q., Wang C., Lui Y., Sun W., Biofabrication 2015, 7, 044106. [DOI] [PubMed] [Google Scholar]
- 12. Müller M., Öztürk E., Arlov Ø., Gatenholm P., Zenobi‐Wong M., Ann. Biomed. Eng. 2017, 45, 210. [DOI] [PubMed] [Google Scholar]
- 13. Blaeser A., Duarte Campos D. F., Puster U., Richtering W., Stevens M. M., Fischer H., Adv. Healthcare Mater. 2016, 5, 326. [DOI] [PubMed] [Google Scholar]
- 14. Kelly B. E., Bhattacharya I., Heidari H., Shusteff M., Spadaccini C. M., Taylor H. K., Science 2019, 363, 1075. [DOI] [PubMed] [Google Scholar]
- 15. Loterie D., Delrot P., Moser C., Nat. Commun. 2020, 11, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bernal P. N., Delrot P., Loterie D., Li Y., Malda J., Moser C., Levato R., Adv. Mater. 2019, 31, 1904209. [DOI] [PubMed] [Google Scholar]
- 17. Cook C. C., Fong E. J., Schwartz J. J., Porcincula D. H., Kaczmarek A. C., Oakdale J. S., Moran B. D., Champley K. M., Rackson C. M., Muralidharan A., McLeod R. R., Shusteff M., Adv. Mater. 2020, 32, 2003376. [DOI] [PubMed] [Google Scholar]
- 18. Lin C.‐C., Ki C. S., Shih H., J. Appl. Polym. Sci. 2015, 132, 41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertlein S., Brown G., Lim K. S., Jungst T., Boeck T., Blunk T., Tessmar J., Hooper G. J., Woodfield T. B. F., Groll J., Adv. Mater. 2017, 29, 1703404. [DOI] [PubMed] [Google Scholar]
- 20. Van Hoorick J., Dobos A., Markovic M., Gheysens T., Van Damme L., Gruber P., Tytgat L., Van Erps J., Thienpont H., Dubruel P., Ovsianikov A., Van Vlierberghe S., Biofabrication 2021, 13, 015017. [DOI] [PubMed] [Google Scholar]
- 21. Dobos A., Van Hoorick J., Steiger W., Gruber P., Markovic M., Andriotis O. G., Rohatschek A., Dubruel P., Thurner P. J., Van Vlierberghe S., Baudis S., Ovsianikov A., Adv. Healthcare Mater. 2020, 9, 1900752. [DOI] [PubMed] [Google Scholar]
- 22. Hoyle C. E., Bowman C. N., Angew. Chem., Int. Ed. 2010, 49, 1540. [DOI] [PubMed] [Google Scholar]
- 23. Kharkar P. M., Rehmann M. S., Skeens K. M., Maverakis E., Kloxin A. M., ACS Biomater. Sci. Eng. 2016, 2, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sawicki L. A., Kloxin A. M., Biomater. Sci. 2014, 2, 1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fairbanks B. D., Schwartz M. P., Halevi A. E., Nuttelman C. R., Bowman C. N., Anseth K. S., Adv. Mater. 2009, 21, 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCall J. D., Anseth K. S., Biomacromolecules 2012, 13, 2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fairbanks B. D., Macdougall L. J., Mavila S., Sinha J., Kirkpatrick B. E., Anseth K. S., Bowman C. N., Chem. Rev. 2021, 121, 6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Hoorick J., Gruber P., Markovic M., Rollot M., Graulus G.‐J., Vagenende M., Tromayer M., Van Erps J., Thienpont H., Martins J. C., Baudis S., Ovsianikov A., Dubruel P., Van Vlierberghe S., Macromol. Rapid Commun. 2018, 39, 1800181. [DOI] [PubMed] [Google Scholar]
- 29. Bryant S. J., Chowdhury T. T., Lee D. A., Bader D. L., Anseth K. S., Ann. Biomed. Eng. 2004, 32, 407. [DOI] [PubMed] [Google Scholar]
- 30. Bian L., Hou C., Tous E., Rai R., Mauck R. L., Burdick J. A., Biomaterials 2013, 34, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wolf K., te Lindert M., Krause M., Alexander S., Te Riet J., Willis A. L., Hoffman R. M., Figdor C. G., Weiss S. J., Friedl P., J. Cell Biol. 2013, 201, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kwon M. Y., Wang C., Galarraga J. H., Puré E., Han L., Burdick J. A., Biomaterials 2019, 222, 119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Den Bulcke A. I., Bogdanov B., De Rooze N., Schacht E. H., Cornelissen M., Berghmans H., Biomacromolecules 2000, 1, 31. [DOI] [PubMed] [Google Scholar]
- 34. Van Hoorick J., Tytgat L., Dobos A., Ottevaere H., Van Erps J., Thienpont H., Ovsianikov A., Dubruel P., Van Vlierberghe S., Acta Biomater. 2019, 97, 46. [DOI] [PubMed] [Google Scholar]
- 35. Klotz B. J., Gawlitta D., Rosenberg A. J. W. P., Malda J., Melchels F. P. W., Trends Biotechnol. 2016, 34, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yue K., Trujillo‐de Santiago G., Alvarez M. M., Tamayol A., Annabi N., Khademhosseini A., Biomaterials 2015, 73, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leucht A., Volz A. C., Rogal J., Borchers K., Kluger P. J., Sci. Rep. 2020, 10, 5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loessner D., Meinert C., Kaemmerer E., Martine L. C., Yue K., Levett P. A., Klein T. J., Melchels F. P. W., Khademhosseini A., Hutmacher D. W., Nat. Protoc. 2016, 11, 727. [DOI] [PubMed] [Google Scholar]
- 39. Fan L., Liu C., Chen X., Zou Y., Zhou Z., Lin C., Tan G., Zhou L., Ning C., Wang Q., ACS Appl. Mater. Interfaces 2018, 10, 17742. [DOI] [PubMed] [Google Scholar]
- 40. Ma X., Qu X., Zhu W., Li Y.‐S., Yuan S., Zhang H., Liu J., Wang P., Lai C. S. E., Zanella F., Feng G.‐S., Sheikh F., Chien S., Chen S., Proc. Natl. Acad. Sci. USA 2016, 113, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nair D. P., Podgórski M., Chatani S., Gong T., Xi W., Fenoli C. R., Bowman C. N., Chem. Mater. 2014, 26, 724. [Google Scholar]
- 42. Lee B. H., Shirahama H., Cho N.‐J., Tan L. P., RSC Adv. 2015, 5, 106094. [Google Scholar]
- 43. Mu˜noz Z., Shih H., Lin C.‐C., Biomater. Sci. 2014, 2, 1063. [DOI] [PubMed] [Google Scholar]
- 44. Perera M. M., Ayres N., Polym. Chem. 2017, 8, 6741. [Google Scholar]
- 45. Vlierberghe S. V., Schacht E., Dubruel P., Eur. Polym. J. 2011, 47, 1039. [Google Scholar]
- 46. Van Hoorick J., Declercq H., De Muynck A., Houben A., Van Hoorebeke L., Cornelissen R., Van Erps J., Thienpont H., Dubruel P., Van Vlierberghe S., J. Mater. Sci.: Mater. Med. 2015, 26, 247. [DOI] [PubMed] [Google Scholar]
- 47. Van Hoorick J., Gruber P., Markovic M., Tromayer M., Van Erps J., Thienpont H., Liska R., Ovsianikov A., Dubruel P., Van Vlierberghe S., Biomacromolecules 2017, 18, 3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin C.‐C., Anseth K. S., Pharm. Res. 2009, 26, 631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. D'souza A. A., Shegokar R., Expert Opin. Drug Delivery 2016, 13, 1257 [DOI] [PubMed] [Google Scholar]
- 50. Benedikt S., Wang J., Markovic M., Moszner N., Dietliker K., Ovsianikov A., Grützmacher H., Liska R., J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 473. [Google Scholar]
- 51.Readily3D, https://readily3d.com/ (accessed: July 2021).
- 52. Trappmann B., Baker B. M., Polacheck W. J., Choi C. K., Burdick J. A., Chen C. S., Nat. Commun. 2017, 8, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guo X., Mittelstaedt R. A., Guo L., Shaddock J. G., Heflich R. H., Bigger A. H., Moore M. M., Mei N., Toxicol. In Vitro 2013, 27, 1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guo X., Seo J.‐E., Bryce S. M., Tan J. A., Wu Q., Dial S. L., Moore M. M., Mei N., Toxicol. Sci. 2018, 163, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Appiah C., Arndt C., Siemsen K., Zeitmann A., Staubitz A., Selhuber‐Unkel C., Adv. Mater. 2019, 31, 1807747. [DOI] [PubMed] [Google Scholar]
- 56. Fleischer S., Tavakol D. N., Vunjak‐Novakovic G., Adv. Funct. Mater. 2020, 30, 1910811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. O'Grady B. J., Balotin K. M., Bosworth A. M., McClatchey P. M., Weinstein R. M., Gupta M., Poole K. S., Bellan L. M., Lippmann E. S., ACS Biomater. Sci. Eng. 2020, 6, 5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu L., Li X., Shi X., Wang Y., RSC Adv. 2018, 8, 22764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Piluso S., Vukicevic R., Nochel U., Braune S., Lendlein A., Neffe A. T., Eur. Polym. J. 2018, 100, 77 [Google Scholar]
- 60. Diba M., Camargo W. A., Brindisi M., Farbod K., Klymov A., Schmidt S., Harrington M. J., Draghi L., Boccaccini A. R., Jansen J. A., van den Beucken J. J. J. P., Leeuwenburgh S. C. G., Adv. Funct. Mater. 2017, 27, 1703438 [Google Scholar]
- 61. Buie T., McCune J., Cosgriff‐Hernandez E., Trends Biotechnol. 2020, 38, 546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Claaßen C., Claaßen M. H., Truffault V., Sewald L., Tovar G. E. M., Borchers K., Southan A., Biomacromolecules 2018, 19, 42. [DOI] [PubMed] [Google Scholar]
- 63. Kessel B., Lee M., Bonato A., Tinguely Y., Tosoratti E., Zenobi‐Wong M., Adv. Sci. 2020, 7, 2001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Video 1
Supplemental Video 2
Data Availability Statement
The data that support the findings of this study are openly available in the ETH Research Collection (https://www.research-collection.ethz.ch/) at https://doi.org/10.3929/ethz-b-000479062.