

Abstract
Tissue regeneration evolves toward the biofabrication of sophisticated 3D scaffolds. However, the success of these will be contingent to their capability to integrate within the host. The control of the mechanical or topographical properties of the implant appears as an ideal method to modulate the immune response. However, the interplay between these properties is yet not clear. Dual‐porosity scaffolds with varying mechanical and topographical features are created, and their immunomodulatory properties in rat alveolar macrophages in vitro and in vivo in a rat subcutaneous model are evaluated. Scaffolds are fabricated via additive manufacturing and thermally induced phase separation methods from two copolymers with virtually identical chemistries, but different stiffness. The introduction of porosity enables the modulation of macrophages toward anti‐inflammatory phenotypes, with secretion of IL‐10 and TGF‐β. Soft scaffolds (<5 kPa) result in a pro‐inflammatory phenotype in contrast to stiffer (>40 kPa) scaffolds of comparable porosities supporting a pro‐healing phenotype, which appears to be related to the surface spread area of cells. In vivo, stiff scaffolds integrate, while softer scaffolds appear encapsulated after three weeks of implantation, resulting in chronic inflammation after six weeks. The results demonstrate the importance of evaluating the interplay between topography and stiffness of candidate scaffolds.
Keywords: 3D printing, dual‐porosity, immunomodulation, macrophage polarization
The immune response to implants can be regulated through the surface topography and mechanical properties. There is an interplay between these two parameters; while stiff scaffolds readily promote a pro‐healing phenotype, macrophages in soft scaffolds require the presence of big surface features or pores. These parameters dictate the surface spread area of the cells that appears to define their phenotype.
1. Introduction
Tissue engineering and regeneration fields generally make use of scaffold materials that support or even guide tissue formation. Since the early 2000s several scaffolds’ properties have been identified as essential to be able to regenerate tissues.[ 2 ] These include mechanical properties matching the targeted tissue, biodegradability matching the tissue formation rate, and a porosity that allows cell infiltration, tissue formation, and nutrient and waste transport. Nowadays, these fields evolve toward the development of more sophisticated scaffolds or implant materials that are designed primarily to drive cell differentiation and tissue formation in situ.[ 1 ] Many other scaffold properties have been defined as key aiding at cell guidance and regulation, including surface biochemistry, topography, and even curvature.[ 3 ] However, the implantation of such engineered scaffolds into the body leads to a foreign body response that will determine the success (allowing host cell–biomaterial interactions) or the failure of the implant (leading to encapsulation and chronic inflammation).[ 4 ] Thus, it is of outmost importance for scaffold development to understand the physicochemical parameters, such as mechanical properties and porosities that can induce a pro‐inflammatory or a pro‐regenerative immune response.
The foreign body response starts with the absorption of proteins on the surface of the scaffold and the formation of a provisional matrix. This matrix presents a milieu rich in growth factor and chemokines that serve to recruit cells from the immune system and start an inflammatory response. This innate immune response is orchestrated primarily by host macrophages that become classically activated (M1) and release a compendium of cytokines, being tumor necrosis factor‐α (TNF‐α) the most characteristic. Thereafter, macrophages undergo dynamic changes that allow them to regulate the inflammation and eventually become alternatively activated (M2), producing large amounts of interleukin‐10 (IL‐10) and transforming growth factor‐β (TGF‐β), resolving inflammation and contributing to tissue formation, repair and remodeling.[ 5 ] Alternatively, the implanted material leads to a strong acute inflammatory response in which the macrophage response has an extended time‐frame, releasing more pro‐inflammatory cytokines and reactive oxygen species (ROS) while trying to phagocytize and degrade the implant material. This frustrated attempt to degradation promotes the fusion of macrophages to form foreign body giant cells (FBGC), the encapsulation of the material on a newly collagenous fibrous matrix formed by attracted fibroblasts and, ultimately, its rejection from the body. Thus, the extent of the inflammation dictates the acceptance or rejection of the implant, in which macrophages play a pivotal role.
The potential to modulate the immune response to implant materials has been extensively studied in the last decade.[ 4 , 6 ] Many have shown the importance of the scaffold's (surface) chemistry,[ 7 ] mechanical properties,[ 8 ] topographical cues,[ 9 ] and porosity,[ 10 ] on macrophage polarization. McWhorter et al. investigated the influence of the substrate on macrophage response, by introducing topography and modulating the cell shape.[ 9b ] Using unpattern and channeled 2D substrates, they modulated the shape of bone marrow derived macrophages obtaining well‐spread or elongated morphologies, respectively. Cell shape dictated the profile of cytokine production with a higher release of IL‐4 and expression of arginase‐1 and CD206 receptor, and hence a more anti‐inflammatory phenotype, in macrophages with a higher degree of elongation. Moreover, they showed that upon pharmacological inhibition of actin or myosin, macrophage polarization via induction of elongation was also inhibited, suggesting that polarization is a cell contractility driven process.
Patel et al. recently showed the influence of substrates stiffness in cell elasticity, actin polymerization, and acquisition of a pro‐inflammatory phenotype in RAW 264.7 rat alveolar macrophages and U937 human promonocytic cells.[ 8a ] Upon culture on soft (1.2 kPa elastic moduli) or stiff (150 kPa) 2D substrates cells acquired a different morphology, with a rounded shape and few filopodia projections found in soft substrates and spread morphology and higher amount of filopodia in stiff substrates. They showed that higher actin polymerization (higher cell elasticity in stiffer substrates) led to a pro‐inflammatory phenotype. Combining the effects of stiffness and porosity Jiang et al. studied the polarization of macrophages when confined within the scaffold's pores, observing a pro‐inflammatory phenotype when cells were cultured inside small pores with low stiffness (20–70 kPa and 30 µm pores), which is in disagreement with the study of Patel et al.[ 8 , 11 ] Cell elongation was also observed to play a role, with stiffer materials (190 kPa) and increasing pore size (80 µm) resulting in higher cell elongation and resulting on a more pro‐regenerative phenotype.
Going one step further, Vijaykumar et al. have recently shown that macrophage polarization is intimately related to the expression and localization of Yes‐associated protein (YAP) within monocytes isolated from whole blood.[ 12 ] They choose two different substrates with different mechanical properties, and found that softer hydrogels led to a higher secretion of M2 phenotype characteristic cytokine IL‐10 and lower secretion of TNF‐α and IL‐6. Moreover, cells cultured on stiff substrates presented nuclear expression of YAP, while on softer substrates YAP expression was cytoplasmic. Culture of monocytes in substrates with gradually increasing stiffness resulted in an increasing nuclear expression of YAP and together with this, increased secretion of TNF‐α.
Altogether, these studies investigated the response of macrophages to either well organized topographies or mechanical properties that ranged from 1 kPa to hundreds of kPa (or even glass substrates), concluding that substrates with low mechanical properties (<5 kPa) or patterned (rather than flat) resulted in an anti‐inflammatory phenotype. However, they also point out to the importance of cytoskeletal organization and actin polymerization in the modulation of the macrophage response and results disagree when cells are on confined environments (pores). Thus, there is a need for a better understanding of the interplay between these parameters where cell elongation seems to override the effects of mechanical properties.
Additive manufacturing technologies are among the most used for the biofabrication of scaffolds for tissue regeneration.[ 13 ] Some specific techniques have been developed for the introduction of topographies or microporosities in electrospun, wet‐spun,[ 14 ] and 3D printed scaffolds that can potentially lead to cell guidance and tissue formation.[ 15 ] However, it is still a challenge to introduce controlled topographies in 3D scaffolds, and thus the immunomodulatory capability of these scaffolds is generally controlled by the chemistry of the material used.
Here, we introduced a novel method for the fabrication of dual‐porosity scaffolds based on a combination of extrusion‐based 3D printing of gels and liquid‐liquid thermally induced phase separation (TIPS). We choose two chemically relevant and biocompatible materials with virtually identical chemistries based on poly(lactide‐co‐caprolactone), one amorphous and one semicrystalline, that presented a Young's modulus that differed two orders of magnitude between copolymers (≈4 kPa vs 400 kPa). Dual‐porosity scaffolds fabricated from these two materials presented interconnected porosities across the printed fibers, with pore diameters that were dependent on the concentration of the starting polymer gel. Thus, we fabricated a library of six different scaffolds with varying porosities and mechanical properties (while keeping a virtually identical surface chemistry) and used them to investigate the interplay between these two parameters on the modulation of macrophage response in vitro and the immune response in vivo.
2. Results
2.1. Fabrication of Dual Porosity Scaffolds with Amorphous, P(l,d)LCL, and Semicrystalline, P(l)LCL Copolymers
Recent studies have brought to evidence the potential of modulating the immune response to implanted biomaterials.[ 4 ] However, the majority of these studies investigate the role of different physicochemical characteristics of biomaterials individually and/or in 2D systems. Here, we seek to investigate the effects of porosity and mechanical properties in 3D printed scaffolds. It is well‐known that the introduction of porosity to a given material reduces its mechanical compressive properties. Thus, we used two different biologically relevant materials, with virtually identical chemistries. P(l)LCL and P(l,d)LCL had a lactide:caprolactone monomer ratio of 70:30 and 68:32, respectively. The two copolymers were chosen on account of their biodegradability and their demonstrated ability to support growth of multiple cell lines.[ 16 ] These polymers, of virtually identical chemical composition, accounted for different crystallinity as a consequence of the enantiomeric mixture of lactide used for their polymerization, and thus also for different degradation rates, having the P(l)LCL a slower degradation rate than the amorphous P(I,d)LCL.[ 17 ] The amorphous P(l,d)LCL copolymer accounted for a 15:85 d‐lactide:l‐lactide enantiomer ratio while the semicrystalline copolymer, P(l)LCL, consisted of only l‐lactide enantiomer. Dynamic scanning calorimetry (DSC) traces revealed that the P(l,d)LCL copolymer presented a T g around 31 °C and no melting transition. P(l)LCL copolymer presented a T g at ≈63 °C and a T m at 158 °C (Figure 1a), which confirmed their amorphous and semicrystalline nature, respectively. To evaluate the impact that a different degree of crystallinity might have on the physicochemical properties of the materials we measured the water contact angle on spin‐coated copolymer films, resulting on angles of 78 ± 2° for P(l,d)LCL and 78 ± 3° for P(l)LCL, showing no statistical difference between the two materials (Figure 1b). Evaluation of the thermal stability of the polymers showed an onset of degradation of 300 °C for P(I,d)LCL and of 350 °C for the amorphous P(l,d)LCL (Figure S1, Supporting Information). This difference was considered negligible for the purpose of this study.
Figure 1.
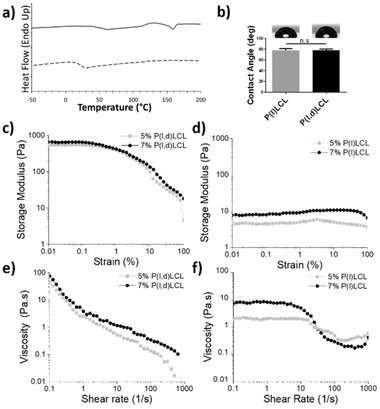
Intrinsic properties of the two copolymers and the formed gels. a) Dynamic scanning calorimetry traces of P(l)LCL (bold line) and P(l,d)LCL (dashed line) showing their semicrystalline and amorphous character, respectively. b) Water contact angle on spin‐coated substrates of the two copolymers. c,d) Storage modulus under varying strains for 5% and 7% gels of P(l,d)LCL and P(l)LCL, respectively. e,f) Viscosity dependence on shear rate for 5% and 7% gels formed from P(l,d)LCL and P(l)LCL, respectively.
Dual‐porosity scaffolds were designed to modulate the innate immune response of implant materials as previously suggested on hydrogel scaffolds.[ 18 ] Dual‐porosity scaffolds were fabricated following a combined technique of 3D plotting and TIPS. We previously showed this combined technique using a solid‐liquid TIPS that allowed us to introduce topographies on 3D printed scaffolds.[ 15a ] Here, we choose a liquid‐liquid phase separation based on 1,4‐dioxane and water, which allowed us to create pores within the printed fibers (Figure 2 ). Scaffolds were produced from P(l)LCL and P(l,d)LCL copolymers starting from gels of 5 or 7 (wt/vol)%. As a control, scaffolds were also fabricated via traditional 3D fiber deposition from the melt, coded here as 100%. While the introduction of porosity on 3D printed objects has already been shown by others, the developed techniques are based on the introduction of a secondary sacrificial compound or porogen that is later washed away.[ 15 , 19 ] Thus, the combination of bioplotting and TIPS represents an easy way to create highly porous scaffolds without the introduction of agents that are not necessarily biocompatible and require extensive washing procedures.
Figure 2.

Structural characterization of dual porosity scaffolds. a) Scanning electron microscopy (SEM) images of scaffolds prepared from P(l)LCL and P(l,d)LCL copolymers as dual porosity scaffolds starting from gels of 5% and 7% polymer concentration and scaffolds fabricated via fused deposition modeling, coded as 100%. Images are shown (left to right) as the overall appearance of the scaffold from the top, a magnification of the scaffold's surface, the overall scaffold cross section and a magnification of the cross section. b) Images of P(l)LCL and P(l,d)LCL dual porosity scaffolds at 5% and 7% gel concentrations obtained with µCT and frequency plots of the pore diameters as measured by SEM. c) Compressive Young's moduli of dual porosity scaffolds at 5% and 7% polymer concentration and of 100% scaffolds. Statistical significance was calculated from one‐way ANOVA with Tukey's multiple comparison test, n = 3, F(DFn, DFd), F(5, 12) = 35,39. P‐adjusted (****) p < 0.0001, (***) p < 0.001, (**) p < 0.01, and (*) p < 0.1.
Gels produced from P(I,d)LCL and P(l)LCL presented very different properties. The storage modulus under shear strain for the amorphous polymer based gels were approximately two orders of magnitude higher than those of the semicrystalline polymer. As expected, a concentration dependence was observed, with gels prepared from 7% concentrations having a higher storage moduli than those at 5%. Both polymers presented a stable behavior up to strains of 0.5%. Measurements of the viscosity over different shear rates demonstrated that all samples showed shear thinning behavior, which was more pronounced on P(l,d)LCL polymer. In the case of P(l)LCL polymer, both gels presented a Newtonian plateau at low shear rates that only started to decrease when shear rates were higher than 10 s−1, traditionally defined with the Carreau‐Yasuda model. This observation would indicate that the physical crosslinks present in P(l,d)LCL gels are negligible in P(l)LCL.
2.2. Structure of Dual Porosity Scaffolds
The fabricated dual porosity scaffolds reproduced well the designed printing patterns while presenting an interconnected porous structure (closed porosity; Table S1, Supporting Information) but were, however, unable to retain the lateral spacing (Figure 2a). Moreover, samples produced from 5% copolymer concentration gels resulted in slightly deformed printed strands of higher diameter (and hence, lower structural pore width). Analysis of the porosity of the fabricated scaffolds via scanning electron microscopy (SEM) revealed a pore mean diameter (±SEM) of 29 ± 0.5 µm with a frequency of 25% for diameters between 20 and 25 µm for 5% gels of P(l)LCL and 20 ± 0.4 µm with frequencies of 17% for 0–5 and 15–20 µm ranges for 5% gels of P(l,d)LCL (Figure 2b; Figure S2, Supporting Information; Tables 1 and 2 ). Increasing the polymer concentration to form gels of 7% resulted on a decreased mean pore diameter of 21 ± 0.3 µm and a frequency of 28% between 10 and 15 µm for P(l)LCL, and 8 ± 0.1 µm and frequencies of 40% between 0 and 5 µm and 35% between 5 and 10 µm for P(l,d)LCL. Thus, increasing the polymer concentration reduced the mean pore diameter but also narrowed the pore size distribution (Figure 2b). Thus, scaffolds fabricated from P(l)LCL at 7% concentration and from P(l,d)LCL at 5% polymer concentration presented a mean pore diameter that was not statistically different and serves as a direct comparison of materials with different mechanical properties at similar porosity (Figure S2, Supporting Information; Table 1).
Table 1.
Pore characteristics and mechanical properties of fabricated scaffolds
| Sample code | Porosity type | Mean pore diameter a) [µm] | Young's modulus b) [kPa] |
|---|---|---|---|
| P(l)LCL nonporous | Structural | N/A | 404 ± 112 |
| P(l)LCL 7% | Structural and in‐strand | 21 ± 0.3 | 40 ± 15 |
| P(l)LCL 5% | Structural and in‐strand | 29 ± 0.5 | 27 ± 5 |
| P(l,d)LCL nonporous | Structural | N/A | 4.9 ± 1.5 |
| P(l,d)LCL 7% | Structural and in‐strand | 8 ± 0.1 | 2.8 ± 0.4 |
| P(l,d)LCL 5% | Structural and in‐strand | 20 ± 0.4 | 2.1 ± 0.1 |
Data shown as mean ± SEM
Data shown as average ± SD.
Table 2.
Pore frequency distribution
| Sample | Main pore diameter and frequency [µm, %] | Secondary pore diameter and frequency [µm, %] | % pores above 10 µm |
|---|---|---|---|
| P(l)LCL 5% | 20–25, 25 | 15–20, 19 | 95 |
| P(l)LCL 7% | 10–15, 28 | 15–20, 22 | 94 |
| P(l,d)LCL 5% | 0–5, 17 | 15–20, 17 | 70 |
| P(l,d)LCL 7% | 0–5, 40 | 5–10, 35 | 25 |
Analysis of the scaffold´s structure via micro‐computed tomography (µCT) revealed the opposite trend, with higher porosity for higher polymer concentration on the starting gels (Figure 2b and Table S1, Supporting Information). Thus, a porosity of 61.5% and 69.1% was measured for P(l)LCL scaffolds with 5% and 7% polymer concentration, respectively. Similarly, gels produced from P(l,d)LCL scaffolds displayed a porosity of 53.3% and 60.9% for starting gel concentrations of 5% and 7%. This slight increase in porosity could be the result of a better structure retention as observed by SEM and µCT scans.
The total surface area, however, demonstrated the high internal porosity (within the fibers) observed also by SEM. A decrease in total surface area was measured with an increasing polymer concentration with values of 3103 and 2020 mm2 for P(l)LCL at 5% and 7%, respectively, and of 3543 and 2548 mm2 for P(l,d)LCL at 5% and 7%, respectively. It is also noteworthy that for a given polymer concentration, scaffolds fabricated from P(l,d)LCL and P(l)LCL accounted both for comparable surface areas (3103 mm2 vs 3543 mm2 for 5% and 2020 mm2 vs 2548 mm2, respectively).
The fabricated dual‐porosity scaffolds presented open porosities that ranged from 53% to 69% depending on the material and the concentration of polymer in the initial gel (Table S1, Supporting Information). These values are in the range of those reported for 3D printed ceramics (up to 59%) using NaCl leaching,[ 15b ] but lower than those reported for polylactide‐co‐glycolide polymers using CuSO4 as porogen (up to 94% porosity). The use of such salts, however, is not convenient for scaffolds aimed at biological applications as failure to remove any reminiscent of the salt can lead to substantial cell dead.
The pore distributions measured for the different scaffolds varied depending on the concentration of the polymer and the material used (Figure 2). Previous studies have analysed the effect of porosity in the foreign body response, showing that pore diameters above 30 µm allowed cell infiltration, promoted a pro‐regenerative phenotype and angiogenesis in vivo[ 10b ] and in vitro with RAW264.7 cells.[ 18a ] However, the average macrophage size is smaller than 15 µm (with a projected area ranging from 113 to 201 µm2, equivalent to 12–16 µm diameter[ 20 ]), and thus we defined this diameter as sufficient to allow cell infiltration.
2.3. Compressive Mechanical Properties of the Scaffolds
Modulating the mechanical properties of cell culture substrates and 3D materials can determine cell fate and also modulate the innate immune response. Thus, we studied the mechanical properties of the scaffolds under compression. As expected, the semicrystalline copolymer P(l)LCL showed higher mechanical properties than the amorphous P(l,d)LCL, with higher compressive Young's modulus for higher polymer concentrations. Scaffolds produced from the melt with traditional fused deposition modeling displayed the highest Young's modulus (E), with values of 404 ± 112 and 4.9 ± 1.5 kPa for P(l)LCL and P(l,d)LCL, respectively. Increasing the fiber porosity resulted in a decreased E, with values of 40 ± 15 and 27 ± 5 kPa for P(l)LCL at 7% and 5% polymer concentration in the gel, respectively. The E measured for the amorphous P(l,d)LCL were lower, of 2.8 ± 0.4 and 2.1 ± 0.1 kPa for the 7% and 5% concentrations, respectively. It has to be noted that while these set of mechanical properties extends the range of other studies that looked at macrophage polarization,[ 8 , 11 ] they are still relatively low for additive manufactured scaffolds.
The designed dual porosity scaffolds fabricated with chemically very similar copolymers presented variations on the fiber internal pore diameter that varied with the polymer concentration and between the different types of copolymers used (Figure 2b and Table S1, Supporting Information). The internal pore diameter of P(l)LCL 7% and P(l,d)LCL 5% scaffolds was, however, not significantly different. The mechanical properties varied greatly with the polymer concentration and also the polymer used. Hence, this system allowed us to evaluate the modulation of the macrophage response on a systematic manner, isolating the parameters of mechanical properties and porosity of the scaffold.
2.4. Cytotoxicity of P(l)LCL and P(l,d)LCL in 2D Substrates
Prior to testing the capability of the dual porosity scaffolds to modulate the immune response, we evaluated the cytotoxicity of the copolymers upon culturing NR8383 rat macrophages on spin‐coated substrates.
Macrophages were cultured for 48 h in P(l)LCL and P(l,d)LCL spin‐coated substrates and in the control tissue culture plate polystyrene (TCP). Attached cells acquired a rounded morphology on the different substrates presenting an elongated or spindle morphology (only few of them) when cultured on P(l,d)LCL substrates (Figure S3, Supporting Information). The viability of the macrophages on the different substrates after 48 h of culture was visualized with live/dead assays with calcein (green, live) and ethidium bromide homodimer (red, dead). Quantification of the viable cells evidenced a good biocompatibility and resulted on values of 95 ± 4%, 98 ± 3%, and 99 ± 2% for P(l,d)LCL, P(l)LCL, and the control tissue culture plate, respectively (Figure 3a,b).
Figure 3.
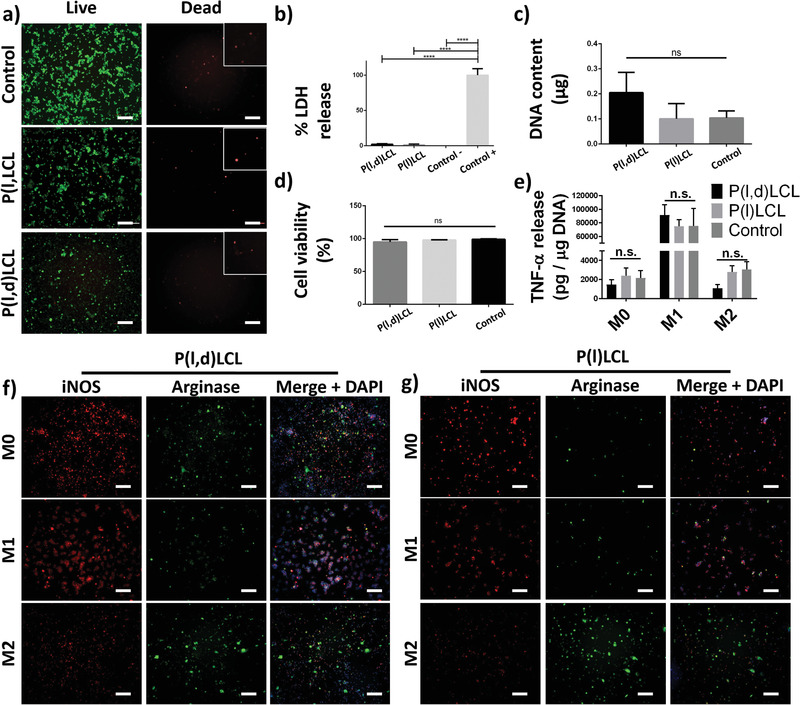
Biocompatibility of P(l)LCL and P(l,d)LCL substrates. a) Fluorescence microscopy images of macrophages cultured on P(l)LCL, P(l,d)LCl, and control TCP substrates and stained for calcein (green, live) and ethidium homodimer (red, death). Scale bar is 200 µm. Insets show higher magnification details of dead cells. Inset size is 120 × 120 µm. b) Percentage of released LDH by macrophages cultured on the copolymer substrates normalized with positive and negative controls of cells cultured on TCP. c) DNA content calculated from cells adhered in the different substrates 48 h after seeding. d) Percentage of viable cells calculated from images in (a). e) Quantification of released TNF‐α per µg of DNA by macrophages cultured on the different substrates in non‐stimulated (M0), M1 (LPS 10 + IFN‐ɣ), and M2 (IL‐13 + IL‐4) stimulated conditions. f,g) Fluorescence microscopy images of macrophages in the different states of stimulation presented in (e) and stained for iNOS (red, M1), arginase (green, M2), and DAPI (nucleus, DNA, blue). Scale bar is 200 µm. Statistical significance was calculated from one‐way ANOVA with Tukey's multiple comparison test. n = 3 for all experiments. Adjusted p‐values (****) p < 0.0001, (***) p < 0.001, (**) p < 0.01, and (*) p < 0.1.
All substrates supported cell attachment, with no significant difference on the number of attached cells or the measured DNA with values of 0.10 ± 0.06, 0.20 ± 0.08, and 0.10 ± 0.02 µg of DNA for P(l)LCL, P(l,d)LCL, and TCP substrates, respectively (Figure 3c). Despite a slight increase on the amount of adhered cells observed on P(l,d)LCL substrates, this was not significantly different from P(l)LCL and control substrates.
To further verify the cytotoxicity of the copolymers, the release of lactase dehydrogenase (LDH) to the media by cultured macrophages was evaluated (and normalized to the positive control) as a measure of cytotoxicity (Figure 3b). The two copolymers showed a very low toxicity with values of 2.1 ± 0.9% and 1.2 ± 1.1% for P(l)LCL and P(l,d)LCL, respectively. The low LDH release measured was well in accordance with cell viability and adhesion studies, supporting altogether the biocompatibility of the substrates.
2.5. Macrophage Polarization in 2D Substrates
Before evaluating the immunomodulatory properties of the dual porosity scaffolds, the potential of the macrophages to polarize toward M1 and M2 phenotypes when stimulated with traditional cytokines was evaluated. As previously reported, we used a combination of lipopolysaccharide (LPS) and interferon gamma (IFN‐ɣ), both at 10 ng mL−1, to push the macrophages toward M1 and a combination of interlukin‐13 (IL‐13) and interlukin‐14 (IL‐14), both at 20 ng mL−1, to drive them toward M2 phenotypes.
Macrophages cultured on the three different substrates proved capable of polarizing toward M1 and M2 phenotypes. Measurement of the tumor necrosis factor‐α (TNF‐α) released to the media by macrophages on the different stimulation states after 48 h of culture showed no significant differences on the cell response in function of the substrate and a clear increase on the production of the protein when cells were stimulated toward M1, thus validating the stimulation capability (Figure 3e). No significant difference was detected on the amount of TNF‐α released by macrophages on the M2 or M0 states, suggesting that none (or few) cells presented an M1 phenotype when cultured on our copolymers without addition of stimulating factors.
The polarization state of stimulated macrophages was further assessed by immunofluorescence staining of characteristic markers inducible nitric oxide (iNOS) and arginase for M1 and M2 phenotypes, respectively (Figure 3f and Figure S4, Supporting Information). Stimulation toward M1 phenotype was evidenced in P(l)LCL, P(l,d)LCL, and control TCP substrates as a high proportion of iNOS+ cells with respect to Arginase+ cells. Contrary, when cells were stimulated toward M2 phenotypes, the amount of iNOS+ cells was limited, with a majority of cells resulting Arginase+, as evidenced by the green staining. This effect was also observed when cells were cultured on the control TCP substrates (Figure S4, Supporting Information).
Non‐stimulated macrophages (M0) cultured on P(l)LCL and P(l,d)LCL substrates presented an intermediate state of those stimulated toward M1 and M2 phenotypes, with similar amounts of iNOS+ and arginase+ cells in P(l,d)LCL substrates and slightly higher amount of iNOS+ cells in P(l)LCL substrates suggesting a more pro‐inflammatory phenotype on macrophages cultured in stiffer substrates.
Observation of the morphology of macrophages revealed that some cells acquired a spindle morphology when cultured on P(l,d)LCL substrates in M0 state, unlike macrophages in any of the other substrates (Figure S4, Supporting Information). When cells were stimulated toward M1 phenotypes, regardless of the chemistry of the substrate used for culture, they appeared forming clumps, as previously reported.[ 9b ] When cells were stimulated toward M2 in P(l)LCL and P(l,d)LCL substrates, they acquired a rounded morphology with few of them presenting the characteristic spindle shape commonly ascribed to this phenotypic state.[ 9b ] Macrophages cultured on control substrates, regardless of the stimulation state, presented a rounded morphology. These differences most likely arise from the different stiffness of the culture substrate, despite all the polymer films being supported by a glass coverslip.[ 12 ]
2.6. Dual‐Porosity Scaffolds Modulate the Immune Response of Macrophages
Having proved the absence of cytotoxicity of the copolymers and their capability to support macrophage adhesion and polarization, next the potential of dual‐porosity scaffolds to modulate the immune response was evaluated in vitro.
Culture of macrophages for 48 h in 3D scaffolds produced via traditional fused deposition modeling, which led to smooth fiber surface, resulted on macrophages presenting a high expression of iNOS and no detectable signal of the M2 characteristic mannose receptor (CD‐206) for both P(l)LCL and P(l,d)LCL copolymers (Figure 4a,b). An increase of the porosity in 7% and 5% dual porosity scaffolds resulted on an increased expression of the mannose receptor that was more intense in P(l)LCL scaffolds, with overall higher mechanical properties and higher porosity, than in P(l,d)LCL scaffolds. Similar to cells cultured on 2D substrates, in dual‐porosity scaffolds macrophages still expressed iNOS but at lower intensities than their counterparts on nonporous materials.
Figure 4.
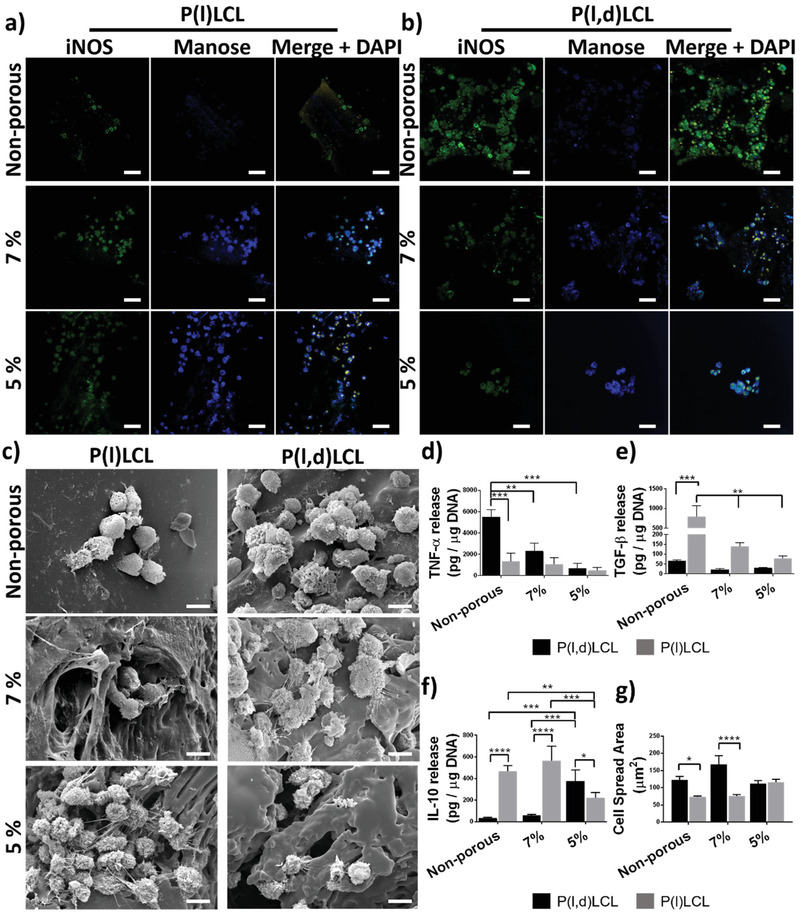
Immunomodulatory properties of dual‐porosity scaffolds. Fluorescence microscopy images of macrophages cultured on a) P(l)LCL and b) P(l,d)LCl scaffolds and stained for iNOS (green, M1), mannose receptor (blue, M2), and DAPI (nucleus, DNA, yellow). Scale bar is 50 µm. c) Scanning electron microscopy images of macrophages cultured in the scaffolds in (a) and (b). Scale bar is 10 µm. Release of characteristic cytokines d) TNF‐α, e) TGF‐β, and f) IL‐10 per µg of DNA by macrophages cultured in nonporous and dual‐porosity scaffolds prepared from 7% and 5% P(l,d)LCL or P(l)LCL copolymer gels. Data are shown as mean ± SD, n = 3 for all experiments. Statistical significance was calculated from two‐way ANOVA with Tukey's multiple comparison test. For TNF‐α: interaction F(2,12) = 5.5; p = 0.02; material type F(2,12) = 10,7, p = 0.002; porosity F(1,12) = 13.84, p = 0.002. For TGF‐β: interaction F(2,9) = 60.79, p < 0.0001; material type F(2,9) = 68.18, p < 0.0001; porosity F(1,9) = 69.28, p < 0.0001. For IL‐10: interaction F(2,9) = 17.07, p = 0.0009; material type F(2,9) = 12.46, p = 0.0026; porosity F(1,9) = 31.41, p = 0.0003. Adjusted p‐values (****) p < 0.0001, (***) p < 0.001, (**) p < 0.01, and (*) p < 0.1. g) Cell spread area of macrophages cultured in 3D scaffolds calculated from individual cells in images in (a) and (b). A total of ten cells per condition were used for statistical analysis. Data are shown as mean ± SEM. Statistical significance was calculated from two‐way ANOVA with Tukey's multiple comparison test. (****) p < 0.0001, (***) p < 0.001, (**) p < 0.01, and (*) p < 0.1.
Together with this differential expression of M1 and M2 markers in the dual‐porosity scaffolds as compared to nonporous scaffolds, a difference in the amount of cells adhered to the materials was also observed (Figure S5, Supplementary Information). Scaffolds fabricated from stiffer P(l)LCL showed a dramatic increase on the amount of adherent cells as the porosity of the scaffolds was increased. Thus, DNA content in nonporous scaffolds was 0.008 ± 0.001 µg and increased to 0.015 ± 0.005 and 0.067 ± 0.016 µg for porous scaffolds of the same material at 7% and 5% concentration, respectively. P(l,d)LCL scaffolds, however, showed the opposite trend with a progressive decrease on the DNA content as the porosity of the scaffolds was increased to values similar of those of P(l)LCL at 5% polymer concentration, the highest one. Nonporous P(l,d)LCL scaffolds had a DNA content of 0.11 ± 0.02 µg, which decreased to 0.10 ± 0.02 and 0.07 ± 0.02 µg for 7% and 5% scaffolds, respectively. These different trends observed on the cell attachment on the two copolymers suggests that there is a balance between the mechanical properties and porosity of the material that has to be met and not the mechanical properties alone, nor the porosity (or topography) are the unique determinants of macrophage adhesion.
The morphology of the cells adhered to the 3D scaffolds was greatly influenced by their topography (Figure 4c). Scaffolds produced via fused deposition modeling and presenting a smooth surface accounted for a rounded morphology in the case of P(l)LCL and rather flattened in the case of P(l,d)LCL copolymer (Figure 4c and Figure S6, Supporting Information). However, an increase in the porosity (and hence, in the surface roughness) resulted on macrophages presenting the projection of filopodia, as observed by SEM. Formation of filopodia was particularly clear for P(l,d)LCL scaffolds at any composition and was accompanied by flattening of the cells. Macrophages cultured in P(l)LCL scaffolds at 5% polymer concentration also presented the projection of abundant filopodia. It is noteworthy that the formation of filopodia appeared to be more influenced by the mechanical properties of the materials than the pore diameter as scaffolds with comparable pore sizes, P(l)LCL 7% and P(l,d)LCL 5%, presented different cell morphologies. These data are in agreement with previously reported studies where a correlation between cell morphology and phenotype was established showing that the projection of filopodia results on an increased cell elasticity and presented an M1‐like phenotype.[ 9 ]
To further study the phenotypic state of macrophages in dual‐porosity scaffolds, their immunomodulation capacity and the relationship between porosity and mechanical properties, the production and release of cytokines was analyzed (Figure 4d–f). Macrophages cultured in the nonporous scaffolds of P(l,d)LCL presented the highest release of TNF‐α as compared to porous scaffolds and scaffolds produced from the stiff P(l)LCL with a release of 5512 ± 1178 and 666 ± 245 pg TNF‐α µg−1 DNA, respectively. This cytokine release was progressively attenuated when the porosity of the softer P(l,d)LCL scaffolds was increased with 2232 ± 1033 pg TNF‐α µg−1 DNA in 7% and 345 ± 264 pg TNF‐α µg−1 DNA in 5% scaffolds, becoming the latter not significantly different than those in P(l)LCL scaffolds. Similarly, in P(l)LCL scaffolds the release of TNF‐α decreased as the porosity increased, with values of 518 ± 243 and 185 ± 65 pg TNF‐α µg−1 DNA for 7% and 5% scaffolds, respectively.
It is noteworthy that these results are in disagreement with previously reported data where it was suggested that high mechanical properties of scaffolds lead preferentially to a M1 pro‐inflammatory phenotype with higher levels of TNF‐α released when covering a range of substrates moduli of 0.3–76.8 kPa.[ 8a ] In line with this, reports on surface roughness as modulator of macrophage polarization also pointed out to a more pro‐inflammatory phenotype as the roughness was increased.[ 21 ] However, these reports make use of substrates that, unlike traditional biodegradable polyesters, are considered to have a good cell adherence; hence, smooth or stiff substrates allow cell attachment and spreading and a reduction in the mechanical properties or increase in the roughness results in a reduced surface area. Nevertheless, this and other studies also pointed out at the importance of actin polymerization and cytoskeletal organization as inductors of M1 phenotypes. Lately, the role of Yes‐associated protein (YAP)‐mediated mechanotransduction has been associated to the modulation of the immune response, prompting cells to acquire an M1‐like phenotype when YAP was localized in the nucleus in stiffer materials.[ 12 ] Considering that YAP nuclear localization has also been associated to cell organization when cultured in patterned substrates,[ 22 ] and that nuclear YAP was most prominent in cells with high surface spread area, it could be hypothesized that the differential spread surface area observed in the different scaffolds and arising from the combined effect of mechanical properties and topography, might have led in this case to the observed M1 polarization.
Macrophages cultured in P(l)LCL scaffolds, however, presented a different phenotype. The concentration of secreted TNF‐α was low and, although not significantly different, decreased with increasing porosity as observed for P(l,d)LCL. The release of pro‐regenerative cytokines such as IL‐10 and of TGF‐β was, overall, higher in all conditions as compared to P(l,d)LCL scaffolds, indicating that stiffer P(l)LCL scaffolds induced a rather anti‐inflammatory phenotype as compared to softer P(l,d)LCL scaffolds.
The release profile of IL‐10 seemed to be dependent on the porosity and presented a maximum in macrophages cultured in 7% P(l)LCL scaffolds with a pore diameter of 21 ± 0.3 µm and release values of 565 ± 133 pg IL‐10 µg−1 DNA, decreasing when the mean pore diameter was increased to 29 ± 0.5 µm for 5% P(l)LCL scaffolds. Surprisingly, at 5% polymer concentration, the release of IL‐10 was higher for the softest materials with a pore diameter of 20 ± 0.4 µm, as compared to their counterparts in the stiff P(l)LCL (374 ± 105 pg IL‐10 µg−1 DNA vs 219 ± 50 pg IL‐10 µg−1 DNA). Macrophages cultured in P(l,d)LCL scaffolds at 7% concentration and displaying a pore diameter of 8 ± 0.1 µm showed the lowest release of IL‐10 only comparable to nonporous scaffolds of the same material.
The amount of TGF‐β released by macrophages was minimum in P(l,d)LCL scaffolds, with values of 34 ± 8, 58 ± 11, and 374 ± 105 pg TGF‐β µg−1 DNA for nonporous, 7% and 5% scaffolds, respectively. TGF‐β release was highest in the stiffer P(l)LCL materials, supporting again the idea that stiffer scaffolds (>40 kPa) presented a more M2‐like pro‐regenerative phenotypes than softer P(l,d)LCL scaffolds. Moreover, a decrease in released TGF‐β was measured when the porosity of the scaffolds was increased independently of the material used. Thus, nonporous P(l)LCL scaffolds released 788 ± 484 pg TGF‐β µg−1 DNA, whereas 7% and 5% scaffolds 140 ± 29 and 78 ± 19 pg TGF‐β µg−1 DNA, respectively.
In the attempt to clarify whether the behavior observed was solely dependent on the cell surface spread area, we measured this area from light scanning microscopy images (Figure 4g). Indeed, we observed a higher surface spread area in macrophages cultured in softer P(l,d)LCL scaffolds as compared to stiffer P(l)LCL scaffolds. Increased porosity only reduced the spread area of macrophages cultured in the soft P(l,d)LCL scaffolds when a mean pore diameter of 20 ± 0.4 µm was reached at 5% polymer concentration. Contrary, macrophages cultured in P(l)LCL scaffolds increased the projected surface area only when a pore diameter of 29 ± 0.4 µm was obtained in 5% scaffolds. This increased surface spread area could partly be the reason of a higher pro‐inflammatory phenotype detected for macrophages cultured in P(l,d)LCL scaffolds.
Cells use filopodia and lamellopodia to sense their microenvironment and find adhesion or anchoring points. When they do find adhesion points, cells can apply traction forces that result in the polymerization of actin and cell spreading. We hypothesized that poorly adherent and stiff P(l)LCL scaffolds led to a low cell spread area when the surface was smooth. Yet, cell spreading was increased as soon as a certain porosity and topography (and hence higher scaffold surface area) was presented to cells. Cells in porous and stiff scaffolds projected filopodia in an attempt to find anchoring points that ultimately resulted on a higher cell projected area. Contrarily, cells on softer P(l,d)LCL scaffolds were readily able to spread when the surface of the filaments was smooth. An increase of the surface porosity and topography in this material also resulted on the projection of filopodia, for which the cell spread area had to be reduced (from an already spread state).
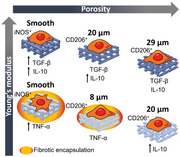
The polarization of macrophages to M2‐like phenotypes has, since some years, being classified into various subsets of macrophages that account for different marker expression and cytokine release profile, namely, M2a, M2b, M2c, and M2d phenotypes, also involved in different aspects of the immune regulation.[ 23 ] Early after implantation, during initial inflammation, macrophages polarize toward M1, presenting a high release of TNF‐α, as we observed in the case of P(l,d)LCL scaffolds, particularly in macrophages cultured in nonporous and 7% scaffolds. After initial inflammation and during wound healing, macrophages polarize toward M2 phenotypes, producing anti‐inflammatory cytokines such as IL‐10. The combinatorial production of IL‐10 and TGF‐β, as we observed for macrophages cultured in P(l)LCL nonporous and 7% scaffolds, has been suggested to be characteristic of M2a and M2b phenotypes responsible for tissue remodeling and phagocytosis of apoptotic cells.[ 24 ] These macrophages are key regulators in the resolution of inflammation and tissue remodeling, and thus we hypothesized that these scaffolds would allow for a better integration in vivo. Further, we observed that macrophages cultured in 5% P(l)LCL and P(l,d)LCL scaffolds accounted for an intermediate anti‐inflammatory release profile, with rather low amount of release TGF‐β but still high production of IL‐10, which we hypothesize that it could be a rather M2d‐like phenotype promoter of angiogenesis (Figure 5 ).
Figure 5.
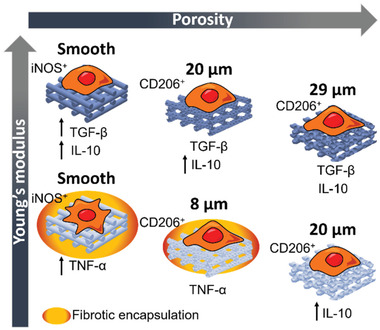
Effect of scaffold's pore diameter size and mechanical properties on the macrophage response.
2.7. Dual‐Porosity Scaffolds Modulate the Immune Response of Macrophages In Vivo
Having proved the immunomodulatory capacity of dual‐porosity scaffolds in vitro, their capability to induce an anti‐inflammatory response in vivo was further evaluated. Dual‐porosity scaffolds fabricated from 7% and 5% gels of P(l)LCL and P(l,d)LCL were, thus, implanted in a nude rat subcutaneous model. Nonporous scaffolds fabricated with the same materials were implanted as a control (Figure 6 ).
Figure 6.

Immune response to dual‐porosity scaffolds in vivo. Histological staining of nonporous and dual‐porosity scaffolds prepared from 7% and 5% copolymer gels of P(l)LCL and P(l,d)LCL after 3 and 6 weeks of implantation on a rat subcutaneous model. Samples were stained for hematoxylin and eosin‐Y (H&E) and for Masson's trichrome (MT). Scale bar in all pictures is 1 mm.
P(l)LCL scaffolds retained the structure during the entire experiment and there was no evidence of acute or chronic inflammation, showing a good biocompatibility of the material. Introduction of porosity resulted on cell infiltration through the fiber of the scaffolds, as observed by hematoxylin and eosin (H&E) staining. MT staining revealed a collagen rich matrix, as visualized in blue, surrounding and infiltrating the scaffolds. After three weeks of implantation, the formation of collagen as bundles of aligned fibers was evident surrounding the implantation site in the case of 7% and 5% dual‐porosity scaffolds and as a dense tissue for nonporous scaffolds (Figures 6 and 7 ). A detailed observation also evidenced the presence of a large amount of cells surrounding the scaffolds. Erythrocytes forming circular patterns in the proximity of the scaffolds and in between the printed strands were also observed, suggesting the formation and infiltration of vasculature. After six weeks, however, this initial fibrous collagen capsule was resolved (reducing to half of the initial measured thickness) into a dense collagenous matrix, indicating that remodeling took place in porous and nonporous P(l)LCL scaffolds (Figure 7). Moreover, a higher presence of cells was detected infiltrating the scaffolds and depositing a collagenous matrix, as evidence by MT stain. Nonporous control samples also showed tissue formation and infiltration. For all three different scaffolds, the presence of vasculature was still observed in the environment of the sample.
Figure 7.
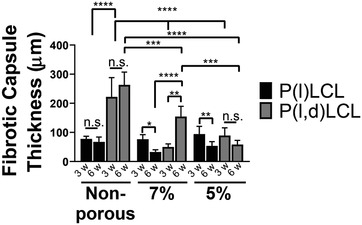
Evolution of the thickness of the formed fibrotic capsule in implanted P(l)LCL and P(l,d)LCL scaffolds after 3 and 6 weeks of implantation. Data are shown as mean ± standard deviation. w denotes weeks. Statistical significance was calculated from two‐way ANOVA with Tukey's multiple comparison test. Five measurements were taken in every sample, n = 4. Interaction F(6,33) = 4.14, p = 0.003; material type F(2,33) = 10.8, p = 0.0002; porosity F(3,33) = 11.99, p < 0.0001. Adjusted p‐values: (****) p < 0.0001, (***) p < 0.001, (**) p < 0.01, and (*) p < 0.1.
P(l,d)LCL scaffolds started to lose the integrity already after three weeks of implantation, being the initial structure only evident in samples prepared from 5% gels. All scaffolds presented the formation of a fibrous collagenous capsule surrounding the material, as evidenced by MT staining. Introduction of porosity, again, resulted on a higher cell infiltration as observed after 3 and 6 weeks of implantation by H&E staining. Samples with smaller pore size (7%) and nonporous samples presented the formation of a thicker fibrous capsule, and thus the cell infiltration in 7% samples was lower and in nonporous scaffolds not visible. The accumulation of erythrocytes and formation of vasculature on the environment of the samples was also noticed in all three scaffold types. After six weeks of implantation the initially formed fibrotic capsule increased size in nonporous P(l,d)LCL scaffolds and 7% scaffolds, becoming chronic (Figure 6). In dual‐porosity scaffolds fabricated from 5% gels, however, the thickness of the fibrotic capsule remained similar to the one observed after three weeks of implantation in P(l,d)LCL scaffolds and to the ones observed in 5% scaffolds of P(l)LCL copolymer. It is noteworthy the observed degradation of the amorphous P(l,d)LCL after six weeks of implantation in nonporous scaffolds, which might have also been responsible of the chronic inflammation.
These data are in good agreement with the results that we observed in vitro where the expression of pro‐regenerative cytokines TGF‐β and IL‐10 was highest in the stiffer scaffolds. Moreover, the release of the pro‐inflammatory cytokine TNF‐α was highest in nonporous P(l,d)LCL scaffolds, in agreement with the chronic inflammation observed in vivo for the same scaffold type. This inflammation was also gradually decreased with the increase in porosity as evidenced by measurement of the thickness of the fibrotic capsule.
To further validate the idea that stiffer scaffolds elicited a lower pro‐inflammatory response as compared to softer materials and that this response was also modulated with the porosity of the scaffolds, the phenotypic state of the macrophages surrounding the scaffolds in animal experiments was evaluated by immunofluorescence assays.
The presence of macrophages was detected in the surroundings of all the scaffolds after 3 and 6 weeks of implantation (Figure 8 and Figure S8, Supporting Information). The presence of macrophages appeared to be higher in P(l,d)LCL scaffolds and 5% P(l)LCL scaffolds as observed by the presence of F4/80+ cells. P(l,d)LCL scaffolds presented a high ratio of iNOS+:F4/80+ cells in all three conditions with barely no CD206+ cells, particularly in nonporous scaffolds, as observed from immunofluorescence assays. As the porosity of the scaffolds increased and the mechanical properties decreased, the ratio of CD206+:F4/80+ cells increased but was still low as compared to iNOS+ cells. P(l)LCL scaffolds presented, at 7% a lower ratio of iNOS+:F4/80+ cells than their counterparts at 7% or nonporous. The presence of CD206+ cells was also higher in these samples and almost nondetectable in 5% and nonporous versions.
Figure 8.
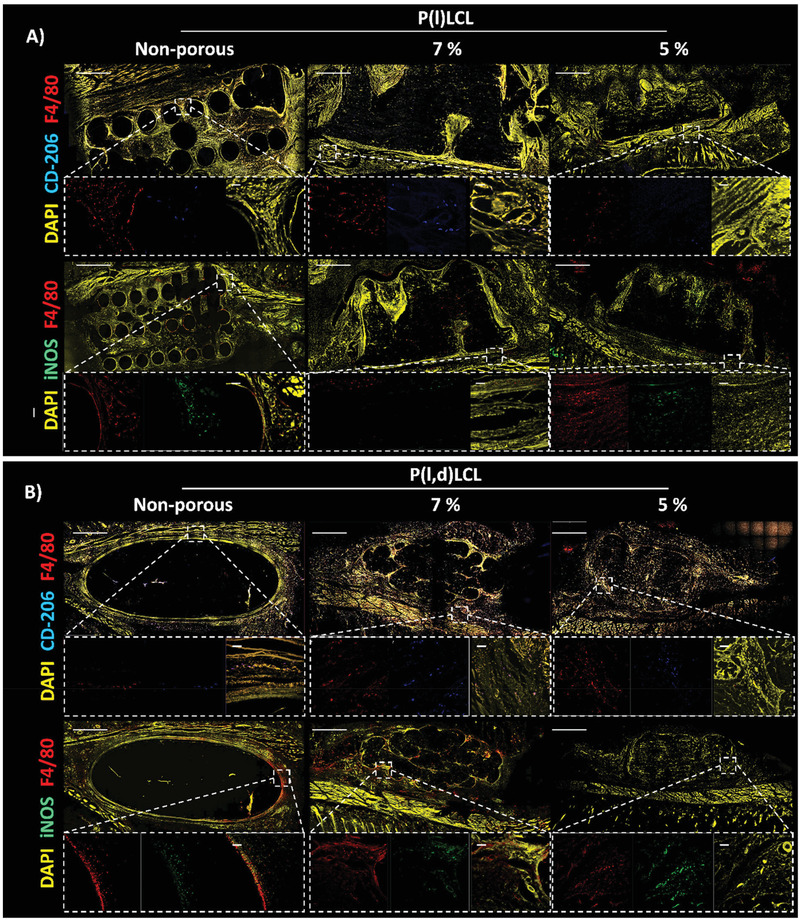
Macrophage polarization state after three weeks of in vivo implantation. Immunofluorescence staining of macrophages in the proximity to nonporous and porous P(l)LCL or P(l,d)LCL scaffolds prepared from 7% and 5% polymer gels. Macrophages were stained for F4/80 (red) and mannose receptor (CD‐206, blue, M2) or iNOS (green, M1). The entire section was also stained for DAPI (nucleus, yellow) to identify other cells. Scale bar in all images is 1 mm. Dashed line boxes indicate areas where a zoom‐in was taken. Scale bar in zoom‐in images is 30 µm.
After six weeks of implantation, the overall amount of F4/80+ cells appeared to be lower in all sample conditions. The majority of F4/80+ cells were also CD206+ for P(l)LCL scaffolds, but still some iNOS+ cells were detected in P(l,d)LCL scaffolds. However, further quantification would be needed to validate these observations.
3. Conclusions
Altogether, we fabricated a library of scaffolds based on two chemically virtually identical copolymers of poly(lacyide‐co‐caprolactone) with a 70:30 ratio of lactide:caprolactone monomers. The two materials differ in the ratio of the lactide enantiomers, and thus presented different crystallinity and mechanical properties. This allowed us to fabricate scaffolds with various porosities and mechanical properties that we used to evaluate the interplay between these two parameters in the modulation of the macrophage phenotype. Within the stiffness range here studied, scaffolds with higher mechanical properties led to a M2‐like macrophage polarization in vitro and to tissue formation and remodeling in vivo. Contrary, softer scaffolds led to a M1 polarization characterized by the production of high amount of TNF‐α. The cytokine production was regulated not only by the mechanical properties of the scaffolds, but also by their porosity. Evaluation of the cell surface spread area revealed that this is a determinant feature in macrophage polarization, resulting in a classical polarization (M1) for cells with a higher surface area and an anti‐inflammatory phenotype for cells with a reduced surface spread area. In vivo, the host immune response was regulated by the porosity and mechanical properties of the scaffolds, but highly conditioned by the degradation rate of the polymers. Thus, stiffer (semicrystalline) materials appeared integrated, prompted tissue formation and, to certain extent, vascularization, while softer and faster degrading materials (amorphous) showed chronic inflammation after six weeks of implantation. These results highlight the need to evaluate not only the stiffness or porosity of potential implant materials, but also the interplay between these to promote scaffold integration and tissue regeneration.
4. Experimental Section
Materials
Poly(d,l‐lactide‐co‐caprolactone) copolymers were kindly obtained from Polyganics B.V. (Groningen, The Netherlands) and Corbion (Amsterdam, The Netherlands) under the trade names of Poly(68/32 [15/85 D/L]) Lactide‐ε‐Caprolactone and PLC7038, respectively. Poly(68/32 [15/85 D/L]) lactide‐ε‐caprolactone was fabricated from a 68/32 lactide to caprolactone monomer ratio, with a 15/85 ratio of the d/l lactide enantiomers as described earlier.[ 25 ] PURASORB PLC7038 was synthesized from a 70/38 lactide to caprolactone monomer ratio with only l‐lactide isomer. These are referred to in the text as P(l,d)LCL and P(l)LCL respectively.
Scaffold Fabrication
Dual porosity scaffolds were prepared by the combination of additive manufacturing and TIPS. Gels of PLCL with polymer concentrations of 5% and 7% (w/v) were first prepared in a 87% (v/v) 1,4‐dioxane (Acros Organics, Thermo Fisher Scientific) solution in deionized water. For that, PLCL based polymers were dissolved by stirring for 1h in 1,4‐dioxane. Next, deionized water was added to the solution and kept stirring at 60 °C for ≈1 h, until the solution became transparent, overcoming the cloud point. Afterward, the gel was immediately placed in the fridge for at least 2 h. Thereafter, the gel was extruded with a pressure‐driven Bioplotter system (SYSENG) with a 90° pattern, a 1000 µm fiber spacing, a 0.2 mm layer thickness, and a travel speed of 200 rpm to form 20 mm × 20 mm × 4 mm scaffolds. The gel was extruded through a needle of 0.8 mm of inner diameter at 0.5–1 and 1.0–1.5 bar of pressure for 5% (w/v) and 7% (w/v) polymer concentrations, respectively. Then the scaffolds were stored at −80 °C and freeze‐dried in a FreeZone 2.5 Liter Benchtop Freeze Dry System (Labconco). As a control, nonporous scaffolds were fabricated via fused deposition modeling with a Bioscaffolder Envisiontec using the same parameters as indicated above with a printing temperature of 195 and 200 °C for P(l,d)LCL and P(l)LCL, respectively, with a G22 needle (400 µm internal diameter).
Contact Angle
Water contact angle was measured using the sessile drop method at room temperature on the surface of polymer films spin‐coated on glass slides as described below. The water contact angle, of a 4 µL drop was measured in five replicas of each condition with Drop Shape Analyzer – DSA30 (KRÜSS GmbH).
Mechanical Analysis
Samples of 4 mm × 4 mm × 4 mm were cut from printed scaffolds with a scalpel and immersed into PBS solution. The compressive modulus of the scaffolds was determined using a TA Eletroforce 3200 mechanical tester equipped with a 450 N load cell at a displacement rate of 0.01 mm s−1 and until ≈50% strain was reached. For data collection and instrument control, WinTest 7 software was used. The Young's modulus was calculated from the elastic regime of the stress–strain curve, between 0.2% and 2% deformation. Measurements were run on triplicate samples.
X‐Ray Microcomputed Tomography
Samples of ≈4 mm × 4 mm × 4 mm were scanned using a Bruker Skyscan 1272 11Mp scanner with cone beam geometry and a 4032 × 2688 detector and an isotropic voxel size of 33 µm3. The alignment, thermal drift, beam hardening, and ring artifacts were corrected using the provided software by Bruker. The acquired data were then 3D reconstructed using FDK implemented in NRecon 1.7.1.0 software and analyzed with CTAn software, both from Bruker.
Dynamic Scanning Calorimetry
The thermal behavior of the two copolymers was analyzed by DSC using a PerkinElmer DSC 8000. The thermal cycle consisted on a heating ramp from −50 to 200 °C at 20 °C min−1, followed by a cooling cycle from 200 to −50 °C at 20 °C min−1 and a final heating cycle from −50 to 200 °C at 20 °C min−1. Data presented correspond to the second heating scan.
Cell Culture
NR8383 cells, a semi‐adherent rat alveolar cell line, were obtained by American Type Culture Collection (ATCC) and cultured in 85% (v/v) of Ham's F12K medium with 2 × 10−3 m l‐glutamine adjusted to contain 1.5 g L−1 sodium bicarbonate and 15% of heat inactivated fetal bovine serum (FBS) at an initial density of 2 × 105 cell mL−1. Fresh media was added every second day and cells were subcultured when the cell density in suspension reached 4 × 105 cell mL−1. Subculture was performed by simply scrapping the cells from the culture plate and collecting all, adherent and floating cells in a centrifugation tube that was then spun at 200 rpm. The formed cell pellet was counted and plated accordingly to obtain again cultures of 2 × 105 cell mL−1.
Cell Culture on Spin‐Coated Polymer Films
For 2D experiments, the two polymers were spin‐coated on top of glass slides to form a thin layer. In brief, 18 mm glass coverslips were placed on the bottom of a 1 L beaker and covered with ≈150 mL of benzyl alcohol (Sigma‐Aldrich). The beaker was covered with aluminum foil and let boiling for 2 h at 200 °C. Afterward, the solution was let to cool down to room temperature and the glass coverslips were placed in a centrifugation tubes filled with absolute ethanol. The coverslips were then sonicated for 10 min after which the ethanol was replaced. The process was repeated three times and the coverslips were dried under a nitrogen stream. Cleaned coverslips were stored on sterile centrifugation tubes until further use. The polymer (either P(l,d)LCL or P(l)LCL) was dissolved in chloroform (Sigma‐Aldrich) at a concentration of 10 mg mL−1 by vigorous stirring and, once dissolved, filtered through a 0.22 µm pore polyethersulfone syringe filter (Acrodisc Supor). A home‐made spin‐coater was used to form a thin layer of polymer on top of the glass coverslips. For that, the glass coverslips were spun at 900 rpm for 10 s, while 50 µL of polymer solution were dispensed. The rotation speed was then increased to 1800 rpm and kept for 30 s to allow solvent evaporation to occur. Spin‐coated coverslips were kept on sterile well‐plates with the polymer film facing up until further use. For cell culture, the coverslips were sterilized by immersing them for 10 min in 70% of ethanol and then washed three times in PBS. Macrophages were seeded on the coverslip at 105 cells mL−1 in normal media with and cultured for 24 h allowing cells to attach. Afterward, media was changed, removing floating macrophages, and experiments were then started.
Cell Culture on Dual Porosity Scaffolds
Additive manufactured scaffolds were cut in cubes of ≈4 mm × 4 mm × 4 mm with the use of a scalpel. For cell culture, the scaffolds were sterilized 15 min in 70% of ethanol, washed three times with PBS and transferred to new nontreated (non‐adherent) 24‐well plates. 4 × 105 cells per scaffold in complete media (25 µL) were added on top of each scaffold and incubated at 37 °C under 5% CO2 for 2 h. The scaffolds were then flipped and incubated for another 2 h favoring a homogeneous cell distribution along the scaffold. After a total of 4 h after seeding, 1 mL of complete media was added to each well. Cells were incubated for 3 d prior to analysis of morphology, cytokine secretion, immunofluorescence, and DNA content.
Macrophage Polarization
Macrophages were cultured on 2D spin‐coated polymer films as described above. After 24 h of initial cell attachment, the media was replaced with media containing 10 ng mL−1 LPS and IFN‐γ for M1 polarization, or with 20 ng mL−1 of IL‐13 and IL‐4 for M2 (all cytokines were acquired from Prepotech). M0 macrophages were non‐stimulated. After 48 h of stimulation, the media was removed and the cells were carefully rinsed with PBS and immediately fixed for 15 min in a 4% formaldehyde (PFA) solution in PBS prepared from a concentrated stock solution (37%, Sigma‐Aldrich) and then subsequently rinsed with PBS. Fixed cells were stored at 4 °C until use.
LDH Release
The cytotoxicity of the materials was measured by the release of LDH to the media by macrophages after 48 h culture (after initial 24 h attachment) on spin‐coated polymer films. The media was recovered and centrifuged to remove any debris or loose cells and stored at 4 °C until use for a maximum of 3 d. The LDH assay (Pierce LDH Assay, Thermo‐Fisher) was performed following the manufacturer's instructions. As negative control (0% LDH release), macrophages were culture on 24‐well plates at the same cell density. For positive controls (100% LDH release) macrophages cultured under identical conditions on 24‐well plates were lysed using the 10× lysis buffer in a 1:100 dilution and incubated at 37 °C, 5% CO2 for 45 min. Samples were prepared in triplicate, and technical measurements were run in triplicate.
Cell Viability
The cell viability was visualized after 48 h of cell culture in 2D substrates by fluorescence microscopy with the calcein/ethidium bromide homodimer Live/Dead staining kit (Thermofisher), which was used following manufacturer instructions. In brief, each sample was washed with PBS and incubated in 1 mL of a solution of 1 × 10−6 m of calcein and 0.25 × 10−6 m of ethidium homodimer‐1 for 30 min at 37 °C under 5% CO2, and imaged with an inverted microscope Nikon Eclipse Ti, equipped with Ander Zyla sCMOS camera. The number of live and dead cells in each of three images per sample and replica were counted and the percentage of viable cells was calculated by dividing the number of live cells by the number of total cells per image (n = 3).
DNA Quantification
Total DNA was quantified to assess cell adhesion. Samples (n = 3), after 48 h of culture, were washed with PBS to remove non‐adhered cells. Adherent cells were scraped from the well, transferred to an Eppendorf tube and centrifuged at 200 rcf for 5 min. Supernatant was removed and cells were frozen at −80 °C. Cells were then frozen in liquid nitrogen and thawed at 56 °C for three times. Samples were then digested with 1 mg mL−1 proteinase K in Tris/EDTA buffer (pH 7.6) overnight at 56 °C. Afterwards, samples were freeze‐thawed again three more times. CyQuant Assay kit (ThermoFisher) was used to quantify total DNA following manufacturer instructions. Fluorescence was measured at emission wavelength 520 nm using a Clariostar (BMG LABTECH) spectrophotometer and DNA concentrations were calculated from a λDNA standard curve.
Scanning Electron Microscopy
The morphology of the scaffolds was characterized by SEM imaging on a FEI/Philips XL30. Samples of ≈4 mm × 4 mm were cut from the full scaffolds using a scalpel and mounted on a SEM pin stub of 12 mm diameter with the use of double‐sided carbon adhesive (PELCO Tabs, Ted Pella, Inc.). The samples were then imaged at a typical acceleration voltage of 10 kV. For macrophage imaging, samples were dried via incubation in an ethanol dilution series in PBS (50%, 70%, 80%, 90%, 96%, and 100%) with 15 min steps for each dilution. Then, 98% hexamethyldisilazane (HMDS, Alfa AesarTM, ThermoFisher) was added to the samples and incubated for 15 min at room temperature. HMDS was then removed and the samples were air‐dried overnight. Dry samples (with or without cells) were then gold sputtered (Cressington Sputter coater 108 Auto) before SEM analysis.
Quantification of Cytokine Release
The secretion of TGF‐β, TNF‐α, and IL‐10 was quantified from the culture media after 3 d of seeding and with an incubation time of 48 h. In brief, the culture media was recovered and centrifuged to separate any loose cells or debris. The recovered supernatant was stored at −30 °C until further use. TGF‐β, TNF‐α, and IL‐10 were quantified using dedicated ELISA kits (Murine TGF‐β1 precoated ELISA kit BGK04202 from Biogems, rat TNF‐α precoated ELISA kit BGK16599 from Biogems and rat IL‐10 Quantikine ELISA kit R1000 from R&D systems), and the absorbance was measured in a multiplate spectrophotometer Clariostar (BMG LABTECH). The concentration of the cytokines was calculated from a standard curve following the manufacturer instructions and was later normalized to the total DNA content of the sample, as calculated from experimental data (Section 2.13). Samples were prepared in triplicate and technical measurements were run in triplicate.
In Vivo Animal Studies
Animal studies were performed on a rat subcutaneous model. Samples of P(l,d)LCL and P(l)LCL at 5%, 7%, and 100% (eight samples of each condition) were implanted and harvested at time points of 3 and 6 weeks after surgery. Implanted samples were cut from additive manufactured full scaffolds with the help of a scalpel in to ≈4 × 4 × 4 mm cubes. The scaffolds were vacuum sealed into bags and sterilized with a 295 nm UV treatment of 4 h (2 h in each side of the cube).
All animals (Crl:NIH‐Foxn1rnu; Charles River) were 8–10 weeks old females with a weight range of 140–212 g and were housed at 21 °C with a libitum access to acidified water and food (10mm Sniff rat/mouse sterilized food in pellets). All animals were acclimatized in the animal research facility for at least one week before receiving the operation. The animals were excluded if there were infections due to surgical procedures or moderate pain that can't be released by pain medications. Prior to the operation, buprenorphine, and carprofen were administrated subcutaneously at 0.05 and 4 mg kg−1 of body weight, respectively. At surgery, animals were anesthetized with isoflurane (IsoFlo, zoeris) at 3–4% (v/v) in oxygen and air. Anesthesia was further maintained during the procedure by 2% isoflurane according to the clinical signs. The dorsal of the animals was shaved and sterilized to create four 1 cm long incisions parallel to the spine on the clean area. Four subcutaneous pockets of 10 × 10 mm were created on the back of the animal (two in each side of the spine) with scissors. A total of 96 samples were allocated into the pockets (one sample per pocket) using simple randomization. Surgeons and animal caretakers were blinded for the allocation during the conduct of the experiment. Thereafter, the incision was closed intracutaneously with Monocryl 4‐0 suture (Ethicon Inc., Johnson&Johnson, Somerville, NJ). 8 h after surgery, buprenorphine was administrated to the animals at 0.03 mg kg−1 bodyweight, and the subsequent 2 d analgesics were administrated at 4 mg kg−1 of body weight of carprofen. The welfare of the animals was evaluated on a daily basis until the animals were sacrificed with gradual CO2 overdoses. No animal was lost during the study.
At the assigned time points, the sample with the surrounding tissue was harvested and process for histology and immunofluorescence assays. All experiments and protocols complied with Dutch Animal Experimental Act and were approved by the local ethical committee and national ethical authority, central committee for animal experiments (in Dutch: centrale commissie dierproeven) (project license number: AVD107002016656).
In Vivo Sample Processing
Harvested samples were processed under sterile conditions. Tissue samples were cut with surgical scissors to a size of ≈2 × 2 cm retaining the scaffold and surrounding tissue. The samples were then rinsed in PBS and transferred immediately to a solution of 4% formaldehyde in PBS and left to fix for 24 h at 4 °C. The day after, the samples were transferred to a solution of 30% sucrose in PBS and changed gradually to a 50:50 solution of 30% sucrose:OCT compound (optimal cutting temperature compound, Scigen) and 100% OCT with 24 h incubation time in each of the solutions and at 4 °C. Thereafter, the samples were inserted on the perpendicular direction (thus, exposing the cross section on the surface) into peel‐A‐way disposable histology molds of 22 × 22 × 20 mm and froze in liquid nitrogen vapor phase. After freezing, the samples were kept at −30 °C until further use. Frozen blocks were cryo microtomed in a Leica 3050S and tissue/scaffold sections collected with the use of cryofilm (Section‐lab Co. Ltd., Japan). The cryofilm was then attached to a glass slide by the extremes with double sided cryotape (3M 9088) and kept at −30 °C until further analysis.
Immunofluorescence of 2D, 3D, and In Vivo Samples
Samples after 48 h of culture in 2D substrates or 3 d in 3D scaffolds were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and then washed twice with PBS. The samples were then permeabilized with 0.1% Triton X‐100 for 10 min and washed twice with PBS. Cells were then blocked with a solution of 3% of bovine serum albumin (BSA) and 0.01% of Triton X‐100 for 1 h at room temperature and washed 3 times with a washing buffer, consisting of 1:10 dilution of the blocking solution describe above. For 2D experiments, cells were incubated overnight at 4 °C with a 1:100 dilution of rabbit anti‐Arginase (PA5‐29645, Invitrogen) or mouse anti‐iNOS 1:500 (ab49999, abcam). Then, samples were washed three times with washing buffer. Samples were later incubated at room temperature for 1 h with the secondary antibodies anti‐rabbit Alexa Fluor‐488 donkey or anti‐mouse Alexa Fluor‐647. All the samples were counterstained with 1 mg mL−1 Hoechst dye at a 1:2000 dilution for 10 min, washed with washing buffer and maintained on PBS, and then imaged with Nikon Eclipse Ti, equipped with Ander Zyla sCMOS camera.
For experiments in dual porosity scaffolds, M2 marker, anti‐arginase was changed to anti‐mannose antibody due to the large amount of background observed for in vivo sections. Cells were stained sequentially with Alexa Fluor 647 anti‐mannose receptor antibody (ab195192, abcam) at 1:100 dilution and FITC conjugated anti‐iNOS at 1:100 dilution (A54235, antibodies.com) for 1 h at room temperature. After rinsing with washing buffer, cells were stained with 1 mg mL−1 Hoechst dye at a 1:2000 dilution for 10 min, rinsed with washing buffer and maintained in PBS until imaging.
Microtomed sample slices from in vivo experiments were first brought to RT for at least 1 h in a PBS solution, to remove the OCT. Then samples were covered with a solution of 0.05% Trypsin and 0.1% CaCl2 in distilled water and incubated for 15 min at 37 °C in a humidified chamber to retrieve antigens. Thereafter, the samples were rinsed in PBS solution and blocked on a 3% BSA solution in PBS for 1 h. Tissue sections were then rinsed with PBS for at least 10 min. Samples were stained either with Alexa 647 conjugated anti‐Mannose receptor antibody (ab195192, Abcam, 1:100 dilution), anti‐F4/80 antibody (ab240946, Abcam, 1:100 dilution), and Hoechst (1 mg mL−1 at 1:2000 dilution) or with FITC conjugated anti‐iNOS at 1:100 dilution (A54235, antibodies.com) and anti‐F4780 and Hoechst. First, samples were stained for 1 h with anti‐F4/80 at RT and then rinsed with washing buffer (described above). Thereafter, secondary antibody Alexa 594 anti‐rabbit was incubated (1:100) for 1 h at RT. After rinsing with washing buffer, conjugated antibody anti‐Mannose or anti‐iNOS were incubated for 1 h at RT and the slices were rinsed again in washing buffer. Last, tissue sections were incubated with Hoechst for 15 min at RT and rinsed thoroughly with PBS. Sections were then dehydrated following a set of 50%, 60%, 70%, 80%, 90%, and 100% ethanol solutions in PBS for 15 min each at RT. Finally, samples were let air dry for 15 min and embedded in DPX media (Sigma‐Aldrich) and mounted between two glass slides following the Kawamoto method.[ 26 ] Samples were imaged on a Leica TCS SP8 confocal microscope.
Histology
Tissue sections were brought to RT in a PBS tank for 1 h to remove the OCT. Samples were then stained for H&E or Masson's trichrome. In brief, samples for H&E were stained on a series of solutions as follows: Lillie Mayer alum hematoxylin (2 min), running tap water (5 min), 0.3% acid alcohol solution (1 min), running tap water (5 min), Scott's tap water (5 min), tap water (2 min), and eosin (2 min). Samples for Masson's trichrome were stained following a series of Weigert's iron hematoxylin (10 min), running tap water (5 min), Biebrich scarlet‐acid fuchsin (10 min), distilled water (5 min), phosphomolybdic‐phosphotungstic acid solution (5 min), anilin blue (5 min), distilled water (1 min), 1% acetic acid solution (30 s), running tap water (5 min). The two stainings were finalized with a dehydration step consisting of a 5 min incubation in a series of solutions with increasing ethanol concentration (50%, 60%, 70%, 80%, 90%, 100%) followed by a 10 s immersion in Histo‐clear solution. Samples were let air dry for 30 min and mounted using DPX media. Sections were mounted following again the Kawamoto method.[ 26 ]
Statistical Analysis
Statistical significance was calculated using GraphPad Prism 8.0.1 software for cell viability measurements, LDH release and DNA content using one‐way ANOVA with Tukey's multiple comparison test; (****) p < 0.0001, (***) p < 0.001, (**) p < 0.01, and (*) p < 0.1. For release of cytokines, cell spread area, mechanical properties, and fibrotic capsule analysis, two‐way ANOVA with Tukey's multiple comparison test; (****) p < 0.0001, (***) p < 0.001, (**) p < 0.01, and (*) p < 0.1.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the ERC starting grant “Cell Hybridge” under the Horizon2020 framework program (Grant No. 637308). S.C.E. acknowledges the supported by the Marie‐Slodowska‐Curie Action “PRIUS‐TE” under the H2020 framework program [845488]. J.F.M. acknowledges the support from the projects CICECO‐Aveiro Institute of Materials (UIDB/50011/2020 and UIDP/50011/2020), financed from the Foundation for Science and Technology/MCTES. The authors are grateful to Andrea Calore for helping with microCT analysis and to Marloes Peters and David Koper for support during the animal experiments.
Camarero‐Espinosa S., Carlos‐Oliveira M., Liu H., Mano J. F., Bouvy N., Moroni L., 3D Printed Dual‐Porosity Scaffolds: The Combined Effect of Stiffness and Porosity in the Modulation of Macrophage Polarization. Adv. Healthcare Mater. 2022, 11, 2101415. 10.1002/adhm.202101415
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Li Y., Xiao Y., Liu C., Chem. Rev. 2017, 117, 4376. [DOI] [PubMed] [Google Scholar]
- 2.a) Hutmacher D. W., Biomaterials 2000, 21, 2529; [DOI] [PubMed] [Google Scholar]; b) Hutmacher D. W., J. Biomater. Sci., Polym. Ed. 2001, 12, 107. [DOI] [PubMed] [Google Scholar]
- 3. Bao M., Xie J., Huck W. T. S., Adv. Sci. 2018, 5, 1800448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith T. D., Nagalla R. R., Chen E. Y., Liu W. F., Adv. Drug Delivery Rev. 2017, 114, 193. [DOI] [PubMed] [Google Scholar]
- 5.a) Sridharan R., Cameron A. R., Kelly D. J., Kearney C. J., O'brien F. J., Mater. Today 2015, 18, 313; [Google Scholar]; b) Chandorkar Y., Ravikumar K., Basu B., ACS Biomater. Sci. Eng. 2019, 5, 19. [DOI] [PubMed] [Google Scholar]
- 6. Huyer L. D., Pascual‐Gil S., Wang Y. F., Mandla S., Yee B., Radisic M., Adv. Funct. Mater. 2020, 30, 1909331. [Google Scholar]
- 7.a) Qian Y., Li L., Song Y., Dong L., Chen P., Li X., Cai K., Germershaus O., Yang L.i, Fan Y., Biomaterials 2018, 164, 22; [DOI] [PubMed] [Google Scholar]; b) Lv L., Xie Y., Li K., Hu T., Lu X., Cao Y., Zheng X., Adv. Healthcare Mater. 2018, 7, 1800675; [DOI] [PubMed] [Google Scholar]; c) Zhang L., Cao Z., Bai T., Carr L., Ella‐Menye J. ‐. R., Irvin C., Ratner B. D., Jiang S., Nat. Biotechnol. 2013, 31, 553. [DOI] [PubMed] [Google Scholar]
- 8.a) Patel N. R., Bole M., Chen C., Hardin C. C., Kho A. T., Mih J., Deng L., Butler J., Tschumperlin D., Fredberg J. J., Krishnan R., Koziel H., PLoS One 2012, 7, e41024; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sadtler K., Wolf M. T., Ganguly S., Moad C. A., Chung L., Majumdar S., Housseau F., Pardoll D. M., Elisseeff J. H., Biomaterials 2019, 192, 405; [DOI] [PubMed] [Google Scholar]; c) Okamoto T., Takagi Y., Kawamoto E., Park E. J., Usuda H., Wada K., Shimaoka M., Exp. Cell Res. 2018, 367, 264. [DOI] [PubMed] [Google Scholar]
- 9.a) Luu T. U., Gott S. C., Woo B. W. K., Rao M. P., Liu W. F., ACS Appl. Mater. Interfaces 2015, 7, 28665; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mcwhorter F. Y., Wang T., Nguyen P., Chung T., Liu W. F., Proc. Natl. Acad. Sci. USA 2013, 110, 17253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Garg K., Pullen N. A., Oskeritzian C. A., Ryan J. J., Bowlin G. L., Biomaterials 2013, 34, 4439; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sussman E. M., Halpin M. C., Muster J., Moon R. T., Ratner B. D., Ann. Biomed. Eng. 2014, 42, 1508; [DOI] [PubMed] [Google Scholar]; c) Tylek T., Blum C., Hrynevich A., Schlegelmilch K., Schilling T., Dalton P. D., Groll J., Biofabrication 2020, 12, 025007. [DOI] [PubMed] [Google Scholar]
- 11. Jiang S., Lyu C., Zhao P., Li W., Kong W., Huang C., Genin G. M., Du Y., Nat. Commun. 2019, 10, 3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meli V. S., Atcha H., Veerasubramanian P. K., Nagalla R. R., Luu T. U., Chen E. Y., Guerrero‐Juarez C. F., Yamaga K., Pandori W., Hsieh J. Y., Downing T. L., Fruman D. A., Lodoen M. B., Plikus M. V., Wang W., Liu W. F., Sci. Adv. 2020, 6, eabb8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Mota C., Camarero‐Espinosa S., Baker M. B., Wieringa P., Moroni L., Chem. Rev. 2020, 120, 10547; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moroni L., Boland T., Burdick J. A., De Maria C., Derby B., Forgacs G., Groll J., Li Q., Malda J., Mironov V. A., Mota C., Nakamura M., Shu W., Takeuchi S., Woodfield T. B. F., Xu T., Yoo J. J., Vozzi G., Trends Biotechnol. 2018, 36, 384. [DOI] [PubMed] [Google Scholar]
- 14. Neves S. C., Mota C., Longoni A., Barrias C. C., Granja P. L., Moroni L., Biofabrication 2016, 8, 025012. [DOI] [PubMed] [Google Scholar]
- 15.a) Di Luca A., Wijn J. R., Blitterswijk C. A., Camarero‐Espinosa S., Moroni L., Macromol. Rapid Commun. 2017, 38, 1700186; [DOI] [PubMed] [Google Scholar]; b) Kleger N., Cihova M., Masania K., Studart A. R., Löffler J. F., Adv. Mater. 2019, 31, 1903783; [DOI] [PubMed] [Google Scholar]; c) Jakus A. E., Geisendorfer N. R., Lewis P. L., Shah R. N., Acta Biomater. 2018, 72, 94; [DOI] [PubMed] [Google Scholar]; d) Moroni L., Licht R., De Boer J., De Wijn J. R., Van Blitterswijk C. A., Biomaterials 2006, 27, 4911; [DOI] [PubMed] [Google Scholar]; e) Prasopthum A., Cooper M., Shakesheff K. M., Yang J., ACS Appl. Mater. Interfaces 2019, 11, 18896. [DOI] [PubMed] [Google Scholar]
- 16.a) Jeong S. I., Kim B.‐S., Kang S. W., Kwon J. H., Lee Y. M., Kim S. H., Kim Y. H., Biomaterials 2004, 25, 5939; [DOI] [PubMed] [Google Scholar]; b) Lee J., Tae G., Kim Y. H., Park I. S., Kim S.‐H., Kim S. H., Biomaterials 2008, 29, 1872. [DOI] [PubMed] [Google Scholar]
- 17. Arias V., Olsén P., Odelius K., Höglund A., Albertsson A.‐C., Polym. Chem. 2015, 6, 3271. [Google Scholar]
- 18.a) Wang Z., Cui Y., Wang J., Yang X., Wu Y., Wang K., Gao X., Li D., Li Y., Zheng X.‐L., Zhu Y., Kong D., Zhao Q., Biomaterials 2014, 35, 5700; [DOI] [PubMed] [Google Scholar]; b) Bartneck M., Heffels K.‐H., Pan Y., Bovi M., Zwadlo‐Klarwasser G., Groll J., Biomaterials 2012, 33, 4136. [DOI] [PubMed] [Google Scholar]
- 19.a) Visscher L. E., Dang H. P., Knackstedt M. A., Hutmacher D. W., Tran P. A., Mater. Sci. Eng., C 2018, 87, 78; [DOI] [PubMed] [Google Scholar]; b) Minas C., Carnelli D., Tervoort E., Studart A. R., Adv. Mater. 2016, 28, 9993. [DOI] [PubMed] [Google Scholar]
- 20. Hoffman E., Patel A., Ball D., Klapwijk J., Millar V., Kumar A., Martin A., Mahendran R., Dailey L. A., Forbes B., Hutter V., Pharm. Res. 2017, 34, 2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Refai A. K., Textor M., Brunette D. M., Waterfield J. D., J. Biomed. Mater. Res., Part A 2004, 70A, 194; [DOI] [PubMed] [Google Scholar]; b) Hotchkiss K. M., Reddy G. B., Hyzy S. L., Schwartz Z., Boyan B. D., Olivares‐Navarrete R., Acta Biomater. 2016, 31, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., Elvassore N., Piccolo S., Nature 2011, 474, 179. [DOI] [PubMed] [Google Scholar]
- 23. Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M., Trends Immunol. 2004, 25, 677. [DOI] [PubMed] [Google Scholar]
- 24.a) Mantovani A., Biswas S. K., Galdiero M. R., Sica A., Locati M., J. Pathol. 2013, 229, 176; [DOI] [PubMed] [Google Scholar]; b) Shapouri‐Moghaddam A., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S.‐A., Mardani F., Seifi B., Mohammadi A., Afshari J. T., Sahebkar A., J. Cell Physiol. 2018, 233, 6425. [DOI] [PubMed] [Google Scholar]
- 25. Hissink C. E., Flipsen T. A. C., Kuijper H. W., Steendam R., Netherlands Patent WO 03/066705, 2003.
- 26. Kawamoto T., Kawamoto K., Methods Mol. Biol. 2014, 1130, 149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.