

Abstract
Ferroptosis as programmed cell death received considerable attention in cancer research. Recently, studies have associated ferroptosis with photodynamic therapy (PDT) because PDT promotes glutathione (GSH) deletion, glutathione peroxidase 4 (GPX4) degradation, and lipid peroxide accumulation. However, PDT‐induced ferroptosis may be potentially prevented by ferroptosis suppressor protein 1 (FSP1). To address this limitation, herein, a novel strategy is developed to trigger ferroptosis by PDT and FSP1 inhibition. For enhancement of this strategy, a photoresponsive nanocomplex, self‐assembled by BODIPY‐modified poly(amidoamine) (BMP), is utilized to stably encapsulate the inhibitor of FSP1 (iFSP1) and chlorin e6 (Ce6). The nanosystem promotes intracellular delivery, penetration, and accumulation of ferroptosis inducers in tumors with light irradiation. The nanosystem presents high‐performance triggering of ferroptosis and immunogenic cell death (ICD) in vitro and in vivo. Importantly, the nanoparticles increase tumor infiltration of CD8+ T cells and further enhance the efficacy of anti‐PD‐L1 immunotherapy. The study suggests the potential of photo‐enhanced synergistic induction of ferroptosis by the photoresponsive nanocomplexes in cancer immunotherapy.
Keywords: BODIPY, charge reversal, ferroptosis, immunotherapy, PAMAM, photodynamic therapy
A photoresponsive nanocarrier is developed to enhance intracellular accumulation of chlorin e6 and inhibitor of ferroptosis suppressor protein 1 (iFSP1) based on light‐induced charge reversal of the nanocarrier. Upon light irradiation, photodynamic therapy and FSP1 inhibition synergistically promote ferroptosis and immunogenic cell death through lipid peroxidation, thereby stimulating the immune system against cancer cells.
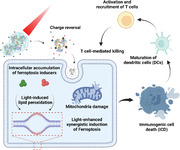
1. Introduction
With improvements in screening, treatment, and prevention, survival rates of cancer patients are improving. However, many types of cancer are still lethal and faced with poor prognosis because of therapy resistance and lack of effective therapeutics.[ 1 ] Therefore, there is still an urgent need to explore novel therapeutics to overcome such issues. Ferroptosis is a form of regulated necrotic cell death marked by oxidative damage to phospholipids, which impairs cell structure and integrity.[ 2 ] Extensive studies show that ferroptosis plays a vital role in tumor suppression, providing new possibilities for cancer therapy.[ 3 ]
Besides tumor suppression, recent studies have revealed that ferroptosis is directly immunogenic and triggers inflammatory responses in the tumor microenvironment.[ 4 ] Release of damage‐associated molecular patterns (DAMPs) caused by ferroptosis can induce maturation of dendritic cells and the cross‐induction of CD8+ T cells, thereby activating the adaptive immune system to mediate anti‐tumor immunity.[ 5 ] Therefore, combining ferroptosis and immunotherapy is promising to enhance anti‐cancer therapeutic efficacy.
Glutathione peroxidase 4 (GPX4), one of the key regulators of ferroptosis, is identified as a phospholipid hydroperoxide glutathione peroxidase that reduces membrane‐bound lipid peroxides by consuming glutathione.[ 6 ] Previously, GPX4 was regarded as the primary ferroptosis protector. Therefore, most of ferroptosis‐based anti‐cancer strategies focused on inhibiting or depleting GPX4.[ 7 ] Triggering ferroptosis through external stimuli, such as light and ultrasound, received much attention since side effects can be avoided by applying stimuli at the specific tumor sites.[ 8 ] Among them, chlorin e6 (Ce6)‐mediated photodynamic therapy (PDT) demonstrated high‐efficiency GSH depletion, lipid peroxide production, and GPX4 degradation to induce ferroptosis.[ 7 , 9 ] However, inhibition of GPX4 fails to trigger ferroptosis in some cancer cell lines.[ 10 ] Discovery of ferroptosis suppressor protein 1 (FSP1) started a new chapter for ferroptosis‐based anti‐cancer therapy.
FSP1, also named AIFM2, cooperates with GPX4 to suppress lipid peroxidation (LPO) as an independent parallel mediator. In the FSP1 pathway, LPO is prevented via reduction of ubiquinone to ubiquinol or regenerating oxidized vitamin E to its non‐radical form.[ 10 , 11 ] Inhibition or loss of FSP1 was found to cause limited cell death but overcome resistance to GPX4 inhibitors in cancer cells.[ 10 , 11 ] No current study was designed to explore the potential synergistic effect between PDT and FSP1 inhibition to efficiently induce ferroptosis.
Herein, we hypothesize that co‐delivery of iFSP1 and Ce6 could mediate photo‐triggered synergistic ferroptosis. BODIPY‐modified poly(amidoamine) (PAMAM) nanosystems[ 12 ] were utilized for photoresponsive and targeted delivery of ferroptosis inducers. Under light irradiation, the nanosystems promoted intracellular delivery of cargo drugs in vitro and drug accumulation in tumors in vivo. Importantly, nanosystems loaded with both Ce6 and iFSP1 were more efficient than those loaded with only Ce6 in inducing ferroptosis in vitro and retarding tumor growth in vivo. Additionally, the strategy led to immunogenic cell death (ICD), further activating the adaptive immune system and enhancing efficacy of anti‐PD‐L1 immunotherapy against 4T1 breast tumors (Scheme 1 ). This proof‐of‐concept study demonstrates a novel system for photo‐enhanced delivery of small molecules to tumor tissues selectively and an efficient photoresponsive strategy to achieve synergistic ferroptosis together with immunotherapy.
Scheme 1.
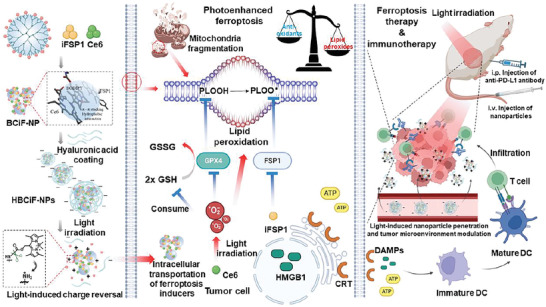
Combination therapy of light‐controlled ferroptosis and immune checkpoint blockade. Light irradiation triggers charge reversal of HA‐coated BMP‐Ce6&iFSP1 nanoparticles (HBCiF‐NPs), thereby promoting intracellular uptake of ferroptosis inducers. Subsequently, photodynamic therapy causes GPX4 downregulation, ROS accumulation, and lipid peroxidation inside cells, whose protection mechanism can be further inhibited by iFSP1. Ferroptosis further promotes immunogenic cell death and activates the immune system against cancer, which is reinforced by anti‐PD‐L1 therapy.
2. Results and Discussion
Generation 5 (G5) PAMAM dendrimer as a hyperbranched polymer processes 128 terminal amino groups which can be conjugated with functional molecules for different applications,[ 13 ] including several ongoing clinical trials for various diseases.[ 14 ] Furthermore, the positive charges from multiple amino groups can enhance cellular uptake and endosomal escape of cargos.[ 15 ] In our previous study, we developed a green light‐responsive nanocarrier by conjugating boron‐dipyrromethene (BODIPY)‐based photoremovable protecting groups (hereinafter referred to as BODIPY) to G5 PAMAM for intracellular protein delivery.[ 12 ] Herein, we investigated the ability of BODIPY‐modified PAMAM (BMP) to form nanoparticles and enhance intracellular delivery of hydrophobic small molecules, Ce6 and iFSP1, for anti‐tumor therapy. The synthesis route of BMP was shown in Figure S1a (Supporting Information). After light irradiation, BODIPY PPGs can be photocleaved from BMP (Figure S1c, Supporting Information). The BODIPY grafting number in one PAMAM molecule was 62.34, measured by UV‐vis spectroscopy (Figure S2a, Supporting Information).
Ce6 and iFSP1 can potentially interact with BODIPY by π‐π stacking and hydrophobic interaction, thereby promoting self‐assembly of BMP and these ferroptosis inducers into nanoparticles (Figure 1a). To demonstrate our hypothesis, we prepared BMP‐Ce6&iFSP1 complexes by flash nanoprecipitation and optimized the nanoparticle formulations (Figure S2b, Supporting Information). The complexes were further coated with hyaluronic acid (HA) to endow the nanoparticles with negative charge and higher stability for less off‐target drug delivery and longer circulation time (Figure S2c, Supporting Information). The size and polydisperse index (PdI) of the nanoparticles were 143.2 nm and 0.154, respectively (Figure 1b). UV‐vis spectroscopy can be used to determine whether Ce6 and BMP interact with each other by π‐π stacking, which was verified by an obvious shift of Ce6 characteristic peak (Figure 1c). The absorption spectrum of BODIPY overlaps the fluorescence emission spectrum of iFSP1. Hence, we determined the interaction between the two molecules through fluorescence resonance energy transfer (FRET) effect. As shown in Figure 1d, the fluorescence intensity of iFSP1 decreased obviously after being mixed with BMP. After optimization and verification, we successfully acquired HA‐coated BMP‐Ce6&iFSP1 nanoparticles (HBCiF‐NPs). The encapsulation efficiencies of Ce6 and iFSP1 were 21.09% and 20.74%, respectively. The loading content of the drugs was 18.64% (Figure S2d, Supporting Information). We further tested the stability of the nanoparticles in DMEM complete medium containing 10% fetal bovine serum at 37 °C. The result showed that the nanoparticles could keep stable for at least 48 h without obvious size change and PdI increase (Figure S3, Supporting Information).
Figure 1.
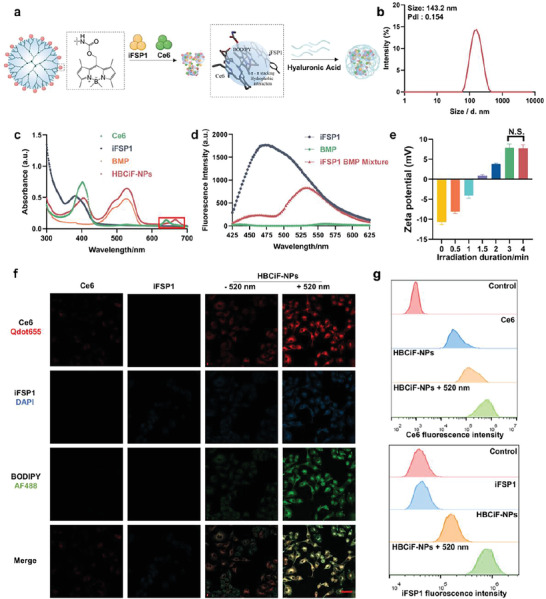
Fabrication/characterization of HBCiF‐NPs and light‐enhanced cellular uptake of Ce6 and iFSP1. a) Structure of BMP and preparation of HBCiF‐NPs. b) Size distribution of HBCiF‐NPs after preparation and centrifugation. c) UV‐vis spectra of Ce6, iFSP1, BMP, and HBCiF‐NPs. The red box highlights the red shift of Ce6 characteristic peak. d) Fluorescence resonance energy transfer between BODIPY and iFSP1, λex = 385 nm. e) Zeta potential change under light irradiation for different durations. (n = 3, mean ± SD). Confocal laser scanning microscopy (CLSM) images f) and flow cytometry analysis g) of living A549 cells incubated with free Ce6, free iFSP1, HBCiF‐NPs, and light irradiated HBCiF‐NPs, separately, for 4 h at 37 °C. Scale bar: 50 µm. Ce6, iFSP1, and BODIPY were visualized using Qdot655 (red), DAPI (blue), and AF488 channels (green), respectively. Light irradiation: Xe lamp, 520 nm, 25 mW cm−2, 3 min.
According to our previous findings, light irradiation could mediate charge reversal of HA‐coated BMP/protein complexes because of photocleavage of BODIPY, thereby enhancing cellular uptake of proteins.[ 12 ] To exploit the potential of BMP for intracellular delivery of hydrophobic small molecules, we characterized photoresponsiveness of HBCiF‐NPs. Before 520 nm light irradiation, the nanoparticles were negatively charged because of HA coating while the nanoparticles gradually changed to be positively charged with the light irradiation duration increasing (Figure 1e). This is because the photocleavage of BODIPY from BMP caused the exposure of amino groups. By transmission electron microscopy (TEM), the morphology investigation was conducted. BCiF‐NPs showed a spherical structure while the size of HBCiF‐NPs slightly increased after HA coating. Upon light irradiation, the nanoparticles dissociated into small complexes (Figure S4, Supporting Information). Such surface charge reversal and morphology change might promote the cellular uptake of cargo drugs. To demonstrate our hypothesis, we incubated A549 cells with free Ce6, free iFSP1, as well as HBCiF‐NPs with or without 520 nm‐light irradiation. As shown in Figure 1f, the confocal microscopy result showed that cellular uptake of both Ce6 and iFSP1 was enhanced greatly when cells were treated with 520 nm light‐irradiated nanoparticles compared with Ce6, iFSP1, and non‐irradiated nanoparticles. A similar result was obtained by flow cytometry analysis. As shown in Figure 1g; Figure S5 (Supporting Information), fluorescence intensity of Ce6 and iFSP1 in the cells treated with light‐irradiated HBCiF‐NPs increased by ≈15 times and 75 times in comparison with those treated with free Ce6 and iFSP1, respectively. In addition, non‐irradiated nanoparticles also increased the cellular uptake of cargo drugs, which could be explained by the interaction between HA and CD44 overexpressed on A549 cell surface.[ 16 ] It should be noticed that the fluorescence intensity of BODIPY also increased in the cells treated with irradiated nanoparticles (Figure 1f). This result indicates that photocleaved BODIPY was not released from the complexes immediately after light irradiation, probably because of its strong hydrophobicity. Hence, cargo drugs were still encapsulated in the positively charged complexes, thereby being transported into cells. In summary, BMP exhibited a robust ability to achieve photo‐enhanced intracellular delivery of ferroptosis inducers.
Clinical translation of nanocarriers for anti‐cancer therapy is faced with challenges of inefficient accumulation and poor penetration in tumors due to elevated interstitial fluid pressure, insufficient blood supply, and intricate stroma barriers.[ 17 ] Developing drug delivery strategies to enhance drug penetration in tumors is important to increasing therapeutic efficacy of nanomedicines. Therefore, we prepared A549 3D spheroids to investigate the penetration ability of the nanoparticles. The 3D tumor spheroids were incubated with non‐irradiated HA‐coated BMP‐Ce6 nanoparticles (HBC‐NPs), pre‐irradiated HBC‐NPs, or free Ce6. As shown in Figure S6 (Supporting Information), the fluorescence intensity of the spheroids treated with free Ce6 and non‐irradiated HBC‐NPs was much weaker than that of the spheroid treated with pre‐irradiated HBC‐NPs. More importantly, non‐irradiated HBC‐NPs were mostly observed at the edge of the spheroids while pre‐irradiated HBC‐NPs distributed in the whole spheroids with stronger fluorescence. The result indicates that non‐irradiated nanoparticles could not penetrate deeply into solid tumors despite higher cellular uptake compared with free drugs. We further quantified the fluorescence intensity of treated spheroids. The results showed high fluorescence intensity was found even in the core of pre‐irradiated HBC‐NPs‐treated spheroids. Such results confirmed enhanced tumor penetration ability of HBC‐NPs after light irradiation.
Ce6 is a traditional PDT photosensitizer that can generate singlet oxygen under NIR light irradiation. It is reported that PDT can trigger autophagy by suppressing AKT‐mTOR signaling.[ 18 ] Additionally, mTOR inhibition leads to GPX4 degradation and elevates ferroptosis.[ 19 ] Thus, PDT not only accumulates reactive oxygen species (ROS) inside cells to promote LPO but also downregulates GPX4 to hinder lipid peroxide elimination. Furthermore, iFSP1 blocks another parallel LPO protection pathway to induce lipid peroxide accumulation. To thoroughly break lipid peroxide elimination homeostasis, Ce6 and iFSP1 were selected as the cargo drugs for anti‐cancer therapy. First, the synergism of Ce6 and iFSP1 was investigated. A549 cells were incubated with Ce6 and iFSP1, followed by light irradiation with 650 nm Xe lamp or not. Without light irradiation, A549 cell viabilities in all groups were nearly 100%, which indicated that both Ce6 and iFSP1 had negligible dark toxicity. In contrast, Ce6 and iFSP1 combination (Ce6&iFSP1) treatment with light irradiation significantly caused more cell death compared with Ce6 or iFSP1 only. At the concentration of 8 µg/mL, single drug treatment induced about 30% and 0% cell death for Ce6 and iFSP1, respectively, while Ce6&iFSP1 resulted in around 80% cell death upon light irradiation (Figure S7a, Supporting Information). We further used three calculation models (Loewe, HSA, and Bliss) to quantify the synergistic effect. All the most synergistic scores were higher than 10 (Figure S7b, Supporting Information), indicating that Ce6 and iFSP1 do have a synergistic effect on induction of cell death.
In order to study if the synergism of Ce6 and iFSP1 can be elevated by our photoresponsive BMP system, 4T1 and A549 cells were incubated with free Ce6, Ce6&iFSP1, HBC‐NPs, 520 nm‐light pre‐irradiated HBC‐NPs, HBCiF‐NPs, and 520 nm‐light pre‐irradiated HBCiF‐NPs for 4 h. Afterward, the cells were irradiated with 650 nm light or not. As shown in Figure 2a,b, all groups displayed neglectable dark toxicity for both cell lines. Under 650 nm light irradiation, cell growth was significantly inhibited by pre‐irradiated HBCiF‐NPs much more robustly than non‐irradiated ones because of enhanced cellular uptake after photoactivation. Moreover, pre‐irradiated HBCiF‐NPs resulted in significantly higher cytotoxicity than pre‐irradiated HBC‐NPs, which reveals that encapsulated Ce6 and iFSP1 still caused cell death synergistically as same as free drugs. More importantly, the viability of the cells treated with pre‐irradiated HBCiF‐NPs was remarkably decreased in comparison with that of Ce6&iFSP1‐treated cells (8% versus 99% and 3% versus 87% for 4T1 and A549, respectively, at 1 µg mL−1 Ce6 and 0.488 µg mL−1 iFSP1), indicating that the synergistic cell death caused by Ce6 and iFSP1 can be promoted with the assistance of our system.
Figure 2.
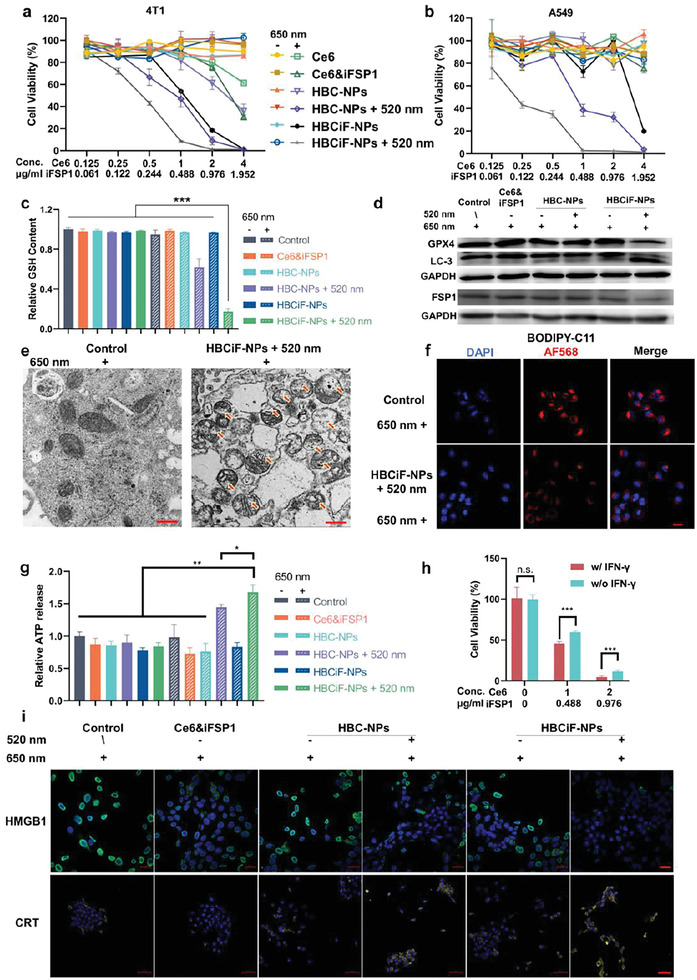
Ferroptosis and immunogenic cell death (ICD) induced by dual light‐responsive HBCiF‐NPs. MTT assays of Ce6/iFSP1 dose‐dependent cytotoxicity on 4T1 cells a) and A549 cells b) treated with Ce6, Ce6&iFSP1, HBC‐NPs, 520 nm light pre‐irradiated HBC‐NPs, HBCiF‐NPs, and 520 nm‐light pre‐irradiated HBCiF‐NPs for 4 h at 37 °C. The 650 nm light irradiation was applied after drug/nanoparticle incubation. c) Intracellular GSH concentrations of A549 cells with different treatments. The average concentration in the control group was set as 1. d) Western blot assay of GPX4, LC‐3, and FSP1 expression in A549 cells with different treatments. The 650 nm light irradiation was applied in all groups. e) Transmission electron microscopy (TEM) image of cross section of A549 cells with or without 520 nm pre‐irradiated HBCiF‐NPs incubation. The 650 nm light irradiation was applied in both groups (scale bar: 500 nm). f) CLSM analysis of lipid ROS generation in normal cells and the cells treated with 520 nm‐light pre‐irradiated HBCiF‐NPs. Cells in both groups were received 650 nm light irradiation (scale bar: 20 µm). g) Extracellular ATP concentrations of A549 cells with different treatments. ATP concentrations were represented by luminescence intensity. And the average luminescence intensity in the control group was set as 1. h) MTT assay of Ce6/iFSP1 dose‐dependent cytotoxicity on A549 cells treated with 520 nm‐light pre‐irradiated HBCiF‐NPs and IFN‐γ (250 ng mL−1) or not. i) CLSM examination of HMGB1 release (scale bar: 20 µm) and CRT exposure (scale bar: 50 µm) on the cell surface of the A549 cells with different treatments. The 650 nm light irradiation was applied in all groups. Light irradiation: Xe lamp, 520 nm, 25 mW cm−2, 3 min; 650 nm, 5 mW cm−2, 30 min. Data are means ± SD, n = 3. n.s., non‐significant. *p<0.5, **p<0.01, ***p<0.001.
To confirm if the nanosystem caused ferroptosis, we further investigated levels of several ferroptosis indicators in treated cells. Intracellular GSH concentration is maintained by a complicated homeostatic system inside cells, which keeps the balance of redox homeostasis.[ 20 ] Once GSH is consumed and ROS are accumulated continuously, lipid peroxides will be generated and cannot be eliminated, thereby breaking the balance and leading to cell ferroptosis.[ 21 ] Therefore, intracellular GSH concentration was measured to indicate whether redox homeostasis was maintained inside cells after different treatments. The result shown in Figure 2c was consistent with the MTT assay. Under 650 nm light irradiation, the cells treated with 520 nm‐light pre‐irradiated HBCiF‐NPs displayed the lowest GSH level among all groups. The decrease of GSH content inside the cells provided the prerequisite for ferroptosis.
Next, the protein level of GPX4 was measured by western blot analysis. Under 650 nm light irradiation, GPX4 level was obviously downregulated by 520 nm‐light pre‐irradiated HBCiF‐NPs as shown in Figure 2d. As mentioned above, PDT can induce autophagy, thereby promoting degradation of GPX4. The transformation of LC‐3, a well‐established marker of autophagy, was subsequently investigated with western blot technique. As expected, under 650 nm light irradiation, the transformation of LC‐3 from type‐I to type‐II was obviously facilitated by pre‐irradiated HBCiF‐NPs, indicating the formation of autophagosome and occurrence of autophagy. Importantly, the FSP1 level was also downregulated after the cells were treated with pre‐irradiated HBCiF‐NPs and 650 nm light irradiation (Figure 2d), indicating elimination of lipid peroxyl radicals by the FSP1‐mediated pathway was also hindered. In addition, we quantified the level of intracellular coenzymer Q10 (CoQ10) in 4T1 cells since FSP1 reduces CoQ10 to trap lipid peroxyl radicals. The result showed that CoQ10 increased significantly after the treatment of HBCiF‐NPs and dual light irradiation compared with other groups (Figure S8, Supporting Information), which can be explained by FSP1 inhibition by iFSP1 and unbalanced redox homeostasis due to PDT‐induced oxidative stress. Taken together, our strategy hindered the FSP1‐mediated ferroptosis protection pathway by directly inhibiting FSP1 and downregulating its protein level.
According to previous studies,[ 22 ] mitochondria undergo fragmentation and cristae alteration during ferroptosis. Therefore, we used TEM to conduct a morphology investigation of mitochondria in the cells with or without HBCiF‐NP treatment. As shown in Figure 2e, the mitochondria in pre‐irradiated HBCiF‐NPs‐treated cells were fragmented and the cristae disappeared or were damaged under 650 nm light irradiation. Because ferroptosis is associated with increased lipid peroxidation, BODIPY‐C11, whose fluorescence at 581/591 nm decreases when it reacts with lipid peroxides, was used as a fluorescent probe to measure the level of LPO. After being treated with pre‐irradiated HBCiF‐NPs, the cells under 650 nm light irradiation displayed lower BODIPY‐C11 fluorescence intensity (Figure 2f), indicating an increased intracellular lipid peroxide level. Taken together, 520 nm‐light pre‐irradiated HBCiF‐NPs could efficiently lead to GSH depletion, GPX4 degradation, and lipid peroxide accumulation in the cells under 650 nm light irradiation.
Numerous studies have reported that ferroptosis directly promotes ICD, during which adenosine triphosphate (ATP) and high mobility group box 1 (HMGB1) protein are released from cells and calreticulin (CRT) is exposed on the surface of cells.[ 23 ] Such DAMPs serve as an “eat me signal”, which mediates dendritic cell (DC) recruitment and activation, thereby promoting T cell infiltration and cytotoxicity.[ 24 ] We first measured the extracellular levels of ATP excreted by cells. As expected, a higher level of ATP was detected after 650 nm light irradiation in the pre‐irradiated HBCiF‐NPs group compared with other groups (Figure 2g). HMGB1 in the control group located in the nuclei of the cells (Figure 2i), while the cells treated with 520 nm‐light pre‐irradiated HBCiF‐NPs displayed less HMGB1 in the nuclei under 650 nm light irradiation, indicating the release of HMGB1 from ferroptotic cells. Furthermore, pre‐irradiated HBCiF‐NPs upregulated the exposure of CRT on cell surface under 650 nm light irradiation. In summary, all results shown above proved that our system could induce enhanced ICD by promoting ferroptosis with light irradiation.
It is reported that IFN‐γ secreted by T cells primes tumor cell LPO.[ 25 ] To mimic in vivo situation and investigate whether the nanoparticles can boost immune responses against tumor cells, A549 cells were incubated with human‐derived IFN‐γ and pre‐irradiated HBCiF‐NPs. The result showed that IFN‐γ alone did not cause cell death while combination of IFN‐γ and pre‐irradiated HBCiF‐NPs reduced cell viability significantly under 650 nm light irradiation (Figure 2h). Without the nanoparticles, IFN‐γ‐initiated LPO did not induce cell death probably because lipid peroxides were eliminated by the actions of GPX4 and FSP1. Whereas, the nanoparticles helped boost the IFN‐γ‐initiated LPO due to the inhibition of GPX4 and FSP1, thus causing significant cell death.
Given the robust ability of HBCiF‐NPs to achieve photo‐enhanced cellular uptake of cargos and ferroptosis, we next explored their in vivo performance. The pharmacokinetic study of Ce6 in plasma was performed by HPLC and fluorescence intensity measurement after free drugs or nanoparticles were administered to BALB/c mice intravenously. Both results showed that the half‐life of Ce6 was extended when Ce6 was encapsulated in the nanoparticles (Figure S9, Supporting Information), indicating that BMP and HA coating could protect cargo drugs from rapid metabolism. Then, we inoculated 4T1 cells in the flank of BALB/c mice. The targeting ability of the nanoparticles in the 4T1 tumor‐bearing BALB/c mice was evaluated by an in vivo imaging system (IVIS). The mice were intravenously injected with PBS, Ce6&iFSP1, or HBCiF‐NPs followed by 530 nm light irradiation at tumor sites or not. The 530 nm light irradiation was applied immediately after administration. After 1, 2, 4, and 24 h, the fluorescence intensity from Ce6 was monitored. The non‐irradiated mice with nanoparticle administration (hereinafter referred to as non‐irradiated mice) showed a slight fluorescence increase at the tumor site compared with the Ce6&iFSP1‐treated mice. Furthermore, after 530 nm light irradiation, significantly more Ce6 accumulated and retained at the tumor sites of the mice treated with HBCiF‐NPs compared with the non‐irradiated mice (Figure 3a). After 24 h, we sacrificed the mice and collected tumors and major organs to measure the distribution of Ce6. With the administration of HBCiF‐NPs and light irradiation at the tumor sites, significantly stronger fluorescence was observed through the IVIS imaging. However, very weak fluorescence in tumors was observed in both the Ce6&iFSP1‐treated mice and the non‐irradiated mice (Figure 3a,b). The fluorescence intensity in kidney, spleen, and heart from the non‐irradiated mice was significantly higher than those from the Ce6&iFSP1‐treated mice, probably because the nanoparticles delayed metabolism and elimination of loaded drugs. Noticeably, there was no significant difference of fluorescence in liver, kidney, spleen, heart, and lung between Ce6&iFSP1 and HBCiF‐NPs + 530 nm light irradiation groups. This result implies that our strategy would not cause enhanced drug accumulation in normal organs.
Figure 3.
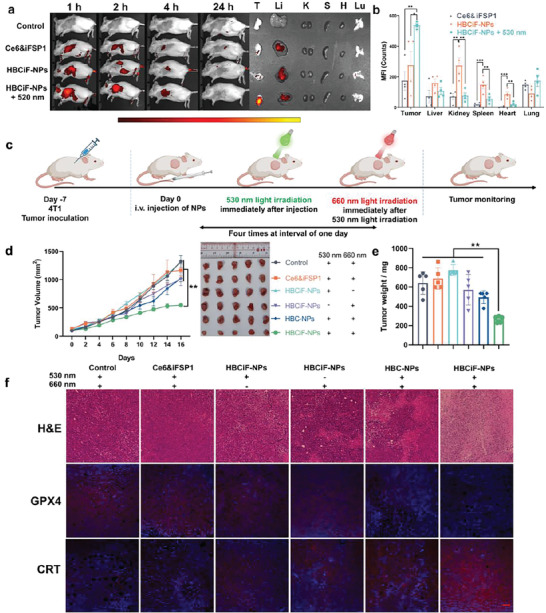
Biodistribution of HBCiF‐NPs and light‐induced ferroptosis of tumor cells in vivo. a) Fluorescence imaging of the biodistribution of Ce6 in the mice treated with saline, Ce6&iFSP1, and HBCiF‐NPs by IVIS. One group of mice treated with HBCiF‐NPs received 530 nm light irradiation on the tumor sites immediately after Ninjection. Ce6 distribution in major organs was determined after 24 h. Ce6 was detected using the AF647 channel. (T: tumor, Li: liver, K: kidney, S: spleen, H: heart, Lu: lung). b) Mean fluorescence intensity (MFI) of Ce6 in major organs ex vivo in diverse groups (n = 4, data were means ± SEM). c) Treatment schedule of ferroptosis therapy by HBCiF‐NPs and dual light irradiation in 4T1 tumor‐bearing BALB/c mice. d) Tumor growth curves and photos of the tumors from the mice with different treatments (n = 5, data were means ± SEM). e) Weight analysis of the above tumors (n = 5, data were means ± SD). f) H&E staining and immunofluorescence of GPX4 and CRT of tumor tissues in different groups (scale bar: 100 µm). Light irradiation: Laser, 530 nm, 80 mW cm−2, 5 min; 660 nm, 20 mW cm−2, 1 min. Ce6 dosage: 1.5 mg kg−1; iFSP1 dosage: 0.75 mg kg−1. *p<0.5, **p<0.01, ***p<0.001.
To investigate anti‐tumor efficacy of HBCiF‐NPs in vivo, 4T1 breast tumor‐bearing mice were randomly divided into 6 groups and received different treatments, including saline, Ce6&iFSP1 + dual light (530 and 660 nm light), HBC‐NPs + dual light, HBCiF‐NPs + 530 nm light, HBCiF‐NPs + 660 nm light, and HBCiF‐NPs + dual light (Figure 3c). In previous reports, the administration of Ce6 normally ranges from 3 mg kg−1 to 5 mg kg−1.[ 7 , 26 ] To highlight the high delivery efficiency of our system, 1.5 mg kg−1 Ce6 dosage was used in this study. According to the IVIS data shown in Figure 3a,b, Ce6 was gradually metabolized after administration. Therefore, 660 nm light was applied right after 530 nm light irradiation. As shown in Figure 3d, both HBCiF‐NPs + 530 nm light and HBCiF‐NPs + 660 nm light did not slow down tumor growth rate without a second light irradiation at the tumor site compared with saline. In contrast, the tumor growth was significantly inhibited when the mice were treated with HBCiF‐NPs + dual light. The comparisons reflected 530 nm light‐enhanced tumor accumulation of drugs and 660 nm light‐triggered ferroptosis in tumors. Notably, the anti‐tumor efficacy of HBCiF‐NPs was superior to HBC‐NPs and Ce6&iFSP1 under dual light irradiation (Figure 3d), further confirming the synergistic anti‐cancer effect of Ce6 and iFSP1 in vivo and photo‐enhanced drug accumulation in tumors. Importantly, all treatments had no significant influences on the body weight in comparison with saline during the treatment period (Figure S10, Supporting Information), indicating good biosafety of our system. After sacrificing the mice, tumors were harvested and analyzed. Consistent with the tumor volume curves (Figure 3d), both tumor weight (Figure 3e) and cell number visualized by H&E staining (Figure 3f) of the mice treated with HBCiF‐NPs + dual light were lower than those of the mice in other groups. Furthermore, immunofluorescence assay of the tumor tissues showed that HBCiF‐NPs + dual light could significantly downregulate GPX4 in vivo, implying ferroptosis induction by the strategy (Figure 3f). Moreover, CRT transportation onto the surface of tumor cell membrane was also observed as similar to the in vitro data (Figure 3f), verifying ICD induction in vivo by HBCiF‐NPs + dual light. In summary, HBCiF‐NPs showed robust photo‐targeting ability and promoted retention in the tumor tissues with 530 nm light activation. Under further 660 nm light irradiation, HBCiF‐NPs could induce synergistic anti‐tumor effect of Ce6 and iFSP1 through ferroptosis.
Interestingly, after sacrificing mice, we found there was significant difference of spleen volumes and weight between the mice treated with HBCiF‐NPs + dual light and the saline‐treated mice (238 mg versus 550 mg) as shown in Figure S11 (Supporting Information). It is reported that spleen weight varies with tumor progression and negatively correlated with CD8+ T cells percentage in the spleen and peripheral blood.[ 27 ] Therefore, we believe that our strategy might activate the immune system against cancer, whose potential on immunotherapy could be further exploited.
For light‐associated therapies, penetration of light is an important parameter to achieve better efficacy of the therapies. Absorption of the light within the "phototherapeutic window" (600–900 nm) by the common physiological chromophores is low, thereby allowing light in this wavelength range to penetrate deeply into body tissues.[ 28 ] HBCiF‐NPs used in the above studies are dual‐light responsive. Green light (520 nm or 530 nm) and NIR light (650 nm or 660 nm) were utilized to achieve photo‐targeting and promote ferroptosis, respectively. Thus, photoresponsive targeting and photo‐activated ferroptosis could be studied separately with HBCiF‐NPs. Nevertheless, green light is obviously out of the "phototherapeutic window" and the efficiency of the strategy needs to be improved. Therefore, the green light‐responsive nanocarrier, BMP, was further upgraded to be NIR‐light responsive (Figure 4a) by conjugating NIR light‐responsive BODIPY to PAMAM with the same method. The synthesis route of NIR light‐responsive BODIPY‐modified PAMAM (NBMP) was shown in Figure S1b (Supporting Information). 1H‐NMR spectroscopy result demonstrates successful synthesis of NBMP (Figure S12–S15, Supporting Information). The grafting number of NIR light‐responsive BODIPY PPGs on each PAMAM was 58.92 as calculated by UV–vis spectroscopy. As expected, NBMP formed nanoparticles with Ce6 and iFSP1. HA was further used to coat the nanoparticles to finally acquire HA‐coated NBMP‐Ce6&iFSP1 nanoparticles (HNCiF‐NPs) with the same method as the preparation of HBCiF‐NPs. The hydrodynamic diameter of HNCiF‐NPs was 157.8 nm with a narrow particle size distribution (PdI: 0.171) (Figure S16, Supporting Information). The 650 nm light irradiation still could reverse the zeta potential of HNCiF‐NPs from negative to positive (Figure S16, Supporting Information), implying 650 nm light might be able to activate HNCiF‐NPs to achieve photo‐enhanced cellular uptake of cargo drugs. Therefore, in vitro anti‐cancer efficiency of HNCiF‐NPs was determined by MTT assay. Prior to being incubated with 4T1 cells, HNCiF‐NPs and HA‐coated NBMP‐Ce6 nanoparticles (HNC‐NPs) were pre‐activated with 650 nm light. After 4 h, the culture medium was changed to fresh medium and the cells were irradiated by 650 nm light again or not. Consistent with the findings mentioned above, the result showed that pre‐irradiated HNCiF‐NPs showed significantly enhanced cell death than Ce6&iFSP1 and pre‐irradiated HNC‐NPs when the cells were irradiated with 650 nm light (Figure S17, Supporting Information), indicating that our system could be used to achieve enhanced cellular uptake and synergistic ferroptosis with single‐wavelength light.
Figure 4.
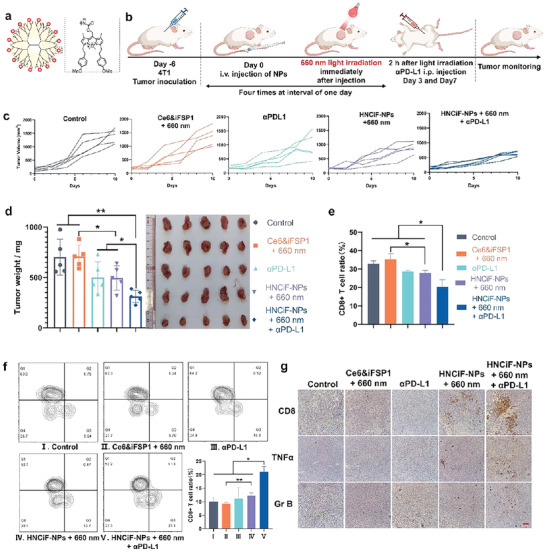
In vivo immune activation and combination therapy by NIR light‐responsive HNCiF‐NPs‐induced ferroptosis and PD‐L1 blockade in vivo a) Structure of NIR‐BODIPY‐modified PAMAM (NBMP). b) Treatment schedule of HNCiF‐NPs and PD‐L1 combination therapy. c) Tumor growth curves of the mice with different treatments (n = 5). d) Tumor weight and photos of the tumors from the mice in distinct groups. e) Percentage of CD8+ cells within the CD3+ cell population of the DLN from the mice with different treatments (n = 5). f) Percentage of tumor infiltrating CD8+ T cells within the CD3+ cells of the mice with different treatments (n = 3). g) CD8, TNFα, and granzyme B (Gr B) immunohistochemical staining of tumor sections from the treated mice (scale bar: 100 µm). Light irradiation: Laser, 660 nm, 60 mW cm−2, 1 min. Ce6 dosage: 0.75 mg kg‐1; iFSP1 dosage: 0.38 mg kg−1. Data were means ± SD. *p<0.5, **p<0.01.
Because HBCiF‐NPs induced obvious ICD and exhibited promising potential on immune system activation, we investigated in vivo anti‐tumor effect of HNCiF‐NPs combined with anti‐PD‐L1 antibody (αPD‐L1). BALB/c mice bearing subcutaneous 4T1 tumor were randomly divided into 5 groups including saline, Ce6&iFSP1 + 660 nm light, αPD‐L1, HNCiF‐NPs + 660 nm light, and HNCiF‐NPs + 660 nm light + αPD‐L1. Light irradiation at the tumor sites was applied immediately after intravenous injection of the formulations. αPD‐L1 was intraperitoneally injected 2 h after light treatment (Figure 4b). The result showed that HNCiF‐NPs inhibited tumor growth more efficiently compared with saline, and Ce6&iFSP1 (Figure 4c,d), indicating that synergism of Ce6 and iFSP1 was enhanced by NBMP and light‐induced drug accumulation and ROS production in tumors. Furthermore, anti‐tumor efficacy of HNCiF‐NPs was further boosted with αPD‐L1 combination therapy. To further confirm that the immune system was stimulated after treatments, we determined DC maturation in spleen by flow cytometry because we found significant spleen volume and weight difference between the HBCiF‐NP‐treated mice and saline‐treated mice as mentioned above (Figure S11, Supporting Information). The result showed that HNCiF‐NPs significantly increased CD80/CD86 expression in DCs (Figure S18, Supporting Information). The combination with αPD‐L1 further enhanced the expression, which might be attributed to nanoparticle‐induced ICD and αPD‐L1 enhanced cytotoxicity of T cells. CD8+ T cells are reported to mediate ferroptosis during cancer immunotherapy.[ 29 ] Therefore, we further quantify CD8+ T cell percentage in spleen, draining lymph nodes (DLN), and tumors. Consistent with DC maturation, the percentage of CD8+ T cells increased in splenic T cells after the mice were treated with HNCiF‐NPs + 660 nm light + αPD‐L1 (Figure S19, Supporting Information), indicating that matured DCs promoted CD8+ T cell differentiation. Interestingly, the overall percentage of CD8+ T cells in the DLN dropped in response to the treatment of HNCiF‐NP + 660 nm light + αPD‐L1 compared with other groups (Figure 4e). Similar results were reported in a previous study and explained by the migration of CD8+ T cells from DLN to tumors.[ 30 ] To demonstrate this hypothesis, we measured CD8+ T cell infiltration in tumors by flow cytometry (Figure 4f) and immunohistochemistry (IHC) (Figure 4g). Flow cytometry results showed that more CD8+ T cells infiltrated into tumors after the treatment of HNCiF‐NPs + 660 nm light, which was further fostered by αPD‐L1 combination therapy. IHC staining of TNFα in the tumors also demonstrated pro‐inflammatory tumor microenvironment in the HNCiF‐NPs + 660 nm light group (Figure 4g). The increase of granzyme B (Gr B) further validated T cell‐mediated anti‐cancer effect (Figure 4g). Thus, better anti‐cancer efficacy of HNCiF‐NPs attributed to not only light‐enhanced ferroptosis but also ferroptosis‐induced immune responses. In summary, HNCiF‐NPs could significantly induce ferroptosis in vitro and in vivo with single 660 nm light irradiation, simplifying our strategy and increasing light penetration into tumors. Furthermore, such a strategy could stimulate the immune system against tumors and enhance anti‐PD‐L1 immunotherapy.
During the treatment period, there was no significant difference in body weight among different groups (Figure S20, Supporting Information). H&E staining analysis showed that no histological damage and morphological abnormality existing in heart, liver, spleen, lung, and kidney by the treatment of HNCiF‐NPs + 660 nm light + αPD‐L1 (Figure 5a). Moreover, the activity of alanine aminotransferase (ALT) (Figure 5b) and aspartate aminotransferase (AST) (Figure 5c) in the HNCiF‐NPs + 660 nm light + αPD‐L1 group was similar with that of the control group and in the normal ranges. [ 22 , 31 ] In a word, our combination therapy did not cause any obvious side effects on non‐tumor tissues.
Figure 5.
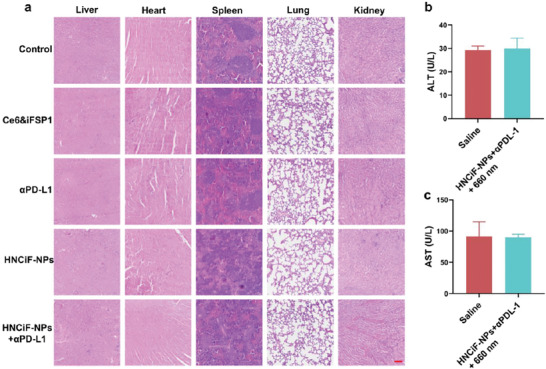
In vivo biosafety evaluation of HNCiF‐NPs&αPD‐L1 combination therapy. a) H&E staining assessment of major organs (liver, heart, spleen, lung, and kidney) after different treatments (scale bar: 100 µm). Blood ALT b) and AST c) activity analysis of the mice treated with saline or HNCiF‐NPs + 660 nm light + αPD‐L1. Light irradiation: Laser, 660 nm, 60 mW cm‐2, 1 min Data are means ± SD.
3. Conclusion
In this study, we developed a green light‐responsive BODIPY‐modified PAMAM for photo‐enhanced cellular uptake and tumor accumulation of cargo drugs. With this carrier, we achieved photo‐enhanced intracellular delivery of Ce6 and iFSP1 to induce synergistic ferroptosis, thereby leading to ICD. The nanoparticles could inhibit 4T1 tumor growth with dual light irradiation. For more efficient light penetration and simpler therapy procedure, we re‐designed the carrier to be NIR light‐responsive, which means we can achieve photo‐enhanced drug accumulation and light‐induced ferroptosis with single‐wavelength light irradiation. We demonstrated that our strategy could activate the immune system against tumors and enhance efficacy of anti‐PD‐L1 immunotherapy. The Ce6 and iFSP1 combination therapy was put forward for the first time. And BODIPY‐modified PAMAM was verified as an efficient drug carrier for clinical applications to increase therapeutic efficacy and decrease systemic side effects.
4. Experimental Section
Materials
2,4‐dimethyl‐1H‐pyrrole, 2‐chloro‐2‐oxoethyl acetate, 4‐nitrophenyl chloroformatedimethyl (4‐NPC), dimethyl sulfoxide (anhydrous, DMSO), dichloromethane (anhydrous), and pyridine (anhydrous) were purchased from J&K Chemical (Beijing, China). N,N‐diisopropylethylamine (DIPEA), generation 5 poly(amidoamine), 4‐anisaldehyde, methylmagnesium bromide (MeMgBr), hyaluronic acid (HA), chlorin e6 were ordered from Dieckmann (Shenzhen, China). Hoechst 33 342, BODIPY 581/591 C11, and glutathione (GSH) colorimetric detection kit were obtained from Thermo Fisher (MA, USA). iFSP1 was purchased from MedChemExpress (MCE) (Shanghai, China). Luminescent ATP detection assay kit was ordered from Abcam (Cambridge, UK). Antibodies used in this study and their origin were listed in Table S1 (Supporting Information).
Cell Lines and Cell Culture
A549 cells (human lung carcinoma cell line) and 4T1 cells (mouse breast tumor cell line) were obtained from American Type Culture Collection (ATCC). Both cell lines were cultured in complete DMEM cell culture medium containing 10% fetal bovine serum (FBS), penicillin (100 µg mL−1), and streptomycin (100 µg mL−1) at 37 °C with 5% CO2.
Synthesis of Green Light‐Responsive BODIPY‐Modified PAMAM
The synthesis of green light‐responsive BODIPY and BODIPY‐modified PAMAM was shown in previous study.[ 12 ]
Synthesis of (5,5‐difluoro‐3,7‐bis((E)−4‐methoxystyryl)−1,9‐dimethyl‐5H‐4λ4,5λ4‐dipyrrolo[1,2‐c:2′,1′‐f][1,3,2]diazaborinin‐10‐yl)methyl acetate (1): 8‐Acetoxymethyl‐1,3,5,7‐tetramethyl pyrromethene fluoroborate (200 mg, 0.625 mmol, 1 eq.) was reacted with 4‐anisaldehyde (4 mL, 32.8 mmol, 53 eq.) at 60 °C for 2 h under a nitrogen atmosphere in the dark. The reaction was monitored by TLC. During the period, the color of the mixture changed from red to purple and then to dark green. The mixture was cooled to room temperature and purified by flash chromatography with a CombiFlash® system (Teledyne ISCO, NE, USA). The product was obtained as a deep red powder (258 mg, 74.2% yield). 1H spectrum of the product was in agreement with published data.[ 32 ]
Synthesis of (5,5‐difluoro‐3,7‐bis((E)−4‐methoxystyryl)−1,9‐dimethyl‐5H‐4λ4,5λ4‐dipyrrolo[1,2‐c:2′,1′‐f][1,3,2]diazaborinin‐10‐yl)methanol (2): A mixture of aqueous NaOH solution (43 mg, 0.5 mL, 1.076 mmol) and methanol (9.5 mL) was dropwise added to a solution of compound 1 (150 mg, 0.269 mmol, 1 eq.) in DCM. The reaction was monitored by TLC and stirred for 2 h in the dark at room temperature. After this time, the solvent was evaporated and re‐dissolved with DCM. Then, the solution was extracted by 0.01 M HCl. The organic layer was collected and hydrated by anhydrous sodium sulfate. The product was obtained as a dark red powder by evaporation without further purification (126 mg, 90.87% yield).
Synthesis of (3,7‐bis((E)−4‐methoxystyryl)−1,5,5,9‐tetramethyl‐5H‐4λ4,5λ4‐dipyrrolo[1,2‐c:2′,1′‐f][1,3,2]diazaborinin‐10‐yl)methanol (3): Compound 3 (100 mg, 0.194 mmol, 1 eq) was dispersed in 5 mL Et2O. Methylmagenesium bromide (3 M, tetrahydrofuran (THF) solution, 0.3 mL, 0.97 mmol) was added dropwise to the compound 3 solution under a nitrogen atmosphere in the dark. The reaction was monitored by TLC. The color of the solution changed from dark green to dark blue. After 2 h stirring, the reaction was stopped by adding 0.5 mL distilled water to the mixture. THF and Et2O were removed by evaporation. The residue was re‐dissolved in DCM. The DCM solution was extracted by saturated sodium chloride aqueous solution three times. The organic layer was collected and dehydrated by anhydrous sodium sulfate. The resulting product was then obtained as a dark blue solid by flash chromatography (48 mg, 48.9% yield). 1H spectrum of the product was in agreement with published data.[ 32 ]
Synthesis of (3,7‐bis((E)−4‐methoxystyryl)−1,5,5,9‐tetramethyl‐5H‐4λ4,5λ4‐dipyrrolo[1,2‐c:2′,1′‐f][1,3,2]diazaborinin‐10‐yl)methyl (4‐nitrophenyl) carbonate (4): Compound 3 (40 mg, 0.079 mmol, 1 eq.) was dissolved in anhydrous DCM (2 mL) with DIPEA 80 µL, 0.39 mmol, 5 eq.) and pyridine (30 µL, 0.312 mmol, 4 eq.) at nitrogen atmosphere. 4‐nitrophenyl chloroformate (158 mg, 0.78 mmol, 10 eq.) in 1 mL DCM was dropwise added to the compound 3 solution in an ice bath. The reaction was stirred in the dark for 4 h. After this time, the mixture was purified by flash chromatography. The product was obtained as a dark powder (41 mg. 77.3% yield).
Synthesis of NIR Light‐Responsive BODIPY‐Modified PAMAM (NMP)
Methanol was evaporated to get PAMAM (10 mg, 0.00034 mmol) as a transparent gel from PAMAM methanol solution (5 wt.%, 250 µL). Anhydrous DMSO (500 µL) with DIPEA (2 µL) was used to dissolve PAMAM. The DCM solution of compound 4 (28 mg, 0.034 mmol, 100 eq.) was dropwise added to the PAMAM solution. The reaction mixture was stirred overnight under a nitrogen atmosphere in the dark. Then the mixture was dialyzed (MW: 3500 Da) against DMSO until there was no obvious BODIPY fluorescence in the outer fluid. The product was then dialyzed against distilled water, lyophilized, and characterized by UV‐vis spectroscopy (SpectraMax® M4, Molecular Devices LLC) and 1H‐NMR (Bruker DX 500 spectrometer at 400 Hz).
Preparation and Characterization of HA Coated B(N)MP‐Ce6&iFSP1 Nanoparticles (HB(N)CiF‐NPs)
The nanoparticles were prepared through flash nanoprecipitation. Briefly, Ce6 and iFSP1 were added to the water under vigorous stirring. B(N)MP was then immediately added to the solution. To further stabilize the nanoparticles and endow them with negative charge, hyaluronic acid (HA) was added subsequently. The nanoparticles without iFSP1 (HBC‐NPs) were prepared following the same protocol only without iFSP1 added. To remove excessive materials or drugs, the nanoparticles were centrifuged at 18 000 g for 30 min and resuspended. The nanoparticles were then sonicated and centrifuged at 5000 rpm to remove aggregates. The stability of the nanoparticles was measured by dynamic light scattering (DLS) using Malvern Zetasizer (Nano ZS90, Malvern, UK) in complete medium at 37 °C for 48 h. The fluorescence and UV‐vis spectra were obtained with SpectraMax® M4 (CA, USA). The changes of morphology and zeta‐potential of the nanoparticles before and after light irradiation were observed by transmission electron microscope (TEM, Philips, CM100) and measured by Malvern Zetasizer, respectively. The light sources were: Xe lamp, CEL‐PE300L, 520 nm, 25 mW cm−2, 3 min for HBCiF‐NPs and NIR laser (635‐830 nm) 50 mW cm−2, 3 min for HNCiF‐NPs.
Cytosolic Delivery of Ce6 and iFSP1 by HBCiF‐NPs
1 × 105 cells were seeded in 24‐well plates and cultured overnight. The as‐prepared nanoparticles or free drugs were pre‐irradiated by 520 nm light (Xe lamp, CEL‐PE300L, 25 mW cm−2, 3 min) and directly added to the cell culture media. Cells without any treatment or with non‐irradiated nanoparticle incubation were regarded as controls for comparison. After 4 h incubation, the cells were trypsinized and washed with PBS. Flow cytometry (ACEA NovoCyte Advanteon BVYG) and confocal laser scanning microscopy (CLSM, Carl Zeiss LSM 980) were used to determine cellular uptake of Ce6 and iFSP1. Ce6, iFSP1, and BODIPY were detected at the Qdot655, DAPI, and AF488 channels, respectively.
MTT Assay of Ce6/iFSP1 Dose‐Dependent Toxicity on A549 and 4T1 Cells
1 × 104 A549 or 4T1 cells were seeded in 96‐well plates and cultured overnight. Cells were incubated with free Ce6, Ce6&iFSP1, HBC‐NPs, 520 nm light (Xe lamp, CEL‐PE300L, 25 mW cm−2, 3 min) preirradiated HBC‐NPs, and HBCiF‐NPs, or 520 nm light preirradiated HBCiF‐NPs with different concentrations of Ce6 and iFSP1. After 4 h incubation, the cell culture media containing drugs were removed. The cells were washed with PBS and cultured in fresh media. Then, the cells were irradiated under 650 nm light (Xe lamp, CEL‐PE300L, 5 mW cm−2, 30 min). After irradiation, the cells were cultured overnight. 10 µL of 5 mg/mL MTT solution was added to each well and incubated with cells for 2 h. Then, the culture media were removed and 100 µL DMSO was added to each well to totally dissolve formazan. The absorbance at 570 nm reflected cell viability and the absorbance of the control group without any treatment was regarded as 100% cell viability. For the synergism analysis of Ce6 and iFSP1, the data were obtained on the SynergyFinder website (http://www.synergyfinder.org/).
Western Blot Analysis
The protein samples were collected with RIPA lysis buffer. After sample collection, protein concentrations were quantified by BCA protein assay. The protein levels of GPX4, LC3, FSP1, and GAPDH were analyzed by polyacrylamide gel electrophoresis.
Intracellular GSH Concentration Determination and ATP Release Detection
1 × 105 A549 cells were seeded in 24‐well plates and cultured overnight. After that, cells were treated as same as the MTT assay mentioned above with 0.5 µg ml−1 Ce6 and 0.244 µg ml−1 iFSP1. After overnight incubation, the supernatants were collected, and ATP level was detected following the protocol provided in the kit. For intracellular GSH concentration determination, the cells were lysed by freeze‐thaw cycling. To make sure the cell numbers in each group were consistent for comparison, the protein concentration was measured by BCA assay. Then, intracellular concentrations of GSH in each group were determined according to the provided protocol in the kit.
Intracellular CoQ10 Content Determination
The contents of intracellular CoQ10 in 4T1 cells were measured by following the instructions of the mouse CoQ10 ELISA kit (JL49867, Jianglai Bio, Shanghai, China). Before conducting CoQ10, the total protein concentrations were determined by a BCA assay. The intracellular CoQ10 content was represented by concentrations of CoQ10/concentrations of total proteins.
High Mobility Group box 1 (HMGB1) Release and Calreticulin (CRT) Cell Surface Exposure Detection
1 × 105 A549 cells were seeded in a 35 mm confocal dish in each group. When ≈80% confluent, the cells were treated as the MTT assay mentioned above with 0.5 µg ml−1 Ce6 and 0.244 µg ml−1 iFSP1. After overnight incubation, HMGB1 was visualized by immunofluorescence staining. Briefly, cells were fixed with 4% paraformaldehyde. For HMGB1 detection, cells were permeabilized with 0.1% Triton X‐100. Afterward, cells were incubated with primary antibodies overnight. To visualize CRT on cell surface, cells were incubated with anti‐CRT antibody overnight without being permeabilized. After washing, cells were incubated with Alexa fluor 568‐conjugated anti‐rabbit goat IgG antibody at room temperature for 1 h. The nucleus was stained with DAPI. Images were taken with a fluorescence microscope (Carl Zeiss LSM980).
TEM Imaging of Mitochondrial Morphology
A549 cells were seeded in 10 cm2 tissue culture dish. When ≈80% confluent, the cells were treated with 520 nm light pre‐irradiated HBCiF‐NPs (Xe lamp, 25 mW cm−2, 3 min, 0.5 µg ml−1 Ce6 and 0.244 µg ml−1 iFSP1) or saline. After 4 h incubation, the medium containing nanoparticles was removed and changed to fresh medium. Then the cells were irradiated with 650 nm light (Xe lamp, 5 mW cm−2, 30 min). After overnight incubation, the cells were trypsinized and collected by centrifugation. After removing the medium, the cell pellet in each group was washed with PBS and fixed in 2.5% glutaraldehyde. The mitochondria morphology was observed by a transmission electron microscope (Philips CM100).
Measurement of Anti‐Tumor Efficacy of HBCiF‐NPs In Vivo
BALB/c mice used in this study were approved and handled in accordance with the guidelines provided by the Committee on the Use of Live Animals in Teaching and Research (CULATR) (CULATR reference number: 5881‐21). The 4T1 tumor‐bearing mouse model was established by subcutaneous injection of 2 × 106 4T1 tumor cells into the flank of mice. When the average tumor volume reached around 100 mm3, mice were randomly divided into 6 groups (n = 5): 1) Saline, 2) free drugs (Ce6&iFSP1 + 530 nm (80 mW cm−2, 5 min) + 660 nm light irradiations (20 mW cm−2, 2 min), 3) HBCiF‐NPs + 530 nm light irradiation, 4) HBCiF‐NPs + 530 nm light irradiation, 5) HBC‐NPs + 530 nm + 660 light irradiation, 6) HBCiF‐NPs + 530 nm + 650 light irradiation. The dosages of Ce6 and iFSP1 were 1.5 mg kg−1 and 0.75 mg kg−1, respectively. Light irradiations were utilized immediately at the tumor site in sequence after administration. The procedure was repeated four times at interval of one day. The tumor volumes and mice body weights were monitored every other day. Tumor volume was calculated in accordance with the following formula:
| (1) |
After sacrificing tumor‐bearing mice, tumors and spleens of mice were harvested, weighed, and photographed. Additionally, the tumor tissues and major organs including heart, liver, spleen, lung, and kidney were paraffin‐embedded and sectioned for H&E staining and immunofluorescence staining.
In Vivo Anti‐Tumor Efficacy Analysis of HNCiF‐NPs
When the tumor volume reached ≈100 mm3, mice were randomly divided into 5 groups (n = 5): 1) Saline, 2) Ce6&iFSP1 + 660 nm light irradiation, 3) αPD‐L1, 4) HNCiF‐NPs + 660 nm light irradiation, 5) αPD‐L1 + HNCiF‐NPs + 660 nm light irradiation. The dosages of Ce6 and iFSP1 were 0.75 mg kg−1 and 0.38 mg kg−1, respectively. Saline, Ce6&iFSP1, and HNCiF‐NPs were administered by intravenous injection. The procedure was repeated 4 times at interval of one day. After injection, 660 nm light irradiation (60 mW cm−2, 1 min) was immediately applied at the tumor site. αPD‐L1 was intraperitoneally injected 2 h after the 2nd and 4th nanoparticle administration. The tumor volumes and mice body weights were monitored every other day. After treatments, blood samples were collected via heart puncture and incubated at room temperature for 30 min. Serum was separated from blood through centrifuging twice at 6000 rpm for 15 min. Afterward, mice were sacrificed, tumors were harvested, weighted, and photographed. The tumor tissues and major organs were paraffin‐embedded and sectioned for H&E staining and immunohistochemical staining.
Splenic T Cell and Dendritic Cell (DC) Analysis
To determine the ratio of CD8+ T cells in splenic T cells, the spleens were cut into small pieces, digested with collagenase IV, and filtered to acquire the single cell suspension. After that, the single cell suspension was stained with mixture of anti‐CD45‐FITC, anti‐CD3‐APC/Cy7, anti‐CD4‐Pacific blue, and anti‐CD8‐Percp/Cy5.5 antibodies for T cells. To quantify dendritic cell maturation, single cells from different groups were stained with mixture of anti‐IA/IE‐FITC, anti‐CD11c‐Pacific blue, anti‐CD80‐APC, and anti‐CD86‐PE/Cy7 antibodies. The immune cell population and mean fluorescence intensity were analyzed via flow cytometry (CytoFLEX, Beckman).
In Vivo Biosafety Evaluation
To evaluate the biosafety of HNCiF‐NPs, major organs including heart, liver, spleen, lung, and kidney and blood samples were collected after sacrificing the treated mice. The morphologies of organs were observed after H&E staining. In addition, serum activities of ALT and AST were determined to evaluate if HNCiF‐NPs caused toxicity on liver.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
Y. Z. and K. C. contributed equally to this work. This work was supported by the National Natural Science Foundation of China (Excellent Young Scientists Fund, No. 82222903), Health and Medical Research Fund of Hong Kong (No. 07181936), Li Ka Shing Faculty of Medicine (Start‐up Fund) of The University of Hong Kong, and Seed Fund for Basic Research of The University of Hong Kong (No. 202111159152).
Zhou Y., Chen K., Lin W. K., Liu J., Kang W., Zhang Y., Yang R., Jin L., Cheng Y., Xu A., Wang W., Photo‐Enhanced Synergistic Induction of Ferroptosis for Anti‐Cancer Immunotherapy. Adv. Healthcare Mater. 2023, 12, 2300994. 10.1002/adhm.202300994
Contributor Information
Aimin Xu, Email: amxu@hku.hk.
Weiping Wang, Email: wangwp@hku.hk.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.a) Mansoori B., Mohammadi A., Davudian S., Shirjang S., Baradaran B., Adv. Pharm. Bull. 2017, 7, 339; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Su Z., Dong S., Zhao S.‐C., Liu K., Tan Y., Jiang X., Assaraf Y. G., Qin B., Chen Z.‐S., Zou C., Drug Resist. Updates 2021, 58, 100777; [DOI] [PubMed] [Google Scholar]; c) Weiss F., Lauffenburger D., Friedl P., Nat. Rev. Cancer 2022, 22, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., Patel D. N., Bauer A. J., Cantley A. M., Yang W. S., Morrison B., Stockwell B. R., Cell 2012, 149, 1060; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Conrad M., Angeli J. P. F., Vandenabeele P., Stockwell B. R., Nat. Rev. Drug Discovery 2016, 15, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Zhang C., Liu X., Jin S., Chen Y., Guo R., Mol Cancer 2022, 21, 47; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao M., Deng J., Liu F., Fan A., Wang Y., Wu H., Ding D., Kong D., Wang Z., Peer D., Zhao Y., Biomaterials 2019, 223, 119486. [DOI] [PubMed] [Google Scholar]
- 4.a) Zhao L., Zhou X., Xie F., Zhang L., Yan H., Huang J., Zhang C., Zhou F., Chen J., Zhang L., Cancer Commun 2022, 42, 88; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yu Y., Huang Z., Chen Q., Zhang Z., Jiang H., Gu R., Ding Y., Hu Y., Biomaterials 2022, 288, 121724. [DOI] [PubMed] [Google Scholar]
- 5.a) Wen Q., Liu J., Kang R., Zhou B., Tang D., Biochem. Biophys. Res. Commun. 2019, 510, 278; [DOI] [PubMed] [Google Scholar]; b) Shi L., Liu Y., Li M., Luo Z., FEBS J 2022, 289, 3655; [DOI] [PubMed] [Google Scholar]; c) Wiernicki B., Maschalidi S., Pinney J., Adjemian S., Vanden Berghe T., Ravichandran K. S., Vandenabeele P., Nat. Commun. 2022, 13, 3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Song X., Wang X., Liu Z., Yu Z., Front Oncol 2020, 10, 597434; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ursini F., Maiorino M., Free Radic Biol Med 2020, 152, 175. [DOI] [PubMed] [Google Scholar]
- 7.a) Song R., Li T., Ye J., Sun F., Hou B., Saeed M., Gao J., Wang Y., Zhu Q., Xu Z., Yu H., Adv. Mater. 2021, 33, 2101155; [DOI] [PubMed] [Google Scholar]; b) Meng X., Deng J., Liu F., Guo T., Liu M., Dai P., Fan A., Wang Z., Zhao Y., Nano Lett. 2019, 19, 7866; [DOI] [PubMed] [Google Scholar]; c) Guo X., Liu F., Deng J., Dai P., Qin Y., Li Z., Wang B., Fan A., Wang Z., Zhao Y., ACS Nano 2020, 14, 14715. [DOI] [PubMed] [Google Scholar]
- 8.a) Li Y., Qin Y., Shang Y., Li Y., Liu F., Luo J., Zhu J., Guo X., Wang Z., Zhao Y., Adv. Funct. Mater. 2022, 32, 2112000; [Google Scholar]; b) Zhu J., Dai P., Liu F., Li Y., Qin Y., Yang Q., Tian R., Fan A., Medeiros S. d. F., Wang Z., Nano Lett. 2020, 20, 6235. [DOI] [PubMed] [Google Scholar]
- 9.a) Zhang P., Liu C., Wu W., Mao Y., Qin Y., Hu J., Hu J., Fu J., Hua D., Yin J., Chem. Eng. J. 2021, 425, 131543; [Google Scholar]; b) Zhu T., Shi L., Yu C., Dong Y., Qiu F., Shen L., Qian Q., Zhou G., Zhu X., Theranostics 2019, 9, 3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doll S., Freitas F. P., Shah R., Aldrovandi M., da Silva M. C., Ingold I., Goya Grocin A., Xavier da Silva T. N., Panzilius E., Scheel C. H., Mourao A., Buday K., Sato M., Wanninger J., Vignane T., Mohana V., Rehberg M., Flatley A., Schepers A., Kurz A., White D., Sauer M., Sattler M., Tate E. W., Schmitz W., Schulze A., O'Donnell V., Proneth B., Popowicz G. M., Pratt D. A., et al., Nature 2019, 575, 693. [DOI] [PubMed] [Google Scholar]
- 11. Bersuker K., Hendricks J. M., Li Z., Magtanong L., Ford B., Tang P. H., Roberts M. A., Tong B., Maimone T. J., Zoncu R., Bassik M. C., Nomura D. K., Dixon S. J., Olzmann J. A., Nature 2019, 575, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou Y., Gao Y., Pang L., Kang W., Man K., Wang W., Nano Res. 2023, 16, 1042. [Google Scholar]
- 13.a) Chen K., Chen Q., Wang K., Zhu J., Li W., Li W., Qiu L., Guan G., Qiao M., Zhao X., Int. J. Pharm. 2016, 509, 314; [DOI] [PubMed] [Google Scholar]; b) Wei G., Chen J., Jing Z., Li Y., Li Z., Zheng W., Sun X., Zhao W., Zhang Z., Wang X., J. Colloid Interface Sci. 2022, 608, 1355. [DOI] [PubMed] [Google Scholar]
- 14. Caminade A. M., J Pers Med 2022, 12, 1334.36013283 [Google Scholar]
- 15.a) Nam H. Y., Nam K., Hahn H. J., Kim B. H., Lim H. J., Kim H. J., Choi J. S., Park J.‐S., Biomaterials 2009, 30, 665; [DOI] [PubMed] [Google Scholar]; b) Jin G.‐w., Koo H., Nam K., Kim H., Lee S., Park J.‐S., Lee Y., Polymer 2011, 52, 339. [Google Scholar]
- 16. Hu B., Ma Y., Yang Y., Zhang L., Han H., Chen J., Oncol Lett 2018, 15, 5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Souri M., Soltani M., Moradi Kashkooli F., Kiani Shahvandi M., J. Controlled Release 2022, 341, 227; [DOI] [PubMed] [Google Scholar]; b) Qian X., Xu X., Wu Y., Wang J., Li J., Chen S., Wen J., Li Y., Zhang Z., J. Controlled Release 2022, 346, 193. [DOI] [PubMed] [Google Scholar]
- 18. Han X., Zhong Z., Kou J., Zheng Y., Liu Z., Jiang Y., Zhang Z., Gao Z., Lin C., Tian Y., Cell. Physiol. Biochem. 2018, 48, 1616. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y., Wang Y., Liu J., Kang R., Tang D., Cancer Gene Ther. 2021, 28, 55. [DOI] [PubMed] [Google Scholar]
- 20. Lushchak V. I., J Amino Acids 2012, 2012, 736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu J., Wang X., Su Y., Shao J., Song X., Wang W., Zhong L., Gan L., Zhao Y., Dong X., Biomaterials 2022, 288, 121704. [DOI] [PubMed] [Google Scholar]
- 22.a) Wang H., Liu C., Zhao Y., Gao G., Eur. J. Cell Biol. 2020, 99, 151058; [DOI] [PubMed] [Google Scholar]; b) Liang H., Wu X., Zhao G., Feng K., Ni K., Sun X., J. Am. Chem. Soc. 2021, 143, 15812. [DOI] [PubMed] [Google Scholar]
- 23. Chen C., Wang Z., Jia S., Zhang Y., Ji S., Zhao Z., Kwok R. T., Lam J. W., Ding D., Shi Y., Adv. Sci. 2022, 9, 2104885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krysko D. V., Garg A. D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P., Nat. Rev. Cancer 2012, 12, 860. [DOI] [PubMed] [Google Scholar]
- 25. Wang W., Green M., Choi J. E., Gijón M., Kennedy P. D., Johnson J. K., Liao P., Lang X., Kryczek I., Sell A., Xia H., Zhou J., Li G., Li J., Li W., Wei S., Vatan L., Zhang H., Szeliga W., Gu W., Liu R., Lawrence T. S., Lamb C., Tanno Y., Cieslik M., Stone E., Georgiou G., Chan T. A., Chinnaiyan A., Zou W., Nature 2019, 569, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Ma T., Zhang Q., Xuan Q., Zhuang J., Zhang W., Li H., Chen C., Wang P., Chem. Eng. J. 2021, 424, 130536; [Google Scholar]; b) Wang Y., Gong N., Ma C., Zhang Y., Tan H., Qing G., Zhang J., Wang Y., Wang J., Chen S., Li X., Ni Q., Yuan Y., Gan Y., Chen J., Li F., Zhang J., Ou C., Zhao Y., Liu X., Liang X.‐J., Nat. Commun. 2021, 12, 4964; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu H., Hu Y., Sun Y., Wan C., Zhang Z., Dai X., Lin Z., He Q., Yang Z., Huang P., Xiong Y., Cao J., Chen X., Chen Q., Lovell J. F., Xu Z., Jin H., Yang K., ACS Nano 2019, 13, 12638. [DOI] [PubMed] [Google Scholar]
- 27. Jiang W., Li Y., Zhang S., Kong G., Li Z., Oncol Lett 2021, 22, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang J., Higgins S. L., Winkel B. S., Brewer K. J., Chem. Commun. 2011, 47, 9786. [DOI] [PubMed] [Google Scholar]
- 29. Wang W., Green M., Choi J. E., Gijon M., Kennedy P. D., Johnson J. K., Liao P., Lang X., Kryczek I., Sell A., Xia H., Zhou J., Li G., Li J., Li W., Wei S., Vatan L., Zhang H., Szeliga W., Gu W., Liu R., Lawrence T. S., Lamb C., Tanno Y., Cieslik M., Stone E., Georgiou G., Chan T. A., Chinnaiyan A., Zou W., Nature 2019, 569, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bahmani B., Gong H., Luk B. T., Haushalter K. J., DeTeresa E., Previti M., Zhou J., Gao W., Bui J. D., Zhang L., Fang R. H., Zhang J., Nat. Commun. 2021, 12, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sher Y.‐P., Hung M.‐C., Bio Protoc 2013, 3, e931. [Google Scholar]
- 32. Peterson J. A., Wijesooriya C., Gehrmann E. J., Mahoney K. M., Goswami P. P., Albright T. R., Syed A., Dutton A. S., Smith E. A., Winter A. H., J. Am. Chem. Soc. 2018, 140, 7343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.