

Abstract
Poly(glycerol sebacate) (PGS) continues to attract attention for biomedical applications owing to its favorable combination of properties. Conventionally polymerized by a two‐step polycondensation of glycerol and sebacic acid, variations of synthesis parameters, reactant concentrations or by specific chemical modifications, PGS materials can be obtained exhibiting a wide range of physicochemical, mechanical, and morphological properties for a variety of applications. PGS has been extensively used in tissue engineering (TE) of cardiovascular, nerve, cartilage, bone and corneal tissues. Applications of PGS based materials in drug delivery systems and wound healing are also well documented. Research and development in the field of PGS continue to progress, involving mainly the synthesis of modified structures using copolymers, hybrid, and composite materials. Moreover, the production of self‐healing and electroactive materials has been introduced recently. After almost 20 years of research on PGS, previous publications have outlined its synthesis, modification, properties, and biomedical applications, however, a review paper covering the most recent developments in the field is lacking. The present review thus covers comprehensively literature of the last five years on PGS‐based biomaterials and devices focusing on advanced modifications of PGS for applications in medicine and highlighting notable advances of PGS based systems in TE and drug delivery.
Keywords: biodegradable polyester, bioelectronics, biomedical applications, composites, poly(glycerol sebacate), tissue engineering
The present review paper covers comprehensively the published literature of the last 5 years on poly (glycerol sebacate) (PGS)‐based biomaterials including advanced preparation and processing techniques. It focuses on the ongoing modification of PGS for medical applications and its remarkable developments in tissue engineering and drug delivery applications.
1. Introduction
Among a large number of different polymeric biomaterials, poly (glycerol sebacate) (PGS) has been applied in versatile applications in recent years (Figure 1 ).[ 1 ] Since 2002 when, for the first time, the synthesis of PGS as a tough biodegradable polyester was reported by Wang et al.,[ 2 ] it became increasingly attractive for tissue engineering (TE) due to its straightforward and controllable production. The synthesis can be performed under appropriate conditions with specific reaction times and temperatures resulting in extraordinary properties.[ 3 ] All methods for the synthesis of PGS are based on the polycondensation reaction of glycerol and sebacic acid leading to a covalently cross‐linked, 3D network of random coils with hydroxyl groups attached to the backbone.[ 2 ] Both precursors used in synthesizing PGS are natural components found in the body; glycerol, the basic building block for lipids, and sebacic acid, a natural metabolic intermediate in ω‐oxidation of medium‐ to long‐chain fatty acids.[ 2 , 4 ] The synthesis parameters enable a tunable degree of esterification, which, in turn, determine the chemical and mechanical properties and the degradation behavior of this polymer. Thus, PGS is a flexible elastomer with a nonlinear stress–strain behavior, which can almost completely recover from large deformations, due to cross‐linking and hydrogen bonding interactions between the hydroxyl groups.[ 2 , 4 , 5 ] Also, shape‐memory properties were observed for this polymer.[ 6 ] These outstanding mechanical properties can be designed and customized by adjustments of the molar ratio of glycerol and sebacic acid, reaction temperature and reaction time.[ 7 , 8 , 9 , 10 ] In in vivo as well as in vitro applications, PGS is a bioresorbable material that under hydrolytic cleavage of ester linkages, loses slowly the mechanical strength relative to mass loss.[ 2 , 11 , 12 ] In‐vitro, dense PGS disks subjected to 2 d of cross‐linking at 120 °C and 40 m torr were found to be degraded by 17 ± 6% after immersion in phosphate buffered saline (PBS) solution at 37 °C for 60 d.[ 2 ] In comparison to in vitro conditions in the absence of enzymes, the degradation rate is accelerated in vivo by enzymes, altering the polymer structure catalyzing the hydrolysis reaction in ester group‐containing polymers like PGS.[ 13 ] When implanted in Sprague–Dawley rats subcutaneously, the same PGS samples were entirely degraded after 60 d.[ 2 ]
Figure 1.
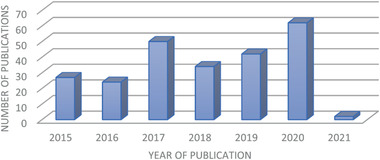
Number of published articles of PGS‐based materials during 2015 and the beginning of 2021. Data from WEB OF SCIENCE using POLY (GLYCEROL SEBACATE) in the field “topic” from 2015 to 2021.
A further advantage of PGS, compared to other frequently used polymers in TE such as poly(ɛ‐caprolactone) (PCL), poly(lactic‐co‐glycolic acid) PLGA and poly(glycolic acid) PGA is the ability to tune its mechanical properties, especially stiffness, by introducing simple changes to the polymerization and curing procedures. For example, Young's moduli of PGS are reported in the 0.77–1.9 MPa range when solely the curing time is changed and values ranging from 0.01 to 5 MPa are reported when the monomer stoichiometry of PGS is altered, respectively.[ 14 ] In comparison, PCL, PGA, and PLGA exhibit less adjustable stiffness values of approximately 0.2–0.3 MPa, ≈7 GPa and 1.4–2.8 MPa, respectively.[ 15 ] In addition to the elastic modulus, the ultimate tensile strength (UTS) and elongation at break of PGS can also be controlled in wide ranges by varying the process parameters. PGS exhibits UTS values of above 5 MPa and an elongation higher than 267%.[ 15 ] In comparison, values of PLGA, PGA and PCL range from 41 to 55 MPa and 3–10%, 70 MPa and below 3% and 21 MPa and 300–500%, respectively.[ 15 ] Thus, a wide range of properties can be covered using PGS, which is relevant to support the wider applications of this polymer in the biomedical field.
PGS is nonimmunogenic, and it was shown to be noncytotoxic in vitro and provoked only a minimal inflammatory response with little fibrous capsule formation in vivo.[ 2 , 16 ] Due to its tailorable mechanical properties and degradation behavior, aiming to fulfil criteria for a particular application, PGS won its place as a relevant scaffold material, especially for the regeneration of soft tissue like the myocardium, blood vessels, cornea, and nerve tissue.[ 1 , 4 , 16 , 17 ] Over the years, PGS‐based biomaterials have been increasingly investigated for drug delivery and TE applications such as cardiac and cardiovascular,[ 18 ] skin and wound healing,[ 19 ] nerve,[ 20 ] corneal and oral tissues,[ 16 ] musculoskeletal,[ 21 ] adipose,[ 22 ] cartilage,[ 23 ] dental and bone as well as for soft bioelectronic applications.[ 24 , 25 , 229 ]
Since biomedical applications of PGS with advancing technological developments, e.g., novel fabrication techniques and PGS modifications are continuously growing, this review aims to consolidate the existing knowledge of the past five years following previous reviews on PGS‐based materials for biomedical applications published in 2012[ 4 ] and later in 2015.[ 26 ] This article focuses on recent modification techniques of PGS and fabrication techniques for PGS‐based biomaterials. Furthermore, latest advances in the main application fields of PGS‐based biomaterials, i.e., cardiac TE (CTE), vascular TE, nerve TE, skin and wound healing TE, bone TE (BTE), and drug delivery, will be closely presented. This article seeks to give a concise overview of the recent developments in the broad field of PGS‐based materials.
The literature search for this review article was conducted in the databases WEB OF SCIENCE as well as SCOPUS using the keyword POLY(GLYCEROL SEBACATE) or POLY GLYCEROL SEBACATE in the field “topic” search box. In the time range from 2015 to 2021, about 241 papers were published (Figure 1).
2. Modification of PGS
Conventionally, PGS follows a two‐step synthesis route via prepolycondensation and cross‐linking.[ 2 , 4 , 27 ] Briefly, prepolycondensed PGS (PGSp) is synthesized by a polycondensation reaction of an equimolar mixture, typically, of glycerol and sebacic acid at 120 °C under a nitrogen or argon atmosphere for 24 h (Figure 2 ). For a subsequent cross‐linking step during the conventional synthesis of PGS, PGSp is kept in a vacuum for at least 48 h at 120 °C. With a kinetic model of the polycondensation of sebacic acid and glycerol created by Matyszczak et al.[ 5 ] a prediction and optimization of the synthesis reaction are possible. The model is used to determine the moment of the desired reaction at which desired product parameters like average molecular weight and polydispersity can be achieved. Perin and Felisberti[ 28 ] on the other hand, developed an alternative polymerization technique for PGS using Candida antarctica lipase B (CALB) as a catalyst for the polycondensation of glycerol and sebacic acid, allowing them to synthesize PGS in acetone at temperatures between 30 and 50 °C with different molecular weights and degrees of branching.[ 28 ] However, due to the harsh cross‐linking conditions (≥120 °C, vacuum) and relatively long reaction times (≥24 h) required for its curing, pure, unmodified PGS exhibits a lack of processability.[ 27 ] Alternative processing strategies are required to address these limitations, mostly achieved by blending PGS prepolymer with other materials or by chemical modification of its pre‐condensed form (Figure 2).
Figure 2.
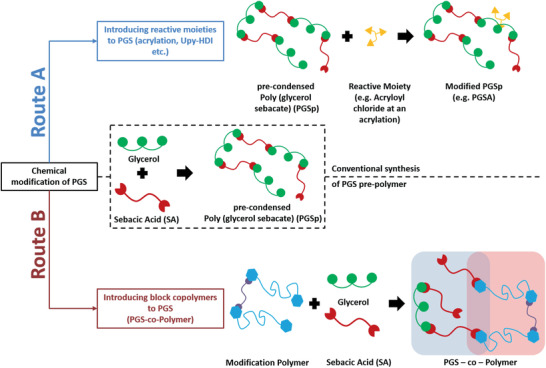
Chemical modification of PGS through introducing reactive moieties (Route A) or block copolymers (Route B).
2.1. Blending of PGS with Other Biopolymers
The processing of thermoset polymers like PGS is typically limited to simple reproduction techniques due to the subsequent required harsh cross‐linking, making it challenging to fabricate complex 3D architectures. A simple way of improving the processability of PGSp is by blending it with other materials.[ 29 , 30 ]
The key is to design compositions containing both the thermoplastic precursors of the thermoset PGS and a sacrificial or non‐sacrificial carrier material, respectively, which serves as an aggregate to provide mechanical support, helping to retain the desired 3D shape before and during cross‐linking.[ 31 ]
So far, blending of PGS with other polymers was mainly conducted to produce suitable solutions for electrospinning. Indeed, neat PGSp exhibits difficulties for electrospinning due to its low viscosity in a solution for example, in common electrospinning solvents like chloroform (CF): ethanol mixtures.[ 32 ] A well‐investigated method to address this problem is to mix PGS prepolymer with PCL.[ 17 , 29 , 33 , 34 ] PCL is a highly used thermoplastic polymer in TE. It is approved by the Food and Drug Administration (FDA), biocompatible, and exhibits a long‐term biodegradability with high stiffness.[ 37 ] For blending PGSp with PCL, both polymers are dissolved in a solvent, mostly tetrahydrofuran (THF) or a CF/ethanol mixture, with varying weight ratios.[ 38 ] After stirring the mixture for a certain amount of time, the solution is either processed directly, as in the case of solution‐electrospinning or used after the solvent has been removed. Mixing PGSp with PCL produces stable scaffolds, which do not need any further post‐processing, such as thermal curing or photo‐cross‐linking. Variation of the ratio of PGS and PCL in the blended solution showed it could alter the mechanical properties of the blend, degradation rate and hydrophilicity of the structure. Studies have shown that increasing the concentration of PGS enhanced the degradation rate and hydrophilicity of PCL and lowered the stiffness of PCL while PCL‐PGS electrospun fibers with 3:1 ratio (PCL: PGS), showed three times lower storage modulus (0.6 MPa) than that of PCL fibers (1.9 MPa).[ 29 , 30 ]
Besides PCL, thermoplastic polyurethane (TPU) has also been used to gain a suitable electrospinnability of PGSp.[ 35 ] TPU pellets and PGSp with polymer ratios of 6:6, 6:4, and 6:2 have been dissolved in a 6:4 CF and N,N‐dimethylformamide (DMF) solution. The concentration of TPU was kept constant at 6% (w/v). In the reported study, either 1,1,1,3,3,3‐hexafluoroisopropanol (HFIP) alone or a binary solvent of 2,2,2‐trifluoroethanol (TFE) with acetic acid in a ratio of 5:5 could serve as a solvent system for TPU/PGSp.[ 35 ]
As an alternative to electrospinning, O'Brien et al. used the electro‐less Substrate Translation and Rotation for Aligned Nanofiber Deposition (STRAND) process.[ 36 ] In order to perform the STRAND process with PGS, the biocompatible polymer polyethene oxide (PEO) was added to act as a strengthening agent. Molar ratios of 1:1, 2:1, and 1:2 of PGS: PEO, ranging from 10 to 25% (w/v), were dissolved in a 1:1 CF: ethanol solution. Several concentrations were tested regarding their effect on fiber diameter and morphology.[ 36 ]
2.2. PGS‐Based Composites with Inorganic Fillers
Apart from blending PGSp with different polymers, another frequently used method is the incorporation of inorganic particles into precondensed PGS to produce composite scaffolds or structures with tailored properties for various TE applications.[ 39 , 40 , 41 , 42 , 43 , 44 , 45 ] The most conventional approach is the combination of PGSp with sodium chloride (NaCl) particles. In this so‐called salt leaching technique, molten PGS prepolymer is mixed with salt particles acting as a sacrificial carrier material, which is then removed after cross‐linking to develop porous PGS structures. By adjusting the weight ratio of PGSp:NaCl, different microporous architectures can be achieved. At weight ratios of 1:2 or 1:3 (PGSp:NaCl), sodium salt granules provided adequate mechanical support and shape retention during the harsh cross‐linking conditions of PGS. Moreover, it was shown that this mixture was suitable for 3D printing applications.[ 31 ] In addition to NaCl, various other particulate materials have been incorporated into PGS as a matrix mainly to control the mechanical properties and the degradation susceptibility of pure PGS. From these particle fillers directly added to the PGS as a matrix, few to name are, i.e., bioactive glass (BG),[ 46 ] silica,[ 47 ] β‐tricalcium phosphate (TCP),[ 48 , 49 , 50 , 51 ] cellulose nanocrystals,[ 52 ] and those added to PGS as a fiber are, i.e., silk fibroin,[ 53 ] chitosan (CH),[ 53 , 54 , 55 ] or BG fibers.[ 42 ]
2.3. Chemical Modification
Chemical modification of PGS is generally undertaken to alter its chemical, mechanical, and degradation properties in order to adapt them closer to the specific target application. Modified PGS is more flexible regarding its cross‐linking conditions without the need of further processing of high temperature or in a vacuum, making it more suitable for a variety of applications. The synthetic strategies can be separated into two main modification routes (Figure 2). Route A describes the introduction of reactive moieties into the PGS matrix, whereas route B focuses on the block copolymerization of PGS with other linear polymers.
2.3.1. Introduction of Reactive Moieties (Route A)
The most widely used modification of PGS, especially for additive manufacturing, is its acrylation or methacrylation.
Nijst et al. reported a photopolymerization approach by chemically modifying the PGS prepolymer by introducing reactive acrylate moieties.[ 27 ] Briefly, PGS prepolymer with anhydrous dichloromethane (DCM) and 4‐(dimethylamino) pyridine (DMAP) was cooled in a reaction flask to 0 °C under nitrogen atmosphere. Acryloyl chloride was then slowly added parallel to an equimolar amount of trimethylamine (Figure 3 ). After reaching room temperature, the mixture was stirred for an additional 24 h. The resulting mixture was dissolved in ethyl acetate, filtered, and dried at 45 °C in a vacuum (5 Pa).[ 27 ] Vinyl bonds in poly (glycerol sebacate) acrylate (PGSA) could be subsequently cross‐linked via redox polymerization or photoinitiated free radical polymerization via ultraviolet (UV) radiation in the presence of a photoinitiator, mostly 2‐dimethoxy‐2‐phenyl acetophenone.[ 27 ] Via photopolymerization, the PGSA cured rapidly within a few minutes, which is more efficient than the previous long curing time (48h) reported in the conventional synthesis process.[ 2 ]
Figure 3.
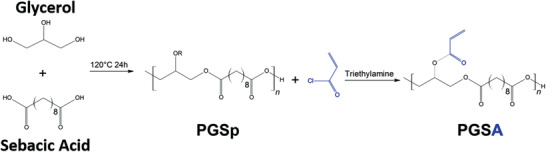
Schematic representation of the chemical synthesis of PGSp and its further modification using trimethylamine to produce acrylated PGS (PGSA). Modified from.[ 27 ]
An alternative approach to introduce reactive moieties besides acryloyl chloride into the PGSp network was proposed by Wu et al.:[ 57 ] 2‐ureido‐4[1H]‐pyrimidone‐hexamethylene diisocyanate (UPy‐HDI) was used to generate chemically cross‐linked supramolecular PGS (PGS‐U). In this process, PGS prepolymer dissolved in THF and HDI were mixed and after reacting for 4 h at 55 °C under a nitrogen atmosphere and evaporation of the solvent, a PGS‐U polymer film formed via strong non‐covalently hydrogen bonding.[ 57 ]
Yeh et al. designed an alternative method to produce photocurable PGS using thiolene click chemistry to control PGS cross‐linking. In short, PGS prepolymer was dissolved in anhydrous DCM containing 500 ppm 4‐methoxy phenol and 0.1 wt% DMAP. The reaction flask was purged with nitrogen for 10 min at 0 °C. 5‐Norbornene‐2‐carbonyl chloride in DCM was added dropwise into the PGS solution parallel to triethylamine, which was subsequently stirred at room temperature overnight. After several filtration and drying steps, the remaining DCM was removed using a rotary evaporator leaving a viscous liquid, which was further dried in an oven at 37 °C overnight, purged with nitrogen afterwards, and then stored at −20 °C. Norbornene modification of PGS (Nor‐PGS) was ≈15% (calculated based on nuclear magnetic resonance (NMR) analysis) with a yield of Nor‐PGS of ≈96%.[ 58 ] Norbornene modification on NorP‐GS and therefore its cross‐linking could also be altered by changing the initial amount of PGSp. Tsai et al.[ 59 ] further modified the Nor‐PGS approach by copolymerizing Nor‐PGS with polyethylene glycol (PEG). As a result, NorPGS‐co‐PEG could be controlled in its mechanical and degradation properties as well as in vitro swelling behavior.[ 59 ]
In general, by controlling the percentage of reactive moieties, like acryloyl chloride, UPy‐HDI, or norbornene in PGS, it is also possible to control mechanical as well as hydrolysis and in vitro enzymatic degradation profiles of the modified PGS to meet specific requirements for various applications.[ 4 , 56 , 57 , 60 , 61 ]
2.3.2. Introduction of Block Copolymers (Route B)
The most common method to alter cross‐linking densities of polymers is to change the molecular weights of their precondensed form. However, this could lead in some polymers to physical entanglements and chain extension when cross‐linked via radical polymerization, limiting their applications in further processing steps.[ 62 ]
An alternative approach is a polycondensation of PGSp with linear polymers (Figure 2 Route B), such as PCL[ 63 ] and PEG[ 62 , 64 , 65 , 66 , 67 ] and their acrylated forms, poly(ethylene glycol) methyl ether methacrylate (PEGMEMA),[ 68 ] poly(tetramethylene oxide) glycol (PTMO),[ 69 ] or simply with gelatin.[ 70 ]
Briefly, precondensed PGS could be added via a two‐step polycondensation approach. In the first step, segments of the linear modifier polymer and sebacic acid are polymerized. Addition of glycerol leads to a polycondensation of PGS, whereby parts of sebacic acid combine with the linear polymer (Figure 2 Route B).[ 64 , 65 , 66 , 67 ] Through altering the modifier polymer content and the ratio of sebacic acid to glycerol, a series of PGS‐co‐polymers can be designed and synthesized.
Wilson et al. investigated the copolymerization of PTMO with PGS. They either synthesized PGS with PTMO or mixed PGS‐co‐PTMO with the thermoplastic polyester elastomer Hytrel 3078.[ 69 ] A two‐step polymerization resulted in the copolymer PGS‐b‐PTMO. In the first step, sebacic acid‐co‐PTMO (SA‐co‐PTMO) was synthesized in molar ratios of 1:0.65/0.75/0.85 with 0.5 wt% of FASCAT 9100 (mono‐n‐butyl tin oxide or butylstannoic acid) as a catalyst. The educts were molten and reacted under reduced vacuum for 8 h at 150 °C. In the second step, glycerol (0.35/0.25/0.15 mol) was added and reacted for another 8 h at 135 °C. Cross‐linking of PGS‐b‐PTMO was carried out using 2 wt% (for glycerol) of 4,4′‐methylene bis (phenyl isocyanate) (MDI). PGS‐b‐PTMO and MDI were then dissolved in CF and stirred for 4 to 5 h. The solution was kept in a fume hood for 12 h to evaporate the solvent and subsequently cross‐linked in a vacuum oven at 170 °C under reduced vacuum for 8 h.[ 69 ]
In the case of gelatin as a modifying polymer, copolymerization is realized via an endothermic reaction step of PGSp and gelatin without the need of any cross‐linking agent. In this manner, gelatin is introduced in the PGSp network via ester and amide linkages rather than extending the polymer network chains.[ 70 ] Therefore, elastomeric properties, water swelling capability, as well as pH‐responsive behaviors of the two distinct materials are maintained.[ 70 ]
Rostamian et al.[ 63 ] demonstrated the successful incorporation of hydroxyapatite nanoparticles in PGS‐co‐PCL, which was synthesized by melt polycondensation. The authors found a beneficial effect of the addition of caprolactone section in PGS‐co‐PCL regarding its viscoelastic properties and wettability. The incorporation of hydroxyapatite nanoparticles enhanced a potential application in BTE.[ 63 ]
3. Fabrication Techniques for PGS‐Based Biomaterials
Since PGS prepolymer can be either melted or dissolved in several solvents including THF, HFIP, DCM, DMF, CF, ethanol, methanol, acetic acid, and formic acid, its processing is relatively uncomplicated and many different fabrication techniques can be used. The fabrication technique has to be chosen according to the target application and specifically the required morphology, e.g., whether a strong solid sheet, macro‐ or microporous membranes or structures with/without anisotropic behavior or injectable scaffolds are desired. Popular fabrication techniques for PGS‐based components are presented in the following sections (Figure 4 ).
Figure 4.
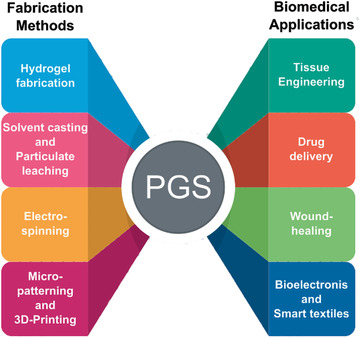
Representative manufacturing methods and biomedical applications with PGS as the primary material. (Figure generated using Freepik.com).
3.1. Solvent Casting/Particulate Leaching
Solvent casting/particulate leaching is a technique used to produce either dense films or porous membranes. It is a simple, reproducible method, which does not require sophisticated lab equipment. The pore size, porosity, and geometry can be easily controlled by the amount, size, and shape of the porogen, e.g., salt particles.[ 71 ] However, this method requires long soaking periods in water to leach all porogens from the membrane, which limits the applications such as drug loading/delivery. Moreover, limited interconnectivity of the pores in the structure affects the mechanical properties and residues of the salt particles or solvents can influence the biocompatibility of the resulted scaffolds.[ 71 ]
Pure PGS,[ 72 , 73 ] modified[ 64 , 74 , 75 , 76 , 77 , 78 , 79 ] (see Section Modification of PGS), or combined with other materials[ 21 , 24 , 47 , 52 , 57 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 ] has been processed into films and porous scaffolds using the salt leaching method. Initially, this technique was used to develop PGS‐based cardiac patches.[ 8 , 89 ] Since then, its application field changed to mainly hard TE. For the application in BTE, inorganic fillers like BG[ 47 ] and nanosilicates[ 24 , 83 ] have been applied to enhance the mechanical properties of the PGS network by improving its stiffness without affecting the elastomeric properties. The in vitro degradation rate of PGS composites has been shown to be tunable by the addition of inorganic fillers. In the case of using nanohydroxyapatite (nHA) as a reinforcement,[ 82 ] the elastic modulus and tensile strength were increased, however, the flexibility of the films decreased, and the degradation rate was accelerated. PGS/nanosilicate porous scaffolds[ 83 ] supported cell adhesion, spreading, proliferation, and osteogenic differentiation, and showed in vivo biocompatibility, whereas PGS–BG composites[ 47 ] showed significantly enhanced proliferation of MC3T3 cells compared to neat PGS films. In contrast to inorganic fillers, also synthetic (e.g., polylactic acid (PLA)[ 80 ]) or natural (cellulose nanocrystals,[ 52 ] ureido‐pyrimidinone[ 57 ]) polymers or metabolic compounds like tyramine[ 84 ] have been successfully blended with PGS to improve the mechanical, hydrophilic, and degradation properties. The addition of electrically conductive compounds like carbon nanotubes,[ 88 ] aniline pentamer,[ 81 , 85 ] graphene,[ 86 ] or plasma polymerization of polypyrrole[ 90 ] has been investigated to create highly tunable conductive films with enhanced cell survival and attachment[ 86 ] and enhanced Schwann cells (SCs) myelin gene expression.[ 81 ]
3.2. Electrospinning
Electrospun fibrous meshes have attracted much attention in TE and regenerative medicine as scaffold constructs as they closely resemble the fibrous native extracellular matrix (ECM) and offer suitable structures for cell attachment and subsequent tissue organization.[ 91 , 92 ] Moreover, electrospun meshes are porous with interconnected porosity made of fibers in nano‐ to micrometer range of diameter and high surface‐to‐volume‐ratio. The fibrous structure can be spun from a variety of synthetic and natural polymers with random and aligned arrangements or core–shell structures.[ 93 , 94 ] In electrospinning briefly, a high voltage is generated between a polymeric solution (or melt) and the grounded collector and a polymeric fiber is ejected and stretched toward the collector if the voltage is sufficiently high and is able to surpass the critical voltage threshold and thereby the surface tension of the polymer solution.[ 94 ] Various parameters, like solution viscosity, processing (e.g., voltage, distance between needle and collector, flow rate) and ambient parameters (e.g., temperature, humidity) influence the electrospinning process and the resulting fiber morphology.[ 95 ]
As mentioned above, PGS itself cannot be electrospun into stable fibers due to its low solution viscosity,[ 32 ] the low glass transition temperature (below room temperature) of precondensed PGS (PGSp) as well as the insolubility of its cross‐linked form in organic solutions. As a result, electrospun fibers of PGSp have fused into a nonporous film instead of a porous fiber mat. During the subsequent high‐temperature cross‐linking, this fusion is even intensified.[ 96 ] As a result, PGSp must be blended with a spinnable carrier polymer to allow non‐destructive fiber formation by electrospinning. According to its specific application ranging from soft to hard TE also including drug delivery, synthetic polymers like PCL,[ 16 , 19 , 21 , 29 , 33 , 38 , 45 , 97 , 98 , 99 , 100 , 101 , 102 , 103 , 104 , 105 , 106 , 107 , 108 , 109 , 110 , 111 ] poly(vinyl alcohol) (PVA),[ 96 , 112 , 113 , 114 ] poly(butylene succinate‐co‐dilinoleic succinate) PBS‐DLS,[ 115 , 116 ] PLA,[ 117 ] poly(l‐lactic acid) (PLLA),[ 118 , 119 ] (PEO),[ 120 ] PLGA,[ 121 , 122 ] polyethersulfone (PSF),[ 123 ] polyhydroxybutyrate (PHB),[ 124 ] polyvinylpyrrolidone (PVP),[ 125 , 126 ] TPU,[ 35 ] and natural polymers like collagen,[ 127 ] gelatin,[ 128 ] fibrinogen,[ 129 ] zein,[ 130 , 131 ] and chitin and lignin[ 132 ] have been used for blending with PGS for successful electrospinning. Additional to blending PGS with another polymer, electrospinning of PGS within a core–shell system has been reported too where the sacrificial polymer is used as a shell which will be removed after PGS cross‐linking. Generally, PVA[ 96 , 112 , 133 ] or a combination of PEO and PLA,[ 120 , 134 ] are commonly used as sacrificial polymers. Pure PGS fiber mats show appropriate mechanical properties (for selected applications) and in vitro cytocompatibility has been proven with 3T3 fibroblasts[ 96 ] and human umbilical artery smooth muscle cells (HUASMCs).[ 112 , 120 , 133 , 134 ] Lang et al.[ 135 ] showed the synthesis of PGS by CALB catalysis, whereby different fractions of glycerol units in PGS were replaced by 1,8‐octanediol units, enabling the fabrication of fibers via electrospinning without the need of a second component.[ 135 ]
Electrospun fiber mats have been further functionalized by incorporation of inorganic particles like β‐TCP,[ 51 ] nHA,[ 100 ] or BG[ 136 ] for increased bioactivity and osteoblast adhesion properties, binding of vascular endothelial growth factor (VEGF),[ 106 , 129 ] or sputtering of electrospun membranes with silver showing excellent pathogenic antibacterial properties.[ 45 , 97 ] Moreover, biomacromolecules or drugs can be loaded in PGS‐based fibers either directly by blending drugs within the fiber material and core‐shell fibers or by covalently immobilizing the drug on the final mats.[ 98 , 99 , 124 , 128 ]
A different approach to produce PGS fibers without applying an electric field can be made using pressurized gyration.[ 137 ] Thus, the polymer solution is forced out a cylindrical aluminum drum via rapid rotation (36 000 rpm) and an applied nitrogen pressure (0.1 MPa N2).[ 137 ]
An overview of all electrospinning approaches involving PGS, which have been reported in the last five years, is shown in Table 1 .
Table 1.
Overview of PGS used in electrospinning approaches. Abbreviations: KGN, kartogenin, mAcr‐PGS, methacrylated PGS; PBS‐DLS, poly(butylene succinate‐co‐dilinoleic succinate); PBMCs, peripheral blood mononuclear cells; PMMA, poly(methyl methacrylate); PSF, polyethersulfone; PVA, poly(vinyl alcohol); AcOH, acetic acid, EtOH, ethanol, FA, formic acid; MeOH, methanol; SRB, sulforhodamine B; TFA, trifluoroacetic acid; YM, Young's modulus
| PGS in combination with | Solvents | Mechanical properties | Other specifics | Cells/animal model | Cellular response | Proposed application | Refs. |
|---|---|---|---|---|---|---|---|
| PCL | CF/EtOH |
YM: 6.3 MPa (random) ‐ 9.6 MPa (aligned) UTS: ‐ Strain: ‐ |
Aligned fibers | HUVECs | Enhanced cellular proliferation and organization by alignment; formation of highly organized endothelial constructs | Vascular TE | [ 105 ] |
| PCL, VEGF | DCM/MeOH |
YM: 8 ± 2 MPa UTS: 3 ± 0.5 MPa Strain: 142 ± 29% |
VEGF functionalization; potential for the formation of vascular tree in fiber mat | C2C12 myoblasts, rat cardiac progenitor cells, rat aortic endothelial cells | Attachment and growth of myogenic and vasculogenic cell lines | CTE | [ 106 ] |
| Sacrificial PVA, PCL | HFIP (PGS/PVA), TFE/H2O (PCL) | YM: 0.67– 0.871 MPa | Removal of PVA leaving neat PGS fibers behind | In vivo mice model | No thrombosis or stenosis; organization of contractile SMCs and neotissue in the inner part of the graft, macrophage driven‐inflammatory response to remaining polymer up to 12 months postimplantation | Vascular TE | [ 107 ] |
| PCL | DCM/MeOH | – | Patterned topography | C2C12 myoblasts, neonatal rat cardiomyocytes | Alignment of both cell types due to the surface topography | CTE | [ 110 ] |
| PCL | CF/EtOH |
YM: 4.25– 7.61 MPa UTS: 1.78– 3.14 MPa Strain: 218 – 453% |
Good anticoagulation property with low hemolysis percentage | Human vascular endothelial cell line EAhy926 | Good biocompatibility | Vascular TE | [ 111 ] |
| PCL, silver | CF/EtOH |
YM: 3.3 MPa (uncoated) –≈8 (with Ag) MPa UTS: ≈1.5 (uncoated) ‐ ≈2.6 (with Ag) MPa Strain: ≈200 (uncoated) ‐ ≈175 (with Ag) % |
Silver coating with antibacterial properties | – | – | Diverse | [ 45 ] |
| PCL | CF/EtOH | – | Aligned fibers | Human corneal epithelial cells, human corneal keratocytes | Good biocompatibility, formation of a confluent cell layer | Corneal TE | [ 38 ] |
| PCL | CF/EtOH |
Surface moduli of 0.26 GPa, 0.29 GPa, 0.12 GPa, and 0.16 GPa for PGS/PCL 4:1, 3:1, 2:1 1:1 |
Aligned fibers | Human corneal endothelial cells, human conjunctival epithelial cells, and PBMCs | Best cell organization, cyto‐ and immunocompatibility in a higher content of PGS in PGS–PCL blend | Corneal TE | [ 16 ] |
| PCL, silver | CF/EtOH | – | Silver coating with pattern | NIH 3T3 fibroblasts | Good biocompatibility | Diverse | [ 97 ] |
| PCL, PEGylated‐CH nanoparticles | CF/EtOH | – | Additional patterning of a bioresorbable metallic heater for thermal stimulation of on‐demand antibiotic release; antibacterial properties | Keratinocytes | Good biocompatibility | Wound healing, drug delivery | [ 19 ] |
| PCL, gelatin, dexamethasone | CF/MeOH (PGS‐PCL‐dex), AcOH/H2O (gelatin) |
YM: 6.3 (wet) ‐7.8 (dry) MPa UTS: 0.74 (dry) ‐1.64 (wet) MPa Strain: 36.8 (dry) ‐ 102 (wet) % |
Sustained drug release for 30 d | Gingival fibroblast cells | No evidence of cytotoxicity; cell proliferation not adversely affected by Dex release | Soft TE | [ 98 ] |
| PCL, heparin | TFE |
YM: ≈3 –≈15.5 MPa UTS: ≈2–≈4 MPa Strain: ≈250 –≈900% |
Heparin functionalization | HUVECs | Improved cell attachment and proliferation after grafting of heparin | Diverse | [ 99 ] |
| PCL, CH, β‐TCP | CF (PGS‐PCL‐β‐TCP), TFA (PGS‐PCL‐CH) |
YM: ‐ UTS: ≈1.1 –≈1.4 MPa Strain: ≈0.8 –≈2.4 |
Bilayered | Human fetal osteoblasts | Optimum cell behavior in blend composition with 10% β‐TCP | BTE | [ 51 ] |
| PCL, hydroxyapatite nanoparticles | d.n.a. | d.n.a. | d.n.a. | Bone marrow‐derived MSCs | Excellent biocompatibility and osteoblast adhesion | Bone and cartilage TE | [ 100 ] |
| PCL | CF/acetone (PGS‐PCL), AcOH/FA (PCL) |
YM: 2.6– 3.1 MPa UTS: 0.8– 1.5 MPa Strain: ≈58 –≈65% |
Nonhemolytic behavior | MSCs | Supported attachment, growth, and infiltration of cells by blend fiber mat | Vascular TE | [ 101 ] |
| PCL | AcOH |
YM: 7– 11 MPa UTS: 4.4– 5 MPa Strain: 304–333% |
Use of mildly cross‐linked PGS for electrospinning | Bone marrow‐derived stroma cell line ST2 | Better cytocompatibility and cell adhesion after sterilization by 70% ethanol compared to UV sterilization | CTE | [ 33 ] |
| PCL | CF/EtOH/DMF |
YM: 8.32 (dry) – 8.62 (wet) MPa UTS: ‐ Strain: 783 (dry)–1133 (wet) % |
Bilayered construct with PU membrane layer | Mouse lung fibroblast cells (L929), HUVECs | No toxic effect of leachable components on fibroblasts; endothelial cell adhesion and proliferation on PGS/PCL fiber mat | Hernia treatment | [ 104 ] |
| PCL, KGN | TFE |
YM: 5.06 (nonaligned)– 11.78 (aligned) MPa UTS: ≈2.3 (non‐aligned) –≈3 (aligned) MPa |
Aligned core–shell fibers | Human bone marrow MSC (hBMSC) | Significant proliferation and chondrogenic differentiation on KGN‐loaded aligned nanofibrous scaffold | Cartilage TE | [ 103 ] |
| Sacrificial PVA | H2O (PVA), THF (PGS) |
YM: ≈3 MPa (wet) –≈33 MPa (dry) UTS: ≈ 1.5 MPa (wet) –≈7 MPa (dry) Strain: 95 % (wet)– ≈130% (dry) |
PGS only fibers | SNL mouse fibroblasts | Good biocompatibility | Soft TE | [ 133 ] |
| Sacrificial PVA | HFIP |
YM: 0.1– 0.8 MPa UTS: ≈1 MPa Strain: 200 –800% |
PGS only fibers | 3T3 fibroblasts, Human cord blood endothelial cells (hcbEC), in vivo mice model |
Round to elongated adhered cell morphology; higher cytocompatibility of highly cross‐linked materials; In vivo: surrounded by collagen‐rich matrix and multinucleated giant cells and fibroblast‐like cells in implant bulk |
Diverse | [ 96 ] |
| PVA | DMF/H2O |
YM: 0.04 (wet)– 7.08 (dry) MPa UTS: 2 (dry)–70 (dry) MPa Strain: 34.5 (dry)–287 (wet) % |
Modified PGS synthesis (1:0.8 molar mixture of glycerol and sebacic acid; 170 °C; 3, 5, and 7 h); Aligned fibers |
Rat pheochromocytoma cells (PC12) | Good biocompatibility | Nerve TE | [ 114 ] |
| PVA, lignin | H2O, DMF |
YM: 0.1– 0.4 MPa UTS: 20.24–72.8 MPa Strain: 51– 179.5% |
– | Rat pheochromocytoma cells PC12 | Cell proliferation and neural cell differentiation in the presence of lignin | Nerve TE | [ 113 ] |
| PLLA | CF/DMF (PLLA), THF (PGS) |
YM: 6.5– 15.8 MPa UTS: 2.2– 4.5 MPa Strain: 25.1–43.4% |
Aligned core–shell fibers | – | – | Diverse | [ 118 ] |
| PLLA | DMF/DMC |
YM: 7.2– 336.2 MPa UTS: 1.1– 6.8 MPa Strain:13– 66% |
During cross‐linking creation of core (PLLA)–shell (PGS) fibers | Hypothalamus A59 nerve cells | Enhanced cell adhesion and proliferation for core–shell samples | Nerve TE | [ 119 ] |
| PLGA | HFIP (PGS), NaCl, SRB dye, HFIP (PLGA) | – | – | – | – | Diverse | [ 121 ] |
| PLGA | HFIP |
YM: 1.3 ± 0.66 MPa UTS: ‐ Strain: – |
– | SIMS salivary ductal epithelial cell, NIH3T3 mesenchymal cells | Cell penetration in the structure and apical localization of tight junction proteins; coculture facilitated epithelial tissue reorganization and significantly increased apical localization of tight junction protein | Soft TE | [ 122 ] |
| PLA | HFIP | – | Different esterification levels of PGS | – | – | Diverse | [ 117 ] |
| Sacrificial PEO/PLA | THF (PGS), THF/DMF (PEO/PLA) |
YM: ≈1– 140 MPa UTS: ≈0.5–6 MPa Strain: ≈1.05–2.5% |
PGS only fibers | HUASMCs | Good biocompatibility | Soft TE | [ 120 ] |
| Sacrificial PEO/PLA | THF (PGS), DCM/DMF (PEO‐PLA) |
YM: ≈0.5 (perpendicular) – ≈ 1 (in alignment) MPa UTS: ≈0.36 (perpendicular) –≈0.72 (in alignment) MPa Strain: ≈160 (perpendicular) – ≈ 170 (in alignment) % |
Aligned core–shell fibers | HUASMCs | Cell adhesion on random and aligned fibers, and cell alignment on aligned fibers | Diverse | [ 134 ] |
| Collagen type I, silk fibroin | HFIP |
YM: 2.3– 5.0 MPa UTS: 0.8– 1.5 MPa Strain: 30–70% |
Low thrombogenic potential | Endothelial cells | Cells adhered, proliferated, and formed cell–cell junctions | Vascular TE | [ 127 ] |
| Fibrinogen, VEGF | HFIP | – | VEGF functionalization | In vivo porcine model | Improvement in ejection fraction (EF) and prevention of LV enlargement; expression of cardiac marker proteins | CTE | [ 129 ] |
| PBS‐DLA | DCM/MeOH |
YM: 1.2– 7.5 MPa UTS: 1.58–2.7 MPa Strain: 85.5–147.6% |
– | C2C12 myoblasts, postnatal rat cardiomyocytes | Cytocompatibility with C2C12 myoblasts, the higher the PBS–DLA content in the fiber mats, the better the cell attachment and proliferation; postnatal rat cardiomyocytes better attachment on higher PGS content fibers with well‐aligned sarcomeres and high amounts of connexin43 | CTE | [ 115 ] |
| PBS‐DLS | DCM/MeOh |
YM: 3.5 (37 °C)–4.7 (RT) MPa UTS: 1.23 (37 °C)– 1.8 (RT) MPa Strain: ≈132% |
Surface modification by exposing carboxylic groups | – | – | Soft TE | [ 116 ] |
| Zein | AcOH |
YM: 6.5– 14 MPa UTS: 0.32–13 MPa Strain: 5–7% |
– | – | – | CTE | [ 130 ] |
| Zein | AcOH |
YM: 7– 32 MPa UTS: 1–‐ 2 MPa Strain: 5– 11 MPa |
Use of mildly cross‐linked PGS for electrospinning | – | – | Soft TE | [ 131 ] |
| PGS–PMMA, gelatin | HFIP | – | Methyl mAcr–PGS | Rat PC12 cells | Fiber mats promoted cell proliferation, elongated cell morphology, potential for neurite outgrowth | Nerve TE | [ 20 ] |
| PSF | THF/DMF | – | Compressible, moldable, 3D, drug delivery | In vivo mice model | – | Diverse | [ 123 ] |
| Chitin, Lignin, PEO | EtOH, NaOH, H2O |
YM: ≈7–≈12 MPa UTS: ≈0.75 –≈3 MPa Strain: ≈20 –≈140% |
Antibacterial and antifungal activity | – | – | Wound healing | [ 132 ] |
| SIM, PHB, CIP | CF/DMF(PGS‐Sim), TFA (PHB‐CIP) | – | Dual drug incorporation in core–shell fibers; burst release of CIP, slower release SIM; antibacterial activity | – | – | Wound healing | [ 124 ] |
| Gelatin, CIP | Aqueous AcOH | – | Antibacterial activity | Fibroblast cell line (L929) | Promoted cell attachment, growth, proliferation, and immigration from the surface into interconnected pores | Wound healing | [ 128 ] |
| TPU |
CF/DMF or HFIP or TFE/AcOH |
YM: 0.83– 1.14 MPa UTS:8.76–9.67 MPa Strain: 339–375% |
Leaf‐like structure observed for HFIP | Swiss mouse NIH 3T3 fibroblasts | Improved biocompatibility compared to TPU only scaffolds | Vocal fold TE | [ 35 ] |
| PVP | HFIP (PGS), DMF/EtOH/H2O (PVP) |
YM: 1.3– 170 MPa UTS: 1.1– 3.5 MPa Strain: 3.1–328% |
Nozzle free electrospinning | Human skin fibroblast cells (HDF‐hTERT) | Good cell viability and proliferation | Skin TE | [ 232 ] |
| Poly(glycerol‐1,8‐octanediol‐sebacate) | CF/EtOH |
YM: 3.61 MPa uncross‐linked, 106.1 MPa cross‐linked UTS: 0.13 MPa uncross‐linked, 4.94 MPa cross‐linked Strain: 8% uncross‐linked, 23% cross‐linked |
PGS synthesized by Candida antartica lipase B catalysis; different fractions of glycerol units replaced by 1,8‐octanediol units | – | – | Diverse | [ 135 ] |
3.3. Hydrogels
Hydrogels have gained increasing interest in recent years thanks to their extraordinary composition and structure, which is similar to the native ECM, thus, providing a desirable framework for cellular behavior.[ 139 , 140 ] Being a 3D network of hydrophilic polymers, where chains are cross‐linked either by covalent bonds or by physical intra‐ and intermolecular attractions, hydrogels can absorb large quantities of fluids and they swell while maintaining their structure without dissolving.[ 139 ] Hydrogels can be processed in various shapes and architectures by using common techniques like emulsification, lyophilization, combinations of both, solvent casting‐leaching, gas foaming‐leaching, photolithography, electrospinning, micromolding, and 3D printing.[ 139 ]
The incorporation of PGS into hydrogels has been shown to positively influence hydrogels’ molecular network structure and elastomeric properties affecting also hydrogels’ biodegradability.[ 70 ] In various studies, it was shown that PGS could also be combined with hydrogels and the most common example is PEG which could be copolymerized with PGS.[ 66 , 141 , 142 , 143 ] Copolymer of PEGMEMA–PGS hydrogels showed tunable storage and loss moduli in a range between cells and human tissue[ 68 , 144 ] and in the case of PEG‐co‐PGS with grafted tyramine, a highly cytocompatible behavior was reported.[ 142 ] Injectable scaffolds based on PEG‐co‐PGS have been developed, which are very attractive from a clinical perspective as they can be minimally invasively applied and thereby reduce patient discomfort, risk of infection, scar formation, and the cost of treatment.[ 145 ] In contrast to these conventional hydrogels, Yoon and Chen[ 70 ] fabricated an elastomeric and pH‐responsive hydrogel, which has been made of PGS and gelatin without using any additional cross‐linking agent. Their proposed polycondensation reaction between PGSp and gelatin hydroxyl and carboxyl groups revealed ester bonds, whereas amine and carboxyl groups led to amide bonds. Afterwards, PGSPp was finally cross‐linked at elevated temperatures. By this process, highly flexible and stretchable films and also 3D scaffolds with an interconnected porous structure with shape recovery ability could be fabricated. Mouse fibroblasts confirmed the good cell cytocompatibility of such scaffolds.[ 70 ] Recently, a physical double‐network hydrogel adhesive consisting of catechol–Fe3+ coordination cross‐linked PGS‐co‐PEG‐g‐catechol and quadruple hydrogen bonding cross‐linked ureido‐pyrimidinone modified gelatin (GTU) was developed. This injectable adhesive hydrogel was designed to treat multidrug‐resistant bacteria infection and full‐thickness skin wound repair.[ 143 ] In contrast to these PGS‐based hydrogels, Eslami et al.[ 146 ] fabricated hydrogels from methacrylated gelatin and hyaluronic acid reinforced with PGS–PCL microfibers. Similarly, high cell viability and metabolic activity were measured for the hydrogel/microfibrous composites compared to neat hydrogels and neat fiber mats, but significantly higher levels of collagen I and elastin gene expression were reported. Additionally, the composite scaffold demonstrated suitable mechanical properties for CTE.
3.4. Freeze‐Drying
A wide range of highly developed technologies (i.e., electrospinning, 3D printing, micropatterning) has been considered for processing PGS as a porous scaffold. However, they all result in relatively small scaffolds with nonsufficient porosities, which, in turn, may limit the full capability and acceptance of PGS in a broader range of TE applications.[ 147 ] A different approach introduced to generate highly interconnected porous scaffolds is freeze‐drying. Freeze‐drying, also known as lyophilization, is a solvent removal process in which the solvent of the polymer solution crystallizes below its triple point, followed by subsequent pressure reduction, and by adding heat, the frozen solvent in the material can sublimate from the solid into the vapor phase.[ 147 ] Freeze drying of PGS requires the addition of a structure‐supporting polymer, e.g., PLLA,[ 22 ] silk fibroin microfibers (mSF), CH,[ 53 ] or poly (glycerol sebacate urethane) (PGSU).[ 148 , 149 ] As mentioned above, the cross‐linking of PGS takes place at high temperatures, which liquefies precondensed PGS. This process annihilates the porous structure from the freeze‐drying process during curing. A structure‐supporting polymer is then needed, that can withstand the harsh conditions of the curing process, protecting the pore structure obtained by freeze‐drying.[ 147 ]
In short, the freeze‐drying of PGS includes pouring a PGS solution into a Teflon mold, which is then frozen at ‐20 °C and placed in a freeze dryer for lyophilization. Solutions are typically cooled down to ‐30 °C to ‐50 °C and held at such low temperatures for several hours. The frozen solutions are then heated up and sublimated under vacuum. The temperature is raised to room temperature and hold again for several hours, completing the freeze‐drying procedure.[ 22 , 50 , 53 , 148 , 150 ]
Frydrych et al.[ 22 ] used a PGS/PLLA mixture to replicate native fatty tissue. Fatty tissue is a specialized connective soft tissue composed of adipocytes. Defects can arise from various causes including complex trauma, tumor resections, and congenital abnormalities.[ 151 ] In their research, Frydrych et al.[ 22 ] dissolved various combinations of PGS and PLLA in either 1,4‐dioxane or DMC, exhibiting two different freezing points of 10–12 °C and 2–4 °C, respectively. Scanning electron microscopy (SEM) showed that the produced scaffolds had different microstructural properties depending on the type of the used organic solvent. The reported elastic modulus was 0.030 MPa, tensile strength 0.007 MPa, elongation at maximum stress of 25% as well as a complete shape recovery upon release of the compressive load were 73 v%. The PGS–PLLA combinations revealed comparable mechanical strength to native high and low stress adapted adipose tissue. Additionally, the highly interconnected open‐pore structure with porosities and pore sizes ranging between 91–92% and 109–141 µm, respectively, confirmed the suitability of PGS/PLLA scaffolds for applications such as adipose TE.[ 22 ]
To avoid heat curing, Frydrych and Chen[ 148 ] and Samourides et al.[ 149 ] used chemically cross‐linkable PGSU to create porous structures. PGSU is an altered form of PGS using hexamethylene diisocyanate (HDI), as described in the chemical modification section (Section 2.3). Despite eliminating the heat cross‐linking step, scaffolds with an interconnected open‐pored microstructure and exhibiting excellent mechanical and cyclic load properties were produced. The as‐produced scaffolds were biodegradable, biocompatible, and conducted proangiogenic properties tissue ingrowth as well as angiogenesis.[ 148 , 149 ]
In a study by Zhang et al.,[ 53 ] PGS has been blended with mSF and CH, respectively, which should act as modifiers for a combined particulate leaching and freeze‐drying technique. NaCl particles, precondensed PGS, and one of the modifiers were mixed in a Teflon mold and cured for 12 h at 140 °C to produce mSF/PGS and CH/PGS composites, respectively. After curing, the salt particles were removed using deionized water and subsequently, the samples were lyophilized for 24 h to obtain a porous structure. Highly cross‐linked and open‐porous scaffolds with an increased cross‐link density as well as improved water absorption capacity compared to neat PGS has been achieved with both, mSF/PGS and CH/PGS scaffolds. Furthermore, the degradation rate could be controlled with different amounts of modifiers. Suitable biocompatibility for the use in skin replacement therapy was evaluated using mouse dermal fibroblasts of C57 BL/6 mice. Cell culture experiments showed suitable cell attachment as well as proliferation and deep ingrowth into the porous structure of the scaffold.[ 53 ]
3.5. Micropatterning
Micropatterning is the creation of specifically patterned and textured surfaces to gain a defined material behavior or to mimic the extracellular microenvironment of tissues or organs. It allows better control over particular constraints in a 3D platform compared to traditional 2D culture in tissue culture plates.[ 152 , 153 ] Cells are forced to adopt a shaped pattern of adhesion, which helps to study the cell behaviors exposed to micro topographic features as well as developing microfluidic devices.[ 153 ]
Future constructions of implantable tissue structures will require novel 3D microfluidic environments with high potential for TE. The biodegradable and biocompatible elastomer PGS is widely used in such approaches. Its transparency and thermoset properties are suitable for microfabrication techniques such as laser ablation.[ 154 ] On the other hand, micropatterning using laser ablation is considered as a green and rapid manufacturing process with low material selection barrier and high adjustability.[ 155 ] As Hsieh et al.[ 155 ] and Yeh et al.[ 156 ] assessed in their work, creating micro‐channels directly on cross‐linked PGS showed no nano/micro‐sized fractures or cracks across the PGS surface compared to silicone polydimethylsiloxane (PDMS), which is often used as a material for a subsequent molding of PGS (Figure 5 ).[ 155 , 156 ]
Figure 5.
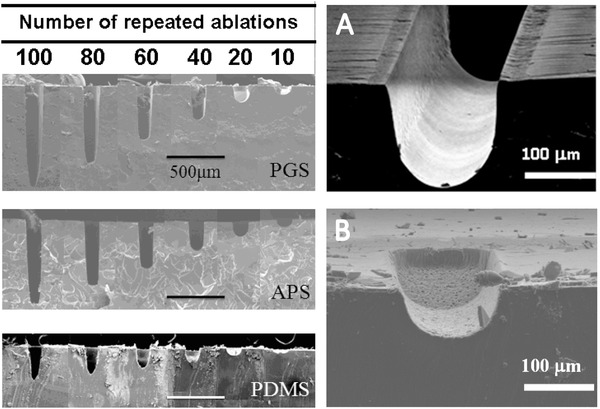
SEM cross‐section images of produced microchannels on PGS, poly(1,3‐diamino‐2‐hydroxypropane‐co‐polyol sebacate) and PDMS with increasing ablation times. A) Laser ablated 25 times on PGS showing high edge quality B) whereas PDMS presented low edge quality with the same ablation time. Adapted from Hsieh et al. Reproduced under the terms of the CC‐BY license.[ 155 ] Copyright 2017, the Authors. Published by MDPI.
A PDMS mold is mostly produced by using epoxy‐based negative masks on silicon wafers generated by photolithography. A mixture of PDMS elastomer is then poured on these patterned surfaces and deformed after drying. These forms allow the fabrication of highly elastic PGS substrates with defined micropatterned channels.[ 154 , 157 , 158 ]
Besides laser ablation and photolithography, Morgan et al.[ 18 ] created a porous structure with primary (macroscale) and secondary (microscale) pores. Therefore, a combination of poly (limonene thioether) (PLT32i) and PGSp has been cast onto sintered spherical particles of poly(methyl methacrylate) (PMMA), which were leached out subsequently.[ 18 ] Those porous structures were intended to create spatially organized, contractile heart tissue. Primary pores were able to control the orientation of heart cells and implement sufficient perfusion, while the secondary pores extended the retention of heart cells and limited the volume fraction of the polymer.[ 18 ]
Hu et al.[ 159 ] developed a degradable micropatterned PGS scaffold copolymerized with aniline trimer (5 wt%, 10 wt., and 15 wt%) to obtain an electroactive property (Figure 6 ). The as‐produced films showed good cell viability as well as increased proliferation of rat cardiomyoblast‐derived H9c2 cells. Additionally, electroactive films with a 10 wt% aniline trimer content significantly enhanced cell–cell interaction, growth, and synchronous calcium transients of neonatal rat primary cardiomyocytes.[ 159 ]
Figure 6.
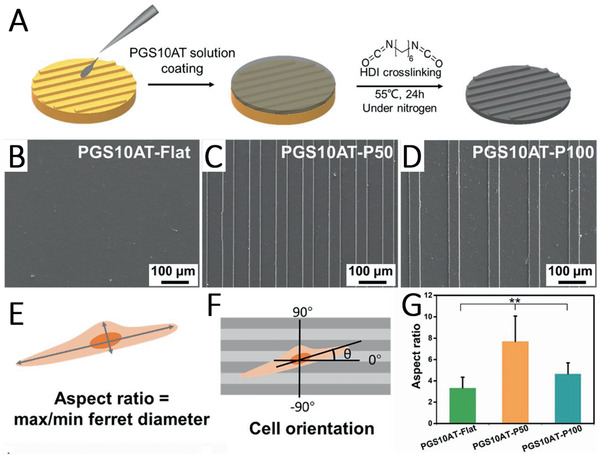
A) Scheme showing the fabrication of micropatterned PGS–aniline trimer (PGS‐AT) films. B) SEM images of flat PGS‐AT films, C) PGS‐AT films with a groove/ridge dimension of 50/50 µm and D) 50/100 µm. E) Scheme of a cellular aspect ratio and F) cellular alignment on the microstructured surface. G) Cellular aspect ratio on different patterned PGS‐AT films. Reproduced with permission.[ 159 ] Copyright 2019, Elsevier.
3.6. 3D Printing
Growing in popularity, another technique to fabricate complex structured scaffolds with a wide variety of design options is additive manufacturing. Unmodified PGS alone, however, exhibits as already mentioned, a lack of processability due to its harsh cross‐linking conditions.[ 27 ] Therefore, alternative strategies than thermal processing of PGS such as chemical modification or blending with other materials are developed.
3.6.1. Extrusion‐Based printing
Extrusion‐based 3D printing is widely used in TE. In this process, a printable material is pushed through a nozzle and the shape of the printed part is stabilized, mostly via cross‐linking of the material, in order to preserve the printed layer‐by‐layer structure. Extrusion‐based systems have the advantage of being easy to use and relatively cost‐effective compared to other 3D printing systems.
In 2016, for the first time, Yeh et al.[ 156 ] demonstrated the 3D printing of photocurable PGS. They used the potential of 3D printing for the production of sophisticated biocompatible, elastomeric tissue substitutes. An extrusion‐based 3D printing set‐up and a biodegradable and photocurable PGSA ink were used. Two different PGSA macromers were produced with two different molecular weights of PGS (low molecular weight PGS with a M n of 5.78 kDa and high molecular weight PGS with a M n of 6.32 kDa). Subsequently, these macromers were blended, which led to photocurable macromer inks with variable viscosities. With optimal ink viscosities, scaffolds could be printed with a high resolution up to ten layers in height. The mechanical properties of the 3D printed scaffolds depended on the respective printing density. Scaffolds with a lower printing density had lower moduli (Young's modulus of 310 kPa) and failure properties (failure strain of 40%) than scaffolds with a higher density (Young's modulus of 480 kPa and failure strain of 70%) compared to cast control samples (Young's modulus of 740 kPa and failure strain of 86%). The cytocompatibility was tested using 3T3 fibroblasts where significant adhesion, cell spreading, and proliferation were observed on PGSA showing the suitability of the elastomeric scaffolds in soft TE.[ 231 ]
The set‐up was further modified by Yeh et al.[ 58 ] using a specifically functionalized photocurable Nor‐PGS with thiol–ene click chemistry to control PGS cross‐linking. Subsequently, after extrusion of the viscous Nor‐PGS macromer paste, it was cross‐linked using UV light to obtain a high shape fidelity of the printed parts. The printed Nor‐PGS structures can be cross‐linked rapidly (<1 min) leading to porous, elastomeric scaffolds with mechanical properties comparable with the formerly produced PGSA scaffolds.[ 58 , 231 ]
Lei et al.[ 31 ] and Yang et al.[ 37 ] investigated a different approach without a chemical modification of PGS by mixing either neat PGS or a PCL/PGS blend with NaCl as a removable thickener for printing and a reinforcer for curing. The combinations were directly printed using an extrusion‐based set‐up. The produced NaCl‐containing scaffolds possessed a stacked construction with regular crisscrossed filaments. Good printability was established with a PGS/salt ratio of 1:2 and a printing temperature of 50 °C. After leaching the salt particles, interconnected micropores formed.[ 31 ] A Young's modulus of 748.5 ± 21.0 kPa, a tensile strength of 302.7 ± 7.7 kPa and a failure strain of 57.3 ± 1.3% confirmed the suitability of this scaffold for CTE. In vivo studies of printed PCL/PGS scaffolds as cardiac patches in adult male Sprague‐Dawley rats showed increased wall thickness, pumping function, angiogenesis, and cell ingrowth after an induced myocardial infarction (MI). Decreased apoptosis and infarction size demonstrated its therapeutic suitability and versatile applicability of these novel 3D printed, cardiac patches.[ 37 ]
In a further innovative approach, Lei et al.[ 138 ] developed a four‐axis printer that applies the print strands to a rotating collector. This enabled the successful printing of hydrogels, thermoplastics, and thermosets. In this way, tubular structures would be produced relatively easily, e.g., for tracheal cartilage applications.[ 138 ]
Recently, Touré et al.[ 46 ] combined an electrospun PCL/PGS fiber mat with a 3D printed PCL/PGS structure, containing BG particles. Three different types of extrusion‐based scaffolds were produced using PCL/PGS as a reference, with 5 wt% BG and with 10 wt% BG. After producing the 3D scaffolds, a 14 wt% polymer solution of PCL/PGS (mixing ratio of 1:1) was electrospun directly on top of these scaffolds. Excellent adhesion between the layers resulted in Young's moduli of 240–310 MPa. The composite materials were biodegradability and a weight loss ≈14% and equilibrium in pH was measured after two months of incubation which was followed by the release of alkali ions from the BG particles. Cytotoxicity tests with fibroblasts proved the biocompatibility of PCL/PGS/BG constructs with a cell viability of over 125% on the second day, making these scaffolds suitable candidates for TE of tendons and ligaments.[ 46 ] Recently, Farizhandi et al.[ 161 ] focused their work on the printing of electronically conductive and flexible bioelectronic devices. For this purpose, they combined lithium phenyl‐2,4,6‐trimethylbenzoylphosphinate as photoinitiator with PGS and zinc (Zn). The resulting 3D printed scaffolds showed remarkable mechanical and adhesive properties as well as noncytotoxic behavior both in vitro and in vivo, rendering it suitable for several biocompatible applications.[ 161 ]
3.6.2. Laser‐Based Printing
Stereolithography (SLA),[ 162 ] direct laser writing (DLW), and digital light processing additive manufacturing (DLP‐AM)[ 62 ] are 3D printing methods, which exploit the cross‐linking between acrylated polymer chains via the exposure to light at various wavelengths involving radical polymerizations. These fabrication methods are considered as highly precise and efficient compared to fused deposition modeling or extrusion‐based 3D printing. Either a single laser beam (SLA, DLW) or a digital mirror device (DMD) (DLP‐AM) is used simultaneously to control the exposure of light in an array.[ 163 , 164 ]
Several research groups have studied the effect of degree of acrylation in PGS and the weight ratio between the pre‐polymers towards mechanical and degradation properties as well as processability.[ 62 , 162 , 165 , 166 ] Chen et al.[ 62 ] copolymerized PGSA either with polycaprolactone diacrylate (PCLDA) or poly(ethylene glycol) diacrylate (PEGDA) to form a polymer network and thus to overcome the relatively low degradation rate of neat PCLDA and PEGDA in vivo. They used a DLP‐AM set‐up to produce scaffold structures with adjustable mechanical and degradation properties. In their study, it was found that blending prepolymers of PGSA with various degrees of acrylation and various weight ratios, the viscosity of the prepolymers remained stable, but became even more suitable for 3D printing than the substances alone. By altering the weight ratios of the used materials, it was possible to control Young's moduli within a range of 0.67 to 10.54 MPa. Moreover, a significantly improved degradability of the scaffolds was achieved.[ 62 ]
Pashneh‐Tala et al.[ 165 ] investigated the effect of the molecular weight and degree of methacrylation of PGS‐methacrylate on the printability using a 2‐photon polymerization technique. The degree of methacrylation was observed to have the most substantial influence on degradability and mechanical properties. Using 2‐photon polymerization, they succeeded in creating 3D scaffolds with a minimum feature size of 10 µm. Cell culture studies showed a sustained growth and proliferation of vascular smooth muscle cells (SMCs), indicating that the material is a promising candidate for TE applications.[ 165 ]
Singh et al.,[ 162 ] on the other hand, assessed the degradation rate and mechanical properties of PGS‐methacrylate and structured nerve guidance conduits (NGC) fabricated via SLA. As produced conduits of PGS‐methacrylate, with a 0.75 degree of methacrylation, were flexible and exhibited an appropriate compressive Young's modulus (3.2 MPa) as well as suture retention strength (12.3 MPa) for peripheral nerve repair. In vivo results in Thy‐1‐YFP‐H mice showed a supported regeneration of axons, neurites aligned toward the topographical conduit grooves and no increase of neuropathic pain, in comparison to native nerve grafts concluding that PGS‐methacrylate is an attractive synthetic alternative to native grafts.[ 162 ]
Wang et al.[ 166 ] used DLP‐based 3D printing of a PGSA/PEGDA combination to generate nature‐inspired double network structures consisting of interconnected segments with different mechanical properties. By assigning distinct exposure times at specific locations, it was possible to create networks with stiff, strong frames to maintain the rough shape of the scaffold and diagonal elastic struts to absorb tensile energy from the same material in a single shot. In this way, finely resolved structures could be printed with less than 100 µm in size and a volume of less than 1 mL as well as structures with a volume of more than 10 mL. Subsequent mechanical tests revealed an expected influence of both exposure time and PEGDA amount on the mechanical properties of the printed structures. The mechanical properties of these scaffolds could be tailored by an alternating double network structure.[ 166 ] Further advances regarding DLP‐based 3D printing of PGSA have been made by Wu et al.[ 167 ] in order to print porous tubular structures. Varying the PGSA concentration, the type of diluent (dimethyl sulfoxide, 2‐butoxyethyl acetate) and the degree of acrylation, PGSA based inks with viscosities below 5 Pa s and exposure time of less than 10 s were found as suitable for DLP fabrication. The final compositions enabled the fabrication of complex 3D models with high mimicry and a minimum feature thickness of 80 µm, which can be used in a variety of TE applications.[ 167 ] An overview of all printing approaches involving PGS of the last five years is shown in Table 2 .
Table 2.
Overview of PGS used in 3D printing approaches. Printing techniques are separated into extrusion‐based and laser‐based methods
| Printing technique | Material combination | Mechanical properties | Cell response | Special features | Proposed application | Ref. | |
|---|---|---|---|---|---|---|---|
| Extrusion‐based | commercially available 3D filament printer (Revolution XL, Quintessential Universal Building Devices) modified for syringe extrusion | Photocurable PGSA combining a ratio of 10:90 of
|
Depending on printing density
a)
:
E‐modulus: 310 kPa Tensile strength: 120 kPa Failure strain: 40%
E‐modulus: 350 kPa Tensile strength: 210 kPa Failure strain: 67%
E‐modulus: 480 kPa Tensile strength: 330 kPa Failure strain: 70% |
No cytotoxic response in contact with NIH 3T3 fibroblasts, increased cell proliferation within 4 d of culture |
|
Complex biocompatible, elastomeric tissue replacements (Soft TE) |
[ 231 ] |
| Specific functionalized photocurable Nor‐PGS with thiol–ene click chemistry | Depending on thiol/norbornene ratios (N):
E‐modulus: 110 kPa Tensile strength: 260 kPa Failure strain: 240%
E‐modulus: 340 kPa Tensile strength: 690 kPa Failure strain: 200%
E‐modulus: 400 kPa Tensile strength: 790 kPa Failure strain: 170% |
No cytotoxic response in contact with NIH 3T3 fibroblasts, increased cell proliferation within 8 d of culture |
|
Elastomeric, biodegradable and cytocompatible scaffolds for soft tissue applications | [ 58 ] | ||
| Commercially available fused depositioning system (HTS‐400; Fochif Mechatronics Technology) | Neat PGS with NaCl (NaCl‐PGS) at a ratio of PGS:NaCl = 1:2 | Depending on used needle gauge (G):
E‐modulus: 150.7 kPa Tensile strength: ≈80 kPa Failure strain: ≈40%
E‐modulus: 239.4 kPa Tensile strength: ≈90 kPa Failure strain: ≈30% |
Cytocompatible, biocompatible and biodegradable in vivo after subcutaneous and epicardial implantation in male Sprague Dawley rats Thickening of the LV wall and attenuated LV dilatation after 28 d postimplantation of 3D‐printed PGS patches |
|
Used in a vapo‐mechanical sensor and soft actuator or as a myocardial patch | [ 31 ] | |
| PGS:PCL combination in a ratio of 9:1 mixed with NaCl (NaCl‐PGS/PCL) at a 1:2 weight ratio (PGS/PCL : NaCl) |
E‐modulus: 190.1 kPa Tensile strength: 62.2 MPa Failure strain: 28.9%
E‐modulus: 31.5 MPa Tensile strength: 3.3 MPa Failure strain: 475.0%
E‐modulus: 748.5 kPa Tensile strength: 302.7 kPa Failure strain: 57.3% |
Epicardial implantation in male Sprague Dawley rats showed preserved heart function, increased LV wall thickness, reduced infarct size, promoted vascularization, induced tissue repair by recruiting M2 macrophages, and inhibited myocardial apoptosis |
|
Potential for treating multiple cardiovascular diseases (CTE) | [ 37 ] | ||
| Combination of a DIW‐3D printer (HTS‐400; Fochif Mechatronics Technology) and a rotary receiver | Neat PGS with NaCl (NaCl–PGS) at a ratio of PGS:NaCl = 1:2 combined with an electrospun gelatin fiber mesh post‐printing | Mechanical properties after 12 weeks in vivo implantation with and without cells:
E‐modulus: ≈2.40 MPa
E‐modulus: ≈8.00 MPa
E‐modulus: ≈11.5 MPa |
Chondrocytes from auricular cartilage of New Zealand white rabbits showed excellent cell proliferation without noticeable apoptosis within 4 d of culture; In vivo results in nude mice showed an increased DNA, GAG as well as collagen content after 12 weeks of implantation |
|
Potential for tracheal cartilage reconstruction | [ 138 ] | |
| Commercially available 3D‐Bioplotter Developer Series (EnvisionTec) | PGS/PCL combination in a ratio of PGS:PCL = 1:1 combined with BG particles and an electrospun PGS/PCL fiber mat postprinting | Depending on the amount of incorporated BG particles:
E‐modulus: 250 MPa Maximum load: 6 N Failure length: 1.5 mm
E‐modulus: 241 MPa Maximum load: ≈4 N Failure length: ≈1.4 mm
E‐modulus: 311 MPa Maximum load: ≈3.5 N Failure length: ≈0.6 mm |
NIH 3T3 fibroblasts showed no cytotoxic response with an improved cell proliferation within 7 d of culture |
|
Potential for tendon and ligament TE applications | [ 46 ] | |
| Commercially available 3D‐Bioplotter Allevi 2 | Photocurable PGSA with chemically sintered Zn powder in various ratios |
Depending on the amount of incorporated Zn powder: Conductivity: up to 11.8 mS m‐1 Compression modulus: up to 1179.5 kPa E‐modulus: up to 1099.9 kPa |
No cytotoxic response in contact with C2C12 myoblasts as well as in in vivo studies using Wistar rat models |
|
Bio‐integrated electronics in wearable electronics, electronic skin, robotics, implantable electronics and human–machine interfacing | [ 161 ] | |
| Laser‐based | DLP‐AM system developed by the Cheng lab at the National Taiwan University of Science and Technology | Photocurable PGSA with an altered degree of acrylation (30, 15, and 7%) combined with PCLDA (PGSA: PCLDA = 2:1) or PEGDA (PGSA: PEGDA = 1:1) | Depending on the degree of acrylation and mixing ratio:
E‐modulus: 0.12 MPa Tensile strength: 0.10 MPa Failure strain: 121.23%
E‐modulus: 1.55 MPa Tensile strength: 0.63 MPa Failure strain: 46.95%
E‐modulus: 5.10 MPa Tensile strength: 1.36 MPa Failure strain: 28.43%
E‐modulus: 18.98 MPa Tensile strength: 3.19 MPa Failure strain: 21.50%
E‐modulus: 4.25 MPa Tensile strength: 0.80 MPa Failure strain: 21.29%
E‐modulus: 7.58 MPa Tensile strength: 0.91 MPa Failure strain: 13.63%
E‐modulus: 10.54 MPa Tensile strength: 1.10 MPa Failure strain: 12.96% |
Not conducted in this study |
|
Potential for multiple TE applications | [ 62 ] |
E‐modulus: 4.35 MPa Tensile strength: 0.58 MPa Failure strain: 15.34%
E‐modulus: 1.42 MPa Tensile strength: 0.19 MPa Failure strain: 22.39%
E‐modulus: 2.85 MPa Tensile strength: 0.20 MPa Failure strain: 11.28%
E‐modulus: 7.00 MPa Tensile strength: 0.69 MPa Failure strain: 14.08% |
|||||||
| Femtosecond DLW using 2PP with a PHAROS femtosecond Yb:KGW laser (Light Conversion Ltd) | Photocurable mAcr‐PGS of
|
Depending on the degree of methacrylation (N) and molecular weight:
Low M w E‐modulus: ≈0.50 MPa Tensile strength: ≈0.80 MPa High M w E‐modulus: ≈0.90 MPa Tensile strength: ≈0.60 MPa
Low M w E‐modulus: ≈1.50 MPa Tensile strength: ≈0.90 MPa High M w E‐modulus: ≈1.50 MPa Tensile strength: ≈0.90 MPa
Low M w E‐modulus: ≈6.80 MPa Tensile strength: ≈3.80 MPa |
Cytocompatible with human dermal fibroblasts, human adipose‐derived stem cells (ADSCs) and human coronary artery SMCs, enhanced cell proliferation during the 7 and 14 d of culture, respectively |
|
Potential for multiple TE applications both in vitro and in vivo | [ 165 ] | |
| In house designed micro SLA set‐up |
Photocurable mAcr‐PGS with altered degree of methacrylation (N = 0.22–1.00) |
Mechanical compression testing of a produced NGC (N = 0.75b): Max compression: 0.57 mm Compression at break: 11.4% Stiffness: 3.2 MPa Suture retention strength: 12.3 MPa |
Neuronal NG108‐15 cells as well as primary SCs from male Wistar rats showed improved neuronal and glial cell growth in vitro |
|
Potential for nerve conduit guidance (nerve TE) | [ 162 ] | |
|
In vivo: an increased regeneration of axons, oriented axonal growth without an increase of neuropathic pain in comparison to native nerve grafts |
|||||||
| DLP‐based 3D printing using a DMD chip (Texas Instruments) | Photocurable PGSA combined with altered amounts of PEDGA (1%, 5% and 10%) | Depending on light exposure time (in s) and printed structure (single or double network):
E‐modulus: 11.91 kPa Tensile strength: 5.92 kPa Tensile strain: 62.58%
E‐modulus: 47.66 kPa Tensile strength: 14.11 kPa Tensile strain: 43.31%
E‐modulus: 32.09 kPa Tensile strength: 15.08 kPa Tensile strain: 64.18% |
HUVECs showed excellent viability (>90%) within 7 d of culture |
|
Specifically optimizable for different biomedical applications (soft TE) | [ 166 ] | |
| DLP‐based 3D printing using an Ember desktop 3D printer (Autodesk) with a built‐in light‐emitting diode projector | Photocurable PGSA with different degrees of acrylation mixed with 1wt% dipehyl (2,4,6‐trimethylbenzoyl) phosphine oxide photoinitiator | Mechanical tensile testing on films:
E‐modulus: 192.3 kPa Tensile strength: 460.2 kPa Tensile strain: 239%
E‐modulus: 859.0 kPa Tensile strength: 326.8 kPa Tensile strain: 39%
E‐modulus: 3668.7 kPa Tensile strength: 919.1 kPa Tensile strain: 26%
E‐modulus: 226.8 kPa Tensile strength: 556.9 kPa Tensile strain: 244%
E‐modulus: 3097.6 kPa Tensile strength: 1273.3 kPa Tensile Strain: 42% |
Not conducted in this study |
|
Potential for multiple TE applications | [ 167 ] | |
Printing density referred to as the amount of interior volume designated to be occupied by material (fill density setting, in Slic3r), including 30% for low density, 45% for medium density, and 60% for high density; b)NGC = nerve guidance conduit, Hounslow mechanical analysis was conducted along the axial direction of the NGC.
4. Biomedical Applications
4.1. Cardiac TE (CTE)
CTE is an important branch of TE aiming at developing cell‐scaffold constructs to induce cardiac tissue repair.[ 160 , 168 ] Today the primary fabrication method for cardiac tissue‐engineered scaffolds is electrospinning. Thus produced fiber mats can be fabricated with continuous fibers in nano‐ to microscale, which show morphological similarities to the native fibrous ECM regarding high surface‐to‐volume ratio, high porosity, and variable pore size.[ 169 ] As already mentioned, due to its low solution viscosity, PGS has been blended with other polymers to be spinnable. Often, PGS blended with PCL and spun as a fiber mat was used for CTE application.[ 32 , 170 ] In recent years, this blend has been further optimized. A patterned fiber mat of PGS/PCL was electrospun on a Teflon‐coated silicon wafer with imprinted topographical features, like squares and grooves (Figure 7 ).[ 110 ] In vitro experiments using C2C12 myoblasts and neonatal rat cardiomyocytes showed a successful alignment of both cell types after 24 h of culture. Furthermore, excellent cell‐to‐cell communication was confirmed by measuring the expression of connexin43 on imprinted scaffolds.[ 110 ] In contrast to topographical cues, Rai et al.[ 106 ] functionalized PGS‐based fiber mats chemically by conjugating VEGF via amide groups. VEGF was homogeneously immobilized on the fibers and the functionalized scaffolds supported the attachment, growth, and proliferation of C2C12, rat cardiac progenitor cells, and rat aortic endothelial cells (rAoECs) compared to unfunctionalized PGS/PCL scaffolds. Additionally, morphological changes of rAoECs confirmed the formation of a vascular tree in the fiber mat.[ 106 ] Compared to previously established work on PGS/PCL electrospun blends, in another study, the solvent system was exchanged from commonly used (harsh) solvents, e.g., CF, methanol, DCM or dimethyl carbonate, to the less toxic solvent acetic acid to lower the level of toxicity during the fabrication process as well as to maintain scaffold biocompatibility.[ 33 ] It could be observed that acetic acid had no negative influence on fiber formation, and a homogenous and defect‐free fiber mat was achieved. Moreover, this study revealed the significant influence of the disinfection method (UV vs immersion in 70% ethanol) on the biocompatibility. When samples were disinfected in ethanol, partially PGS and its unreacted monomers were leached out from the fiber mat, and created pores in the fibers leading to higher cell viability and cell proliferation compared to UV disinfected samples.[ 33 ] This behavior has also been reported by Apsite et al.[ 29 ] after incubation of PCL/PGS fibers in PBS where they also obtained porous fibers of PCL after 4 weeks of incubation in 37 °C. Modified PGS, i.e., poly(1,3‐diamino‐2‐hydroxypropane‐co‐glycerol sebacate)‐co‐PEG, has also been electrospun together with PCL in order to obtain fiber mats exhibiting uniaxial mechanical properties required for human aortic valve leaflets.[ 171 ]
Figure 7.
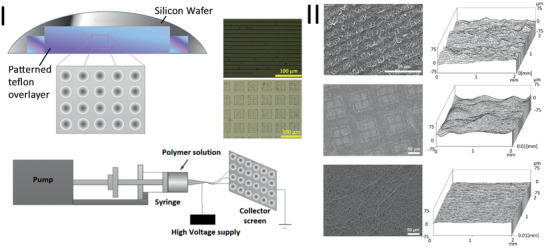
Scheme showing the microcasting process of PGS films via a patterned silicon wafer and its Teflon overlayer as well as the electrospinning setup, where the silicon wafer replaces the conventional collector (I). SEM images showing different topographical PGS/PCL fiber mats (left) and their corresponding surface roughness profiles (right) (II). Reproduced with permission.[ 110 ] Copyright 2016, Elsevier.
Furthermore, PGS/PCL electrospun fibers have been used as reinforcement for methacrylated gelatin/hyaluronic acid hydrogels via immersion technique.[ 146 ] PGS/PCL fiber/hydrogel composite scaffolds showed higher metabolic activity of valvular interstitial cells compared to the fiber mat and the hydrogel alone. Additionally, collagen type I and elastin expression significantly increased for PGS/PCL fiber‐reinforced composite scaffolds. Due to the optimal expression levels of collagen and elastin genes achieved with the composite scaffolds, the authors concluded the scaffold's suitability for treating congenital heart defects.[ 146 ]
PGS has also been blended with poly(butylene succinate‐butylene dilinoleate) (PBS‐DLA), a multiblock thermoplastic elastomer,[ 115 , 172 ] which led to suitable mechanical and degradation properties for cardiac patches and exhibited higher C2C12 cell viability on fiber mats with increasing PBS–DLA content.
On the other hand, PGS has also been combined with natural polymers as they 1) provide living cells important ligands required for adhesion and proliferation, 2) mainly degradation byproducts are nontoxic and 3) initiate a low immune response.[ 29 ] The combination of PGS with collagen and silk fibroin (1:4.5:4.5) has demonstrated mechanical properties which were close to those of heart valve tissue. Furthermore, it presented a low rate degradation and thrombogenic platelet adhesion compared with collagen hydrogels or fiber mats was reduced from 220% to 20%.[ 127 ]
The further potential of PGS/fibrinogen core/shell fibers was investigated in an in vivo porcine model with MI.[ 129 ] The results revealed that a combination of PGS/fibrinogen/VEGF and mesenchymal stem cells (MSCs) promoted recovery of left ventricle function after MI, proven by the improvement in EF and prevention of left ventricle enlargement. Furthermore, cardiac marker proteins troponin and actinin, as well as endothelial cell marker protein CD31 were expressed by the MSCs in contact with the core–shell fibrous scaffold showing differentiation of human bone marrow MSCs into cardiac and endothelial cells.[ 129 ] Functional and structural integrity, as well as a suitable matrix for entrapment of MSCs, was successfully obtained using these PGS/fibrinogen/VEGF fiber mats.
A vegetable protein, namely zein, the primary storage protein in corn,[ 173 ] was also chosen as blending material for PGS.[ 130 ] Compared to proteins from animal sources, vegetable proteins are readily available with low‐cost and as coproduct cereal grains can be processed as food or fuel. Moreover, they are biodegradable and exhibit less immunogenicity.[ 174 ] Bead free PGS/zein fiber mats were produced from an acetic acid solvent system and they were stable in PBS over 28 d, which could be beneficial for cardiac patch application.[ 130 ]
Apart from electrospinning, solvent casting,[ 87 ] micropatterning,[ 157 , 159 ] micromolding[ 18 ] and 3D printing[ 37 ] have also been used to create CTE scaffolds. As cardiac tissue is an electromechanical tissue, drug‐loaded conductive PGS/nanoparticle‐based composite films have been developed via solvent casting.[ 87 ] These composite patches made of PGS, collagen type I, polypyrrole, and a small molecule as a model drug (3i‐1000, a small molecule inhibitor of GATA4‐NKX2‐5 interaction) exhibited excellent tensile properties, suitable conductivity, and a sustained release of the model drug. The combination of the films proved to be a suitable matrix for cardiomyoblast attachment and infiltration and stayed viable for 21 d. Hu et al.[ 159 ] created an electroactive and biodegradable PGS‐co‐aniline trimer elastomeric scaffold via micropatterning. The films showed electroactive properties and elastic modulus comparable to those of the native heart tissue and good viability and proliferation of rat cardiomyoblast‐derived H9c2 cells. 10 wt% of aniline trimer was found to be the optimum concentration, which led to significantly enhanced cell–cell interaction, maturation and synchronous calcium transients of neonatal rat primary cardiomyocytes. The groove/ridge surface characteristics guided CM's alignment and elongation.
A three‐layered scaffold made of PLT32i and PGS with multiscale, hierarchical pore architecture was developed via micromolding and porogen leaching for CTE (Figure 8 ).[ 18 ] Porous grids with microchannels and porous films were prepared from PLT32i, whereas the porous vascular‐parenchymal interface was made of PGS. After assembly and perfusion of cells in the structure, a contractile, prevascularized heart tissue was obtained. The PLT32i offered a hierarchical architecture of macroscale pore channels, which enabled robust perfusion, guided heart cell alignment, and microscale pores that increased heart cell retention while reducing polymer volume fraction. At the same time, PGS served as a microporous vascular–parenchymal interface providing high oxygen permeance.
Figure 8.
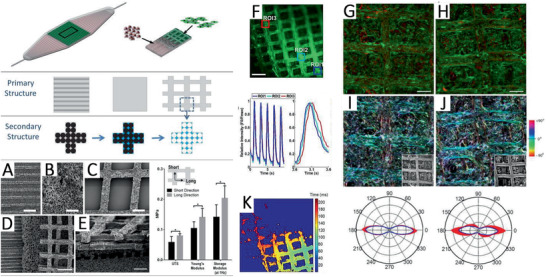
Hierarchical architecture of scaffolds for coculturing vascularized cardiac tissue. Consisting of perfusable channels for human umbilical vein endothelial cells (HUVECs) (red), a vascular–parenchymal interface, and two offset grids with rectangular through‐pores for heart cells (green). Primary and secondary pore structures were generated using micromolding and porogen leaching. A‐E) SEM images show the different porous interfaces (scale bars: A–C), E) 200 µm, D) 500 µm). F) The intensity of Ca2+ signal over time in the selected regions of interest (Construct stained with Fluo‐4AM). Time activation maps showing excitation (K). Heart cell orientation on day 5 shown in confocal micrographs G,H) before and I,J) after pixel‐by‐pixel image analysis. Reproduced with permission.[ 18 ] Copyright 2016, Wiley‐VCH GmbH.
Recently, mixtures of PCL, PGS, and NaCl particles were 3D printed to achieve a stacked construction with regular crisscrossed filaments and interconnected micropores (Figure 9 ).[ 37 ] The scaffold showed superior mechanical properties compared to its single components and favorable biodegradability and biocompatibility. In a rat model, 3D printed PGS/PCL scaffolds showed improved and preserved heart function after MI as it increased left ventricle wall thickness, reduced infarct size, promoted vascularization, and increased M2 macrophage infiltration. Due to its formability to any desired size or shape, it is expected that 3D printing will experience an upward trend in biomedical applications, including for developing PGS‐based scaffolds for CTE.
Figure 9.
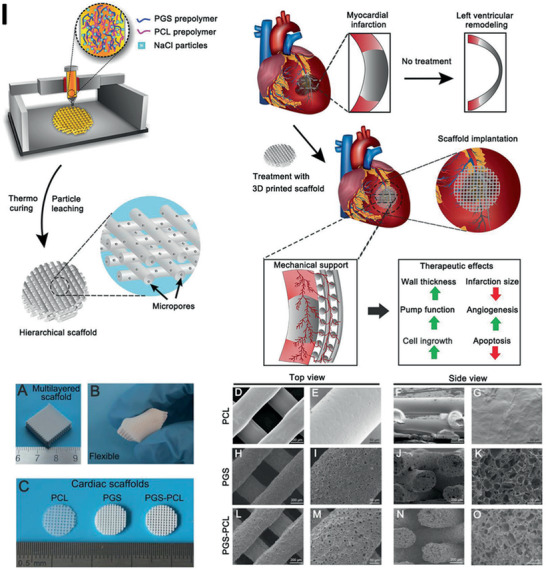
Scheme showing the 3D printing set‐up and mixed ink of PGS and PCL prepolymer with salt particles as well as the therapeutic effects of infarcted hearts when treated with the PGS–PCL scaffolds on infarcted myocardium. A) Images showing a 3D‐printed PGS–PCL scaffold with the multilayer structure as well as the flexibility of B) PGS–PCL scaffolds. C) Different 3D‐printed cardiac patches. Top‐view and cross‐sectional SEM images showing the morphology of D–G) 3D printed PCL, H–K) PGS and L–O) PGS‐PCL scaffolds (scale bars: D), F), H), J), L), N)‐C), E) 200 µm, E), I), M), 50 µm, G), K), O) 20 µm). Reproduced with permission.[ 37 ] Copyright 2019, Wiley‐VCH GmbH.
Ruther et al.[ 39 ] investigated the adhesion of electrospun gelatin fibers on porous PGS substrates forming bilayered scaffolds,[ 39 ] whereby direct electrospinning on the PGS substrate versus gelatin gluing of the two layers was compared. The authors concluded (according to adhesion tests) that gelatin gluing, which resulted in chemical bonding of the components, led to the best results even after two weeks immersion in phosphate‐buffered saline, thus demonstrating the possibility of using porous PGS substrates in the design of layered cardiac patches in combination with electrospun biopolymer (e.g., gelatin) fibers.
In contrast to the use of preformed scaffolds by electrospinning, solvent casting, micropatterning, micromolding, and 3D printing, injectable and photo‐crosslinkable versions of PGS have been developed. Recently, Hamada et al.[ 175 ] investigated a therapeutic strategy to deliver paracrine secretions, such as extracellular vesicles (EV), to damaged heart muscles.[ 175 ] In this approach, poly (glycerol‐co‐sebacate) acrylate‐ethylene glycol (PGSA‐g‐EG) was mixed with induced pluripotent stem progenitor cell‐derived EVs and delivered directly via a syringe to the affected myocardium.[ 175 ] PGSA‐g‐EG was then photocrosslinked with a LED lamp in situ.[ 175 ] The polymer was shown to release EV during a two‐week in vitro degradation study continuously and to adhere to the epicardium for at least one month in an in vivo rat model.[ 175 ]
Overall, PGS structures in various combinations with other polymers have been fabricated mainly by electrospinning and such systems have shown promising results for CTE. However, modern fabrication techniques like micropatterning and 3D printing are gaining ground, especially as they enable the fabrication of thicker constructs (in comparison to electrospinning). In future, combinations of both nano and microscale fabrication techniques will be considered, as the scaffold properties will be enhanced on the basis of developing robust, hierarchical 3D structures.
4.2. Vascular TE
As mentioned above, cardiovascular diseases remain one of the leading causes of mortality and morbidity worldwide. In the event of atherosclerosis, plaque deposits and the arterial wall hardens, which results in stenosis or blockage of the blood vessel. Lack of oxygen and nutrient supply can lead to severe consequences like MI, stroke, and other health complications.[ 176 ] Usually, bypass surgery with autologous venous or arterial grafts is performed which has limited access to available grafts, requires additional surgeries, and has a risk of donor site morbidity. On the other side, synthetic vascular grafts can only be used for large‐diameter blood vessels due to thrombus formation and frequent failure in small‐diameter grafts nowadays.[ 176 ] When designing a tissue‐engineered vascular graft (TEVG), the essential requirements are its biocompatibility, mechanical strength, and nonthrombogenicity. Commonly, scaffolds for vascular grafts are fabricated via sheet rolling, matrix molding, direct scaffolding like electrospinning or 3D printing, as well as further processing of decellularized xenogeneic grafts. Regarding PGS, electrospinning and salt leaching or combinations of both have been mostly investigated for vascular TE applications.
The material combination of PGS and PCL also proved to be favorable for the fabrication of vascular grafts. Small‐diameter tubular PGS/PCL grafts with an internal diameter of 2 mm were fabricated by electrospinning.[ 111 ] A 1:1 PGS: PCL ratio with a relatively large average fiber diameter of 3.94 ± 1.39 and 5.57 ± 1.55 µm revealed comparable mechanical properties and suture retention to the native human artery. The introduced materials presented acceptable cytocompatibility to human vascular endothelial cell line EAhy926 and the fabricated vascular grafts offered suitable anticoagulation property with a low hemolysis percentage compared to pure PCL scaffolds. The effect of PGS/PCL fiber organization on the behavior of endothelial cells was investigated by Gaharwar et al.[ 105 ] Aligned PGS/PCL fibers not only led to higher elastic moduli compared to random fibers but also induced enhanced endothelial cell proliferation and cell organization in response to the topographical cues.[ 105 ] To improve the mechanical properties of the graft, sequential electrospinning was also developed to enclose the inner PGS/PCL layer with a layer of neat PCL.[ 101 ] The two layers firmly adhered, and the two distinct fiber mats revealed a fiber diameter gradient from the outside (139 ± 29 nm) to the inside (370 ± 73 nm). The contact angle of PGS/PCL decreased to 51 ± 3° compared to pure PCL with 117 ± 5°. The PGS/PCL layer was capable of mimicking antithrombotic features of the native intima and supported attachment, growth, and infiltration of MSCs. Similarly, a bilayered tubular graft was fabricated, which presented a thicker inner layer of PGS microfibers and a thin outer layer made of PCL (Figure 10 ).[ 107 ] For neat PGS fibers, PGS was blended with PVA, which was removed with water and ethanol after cross‐linking of PGS. Subsequently, the PGS core was hardened by freeze‐drying in a NaCl solution that filled the pores with salt crystals. PCL fibers were electrospun onto the PGS core, and the final vascular graft was washed in water for NaCl removal. The bilayered tubular grafts were implanted as infrarenal aortic interposition grafts in mice, where they remained up to 12 months without any sign of thrombosis or stenosis in the absence of anti‐platelet therapy and anti‐coagulation. The long‐term strength of the graft was confirmed as no rupture or catastrophic failure was detected and PCL sheath supported this stability. After 12 months of in vivo test, the remaining polymer of outer regions of the graft was still remained, whereas in the inner region organized contractile SMCs were found and neo‐tissue was formed. No graft‐related mortality was observed, and remodeling of graft and the adjacent aorta had taken place.[ 107 ] Stowell et al.[ 177 ] evaluated an electrospun PGS/PCL core/shell combination for potential use as a small‐diameter vascular graft. However, the highly porous, bilayered vessels showed almost complete degradation and inflammatory response after 15 d in vivo in a sheep model, rendering it insufficient as remodeling grafts in large animals.[ 177 ] Moreover, Wu et al.[ 178 ] showed in their in vivo work that the thickness of a PGS/PCL core/shell vascular implant influences remodeling. They created PGS grafts with high interconnected porosity and varying vessel wall thickness. However, all of the scaffolds suffered high rates of occlusion and rupture, requiring further research work.[ 178 ] On the other hand, a combination of a salt‐leached palmitic acid‐modified PGS core and a PCL electrospun outer layer showed reduced inflammation, robust mechanical properties, and a layered distribution of SMCs and ECM proteins in vivo, improving the overall performance of PGS‐based vascular grafts.[ 179 ]
Figure 10.
Scheme of the electrospinning process for the production of artificial vascular grafts. In brief, a polymer solution of PGSp (blue) and PVA (red) is deposited by an applied voltage on a plastic rod (1) and then drawn off on a rotating stainless‐steel mandrel (2). The PGSp–PVA fibers are then thermally cross‐linked and cleaned. Eventually, a PCL solution (violet) is deposited by electrospinning first on an aluminum plate anode (1) and then drawn off onto the rotating cross‐linked PGS core (2). The final result is a composite of an electrospun PGS microfiber core (green) and an electrospun PCL fiber coating (violet) (a). Macroscopic and inset transverse view of a finished graft before implantation, which is shown next to an American dime for size comparison (b). In situ view of a TEVG that conducts blood flow on the day of implantation (c). Representative Doppler ultrasound images of transplanted TEVGs with (top) and without dilatation (bottom) 12 months after implantation. Yellow arrows indicate the proximal anastomosis with the adjacent proximal infrarenal abdominal aorta on the left and the implanted electrospun TEVG on the right (d). 3D reconstruction of a microcomputed tomography image of the same graft. The graft is highlighted in red. The adjacent proximal and distal abdominal aorta is stained white (e). Reproduced with permission.[ 107 ] Copyright 2016, Springer Nature.
As a reasonably simple method, salt leaching was also used to create vascular grafts of neat PGS. PGS was mixed with NaCl particles and filled in a mold, cross‐linked, either according to the standard procedure[ 180 ] or microwave‐assisted,[ 72 ] and later purified. Microwave‐assisted cross‐linking led to similar porosity, mechanical properties, and biocompatibility compared to conventional cross‐linking, however, the process was eight times faster.[ 72 ] Porous PGS grafts were exposed to enzymatic degradation to understand the interplay between degradation and graft erosion.[ 180 ] Despite their porous structure, the scaffolds lost the mass exponentially via surface erosion, which led to a 20% reduction of the outer diameter but no significant change in apparent density. Furthermore, the mechanical properties of the graft were not affected by the mass loss.[ 180 ]
Fabricated synthetic vascular grafts from PGS core and electrospun PCL shell have also been reported in several studies using a combination of electrospinning and salt leaching.[ 21 , 102 , 181 , 182 ] These acellular grafts degraded rapidly and supported the regeneration of neoarteries nearly free of foreign body reaction in rat abdominal aorta.[ 21 ] To enhance mechanical properties suitable for human application, the thickness of the PCL sheath was increased, which significantly reinforced the whole grafts. This led to a significantly decreased risk of dilation, rupture, as well as enabled long‐term muscular remodeling. Later, the superiority of PGS/PCL grafts over autologous jugular vein grafts was observed regarding the reconstruction of the carotid artery.[ 182 ] The comparison showed that PGS/PCL grafts were remodeled into neoarteries with smooth and even vessel wall comparable to native carotid arteries within 12 months after implantation, whereas autologous grafts remodeled into neoarteries with dilated vessel cavity and thickening of the vessel wall. Additionally, in PGS/PCL neoarteries, muscular components were well organized, and fibers with higher flexibility and elasticity were observed. The synthetic scaffolds also enabled the growth of reinnervated adventitia for adequate nerve responsiveness. These results were confirmed by investigations reported by Lee et al.[ 181 ]
In a related study, anisotropic PGS membranes have been fabricated via the help of sacrificial fibers by Hsu et al. and Li et al.[ 112 , 183 ] Electrospun‐aligned PVA fiber mats were immersed in PGS solution with subsequent removal of PVA fibers after PGS cross‐linking.[ 112 ] The resulting PGS membranes showed aligned grooves on the surface and mechanical tests revealed an anisotropic behavior, whereas porosity was below 20%. Moreover, as PGS seemed to react with PVA, PLA/PEO electrospun fibers were used as spacers.[ 183 ] Successful removal of PEO at elevated temperature during cross‐linking and washing with water and PLA by washing with CF was confirmed by FTIR analysis and cross‐section observations. The groove width (1–3 µm) was significantly larger than the PGS‐PVA membrane (200 nm). In vitro biocompatibility and the ability of the PGS membrane to guide A10 smooth muscle cell orientation were demonstrated. A possible application in vascular TE of the anisotropic PGS membrane was demonstrated by the fabrication of a tubular scaffold, whose compliance was comparable to that of native arteries (2–8% per 100 mmHg, depending on the orientation of the sacrificial fibers).
As the application of TEVGs is often limited due to the slow degradation rate, causing a prolonged inflammatory response or alter remodeling, rapidly degrading PGA braids were dip‐coated with PGS and cured in a vacuum oven (120°, 40 h).[ 184 , 185 ] The graft was almost completely degraded after six months in vivo (rat abdominal aorta model) and remodeled to a new artery with continuous endothelium, contractile SMCs, ECM deposition, and similar mechanical properties to those of the native aorta were obtained. Additionally, PGS coating decreased the inflammation and calcification in the new artery.
In contrast to purely laboratory made TEVGs, the use of decellularized tissues should be considered as well. Despite the high potential of decellularized tissues and organs to maintain unique conformation and composition as native equivalents, the process of decellularization may weaken the tissue extensively. To overcome this limitation, freshly slaughtered sheep aortae were decellularized and then combined with PGS synthesized via microwave irradiation.[ 186 ] The vascular hybrid graft preserved porosity in the medial layer of the vessel. Tensile strength and Young's modulus decreased by a factor of three when PGS was introduced to the decellularized graft, whereas the elongation at breaking point increased threefold. No cytotoxic effects of human abdominal aorta SMCs could be observed in hybrid graft extracts. The study showed that introducing PGS in a native decellularized ECM structure can provide sufficient strength, recellularization capacity, and handling properties.
In summary, it can be stated that TEVGs have been mainly fabricated by electrospinning and porogen leaching or a combination of both, whereby material choices involving both PGS and PCL have shown promising features for vascular TE.
4.3. Nerve TE
In case of a traumatic injury of peripheral nerves, medical therapy has to be undertaken as the intrinsic regeneration capability of the adult mammalian peripheral nervous system is always incomplete and deficient in functional recovery, which would significantly affect the patient's quality of life.[ 187 ] The standard therapy is still autologous nerve grafting. However, this technique is constrained due to a restricted supply of donated nerves, the necessity for a second surgery, donor site morbidity, loss of function, a mismatch between donor nerve and recipient site and clinically functional recovery rates of only 80%.[ 187 , 188 , 189 ] Research within a variety of biological or artificial nerve grafts has been carried out to supplement or even substitute autologous nerve grafts. Nerve grafts have to fulfill several requirements as biocompatibility, biodegradability, permeability, biochemical properties, as well as macro and microarchitecture.[ 187 ] To fabricate structures for nerve regeneration, conventional methods like particulate leaching, extrusion, mesh or film rolling, and molding followed by freeze‐drying are nowadays extended by additive manufacturing techniques and submicron and nanoscale techniques like electrospinning, phase separation, and self‐assembly.[ 187 ]
A simple, porous PGS film proved to promote motor function recovery through nerve generation by enhancing axon growth and neuron sprouting in a rat model, where the complete transection of the spinal cord was employed.[ 189 ] These results were even improved if combined with chondroitinase ABC. For nerve TE, electrically conductive grafts have shown great potential. Wu et al.[ 81 ] fabricated conductive films of PGS‐co‐aniline pentamer cross‐linked with HDI. The electroactivity of the films was proven by UV spectroscopy and cyclic voltammetry, while the conductivity of the films was determined by four‐probe measurement to be between 1.4 × 10−6 and 8.5 × 10−5 S cm‐1, depending on the amount of aniline pentamer; sufficient for conduction of electrical signals in vivo. Mechanical tests revealed higher tensile stress but lower tensile strain for higher aniline pentamer concentration. As the cross‐linking density increased with aniline pentamer concentration, the authors suggested that the strong physical cross‐linking of aniline pentamer segments significantly contributed to the improved mechanical properties. Furthermore, aniline pentamer was also responsible for slower degradation rates in in vitro experiments. SC's myelin gene expression, as well as neurotrophin secretion, were significantly increased and the polymer films were able to induce SC's myelination.
In order to achieve a similar effect, PGS was blended with calcium titanate (CaTiO3), and the effect of calcium ion release on axon outgrowth was investigated.[ 190 ] CaTiO3 has been shown to possess conductive properties and piezoelectric effects.[ 191 , 192 , 193 ] Furthermore, it was demonstrated that calcium is a critical ion for nerve regeneration as it develops single nerve cells and functional connections between them. Additionally, the axonal outgrowth rate is regulated by calcium signaling.[ 194 ] The elastic modulus and tensile strength of the composite film increased dramatically when CaTiO3 nanoparticles were added. The authors related this effect to the enhanced cross‐linking density and the bonds between CaTiO3 particles and PGS, the physical barrier of CaTiO3 particles in the polymer chains acting against slippage of elastomer chains, and the bound rubber effect (structural units of elastomer are absorbed onto the surface of the reinforcing particles). Higher CaTiO3 content led to slightly increased calcium ion release, with an upward trend from 1 to 18 d. However, high calcium ion concentrations might be harmful to nerve and axonal regeneration as well as on metabolic activities.[ 195 ] However, PC12 cell studies revealed good cell adhesion and proliferation and also axon outgrowth and extension on such composite samples.[ 190 ]
A related study showed that an NGC of PCL, PGS, and graphene nanosheets showed promising properties for its application in nerve regeneration.[ 86 ] At an optimal concentration of PCL‐PGS (50:50), the addition of 1 wt% graphene nanosheets resulted in a 31‐fold increase of electrical conductivity to a final value of 5.78 × 10–3 ± 0.86 × 10–3 Ohm m‐1. Besides that, the addition of 1 wt% graphene led to enhanced cell survival and cell attachment. Furthermore, PGS positively influenced cellular behavior probably due to enhancing the wettability. PGS‐maleate with integrated magnesium (Mg) ions has also been proposed for nerve TE.[ 196 ] Other cations like Ca2+, Mg2+ can also mediate diverse cell–material interactions.[ 196 , 197 , 198 ] Whereas the addition of Mg ions did not considerably influence the physical and mechanical properties compared to pure PGS‐maleate, the proliferation and neural‐specific gene expression of SCs were significantly improved. Additionally, the authors claimed that this hybrid of magnesium ion (Mg2+) and poly(glycerol‐sebacate‐maleate) is a soft material with desirable injectability, but it can also be used to design 3D elastic constructs.
To achieve highly porous scaffolds, electrospinning has also been applied in nerve TE. PGS‐PMMA/gelatin fiber mats production with average fiber diameter increasing from 167 ± 33 nm up to 631 ± 156 nm by addition of gelatin was reported.[ 20 ] In contrast, a reduced contact angle of fiber mats by increasing gelatin content was reported, which improved the PC12 cell proliferation values significantly compared to PGS–PMMA scaffolds. Interestingly, this blend induced the differentiation of PC12 cells into neuron‐like cells even in the absence of any nerve growth factor (NGF) or chemical treatment, demonstrating the material suitability for nerve regeneration. PGS–PLLA electrospun fibers developed a core–shell behavior with PLLA material concentrated in the core after cross‐linking at 120 °C in vacuum for 48 h.[ 119 ] An increasing PGS concentration led to a reduction in average fiber diameter to a minimum of 332 ± 103 nm. The ductility of the blend fiber mats with up to 40% PGS was improved in comparison to PLLA scaffolds. Hypothalamus A59 nerve cells compared to the polystyrene control showed lower proliferation. However, the differentiation of A59 cells into neuron‐like cells without the help of growth factors or chemical treatments was detected. Saudi et al.[ 114 ] electrospun aligned PGS–PVA fibers from the slightly modified synthesized PGS. Mechanical tests of the fiber mats showed suitable mechanical properties, as Young's moduli were within the range of peripheral nerve (between several tens of kPa to 0.5 MPa). Due to the higher fiber fusion in a higher concentration of PGS, the higher ultimate tensile strength and Young's modulus were measured. Rat pheochromocytoma cells PC12 tests proved that PVA–PGS 50:50 and 40:60 were the better combinations in terms of cell proliferation. Later, the same group[ 113 ] improved their system by adding lignin, which has been recently indicated for nerve tissue application. Lignin, a natural polymer, showed antioxidant property, oxygen‐free radical scavenger critical for nerve regeneration.[ 113 , 199 , 200 ] With increasing lignin concentration in the blend fibers, Young's moduli increased up to 0.4 MPa and PC12 cell proliferation was enhanced. Furthermore, lignin showed neural cell differentiation potential proven by the mRNA expression level for Gfap, β‐Tub III, and Map2 and immunocytochemistry.
4D biofabrication approach using electrospun bilayer has also been introduced for the fabrication of NGCs. This bilayer was made of an aligned electrospun mesh of PCL/PGS blend and random electrospun mesh of hyaluronic acid. Aligned fibers of PCL/PGS with 70/30 ratio presented a modified stiffness and wettability in comparison with pure PCL and was introduced as an optimum composition for nerve regeneration. The random nanofibrous mesh of hyaluronic acid showed a great adhesion to the PCL/PGS layer due to the modified wettability and different swelling degrees of the hydrogel‐based layer and PCL/PGS. This different swelling ratio resulted in self‐folding of the bilayer mesh, which was reversible by choosing a different water‐based media. The diameter of the tubular construct could be well controlled by varying the thickness of each layer as well as the overall thickness of the bilayer. PC12 cells cultured on the aligned fibers of the PCL/PGS showed enhanced alignment even after one day of culture and after 4 days of differentiation in the presence of 100 ng mL‐1 NGF, PC12 cells showed outgrowth and formation of neurites.[ 29 ]
Laser ablation was used to create a PGS NGCs with microscaled patterns of channels and chambers mimicking the ultrastructure of neural tissue.[ 156 ] The channels were fabricated for neuronal axons guiding, while the chambers were designed for Schwann cell attachment. Additionally, a continuous gradient distribution of NGF 7S was successfully immobilized in a gelatin coating. Differentiated neural stem cells (NSCs) attached and directionally extended as well as neurite length and neuronal gene expression significantly increased with higher NGF 7S concentrations. In coculture with SCs, NSCs were able to differentiate into neuronal cells with strong expression of mature neuronal markers as well as myelin basic protein. By combining physical, biological, and chemical cues, this device could serve as an advanced artificial NGC for peripheral nerve regeneration.
Singh et al.[ 162 ] extensively studied methacrylated PGS (mAcr‐PGS) tubes for nerve regeneration in mice (Figure 11 ). The NGC tubes were fabricated by SLA and postprocessed via laser cutting to obtain a wall thickness of 0.35 mm, a length of 5 mm and a diameter of 1 mm. The mAcr‐PGS tubes were flexible, resistant to kinking, and could withstand suturing, which makes them applicable in larger gap models. Furthermore, in vitro analysis showed that neurites lengthened and aligned within the grooves of the NGC as well as neuronal and glial cell growth was observed. In in vivo experiments, the regeneration of axons, directed axonal growth and no increase of neuropathic pain by mAcr‐PGS conduits were found compared to native nerve grafts.
Figure 11.
Computer‐aided model (Google Sketchup) of the micro‐SLA set‐up by Singh et al. for the production of NGCs. The setup consists of a 405 nm laser [A], a DMD [B], a motorized Z‐table [C] and a container with liquid polymer [D]. a,b) The elastic properties of the mAcr‐PGS NGCs. c) A CAD model (Maya, Autodesk) of an ideal 3D printed NGC. d) A final 3D‐printed and finished product of the NGC, ready for implantation. Scanning electron microscope images show the z‐translation speed of 0.03 mm s‐1 and various laser powers of e) 80 mW, f) 65 mW, g) 30 mW and h) 10 mW. Reproduced under the terms of the CC‐BY 4.0 license.[ 162 ] Copyright 2018, the Authors. Published by Elsevier.
Overall, from very simple porous and pristine films, to electroactive, conductive scaffolds with diverse conducting materials and electrospun fibers in combination with synthetic and natural polymers, PGS has shown promising results as NGCs, promoting neuronal cell growth as well as axon outgrowth and extension.
4.4. Skin TE/Wound Healing
Injury to the skin or tissues in general due to external (e.g., surgical interventions, operations) or endogenous influences (e.g., chronic diseases, acute or thermal trauma) can lead to a significant impairment of physiological activities or even death. In recent years, tissue‐engineered skin constructs and wound dressings have shown great potential in the treatment of various skin‐related diseases. Treatment methods have evolved from previous pure films to more sophisticated, multilayered skin replacement materials.[ 201 ] Wound dressings or artificial skin generally represent a protective barrier that must support the different stages of the healing process and therefore have suitable properties to control fluid loss, infections, contractures, and scarring.[ 202 ]
Thanks to the stable batch‐to‐batch consistency and easy tailorability, PGS is utilized for skin TE and wound healing applications.[ 201 ] However, unlike natural polymers, synthetic polymers show a lack of cellular recognition that limits their use as a stand‐alone material in skin TE. Therefore, in this application also various modifications of PGS in combination with natural polymers, such as collagen or gelatin, or growth factors and antibiotics were reported. Similarly, electrospun mesh with a 3D porous structure was an attractive substrate with highly interconnected porosity resulting in high oxygen permeability and adequate transportation of wound secretions away from injured areas.[ 128 ] For example, a combination of ciprofloxacin (CIP) loaded gel/PGS fiber membranes was reported by Ayati Najafabadi et al.[ 128 ] for skin TE application, where they examined variable ratios of sebacic acid to glycerol and evaluated their biological and physical properties.[ 128 , 202 ] Similar to the study of Shirazaki et al.,[ 202 ] they used 1‐Ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide/N‐hydroxysuccinimide cross‐linking reaction of gelatin to control the release of CIP over time. Highly porous fiber mats with a porosity of over 80% resulted in an adequate degradation rate in an aqueous environment, together with an appropriate prolonged antibacterial release by swelling.[ 202 ] Antimicrobial tests showed that CIP could effectively combat both Gram‐negative (Escherichia coli) and Gram‐positive (Staphylococcus aureus) bacteria over time, but it does not affect cells (fibroblast L929) negatively, which is an essential feature of an ideal wound dressing.[ 128 ]
Another way of embedding antimicrobial substances into a fibrous PGS structure was researched by Heydari et al.[ 124 ] In their study, a novel biodegradable PGS/PHB wound dressing with simvastatin (SIM) and CIP was successfully produced by coaxial electrospinning. Therefore, PHB/CIP was spun as the outer shell and PGS/SIM as core material and the produced core‐shell samples showed controlled release of the embedded antibiotics. CIP loaded into the surrounding PHB part of the fiber showed a burst release during the first 24 h, which facilitated the control of wound infections in the initial state. SIM loaded in the PGS core part, however, showed a slow rate of release, allowing sufficient time for wound healing.[ 124 ]
Besides the commonly used antibiotics CIP and SIM, Abudula et al.[ 132 ] used a chitin–lignin composition as antibacterial substances incorporated in PGS fibers. Chitin and lignin occur mainly as biowaste, produced as by‐products of crustaceans and plant biomass. The authors ascertained in the course of their work that the best mechanical properties of the hybrid fibers were achieved at a volume ratio of the chitin–lignin sol–gel solution and PGS solution of 85/15, whereas higher amounts of PGS mediated the best antimicrobial properties, with activity both against bacteria and fungi.[ 132 ]
Most of the treatment strategies for skin tissue replacements and wound healing are currently based on systemic, poorly controlled release of high doses of antibiotics, which can lead to side effects and drug resistance in the long run. Thus, a controlled temporal release of the drug would be desirable for future wound healing treatment. Tamayol et al.[ 19 ] developed thermally controllable, antibiotic‐releasing fiber mats using a PGS‐PCL blend embedded with drug‐loaded thermo‐responsive nanoparticles of PEGylated‐CH.[ 19 ] A bioresorbable metallic heating element made of Zn, Mg, and silver (Ag), respectively, was patterned directly onto the nanofiber substrate to apply thermal stimulation to release antibiotics whenever required (Figure 12 ). In the case of the bioresorbable metals Zn and Mg, the degradation rate could be controlled, with drug release being proportional to the voltage applied to the heating element. In vitro studies confirmed the biocompatibility as well as biodegradability of the fiber complex, which was able to effectively release antibiotics against various strains of bacteria (Staphylococcus aureus and Escherichia coli) on demand. Therefore, this smart platform could be further investigated in the future as smart surgical networks, TE scaffolds, and wound dressings that can release drugs on demand in response to external stimuli.[ 19 ]
Figure 12.
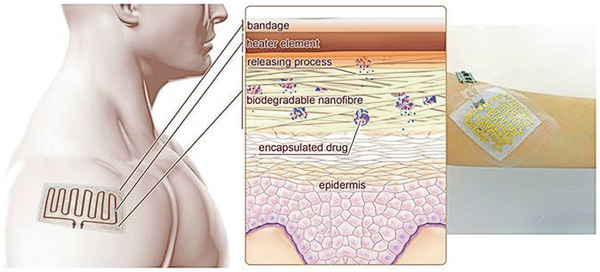
The functional principle of a drug delivery system with integrated heating element and electronics developed by Tamayol et al.[ 19 ] Heat‐sensitive drug nanocarriers were incorporated into nanofibers of technical fabric and were able to deliver their drugs at temperature increase initiated by the integrated, flexible heating element. Reproduced under the terms of the CC‐BY license.[ 19 ] Copyright 2017, the Authors. Published by Springer Nature.
Further applications of a PGS/PCL nanofibrous mesh as a suture/adhesive material were recently studied [ 203 ] The study involved an electrospun PGS/PCL mesh in combination with a photo‐crosslinkable glue composed of 5% (w/v) GelMA prepolymer solution to fix the material in a volume muscle loss model. A high adherence to the tissue and under large dynamic movement post‐surgery stretching was reported and high mechanical stresses did not result in fracture at the suture point.
Besides electrospinning, Rosenbalm et al.[ 82 ] assessed a PGS/nHA combination for wound closure via tissue transport. They mixed different amounts of nHA (3 wt% and 5 wt%) with PGS and cast it into thin rectangular films. They found out that the addition of 5 wt% nHA to PGS showed accelerated degradation combined with reduced flexibility and tensile strength due to an interruption in the cross‐linking of PGS. When the PGS was doped with 3 wt% nHA, no reduction in flexibility and elongation was observed, but a simultaneous increase in tensile strength was measured within the desired force range for the closure of soft tissue defects.[ 82 ] Based on these findings, adding 3 wt% nHA to the shape memory polymer PGS could serve as a right candidate for the closure of nonhealing large soft tissue wounds.
Zhao et al.,[ 143 ] on the other hand, developed an injectable physical double network removable hydrogel adhesive for treating multidrug‐resistant bacterial infection as well as full‐thickness skin incisions. The hydrogel system was composed of catechol–Fe3+ cross‐linked poly(glycerol sebacate)‐co‐poly(ethylene glycol)‐g‐catechol prepolymer (PEGSD) and GTU. PEGSD‐GTU was prepared by merely mixing solutions of PEGSD, iron(III) chloride, and GTU in physiological conditions. The double network hydrogel developed in this way showed rapid shape adaptation as well as self‐healing properties. Such hydrogel could be dissolved again by near‐infrared irradiation or change of the acidic milieu (pH) via an accompanying sol–gel transition or dissolution, respectively. In vivo experiments proved that PEGSD‐GTU showed adequate hemostasis of skin lesions and a high degree of the killing of methicillin‐resistant Staphylococcus aureus (MRSA) compared to commercially available biomedical adhesives or surgical sutures. Besides, the material showed a regulated inflammatory response, accelerated collagen deposition and vascularization. Together with its dissolvable and antioxidant properties, PEGSD‐GTU is considered an excellent multifunctional dressing for the treatment of in vivo MRSA infections, wound closure as well as wound healing,[ 143 ] which shows yet another attractive application of PGS in the wound healing sector.
Not only the incorporation of commercially available antibiotics like CIP and SIM into electrospun PGS fiber mats has shown potential for skin/wound TE, but also the addition of nHA to PGS demonstrated the wound closure capability of nonhealing large soft tissue wounds. Furthermore, injectable hydrogel systems with PEGSD proved the usefulness of PGS in skin/wound TE applications due to rapid shape adaption, self‐healing properties, and high rate of MRSA killing.
4.5. Bone TE (BTE)
In recent decades, the number of bone injuries has increased steeply upward because of the aging population and increasing incidence, bone necrosis, bone cancer, trauma, or other diseases. BTE evolved as a potential alternative compared to conventional bone grafts.[ 204 ] Many different materials from ceramics to polymers and composites have been investigated, but the most promising group are osteoinductive biomaterials, which can induce ectopic bone formation by instructing its surrounding in vivo environment to form bone.[ 204 , 205 , 206 , 207 ] This ability has been demonstrated for a family of biomaterials of natural and synthetic ceramics like hydroxyapatite, various calcium phosphate compositions, and BGs.[ 204 ] In general, scaffolds for BTE should offer excellent biocompatibility, biodegradability, osteoinductivity, and appropriate mechanical integrity.[ 208 ] As natural bone is a composite of inorganic hydroxyapatite crystals and organic collagen fibers, polymer‐ceramic composites represent an ideal approach for BTE. Besides PHB, PCL, PLA, PLGA, gelatin, CS, and collagen,[ 209 ] PGS came into focus for BTE. Even though PGS is a relatively soft and elastic polyester with mechanical properties usually in the range of 0.25 to 1.45 MPa for the elastic modulus and 0.3 to 1.5 MPa for the tensile strength,[ 4 , 8 , 47 ] the incorporation of ceramic particles or the infusion of PGS into a ceramic scaffold is convenient approaches to enhance mechanical properties of the scaffold suitable for bone regeneration.
For example, PGS‐based scaffolds have been fabricated by solvent casting or salt leaching with the addition of ceramic particles. Zhao et al.[ 47 ] added silica glass particles to PGS, resulting in a hybrid porous elastomer with tunable elastic properties. By changing the silica phase content, the tensile strength ranged between 1 to 5 MPa and 2 to 32 MPa, respectively. Furthermore, the scaffold's hydrophilicity was enhanced by the inclusion of silica particles. The hybrid PGS–silica scaffolds showed a significantly elevated proliferation of MC3T3 compared to PGS scaffolds. Similar results were found by Kerativitayanan et al.,[ 24 ] who incorporated nanosilicates into a PGS matrix. The mechanical stiffness was increased without affecting the elastomeric properties. They also observed that the addition of nanosilicates significantly enhanced cell adhesion, supported preosteoblast cell proliferation, upregulated alkaline phosphates, and mineralized matrix production. It was found that by the addition of nanosilicates, the degree of cross‐linking, hydrophilicity, and thermal and structural stability could be tailored.[ 24 ] The same group[ 83 ] further investigated the PGS–nanosilicate composite with pores induced by salt leaching. The addition of pores did not change the effect of nanosilicates on the PGS matrix. Similarly, nanosilicates increased mechanical stiffness without comprising the elastomeric properties. The modulus was increased 4.5‐fold by the addition of 10% nanosilicates to PGS. Furthermore, the recovery after eight cycles of compression was determined to be 98.4 ± 1.4% and 97.6 ± 3.4% for PGS and PGS with 10% nanosilicates, respectively. The authors suggested that the mechanical properties of PGS/nanosilicate scaffolds were tunable with the incorporation of nanosilicates. Moreover, the elasticity of the scaffold could be favorable as some flexibility is necessary during the early stage of fracture repair involving cartilage formation before bone calcification. The incorporation of nanosilicates could also modulate the degradation rate of nanocomposite PGS scaffolds, and the cell adhesion, spreading, and proliferation was supported by the addition of nanosilicates as well as the osteogenic differentiation of preosteoblasts was promoted. Furthermore, the alkaline phosphatase activity and production of matrix mineralization were increased. In vivo experiments in mice, where the nanocomposite scaffolds were implanted between femur bone and quadriceps, indicated biodegradability and biocompatibility of the scaffolds 21 d after implantation. No significant signs of inflammation could be observed post‐implantation.
Instead of silicates, β‐TCP particles have also been used as a filler material for PGS matrices. Tevlek et al.[ 49 ] fabricated a bilayered guided bone regeneration construct by adding β‐TCP particles inside PGS prepolymer and during the cross‐linking process, the particles concentrated in the lower part of the PGS matrix. With this approach, they fabricated a gradient construct for bone‐soft tissue interface applications, which has a porous ceramic phase for bone regeneration, a transition zone and an impermeable elastomer side preventing soft tissue ingrowth. It was found out that the β‐TCP particles were homogeneously distributed within the PGS matrix, obtaining an overall open porosity of 35%. The addition of β‐TCP particles led to a fivefold increased compression strength (up to 14 ± 2.3 MPa) without a visible increase of elongation. In vitro cell studies showed a biocompatible behavior of the neat PGS side and an excellent bone‐side cell attachment. Biological properties were enhanced, and osteoblast morphologies were more pronounced in comparison to the control when a bone morphogenetic protein (BMP)‐2 or BMP‐2 and transforming growth factor‐β1 were applied on the constructs. The same group[ 210 ] later showed that this elastomeric composite of PGS/β‐TCP particles could also be fabricated by extrusion. The process did not negatively influence the mechanical flexibility as well as cytocompatibility of the final scaffolds, which showed shape‐memory features and could be shaped into the desired size and various forms via temperature stimuli. β‐TCP particles also demonstrated a reinforcing effect on the mechanical properties of PEGylated PGS/ β‐TCP particles composite scaffolds.[ 48 ] The most promising formulation proved to be a 50% weight ratio of β‐TCP to PEGS20 prepolymer as the maximum tensile strength was 9.58 ± 0.02 MPa, being 1.5‐fold higher than that of neat PEGS membranes. Additionally, this composition showed the highest bone volume to tissue volume ratio of 17.26 ± 1.49% after 4 weeks and 23.24± 2.85% after 8 weeks postsurgery.
To alter osteoblast cell adhesion, proliferation, and differentiation, maleic anhydride has been grafted onto PGS and mixed with nHA in different weight ratios in a salt leaching fabrication process.[ 211 ] Interconnected porous scaffolds with an average pore size of 150 to 300 µm and porosities between 84% and 90% were achieved. The compressive strength of the scaffolds improved 10‐fold when the hydroxyapatite content was increased. The addition of nHA to PGS‐g‐M resulted in elevated human adipose‐derived stem cell proliferation and enhanced expression of osteogenic related genes like Runt‐related transcription factor 2, osteocalcin, and alpha‐1 type I collagen, while causing little inflammatory response.
PGS has also been used to infiltrate ceramic‐based scaffolds. Pure ceramic scaffolds are commonly fabricated by a porogen‐based process, foam replica method, or 3D printing technique. They fulfill a variety of essential requirements for BTE scaffolds like inherent biocompatibility, osteoconductivity, and degradation rate, but they usually lack sufficient mechanical strength. Polymer coatings or the infiltration of polymers, being either natural polymers like gelatin[ 212 ] or biotechnology‐derived polymers like poly(3‐hydroxybutyrate‐co‐3‐hydroxyvalerate),[ 213 ] onto ceramic scaffolds have been investigated before and the approach has been shown to be sufficient to enhance the mechanical strength of the scaffolds. As a reinforcement material in bone repair, PGS has several advantages over other conventional bone repair biomaterials like its long‐lasting and tunable elastomeric properties, as well as its adjustable degradation kinetics,[ 2 ] and its acidic behavior, which enables a reaction with alkaline bioceramics and BGs via metallic carboxylation.[ 214 , 215 ] Lin et al.[ 216 ] fabricated multiparameter‐adjustable interconnected macro‐ and mesoporous bioactive glass (MBG) scaffolds by PU foam templating and impregnated those with dissolved PGS of different mass ratios (between 0.2 up to 3.2 g mL‐1) in ethanol. The PGS in the final scaffolds remained uncross‐linked. PGS impregnation led to mechanical strength and toughness enhancement in comparison to pure MBG scaffolds and resulted in a broad range of adjustable mechanical strength values covering that of human trabecular bone. Whereas noncoated MBG scaffolds were crushed into particles, the PGS coating slowed down the crack propagation in MBG struts, and the PGS reinforced scaffolds maintained their shape and integrity. Furthermore, the PGS coating could tailor the degradation rate of the composite scaffold. The dissolution of MBG could lead to elevated pH values, which were counteracted by the dissolution of PGS, keeping the pH value in a suitable range. The PGS coating was also found to promote cell attachment and proliferation in a dose‐dependent manner, without negatively affecting the osteogenic induction capacity of the MBG substrate. This system was further improved by loading recombinant human bone morphogenetic protein‐2 (rhBMP‐2), a potent osteoinductive growth factor, into the MBG substrate and infiltrating it with uncross‐linked PEGylated PGS.[ 217 ] While PGS coating supported the proliferation of rat bone marrow stem cells, PGS coating exhibited an improvement in osteogenic differentiation. Finally, in vivo experiments proved that rhBMP‐2‐loaded MBG/PEGS scaffolds showed a rapid bone‐forming capacity. PGS has also been used to infiltrate β‐TCP foam replica scaffolds with subsequent cross‐linking of PGS, which enhanced the mechanical strength of the composite scaffold (3.7 fold increased elongation at break and 200 fold increased compressive strength) in comparison to pristine β‐TCP scaffolds.[ 50 ] Additionally, the composite scaffolds exhibited a complete and rapid recovery of their original shape after an abrupt drop of compressive load. It was also shown that PGS could direct the scaffold biomineralization, from a Ca/P particulate shape into a nanofiber‐interweaved structure. As shown by Lin et al.,[ 216 ] the scaffolds demonstrated pH autoregulation. HUVEC and rat bone marrow stromal cells showed efficient cell penetration into the scaffolds and suitable cell proliferation.[ 50 ] Also, calcium phosphate scaffolds infiltrated with PEGylated PGS have shown optimal mechanical properties and synchronously bone marrow‐derived stem cell attachment and proliferation in a dose‐dependent means.[ 218 ] Moreover, PEGS/calcium phosphate cement (CPC) scaffolds were shown to induce osteogenesis compared to pure CPC scaffolds in in vivo experiments.
A ceramic‐free concept for BTE was developed by Shi et al.[ 80 ] and Zaky et al.[ 219 ] In an earlier study,[ 80 ] PGS was blended with PLA, as PLA itself has high mechanical properties but also inherent brittleness. The superelasticity of PGS was used to counteract the brittleness of PLA. It was found[ 80 ] that PGS could effectively modify PLA‐based salt leached scaffolds as it improved the hydrophilicity, toughness, ductility and bioactivity of the scaffolds. Especially with oxygen plasma pre‐treatment, the scaffolds supported cell adhesion and proliferation and showed the highest osteogenic markers ALP Runt‐related transcription factor 2 and osteocalcin mRNA expression. In another study[ 219 ] neat PGS porous scaffolds were compared to PLA/PGA porous scaffolds. In vivo experiments showed a full gap bridging with newly formed bone by PGS elastomer within eight weeks, whereas negative controls developed only minimal bone formation. The study implied that PGS had osteoconductive properties as it contributed to bone regeneration by recruiting host progenitor/stem cell populations. Furthermore, mechanical signals could be transmitted via the load‐transducing substrate to the populated cells, which promoted differentiation and matrix maturation. It was concluded that the material properties of PGS are closer to osteoid tissue rather than mineralized bone.[ 80 , 219 ]
Last, also nanostructured scaffolds by electrospinning have been developed for applications in BTE, as it is well known that nanotopography influences osteoinductivity and osseointegration.[ 204 , 220 , 221 , 222 , 223 ] The group of Wang et al.[ 100 ] electrospun PCL/PGS blend nanofibers and successfully incorporated nanoscaled hydroxyapatite particles into the fibers. The fiber mat showed excellent biocompatibility and osteoblast adhesion properties with respect to bone marrow‐derived MSCs. However, these scaffolds are intended to be used at the interface between bone and cartilage. Also for guided bone regeneration but in the dental area, a PCL/PGS with 10% β‐TCP electrospun fiber mat was chosen as the best membrane in respect to physical, mechanical, and biological behavior.[ 51 ]
Generally, in BTE, PGS–ceramic composites have been successfully used, whereby either the incorporation of ceramic particles into the PGS scaffold or the infusion of PGS into a ceramic scaffold can enhance the mechanical strength and toughness to reach values suitable for bone regeneration. Additionally, ceramic‐free concepts have been developed which have also shown appropriate properties for bone substituting materials.
4.6. Drug delivery
Controlled drug delivery systems able to administer medications at pre‐destined rates and periods are becoming increasingly important in medicine. The goal of a controlled release is to transport an appropriate amount of drugs to affected areas of the patient's body without causing adverse side effects on other tissues. New developments in intelligent drug delivery systems respond directly to pathophysiological diseases without harming surrounding healthy cells and tissues.[ 224 ] One of the most common applications for drug delivery systems is in the already mentioned field of wound healing. However, they are also increasingly being used to combat cancer as well as treatment of other diseases like periodontal or ocular infections, including diabetic retinopathy.[ 225 ]
In order to effectively incorporate hydrophobic pharmaceuticals into drug delivery systems in general, the carrier material should be stable in an aqueous environment. By increasing the solubility of a drug in an aqueous medium, its toxicity can be reduced and its pharmacokinetic release can be controlled by allowing hydrophobic drug molecules to interact noncovalently with the hydrophobic domains of the release material.[ 226 ]
Louage et al.[ 226 ] have shown that well‐defined nanoparticles can be formed using PGS by a straightforward method of solvent displacement. In this technology, a PGS stock solution is mixed under stirring in ethanol dropwise with water. The as‐produced PGS nanoparticles, with sizes between 112 and 209 nm, were stable in aqueous media within a period of 96 h. The measured critical aggregation concentration of PGS indicated that the nanoparticles were unlikely to disassemble upon dilution rapidly. Thus, paclitaxel (PTX) and flubendazole (FLU), both drugs known for their antimitotic effect, were encapsulated in the PGS particles and their effect on cancer cells was evaluated in vitro. It was shown that the physically encapsulated amount of PTX and FLU was sufficient to significantly reduce the survival rate of SKOV‐3 cancer cells.[ 226 ]
Another PGS‐based system for administering anticancer drugs to a defined target was developed by Naghizadeh et al.[ 54 ] They embedded fluorouracil (FU) (5‐FU) in a PGS‐PEG/CS‐co‐PEG matrix and coated it subsequently with iron oxide (PGS‐PEG/CS‐PEG@Fe3O4). CS‐PEG was produced under mild conditions by binding PEG to the hydroxyl group of CS. To realize a release of 5‐FU with two different rates, CS‐PEG was mixed with PGS‐PEG. PGS‐PEG/CS‐PEG@Fe3O4 showed a controlled release of 5‐FU in simulated physiological fluids, suggesting a model of the retention mechanism. The retention mechanism assumed that hydrophobic‐hydrophobic interactions interact more strongly with the drug than pure CS or PGS without a hydrophobic group. Cytotoxicity studies on HT29 cell lines showed that 5‐FU‐loaded PGS‐PEG/CS‐PEG@Fe3O4 nanoparticles successfully accumulated around the cells and induced enhanced cytotoxicity on cancer cells compared to pure 5‐FU, confirming that the released drug remained active.[ 54 ]
Yang et al.[ 227 ] showed in their work the encapsulation of berberine and chlorhexidine intending to treat periodontal diseases. They initiated a PGS swelling technique, using ethanol as a swelling agent due to its low toxicity, dissolving berberine chloride and chlorhexidine in it to produce a saturated solution. Subsequently, PGS cubes were immersed in the saturated drug/ethanol solution to load the drug at 37 °C for 48 h. The drug‐loaded PGS cubes were then washed with distilled water to remove surface‐adhered drugs.[ 227 ] Loading PGS with drugs like berberine and chlorhexidine showed a change neither in Young's modulus nor in the maximum strain compared to neat PGS. However, the surface wettability was slightly reduced, and the cell compatibility improved. Loaded PGS showed a constant drug release in vitro with an antibacterial effect. A general antimicrobial test proved that chlorhexidine‐loaded PGS groups were functional against typical Gram‐positive, Gram‐negative, and antibiotic‐resistant bacteria. Berberine‐loaded PGS groups, on the other hand, showed good antibacterial activity against periodontal pathogens. Thus, the obtained data indicated berberine and chlorhexidine loaded PGS to be an attractive candidate for the use as an implant material to treat periodontal diseases.[ 227 ]
Silva et al.[ 103 ] used a coaxial electrospinning technique to produce PGS/PCL aligned nanofibers (core:PGS; shell:PCL) for cartilage TE applications. Kartogenin (KGN), a small bioactive molecule shown to promote chondrogenesis of MSCs, was incorporated into the PGS core solution to create coaxial PGS‐KGN/PCL nanofibers. KGN release kinetics and biological performance of the scaffolds were evaluated in comparison to KGN‐loaded monoaxial fibers and corresponding unloaded controls. It was found that coaxial PGS‐KGN/PCL nanofibers showed more controlled and sustained KGN release than monoaxial PCL‐KGN nanofibers in a period over 21 d. Elevated sGAG levels and expression of genes for chondrogenic markers indicated that KGN‐loaded scaffolds could significantly enhance cell proliferation and chondrogenic differentiation of MSCs from human bone marrow in incomplete chondrogenic medium (without TGF‐β3) underlining the potential of coaxial PGS‐KGN/PCL‐oriented nanofibers as bioactive scaffolds for cartilage TE applications.[ 103 ]
Another technology being considered for implantable scaffolds is based on hydrogels. Ye et al.[ 144 ] invented a drug delivery vehicle made of a “self‐healing” PGS‐PEGMEMA/α cyclodextrin (αCD) hydrogel.[ 144 ] Their thixotropic hydrogel can be sheared into a liquid during injection and is capable of quickly “healing” back into the gel‐state after injection. By variation of the αCD concentration, the storage and loss moduli of the hydrogels can be controlled between a few kPa up to a few 100 kPa, a range that corresponds to the moduli of cells and human soft tissues. In in vitro experiments, it was found that the hydrogel had a linear mass erosion profile as well as a biphasic drug release profile of incubated anticancer drug doxorubicin.[ 144 , 228 ] The first phase was driven primarily by diffusion of the medication as the second phase was induced by hydrogel erosion, making it a suitable material for a controlled drug release.[ 144 ]
Besides incorporating medications directly into the polymer, Lee et al.[ 123 ] developed superhydrophobic, reversibly elastic, moldable, and electrospun (SupREME) fibers using the core–shell electrospinning technique. They used hydrophobic polysulfone as a shell material and elastic PGS as a core. These two‐component fibers act as excipients to temporarily increase the local residence time of drugs by sealing them off from the liquid environment (Figure 13 ). The fibers support sustained drug delivery by physically delaying a burst release into the fluidic environment or preventing an immediate spreading to off‐target areas. Another advantage of the fibers is that their reversible elastic properties would also make them suitable for minimally invasive administration using a laparoscope or endoscope.[ 123 ]
Figure 13.
SupREME fibers as an adjuvant for local drug delivery. SupREME fibers facilitate local drug delivery through minimally invasive approaches by shielding the drug from the aqueous environment. This allows the drug to diffuse slowly into the environment and is not immediately washed away by the bloodstream. Reproduced with permission.[ 123 ] Copyright 2017, Wiley‐VCH GmbH.
Concluding this section, by incorporation of drugs into PGS nanoparticles, hydrogels or electrospun fiber mats, controlled drug release systems with constant or biphasic release profiles have been developed for a variety of applications in TE.
4.7. Bioelectronics and Smart Textiles
Application of sensitive and elastic devices with high elasticity as wearable electronics, smart textile and flexible sensors has grown in the last years. The wearable technologies require sensors, which match the mechanical properties of host skin while they are biocompatible. Therefore the stretchability and compressibility of the introduced elastomers are essential. Commercial elastomers for skin mostly show a high stiffness, which brings discomfort to the skin. Therefore, development of bioelastomers is significant and PGS has been recently introduced to this topic as it can be processed as seamlessly integrated wearable sensors to monitor the vital biosignals such as blood oxygen, glucose, pressure, and heart rate. Sencadas et al.[ 229 ] showed recently that degradable elastomeric porous PGS could be used as a sensitive piezoresistive sensor with enhanced electromechanical performance. PGS was blended with multiwalled carbon nanotubes (MWCNTs) and NaCl to generate a porous structure, which could handle an extensive range of pressures (<8 kPa). The sensitivity of the pressure sensor after eight weeks of incubation in simulated body fluid has increased from 0.12 ± 0.03 kPa –1 up to 8 ± 0.20 kPa –1. Moreover, low pressure like 100 Pa could be detected by this flexible sensor which proved the high sensitivity of the sensor within a short response time <20 ms. The PGS sensors with foam structure showed sensing characteristics comparable to that of a human finger. PGS composite containing MWCNTs showed a higher conductivity and potential as a wearable sensor.
To generate the electrical conductivity, Memic et al.[ 97 ] and Kalakonda et al.[ 45 ] reported the production of a micro‐ and nanofibrous electrospun mesh of PGS–PCL with high stiffness which was coated by silver using a custom radio frequency sputtering method. These patches were also very flexible and electrically conductive, and the thickness of the Ag coating could be controlled to tailor the conductivity. Moreover, the stretchable patch showed a comfortable contact with skin and possessed excellent pattern‐substrate fidelity on nonflat surfaces. The authors showed the PGS–PCL substrate was similar in structure to paper composed of a mesh of thin fibers, which can be beneficial for applications like paper electronics. The advantage of PGS–PCL mesh to paper was higher elasticity as well as more extended stability in aqueous environments. Therefore, fabrication of elastomeric electronics compatible for nonflat surfaces was also reported after inkjet printing the metallic microstrips on degradable elastic PCL–PGS.[ 230 ] Different conductive patterns using silver ink were printed on the surfaces of the fabricated PGS–PCL sheets to engineer flexible electronics which was evaluated as a biocompatible heater, temperature sensor, and as a wireless detector to monitor strain signals. For example, to show the application of this sensor as a temperature sensor a patterned silver ink in a shape of spiral deposited on the surface of PGS–PCL could detect the changes in the surrounding temperature with electrical resistance. After placing the patterned sheets on the surface of a heater, the measured electrical resistance was calibrated with the surface temperature. A linear relationship between the electrical resistance and range of temperature between 27 °C and 40 °C relevant to the biological application was detected. Furthermore, this paper‐like mesh was tested as a wireless strain sensor, which can detect the motion of the muscle tissue in electromyography.
5. Concluding Remarks
As elaborated in this review, PGS shows great potential for current and future biomedical applications. In the last five years, mechanical as well as chemical properties of PGS have been further developed by new synthesis reactions and co‐polymerization strategies so that a large number of PGS‐based polymeric systems have become available. Furthermore, the processability of PGS has been improved by new processing techniques. Such advances in PGS‐related materials and technologies represent a very promising basis to maintain the future potential of PGS in the biomedical field.
Since PGS is an extremely versatile, biocompatible, and bioresorbable polymer with adjustable mechanical properties and degradability, it is becoming a favorable biomaterial for a variety of technologies suitable for TE applications. Initially developed for the use in soft TE, its study is extended for utilization in bone tissue regeneration, dental applications or for the administration of drugs. Although PGS itself already offers desirable properties for many purposes in TE, chemical modification of PGS is a common approach being exploited to increase PGS's fields of application significantly. Photo‐cross‐linkable forms of the polyester enable the use of advanced technologies such as rapid prototyping, solid‐free form fabrication, micro‐ablation, or extrusion‐based printing to produce sophisticated porous and structured 3D PGS scaffolds. Blending neat, untreated PGS with other synthetic or natural polymers is also used to enable scaffolds with modified physical properties and biocompatibility using techniques such as electrospinning or hydrogel injection. The addition of conductive polymers/components has also been explored to obtain electrically conductive composites, mainly used for application in cardiac or nerve TE. The combination of PGS with inorganic particles, which is an exciting approach for BTE applications, has been investigated to improve the mechanical properties of scaffolds and their bioactivity. Increased research in the last five years and the application of state‐of‐the‐art technologies have shown an ongoing and continuous interest in this adjustable polymer, which will not diminish in the future as new possibilities for exploiting PGS properties in the biomedical field are found. We hope that the present review paper will serve the community as a comprehensive update about PGS and will promote further interest in this versatile polyester for biomedical applications.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
L.V. and F.R. contributed equally to this work. This work was supported by DFG (German Research Foundation) (Grant BO 1191/14‐1). L.V. and R.F. designed and wrote the first draft and have participated equally in the writing of the manuscript. S.S. helped in proof‐reading and wrote Section 4.7. A.R.B. conceived the idea of the paper, contributed to the literature search, designed and proof‐read the manuscript.
Open access funding enabled and organized by Projekt DEAL.
Biographies
Lena Vogt is currently a Ph.D. candidate in the lab of Prof. Aldo R. Boccaccini at the Institute of Biomaterials, University of Erlangen‐Nuremberg (FAU), Germany. She holds Bachelor's and Master's degrees in medical engineering from FAU. In her Ph.D. project, she focuses on the development and characterization of fibrous scaffolds obtained by electrospinning for cardiac tissue engineering applications.

Florian Ruther is a Ph.D. student at the Institute of Biomaterials, University of Erlangen‐Nuremberg, Germany. He received his Bachelor's and Master's degrees in medical engineering and is currently doing his Ph.D. on biomedical materials. His research focuses on the development and characterization of biopolymers and composites, biofabrication, electrospinning as well as tissue engineering of myocardial tissues.

Sahar Salehi is a leader of the research group “Biomaterials for Tissue Regeneration” at the Department of Biomaterials, University of Bayreuth (UBT), Germany. She received her Bachelor's and Master's degree in materials engineering and Ph.D. in biomaterials. After her postdoc at Tohoku University, Japan, she established her research group at UBT. Her research focuses on biomaterials development, micro/nanoengineering technologies, biofabrication, tissue engineering of musculoskeletal tissues, and tissue‐like constructs with gradient structures.

Aldo R. Boccaccini is professor of biomaterials and head of the Institute of Biomaterials at University of Erlangen‐Nuremberg, Germany. He is also a visiting professor at Imperial College London (UK). He received his doctoral degree in materials science from RWTH Aachen University (Germany) in 1994 and has held postdoctoral appointments at University of Birmingham (UK), University of California San Diego (USA), and Ilmenau Technical University (Germany). His research activities are in the broad area of glasses, ceramics, and composites for biomedical applications. He has pioneered the use of bioactive composite scaffolds incorporating bioactive glasses for tissue engineering.

Vogt L., Ruther F., Salehi S., Boccaccini A. R., Poly(Glycerol Sebacate) in Biomedical Applications—A Review of the Recent Literature. Adv. Healthcare Mater. 2021, 10, 2002026. 10.1002/adhm.202002026
Contributor Information
Sahar Salehi, Email: sahar.salehi@bm.uni-bayreuth.de.
Aldo R. Boccaccini, Email: aldo.boccaccini@fau.de.
References
- 1. Gadomska‐Gajadhur A., Wrzecionek M., Matyszczak G., Piętowski P., Więcław M., Ruśkowski P., Org. Process Res. Dev. 2018, 22, 1793. [Google Scholar]
- 2. Wang Y., Ameer G. A., Sheppard B. J., Langer R., Nat. Biotechnol. 2002, 20, 602. [DOI] [PubMed] [Google Scholar]
- 3. Martín‐Cabezuelo R., Rodríguez‐Hernández J. C., Vilariño‐Feltrer G., Vallés‐Lluch A., Polymers 2021, 13, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rai R., Tallawi M., Grigore A., Boccaccini A. R., Prog. Polym. Sci. 2012, 37, 1051. [Google Scholar]
- 5. Matyszczak G., Wrzecionek M., Gadomska‐Gajadhur A., Ruśkowski P., Org. Process Res. Dev. 2020, 24, 1104. [Google Scholar]
- 6. Cai W., Liu L., Mater. Lett. 2008, 62, 2171. [Google Scholar]
- 7. Kemppainen J. M., Hollister S. J., J. Biomed. Mater. Res., Part A 2010, 94A, 9. [DOI] [PubMed] [Google Scholar]
- 8. Chen Q.‐Z., Bismarck A., Hansen U., Junaid S., Tran M. Q., Harding S. E., Ali N. N., Boccaccini A. R., Biomaterials 2008, 29, 47. [DOI] [PubMed] [Google Scholar]
- 9. Liu Q., Tian M., Shi R., Zhang L., Chen D., Tian W., J. Appl. Polym. Sci. 2007, 104, 1131. [Google Scholar]
- 10. Salehi S., Fathi M., Javanmard S., Barneh F., Moshayedi M., Adv. Biomed. Res. 2015, 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y., Kim Y. M., Langer R., J. Biomed. Mater. Res. 2003, 66A, 192. [DOI] [PubMed] [Google Scholar]
- 12. Jaafar I. H., Ammar M. M., Jedlicka S. S., Pearson R. A., Coulter J. P., J. Mater. Sci. 2010, 45, 2525. [Google Scholar]
- 13. Smith R., Oliver C., Williams D. F., J. Biomed. Mater. Res. 1987, 21, 991. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y., Lu S., Gabriele P., Harris J. J., Mater. Matters 2016, 11, 3. [Google Scholar]
- 15. Barrett D., Yousaf M., Molecules 2009, 14, 4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salehi S., Czugala M., Stafiej P., Fathi M., Bahners T., Gutmann J. S., Singer B. B., Fuchsluger T. A., Acta Biomater. 2017, 50, 370. [DOI] [PubMed] [Google Scholar]
- 17. Salehi S., Fathi M., Javanmard S. H., Bahners T., Gutmann J. S., Ergün S., Steuhl K. P., Fuchsluger T. A., Macromol. Mater. Eng. 2014, 299, 455. [Google Scholar]
- 18. Morgan K. Y., Sklaviadis D., Tochka Z. L., Fischer K. M., Hearon K., Morgan T. D., Langer R., Freed L. E., Adv. Funct. Mater. 2016, 26, 5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tamayol A., Hassani Najafabadi A., Mostafalu P., Yetisen A. K., Commotto M., Aldhahri M., Abdel‐wahab M. S., Najafabadi Z. I., Latifi S., Akbari M., Annabi N., Yun S. H., Memic A., Dokmeci M. R., Khademhosseini A., Sci. Rep. 2017, 7, 9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu J., Kai D., Ye H., Tian L., Ding X., Ramakrishna S., Loh X. J., Mater. Sci. Eng. C 2017, 70, 1089. [DOI] [PubMed] [Google Scholar]
- 21. Yang X., Wei J., Lei D., Liu Y., Wu W., Biomaterials 2016, 88, 34. [DOI] [PubMed] [Google Scholar]
- 22. Frydrych M., Román S., MacNeil S., Chen B., Acta Biomater. 2015, 18, 40. [DOI] [PubMed] [Google Scholar]
- 23. Lin D., Cai B., Wang L., Cai L., Wang Z., Xie J., Lv Q., Yuan Y., Liu C., Shen S. G., Biomaterials 2020, 253, 120095. [DOI] [PubMed] [Google Scholar]
- 24. Kerativitayanan P., Gaharwar A. K., Acta Biomater. 2015, 26, 34. [DOI] [PubMed] [Google Scholar]
- 25. Tallá Ferrer C., Vilariño‐Feltrer G., Rizk M., Sydow H. G., Vallés‐Lluch A., Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 761. [Google Scholar]
- 26. Loh X. J., Abdul Karim A., Owh C., J. Mater. Chem. B 2015, 3, 7641. [DOI] [PubMed] [Google Scholar]
- 27. Nijst C. L. E., Bruggeman J. P., Karp J. M., Ferreira L., Zumbuehl A., Bettinger C. J., Langer R., Biomacromolecules 2007, 8, 3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perin G. B., Felisberti M. I., Macromolecules 2020, 53, 7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Apsite I., Constante G., Dulle M., Vogt L., Caspari A., Boccaccini A. R., Synytska A., Salehi S., Ionov L., Biofabrication 2020, 12, 035027. [DOI] [PubMed] [Google Scholar]
- 30. Salehi S., Bahners T., Gutmann J. S., Gao S.‐L., Mäder E., Fuchsluger T. A., RSC Adv. 2014, 4, 16951. [Google Scholar]
- 31. Lei D., Yang Y., Liu Z., Chen S., Song B., Shen A., Yang B., Li S., Yuan Z., Qi Q., Sun L., Guo Y., Zuo H., Huang S., Yang Q., Mo X., He C., Zhu B., Jeffries E. M., Qing F.‐L., Ye X., Zhao Q., You Z., Mater. Horiz. 2019, 6, 394. [Google Scholar]
- 32. Sant S., Hwang C. M., Lee S.‐H., Khademhosseini A., J. Tissue Eng. Regener. Med. 2011, 5, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vogt L., Rivera L. R., Liverani L., Piegat A., El Fray M., Boccaccini A. R., Mater. Sci. Eng. C 2019, 103, 109712. [DOI] [PubMed] [Google Scholar]
- 34. Liu Y., Tian K., Hao J., Yang T., Geng X., Zhang W., J. Mater. Sci. Mater. Med. 2019, 30, 53. [DOI] [PubMed] [Google Scholar]
- 35. Jiang L., Jiang Y., Stiadle J., Wang X., Wang L., Li Q., Shen C., Thibeault S. L., Turng L.‐S., Mater. Sci. Eng. C 2019, 94, 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Brien D., Hankins A., Golestaneh N., Paranjape M., Biomed. Microdevices 2019, 21, 53. [DOI] [PubMed] [Google Scholar]
- 37. Yang Y., Lei D., Huang S., Yang Q., Song B., Guo Y., Shen A., Yuan Z., Li S., Qing F., Qing F.‐L., Ye X. F., You Z., Zhao Q., Adv. Healthcare Mater. 2019, 8, 1900065. [DOI] [PubMed] [Google Scholar]
- 38. Stafiej P., Küng F., Thieme D., Czugala M., Kruse F. E., Schubert D. W., Fuchsluger T. A., Mater. Sci. Eng. C 2017, 71, 764. [DOI] [PubMed] [Google Scholar]
- 39. Ruther F., Zimmermann A., Engel F. B., Boccaccini A. R., Adv. Eng. Mater. 2019, 22, 1900986. [Google Scholar]
- 40. Vilariño‐Feltrer G., Muñoz‐Santa A., Conejero‐García Á., Vallés‐Lluch A., Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 938. [Google Scholar]
- 41. Wang M., Lei D., Liu Z., Chen S., Sun L., Lv Z., Huang P., Jiang Z., You Z., J. Biomater. Sci., Polym. Ed. 2017, 28, 1728. [DOI] [PubMed] [Google Scholar]
- 42. Souza M., Tansaz S., Zanotto E., Boccaccini A., Materials 2017, 10, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang X., Jia C., Qiao X., Liu T., Sun K., Polym. Test. 2016, 54, 118. [Google Scholar]
- 44. Theerathanagorn T., Klangjorhor J., Sakulsombat M., Pothacharoen P., Pruksakorn D., Kongtawelert P., Janvikul W., J. Biomater. Sci., Polym. Ed. 2015, 26, 1386. [DOI] [PubMed] [Google Scholar]
- 45. Kalakonda P., Aldhahri M. A., Abdel‐wahab M. S., Tamayol A., Moghaddam K. M., Ben Rached F., Pain A., Khademhosseini A., Memic A., Chaieb S., RSC Adv. 2017, 7, 34331. [Google Scholar]
- 46. Touré A. B. R., Mele E., Christie J. K., Nanomaterials 2020, 10, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao X., Wu Y., Du Y., Chen X., Lei B., Xue Y., Ma P. X., J. Mater. Chem. B 2015, 3, 3222. [DOI] [PubMed] [Google Scholar]
- 48. Yu S., Shi J., Liu Y., Si J., Yuan Y., Liu C., J. Mater. Chem. B 2019, 7, 3279. [Google Scholar]
- 49. Tevlek A., Hosseinian P., Ogutcu C., Turk M., Aydin H. M., Mater. Sci. Eng. C 2017, 72, 316. [DOI] [PubMed] [Google Scholar]
- 50. Yang K., Zhang J., Ma X., Ma Y., Kan C., Ma H., Li Y., Yuan Y., Liu C., Mater. Sci. Eng. C 2015, 56, 37. [DOI] [PubMed] [Google Scholar]
- 51. Masoudi Rad M., Nouri Khorasani S., Ghasemi‐Mobarakeh L., Prabhakaran M. P., Foroughi M. R., Kharaziha M., Saadatkish N., Ramakrishna S., Mater. Sci. Eng. C 2017, 80, 75. [DOI] [PubMed] [Google Scholar]
- 52. Zhou L., He H., Jiang C., He S., J. Appl. Polym. Sci. 2015, 132, 42196. [Google Scholar]
- 53. Zhang X., Jia C., Qiao X., Liu T., Sun K., Polym. Test. 2017, 62, 88. [Google Scholar]
- 54. Naghizadeh S., Hassanzadeh Nemati N., Hassani Najafabadi A., Niknejad H., Khani M.‐M., Int. J. Polym. Mater. Polym. Biomater. 2018, 67, 212. [Google Scholar]
- 55. Nguyen A. H., Marsh P., Schmiess‐Heine L., Burke P. J., Lee A., Lee J., Cao H., J. Biol. Eng. 2019, 13, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Farr N., Pashneh‐Tala S., Stehling N., Claeyssens F., Green N., Rodenburg C., Macromol. Rapid Commun. 2020, 41, 1900484. [DOI] [PubMed] [Google Scholar]
- 57. Wu Y., Wang L., Zhao X., Hou S., Guo B., Ma P. X., Biomaterials 2016, 104, 18. [DOI] [PubMed] [Google Scholar]
- 58. Yeh Y.‐C., Ouyang L., Highley C. B., Burdick J. A., Polym. Chem. 2017, 8, 5091. [Google Scholar]
- 59. Tsai Y.‐T., Chang C.‐W., Yeh Y.‐C., Biomater. Sci. 2020, 8, 4728. [DOI] [PubMed] [Google Scholar]
- 60. Wang L., Xu K., Hou X., Han Y., Liu S., Wiraja C., Yang C., Yang J., Wang M., Dong X., Huang W., Xu C., ACS Appl. Mater. Interfaces 2017, 9, 9528. [DOI] [PubMed] [Google Scholar]
- 61. Pashneh‐Tala S., Moorehead R., Claeyssens F., J. Biomater. Appl. 2020, 34, 1114. [DOI] [PubMed] [Google Scholar]
- 62. Chen J.‐Y., Hwang J., Ao‐Ieong W.‐S., Lin Y.‐C., Hsieh Y.‐K., Cheng Y.‐L., Wang J., Polymers 2018, 10, 1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rostamian M., Kalaee M. R., Dehkordi S. R., Panahi‐Sarmad M., Tirgar M., Goodarzi V., Eur. Polym. J. 2020, 138, 109985. [Google Scholar]
- 64. Jia Y., Wang W., Zhou X., Nie W., Chen L., He C., Polym. Chem. 2016, 7, 2553. [Google Scholar]
- 65. Wang Y., Wu H., Wang Z., Zhang J., Zhu J., Ma Y., Yang Z., Yuan Y., Polymers 2019, 11, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhao X., Wu H., Guo B., Dong R., Qiu Y., Ma P. X., Biomaterials 2017, 122, 34. [DOI] [PubMed] [Google Scholar]
- 67. Wang Z., Ma Y., Wang Y., Liu Y., Chen K., Wu Z., Yu S., Yuan Y., Liu C., Acta Biomater. 2018, 71, 279. [DOI] [PubMed] [Google Scholar]
- 68. Ye H., Owh C., Loh X. J., RSC Adv. 2015, 5, 48720. [Google Scholar]
- 69. Wilson R., Divakaran A. V., S K., Varyambath A., Kumaran A., Sivaram S., Ragupathy L., ACS Omega 2018, 3, 18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yoon S., Chen B., Polym. Chem. 2018, 9, 3727. [Google Scholar]
- 71. Garg T., Singh O., Arora S., Murthy R. S. R., Crit. Rev. Ther. Drug Carrier Syst. 2012, 29, 1. [DOI] [PubMed] [Google Scholar]
- 72. Lee S. H., Lee K.‐W., Gade P. S., Robertson A. M., Wang Y., J. Biomater. Sci., Polym. Ed. 2018, 29, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sencadas V., Sadat S., Silva D. M., J. Mech. Behav. Biomed. Mater. 2020, 102, 103474. [DOI] [PubMed] [Google Scholar]
- 74. Oh S. J., Woo J. O., Park J.‐E., Son K., Mater. Lett. 2015, 143, 219. [Google Scholar]
- 75. Moorhoff C., Li Y., Cook W. D., Braybrook C., Chen Q.‐Z., Polym. Int. 2015, 64, 668. [Google Scholar]
- 76. Aydin H. M., Salimi K., Yilmaz M., Turk M., Rzayev Z. M. O., Pişkin E., J. Tissue Eng. Regener. Med. 2016, 10, E14. [DOI] [PubMed] [Google Scholar]
- 77. Huang P., Bi X., Gao J., Sun L., Wang S., Chen S., Fan X., You Z., Wang Y., J. Mater. Chem. B 2016, 4, 2090. [DOI] [PubMed] [Google Scholar]
- 78. Natarajan J., Madras G., Chatterjee K., RSC Adv. 2016, 6, 61492. [Google Scholar]
- 79. Wang S., Jeffries E., Gao J., Sun L., You Z., Wang Y., ACS Appl. Mater. Interfaces 2016, 8, 9590. [DOI] [PubMed] [Google Scholar]
- 80. Shi H., Gan Q., Liu X., Ma Y., Hu J., Yuan Y., Liu C., RSC Adv. 2015, 5, 79703. [Google Scholar]
- 81. Wu Y., Wang L., Guo B., Shao Y., Ma P. X., Biomaterials 2016, 87, 18. [DOI] [PubMed] [Google Scholar]
- 82. Rosenbalm T. N., Teruel M., Day C. S., Donati G. L., Morykwas M., Argenta L., Kuthirummal N., Levi‐Polyachenko N., J. Biomed. Mater. Res., Part B 2016, 104, 1366. [DOI] [PubMed] [Google Scholar]
- 83. Kerativitayanan P., Tatullo M., Khariton M., Joshi P., Perniconi B., Gaharwar A. K., ACS Biomater. Sci. Eng. 2017, 3, 590. [DOI] [PubMed] [Google Scholar]
- 84. Ding X., Wu Y.‐L., Gao J., Wells A., Lee K.‐W., Wang Y., J. Mater. Chem. B 2017, 5, 6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dong R., Zhao X., Guo B., Ma P. X., Biomacromolecules 2017, 18, 2808. [DOI] [PubMed] [Google Scholar]
- 86. Ghafaralahi S., Ebrahimian‐Hosseinabadi M., Zargar Kharazi A., J. Bioact. Compat. Polym. 2018, 33, 529. [Google Scholar]
- 87. Zanjanizadeh Ezazi N., Ajdary R., Correia A., Mäkilä E., Salonen J., Kemell M., Hirvonen J., Rojas O. J., Ruskoaho H. J., Santos H. A., ACS Appl. Mater. Interfaces 2020, 12, 6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gaharwar A. K., Patel A., Dolatshahi‐Pirouz A., Zhang H., Rangarajan K., Iviglia G., Shin S.‐R., Hussain M. A., Khademhosseini A., Biomater. Sci. 2015, 3, 46. [DOI] [PubMed] [Google Scholar]
- 89. Chen Q.‐Z., Ishii H., Thouas G. A., Lyon A. R., Wright J. S., Blaker J. J., Chrzanowski W., Boccaccini A. R., Ali N. N., Knowles J. C., Harding S. E., Biomaterials 2010, 31, 3885. [DOI] [PubMed] [Google Scholar]
- 90. Martín‐Pat G. E., Rodriguez‐Fuentes N., Cervantes‐Uc J. M., Rosales‐Ibáñez R., Carrillo‐Escalante H. J., Ku‐Gonzalez A. F., Avila‐Ortega A., Hernandez‐Sanchez F., J. Biomater. Appl. 2020, 35, 485. [DOI] [PubMed] [Google Scholar]
- 91. Sell S., Barnes C., Smith M., McClure M., Madurantakam P., Grant J., McManus M., Bowlin G., Polym. Int. 2007, 56, 1349. [Google Scholar]
- 92. Wang X., Ding B., Li B., Mater. Today 2013, 16, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu H., Ding X., Zhou G., Li P., Wei X., Fan Y., J. Nanomater. 2013, 2013, 495708. [Google Scholar]
- 94. Sill T. J., von Recum H. A., Biomaterials 2008, 29, 1989. [DOI] [PubMed] [Google Scholar]
- 95. Li Z., Wang C., in One‐Dimensional Nanostructures (Ed: Wang Z. M.), Springer, Berlin: 2013, pp. 15–28. [Google Scholar]
- 96. Jeffries E. M., Allen R. A., Gao J., Pesce M., Wang Y., Acta Biomater. 2015, 18, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Memic A., Aldhahri M., Tamayol A., Mostafalu P., Abdel‐wahab M., Samandari M., Moghaddam K., Annabi N., Bencherif S., Khademhosseini A., Nanomaterials 2017, 7, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Nadim A., Khorasani S. N., Kharaziha M., Davoodi S. M., Mater. Sci. Eng. C 2017, 78, 47. [DOI] [PubMed] [Google Scholar]
- 99. Hou L., Zhang X., Mikael P. E., Lin L., Dong W., Zheng Y., Simmons T. J., Zhang F., Linhardt R. J., ACS Omega 2017, 2, 6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang Y., Xiao Y., Chen Z., Leng X., Ren J., Jiang X., Sci. Adv. Mater. 2018, 10, 1140. [Google Scholar]
- 101. Kharazi A. Z., Atari M., Vatankhah E., Javanmard S. H., Polym. Adv. Technol. 2018, 29, 3151. [Google Scholar]
- 102. Wu W., Jia S., Chen W., Liu X., Zhang S., Mater. Sci. Eng. C 2019, 101, 1. [DOI] [PubMed] [Google Scholar]
- 103. Silva J. C., Udangawa R. N., Chen J., Mancinelli C. D., Garrudo F. F. F., Mikael P. E., Cabral J. M. S., Ferreira F. C., Linhardt R. J., Mater. Sci. Eng. C 2020, 107, 110291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kaya M., Ahi Z. B., Ergene E., Yilgor Huri P., Tuzlakoglu K., J. Tissue Eng. Regener. Med. 2020, 14, 347. [DOI] [PubMed] [Google Scholar]
- 105. Gaharwar A. K., Nikkhah M., Sant S., Khademhosseini A., Biofabrication 2014, 7, 015001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rai R., Tallawi M., Frati C., Falco A., Gervasi A., Quaini F., Roether J. A., Hochburger T., Schubert D. W., Seik L., Barbani N., Lazzeri L., Rosellini E., Boccaccini A. R., Adv. Healthcare Mater. 2015, 4, 2012. [DOI] [PubMed] [Google Scholar]
- 107. Khosravi R., Best C. A., Allen R. A., Stowell C. E. T., Onwuka E., Zhuang J. J., Lee Y.‐U., Yi T., Bersi M. R., Shinoka T., Humphrey J. D., Wang Y., Breuer C. K., Ann. Biomed. Eng. 2016, 44, 2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Behtaj S., Karamali F., Masaeli E., Anissimov Y. G., Rybachuk M., Biochem. Eng. J. 2021, 166, 107846. [Google Scholar]
- 109. Fakhrali A., Semnani D., Salehi H., Ghane M., Polym. Adv. Technol. 2020, 31, 3134. [Google Scholar]
- 110. Tallawi M., Dippold D., Rai R., D'Atri D., Roether J. A., Schubert D. W., Rosellini E., Engel F. B., Boccaccini A. R., Mater. Sci. Eng. C 2016, 69, 569. [DOI] [PubMed] [Google Scholar]
- 111. Ferrari P. F., Aliakbarian B., Lagazzo A., Tamayol A., Palombo D., Perego P., Int. J. Polym. Mater. Polym. Biomater. 2017, 66, 635. [Google Scholar]
- 112. Hsu C.‐N., Lee P.‐Y., Tuan‐Mu H.‐Y., Li C.‐Y., Hu J.‐J., J. Biomed. Mater. Res., Part B 2018, 106, 760. [DOI] [PubMed] [Google Scholar]
- 113. Saudi A., Amini S., Amirpour N., Kazemi M., Zargar Kharazi A., Salehi H., Rafienia M., Mater. Sci. Eng. C 2019, 104, 110005. [DOI] [PubMed] [Google Scholar]
- 114. Saudi A., Rafienia M., Zargar Kharazi A., Salehi H., Zarrabi A., Karevan M., Polym. Adv. Technol. 2019, 30, 1427. [Google Scholar]
- 115. Tallawi M., Zebrowski D. C., Rai R., Roether J. A., Schubert D. W., El Fray M., Engel F. B., Aifantis K. E., Boccaccini A. R., Tissue Eng., Part C 2015, 21, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Liverani L., Piegat A., Niemczyk A., El Fray M., Boccaccini A. R., Eur. Polym. J. 2016, 81, 295. [Google Scholar]
- 117. Denis P., Wrzecionek M., Gadomska‐Gajadhur A., Sajkiewicz P., Polymers 2019, 11, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xu B., Cook W. D., Zhu C., Chen Q., Polym. Int. 2016, 65, 423. [Google Scholar]
- 119. Yan Y., Sencadas V., Jin T., Huang X., Chen J., Wei D., Jiang Z., J. Colloid Interface Sci. 2017, 508, 87. [DOI] [PubMed] [Google Scholar]
- 120. You Z.‐R., Hu M.‐H., Tuan‐Mu H.‐Y., Hu J.‐J., J. Mech. Behav. Biomed. Mater. 2016, 63, 220. [DOI] [PubMed] [Google Scholar]
- 121. Sfakis L., Sharikova A., Tuschel D., Costa F. X., Larsen M., Khmaladze A., Castracane J., Biomed. Opt. Express 2017, 8, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sfakis L., Kamaldinov T., Khmaladze A., Hosseini Z., Nelson D., Larsen M., Castracane J., Int. J. Mol. Sci. 2018, 19, 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lee S., Kim B., Kim S.‐H., Kim E., Jang J.‐H., Adv. Funct. Mater. 2017, 27, 1702310. [Google Scholar]
- 124. Heydari P., Varshosaz J., Zargar Kharazi A., Karbasi S., Polym. Adv. Technol. 2018, 29, 1795. [Google Scholar]
- 125. Memic A., Aldhahri M., Tamayol A., Mostafalu P., Abdel‐Wahab M. S., Samandari M., Moghaddam K. M., Annabi N., Bencherif S. A., Khademhosseini A., Nanomaterials 2017, 7, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Flaig F., Faria Bellani C., Uyumaz Ö., Schlatter G., Hébraud A., Mater. Adv. 2021, 2, 1284. [Google Scholar]
- 127. Wang R., Levi‐Polyanchenko N., Morykwas M., Argenta L., Wagner W. D., J. Biomed. Mater. Res., Part A 2015, 103, 1150. [DOI] [PubMed] [Google Scholar]
- 128. Ayati Najafabadi S. A., Shirazaki P., Zargar Kharazi A., Varshosaz J., Tahriri M., Tayebi L., Asia‐Pac. J. Chem. Eng. 2018, 13, e2255. [Google Scholar]
- 129. Ravichandran R., Venugopal J. R., Mukherjee S., Sundarrajan S., Ramakrishna S., Tissue Eng., Part A 2015, 21, 1288. [DOI] [PubMed] [Google Scholar]
- 130. Dippold D., Tallawi M., Tansaz S., Roether J. A., Boccaccini A. R., Eur. Polym. J. 2016, 75, 504. [Google Scholar]
- 131. Vogt L., Liverani L., Roether J., Boccaccini A., Nanomaterials 2018, 8, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Abudula T., Gzara L., Simonetti G., Alshahrie A., Salah N., Morganti P., Chianese A., Fallahi A., Tamayol A., Bencherif S., Memic A., Materials 2018, 11, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Xu B., Li Y., Zhu C., Cook W. D., Forsythe J., Chen Q., Eur. Polym. J. 2015, 64, 79. [Google Scholar]
- 134. Wu H.‐J., Hu M.‐H., Tuan‐Mu H.‐Y., Hu J.‐J., Mater. Sci. Eng. C 2019, 100, 30. [DOI] [PubMed] [Google Scholar]
- 135. Lang K., Bhattacharya S., Ning Z., Sánchez‐Leija R. J., Bramson M. T. K., Centore R., Corr D. T., Linhardt R. J., Gross R. A., Biomacromolecules 2020, 21, 3197. [DOI] [PubMed] [Google Scholar]
- 136. Luginina M., Schuhladen K., Orrú R., Cao G., Boccaccini A. R., Liverani L., Nanomaterials 2020, 10, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gultekinoglu M., Öztürk Ş., Chen B., Edirisinghe M., Ulubayram K., Eur. Polym. J. 2019, 121, 109297. [Google Scholar]
- 138. Lei D., Luo B., Guo Y., Wang D., Yang H., Wang S., Xuan H., Shen A., Zhang Y., Liu Z., He C., Qing F.‐L., Xu Y., Zhou G., You Z., Sci. China Mater. 2019, 62, 1910. [Google Scholar]
- 139. El‐Sherbiny I. M., Yacoub M. H., Global Cardiol. Sci. Pract. 2013, 2013, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mohamed M. A., Fallahi A., El‐Sokkary A. M. A., Salehi S., Akl M. A., Jafari A., Tamayol A., Fenniri H., Khademhosseini A., Andreadis S. T., Cheng C., Prog. Polym. Sci. 2019, 98, 101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Frydrych M., Román S., Green N. H., MacNeil S., Chen B., Polym. Chem. 2015, 6, 7974. [Google Scholar]
- 142. Choi S. M., Lee Y., Son J. Y., Bae J. W., Park K. M., Park K. D., Macromol. Res. 2017, 25, 85. [Google Scholar]
- 143. Zhao X., Liang Y., Huang Y., He J., Han Y., Guo B., Adv. Funct. Mater. 2020, 30, 1910748. [Google Scholar]
- 144. Ye H., Owh C., Jiang S., Ng C., Wirawan D., Loh X., Polymers 2016, 8, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hou Q., De Bank P. A., Shakesheff K. M., J. Mater. Chem. 2004, 14, 1915. [Google Scholar]
- 146. Eslami M., Javadi G., Agdami N., Shokrgozar M. A., Cell J. 2015, 17, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Frydrych M., Chen B., J. Mater. Chem. B 2013, 1, 6650. [DOI] [PubMed] [Google Scholar]
- 148. Frydrych M., Chen B., Polymer 2017, 122, 159. [Google Scholar]
- 149. Samourides A., Browning L., Hearnden V., Chen B., Mater. Sci. Eng. C 2020, 108, 110384. [DOI] [PubMed] [Google Scholar]
- 150. Liang B., Shi Q., Xu J., Chai Y.‐M., Xu J.‐G., Front. Chem. 2020, 8, 603577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Choi J. H., Gimble J. M., Lee K., Marra K. G., Rubin J. P., Yoo J. J., Vunjak‐Novakovic G., Kaplan D. L., Tissue Eng., Part B 2010, 16, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Quist A. P., Oscarsson S., Expert Opin. Drug Discovery 2010, 5, 569. [DOI] [PubMed] [Google Scholar]
- 153. Martinez‐Rivas A., González‐Quijano G., Proa‐Coronado S., Séverac C., Dague E., Micromachines 2017, 8, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Mogosanu D.‐E., Verplancke R., Dubruel P., Vanfleteren J., Mater. Des. 2016, 89, 1315. [Google Scholar]
- 155. Hsieh Y.‐K., Chen S.‐C., Huang W.‐L., Hsu K.‐P., Gorday K., Wang T., Wang J., Polymers 2017, 9, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Yeh C.‐W., Wang L.‐W., Wu H.‐C., Hsieh Y.‐K., Wang J., Chen M.‐H., Wang T.‐W., Biofabrication 2017, 9, 015024. [DOI] [PubMed] [Google Scholar]
- 157. Zhu C., Rodda A. E., Truong V. X., Shi Y., Zhou K., Haynes J. M., Wang B., Cook W. D., Forsythe J. S., ACS Biomater. Sci. Eng. 2018, 4, 2494. [DOI] [PubMed] [Google Scholar]
- 158. Ladd M. R., Costello C. M., Gosztyla C., Werts A. D., Johnson B., Fulton W. B., Martin L. Y., Redfield E. J., Crawford B., Panaparambil R., Sodhi C. P., March J. C., Hackam D. J., Tissue Eng., Part A 2019, 25, 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hu T., Wu Y., Zhao X., Wang L., Bi L., Ma P. X., Guo B., Chem. Eng. J. 2019, 366, 208. [Google Scholar]
- 160. Ruvinov E., Sapir Y., Cohen S., Synth. Lect. Tissue Eng. 2012, 4, 200. [Google Scholar]
- 161. Kazemzadeh Farizhandi A. A., Khalajabadi S. Z., Krishnadoss V., Noshadi I., J. Mech. Behav. Biomed. Mater. 2020, 110, 103960. [DOI] [PubMed] [Google Scholar]
- 162. Singh D., Harding A. J., Albadawi E., Boissonade F. M., Haycock J. W., Claeyssens F., Acta Biomater. 2018, 78, 48. [DOI] [PubMed] [Google Scholar]
- 163. Pereira R. F., Bártolo P. J., Engineering 2015, 1, 090. [Google Scholar]
- 164. Ostrovidov S., Salehi S., Costantini M., Suthiwanich K., Ebrahimi M., Sadeghian R. B., Fujie T., Shi X., Cannata S., Gargioli C., Tamayol A., Dokmeci M. R., Orive G., Swieszkowski W., Khademhosseini A., Small 2019, 15, 1805530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Pashneh‐Tala S., Owen R., Bahmaee H., Rekštytė S., Malinauskas M., Claeyssens F., Front. Phys. 2018, 6, 41. [Google Scholar]
- 166. Wang P., Berry D. B., Song Z., Kiratitanaporn W., Schimelman J., Moran A., He F., Xi B., Cai S., Chen S., Adv. Funct. Mater. 2020, 30, 1910391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wu Y., D'Amato A. R., Yan A. M., Wang R. Q., Ding X., Wang Y., ACS Appl. Bio Mater. 2020, 3, 7575. [DOI] [PubMed] [Google Scholar]
- 168. Rodrigues I. C. P., Kaasi A., Maciel Filho R., Jardini A. L., Gabriel L. P., Einstein 2018, 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kitsara M., Agbulut O., Kontziampasis D., Chen Y., Menasché P., Acta Biomater. 2017, 48, 20. [DOI] [PubMed] [Google Scholar]
- 170. Sant S., Iyer D., Gaharwar A. K., Patel A., Khademhosseini A., Acta Biomater. 2013, 9, 5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Xue Y., Ravishankar P., Zeballos M. A., Sant V., Balachandran K., Sant S., Polym. Adv. Technol. 2020, 31, 94. [Google Scholar]
- 172. Merle B., Kraus X., Tallawi M., Scharfe B., El Fray M., Aifantis K. E., Boccaccini A. R., Göken M., Mater. Lett. 2018, 221, 115. [Google Scholar]
- 173. Shukla R., Cheryan M., Ind. Crops Prod. 2001, 13, 171. [Google Scholar]
- 174. Reddy N., Yang Y., Trends Biotechnol. 2011, 29, 490. [DOI] [PubMed] [Google Scholar]
- 175. Hamada T., Dubois J. L. N., Bellamy V., Pidial L., Hagège A., Pereira M. N., Menasché P., Acta Biomater. 2020, 115, 92. [DOI] [PubMed] [Google Scholar]
- 176. Li S., Sengupta D., Chien S., Wiley Interdiscip. Rev.: Syst. Biol. Med. 2014, 6, 61. [DOI] [PubMed] [Google Scholar]
- 177. Stowell C. E. T., Li X., Matsunaga M. H., Cockreham C. B., Kelly K. M., Cheetham J., Tzeng E., Wang Y., J. Tissue Eng. Regener. Med. 2020, term. 3128, 14, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Wu Y.‐L., Szafron J. M., Blum K. M., Zbinden J. C., Khosravi R., Best C. A., Reinhardt J. W., Zeng Q., Yi T., Shinoka T., Humphrey J. D., Breuer C. K., Wang Y., Tissue Eng., Part A 2020, ten. tea.2020.0166. [Google Scholar]
- 179. Fu J., Ding X., Stowell C. E. T., Wu Y.‐L., Wang Y., Biomaterials 2020, 257, 120251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Gade P. S., Lee K., Pfaff B. N., Wang Y., Robertson A. M., J. R. Soc., Interface 2017, 14, 20170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Lee K.‐W., Gade P. S., Dong L., Zhang Z., Aral A. M., Gao J., Ding X., Stowell C. E. T., Nisar M. U., Kim K., Reinhardt D. P., Solari M. G., Gorantla V. S., Robertson A. M., Wang Y., Biomaterials 2018, 181, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Yang X., Gao Z., Liu H., Wu W., J. Tissue Eng. Regener. Med. 2019, 13, 2055. [DOI] [PubMed] [Google Scholar]
- 183. Li C.‐Y., Hu M.‐H., Hu J.‐J., Polymers 2019, 11, 1492.31547419 [Google Scholar]
- 184. Fukunishi T., Ong C. S., Lui C., Pitaktong I., Smoot C., Harris J., Gabriele P., Vricella L., Santhanam L., Lu S., Hibino N., Tissue Eng., Part A 2019, 25, 632. [DOI] [PubMed] [Google Scholar]
- 185. Zbinden J. C., Blum K. M., Berman A. G., Ramachandra A. B., Szafron J. M., Kerr K. E., Anderson J. L., Sangha G. S., Earl C. C., Nigh N. R., Mirhaidari G. J. M., Reinhardt J. W., Chang Y. C., Yi T., Smalley R., Gabriele P. D., Harris J. J., Humphrey J. D., Goergen C. J., Breuer C. K., Adv. Healthcare Mater. 2020, 9, 2001093. [Google Scholar]
- 186. Guler S., Hosseinian P., Aydin H. M., Tissue Eng., Part C 2017, 23, 21. [DOI] [PubMed] [Google Scholar]
- 187. Gu X., Ding F., Yang Y., Liu J., in Neural Regeneration (Eds: Kwok‐Fai S., Xiao‐Ming X.), Elsevier, Amsterdam: 2015, p. 73. [Google Scholar]
- 188. Gao Y., Wang Y., Kong D., Qu B., Su X., Li H., Pi H., Neural Regener. Res. 2015, 10, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Pan Q., Guo Y., Kong F., J. Biomed. Mater. Res., Part B 2018, 106, 1770. [DOI] [PubMed] [Google Scholar]
- 190. Zargar Kharazi A., Dini G., Naser R., J. Biomed. Mater. Res., Part A 2018, 106, 2181. [DOI] [PubMed] [Google Scholar]
- 191. Li G., Xiao Q., McNaughton R., Han L., Zhang L., Wang Y., Yang Y., Colloids Surf., B 2017, 158, 57. [DOI] [PubMed] [Google Scholar]
- 192. Zhang X. F., Kehoe S., Adhi S. K., Ajithkumar T. G., Moane S., O'Shea H., Boyd D., Mater. Sci. Eng. C 2011, 31, 669. [Google Scholar]
- 193. Ciofani G., Danti S., Ricotti L., D'Alessandro D., Moscato S., Mattoli V., in Piezoelectric Nanomaterials for Biomedical Applications (Eds: Ciofani G., Menciassi A.), Springer, Berlin, 2012, p. 213. [Google Scholar]
- 194. Rosenberg S. S., Spitzer N. C., Cold Spring Harbor Perspect. Biol. 2011, 3, a0a004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Adalbert R., Morreale G., Paizs M., Conforti L., Walker S. A., Roderick H. L., Bootman M. D., Siklós L., Coleman M. P., Neuroscience 2012, 225, 44. [DOI] [PubMed] [Google Scholar]
- 196. Sun L., Wang M., Chen S., Sun B., Guo Y., He C., Mo X., Zhu B., You Z., Acta Biomater. 2019, 85, 310. [DOI] [PubMed] [Google Scholar]
- 197. Kang H., Zhang K., Jung H. J., Yang B., Chen X., Pan Q., Li R., Xu X., Li G., Dravid V. P., Bian L., Adv. Mater. 2018, 30, 1803591. [DOI] [PubMed] [Google Scholar]
- 198. Zreiqat H., Howlett C. R., Zannettino A., Evans P., Schulze‐Tanzil G., Knabe C., Shakibaei M., J. Biomed. Mater. Res. 2002, 62, 175. [DOI] [PubMed] [Google Scholar]
- 199. Wang J., Tian L., Luo B., Ramakrishna S., Kai D., Loh X. J., Yang I. H., Deen G. R., Mo X., Colloids Surf., B 2018, 169, 356. [DOI] [PubMed] [Google Scholar]
- 200. Ugartondo V., Mitjans M., Vinardell M. P., Ind. Crops Prod. 2009, 30, 184. [Google Scholar]
- 201. Bhardwaj N., Chouhan D., Mandal B. B., Curr. Pharm. Des. 2017, 23, 3455. [DOI] [PubMed] [Google Scholar]
- 202. Shirazaki P., Varshosaz J., Kharazi A., Adv. Biomed. Res. 2017, 6, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Jodat Y. A., Zhang T., Al Tanoury Z., Kamperman T., Shi K., Huang Y., Panayi A., Endo Y., Wang X., Quint J., Arnaout A., Kiaee K., Hassan S., Lee J., Huidobro Martinez A. F., Ochoa S. L., Lee K., Calabrese M., Carlucci A., Tamayol A., Sinha I., Pourquié O., Shin S. R., PREPRINT available at Research Square, Version 1, posted 20 Jan. 2021, submitted, 10.21203/rs.3.rs-146091/v1. [DOI] [Google Scholar]
- 204. Amini A. R., Laurencin C. T., Nukavarapu S. P., Crit. Rev. Biomed. Eng. 2012, 40, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Blokhuis T. J., Arts J. J. C., Injury 2011, 42, S26. [DOI] [PubMed] [Google Scholar]
- 206. Barradas A., Yuan H., van Blitterswijk C., Habibovic P., Eur. Cells Mater. 2011, 21, 407. [DOI] [PubMed] [Google Scholar]
- 207. Habibovic P., de Groot K., J. Tissue Eng. Regener. Med. 2007, 1, 25. [DOI] [PubMed] [Google Scholar]
- 208. Ghassemi T., Shahroodi A., Ebrahimzadeh M. H., Mousavian A., Movaffagh J., Moradi A., Arch. Bone Jt. Surg. 2018, 6, 90. [PMC free article] [PubMed] [Google Scholar]
- 209. Shi C., Yuan Z., Han F., Zhu C., Li B., Ann. Jt. 2016, 1, 27. [Google Scholar]
- 210. Tevlek A., Agacik D. T., Aydin H. M., J. Appl. Polym. Sci. 2020, 137, 48689. [Google Scholar]
- 211. Wang Y., Sun N., Zhang Y., Zhao B., Zhang Z., Zhou X., Zhou Y., Liu H., Zhang Y., Liu J., Sci. Rep. 2019, 9, 7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Bellucci D., Sola A., Gentile P., Ciardelli G., Cannillo V., J. Biomed. Mater. Res., Part A 2012, 100A, 3259. [DOI] [PubMed] [Google Scholar]
- 213. Li W., Nooeaid P., Roether J. A., Schubert D. W., Boccaccini A. R., J. Eur. Ceram. Soc. 2014, 34, 505. [Google Scholar]
- 214. Chen Q.‐Z., Quinn J. M. W., Thouas G. A., Zhou X., Komesaroff P. A., Adv. Eng. Mater. 2010, 12, B642. [Google Scholar]
- 215. MA L.‐J., WU Y., Anal. Sci. 2007, 23, 799. [DOI] [PubMed] [Google Scholar]
- 216. Lin D., Yang K., Tang W., Liu Y., Yuan Y., Liu C., Colloids Surf., B 2015, 131, 1. [DOI] [PubMed] [Google Scholar]
- 217. Chai Y., Lin D., Ma Y., Yuan Y., Liu C., J. Mater. Chem. B 2017, 5, 4633. [DOI] [PubMed] [Google Scholar]
- 218. Ma Y., Zhang W., Wang Z., Wang Z., Xie Q., Niu H., Guo H., Yuan Y., Liu C., Acta Biomater. 2016, 44, 110. [DOI] [PubMed] [Google Scholar]
- 219. Zaky S. H., Lee K. W., Gao J., Jensen A., Verdelis K., Wang Y., Almarza A. J., Sfeir C., Acta Biomater. 2017, 54, 95. [DOI] [PubMed] [Google Scholar]
- 220. Laurencin C. T., Kumbar S. G., Nukavarapu S. P., Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 6. [DOI] [PubMed] [Google Scholar]
- 221. Christenson E. M., Anseth K. S., van den Beucken J. J. J. P., Chan C. K., Ercan B., Jansen J. A., Laurencin C. T., Li W.‐J., Murugan R., Nair L. S., Ramakrishna S., Tuan R. S., Webster T. J., Mikos A. G., J. Orthop. Res. 2007, 25, 11. [DOI] [PubMed] [Google Scholar]
- 222. Porter J. R., Ruckh T. T., Popat K. C., Biotechnol. Prog. 2009, 25, 1539. [DOI] [PubMed] [Google Scholar]
- 223. Ward B. C., Webster T. J., Biomaterials 2006, 27, 3064. [DOI] [PubMed] [Google Scholar]
- 224. Safari J., Zarnegar Z., J. Saudi Chem. Soc. 2014, 18, 85. [Google Scholar]
- 225. Chegini S. P., Varshosaz J., Sadeghi H. M., Dehghani A., Minayian M., IET Nanobiotechnol. 2019, 13, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Louage B., Tack L., Wang Y., De Geest B. G., Polym. Chem. 2017, 8, 5033. [Google Scholar]
- 227. Yang B., Lv W., Deng Y., RSC Adv. 2017, 7, 37426. [Google Scholar]
- 228. Arcamone F., Doxorubicin: Anticancer Antibiotics, Elsevier Science, Amsterdam: 2012. [Google Scholar]
- 229. Sencadas V., Tawk C., Alici G., ACS Appl. Mater. Interfaces 2020, 12, 8761. [DOI] [PubMed] [Google Scholar]
- 230. Najafabadi A. H., Tamayol A., Annabi N., Ochoa M., Mostafalu P., Akbari M., Nikkhah M., Rahimi R., Dokmeci M. R., Sonkusale S., Ziaie B., Khademhosseini A., Adv. Mater. 2014, 26, 5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Yeh Y. C., Highley C. B., Ouyang L., Burdick J. A., Biofabrication 2016, 8, 045004. [DOI] [PubMed] [Google Scholar]
- 232. Keirouz A., Fortunato G., Zhang M., Callanan A., Radacsi N., Medical Engineering & Physics 2019, 71, 56. [DOI] [PubMed] [Google Scholar]