

Abstract
Design criteria for tissue‐engineered materials in regenerative medicine include robust biological effectiveness, off‐the‐shelf availability, and scalable manufacturing under standardized conditions. For bone repair, existing strategies rely on primary autologous cells, associated with unpredictable performance, limited availability and complex logistic. Here, a conceptual shift based on the manufacturing of devitalized human hypertrophic cartilage (HyC), as cell‐free material inducing bone formation by recapitulating the developmental process of endochondral ossification, is reported. The strategy relies on a customized human mesenchymal line expressing bone morphogenetic protein‐2 (BMP‐2), critically required for robust chondrogenesis and concomitant extracellular matrix (ECM) enrichment. Following apoptosis‐driven devitalization, lyophilization, and storage, the resulting off‐the‐shelf cartilage tissue exhibits unprecedented osteoinductive properties, unmatched by synthetic delivery of BMP‐2 or by living engineered grafts. Scalability and pre‐clinical efficacy are demonstrated by bioreactor‐based production and subsequent orthotopic assessment. The findings exemplify the broader paradigm of programming human cell lines as biological factory units to engineer customized ECMs, designed to activate specific regenerative processes.
Keywords: BMP2, bone grafts, endochondral ossification, extracellular matrices, regenerative medicine
Custom design of off‐the‐shelf tissue grafts by genetically programmed human cell lines is exemplified in the bone regeneration context. Human cartilage tissue is engineered and devitalized within a 3D culture bioreactor system, enabling scalable graft manufacturing. Following lyophilization, the designed acellular template retains regenerative cues, instructing bone repair at unprecedented efficiency, compared to growth factor delivery or living engineered grafts.
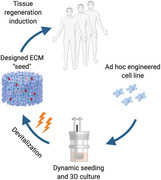
1. Introduction
Over two million individuals need a bone transplantation annually. In order to overcome the limited availability and severe donor site morbidity associated with autologous bone grafting, an increasing number of synthetic or biologically derived off‐the‐shelf materials are being proposed.[ 1 ] Yet none has matched the biological performance of autologous bone transplantation, remaining the clinical gold standard. Osteoinductive materials based on the delivery of bone morphogenetic protein 2 (BMP‐2) have not reached broad clinical adoption, since the supraphysiological dose required for a therapeutic effect can lead to ectopic bone formation or tumorigenicity. Tissue engineered grafts may offer a promising alternative,[ 2 , 3 ] although challenges related to degree and reproducibility of performance need to be overcome to justify the complexity and costs of manufacturing.[ 4 ] In order to gain effectiveness and robustness, “developmental engineering” principles[ 5 ] were proposed for the design of cellular grafts capable of instructing regeneration through activation of the processes by which tissues naturally form or repair. During embryogenesis as well as in fracture healing,[ 6 ] new bone forms by endochondral ossification,[ 7 ] whereby mesenchymal cells generate hypertrophic cartilage (HyC) as a transient template capable to attract endothelial, osteogenic and hematopoietic progenitors. As blood vessels invade the HyC, the tissue is progressively remodeled into a bone organ, including a bone marrow compartment. With the goal to recapitulate the endochondral ossification process, different groups have demonstrated that HyC can be engineered using human bone marrow‐derived mesenchymal stromal cells (hBM‐MSCs),[ 8 , 9 ] periosteum‐ or fat‐derived cells,[ 10 , 11 ] and indeed efficiently remodel into bone upon ectopic or orthotopic implantation.
The promise of engineered HyC as a bone graft substitute has further developed following the proof‐of‐principle demonstration that it can exhibit osteoinductive properties upon suitable devitalization,[ 12 ] thanks to the signaling cues embedded in the extracellular matrix (ECM) and physiologically presented[ 13 ] at the site of implantation. These findings open the possibility to bypass dependence upon autologous cells, associated with inter‐individual variability and logistic complexity, and to generate off‐the‐shelf materials as “seeds” instructing developmentally inspired bone regeneration. However, this vision is currently hampered by the rather limited performance of HyC following devitalization, as well as by the lack of a universal chondrogenic cell source enabling a scalable HyC production of standardized quality.
In this study, we aim at manufacturing human HyC as a cell‐free biomaterial harboring the necessary signals for standard and effective bone formation. Our strategy relies on the design of a human mesenchymal cell line capable of HyC formation, through constitutive BMP‐2 expression. We hypothesize that this will confer robust chondrogenic capacity, but also result in BMP‐2‐enrichment of the ECM. Following apoptosis‐driven devitalization, lyophilization and storage, we target the retention of the resulting ECM properties, while demonstrating bioreactor‐based scalability and pre‐clinical validation of performance.
2. Results and Discussion
Identifying a cell source capable of reproducible cartilage and subsequent bone formation is a critical challenge. Beyond a limited possibility for large‐scale expansion, primary cell sources typically used for cartilage engineering, namely hBM‐MSCs and human nasal chondrocytes (hNCs),[ 14 ] displayed substantial interdonor heterogeneity in chondrogenic potential (Figure S1a, Supporting Information). Moreover, immortalization by human telomerase catalytic subunit (hTERT)[ 15 ] of chondrogenic batches of hBM‐MSCs and hNCs severely reduced their chondrogenic capacity (Figure S1b, Supporting Information). We further assessed the mouse ATDC5 line, which displays limited but repeatable chondrogenesis in vitro (Figure S1c, Supporting Information). However, ectopic implantation of the resulting engineered cartilage did not initiate the process of endochondral ossification (Figure S1ac, Supporting Information).
As a means to rescue the chondrogenic potential of immortalized hBM‐MSCs, we postulated the constitutive expression of BMP‐2, a key member of the TGF‐β family involved in skeletal development and repair.[ 16 ] As starting material, we exploited a hTERT‐immortalized human mesenchymal line, endowed with a caspase‐based apoptotic inducible system (Mesenchymal Sword of Damocles, MSOD).[ 17 ] The MSOD line was lentivirally transduced for constitutive expression of the human BMP‐2 transgene and truncated nerve growth factor receptor (ΔNGFR) as surface marker reporter (Figure 1a). From the resulting heterogenous population, 48 single clones were generated, out of which one was capable of cartilage formation in micromass pellet culture (Figure S2, Supporting Information). The resulting clonal MSOD‐BMP2 (MSOD‐B) line exhibited a steady proliferation capacity and a marked increase in BMP‐2 protein secretion (Figure S3, Supporting Information), while maintaining a mesenchymal stromal phenotype (positivity for CD29, CD90, CD146, CD73, and negativity for CD34 and CD45, Figure 1b). A trilineage differentiation assay in monolayer culture highlighted the restoration of chondrogenic capacity by MSOD‐B cells (otherwise absent in MSOD), along with an increased osteogenic capacity without reduction of adipogenic differentiation (Figure 1c). In contrast to MSOD cells, chondrogenic culture of MSOD‐B cells in collagen sponges as scaffolding material resulted in frank cartilaginous tissue with hypertrophic features (Safranin‐O and collagen types II and X stainings), as well as matrix enrichment in BMP‐2 (Figure 1d). Tissues generated by MSOD‐B contained less glycosaminoglycans (GAG) and reached lower Bern scores than those formed by selected highly chondrogenic hBM‐MSC batches (Figure 1e), but accumulated significantly higher BMP‐2 amounts (40 ng per engineered tissue, Figure 1e). Upon ectopic implantation, MSOD‐B HyC underwent a complete remodeling into bone and bone marrow tissues, with superior speed and efficiency than HyC from the highly chondrogenic primary hBM‐MSCs (Figure S4, Supporting Information). Conversely, consistent with the absence of cartilage formation in vitro (Figure 1d,e), MSOD tissues did not form any bone in vivo (Figure S4, Supporting Information). We thus report the first human mesenchymal line with robust in vitro chondrogenic potential, and further capable of recapitulating the endochondral ossification process. Importantly, the chondrogenicity of MSOD‐B cells was critically dependent upon the BMP‐Smad 1/5/9 pathway activation. This was demonstrated by the targeted inhibition of BMP type I receptor using the LDN193189 compound,[ 18 ] resulting in effective blocking of Smad 1/5/9 phosphorylation while maintaining active the TGFβ‐Smad 2/3 pathway (Figure 1f). This BMP pathway inhibition did not affect the BMP‐2 secretion (Figure S5a, Supporting Information) and was associated with a clear impairment of MSOD‐B cartilage formation (Figure 1f; and Figure S5b, Supporting Information).
Figure 1.
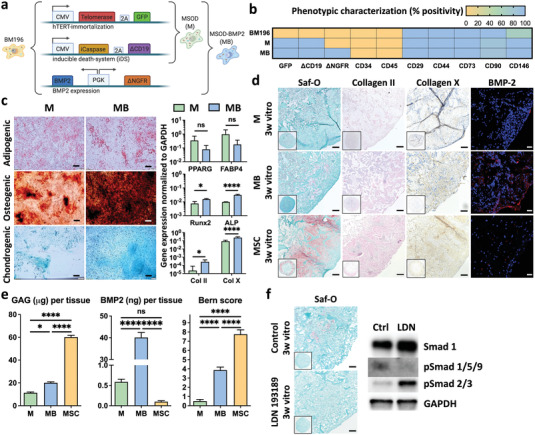
Engineering of a human mesenchymal cell line with robust chondrogenic capacity. a) Generation of mesenchymal cell lines by genetic engineering of primary human bone marrow derived mesenchymal stromal cells (hBM‐MSCs) from a single donor (BM196). Immortalization and death‐inducibility was conferred by human telomerase reverse transcriptase (hTERT) and inducible caspase 9 (iCaspase) expression respectively, leading to the MSOD (M) line. The MSOD‐B (MB) cell line resulted from the subsequent implementation of constitutive bone morphogenetic protein‐2 (BMP‐2) expression. b) The M and MB lines maintain a mesenchymal stromal phenotype when compared to their primary hBM‐MSC counterpart (BM196). c) The MB line displays in standard 2D assays an increased osteogenic and chondrogenic differentiation capacity as compared to the M line (scale bar = 200 µm) (n ≥ 3). Oil‐Red‐O, Alizarin red, and Alcian blue stainings were used, respectively, for adipogenesis, osteogenesis, and chondrogenesis. Gene expression was assessed after 2 (osteogenic) or 3 weeks (adipogenic and chondrogenic) of in vitro differentiation. The graphs represent mean + standard deviation (SD), **p ≤ 0.01, ***p ≤ 0.001, determined by two‐tailed unpaired t‐test. d) The BMP‐2 overexpression confers robust in vitro chondrogenic capacity in MB cells, leading to the formation of hypertrophic cartilage (positively stained for Safranin‐O and for Collagen types II and X), enriched in BMP‐2. In contrast, M cells are not chondrogenic. As a positive control, primary hBM‐MSCs from selected chondrogenic batches (here MSC) form mature cartilage exhibiting a limited BMP‐2 presence (scale bars = 100 µm). e) Quantitative assessments of glycosamynoglycans (GAG), BMP‐2, and Bern scoring (see ref. [ 29 ]) confirming the formation of frank cartilage by MB cells, with a net BMP‐2 enrichment remaining within physiologic ranges. MSC = highly chondrogenic hBM‐MSC batches. f) The MB chondrogenic capacity is critically dependent on the BMP‐2 Smad1/5/9 signaling pathway, since its targeted inhibition by LDN193189 molecule, despite maintenance of the TGFβ‐Smad 2/3 activation, prevents the formation of cartilage (Saf‐O staining). Phosphorylated Smad: pSmad. Scale bars = 100 µm (n = 3). The graphs represent mean + SD, *p ≤ 0.05, ****p ≤ 0.0001 determined by analysis of variance (ANOVA) test.
Figure 4.
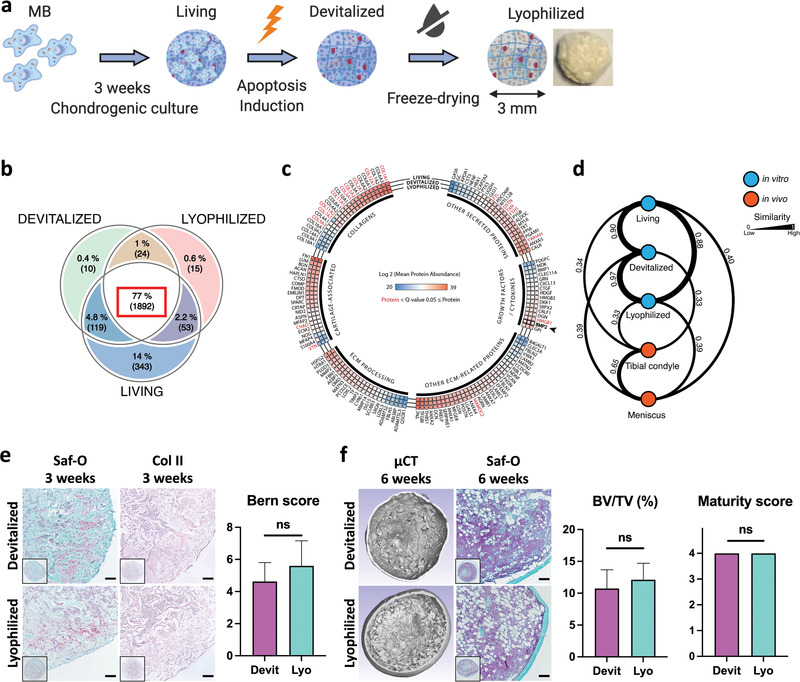
Devitalization and lyophilization of MSOD‐B cartilage result in an off‐the‐shelf graft material with preserved composition and bone formation capacity. a) Cartilage tissues were engineered by chondrogenic culture of MSOD‐B (MB) (Living group), subsequently devitalized by apoptosis induction (Devitalized) and further lyophilized (Lyophilized) through a freeze‐dry process. The impact of the devitalization and lyophilization processes was analyzed by mass‐spectrometry and functional in vivo implantation. b) Venn diagram displaying the shared or unique identified proteins within the respective groups. A total of 2456 protein—present in at least 3 biological replicates from the respective groups—were identified. The large majority of detected proteins was common to the three groups (77%, 1892 proteins) (n = 5). c) The devitalization and lyophilization of MB cartilages minimally affected their composition, as assessed through mean abundance analysis of extracellular proteins detected in each biological groups. Proteins displaying a significantly different abundance (between at least 2 groups) are highlighted in red (q‐value ≤ 0.05) (n = 5). d) Similarity score between Living, Devitalized, Lyophilized tissues, and human primary tissues (tibial condyle and meniscus). The score is derived from the abundance ranking of 34 cartilaginous proteins (see ref. [ 21 ]) and highlights the compositional similarity of MB engineered tissues. e,f) The devitalization and lyophilization did not affect the bone formation capacity of MB cartilage tissues, as assessed by histological (Saf‐O, Collagen type II) and µCT analysis of in vitro and subcutaneously implanted grafts. Scale bars = 100 µm. Bern score (n ≥ 4) quantification of in vitro tissues. Bone volume (BV) as a percentage of total volume (TV) (n = 6) and ossicle maturity scoring (n = 6) analyzed from in vivo implanted tissues. The graphs represent mean + SD. Statistical analysis based on two‐tailed unpaired t‐tests.
We next investigated the possibility to exploit the HyC engineered by MSOD‐B as an osteoinductive cell‐free material, thereby relying on cues embedded in the deposited ECM to instruct bone formation. Tissues generated under chondrogenic conditions using MSOD‐B cells or MSOD as control (Figure 2a), were efficiently devitalized by apoptosis‐induction (Figure 2b), through activation of the iCas9 suicide system (Figure 1a). This method was developed to selectively eliminate the cellular fraction while minimally impacting the deposited ECM.[ 19 ] Following subcutaneous implantation in mice, devitalized ECM materials produced by MSOD displayed poor recruitment of osteoblastic and osteoclastic cells (Osterix and TRAP stainings, Figure 2c), resulting in absence of bone formation up to 6 weeks postimplantation (microcomputed tomography (µCT) and Safranin‐O, Figure 2c). Conversely, devitalized MSOD‐B HyC underwent a highly efficient remodeling into mature ossicles with extensive bone and marrow formation as early as 3 weeks postimplantation (Figure 2c). Most remarkably, devitalized MSOD‐B HyC outperformed in time and efficiency the bone remodeling observed in living HyC generated from highly chondrogenic primary hBM‐MSCs. The latter displayed only partial bone and marrow formation at 6 weeks and persistent areas of cartilage, consistent with previous studies.[ 8 ] The more efficient remodeling of the MSOD‐B HyC was mirrored by a more vigorous osteoclast recruitment, already evident one‐week postimplantation, and by a substantially higher presence of osteoblastic cells (Figure 2c,d). The superiority of devitalized MSOD‐B tissues was confirmed by µCT and histological quantification of the bone/bone marrow structures (“ossicle maturity” scoring, Figure S6, Supporting Information) (Figure 2d). Taken together, these data demonstrate the unprecedented possibility to robustly recapitulate endochondral ossification using a devitalized HyC tissue engineered by a human cell line.
Figure 2.
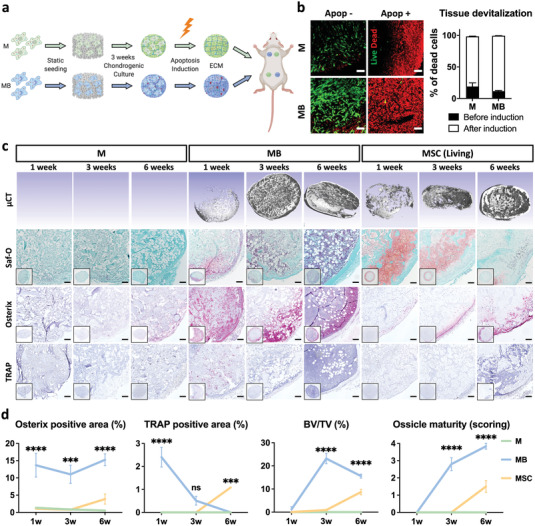
Engineered MSOD‐B cartilage is highly osteoinductive following apoptosis‐driven devitalization. a) Tissues were engineered by chondrogenic culture of MSOD (M) or MSOD‐B (MB) cells on collagen sponges, followed by devitalization through apoptosis induction. The osteoinductivity of the resulting extracellular matrices (ECM) was assessed by subcutaneous implantation in mice. b) Activation of apoptosis led to efficient tissue devitalization as assessed by live (calcein, green)/dead (ethidium bromide, red) staining and flow cytometry Annexin V/PI quantification (n = 3). c) Devitalized MB tissues underwent a fast and efficient remodeling into fully mature bone organs (microtomography, µCT), through massive osteoblastic (Osterix) and osteoclastic (tartrate resistant acid phosphatase (TRAP)) cell recruitment. The activated endogenous endochondral process bypassed in speed and final tissue maturity the performance of living cartilage generated from selected chondrogenic batches of primary hBM‐MSCs (here MSC). In strong contrast, devitalized M tissues did not form any bone. Scale bars = 100 µm. d) Histological quantifications as well as microtomography analysis (bone volume, BV, in percentage of total volume, TV) confirmed the superior and faster bone formation of devitalized MB cartilage tissues, associated with a higher host cell recruitment. Methodology for ossicle maturity score is described in Figure S6, Supporting Information (n ≥ 5). The graphs represent mean ± SD, ***p ≤ 0.001, ****p ≤ 0.0001 determined by two‐way ANOVA between MB and MSC.
We further addressed whether the efficiency of osteoinduction by the MSOD‐B HyC was related to the extent of the cartilage development or to the BMP‐2 amount in the ECM. Through time‐dependent exposure to transforming growth factor β3 (TGFβ3, for 0, 1 or 3 weeks) during MSOD‐B chondrogenic culture, we could regulate the cartilage maturation in the resulting tissues (Safranin‐O and collagen type II stainings, Figure 3a, as well as GAG content and Bern scores, Figure 3b) in a way that was decoupled from the BMP‐2 content (Figure S7, Supporting Information). Upon devitalization and in vivo implantation, the efficiency of bone formation was assessed by µCT and histological analysis (Figure 3a). The quantified bone volume and ossicle maturity scores (Figure 3b) were both positively and statistically significantly correlated with the Bern score of the implanted HyC (respectively p = 0.0009, R 2 = 0.95; p = 0.004, R 2 = 0.91; n = 12), but not with their BMP‐2 content (respectively p = 0.982, R 2 = 0.01; p = 0.717; R 2 = 0.17; n = 11). We next compared the osteoinductivity of devitalized MSOD‐B HyC to that of synthetic materials loaded with BMP‐2. We tested both a hydrogel delivery system (polyethylene glycol, PEG) and the commercially available Infuse kit (Medtronic, Dublin, Ireland), consisting in a collagen‐based scaffold and BMP‐2. When PEG‐ or Infuse materials were loaded with 40 ng of recombinant human BMP‐2, in the range of the amount measured in MSOD‐B HyC, no mineralization was observed after in vivo implantation (Figure 3c). By increasing the BMP‐2 amount by 10‐ to 100‐fold (400 and 4000 ng, respectively), we observed a dose‐dependent increase in bone formation, which however never matched the bone volume and maturity obtained using devitalized MSOD‐B HyC (Figure 3d) and suffered from limited reproducibility. Collectively, these findings indicate that the osteoinductive potency of the ECM engineered by MSOD‐B is critically dependent on its cartilaginous nature, offering the possibility to overcome the safety and performance issues associated with the high BMP‐2 doses otherwise required to induce bone formation.[ 20 ]
Figure 3.

The osteoinductivity of devitalized MSOD‐B tissues critically relies on its cartilage maturity and outperforms the delivery of supra‐physiological BMP‐2 doses. a,b) MB‐derived tissues require a constant exposure to TGFβ3 (3 weeks) to reach the best in vitro cartilage maturity (GAG and Bern score quantification, n = 4 & 6, respectively). The successful in vivo bone formation by MSOD‐B (MB) devitalized tissues is critically dependent and positively correlated to the in vitro cartilage maturity. Tissues were analyzed by µCT and histologically for the presence of bone and cartilage (Saf‐O, collagen II, and Bern score), 6 weeks after implantation (n = 6). Scale bars = 100 µm. Bone volume (BV) as a percentage of total volume (TV) (n = 6) and ossicle maturity scoring (n = 9) were analyzed from in vivo implanted tissues. c,d) The delivery of BMP‐2 through hydrogel (PEG) or collagen‐based scaffold (Infuse) does not match the bone formation capacity of MB devitalized cartilage. Tested BMP‐2 doses range from the amount measured in MB cartilage (≈40 ng), to 10‐fold (≈400 ng), and 100‐fold higher (≈4000 ng). Histology and bone quantification analysis was performed on tissues implanted subcutaneously in mice and retrieved after 6 weeks (n ≥ 3). Scale bars = 100 µm. The graphs represent mean + SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 determined by ANOVA between each group (b) or to the control (MB) (d).
Toward exploiting the MSOD‐B HyC as an off‐the‐shelf osteoinductive material, we introduced a lyophilization step in the production process. The resulting lyophilized tissue consisted of a complex ECM biomaterial displaying an average pore size of 33.28 ± 19.99 µm (scanning electron microscopy (SEM), 101 pores measured, Figure S8, Supporting Information) and a Young's modulus of 37.13 ± 8.69 kPa (unconfined compression testing, compression rate 5 µm s−1, n = 8). Using a liquid chromatography mass spectrometry (LC‐MS)‐based proteomic analysis, we compared unprocessed MSOD‐B HyC (Living), devitalized by apoptosis‐induction (Devitalized), and lyophilized through a freeze‐drying process (Lyophilized) (Figure 4a), in order to assess the protein abundance and diversity, as well as to identify possible compositional changes. After confirming the similar protein distribution in the different groups, consistent across biological replicates (Figure S9a, Supporting Information), a total of 2456 proteins could be identified (full list detailed in Table S1, Supporting Information). Out of these proteins, 77% (1892 proteins, Figure 4b) were common to all three groups. The biggest portion of nonshared proteins was specific to the Living group (14%, Figure 4b) and predominantly accounted for intracellular or membrane‐bound proteins (95%, Table S2, Supporting Information). Within the shared 1892 proteins, 1760 (93%) displayed an identical relative abundance in all three groups (Table S3, Supporting Information), while 132 (7%) were reported as significantly different (Table S4 and Figure S9b, Supporting Information; q‐value ≤ 0.05). We further identified 140 key ECM elements arbitrarily classified as collagens, growth factors/cytokines, cartilage‐associated, ECM processing and other secreted factors (Figure 4c). Out of these 140 selected proteins, 123 displayed a similar abundance in all groups. The most abundant proteins consisted of different collagen chains (type‐1, ‐3, ‐6, and ‐12), including the cartilage‐specific one (type‐2), along with typical cartilaginous proteins (Aggrecan ACAN, Cartilage oligomeric matrix protein COMP). Other abundant proteins included fibronectin (FN1), Tenascin C (TNC), Transforming Growth Factor Beta Induced (TGFBI) and BMP‐2. Importantly, these abundant proteins were detected to similar amounts in all groups, without any significant loss following devitalization or lyophilization. In order to better compare the extent of similarity between engineered cartilage tissues, we established a “similarity score” (ss) based on the “abundance ranking” of a given protein set, rather than on their absolute content (c.f. method section). Based on a list of 34 proteins, reported as main constituents of native cartilage tissues[ 21 ] (Figure S9c, Supporting Information), we confirmed the high similarity between the three groups of MSOD‐B engineered HyC samples (ss > 0.88, with 1 indicating identical ranking), but also their distinct nature from native hyaline or fibrous cartilage tissues (from healthy human tibial condyle and meniscus, respectively) (ss < 0.40) (Figure 4d).[ 21 ] Remarkably, the minimal compositional changes introduced by the devitalization and lyophilization processes had no functional impact, since a full remodeling into mature ossicles was observed following ectopic implantation (Figure 4e,f), even after storage at 4 °C for over 3 months (Figure S10, Supporting Information). Taken together, these findings indicate the possibility to introduce a lyophilization step in the production of devitalized HyC, enabling its exploitation as an off‐the‐shelf material, and provide the basis for the definition of a compositional identity.
The clinical manufacturing of engineered grafts may largely benefit from the adoption of automated culture systems, toward reduction of contamination risks, operator‐dependency, batch‐to‐batch variability and possibly production costs.[ 4 ] We thus adapted a previously developed bioreactor system (Figure S11, Supporting Information) inducing perfusion flow directly through large collagen sponges placed within a plunger.[ 22 ] The device enabled homogenous MSOD‐B distribution throughout the scaffolds, prolonged culture and devitalization of the resulting graft within the same chamber in a streamlined process. The bioreactor‐engineered tissues exhibited similar cartilaginous features than the conventional statically cultured HyC, but with a net 20‐fold increase in volume (discs of 25 mm diameter and 2 mm thickness, Figure 5a,b). Devitalized HyC discs (12 mm diameter), punched out of the larger grafts and ectopically implanted, underwent complete remodeling into bone and marrow tissue within 6 weeks, thus matching the performance of the small‐scale material (Figure 5c). We finally evaluated the bone regenerative capacity of the bioreactor‐engineered HyC in the orthotopic and weight‐bearing model of rat critical sized mandibular defects, mimicking a challenging clinical scenario of maxillofacial reconstructive surgeries.[ 23 ] A material typically used in corresponding clinical settings for its mechanical stability (SmartBone) was implanted in the bone defects, alone or combined with lyophilized HyC beams used as a bandage (Figure 5d). Following 6 weeks in vivo, all implants were still at the implantation site and led to new bone formation. However, de novo bone tissue reached the inner pores of the SmartBone material only if implanted with the HyC bandage (Figure 5e). In the presence of the HyC, histological quantifications revealed a remarkable twofold increase in both the amount of new bone tissue formed at the defect site as well as in the implant osteointegration (Figure 5f). Although the modality of osteointegration based on different possible graft shapes and positioning strategies has not been investigated, here we validated the possibility to achieve it by combining the HyC with other materials. This bears high relevance toward matching specific implantation site requirements (e.g., mechanical stability) while enhancing bone regeneneration.
Figure 5.
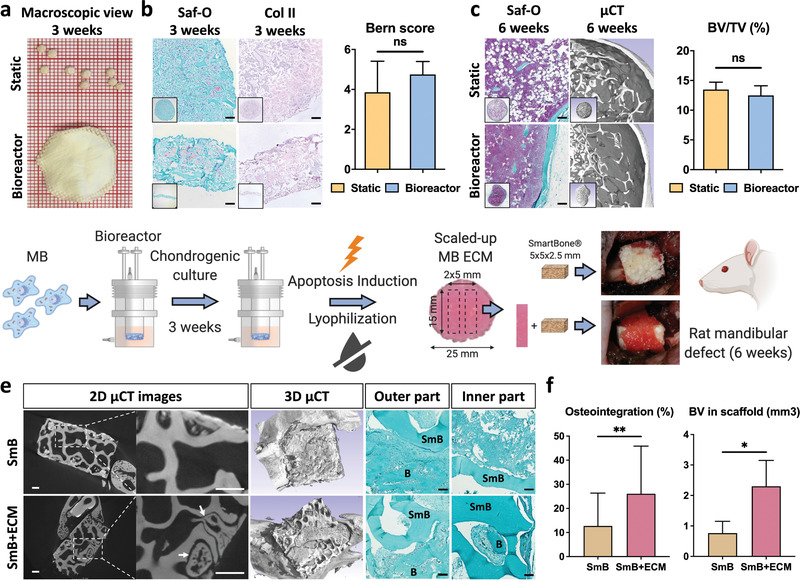
Scaled‐up, bioreactor‐manufactured human cartilage grafts enhance bone repair at an orthotopic site. a,b) Lyophilized tissues resulting from the static (top) or bioreactor culture (bottom) displayed a similar cartilage quality, as assessed histologically (Saf‐O and collagen II stainings) and by Bern scoring (n ≥ 4). c) The static and bioreactor‐generated tissues exhibited a similar osteoinductive performance in an ectopic subcutaneous model (histology analysis and µCT quantification of bone volume). To ensure a reliable bone‐to‐volume comparison, tissues of a similar dimension were implanted by punching out samples from the bioreactor‐derived cartilage discs. d) MSOD‐B (MB) cells are dynamically seeded, chondrogenically differentiated and subsequently induced to apoptosis within the bioractor device. Resulting tissues are lyophilized prior to their in vitro and in vivo evaluation. The bioreactor‐engineered cartilage repair capacity was evaluated in a nude rat mandibular defect model. The SmartBone (SmB) material was implanted alone or in combination with 5 × 15 mm bands cut out of the large 25 mm cartilage disc (SmB + ECM). e) After 6 weeks in vivo, extensive new bone formation (B) in both the inner and outer regions could be observed in the SmB + ECM implants. In constrast, SmB displayed a limited new bone formation, confined in the outer region. Scale bars = 500 µm for µCT and 100 µm for Saf‐O. f) Quantitative assessment of osteointegration and new bone volume formation confirmed the superior repair resulting from the SmB + ECM grafts (n = 3). The graphs represent mean + SD. *p ≤ 0.05, **p ≤ 0.01 determined by two‐tailed unpaired t‐tests.
3. Conclusion
We report the engineering of human HyC as off‐the‐shelf material capable to induce resident cells toward developmentally inspired processes leading to de novo bone formation. The unprecedented standardized osteoinductive performance, unmatched by commercially available materials or even living grafts, was critically dependent on a newly established cell line, whereby constitutive BMP‐2 expression enabled activation of chondrogenesis and deposition of a morphogen‐enriched ECM. Robustness, scalability and partial automatization of the manufacturing process in a bioreactor system paves the way toward clinical translation, which now requires in‐depth safety assessment and validation in large animal models. Beyond the specific context of implementation, the study introduces an important conceptual shift, from the initial paradigm of engineering living grafts through autologous or allogeneic cells[ 24 ] to the manufacturing of devitalized tissue “seeds”, orchestrating an endogenous regenerative response (Figure S12, Supporting Information). The use of cell lines facilitates not only process standardization, but also product customization. In fact, cells can be genetically modified to overexpress specific factors and then used as modular components of a molecular systems engineering approach, toward the design of ECM materials with defined biological, biochemical and biomechanical properties.[ 25 ] These matrices could thus be customized to have “beyond‐natural” functions and matched to specific requirements not only of different indications, but also of patient phenotypes and endotypes, in line with the emerging concepts of personalized medicine.[ 26 ]
4. Experimental Section
Cell Culture
Human bone marrow derived mesenchymal stromal cells (hBM‐MSCs) were isolated from human bone marrow aspirates obtained from routine orthopedic surgical procedures involving exposure of the iliac crest, after ethical approval (Ethikkommission beider Basel, Ref.78/07) and informed donor consent. Briefly, marrow aspirates (20 mL volume) were harvested from healthy donors using a bone marrow biopsy needle inserted through the cortical bone and immediately transferred into plastic tubes containing 15 000 IU heparin. After diluting the marrow aspirate with phosphate buffered saline (PBS) at a ratio of 1:4, nucleated cells were counted and seeded at a density of 3 × 106 cells cm−2 in complete medium (CM, α‐minimum essential medium (αMEM) with 10% fetal bovine serum, 1% HEPES (1 m), 1% sodium pyruvate (100 × 10−3 m) and 1% of penicillin–streptomycin–glutamine (100X) solution (all from Gibco)) supplemented with 5 ng mL−1 of fibroblast growth factor‐2 (FGF‐2, R&D Systems) and cultured in a humidified 37 °C/5% CO2 incubator. hBM‐MSCs were selected based on adhesion and proliferation on the plastic substrate one week after seeding. Cells were then detached using trypsin‐EDTA 0.05% (Gibco), counted and seeded at a density of 3000 cells cm−2 for expansion. MSOD and MSOD‐BMP2 (MB) cells were cultured in the same condition as hBM‐MSCs with a seeding density of 6000 cells cm−2.
Lentivirus Production and Cellular Transduction
The lentivirus BMP‐2/ΔNGFR was generated from the bidirectional self‐inactivating backbone vector pCCLsin.cPPT.ΔLNGFR.minCMV.hPGK.eGFP.Wpre.[ 27 ] Briefly, human codon optimized BMP‐2 cDNA, taken from Ensembl, was flanked by AgeI and SalI restriction sites. The artificial sequence was cloned in a plasmid by Gene art (Invitrogen). Then, the BMP‐2 plasmid was digested with SalI and AgeI (New England Biolabs, NEB) and the resulting fragment was ligated into the digested backbone vector in place of the eGFP driven by the human PGK (phosphor‐glycerate kinase 1) promoter. The resulting BMP‐2‐ΔNGFR vector was packaged in 293T cells by an integrase‐competent third‐generation construct and pseudotyped by the vesicular stomatitis virus G protein (VSV‐G). The three packaging plasmids were driven by the CMV promoter and included the GAG and POL gene (Plasmid #12 251, Addgene), the REV gene (Plasmid #12 253, Addgene), and the VSV‐G gene (Plasmid #12 259, Addgene). The FuGENE 6 (Promega) was used as transfection reagent. Cell culture medium was changed 16 h post‐transfection. Viral supernatant was harvested after 48 h, passed through a 0.45 µm filter and concentrated by ultracentrifugation at 70 000 g for 2 h. The concentrated virus was re‐suspended in sterile phosphate buffered saline (PBS) and stored at −80 °C until use. Viral titer was determined by transduction of 293T cells. Transduced cells were analyzed 72 h later by flow cytometry to detect ΔNGFR. Viral titers were typically in the range of 3 × 109 UI mL−1.
MSOD cells at passage 8 were plated at 6000 cells cm−2 in 150 mm dishes the day preceding the transduction. Cells were transduced overnight by incubation with BMP‐2‐ΔNGFR lentivirus vector at a multiplicity of infection of 1, followed by fresh medium replacement. Cells stably expressing hTERT‐GFP, inducible caspase 9 (iC9)‐ΔCD19 and BMP‐2‐ΔNGFR were purified using a FACS‐SOPR Aria III cell sorter (Becton Dickinson, Basel, Switzerland). Sorted cells were plated at single cell density and 48 colonies were selected from which 10 clones could be further expanded and assessed for chondrogenic micromass pellet culture.
Phenotypic Analysis
The immunophenotypic analysis of hBM‐MSCs, MSOD, and MSOD‐B lines was performed using LSR II FORTESSA SORP (BD Biosciences) cell analyzer. Cells were harvested by regular trypsinization step and labeled at 4 °C for 20 min with following fluorochrome‐conjugated antibodies diluted in PBS supplemented with 2% FBS and 0.5 × 10−3 m EDTA: human anti‐CD271 (for ΔNGFR detection, BD BIOSCIENCES cat#562 123), human anti‐CD19 (BioLegend, cat#119 520), human anti‐CD34 (BioLegend cat# 343 512), human anti‐CD45 (BD BIOSCIENCES cat#560 973), human anti‐CD29 (BioLegend, cat#303 014), human anti‐CD44 (BD BIOSCIENCES cat# 559 942), human anti‐CD73 (BD BIOSCIENCES cat# 561 014), human anti‐CD90 (BD BIOSCIENCES cat# 559 869), human anti‐CD146 (BioLegend, cat#342 003). Positive expression was defined based on superior fluorescence intensity than the respective unstained and isotype controls.
In Vitro Differentiation
MSOD and MSOD‐B lines were seeded at 6000 cells cm−2 and differentiated for 3 weeks toward the osteogenic, adipogenic and chondrogenic lineages upon reaching confluency. Osteogenic medium (OM) consisted of CM supplemented with 10 × 10−9 m dexamethasone, 0.1 × 10−3 m ascorbic acid‐2‐phosphate and 10 × 10−3 m β‐glycerophosphate. Adipogenic medium consisted of CM supplemented with 10−6 m dexamethasone, 10 µg mL−1 methyl‐iso buthylxantine, 0.1 × 10−3 m Indomethacin and 10 µg mL−1 Insulin ACTRAPID HM. Chondrogenic medium consisted of serum‐free CM supplemented with 10 ng mL−1 TGFβ3, 10−6 m Dexamethasone and 0.1 × 10−3 m ascorbic acid‐2‐phosphate. The medium was changed twice per week in each condition.
Generation of In Vitro 3D Cartilage Tissues
Tissues were cultured statically in standard 12 well culture plates coated with 2% agarose to prevent cells from attaching and proliferating at the bottom. Cells (MSOD, MSOD‐B, hBM‐MSC, human nasal chondrocytes (hNCs), and ATDC5 (99 072 806, Sigma‐aldrich)) were seeded at a density of 2 million cells in 35 µL on type I collagen sponges previously punched to a diameter of 6 mm (Avitene Ultrafoam, BD). After a 1h incubation time at 37 °C to allow cell attachment, constructs were primed toward chondrogenic differentiation for 3 weeks to achieve cartilage tissue formation. Chondrogenic medium consisted of DMEM supplemented with penicillin‐streptomycin‐glutamine (Gibco), HEPES (Gibco), sodium pyruvate (Gibco), ITS‐A (insulin, transferrin, selenium) (Gibco), human serum albumin 0.12% (CSL Behring), 0.1 × 10−3 m ascorbic acid (A5960, Sigma), 10−7 m dexamethasone (D4902, Sigma) and 10 ng mL−1 TGF‐β3 (Novartis). ATDC5 seeded tissues were differentiated using DMEM/F12 Glutamax (31 331, Gibco), 5% FBS, 100 U mL−1 penicillin and 100 µg mL−1 streptomycin (15 140 122, Gibco), 0,2 × 10−3 m ascorbic acid and ITS (41400‐045, Gibco), 10 ng mL−1 TGFβ3 (Novartis), 10−7 m dexamethasone (D4902, Sigma) for 3 weeks followed by 1 week of αMEM (22 571, Gibco), 5% FBS, 100 U mL−1 penicillin and 100 µg mL−1 streptomycin (15 140 122, Gibco), 0,2 × 10−3 m ascorbic acid (A5960, Sigma) and ITS (41400‐045, Gibco), 10 × 10−9 m β‐glycerophosphate (50 020, Sigma). The last 2 days, 50 × 10−6 m of phenanthroline (P9375‐5G, Sigma) were added in addition to the medium.
For perfusion bioreactor culture (Figure 11, Supporting Information), collagen sponges of 27 mm diameter and 2 mm thickness were placed in the bioreactor chamber stabilized by an EFTE nylon mesh (Fluorotex Sefar, 09‐590/47) and a 1 mm thick fixation ring, leading to a 25 mm diameter perfusable disc. Before injecting the cells, 35 mL of CM were injected from the bottom valve to wet the scaffold, then 40 million cells were resuspended in 5 mL of CM and injected within the bioreactor system from the top valve. After 1h static incubation to allow cell adhesion to the scaffold, additional 10 mL of CM were injected in the outside chamber and the perfusion was initiated overnight with a speed of 1.2 mm min−1. The day after, medium was changed to chondrogenic medium and replaced twice a week.
Tissue Devitalization
Following the 3 weeks in vitro differentiation, medium was changed and complemented with the AP20187 dimerizer at a concentration of 100 × 10−9 m (ApexBio) to induce apoptosis overnight. Retrieved devitalized tissues were washed once with PBS before assessment, further lyophilization or in vivo implantation. Devitalization was assessed both by confocal image analysis after live/dead staining (ThermoFischer scientific, L3224) and by flow cytometry using AnnexinV‐APC (BD Biosciences, cat#550 475) and Propidium Iodide (PI, BD Biosciences, cat# 51‐66211E) in Annexin‐V binding buffer (BD Biosciences, cat# 556 454).
Tissue Lyophilization
Tissues were transferred to screw cap eppendorf tubes and snap frozen in liquid nitrogen (LN2) prior to lyophilization. Lyophilizer (Martin Christ, Alpha 2–4 LSCplus) was set at a temperature and pressure respectively lower than −40 °C and 0.05 mbar overnight with the tubes slightly unscrewed to allow for tissue water sublimation. Upon retrieval, tissues were either stored at 4 °C, implanted in vivo after rehydration or analyzed.
Glycosaminoglycans (GAG) Measurements
GAG content was assessed using the Barbosa method.[ 28 ] Briefly, samples were digested overnight at 56 °C in 1 mL of proteinase K solution (Sigma Aldrich, P2308), and 100 µL of the resulting digested solution was incubated with 1 mL of DMMB solution (16 mg L−1 dimethylmethylene blue, 6 × 10−3 m sodium formate, 200 × 10−3 m GuHCL, all from Sigma‐Aldrich, pH 3.0) on a shaker at room temperature for 30 min. After centrifugation, precipitated DMMB‐GAG complexes were dissolved in decomplexion solution (4 m GuHCL, 50 × 10−3 m Na‐Acetate, 10% Propan‐1‐ol, all from Sigma‐Aldrich, pH 6.8) at 60 °C for 15 min. Absorption was measured at 656 nm and corresponding GAG concentrations were calculated using a standard curve prepared with purified bovine chondroitin sulfate (Sigma‐Aldrich).
BMP‐2 Quantification
BMP‐2 protein content within supernatant and tissues was assessed using the human BMP‐2 DuoSet ELISA (R&D Systems) according to the manufacturer's instructions.
Histological Analysis
Samples used for histology were embedded in paraffin and sections of 5 µm thickness were prepared using a microtome (Microm, HM430, Thermo Scientific). Safranin‐O, Alizarin red, hematoxylin/eosin, TRAP, osterix, collagen type II, and X stainings were performed as previously described.[ 8 ]
BMP‐2 staining was performed using the goat anti‐BMP‐2 antibody (Santa Cruz, sc‐6895) at a dilution 1:200 in 2% bovine serum albumin (BSA) in phosphate buffer saline (PBS), overnight at 4 °C, following a step of deparaffinization and antigen retrieval for 60 min at 95 °C (KOS microwave tissue processor, Milestone) in citrate buffer pH6.0. Secondary donkey anti‐goat IgG alexa 494 antibody (Life Technologies, A11058) at a concentration 1:200 in 2% PBS/BSA was incubated for 90 min at room temperature. DAPI was applied for 5 min at the end of the staining. Washing steps were performed with PBS and slides were mounted using Dako Faramount Aqueous mounting medium Ready to use (S3025).
Bern Score
Bern score quantification was based on cartilage tissue quality and was performed as previously published.[ 29 ] Briefly, each tissue section is scored from 0 to 3 using three different categories, namely: uniformity and darkness of Safranin‐O‐fast green stain, distance between cells/amount of matrix accumulated, and cellular morphology. Score from each category of each tissue section was then added up for a maximum score of 9. Scoring was performed blindly by two different experimenters on at least three tissue sections from different tissues representing a middle section of tissue pellets/ossicles. Each tissue section's average is transposed into a single value.
Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT‐PCR)
Total RNA was extracted from in vitro engineered tissues using the Quick‐RNA extraction kit (Zymo Research, R1055), according to manufacturer's instructions. Following cDNA synthesis (Invitrogen 18 080 044 & Promega, C1181), qRT‐PCR was performed using assay on demand (Applied Biosystems) for the following genes: glyceraldehyde 3‐phosphate dehydrogenase (GAPDH, Hs02758991_g1), fatty acid binding protein 4 (FABP4, Hs01086177_m1), peroxisome proliferator‐activated receptor gamma (PPARG, Hs00234592_m1), runt‐related transcription factor 2 (Runx2, Hs00231692_m1), alkaline phosphatase (ALP, Hs01029144_m1), collagen type II (ColII, Hs00264051_m1), collagen type X (ColX, Hs00166657_m1). BMP‐2 optimized sequence was submitted to TaqMan gene expression assay (Thermofisher scientific) for custom gene expression assay.
BMP‐2 Gels Formation
PEG hydrogels were generated as previously described.[ 30 ] Briefly, an equimolar ratio of 8‐armed PEG equipped with peptides that allow for cross‐linkage by transglutaminase factor XIII and cell mediated degradation (PEG‐MMP sensitive‐Lys: FKGG‐GPQGIWGQ‐ERCG and PEG‐Gln: NQEQVSPL‐ERCG) was cross‐linked by addition of 10 U mL−1 of factor XIII in 50 × 10−3 m Tris buffer, pH7.6 in presence of 50 × 10−3 m CaCl2, to generate hydrogels with 1.5% w/v PEG. BMP‐2 was added to the pregel mixture and 35 µL gels were made.
BMP‐2 Collagen Sponges Generation
The in vivo bone forming capacity of the MSOD‐B cartilage tissue was compared with the FDA approved absorbable collagen sponge loaded with different doses of recombinant human BMP‐2 (ACS, supplied with the Medtronic InductOs kit, Medtronic, Dublin, Ireland), prepared according to the manufacturer instructions. Briefly, rhBMP‐2 loaded ACS scaffolds (ϕ = 6 mm, h = 5 mm, dimensions of the dry scaffold) were prepared by soak loading of the protein (stock: 0.2 mg mL−1 in PBS) by pipetting the required protein amount in 20 µL PBS. All samples were incubated with the protein at room temperature for at least 30 min prior to implantation. Three different BMP‐2 doses per scaffold were assessed; 40, 400,g and 4000 ng. Five constructs were generated per condition and subsequently implanted in subcutaneous pockets in athymic nude mice (8 weeks old) for in vivo evaluation.
In Vivo Implantation
All animal studies were approved by the corresponding ethical authorities in Sweden or Switzerland. In Sweden, mouse experiments were approved by the Swedish Board of Agriculture (animal ethical permit 15485‐18). In Switzerland, experiments were approved by the Swiss Federal Veterinary Office (permit 1797 for the mice and 3005 for the rats). Analgesia was provided 1 h prior to surgery by subcutaneous injection of Buprenorphin (0.1 mg kg−1 body weight). Anesthesia with isofluran (2.5%) was maintained on demand with oxygen as a carrier (0.6 L min−1). Prior to surgery the fur was shaved if necessary and disinfected with 70% ethanol. For subcutaneous implantation in mice, 6–10 weeks old female CD‐1 nude mice were used. Two midline incisions (≈5 mm) of the dorsal skin were performed using scissors under sterile conditions and up to four tissues were implanted per animal.
For the rat mandibular defects, surgery was performed on 12 weeks old nude male rats. A dose of Buprenorphin 0.05 mg kg−1 was administered subcutaneously prior to the surgery. An eye ointment was applied to prevent eyes desiccation. In order to avoid any eventual respiratory depressions that may occur during the procedure, the animals were monitored for anesthetic depth by tail pinch reflex and respiratory frequency on a regular basis. The animals were placed on a heating pad to prevent hypothermia. The jaw was shaved on the surgical side and disinfected using Octeniderm. A 20 mm skin incision was made using a scalpel, and the mandibula was exposed at the area of the angle following blunt detachment of masseter and pterygoideus medialis muscles. Battery‐operated electrocautery was used to cauterize small and superficial venous structures when necessary. The segmental defect was created in the mandible with a piezo‐electric instrument. After creating a 5 × 5 mm2 bony defect, the mandibular segment was removed. In the defect, a specifically designed SmartBone (Industrie Biomediche Insubri SA, Mezzovico‐Vira, Switzerland) implant of 5 × 5 × 2.5 mm was implanted either alone or wrapped in a pre‐cut band of 14 mm length and 5 mm width of engineered cartilage tissue, to assess bone formation enhancement in the jaw. The implant was press‐fitted and the wound was carefully closed using 4.0 Vicryl resorbable sutures (Johnson & Johnson, St. Stevens‐Woluwe, Belgium). The sutures were removed after 10 days. Analgesia was given every 8 h for the first 48 h by subcutaneous injection of Buprenorphin 0.05 mg kg−1 body weight and in the drinking water during the night. Meloxicam 1 mg kg−1 was administered subcutaneously at the end of the procedure and every 24 h thereafter for the first week. At the end of the experiment (6 weeks), all animals were sacrificed by CO2 inhalation.
Western Blot
Protein retrieval from tissues was achieved by placing them in lysing matrix M tubes (MP) with 300 µL Radio‐Immunoprecipitation Assay Buffer (RIPA, Sigma, R0278) supplemented with PhosSTOP (Roche) and cOmplete (Roche) according to manufacturer's instructions. Tissues were lysed using the FastPrep‐24 Classic bead beating grinder and lysis system for twice 30 s. The samples were transferred into a new tube and frozen (−80 °C) or used directly. After centrifugation (5 min, 20 000 rcf, 4 °C), the supernatants were collected and the protein concentration was determined using a BCA protein assay kit (ThermoFisher). 5 µg of protein from each sample were subjected to 7.5% SDS/PAGE and then transferred onto PVDF membranes (Millipore). After blocking (5% fat‐free powdered milk in TBS with 0.1% tween for 1 h), membranes were probed with primary antibodies against phospho‐Smad1/5/9, phosphor‐Smad2/3, Smad1, and Smad2/3 (Cell Signalling). Anti‐GAPDH antibody (Origen) was used as loading control. Blots were then exposed to appropriate peroxidase‐coupled secondary antibodies (Southern Biotech). Protein detection was done using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and signals were acquired with Fusion FX imaging system (Vilber).
Scanning Electron Microscopy
SEM was carried out on lyophilized sample after three weeks of chondrogenic differentiation. Tissues was mounted and examined in a Jeol JSM‐7800 FEG‐SEM at Lund University Bioimaging Centre (LBIC). Pore size was calculated by analyzing 101 pores length with ImageJ.
Young's Modulus Measurement
After three weeks of chondrogenic differentiation, the HyC modulus was evaluated using unconfined compression testing. The samples (n = 8) were placed between the two impervious metal plates of the mechanical tester (Instron 8511.20, Canton, MA, USA) and compressed at a rate of 5 µm s−1 using a cell load of 250N (Interface SMT1, Scottsdale, AZ, USA). The Young's modulus was calculated by applying a linear fit on stress–strain curves of samples in the range between 2% and 8% of strain.
Mass Spectrometry Analysis
Sample Preparation: Tissues were processed as previously described.[ 21 ] Briefly, tissue pellets were frozen by liquid nitrogen and pulverized using a pestle and mortar technique. After weighing, proteins were extracted using 15 volumes v/w of chaotropic buffer (4 m GdnHCl, 50 × 10−3 m NaAc, 100 × 10−3 m 6‐aminocaproic acid, 5 × 10−3 m benzamidine, 5 × 10−3 m N‐ethylmaleimide, pH 5.8) for 24 h on an orbital shaker at 4 °C. Samples were centrifuged (13 200g for 30 min) and extracts were precipitated with 9 volumes of ethanol overnight at 4 °C, followed by centrifugation (13 200g for 30 min). The precipitation step was repeated and resulting pellets were dissolved in 0.1 m ammonium bicarbonate pH 8.5. All samples were digested using 1 mg trypsin (Promega, sequencing grade) by incubating on a shaker at 37 °C for 16 h. Peptide concentrations of the digests were determined using a colorimetric assay (Pierce). Samples were diluted 2x using 1 m NaCl and cleaned up by ultrafiltration to remove peptides with glycosaminoglycan (GAG) chains (Nanosep 30 K, PALL). Samples were subsequently desalted using C18 spin columns (UltraMicro Spin, HarvardApparatus) and eluted using 50% acetonitrile in 0.1% formic acid. Samples were dried in a Speedvac and dissolved in 0.1% formic acid before running MS.
Instrument Settings: MS was performed on an orbitrap Fusion mass spectrometer, equipped with a EASY spray ion source, coupled with an EASY‐nLC 1000 pump (all Thermo Fischer Scientific). For the global analysis, 1 µg of peptide digest was injected and concentrated on an Acclaim PepMap 100 C18 precolumn (75 µm × 2 cm, Thermo Scientific) before separation on an Acclaim PepMap RSLC column (75 µm × 25 cm, Thermo Scientific) at 45 °C with a flow rate of 300 nL min−1. Solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) were used to create a nonlinear binary gradient. The percentage of solvent B was maintained at 3% for 3 min, increased from 3% to 30% for 90 min and then increased to 60% for 15 min and then increased to 90% for 5 min and then kept at 90% for another 7 min to wash the column. The MS was operated in the positive data‐dependent acquisition (DDA) mode. Peptides were ionized with a spray voltage of 2 kV and the capillary temperature was set 275 °C. Full MS survey scans from m/z 350–1350 with a resolution of 120 000 were used and the automatic gain control (AGC) target was set to 4 × 105 with an injection time of 50 ms. The most intense ions (top 20) with charge states 2–5 from the full scan MS were selected for fragmentation. The MS2 precursors were isolated with a quadrupole mass filter set to a width of 1.2 m z−1. Precursors were fragmented by high‐energy collision dissociation (HCD) at a normalized collision energy (NCE) of 30%. In MS/MS, resolution was fixed at 30 000, and the AGC target and injection time were 5 × 104 and 54 ms, respectively. The duration of dynamic exclusion was set to 45 s and the mass tolerance was 10 ppm.
Protein Identification and Quantification
Identification was performed using the UniProt human database (UP5640, 2018‐10) with Proteome Discoverer 2.2 (Thermo Scientific). Label‐free protein abundance quantification was obtained by summing peak area intensities from multiple unique peptides for each protein. The processing workflow consisted of the following nodes: Spectrum Selector for spectra pre‐processing (precursor mass range: 350–5000 Da; S/N Threshold: 1.5), Sequest‐HT search engine (Enzyme: Trypsin; Max. missed cleavage sites: 2; peptide length range 6–144 amino acids; precursor mass tolerance: 10 ppm; fragment mass tolerance: 0.02 Da; static modification: cysteine carbamidomethylation; dynamic modification: methionine and proline oxidation, protein N‐terminal acetylation, and Percolator for peptide validation (false discovery rate (FDR) <1% based on peptide q‐value). Results were filtered to keep only the Master protein with at least one unique peptide, and protein grouping was allowed according to the parsimony principle. The protein FDR was set to 0.01. Multiple peptides were measured for each protein using discovery proteomics, label‐free quantification was obtained by summing up peak area intensities from unique peptides for each protein. Peptide intensities were quantified using an algorithm with feature detection and matching in Proteome Discoverer 2.2. Only proteins that were detected in all replicates of at least one sample group were kept for statistical comparison. Quantitative results were normalized against the total peptide amount. The differential expression was analyzed by ANOVA (background based) and adjusted p‐values were calculated using Benjamini–Hochberg method.
Proteomic Similarity Score
A similarity score (ss) has been set in order to compare the extent of similarity between two tissues based on the “abundance ranking” of a given protein set. The selected set corresponds to 34 proteins reported as main constituents of native cartilage tissues[ 21 ] (Figure S8c, Supporting Information). A: The equation first sums the absolute differences of ranking (R) of each protein (P) from the given protein set (N), between two tissues (T1 and T2). B: In order to normalize the obtained value and set a score comprised between 0 and 1, the sum is further divided by the maximum possible ranking sum for a given set of N proteins. C: In order to set 0 and 1 as lowest and highest similarity respectively, a final subtraction has been introduced.

S T1/T2 = Similarity coefficient of Tissue 1 (T1) and Tissue 2 (T2) based on a selected number N of proteins. The value ranges from 0 for absence of similarity, to 1 for full similarity. (Note that S T1/T2 = S T2/T1)
N = total number of proteins used for the similarity assessment
R T1 >Pi = Rank of protein abundance for “Protein i” (Pi) in Tissue 1 (T1), ranges from 1 to N
R T2 >Pi = Rank of protein abundance for “Protein i” (Pi) in Tissue 2 (T2), ranges from 1 to N.
Microcomputed Tomography
Samples were retrieved and fixed overnight in 4% formalin solution at 4 °C. Microtomography of the explants was performed using a tungsten X‐ray source at 70 kV and 260 µA with an aluminum filter of 0.5 mm (Nanotome, GE). Transmission images were acquired for 360° with an incremental step size of 0.25°. Volumes were reconstructed using a modified Feldkamp algorithm (software supplied by manufacturer) at a voxel size of 10 µm. Thresholding, segmentation and 3D measurements were performed using the VG Studio Max software. After microtomography, samples were decalcified in 15% EDTA solution (Sigma‐Aldrich) before histology.
The experimental comparison between the MSOD‐B HyC and the Infuse samples was performed in Lund. Following 6 weeks in vivo, constructs were explanted and scanned on a NanoScan PET/CT scanner (Mediso Medical Imaging Systems, Budapest, Hungary). Micro‐CT was performed at an x‐ray source voltage of 65 kV with an exposure time of 1300 ms and a total of 480 projections were acquired. Postimaging, the raw CT projections were reconstructed to an isotropic voxel size of 19 µm using Nucline software program (inviCRO, Boston, MA, USA). Reconstructed DICOM images were imported into Seg3D2 (v2.2.1, National Institutes of Health, NCRR CIBC, University of Utah, USA) and samples were individually cropped before further processing. A uniform threshold of HU 2700 corresponding to a density of 0.18 g cm−3 hydroxyapatite was applied to the individual specimens to quantify the volume of mineralized tissue, i.e., bone volume (BV). BV among different treatment groups was compared using a nonparametric statistical test (Kruskal Wallis test with Dunn's post‐hoc method) using GraphPad Prism (Prism 8 for MacOS, v8.3.1, GraphPad Software LLC, USA).
Regarding the osteointegration quantification of samples implanted in the mandibular defect, each constructs (n ≥ 3 per condition) were analyzed for the presence of adjacent mandibular bone formation (within 0.5 mm of the SmartBone scaffold edges), using four equidistant zones separated from each other by 1 mm. Length of adjacent bone (osteointegration) was represented as a percentage of total scaffold length around the edges of the SmartBone.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
P.E.B. and I.M. contributed equally to this work. The authors would like to thank Prof. Giuseppe Perale from Industrie Biomediche Insubri SA, Mezzovico‐Vira, Switzerland (IBI‐SA) for kindly providing us the SmartBone material used for the orthotopic implantation in rats. They also thank Dr. Laura Power and Davide Mogentale for their contribution, respectively, to the statistical analysis and to use of the bioreactor system. Figures were created with BioRender.com. Funding for the work was received by the Swiss National Science Foundation (Grant No. 310030‐133110 to I.M.), the Arbeitsgemeinschaft Osteosynthese (AO) Foundation (Development Incubator grant to P.B. and I.M.), the Knut and Alice Wallenberg Foundation, the Medical Faculty at Lund University and Region Skåne (grants to P.E.B), and the Swedish Research Council (Starting Grant No. 2019‐01864 to P.E.B). [Correction added on 4 May 2022, after first online publication: CSAL funding statement has been added.]
Open Access Funding provided by Universitat Basel.
Note: The author associated with the last affiliation, Department of Biomedical Engineering, Lund University, was corrected to Prof. H Isaksson on October 28, 2021, after initial publication online. The wrong author was initially credited.
Pigeot S., Klein T., Gullotta F., Dupard S. J., Garcia A. Garcia, García‐García A., Prithiviraj S., Lorenzo P., Filippi M., Jaquiery C., Kouba L., Asnaghi M. A., Raina D. B., Dasen B., Isaksson H., Önnerfjord P., Tägil M., Bondanza A., Martin I., Bourgine P. E., Manufacturing of Human Tissues as off‐the‐Shelf Grafts Programmed to Induce Regeneration. Adv. Mater. 2021, 33, 2103737. 10.1002/adma.202103737
Contributor Information
Ivan Martin, Email: ivan.martin@usb.ch.
Paul E. Bourgine, Email: paul.bourgine@med.lu.se.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Koons G. L., Diba M., Mikos A. G., Nat. Rev. Mater. 2020, 5, 584. [Google Scholar]
- 2. Okuchi Y., Reeves J., Ng S. S., Doro D. H., Junyent S., Liu K. J., El Haj A. J., Habib S. J., Nat. Mater. 2021, 20, 108. [DOI] [PubMed] [Google Scholar]
- 3. Chen D., Wu J. Y., Kennedy K. M., Yeager K., Bernhard J. C., Ng J. J., Zimmerman B. K., Robinson S., Durney K. M., Shaeffer C., Vila O. F., Takawira C., Gimble J. M., Guo X. E., Ateshian G. A., Lopez M. J., Eisig S. B., Vunjak‐Novakovic G., Sci. Transl. Med. 2020, 12, eabb6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin I., Simmons P. J., Williams D. F., Sci. Transl. Med. 2014, 6, 232fs16. [DOI] [PubMed] [Google Scholar]
- 5. Lenas P., Moos M., Luyten F. P., Tissue Eng., Part B 2009, 15, 395. [DOI] [PubMed] [Google Scholar]
- 6. Deschaseaux F., Sensébé L., Heymann D., Trends Mol. Med. 2009, 15, 417. [DOI] [PubMed] [Google Scholar]
- 7. Kronenberg H. M., Nature 2003, 423, 332. [DOI] [PubMed] [Google Scholar]
- 8. Scotti C., Piccinini E., Takizawa H., Todorov A., Bourgine P., Papadimitropoulos A., Barbero A., Manz M. G., Martin I., Proc. Natl. Acad. Sci. USA 2013, 110, 3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McDermott A. M., Herberg S., Mason D. E., Collins J. M., Pearson H. B., Dawahare J. H., Tang R., Patwa A. N., Grinstaff M. W., Kelly D. J., Alsberg E., Boerckel J. D., Sci. Transl. Med. 2019, 11, eaav7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guerrero J., Pigeot S., Müller J., Schaefer D. J., Martin I., Scherberich A., Acta Biomater. 2018, 77, 142. [DOI] [PubMed] [Google Scholar]
- 11. Nilsson Hall G., Mendes L. F., Gklava C., Geris L., Luyten F. P., Papantoniou I., Adv. Sci. 2020, 7, 1902295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bourgine P. E., Scotti C., Pigeot S., Tchang L. A., Todorov A., Martin I., Proc. Natl. Acad. Sci. USA 2014, 111, 17426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martino M. M., Briquez P. S., Güç E., Tortelli F., Kilarski W. W., Metzger S., Rice J. J., Kuhn G. A., Mul�ler R., Swartz M. A., Hubbell J. A., Science 2014, 343, 885. [DOI] [PubMed] [Google Scholar]
- 14. Mumme M., Barbero A., Miot S., Wixmerten A., Feliciano S., Wolf F., Asnaghi A. M., Baumhoer D., Bieri O., Kretzschmar M., Pagenstert G., Haug M., Schaefer D. J., Martin I., Jakob M., Lancet 2016, 388, 1985. [DOI] [PubMed] [Google Scholar]
- 15. Simonsen J. L., Rosada C., Serakinci N., Justesen J., Stenderup K., Rattan S. I. S., Jensen T. G., Kassem M., Nat. Biotechnol. 2002, 20, 592. [DOI] [PubMed] [Google Scholar]
- 16. Salazar V. S., Gamer L. W., Rosen V., Nat. Rev. Endocrinol. 2016, 12, 203. [DOI] [PubMed] [Google Scholar]
- 17. Bourgine P., Le Magnen C., Pigeot S., Geurts J., Scherberich A., Martin I., Stem Cell Res. 2014, 12, 584. [DOI] [PubMed] [Google Scholar]
- 18. Brazil D. P., Church R. H., Surae S., Godson C., Martin F., Trends Cell Biol. 2015, 25, 249. [DOI] [PubMed] [Google Scholar]
- 19. Bourgine P. E., Pippenger B. E., Todorov A., Tchang L., Martin I., Biomaterials 2013, 34, 6099. [DOI] [PubMed] [Google Scholar]
- 20. James A. W., LaChaud G., Shen J., Asatrian G., Nguyen V., Zhang X., Ting K., Soo C., Tissue Eng., Part B 2016, 22, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Folkesson E., Turkiewicz A., Englund M., Önnerfjord P., BMC Musculoskelet. Disord. 2018, 19, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vukasovic A., Asnaghi M. A., Kostesic P., Quasnichka H., Cozzolino C., Pusic M., Hails L., Trainor N., Krause C., Figallo E., Filardo G., Kon E., Wixmerten A., Maticic D., Pellegrini G., Kafienah W., Hudetz D., Smith T., Martin I., Ivkovic A., Wendt D., Cell Proliferation 2019, 52, e12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deconde A. S., Lee M. K., Sidell D., Aghaloo T., Lee M., Tetradis S., Low K., Elashoff D., Grogan T., Sepahdari A. R., John M. S., JAMA Otolaryngol. – Head Neck Surg. 2014, 140, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langer R., Vacanti J. P., Science 1993, 260, 920. [DOI] [PubMed] [Google Scholar]
- 25. Petersen A., Princ A., Korus G., Ellinghaus A., Leemhuis H., Herrera A., Klaumünzer A., Schreivogel S., Woloszyk A., Schmidt‐Bleek K., Geissler S., Heschel I., Duda G. N., Nat. Commun. 2018, 9, 4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aguado B. A., Grim J. C., Rosales A. M., Watson‐Capps J. J., Anseth K. S., Sci. Transl. Med. 2018, 10, eaam8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Amendola M., Venneri M. A., Biffi A., Vigna E., Naldini L., Nat. Biotechnol. 2005, 23, 108. [DOI] [PubMed] [Google Scholar]
- 28. Barbosa I., Garcia S., Barbier‐Chassefière V., Caruelle J. P., Martelly I., Papy‐García D., Glycobiology 2003, 13, 647. [DOI] [PubMed] [Google Scholar]
- 29. Grogan S. P., Barbero A., Winkelmann V., Rieser F., Fitzsimmons J. S., O'Driscoll S., Martin I., Mainil‐Varlet P., Tissue Eng. 2006, 12, 2141. [DOI] [PubMed] [Google Scholar]
- 30. Stüdle C., Vallmajó‐Martín Q., Haumer A., Guerrero J., Centola M., Mehrkens A., Schaefer D. J., Ehrbar M., Barbero A., Martin I., Biomaterials 2018, 171, 219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.