

Abstract
To streamline the drug discovery pipeline, there is a pressing need for preclinical models which replicate the complexity and scale of native tumors. While there have been advancements in the formation of microscale tumor units, these models are cell‐line dependent, time‐consuming and have not improved clinical trial success rates. In this study, two methods for generating 3D tumor microenvironments are compared, rapidly fabricated hydrogel microspheres and traditional cell‐dense spheroids. These modules are then bioassembled into 3D printed thermoplastic scaffolds, using an automated biofabrication process, to form tumor‐scale models. Modules are formed with SKOV3 and HFF cells as monocultures and cocultures, and the fabrication efficiency, cell architecture, and drug response profiles are characterized, both as single modules and as multimodular constructs. Cell‐encapsulated Gel‐MA microspheres are fabricated with high‐reproducibility and dimensions necessary for automated tumor‐scale bioassembly regardless of cell type, however, only cocultured spheroids form compact modules suitable for bioassembly. Chemosensitivity assays demonstrate the reduced potency of doxorubicin in coculture bioassembled constructs and a ≈five‐fold increase in drug resistance of cocultured cells in 3D modules compared with 2D monolayers. This bioassembly system is efficient and tailorable so that a variety of relevant‐sized tumor constructs could be developed to study tumorigenesis and modernize drug discovery.
Keywords: drug screening, microspheres, modular assembly, spheroids, tumor microenvironment, tumor‐scale models
This work introduces a novel spheroid bioassembly approach for the development of in vitro tumor models which are more representative of the dimensions of clinical tumors. Coculture spheroid and microtissue modules are characterized, compared, and assembled into printed scaffolds using an automated device. Proof‐of‐concept chemosensitivity assays demonstrate compatibility of the constructs with current in vitro screening techniques.
1. Introduction
Cancer drug discovery is an inherently unpredictable and laborious process with clinical success rates showing a downward trajectory over the last decade.[ 1 , 2 ] Of the oncology drugs which progress to Phase 1 trials, only 6.7% are successful, exposing a need for new technologies which contribute to a better understanding of tumorigenesis and improve the precision of preclinical drug discovery.[ 1 , 3 , 4 ] Consequently, considerable effort has been assigned to developing in vitro models that emulate the in vivo tumor microenvironment with greater accuracy.[ 3 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 ] However, with increasing model complexity comes a reduction in capacity for high‐throughput screening, a key step in the drug discovery process.[ 3 , 13 ]
Some of the more promising in vitro models have been developed using 3D culture, including multicellular spheroids or tumor organoids which better mimic the architectural complexity of native tumors.[ 3 , 13 , 14 ] While patient‐derived tumor organoids are able to maintain the intra‐tumor heterogeneity present in individual patients and have substantial utility for personalized treatments,[ 14 , 15 , 16 ] they are a limited resource, and the long cultivation times (up to 4–6 weeks) render them impractical for preclinical screening of new drug candidates.[ 17 ] These constraints are not shared by multicellular spheroids; which have the ability to mimic biomolecular and hypoxia gradients, homotypic and heterotypic cell interactions, as well as nurture native cell polarity and architectural arrangements (Figure 1a).[ 3 ] The established microenvironments of multicellular spheroids allow changes in cell behavior (e.g., migration and proliferation), drug response, gene expression and protein synthesis, which are more indicative of native tumors.[ 3 ] Nevertheless, the formation of compact and reproducible spheroids is only limited to a small subset of cancer cell lines and is highly dependent on the cell phenotype.[ 18 ]
Figure 1.
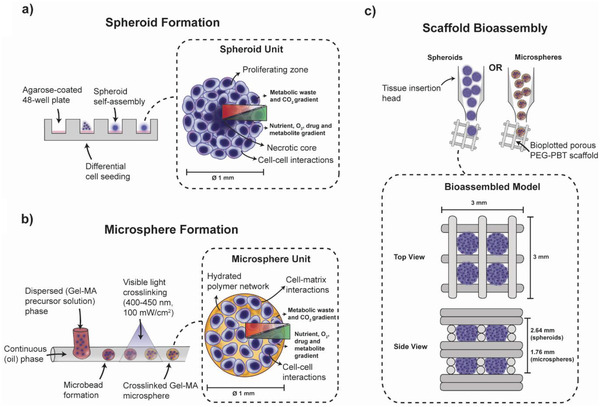
Cancer 3D in vitro model overview. a) Spheroid module formation using the liquid‐overlay technique to produce tailorable multicellular spheroids of tunable size through differential cell seeding. The resulting spheroids have distinct biomolecular gradients and model cell–cell interactions. b) Microsphere module formation using a droplet microfluidics system, coupled with cell‐friendly visible‐light photo‐polymerization. Cancer cells are encapsulated within a spherical hydrated polymer network (e.g., Gel‐MA hydrogel) and demonstrate both cell–cell and cell–matrix interactions. Microsphere and spheroid modules can replicate heterotypic interactions in the tumor microenvironment through modifying the cellular make‐up during assembly (e.g., cocultures with fibroblastic cells). c) Spheroid and microsphere tumor modules can be bioassembled into tumor‐scale composite constructs for screening, drug discovery, or personalized medicine using an automated biofabrication platform which singularizes and inserts tumor modules into bioplotted thermoplastic polymer framework.
With the evolution of novel biomaterials and biofabrication strategies, contemporary cancer models have focused on the encapsulation of tumor cells within a hydrated polymeric matrix which mimics the extracellular matrix of the tumor microenvironment.[ 3 , 11 , 19 , 20 , 21 , 22 , 23 ] As the cells are encapsulated, these hydrogel platforms are not dependent on the self‐assembly of cells into compact spheres and therefore have wider utility for drug screening on cell lines with different phenotypes. Naturally‐derived materials, ranging from single components to complex compositions (e.g., collagen and fibrin, and decellularized matrix and Matrigel respectively),[ 19 , 21 , 22 ] are advantageous as they provide increasingly more accurate representations of the native extracellular matrix components (with Matrigel as the gold‐standard due to the diversity of macromolecules present). However these materials are restricted by high batch‐to‐batch variation, limited control over structure and mechanics, and poor compatibility with biofabrication techniques (e.g., light‐mediated crosslinking, bioprinting, microfluidic approaches). Toward improved tunability, increased structural complexity, control over hydrogel properties and therefore compatibility with biofabrication techniques, semi‐synthetic and synthetic materials (e.g., poly(lactic‐co‐glycolic) acid, polyethylene glycol, peptide and gelatin‐based hydrogels) have gained interest for tumor modeling.[ 11 , 24 , 25 ] These materials allow for the physicochemical properties (e.g., mechanics, porosity, degradation profiles) of the material to be fine‐tuned to obtain increased control over specific microenvironmental cues within tumor models.[ 25 ] Specifically, gelatin‐methacrylol (Gel‐MA) is a commonly used functionalized biomaterial and bioink for 3D bioprinting because it is inexpensive, water‐soluble and can be crosslinked with visible‐light to form a polymer network with tailorable physicochemical properties.[ 11 , 26 , 27 ] Gelatin, as a product of collagen hydrolysis (the most common protein found in the extracellular matrix), can be remodeled by cell‐mediated matrix metalloproteinases and contains the necessary cell‐binding motifs to enable cell survival and function.[ 25 , 64 ] To this end, cell‐encapsulated Gel‐MA microspheres of tailorable size can be rapidly fabricated in high‐throughput, through adopting a droplet microfluidics system, coupled with cell‐friendly visible‐light photopolymerization (Figure 1b).[ 28 , 29 ] The 3D hydrogel microspheres provide enhanced control over the tumor microenvironment (compared to cell‐based spheroids) through introduction of cell–matrix interactions, and thereby are a tailorable system for screening or probing the tumor microenvironment at scale. Another key advantage of hydrogel microspheres, is that the strategy permits near instantaneous fabrication of multicellular cancer microenvironments without reliance on multi‐day self‐assembly.
While these spheroid and microsphere modules better represent the in vivo tumor microenvironment, their small scale is unrealistic compared to the size of native tumors (e.g., 1.0–4.3 cm in breast cancer; 2.5 mm–11.9 cm in ovarian cancer).[ 30 , 31 , 32 ] Advancements in biofabrication and bioprinting have resulted in the formation of larger tissue constructs through bioassembly of prefabricated modules for tissue regeneration applications;[ 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 ] but few studies have attempted bioassembly of tumor modules to produce 3D models capable of replicating the tumor microenvironment at size scales similar to native tumors.[ 45 , 46 , 47 ] Similarly, no comparisons have been made between 3D spheroid and cell‐encapsulated hydrogel models for high‐throughput screening. Modular bioassembly provides the means to upscale models toward biologically‐relevant sizes with precise control over the architectural arrangement of modules and spatial compartmentalization of cell populations, biomaterials and native extracellular matrix microenvironments (Figure 1c).[ 41 ] Furthermore, these modular tumor‐scale models provide opportunities to understand drug/nutrient diffusion profiles which will advance preclinical drug screening capacities, however also bring challenges with compatible analysis tools and technical difficulties. To prove valuable for high‐throughput screening, the formation of these constructs must be reproducible, scalable and automated.[ 41 ]
This study aims to demonstrate a novel approach for the fabrication of well‐defined and reproducible tumor models based on modular assembly; where the direct spatial position of versatile prefabricated modules can be controlled to form constructs of clinically‐relevant sizes. Specifically, the flexibility of an automated bioassembly system is demonstrated through the formation of scalar in vitro tumor models using a range of prefabricated multicellular tumor modules (e.g., hydrogels, spheroids) now commonly used in the literature. To demonstrate the flexibility of this bioassembly strategy, multicellular ovarian carcinoma spheroid and microsphere modules were evaluated based on their fabrication efficiency and compatibility with formation of modular constructs using a medium‐throughput automated 3D bioassembly system. The cell behavior and drug response profiles, comparing standard 2D monolayers versus multicellular 3D spheroid and hydrogel microsphere models, were systematically investigated in a proof‐of‐concept ovarian carcinoma application. This study works to address the throughput versus complexity dichotomy which exists within the field,[ 48 ] by generating more automated systems for the development of tumor‐scale 3D cancer models for in vitro drug testing and personalized medicine. The systems introduced in this study demonstrate further significant potential for in vitro modeling approaches to increase the predictive power of preclinical 3D models of the tumor microenvironment by more accurately anticipating the outcome of candidate drugs before progression to animal studies or clinical trials.
2. Experimental Section
Bioassembled ovarian carcinoma tumor models were constructed by first fabricating and characterizing cell‐laden spheroid and microsphere modules, and then assembling them into printed scaffolds using an automated device. The chemosensitivity of these in vitro models was then assessed through undertaking dose‐response studies with doxorubicin.
2.1. Cell Culture and Fluorescence Labeling
Human ovarian adenocarcinoma cells (SKOV3; ATCC, Virginia, USA) and human foreskin fibroblasts (HFF; ATCC, Virginia, USA) were routinely cultured in Dulbecco's modified eagle's medium (high glucose, GlutaMAX supplement, pyruvate), supplemented with 5% fetal bovine serum (FBS, Gibco, USA), and 1% penicillin‐streptomycin. Cell lines were maintained in tissue culture flasks at 3000 cells cm‐2 and incubated in a humidified environment at 37 °C and 5% CO2. All cell lines were used within 10 passages and were mycoplasma negative.
To identify and track each cell line within the coculture models, SKOV3 and HFF cells were labeled with the Qtracker 655 and Qtracker 800, respectively (Life Technologies, USA). Briefly, cells at 10 million cells mL‐1 were incubated with 10 × 10‐9 m labeling solution for 1 h and washed twice with media before use, as per manufacturer's instructions.
2.2. Spheroid Fabrication and Assessment
To ensure the formation of reproducible spheroids using the liquid‐overlay method, sphericity and spheroid size were investigated on day 7. To assess sphericity, SKOV3 and HFF cells were seeded at 80 000 cells per well at different percentage ratios (100:0, 75:25, 50:50, 25:75 and 0:100, SKOV3:HFF) into 2% agarose coated 48‐well plates (Figure 1a). To determine the relationship between the cell seeding density and spheroid size, labeled SKOV3 and HFF (at a percentage ratio of 75:25 (SKOV3:HFF)) were seeded at varying cell densities (60 000, 80 000, 100 000, 120 000, 140 000, and 160 000 cells).
Spheroids were fixed in 10% neutral buffered formalin overnight and imaged using a Zeiss Axioimager Z1 (Zeiss, Germany). The major and minor diameters of spheroids were recorded by undertaking image analysis using ImageJ software. Axial measurements (Z‐axis) were used to assess the height from the center to the periphery on each side of the spheroid. Sphericity was calculated using:[ 49 ]
| (1) |
where a = major diameter, b = minor diameter and c = spheroid diameter in Z‐axis.
To investigate the distribution of different cell types within spheroids, labeled spheroids were embedded in optimal cutting temperature compound (OCT) and cryosectioned into 15 µm thick sections. Slides were visualized using a Leica TCS SP5 confocal microscope.
2.3. Gel‐MA Microsphere Fabrication and Characterization
Gel‐MA was synthesized as described previously.[ 26 ] Briefly, gelatin was dissolved in PBS (10 wt%, 50 °C) prior to addition of methacrylic anhydride to 60 wt% for 1 h. The product was dialyzed against deionised water and filtered through a 0.22 µm sterile filter; then lyophilized under sterile conditions.
Microspheres were generated using an adapted droplet microfluidics system, coupled with visible‐light photopolymerization as described previously.[ 28 , 35 , 41 ] Dried Gel‐MA (10% w/v) was dissolved in PBS at 37 °C and SKOV2 and HFF cells were encapsulated at 10 million cells per mL with 0.2 × 10‐3 m tris(2,2′‐bipyridyl)dichlororuthenium(II) hexahydrate (Ru) and 2 × 10‐3 m sodium persulfate (SPS). To allow for direct comparisons with spheroids, SKOV3 and HFF coculture microspheres were formed at percentage ratios of 75:25 (SKOV3:HFF). For droplet formation, a precursor solution containing cells was loaded into a 1 mL syringe, as the dispersed phase (flow rate 40 µL min‐1), and sunflower oil was used as the continuous phase (flow rate 1 mL min‐1) (Figure 1b). The microsphere droplets formed were then exposed to visible light to initiate photocrosslinking (OmniCure S1500, Excelitas Technologies with a Rosco IR/UV filter 400–450 nm: 3 min duration; 100 mW cm‐2).[ 27 ] Crosslinked microspheres were collected and washed twice in PBS for 5 min at 300 × g to remove excess oil. Microspheres were dispensed to single wells within 96‐well plates and cultured over 7 days.
To ensure formation of reproducible microspheres, the size distribution of SKOV3, HFF and coculture encapsulated microspheres was assessed. Fabricated microspheres (n = 50) were stained with Coomassie brilliant blue (Thermo Fisher Scientific, USA) for 15 min and imaged using a Zeiss Axioimager Z1 microscope. The major and minor diameters of the microspheres were recorded by undertaking image analysis using ImageJ software and the normal distribution was recorded.
2.4. Cell Viability, Metabolic Activity, and DNA Content Analysis
To assess cell viability within fabricated spheroids and microspheres, samples were washed with PBS and incubated with 1 × 10‐6 m propidium iodide and 1 × 10‐6 m calcein‐AM (Molecular Probes, USA) in PBS for 15 min at room temperature. The samples were washed with PBS and imaged on a fluorescent microscope (Zeiss Axioimager Z1 microscope, Germany).
To ensure consistent cell encapsulation and to monitor cell growth within microspheres, total DNA quantification was assessed on days 0, 7 and 12. Cell‐laden microspheres (n = 3, 4 microspheres/sample) were digested at 56 °C in 1 mg mL‐1 proteinase K dissolved in a 10−2 m Tris‐HCl and 10−3 m disodium EDTA solution, following treatment with DNase‐free RNase A. Total DNA was quantified using the CyQUANT Cell Proliferation Assay Kit according to manufacturer's instructions. A standard curve was constructed using monocultures of SKOV3 and HFF cells, and 75% SKOV3 and 25% HFF cocultures to accurately determine cell number.
The metabolic activity of encapsulated cells on day one was monitored and compared using an alamarBlue assay. Briefly, each sample was incubated with media containing 10% alamarBlue solution for 24 h at 37 °C. Absorbance readings at 570 and 600 nm were used to evaluate dye reduction rates according to manufacturer's instruction.
2.5. Scaffold Fabrication and Bioassembly of Tumor‐Scale Constructs
Biodegradable poly(ethylene glycol)‐terephthalate‐poly(butylene terephthalate) PEG‐PBT block copolymers (55:45 wt% PEGT:PBT and PEG at 300 g mol‐1) were used to fabricate porous scaffolds with a BioScaffolder system (SYS ENG, Germany) (Figure 1c). Fibers, with a diameter of ≈220 µm, were oriented in a repeating 0‐90°‐90°‐0° pattern in order to provide porosity in both the x‐y and z planes for assembly of 1 mm diameter modules.[ 41 ] PEG‐PBT was extruded using a computer‐aided syringe dispenser with a 25 G needle at a temperature of 200 °C, XY‐plane speed of 500 mm/min, auger speed 63 RPM and a fiber height offset of 0.22 mm. Constructs were fabricated with modifications depending on tumor module (Table 1 ) and were further processed in the x‐y plane to achieve 3×3 mm constructs (Figure 1c).
Table 1.
Assembled construct properties depending on tumor module
| Assembled Construct | ||
|---|---|---|
| Spheroid | Microsphere | |
| Dimensions | 3 × 3 × 2.64 mm | 3 × 3 × 1.76 mm |
| Fiber spacing | 0.7 mm | 1.0 mm |
| Fiber layers | 8 (1st layer); 4 (2nd layer) | 6 (1st layer); 2 (2nd layer) |
A custom‐made singularization device, developed in‐house and described previously,[ 41 ] was used for medium‐throughput bioassembly. To determine the reliability of the singularization device to successfully handle individual spheroids and microspheres, tumor modules (n = 10) were loaded into the singularization device and the success or failure in singularization was recorded over 10 cycles. The total number of successful singularization cycles was used to determine the overall efficiency of the singularization system.
To ensure the singularization device was benign and accurate for the deposition of a variety of diverse tumor modules, spheroid and microsphere constructs were assembled and compared to more laborious manual bioassembly. Spheroid and microsphere tumor modules were inserted via a layer‐by‐layer approach into thermoplastic PEG‐PBT scaffolds using the module injection head. For each of two layers, four tumor modules were inserted within the pores of the scaffold using the automated assembly system (Figure 1c). For manually assembled scaffolds, the whole scaffold was 3D plotted synchronously and the tumor modules were inserted manually into the pores and press‐fitted into place.
Spheroids (on day 7) and microspheres (on day 1) were assembled into scaffolds and cultured in 24‐well plates over 12 d. Live/dead staining was performed on days 1, 7 and 12 and metabolic activity was measured (as detailed above) on day 1 to assess cell survival.
2.6. Chemosensitivity Assays
To evaluate the chemosensitivity of the in vitro fabricated tumor models (spheroids, microspheres and assembled tumor‐scale constructs), cytotoxicity assays were undertaken with doxorubicin. As many previous studies have demonstrated the limited response of 3D ovarian cancer models to first‐line cytotoxic drugs, as a proof‐of‐concept an alternative anti‐cancer drug was modeled in this system.[ 50 , 51 , 52 ] Baseline cytotoxicity measurements were taken in 2D monoculture formats. Briefly, SKOV3s, HFFs and co‐culture (SKOV3:HFF 75:25) cells (31 600 cells cm‐2) were seeded in culture media, in 48‐well plates and incubated for 24 h before addition of drug. Spheroids (matured for 7 days), microspheres (on day 1) and assembled constructs were formed as described above (Section 2.3 and 2.4), and doxorubicin was added to triplicate samples at varying concentrations up to 10 × 10‐6 m (with a final concentration of 0.2% DMSO) and then incubated for 4 days in regular culture conditions (media and drug changed every 2 days) (Figure 2 ). DMSO‐only controls were used to normalize data (100% cell viability). Subsequently, the metabolic activity of encapsulated cells was assessed using the alamarBlue assay as described in Section 2.5. Reagent incubation times were 20 h for all 3D tumor models and 3.5 h for 2D culture. The cytotoxicity of doxorubicin was calculated using GraphPad Prism v9.0 (GraphPad Software, USA) and expressed in terms of IC50 values after normalizing to control cells. The IC50 values were calculated from log(inhibitor) dose–response curves generated by plotting cell viability, as a percent DMSO‐treated controls, against log10[drug] formulations.
Figure 2.
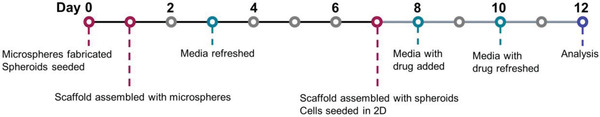
Experimental protocol and chronology.
2.7. Histology and Immunofluorescence Staining
Tumor models were fixed in 10% neutral buffered formalin for 1 h and immersed in 30% (w/v) sucrose in PBS at 4 °C overnight. Samples were embedded in OCT and cryosectioned into 15 µm thick sections. Slides were permeabilized with 0.25% (v/v) Triton X‐100 and blocked in 5% (w/v) BSA for 60 min at room temperature. Sections were stained with anti‐Ki67 (1:100 dilution; Abcam, USA) and anti‐gamma H2AX (1:5000 dilution; Abcam, USA) at 4 °C overnight and then incubated with goat Alexa Fluor 488 anti‐mouse (1:500 dilution; Thermofisher, USA) and goat Alexa Fluor 555 anti‐rabbit (1:500 dilution; Abcam, USA) at 37 °C for 1 h. Counterstaining was performed with 4.5 × 10‐6 m Hoechst 33342 (Invitrogen, USA) and the slides were washed with 0.05% Tween‐20 (Sigma, USA) and imaged using a Leica TCS SP5 confocal microscope. γ‐H2AX and Hoechst 33342‐pixel counts were performed using ImageJ and γ‐H2AX was expressed as the percentage of the pixel count of Hoechst 33342 staining.
2.8. Statistical Analysis
Data are expressed as mean ± standard deviation. Using GraphPad Prism v.9.0, statistical groups of data were compared by one‐way ANOVA or two‐way ANOVA (P < 0.05). Tukey's HSD tests were used to determine which groups accounted for any differences detected.
3. Results and Discussion
3.1. Developing Heterotypic Cell Interactions: Cancer Spheroid Formation and Evaluation
While replicating the cell populations present in solid tumors is important from a biological and treatment perspective,[ 53 , 54 ] the reproducibility and compatibility of the spheroids are also crucial for automated bioassembly of complex tumors and accurate drug efficacy studies.[ 41 , 55 ] To emulate the elaborate, cell‐dense, heterotypic tumor microenvironment with greater precision, monoculture and coculture ovarian carcinoma spheroids were fabricated and the sphericity and diameter of spheroids formed with differing cell ratios and seeding densities of SKOV3 and HFF cells was investigated. The sphericity of the spheroids formed was significantly increased to 0.71 with the addition of 25% HFF cells (P < 0.0001) and to 0.83 at 50% HFF cells (P < 0.01), with no significant differences detected at higher percent ratios (Figure 3a). To maximize the number of cancer cells within the tumor modules, further experiments were conducted using spheroids of 75:25 (SKOV3:HFF). Furthermore, the sphericity of the spheroids was highly consistent across replicates, providing confidence of the reproducibility of the formed spheroids using this method.[ 33 , 41 , 56 ]
Figure 3.
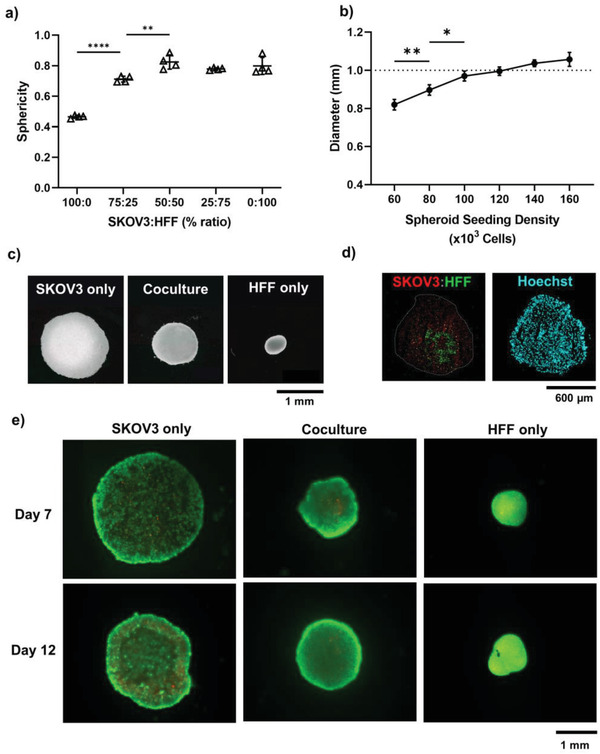
Generation of reproducible ovarian carcinoma spheroids on day 7 using the liquid overlay method. a) Sphericity of spheroids formed with varying percentage ratio of SKOV3 and HFF cells (n = 4). b) Coculture spheroid diameter formed with varying cell seeding density at fixed cell ratios (SKOV3:HFF 75:25) (n = 4). Dotted line at 1 mm represents optimal spheroid size. Error bars represent ± standard deviation. Significant differences are presented between neighboring values (**** p < 0.0001, ** p < 0.01, * p < 0.05). c) Darkfield images of monoculture and coculture spheroids seeded at 120000 cells/well. d) Cell distribution of SKOV3 (Qtracker 800, red) and HFF (Qtracker 655, green) within coculture spheroids. Total nuclei visible via Hoechst 33342 (blue). e) Live (Calcein‐AM, green) and dead (propidium iodide, red) staining of monoculture and coculture spheroids.
Modulating the cellular make‐up of the microspheres allowed for fine‐tuning of spheroid density and size. The HFF monocultures formed spheroids which were smaller with higher cell density, while the SKOV3 monocultures formed discoid, loosely‐packed spheroids (Figure 3c). The reduced capability of SKOV3 cells to form compact spheroids has been attributed to low E‐cadherin expression and low β1‐integrin levels,[ 57 , 58 , 59 ] and hence the highly contractile, cadherin‐expressing HFFs are able to provide increased tight‐junctions to achieve compact coculture spheroids.[ 60 ] As automated bioassembly systems rely on compact spheroidal modules; for further applications, incorporating cadherin‐expressing fibroblastic cell types into spheroids may be key to drive the necessary compression of less‐aggregating cell types.
The diameter of the spheroids (75:25, SKOV3:HFF) could be dynamically tuned by increasing the seeding density, resulting in a range of diameters from 0.82 to 1.06 mm (Figure 3b). Cocultures seeded at 120 000 cells per well reproducibly formed Ø1 mm microspheres which were used for the remaining experiments to make multi‐module cancer constructs (Figure 3b,c). Spherical, compact and reproducible spheroids are important for the successful operation of the automated bioassembly system, where previous studies have shown that spheroids close to the upper‐limit (Ø1 mm) of the device have lower rates of error during construct assembly.[ 41 ]
Assessment of the location of different cell populations within the coculture microspheres showed that the cell distribution was even throughout the spheroids, however, interestingly, HFF cells self‐assembled by migrating to the center of the spheroid while the SKOV3 cells formed a shell around this fibroblastic core (Figure 3d). The formation of a fibroblastic core minimizes free energy as cells with higher cohesion (e.g., higher cadherin expression—such as fibroblasts[ 60 ]) segregated to the middle—a phenomenon not seen with cell lines of similar expression levels.[ 61 , 62 ] Furthermore, SKOV3 DNA is reported to have high methylation of E‐cadherin, with hypoxia further reducing E‐cadherin expression[ 58 , 63 ]–explaining the tendency in our study for SKOV3 to form less compact spheroids and segregate to the outside of coculture spheroids (Figure 3d). This core–shell architectural arrangement of cancer and non‐malignant cells has been shown to regulate tumor cells invasiveness and may explain both the increased sphericity and compactness of the coculture spheroids, and the prevalent directed cell invasion onto the scaffolded construct as seen below in Section 3.3.[ 12 , 64 , 65 , 66 ]
Both the monoculture and coculture spheroids had high cell viability when cultured over 12 days, with dead cells present at the center of the spheroids in each group (Figure 3e). These non‐viable cell populations demonstrate the formation of a hypoxic core as in the native solid tumor microenvironment, whereby diffusion is insufficient to supply the central cells adequately.[ 13 , 54 , 67 ] The benefit of culturing large Ø1 mm spheroids described in this study, and that of larger bioassembled models, is that nutrient and gas exchange gradients are established automatically and better mimic that of the native tumor microenvironment,[ 13 , 54 ] where the outer cells continue to proliferate, migrate and disseminate away from the origin. This observation is supported by the expression of the Ki67 proliferation marker predominantly at the edge of SKOV3 monoculture and coculture spheroids (Figure S1, Supporting Information).
3.2. Introducing the Extracellular Matrix: Cancer Microsphere Fabrication and Evaluation
It has become increasingly known that the extracellular matrix and surrounding stromal cells produce major parts of the tumor matrix and provide the biochemical and biophysical cues that dictate cell behavior and shape the tumor microenvironment.[ 68 , 69 ] In recognition of this, Gel‐MA microspheres were rapidly fabricated using a microfluidics approach to represent a proxy tumor extracellular matrix, inaugurate cell–matrix interactions, and further establish biomolecular gradients. The monoculture and coculture microspheres were successfully optimized to achieve the Ø1 mm target that enables direct comparisons with cellular spheroid modules and for compatibility with downstream approaches, and had a narrow size distribution with a low coefficient of variation (Figure 4a,b). This low dispersion of microsphere size demonstrates the suitability of this microfluidic approach for rapid, high‐throughput production of reproducible multicellular microspheres.[ 28 ] Unlike the formation of spheroids, which cell‐permitting may take anywhere from 24 h to several days to form,[ 18 ] 500 microspheres could be fabricated in under 1 h using this microfluidic approach without reliance on the self‐assembly of cancer cells into compact spheres.
Figure 4.
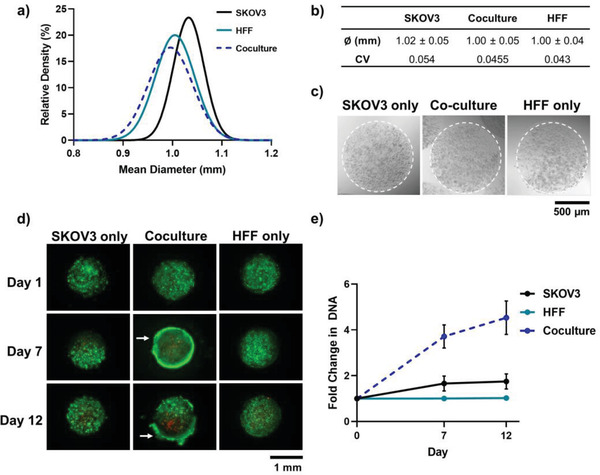
Generation of reproducible ovarian carcinoma Gel‐MA (10%) microspheres using a visible‐light microfluidic approach. a) Size distribution and b) mean diameter (± standard deviation) and coefficient of variation, of microspheres with encapsulated SKOV3 and HFF monocultures, or coculture cells (n = 50). c) Representative bright‐field images of monoculture and coculture microspheres on day 0. d) Live (Calcein‐AM, green) and dead (propidium iodide, red) staining of monoculture and coculture microspheres on days 1, 7 and 12. White arrows indicate positioning of high‐density cell carapace. e) Fold‐change in DNA content (n = 3) for spheroids cultured over 12 days. Error bars represent ± standard deviation.
The microsphere fabrication process was sufficiently precise to avoid significant differences in cell numbers between different cellular groups, with each microsphere containing an average of 5869 ± 281 cells (Table S1, Supporting Information). The HFF and SKOV3 monoculture cells were evenly distributed throughout the Gel‐MA hydrogel microspheres (Figure 4c) and showed high cell viability over 12 days of culture (Figure 4d). As the cells situated at the core of the microspheres remained viable, the Gel‐MA appeared to provide a highly‐hydrated environment which was more diffusion‐permissive than the dense and purely cellular spheroid modules, despite the large module diameters. However, in the coculture microspheres, while the cells were evenly distributed throughout the microsphere on day 1 (Figure S2, Supporting Information), a high cell‐density shell was formed around the periphery of the microspheres at day 7, with a concurrent increase in non‐viable cells at the core (Figure 4d). This resultant cell carapace may be accredited to heightened cancer cell migration towards normoxic and nutrient‐rich areas within the microspheres.[ 70 ] These enhanced migration capabilities may have been enabled by both extracellular matrix and fibroblastic interactions,[ 12 , 53 , 65 , 71 ] and have caused amplification of biomolecular gradients and the formation of a necrotic zone unseen in other conditions.[ 54 , 67 , 72 ] Nevertheless, coculture microspheres continued to grow over 12 days, with a 4.5‐fold increase in DNA content from day 0 (Figure 4d). SKOV3 monoculture microspheres grew 2.8‐fold slower than the coculture microspheres, and HFF monoculture microspheres showed no increase in DNA content over time (Figure 4d). In support of these findings, there is abundant evidence of fibroblasts causing activation and stimulating the proliferation of tumor cells through paracrine signaling.[ 53 , 65 , 73 , 74 ] Interestingly, despite large differences in DNA content and a clear increase in cell number at the microsphere edge, Ki67 markers were localized to the bulk of the microspheres (not the periphery) with equal expression between SKOV3 monoculture and coculture microspheres (Figure S1, Supporting Information). The lack of cell proliferation at the edge of the microsphere is consistent with studies that show greater Ki67 in areas of lower confluence rather than highly‐confluent monolayers.[ 75 ]
3.3. Scaling up: Fabrication and Automated 3D Bioassembly of Tumor‐Scale Models
There are many salient features of the tumor microenvironment that are desirable to replicate when developing in vitro cancer models for drug screening. A multi‐module construct which can be assembled using automated processes will allow for higher‐throughput testing of these complex tumor models at more biologically‐relevant sizes. For the formation of large tumor constructs (i.e., representative of native tumor‐scale), a custom‐made singularization device was utilized for assembly of smaller spheroid and microsphere modules into a bioprinted PEG‐PBT support scaffold framework (Figure 5a).[ 41 ] The custom‐made injection device had high singularization efficiency of 97% and 93% for tumor spheroids and microspheres respectively (Table 2 ) and was comparable to previous studies with chondrocyte spheroids.[ 41 ] The average insertion efficiency was higher for microspheres (≈96%) compared with spheroids (≈60%), which demonstrated a large decrease in efficiency with the second layer (≈33%) (Table 2). The insertion efficiency of the spheroids was 19.2% lower than previously observed,[ 41 ] and these compounding layer errors may be due to the lower sphericity than cartilage spheroids.[ 33 ] Further optimization of the insertion head throughout the z‐plane may increase the efficiency and reduce the error reported in both spheroid and microsphere insertion in ensuing layers.
Figure 5.
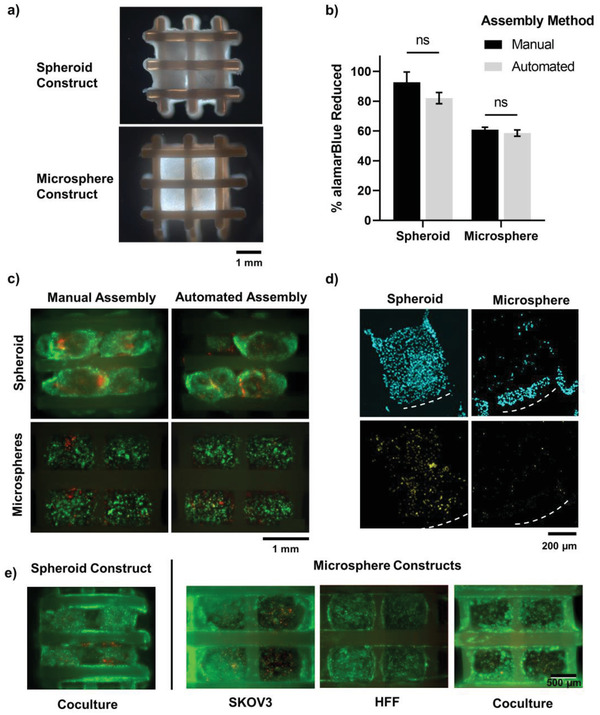
Ovarian carcinoma coculture construct bioassembly into PEGT:PBT scaffolds. a) Darkfield images of spheroid and microsphere bioassembled constructs. b) Metabolic activity (alamarBlue reduction) of manual and automated bioassembled ovarian cancer tumor modules (day 1; n = 3). Errors bars denote ± standard deviation. c) Live (Calcein‐AM, green) and dead (propidium iodide, red) staining of bioassembled SKOV3:HFF coculture spheroids microspheres using manual and automated bioassembly (day 1). d) Immunofluorescence imaging of Ki67 expression (yellow) and cell nuclei (blue) in spheroid and microsphere assembled constructs. e) Live (Calcein‐AM, green) and dead (propidium iodide, red) staining of bioassembled constructs cultured long‐term (day 12) in vitro.
Table 2.
Singularization (n = 100) and insertion efficiency (n = 6 scaffolds, 8 modules per scaffold) of spheroid and microsphere modules (mean ± standard deviation)
| Spheroids | Microspheres | |
|---|---|---|
| Singularization efficiency | 97% ± 4.8 | 93% ± 6.8 |
| Insertion efficiency | ||
| • First Layer | 87.5% ± 13.7 | 100.0% ± 0.0 |
| • Second Layer | 33.3% ± 25.8 | 91.7% ± 12.9 |
| • Both layers | 60.4% ± 12.3 | 95.8% ± 6.5 |
Due to the aspherical and inconsistently sized monoculture spheroids, only coculture spheroids were compatible with the automated bioassembly approach. This approach surpassed the manual approach as it maintained cellular health during bioassembly. As expected, there was no significant difference in the metabolic activity of coculture spheroids or microspheres with the manual or automated assembly strategy (Figure 5b). After 24 h of culture, high cell viability was detected throughout the constructs, however distinct regions of non‐viable cells were visible in the manual assembly of both microspheres and spheroids loaded into the scaffolds (Figure 5c). These distinct regions of cell death were consistent with areas compressed or pinched by insertion forceps during manual assembly, and highlight the superiority of the automated system for not only time‐efficiency and throughput, but specifically for maintaining cellular health during 3D bioassembly of larger tumor‐scale cancer constructs. This is a particularly important consideration for future bioassembled models containing sensitive cell types, such as adipocytes[ 76 ] in breast cancer models.
Over culture time, the assembled spheroids matured and fused to fill the square scaffold pores, with clear evidence of cell proliferation throughout (Figure 5d and Figure S4, Supporting Information). This change in shape and fusion of spheroids has been shown in previous studies,[ 41 , 44 ] and is consistent with the cancer cells migrating towards vacant space. The assembled microspheres retained their spherical shape due to the crosslinked Gel‐MA support material, with cells migrating to the surface of the sphere and onto the PEG‐PBT scaffold, which has been shown extensively to support cell adhesion, proliferation and differentiation in multiple cell and stromal cell populations,[ 77 , 78 , 79 ] resulting in more dispersed cell proliferation staining (Figure 5d,e). Migration of cells onto the thermoplastic PEG‐PBT scaffold fibers was expected from previous studies and is beneficial for the fusion of individual tumor modules in future applications.[ 41 , 72 ] After 12 days of culture both the automated assembled spheroid and microsphere constructs had high viability with cells migrating throughout the whole construct and onto PEG‐PBT support scaffold (Figure 5e).
3.4. Testing the Model: Cytotoxic Efficacy of Doxorubicin
Having developed an automated modular bioassembly system to fabricate tumor‐scale cancer models, proof‐of‐concept drug efficacy studies were performed with a common clinically adopted chemotherapy drug—doxorubicin. Comparisons were made across single module types (i.e., spheroids v. microspheres), assembled constructs and 2D cultured cell controls; comparing both monoculture and coculture conditions (Figure 6 ; Figure S3, Supporting Information). In general, HFF monocultures had higher IC50 values and lessened the effect of doxorubicin on heterologous coculture models (Figure 6a,c; Figure S3, Supporting Information). Furthermore, there were no increases in DNA double‐strand breaks (γ‐H2AX) in HFF monoculture spheroids at any concentration of doxorubicin (Figure 7a). The resistance of HFF monoculture models to doxorubicin is expected as the anthracycline compound targets replicating cells by intercalating with DNA and inhibiting topoisomerase II,[ 80 ] and Ki67 staining showed low levels of proliferation in both HFF spheroids and microspheres (Figure S1, Supporting Information). Furthermore, fibroblasts are known to induce drug resistance in vitro and in vivo,[ 55 , 71 , 81 , 82 ] both through cell signaling and secretion of extracellular matrix molecules which may prevent drug penetration throughout the tumor modules.[ 83 , 84 , 85 ] However, these data may also be explained by the proportion of HFFs (25%) present in the tumor module.
Figure 6.
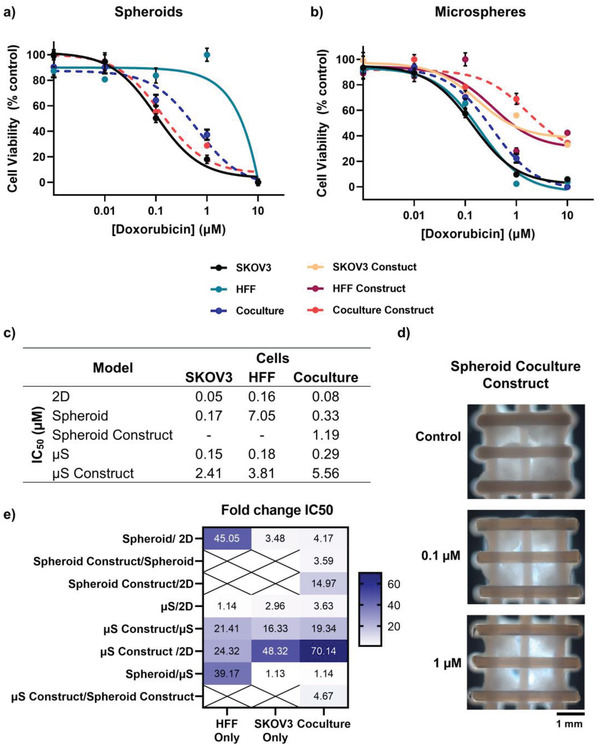
Chemosensitivity of ovarian carcinoma in vitro models to doxorubicin. Dose–response curves of cell viability within fabricated a) spheroids and b) microspheres. Error bars represent ± standard deviation. c) IC50 values of doxorubicin in monoculture and coculture models. d) Darkfield images of assembled coculture construct imaged after 4 days of exposure to doxorubicin. e) Fold‐change in IC50 between different in vitro cancer models. Color intensity denotes degree of fold‐change. Crossed boxes highlight where comparisons could not be made because of an inability to form monoculture assembled constructs. µT = spheroid, µS = microsphere.
Figure 7.
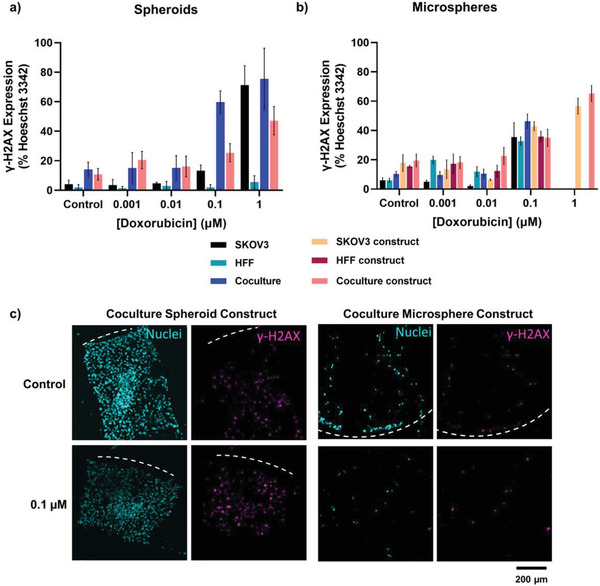
DNA double‐stranded break analysis in doxorubicin treated ovarian cancer models. Pixel count analysis of γ‐H2AX as a percentage of Hoechst 33342 expression for models and constructs containing a) spheroids and b) microspheres. Error bars represent ± standard deviation. c) Representative immunofluorescence images of γ‐H2AX expression (pink) and cell nuclei (blue) in spheroid and microsphere assembled constructs.
While traditional 2D culture can provide high‐throughput screening capacity to progress experiments toward higher complexity formats, they are not representative of the pathophysiology of native tumors and, hence, it is well established that the efficacy and potency of drugs differ depending on the in vitro model used.[ 85 , 86 , 87 , 88 , 89 , 90 ] In this study, 3D models had higher IC50 values compared with 2D models, and all bioassembled constructs had higher IC50 values than their respective individual module counterparts (Figure 6a,b,e). Interestingly, without drug treatment the coculture spheroids and their bioassembled counterparts had the highest number of DNA double‐strand breaks and these (with all other module/construct groups) increased from <0.1 × 10‐6 m doxorubicin incubation as expected (Figure 7a,c and Figure S4, Supporting Information). Similar trends were observed for bioassembled microsphere constructs (Figure 7b). These differences observed in the spheroid and microsphere constructs, may be due to a range of biological mechanisms including the increased complexity of the tumor microenvironment (including the formation of biomolecule gradients), cell morphology differences and architectural organization changes, reduced proliferation rates in 3D culture systems and poor drug penetration throughout the large diameter and tumor‐scale model.[ 13 , 85 , 87 , 88 , 89 , 91 ] However, both individual spheroid and microsphere modules had similar IC50 values at approximately 0.3 × 10‐6 m with moderate DNA‐damage (Figures 6c,e and 7a,b), suggesting that the equal size of the tumor modules may result in similar drug penetration and efficacy despite the difference of an encapsulating extracellular matrix component.
When a bioplotted scaffold was introduced to form the assembled constructs, microspheres had a ≈4.7‐fold higher IC50 than the spheroids (Figure 6c,e). This may be due to the greater migration of cells from the spheroids onto the scaffold whereby an altered morphology was adopted, with greater exposure to doxorubicin than when encapsulated in microspheres.[ 89 ] This reduced cell density in response to increasing doxorubicin concentration was visible throughout the bioassembled spheroid culture construct (Figure 6d and Figure S4, Supporting Information). Overall, a step‐wise increase in tumor microenvironment complexity (through incorporation of extracellular matrix and bioassembly into a scaffold) resulted in vastly different drug efficacy and IC50 curves compared with traditionally‐adopted 2D culture (Figure 6e). Furthermore, these bioassembled tumor‐scale models have since been further applied within in a microphysiological bioreactor perfusion system to improve the tumor microenvironment model by recreating drug diffusion gradients at relevant blood flow rates.[ 92 ] The overall approach taken here reaffirms the utility of modular tissue engineering approaches, involving medium‐throughput bioassembly, automated biofabrication and convergence with microphysiological/perfusion systems,[ 93 ] for developing rapid tumor‐scale models of the tumor microenvironment to expedite drug discovery and personalized medicine.
4. Conclusions
This study demonstrates the advantage of using an automated and integrated biofabrication system for the development of modular constructs toward biologically relevant sizes, which mimic the complex extracellular organization and architecture of the tumor microenvironment. Heterotypic coculture spheroid (cellular) and microsphere (containing hydrogel matrix) modules (Ø1 mm) were reproducibly fabricated and bioassembled to form a multilayer tumor‐scale cancer construct. Microsphere modules could be rapidly generated using a range of cell types and demonstrated higher‐throughput fabrication compared with spheroids. By modulating the cell populations and matrix molecules present in this model, the tumor microenvironment could be further fine‐tuned to mimic the biology of in vivo tumors—resulting in the formation of biomolecular gradients, varied cell densities and quiescent and necrotic regions. Doxorubicin chemosensitivity assays conducted on the assembled spheroid and microsphere constructs showed decreased drug sensitivity in cancer‐fibroblast coculture modules, and with increasing complexity and size of the tumor model (from 2D to individual 3D spheroids to complex tumor‐scale bioassembled construct). Given our oncological model can be fabricated in a medium‐throughput and flexible fashion, there is the potential for the development of a large number of tumor constructs (e.g., with cell types compartmentalized in separate modules) for mechanistic studies or for improved pre‐clinical screening.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
N.V.M. and G.M. contributed equally to this work. The authors acknowledge funding from the Royal Society of New Zealand Rutherford Discovery Fellowship (RDF‐UOO1204; T.W.), the AO Foundation (S‐08‐81 W; T.W.), the New Zealand Health Research Council (Sir Charles Hercus Fellowship 19/135, K.L.), and the Medical Technologies Centre of Research Excellence (MedTech CoRE). The authors also acknowledge Dr. Jonathon Parrish and Assoc Prof Margaret Currie for their scientific discussions and technical input to the manuscript.
Open access publishing facilitated by University of Otago, as part of the Wiley ‐ University of Otago agreement via the Council of Australian University Librarians.
Mekhileri N. V., Major G., Lim K., Mutreja I., Chitcholtan K., Phillips E., Hooper G., Woodfield T., Biofabrication of Modular Spheroids as Tumor‐Scale Microenvironments for Drug Screening. Adv. Healthcare Mater. 2023, 12, 2201581. 10.1002/adhm.202201581
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Smietana K., Siatkowski M., Møller M., Nat. Rev. Drug Discovery 2016, 15, 379. [DOI] [PubMed] [Google Scholar]
- 2. Wong C. H., Siah K. W., Lo A. W., Biostatistics 2019, 20, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Langhans S. A., Front. Pharmacol. 2018, 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hay M., Thomas D. W., Craighead J. L., Economides C., Rosenthal J., Nat. Biotechnol. 2014, 32, 40. [DOI] [PubMed] [Google Scholar]
- 5. Asghar W., El Assal R., Shafiee H., Pitteri S., Paulmurugan R., Demirci U., Mater. Today 2015, 18, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Micek H. M., Visetsouk M. R., Masters K. S., Kreeger P. K., iScience 2020, 23, 101742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meng F., Meyer C. M., Joung D., Vallera D. A., McAlpine M. C., Adv. Mater. 2019, 31, 1806899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Swaminathan S., Hamid Q., Sun W., Clyne A. M., Biofabrication¸2019, 11, 025003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mao S., Pang Y., Liu T., Shao Y., He J., Yang H., Mao Y., Sun W., Biofabrication 2020, 12, 042001. [DOI] [PubMed] [Google Scholar]
- 10. Gadde M., Phillips C., Ghousifam N., Sorace A. G., Wong E., Krishnamurthy S., Syed A., Rahal O., Yankeelov T. E., Woodward W. A., Rylander M. N., Biotechnol. Bioeng. 2020, 117, 3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang M., Tiwari S. K., Agrawal K., Tan M., Dang J., Tam T., Tian J., Wan X., Schimelman J., You S., Xia Q., Rana T. M., Chen S., Small 2021, 17, 2006050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang Y. L., Shiau C., Wu C., Segall J. E., Wu M., Biology S., Biophys. Rev. Lett. 2020, 15, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kunz‐Schughart L. A., Freyer J. P., Hofstaedter F., Ebner R., J. Biomol. Screening 2004, 9, 273. [DOI] [PubMed] [Google Scholar]
- 14. Fiorini E., Veghini L., Corbo V., Front. Cell Dev. Biol. 2020, 8, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maru Y., Tanaka N., Itami M., Hippo Y., Gynecol. Oncol. 2019, 154, 189. [DOI] [PubMed] [Google Scholar]
- 16. Girda E., Huang E. C., Leiserowitz G. S., Smith L. H., Int. J. Gynecol. Cancer 2017, 27, 1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu J., Huang W., Int. J. Stem Cells 2020, 13, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin R. Z., Chang H. Y., Biotechnol. J. 2008, 3, 1172.18566957 [Google Scholar]
- 19. Wang C., Sinha S., Jiang X., Murphy L., Fitch S., Wilson C., Grant G., Yang F., Tissue Eng., Part A 2020, 27, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koch M. K., Jaeschke A., Murekatete B., Ravichandran A., Tsurkan M., Werner C., Soon P., Hutmacher D. W., Haupt L. M., Bray L. J., Acta Biomater. 2020, 114, 256. [DOI] [PubMed] [Google Scholar]
- 21. Geiger F., Rüdiger D., Zahler S., Engelke H., PLoS One 2019, 14, e0225215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J., Tan Y., Zhang H., Zhang Y., Xu P., Chen J., Poh Y.‐C., Tang K., Wang N., Huang B., Nat. Mater. 2012, 11, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J., Zhou Y., Chen W., Yuan Z., You B., Liu Y., Yang S., Li F., Qu C., Zhang X., ACS Appl. Mater. Interfaces 2018, 10, 36641. [DOI] [PubMed] [Google Scholar]
- 24. Vasudevan J., Lim C. T., Fernandez J. G., Adv. Funct. Mater. 2020, 30, 2005383. [Google Scholar]
- 25. Carvalho M. R., Lima D., Reis R. L., Correlo V. M., Oliveira J. M., Trends Biotechnol. 2015, 33, 667. [DOI] [PubMed] [Google Scholar]
- 26. Van Den Bulcke A. I., Bogdanov B., De Rooze N., Schacht E. H., Cornelissen M., Biomacromolecules 2000, 1, 31. [DOI] [PubMed] [Google Scholar]
- 27. Lim K. S., Schon B. S., Mekhileri N. V., Brown G. C. J., Chia C. M., Prabakar S., Hooper G. J., Woodfield T. B. F., ACS Biomater. Sci. Eng. 2016, 2, 1752. [DOI] [PubMed] [Google Scholar]
- 28. Young C., Rozario K., Serra C., Poole‐Warren L., Martens P., Biomicrofluidics 2013, 7, 044109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Serra C., Berton N., Bouquey M., Prat L., Hadziioannou G., Langmuir 2007, 23, 7745. [DOI] [PubMed] [Google Scholar]
- 30. Horvath L. E., Werner T., Boucher K., Jones K., Med. Hypotheses 2013, 80, 684. [DOI] [PubMed] [Google Scholar]
- 31. Kasangian A. A., Gherardi G., Biagioli E., Torri V., Moretti A., Bernardin E., Cordovana A., Farina G., Bramati A., Piva S., Dazzani M. C., Paternã2 E., La Verde N. M., PLoS One 2017, 12, e0189127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown P. O., Palmer C., PLoS Med. 2009, 6, e1000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schon B. S., Schrobback K., Der V. M. V., Stroebel S., Hooper G. J., Woodfield T. B. F, Cell Tissue Res. 2012, 347, 629. [DOI] [PubMed] [Google Scholar]
- 34. Kelm J. M., Djonov V., Ittner L. M., Fluri D., Born W., Hoerstrup S. P., Fussenegger M., Tissue Eng. 2006, 12, 2151. [DOI] [PubMed] [Google Scholar]
- 35. Cui X., Alcala‐Orozco C. R., Baer K., Li J., Murphy C. A., Durham M., Lindberg G., Hooper G. J., Lim K. S., Woodfield T. B. F., Biofabrication 2022, 14, 034101. [DOI] [PubMed] [Google Scholar]
- 36. Dalton P. D., Woodfield T. B. F., Mironov V., Groll J, Adv. Sci. 2020, 7, 1902953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yeh J., Ling Y., Karp J. M., Gantz J., Chandawarkar A., Eng G., Blumling J. III, Langer R., Khademhosseini A., Biomaterials 2006, 27, 5391. [DOI] [PubMed] [Google Scholar]
- 38. Du Y., Lo E., Ali S., Khademhosseini A., Proc. Natl. Acad. Sci. USA 2008, 105, 9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leung B. M., Sefton M. V, Tissue Eng., Part A 2010, 16, 3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Babur B. K., Futrega K., Lott W. B., Klein T. J., Cooper‐White J., Doran M. R., Cell Tissue Res. 2015, 361, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mekhileri N. V., Lim K. S., Brown G. C. J., Mutreja I., Schon B. S., Hooper G. J., Woodfield T. B. F., Biofabrication 2018, 10, 024103. [DOI] [PubMed] [Google Scholar]
- 42. Dikina A. D., Strobel H. A., Lai B. P., Rolle M. W., Biomaterials 2015, 52, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Itoh M., Nakayama K., Noguchi R., Kamohara K., Furukawa K., Uchihashi K., Toda S., Oyama H., Node K., Morita S., PLoS One 2015, 10, e0145971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindberg G. C. J., Cui X., Durham M., Veenendaal L., Schon B. S., Hooper G. J., Lim K. S., Woodfield T. B. F., Adv. Sci. 2021, 8, 2103320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rago A. P., Dean D. M., Morgan J. R., Biotechnol. Bioeng. 2009, 102, 1231. [DOI] [PubMed] [Google Scholar]
- 46. Bruzewicz D. A., McGuigan A. P., Whitesides G. M., Lab Chip 2008, 8, 663. [DOI] [PubMed] [Google Scholar]
- 47. Chen P., Luo Z., Güven S., Tasoglu S., Ganesan A. V., Weng A., Demirci U., Adv. Mater. 2014, 26, 5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Parrish J., Lim K., Zhang B., Radisic M., Woodfield T. B. F., Trends Biotechnol. 2019, 37, 1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krumbein W. C., J Sedimentry Petrol. 1941, 11, 64. [Google Scholar]
- 50. Yee C., Dickson K. A., Muntasir M. N., Ma Y., Marsh D. J., Front. Bioeng. Biotechnol. 2022, 10, 836984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raghavan S., Ward M. R., Rowleym K. R., Wold R. M., Takayama S., Buckanovich R. J., Mehta G., Gynecol. Oncol. 2015, 138, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nowacka M., Sterzynska K., Andrzejewska M., Nowicki M., Januchowski R., Biomed. Pharmacother. 2021, 138, 111536. [DOI] [PubMed] [Google Scholar]
- 53. Sun Y., Fan X., Zhang Q., Shi X., Xu G., Zou C., Tumor Biol. 2017, 39. [DOI] [PubMed] [Google Scholar]
- 54. Cui X., Hartanto Y., Zhang H., J. R. Soc., Interface 2017, 14, 20160877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bu L., Baba H., Ishimoto T., Cancer Sci. 2020, 1111, 3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lindberg G. C. J., Longoni A., Lim K. S., Rosenberg A. J., Hooper G. J., Gawlitta D., Woodfield T. B. F., Acta Biomater. 2019, 85, 117. [DOI] [PubMed] [Google Scholar]
- 57. Sodek K. L., Ringuette M. J., Brown T. J., Int. J. Cancer 2009, 124, 2060. [DOI] [PubMed] [Google Scholar]
- 58. Yuecheng Y., Hongmei Æ. L., Xiaoyan Æ. X., Clin. Exp. Metastasis 2006, 23, 65. [DOI] [PubMed] [Google Scholar]
- 59. Ivascu A., Kubbies M., Int. J. Oncol. 2007, 31, 1403. [PubMed] [Google Scholar]
- 60. Matsuyoshi N., Imamura S., Biochem. Biophys. Res. Commun. 1997, 235, 355. [DOI] [PubMed] [Google Scholar]
- 61. Napolitano A. P., Chai P., Dean D. M., Morgan J. R., Tissue Eng. 2007, 13, 2087. [DOI] [PubMed] [Google Scholar]
- 62. Foty R. A., Steinberg M. S., Int. J. Dev. Biol. 2004, 48, 397. [DOI] [PubMed] [Google Scholar]
- 63. Imai T., Horiuchi A., Wang C., Am. J. Pathol. 2003, 163, 1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lazzari G., Nicolas V., Matsusaki M., Akashi M., Couvreur P., Mura S., Acta Biomater. 2018, 78, 296. [DOI] [PubMed] [Google Scholar]
- 65. Plaster M., Singh S., Tavana H., Adv. Ther. 2019, 2, 1900121. [Google Scholar]
- 66. Tsai C. W., Wang J. H., Young T. H., Artif. Cells, Nanomed., Biotechnol. 2018, 46, 651. [DOI] [PubMed] [Google Scholar]
- 67. Thoma C. R., Zimmermann M., Agarkova I., Kelm J. M., Krek W., Adv. Drug Delivery Rev. 2014, 69‐70, 29. [DOI] [PubMed] [Google Scholar]
- 68. Elgundi Z., Papanicolaou M., Major G., Cox T. R., Melrose J., Whitelock J. M., Farrugia B. L., Front. Oncol. 2020, 9, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pearce O. M. T., Delaine‐Smith R. M., Maniati E., Nichols S., Wang J., Rajeeve V., Ullah D., Chakravarty P., Jones R. R., Montfort A., Dowe T., Gribben J., Jones J. L, Kocher H. M., Serody J. S., Vincent B. G., Connelly J., Brenton J. D., Chelala C., Cutillas P. R., Lockley M., Bessant C., Knight M. M., Balkwill F. R., Cancer Discovery 2018, 8, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gilmore A. C., Flaherty S. J., Somasundaram V., Scheiblin D. A., Lockett S. J., Wink D. A., Heinz W. F., Commun. Biol. 2021, 4, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tao L., Huang G., Song H., Chen Y., Chen L., Oncol. Lett. 2017, 14, 2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Malda J., Woodfield T. B. F., Van Der Vloodt F., Wilson C., Martens D. E., Tramper J., van Blitterswijk C. A., Riesle J., Biomaterials 2005, 26, 63. [DOI] [PubMed] [Google Scholar]
- 73. Subramaniam K. S., Tham S. T., Mohamed Z., Woo Y. L., PLoS One 2013, 8, e68923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Izar B., Joyce C. E., Goff S., Cho N. L., Shah P. M., Sharma G., Li J., Ibrahim N., Gold J., Hodi F. S., Garraway L. A., Novina C. D., Pigm. Cell Melanoma Res. 2016, 29, 656. [DOI] [PubMed] [Google Scholar]
- 75. Endl E., Steinbach P., Knüchel R., Hofstädter F., Cytometry 1997, 29, 233. [PubMed] [Google Scholar]
- 76. Huber B., Borchers K., Tovar G. E. M., Kluger P. J., J. Biomater. Appl. 2016, 30, 699. [DOI] [PubMed] [Google Scholar]
- 77. Woodfield T. B. F., Miot S., Martin I., Van Blitterswijk C. A., Riesle J., Biomaterials 2006, 27, 1043. [DOI] [PubMed] [Google Scholar]
- 78. Woodfield T. B. F., Van Blitterswijk C. A., De Wijn J., Sims T. J., Hollander A. P., Riesle J., Tissue Eng. 2005, 11, 1297. [DOI] [PubMed] [Google Scholar]
- 79. Woodfield T. B. F., Malda J., De Wijn J., Péters F., Riesle J., Van Blitterswijk C. A, Biomaterials 2004, 25, 4149. [DOI] [PubMed] [Google Scholar]
- 80. Nicoletto R. E., Ofner C. M., Cancer Chemother. Pharmacol. 2022, 89, 285. [DOI] [PubMed] [Google Scholar]
- 81. Tiago M., de Oliveira E. M., Brohem C. A., Pennacchi P. C., Paes R. D., Haga R. B., Campa A., de Moraes Barros S. B., Smalley K. S., Maria‐Engler S. S., Tissue Eng., Part A 2014, 20, 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li L., Lu Y., J. Cancer 2011, 2, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Netti P. A., Berk D. A., Swartz M. A., Grodzinsky A. J., Jain R. K., Cancer Res. 2000, 60, 2497. [PubMed] [Google Scholar]
- 84. Kalluri R., Zeisberg M., Nat. Rev. Cancer 2006, 6, 392. [DOI] [PubMed] [Google Scholar]
- 85. Lovitt C. J., Shelper T. B., Avery V. M., BMC Cancer 2018, 18, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shamir E. R., Ewald A. J., Nat. Rev. Mol. Cell Biol. 2014, 15, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Patra B., Lateef M. A., Brodeur M. N., Provencher D., Fleury H., Carmona E., Péant B., Provencher D., Mes‐Masson A.‐M., Gervais T., PLoS One 2020, 15, e0244549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Brodeur M. N., Simeone K., Leclerc‐Deslauniers K., Fleury H., Carmona E., Provencher D. M., Mes‐Masson A.‐M., Sci. Rep. 2021, 11, 18183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Breslin S., Driscoll L. O., Oncotarget 2016, 7, 45745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhao Y., Yao R., Ouyang L., Ding H., Zhang T., Zhang K., Cheng S., Sun W., Biofabrication 2014, 6, 035001. [DOI] [PubMed] [Google Scholar]
- 91. Minchinton A. I., Tannock I. F, Nat. Rev. Cancer 2006, 6, 583. [DOI] [PubMed] [Google Scholar]
- 92. Parrish J., Lim K. S., Baer K., Hooper G. J., Lab Chip 2018, 18, 2757. [DOI] [PubMed] [Google Scholar]
- 93. Parrish J., Lim K., Zhang B., Radisic M., Woodfield T. B. F., Trends Biotechnol. 2019, 37, 1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.