

Abstract
Subcutaneous (SC) administration is a desired route for monoclonal antibodies (mAbs). However, formulating mAbs for small injection volumes at high concentrations with suitable stability and injectability is a significant challenge. Here, this work presents a platform technology that combines the stability of crystalline antibodies with injectability and tunability of soft hydrogel particles. Composite alginate hydrogel particles are generated via a gentle centrifugal encapsulation process which avoids use of chemical reactions or an external organic phase. Crystalline suspension of anti‐programmed cell death protein 1 (PD‐1) antibody (pembrolizumab) is utilized as a model therapeutic antibody. Crystalline forms of the mAb encapsuled in the hydrogel particles lead to stable, high concentration, and injectable formulations. Formulation concentrations as high as 315 mg mL−1 antibody are achieved with encapsulation efficiencies in the range of 89–97%, with no perceivable increase in the number of antibody aggregates. Bioanalytical studies confirm superior maintained quality of the antibody in comparison with formulation approaches involving organic phases and chemical reactions. This work illustrates tuning the alginate particles’ disintegration by using partially oxide alginates. Crystalline mAb‐laden particles are evaluated for their biocompatibility using cell‐based in vitro assays. Furthermore, the pharmacokinetics (PK) of the subcutaneously delivered human anti‐PD‐1 mAb in crystalline antibody‐laden alginate hydrogel particles in Wistar rats is evaluated.
Keywords: antibody delivery, bioavailability, biologics formulation, biomaterials, crystalline formulations, hydrogel particles
Subcutaneous administration is a desired route for monoclonal antibodies (mAbs). However, formulating mAbs for small injection volumes at high concentrations with suitable stability and injectability is a significant challenge. Here, a platform technology that combines the stability of crystalline antibodies with injectability and tunability of soft hydrogel particles is presented to achieve stable, high concentration, and injectable formulations.
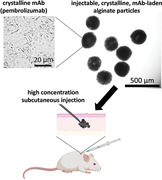
1. Introduction
Many recent innovative therapies are based on monoclonal antibody (mAb) treatments.[ 1 , 2 , 3 ] For example, antibodies against programmed cell death protein 1 (PD‐1) and its ligand, have revolutionized the clinical treatment of cancer.[ 4 ] While all immune‐oncology mAb treatments are administrated intravenously (IV), recently subcutaneous (SC) administration of mAbs has gained significant attention.[ 5 ] Medications such as rituximab and trastuzumab are being clinically evaluated for SC administration.[ 5 ] SC or local injection of therapeutic agents can bring clinically meaningful benefits to patients[ 6 ] and potentially reduced side effects as compared to IV administration.[ 7 ] One of such benefits can be the reduced time of administration. For example, for rituximab the total time at clinic for IV injection is ≈3.7 h which can be reduced to ≈0.8 h for SC administration.[ 8 ] Formulating mAb for SC or local administration requires small volume (ideally ≤ 2 mL), high concentration (ideally ≥ 300 mg mL−1), and moderate viscosity for injectability (ideally ≤ 0.025 Pa s). Immunotherapy mAbs require a significant amount of the antibody for the treatment. For instance, anti‐PD‐1 antibody pembrolizumab dosing regimen is 400 mg every 6 weeks. Considering the limitation in the volume of a local injection, formulating the mAb at required high concentrations can be a major challenge as it exceeds the concentration of any commercially available antibody formulation. As importantly, liquid mAbs at high concentrations become increasingly more prone to aggregation, unfolding, and degradation.[ 9 ] Another challenge related to formulation of biologics arises from the current trend in the field of immune oncology toward combinatorial therapies as combination of mAbs or mAbs and cytokines can even be more challenging to formulate for liquid injections. Considering alternative strategies to enhance protein colloidal stability in such conditions can be crucial in formulating high concentration formulations. Formulating mAbs as solid (e.g., crystalline and amorphous) suspensions in injectable forms can overcome these challenges. Similarly, using biodegradable particulate delivery carriers such as injectable microspheres can play a role by enabling new modes of therapy through carrier design.
Protein crystals are hydrated (≈50% w/w water content) nonstoichiometric structures that have protein molecules within them. Proteins in a crystal are densely packed (>500 mg mL−1), folded (hence functional),[ 10 ] and highly stable in this immobilized state. Protein crystals are formed in the presence of certain salts or other excipients additives. Suspensions of protein crystals can be concentrated by centrifugation or sedimentation and can dissolve outside of the crystallization conditions. These crystals may be formed in a variety of sizes, but suspensions still exceed desirable viscosities for injection.[ 11 ] Conditions can be tuned to produce micron‐scale protein crystals[ 12 ] which can enable encapsulation in hydrogel particles. In our previous work, we have shown that encapsulating such solid suspensions in soft hydrogel particles improves injectability properties compared to a mAb solution or crystal suspensions.[ 13 ]
Hydrogels (cross‐linked networks of hydrophilic polymers) are seen as viable candidates for a range of drug delivery applications.[ 14 , 15 ] This approach can be extended to SC or local delivery of mAbs.[ 16 , 17 , 18 , 19 ] Hydrogels are capable of holding significant amounts of water, biomacromolecules or suspensions within them because of their low polymer content (high theoretical drug loadings). Hydrogels’ modular chemistry, structure, and functionality enable new therapies through carrier design with tunable mesh size and drug release.[ 15 ] Furthermore, hydrogel particles are shear thinning even at high volume fractions and have favorable flow properties.[ 20 ]
While hydrogels can be particularly suitable for unstable drugs (e.g., labile biomacromolecules), hydrogels encapsulating antibodies have not reached clinal studies or commercialization.[ 21 ] There are major difficulties that can act as deterrents in the development of such strategies.[ 22 , 23 ] These can be attributed to shortcomings in materials and processes that are considered that can make translational research unfeasible or are making the technology impractical from the manufacturing perspective. It has been shown that some polymeric networks can cause protein aggregation and denaturation which can take place during the encapsulation or storage.[ 24 ] This can be attributed to the harsh conditions for encapsulation and particle formation that promote protein destabilization. Furthermore, many of the studies using hydrogels for encapsulation of biologics produce the hydrogel through chemical reactions in the presence of the therapeutic biologics[ 25 , 26 , 27 ] which can cause denaturation, unwanted chemical modifications remnant impurities. Such impurities or unwanted chemical/physical alternations to the mAb are a major concern in clinical studies. Furthermore, particle formation through emulsion‐based methods requires an organic phase that can adversely contribute to protein denaturing and also lower encapsulation efficiencies. This generates the need for processes that are gentle and are fully compatible with labile nature of the therapeutic molecules and clinical research.
Sodium alginate (NaALG) is a natural polysaccharide that can form a hydrogel by ionic cross‐linking in mild conditions without use of toxic reactants, making it widely used for biomedical applications.[ 28 , 29 , 30 , 31 , 32 ] NaALG linear polysaccharide chain is comprised of mannuronic (M‐blocks) and guluronic group (G‐blocks) in which only G‐blocks are assumed to take part in the intermolecular ionic cross‐linking with divalent cations such as Ca2+.[ 33 ] While ALG has excellent biocompatible properties, is not enzymatically broken down in vivo hence its slow and uncontrollable degradation can be a cause of concern for bioaccumulation.[ 30 ] Introduction of hydrolytic labile groups on the ALG backbone using partial oxidation has been proposed as a method to enhance biodegradability.[ 34 ] Yet ability of these modified ALGs to form hydrogel particles and to be benign to the mAb chemical/physical stability requires investigation. ALG hydrogel particles can be produced if NaALG droplets are formed and later cross‐linked in a calcium containing bath. Such process can avoid the need for using emulsion‐based methods which can cause protein aggregation/denaturing. We have previously described the governing principles for production of ALG particles via centrifugal extrusion without use of an organic phase.[ 35 ] Yet manufacturing crystalline mAb‐laden ALG particles by extrusion poses unique challenges caused by the presence of shear‐thinning crystals, the need to achieve high antibody encapsulation efficiency and maintain antibody stability throughout the formulation process.
Here, we report a platform that enables highly efficient, densely packed, injectable formulations of biologics. We establish a process to encapsulate crystalline mAbs in ALG hydrogel particles to form high‐concentration, stable formulations and use an anti‐PD‐1 antibody as a model therapeutic. We assess centrifugal extrusion for particle formation and study the design consideration for this process to prevent premature dissolution throughout processing. We evaluate the biodegradation of chemically modified ALG. Furthermore, we evaluate the formulation stability the quality of released mAb from the particles using bioanalytical methods. Our in vitro studies evaluate the biocompatibility (biodegradability, toxicity, and immunogenicity) of the crystalline mAb‐laden particles. Our approach can prevent unwanted chemical reactions and biomacromolecule aggregation and denaturing. Our in vivo rat studies evaluate the technology for SC delivery of mAbs.
2. Results and Discussion
2.1. Crystalline mAb‐Laden ALG Particles
Figure 1 illustrates the process for production of crystalline mAb‐laden ALG particles. This gentle process enables the encapsulation without use of chemical reactions or an external organic phase. Briefly, antibody crystals were formed by precipitation, crystals were resuspended in NaALG, concentrated, and crystalline mAb‐laden ALG particles were formed by centrifugal extrusion. For the studied mAb, crystals were generated by crystallization of mAb in the presence of polyethylene glycol (PEG) and caffeine at 6% and 0.25% w/v respectively (Figure 1a). Caffeine was used as a small molecule ligand that noncovalently bonds to the anti‐PD‐1 antibodies through hydrophobic interaction to enhance crystal nucleation and growth based on our recent findings on anti‐PD‐1 crystal.[ 36 ] Crystallization recovery of up to 98% was achieved during this process. This crystallization condition resulted in antibody crystals 2–7 µm in size which are large enough for centrifugal separation and are suitable for encapsulation (Figure 1b). The mAb crystal suspensions were concentrated and mixed with 10 mg mL−1 NaALG solution to form the pre‐gel suspension and was further concentrated to the desired concentration (Figure 1d). The highest suspension concentration achieved was measured at 476 ± 26 mg mL−1. The encapsulation process was evaluated for low (50 mg mL−1) to high (400 mg mL−1) mAb concentration in the pre‐gel. Later, using centrifugal acceleration, the pre‐gel suspension was extruded through a dispenser to form droplets that cross‐linked upon impact with the CaCl2 bath (Figure 1e). The key advantage of this device is that the droplets are formed in air (continuous phase), eliminating the need for an organic continuous phase or use of surfactant in the continuous or dispersed phase. Both the pre‐gel suspension and the CaCl2 bath contained 10% w/v PEG to avoid dissolution of the crystals. Afterwards the particles were resuspended in 6% w/w PEG solution for injection (Figure 1f).
Figure 1.
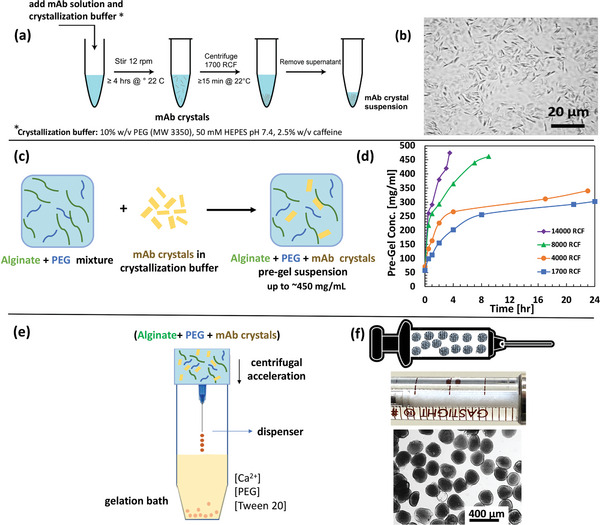
Illustration of the platform for crystal monoclonal antibody (mAb) laden alginate hydrogel particles for high‐concentration antibody formulations evaluated for anti‐PD‐1 antibody (pembrolizumab) crystals as a model antibody. a) mAb crystallization in polyethylene glycol (PEG) solution. b) Micrograph of mAb crystals. c) Preparation of alginate and mAb crystals pre‐gel suspension. d) Concentrating the mAb + alginate suspension using centrifugation. e) Device for preparation of the crystalline mAb‐laden hydrogel particles formed by centrifugal extrusion and hydrogel formation upon impact with Ca2+ bath. f) Appearance of the crystalline mAb‐laden alginate hydrogel particles for subcutaneous (SC) administration loaded in a syringe for SC injection.
2.2. Process Design Considerations
Manufacturing the (ALG + mAb crystals) particles by extrusion (Figure 1e) poses unique challenges. In prior work we have studied the principles of forming ALG particles with use of centrifugal forces.[ 35 ] However, in our current study the presence of solid particles in the ALG suspension along with their shear‐thinning nature can impact particle formation. Furthermore, while high mAb recovery can be achieved during the crystallization process, crystalline mAbs require specific conditions to prevent premature dissolution throughout the hydrogel encapsulation and recovery processes to achieve a high mAb encapsulation efficiency.
Here, we investigated the process design considerations for centrifugal extrusion of crystalline mAb‐laden ALG particles. The goal is to produce uniform spherical particles containing high mAb loadings while having minimal loss of the mAb to the surrounding bath. Furthermore, the particles need to be injectable using a 27‐gauge needle (with internal dimeter of 210 µm) which is commonly used for SC injection.
2.2.1. Physical Process Variables
Physical variables that control the centrifugal extrusion and consequently the product are described in Figure 2a. The pre‐gel suspension flows downwards due to the centrifugal acceleration. Droplets are formed at the tip of the dispenser in the “dripping regime” (refer to the Supporting Information for the dimensionless numbers that dictate the dripping regime). Droplets then travel the distance to the bath (while accelerating) and are ionically cross‐linked during the “impact/gelation” phase.
Figure 2.
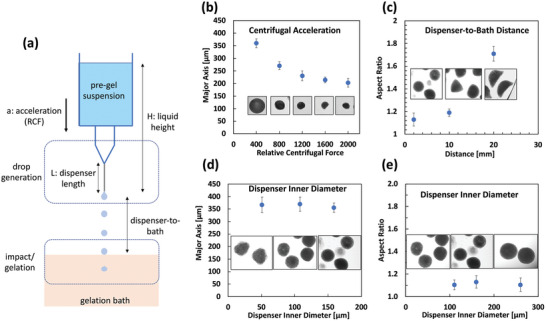
Effect of physical process variables on the properties (particle size and aspect ratio) of the hydrogel particles. a) Droplets are formed at the tip of the dispenser and are cross‐linked upon impact with the calcium chloride bath. Particle formation is dictated by the drop generation and the impact/gelation process. b) Relative centrifugal force (RCF) dictates the size of the particles (particle monoclonal antibody (mAb) loading of 160 mg mL−1). c) Distance between the dispenser and the point of impact affects the impact speed and consequently the particle shape. Particles flatten as collection distance increases. d,e) Dispenser inner diameter has minimal impact on the particle size (d) and shape (e) within the studied range. Experiments at particle mAb loading of 200 mg mL−1 using 1% w/v guluronic rich alginate (MVG), 2 mm dispenser‐to‐bath, 200 relative centrifugal force (RCF) spin rate, 30G needle, and 25 × 10−3 m CaCl2 unless otherwise noted. Sample size was n = 15.
Figure 2b–e illustrates the effects of centrifugal acceleration, dispenser size, and collection distance (dispenser‐to‐bath) on the particles. Experiments were carried out at particle mAb loading of 200 mg mL−1. While centrifugal acceleration reported in relative centrifugal force (RCF) dictates the particle size, shape of the particles is dictated by the dispenser‐to‐bath distance which controls the particle impact speed. Figure 2c indicates that at higher accelerations particles flatten due to inertia caused by the higher accelerations. Interestingly the dispenser inner diameter is less significant in controlling the particle size.
2.2.2. Physiochemical Process Variables
There are physiochemical variables that also control this process, including Ca2+ concentration (in the bath), ALG type/concentration, and additives/surfactant (Figure 3 ). The experiments for the physicochemical process variable were also carried out at particle mAb loading of 200 mg mL−1. In the proposed process the impact/gelation phase has an extra level of complexity caused by the presence of the mAb crystals. The CaCl2 bath needs prevent the mAb crystal dissolution during the hydrogel cross‐linking stage. Furthermore, the amount of CaCl2 in the product should not exceed the limit at which it negatively affects the mAb. Figure 3a shows the unwanted dissolution of the mAb crystals in the cross‐linking bath can be avoided by keeping the CaCl2 concentration at or below 20 × 10−3 m. Furthermore, Figure 3b shows how at higher Ca2+ concentrations not only the mAb crystals dissolve but also particles develop teardrop shapes.
Figure 3.
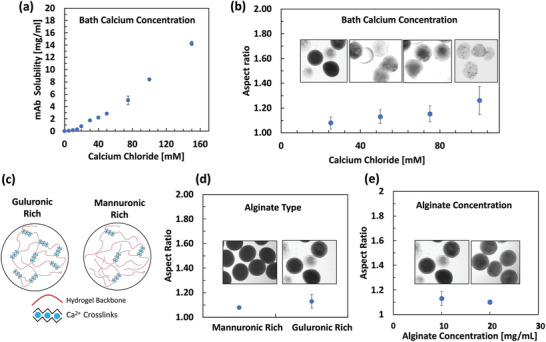
Effect of physiochemical process variables on the properties of the of the hydrogel particles. a) Excess calcium concentration resulted in the unwanted dissolution of the monoclonal antibody (mAb) crystal (samples in triplicate). b) Particles at lower CaCl2 concentration are rounder. c) Guluronic moieties in the sodium alginate are cross‐linked with Ca2+ and as a result, the guluronic rich alginate are more cross‐linked. d) Mannuronic rich alginate forms more spherical particles. e) Spherical crystalline mAb‐laden hydrogel particle can be formed with alginate concentration as low as 10 mg mL−1 alginate. Experiments at particle mAb loading of 200 mg mL−1 using 1% w/v guluronic rich alginate (MVG), 2 mm dispenser‐to‐bath, 200 relative centrifugal force (RCF) spin rate, 30G needle, and 25 × 10−3 m CaCl2 unless otherwise noted. b,d,e) Sample size was n = 15.
A wide variety of NaALGs have been used for biomedical applications, varying in their molecular weights (MWs) and guluronic (G) to mannuronic (M) ratios. G‐blocks are assumed to take part in the intermolecular cross‐linking with divalent cations.[ 28 ] We also evaluated guluronic rich (G/M < 1) and mannuronic rich ALGs (G/M >1.5) for particle formation (and in section 2.3 for their degradation profiles). Our observations indicated that mannuronic‐rich ALG tend to form more spherical particles. Also, we observed that hydrogel particles encapsulating crystalline mAbs can be formed at ALG concentrations as low 10 mg mL−1. To improve the shape of the particles formed by very low viscosity ALG, nonionic surfactant Tween 20 was added to the collection bath to lower the interfacial tension during droplet impact with the bath. For the data on the effect of ALG type and effect of Tween 20 on particle shape refer to Figures S1 and S2 (Supporting Information).
2.2.3. Characterization of Anti‐PD‐1 Crystalline mAb‐Laden ALG Particles
As discussed earlier, in our method suspensions of ALG and mAb crystals are formed and concentrated to the desired concentrations. Later the particles are formed through centrifugal extrusion. Figure 4a illustrates the particle loading (concentration of the mAb within particles) as a function of the pre‐gel suspension. The experiments were carried out from low (50 mg mL−1) to high (400 mg mL−1) mAb concentration in the pre‐gel. To measure particle loading, the concentration of the antibody in a small aliquot of the pre‐gel suspension was measured. After producing the hydrogels, the mAb was released from the hydrogel and the concentration of the mAb within the hydrogel particles was estimated. Our results indicated that at low concentrations the particle loadings were slightly higher than the pre‐gel suspension and the opposite trend at higher concentrations. This can be attributed to the change in the water content of the hydrogel (swelling/deswelling) upon gelation and resuspension.
Figure 4.
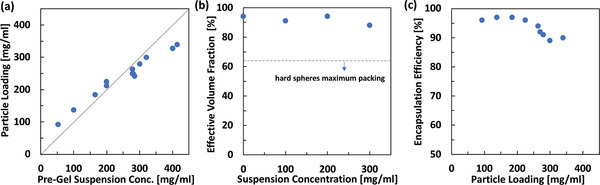
Characterization of the monoclonal antibody (mAb) crystalline mAb‐laden alginate hydrogel particles. a) Relationship between the mAb concentration in the pre‐gel suspension and the particle loading. b) Soft deformable hydrogel particles allow high effective volume fractions even at high antibody loadings. c) Encapsulation efficiency defined as the amount of mAb encapsulated relative to the total amount of mAb crystals in the pre‐gel suspension. Hydrogel particles were formed using 1% w/v mannuronic rich alginate (VLVM). The experiments were carried out from low (50 mg mL−1) to high (400 mg mL−1) mAb concentration in the pre‐gel.
The formulation concentration of the crystalline mAb‐laden particles can be estimated as:
| (1) |
The hydrogel particles’ soft nature affords high effective volume fraction formulations while maintaining shear thinning properties.[ 20 ] To measure the effective volume fraction, the hydrogel particles were centrifuged at 3000 RCF for 10 min and the excess PEG 10% w/v solution was removed (from the top) using a micropipette to achieve the measured particle volume fraction. The effective volume fraction was measured gravimetrically (refer to Equation S1, Supporting Information). In our work, ALG hydrogel particles’ soft nature allows packing of these particle to effective volume fractions as high as 0.9 (Figure 4b). Considering the highest particle concentrations in Figure 4a, formulation concentrations as high as 315 mg mL−1 in mAb were achieved. Comparably hard spheres have a maximum volume packing fraction in the range of 0.58–0.64. As such, for hard spheres achieving similar formulation concentrations of up to 315 mg mL−1 will require significantly higher particle mAb loadings. To put the achieved 315 mg mL−1 formulation into perspective, the majority of high concentration formulations of therapeutic mAbs are at 100–150 mg mL−1, while the highest concentration available is 200 mg mL−1.[ 37 ] Refer to Videos S1 and S2 (Supporting Information) for the ejection of particles from a 27‐gauge needle. The ejected particles were intact when visually compared to the particles before injection (Figure S3, Supporting Information).
The encapsulation efficiency was defined as the mass of antibody encapsulated over the total mass of the mAb crystal in the pre‐gel suspension. To evaluate the encapsulation efficiency, the concentration of the mAb in the CaCl2 bath, resuspension PEG buffer, and the produced particles were measured. The encapsulation efficiency varied between 89% and 97% w/w (Figure 4c) which is much higher than typical encapsulation efficiencies reported for biomacromolecules in microspheres.[ 32 , 38 ] In comparison, prior studies reported encapsulation efficiency of 60% for solid‐in‐oil‐in‐water method,[ 38 ] up to 75% for spray drying method,[ 39 ] and 75% for double emulsion and solvent evaporation technique.[ 32 ]
Figure 5a–c shows the microscopy images of the anti‐PD‐1 crystalline mAb‐laden particles. Figure 5a,b shows particles at concentration of 100 mg mL−1 (low, yet clinically relevant concentration). Particles in Figure 5b,c are at 350 mg mL−1 particle mAb loading (the higher limit of concentration for our proposed process). Particles appear opaque in bright‐field microscopy and appear bright under cross‐polarized microscopy due to the presence of the mAb crystals. Figure 5c demonstrates the colocalization of opaque particles in brightfield, two‐photon excited UV fluorescence from the mAb, and second‐order nonlinear optical imaging of chiral crystals (SONICC) indicating the expected chiral protein crystal structure of the encapsulated antibody.
Figure 5.

Microscopy images of the crystalline monoclonal antibody (mAb) laden alginate hydrogel particles. a) Bright‐field micrograph (left) and cross‐polarized microscopy (right) at 100 mg mL−1 particle loading. b) Bright‐field micrograph (left) and cross‐polarized microscopy (right) at 350 mg mL−1 particle loading. The cross‐polarized microscopy images of the crystalline mAb‐laden hydrogel particles indicated the crystals loaded within the hydrogel particles. c) Bright‐field microscopy (left), ultraviolet two‐photon excited fluorescence (center), and second‐order nonlinear optical imaging of chiral crystals (SONICC, right) imaging of chiral crystals at 250 mg mL−1 particle loading. Scale bars are 400 µm.
2.3. Tuning the Disintegration of ALG Particles by Using Partially Oxidized ALG
Having degradable microgels is essential to eliminate the need for surgical removal after drug delivery is accomplished.[ 40 ] ALG is a biopolymer with excellent gel forming properties, however ALG polymer chains does not enzymatically degrade which can cause bioaccumulation. Additionally, it is important to consider that body fluids also contain calcium ions which can slow down the dissolution of ALG hydrogel. Using partial oxidation of the uronic groups in NaALG to improve degradation has been proposed for improving the biodegradation.[ 34 ] We concentrated on using very low MW ALGs (≤75 KDa) with the rationale that the lower MW ALGs can get cleared from the injection site and body faster. We evaluated the effect of partial oxidation on the disintegration of hydrogel particles formed in this work. Effect of ALG type and extent of partial oxidization on the ALG particle degradation in simulated body fluid (SBF) were measured. SBF was prepared according to the standard and contained among other salts 7.996 mg mL−1 NaCl an 0.278 mg mL−1 CaCl2 to simulate the effect of Na+ and Ca2+ concentration on the particles disintegation.[ 41 ] To prepare partially oxidized ALG, NaALG was reacted with sodium periodate and subsequently the modified ALG was purified. In this reaction one molecule of periodate is consumed by each uronic (monomeric) group in the ALG. Evaluating effect of partial oxidation was carried out for 0% (not modified), 1.5% and 3% oxidation of the uronic units in the ALG. To put the extent of oxidation in perspective, ALG of 75 KDa in MW is consists of ≈350 uronic units. A partial oxidation of 1. 5% or 3% corresponds to ≈5 or ≈10 hydrolyzable units on each polymer chain respectively. Here, we have prepared partially oxidized ALG of both mannuronic rich (ALG M) and guluronic rich (ALG G). The ability of the modified NaALGs to form particles using the centrifugal extrusion method was confirmed. Our results indicated that particles can be formed from both the ALG M and ALG G of up to 3% uronic oxidized (refer to Figure S4, Supporting Information).
Using a gravimetric assay, the disintegration of particles in SBF was measured (Figure 6 ). Firstly, nonmodified ALGs have slow degradation rates in SBF which was in line with the previously reported ALG disintegration in buffers supplemented with calcium.[ 29 ] At the end of 45 days period, 63% and 45% w/w of the of the ALG M of ALG G particles were retained, indicating slow rate of degradation. Furthermore, we observed that partial oxidation affects the particle degradation more for the ALG M particles. To better understand the disintegration rates, the swelling ratio of the hydrogel particles in SBF was measured (refer to Figure S5, Supporting Information). Interestingly swelling ratio of the different hydrogel particles correlated with their disintegration rates. For ALG G we observed an onset of erosion (i.e., the point at which the degradation caused by hydrolysis differentiate between the ALG and modified partially oxidized ALG hydrogel particles). In accordance with the previous literature, we observed improved degradation rates for oxidized ALG particles. Our results indicated that the ALG M hydrogel particles with partial oxidation of 1.5% favorable degradation properties. The better particle shape (Figure 3d) and the tunable degradation properties of ALG M make it the more preferred type of ALG for encapsulation of antibody crystals over the Alg G.
Figure 6.
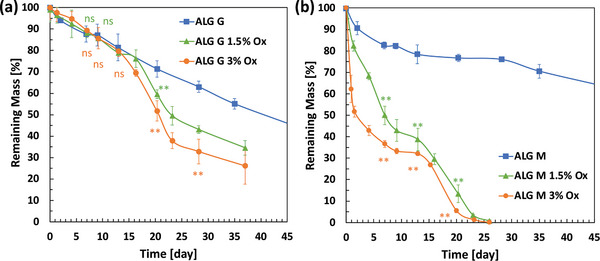
Degradability of the alginate particles formed using low molecular weight alginates (MW ≤ 75 KDa) for a) guluronic rich (ALG G) and b) mannuronic rich (ALG M) alginates with different degrees of partial oxidation in stimulated body fluid (SBF). Partially oxidized ALG G (G/M > 1.5) disintegrates slower that the ALG M (G/M < 1). Mannuronic rich alginate particles with 1.5% and 3% uronic partial oxidation have significantly improved degradability. Samples in triplicate. The t‐test statistical analysis compared each of the partially oxidized ALG samples with the nonoxidized ALG samples. Adjusted p values were indicated as: **p < 0.01, *p < 0.05, ns > 0.05.
2.4. Release of the mAb from ALG Particles
In our formulation anti‐PD‐1 antibody crystals are stabilized within the hydrogel in the presence of PEG. After injection, it is speculated that the PEG dilution from the injection site causes the mAb crystals to dissolve in body fluids. Figure 7a shows cross‐ polarized microscopy images of the crystalline mAb‐laden ALG particles immersed in SBF. At early stage particles are bright, indicating that the mAb remains encapsulated in a crystalline form. Within a few seconds the PEG concentration decreases within the hydrogel, and the crystals start to locally dissolve and are fully dissolved within 120 s. The mAb molecules will later diffuse out from the particle. Particles at different mAb loading of 100 to 310 mg mL−1 were studied and displayed similar release profiles. Furthermore, we compare the release profile for the ALG M and ALG G particles at 200 mg mL−1 particle mAb loading in Figure 7b. For both ALGs the mAb is completely released from the hydrogel within minutes. Similar trends were found for all particle loadings investigated (Figure 7c). The release of mAb from partially oxidized alginates also followed the same trend (refer to Figure S6, Supporting Information). To assure that the encapsulated antibody was completely released, and no quantifiable amount of the antibody was permanently trapped, the SBF was replaced with phosphate buffered saline (PBS, no calcium). In the absence of calcium, the particles were dissolved within days and the concentration of antibody was in this solution was measured to confirm that the antibody was not trapped. This relatively fast complete release suggests that the formulation would not substantially affect the pharmacokinetics (PK) of the drug considering that the antibody absorption by the lymphatic system typically takes 2 or more days.[ 42 , 43 ]
Figure 7.
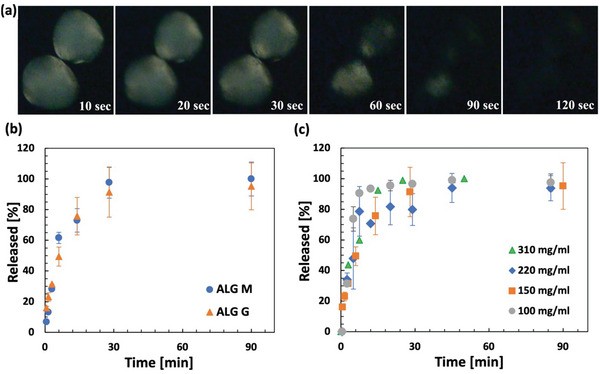
Dissolution and release of the anti‐PD‐1 antibody from the alginate hydrogel particles in simulated body fluid (SBF) at 37 °C. a) Time lapse of cross‐polarized microscopy imaging of the hydrogel particles immersed in SBF which indicates that the antibody crystals are present (bright) at t = 10 s and are dissolved (dark) at t = 120 s. The sample was prepared at 200 mg mL−1 particle loading using ALG M. b) Release profiles of monoclonal antibody (mAb) from guluronic rich (ALG G) and mannuronic rich (ALG M) hydrogel particles at 150 mg mL−1 particle loading. c) Release profiles of the mAb from particles with varying particle loadings indicating that the mAb is completely released from the particles within minutes. Samples in triplicate.
2.5. Quality of the Released mAb Evaluated by Bioanalytical Studies
Our work is aimed at developing an encapsulation process that not only facilitates high particle loadings but also does not affect the protein structure/function which is important in developing formulation of biologics. We evaluated the compatibility of the process, including crystallization, encapsulation, storage, and release, on the mAb quality. For this purpose, the crystalline mAb‐laden ALG particles were prepared at 200 mg mL−1 mAb loading, suspended in the PEG 10% w/v solution, and stored at 4 °C for 5 months.
After the duration of the storage, the release experiments were carried out. For this purpose, the storage buffer (10% w/v PEG solution in 50 × 10−3 m HEPES) was removed. Later the mAb‐laden particles were dispersed in PBS at room temperature for the mAb to release by dissolution and diffusion (the release profile of the mAb in PBS is identical to release in SBF, refer to Figure S7, Supporting Information). The quality of the released mAb was assessed and summarized in Table 1 . The size exclusion chromatography (SEC) data indicates that the entire process has not resulted in any perceivable increase in the number of aggregates and is an indicative of the compatibility of the proposes process with the mAb. Refer to Figure S8 (Supporting Information) for the overlay of SEC of the mAb released from different ALG particles. To assess the function of anti‐PD‐1 antibody subjected to hydrogel processing, samples were dissolved in PBS and were analyzed by an enzyme‐linked immunosorbent assay (ELISA). The ELISA results were within the confidence levels of maintaining bioactivity for all the samples tested, demonstrating that the overall process (precipitation, encapsulation, dissolution, and subsequent handling) did not negatively affect the competitive binding functionality of the anti‐PD‐1 antibody.
Table 1.
Activity and % monomer of the released anti‐PD‐1 antibody from different monoclonal antibody (mAb) crystalline mAb laden alginate particles
| # | Sample | ELISA | Size exclusion chromatography | |
|---|---|---|---|---|
| % Normalized activity | Geometric standard deviation | % Monomer | ||
| 1 | Control (mAb liquid stock) | – | – | 98.0 |
| 2 | Dissolved crystals | 81 | 1 | 97.7 |
| 3 | Alg M | 95 | 1 | 97.3 |
| 4 | Alg G | 97 | 2 | 98.1 |
| 5 | Alg M 1.5%Ox | 89 | 2 | 97.9 |
| 6 | Alg G 1.5%Ox | 106 | 10 | 98.9 |
| 7 | Alg M 3%Ox | 102 | 4 | 97.5 |
| 8 | Alg G 3%Ox | 70 | 4 | 98.3 |
Activity indicates the ability of the mAb in binding with the antibody receptors. Experiments were carried out at particle mAb loading of 200 mg mL−1. Samples 3–8 are the mAbs released from alginate hydrogel particles by dissolution and diffusion in phosphate buffered saline (PBS).
Furthermore, the released mAb was analyzed using ion‐exchange chromatography (IEX) and liquid chromatography mass spectrometry (LC–MS). Our results indicated no charge variants were generated by our process which is important considering the attention in the biopharmaceutical industry to potential influence on the biological activity caused by subtle differences. Furthermore, no mass shifts indicating chemical modification or complexations were detected by LC–MS (refer to Figures S9 and S10, Supporting Information, for the chromatograms). The maintained quality of the mAb indicated the success of the process by avoiding chemical reactions and no use of an organic phase. To confirm this hypothesis, we formed the crystalline mAb laden hydrogel particles using two other chemistries and methods. In Table 2 the bioanalytical studies of the anti‐PD‐1 antibody released from these two methods encapsulating crystalline mAb‐laden hydrogel‐based systems were compared to the ionically cross‐linked alginate hydrogel particles (refer to the Supporting Information for the experimental details). For the free radical polymerization (Case 1), the released mAb showed 6% aggregation and mass shifts in the mAb indicates unwanted chemical modification to the mAb structure. In Case 2, using the more specific click chemistry type reaction the antibody modification was successfully avoided but aggregation number was still 6%. In contrast, our process (Case 3) was able to maintain mAb quality with no increase in the aggregation. Extent of aggregation formation at room temperature and elevated temperature of 37 °C for the crystalline mAb‐laden particles was also quantified and illustrated in Figure S11 (Supporting Information).
Table 2.
Comparison between the technology developed here using ionic cross‐linking of alginate with other chemistries in the monoclonal antibody quality
| Crystalline mAb‐laden hydrogel | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Cross‐linking | Free radical reaction | Click chemistry | Ionic cross‐linking |
| Hydrogel monomer | PEGDA | PEG vinyl sulfone ‐dithiol chemistry | Alginate |
| Production method | Microfluidic | Batch emulsion | Centrifugal extrusion |
| Initiator | UV |
None (slow polymerization) |
Ca2+ |
| Crystallinity | Y | Y | Y |
| Decrease in activity? | N | – | N |
| Aggregation | 6% | 6% | <1% |
| Charge variants detected? | N | N | N |
| Mass shift detected | Y | N | N |
2.6. In Vitro and In Vivo Studies
2.6.1. In Vitro Cytotoxicity and Immunogenicity
Biocompatibility of the particles was examined in vitro by cell viability assay and also monitoring the secretion of proinflammatory cytokines (Figure 8 ). Firstly, viability of the cells in contact with particles (blank) and crystalline mAb‐laden were examined. For this purpose, particles were added to RAW 264.7 macrophage‐like cells (25 000 cells per well) and stored for 20 h. Complete cell viability was observed for all the samples. Also, the assay supernatants were collected and evaluated for the interleukin‐6 (IL‐6) and tumor necrosis factor α (TNFα). For all the samples the IL‐6 was below level of detection and TNFα concentration was minor. Figure 8c indicates that while ALG is not immunogenetic, the ALG M and ALG G particles caused a small statistically significant increase in the TNFα secretion. Interestingly this was not true for the oxidized ALG particles and were confirmed by repeated experiments (refer to Figure S12, Supporting Information). This is in line with previously reported observation of anti‐inflammatory activity of guluronate oligosaccharides obtained by oxidative degradation from ALG in of RAW 264.7 cells.[ 44 ] Endotoxin levels were analyzed using HEK‐Blue mTLR4 cells and Quanti‐Blue reagents and acceptable endotoxin levels were confirmed in the materials and the final product and endotoxin levels were ≈0.01–0.05 EU mL−1 (see Table S1, Supporting Information).
Figure 8.
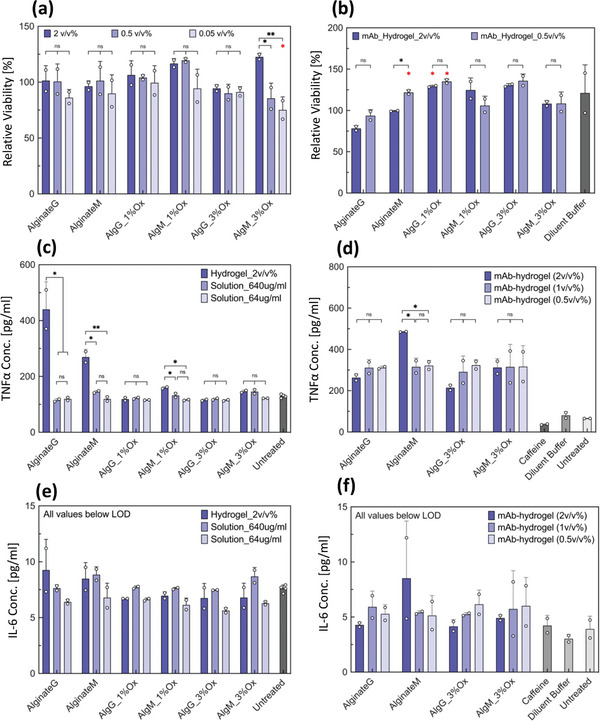
In vitro cell based studies using RAW 264.7 mouse macrophage cells in the presence of alginate hydrogel particles. a) Cell viability after exposure to alginate hydrogel particles (blank). b) Cell viability after exposure to crystalline monoclonal antibody (mAb) laden hydrogel particles. Viability of cells treated with each alginate type was compared with that of untreated cells for statistical analysis. c) Tumor necrosis factor α (TNFα) concentration in the supernatant of alginate hydrogel particles. d) TNFα concentration in the supernatant of crystalline mAb‐laden alginate hydrogel particles. e) Interleukin‐6 (IL‐6) concentration in the supernatant of alginate hydrogel particles. f) IL‐6 concentration in the supernatant of crystalline mAb‐laden alginate hydrogel particles. Crystalline mAb‐laden hydrogel particles were prepared at 100 mg mL−1 particle loading. Up to 2 v/v % of alginate particles per cell media was tested. For (c) and (e) solution Solution of sodium alginate was also tested at 640 and 64 µg mL−1 to simulate an exaggerated effect of degradation and dissolution of the hydrogel particles on the cells. For all (e) and (f) samples, the concentration was below the limit of detection of the ELISA. Samples in duplicate. Data were compared using one‐way ANOVA with Tukey's multiple comparisons test. Adjusted p values were indicated as: **p < 0.01, *p < 0.05, ns > 0.05. Statistical information shown in red indicates comparison with untreated cells; only significant differences are depicted.
2.6.2. In Vivo Studies
Low stability, short half‐life, and low bioavailability of orally administered mAbs limit their administration to mostly intravenous (IV) therapy,[ 45 ] and there is a need for other methods such as SC.[ 5 ] We evaluated the PK of human crystalline anti‐PD‐1 hydrogel formulation in naïve male Wistar rats. For this purpose, samples of human anti‐PD‐1 antibody laden alginate particles were prepared. PK of anti‐PD‐1 crystals (crystalline formulation) and crystalline mAb‐laden alginate particles (hydrogel formulation) were subcutaneously administrated. For both formulations. The rats were injected with 100 µL of sample (Figure 9 and Table 3 ). Analysis of the PK dosing groups indicated relative bioavailability of mAb‐laden hydrogel is similar to anti‐PD‐1 crystalline suspension. Based on the concentration‐time profiles, no indication of fast irregular clearance was detected in these two dosing groups (Figure 9).
Figure 9.
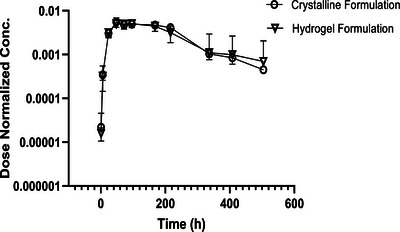
In vivo studies. Pharmacokinetics (PK) of human anti‐PD‐1 antibody pembrolizumab naïve male Wistar rats evaluated for the crystalline formulation and monoclonal antibody (mAb) laden hydrogel formulations. The sample size for the crystalline formulation and the hydrogel formulation was n = 2 and 5, respectively.
Table 3.
Pharmacokinetics (PK) parameters of pembrolizumab (human anti‐PD‐1) administrated subcutaneously in Wistar Han rats using crystalline formulation and monoclonal antibody (mAb) laden alginate hydrogel particle formulation
| Formulation | Crystalline formulation | mAb‐laden ALG particles formulation |
|---|---|---|
| C max/Dose | 0.005 | 0.005 |
| T max [h] | 48 | 48 |
| AUC/Dose | 1.4 | 1.4 |
| Vz‐F [mL kg−1] | 94 | 123 |
| CL [mL h−1 kg−1] | 0.7 | 0.7 |
| Mean residence time [h] | 171 | 174 |
| AUCinf [h*µg mL−1] | 62 222 | 57 495 |
Vz/F: apparent volume of distribution. AUC: area under the curve. CL: apparent total body clearance.
3. Conclusions
The aim of this study was to explore using hydrogel particles and crystalline mAbs to enable SC or local administration of biologics at high concentrations. Here, we established an encapsulation process based on centrifugal extrusion of crystalline mAbs and sodium alginate to form hydrogel particles with high encapsulation efficiency and antibody loadings. Our process has the benefits of encapsulating antibodies in a highly stable crystalline state while avoiding chemical reactions in the presence of the antibody and does not require or use an organic dispersing phase for formation of the droplets. Hydrogel particles of up to 350 mg mL−1 loading were prepared. These hydrogel particles’ soft nature allowed high particle packings that enabled injectable formulations of up to 315 mg mL−1 anti‐PD‐1 antibody. We studied the physical and physiochemical parameters to control this process. While centrifugal acceleration dictates particle size, particle shapes were mostly dictated by the bath conditions. Our results indicated that mannuronic rich alginate can form more spherical particles compared to guluronic rich alginates. Comprehensive bioanalytical studies including competitive binding ELISA, SEC, IEX, and LC–MS provided evidence on the maintained quality of the released antibody. The process of encapsulation and release did not induce a quantifiable amount of aggregation, mass shift or charge variants. The proposed technology was compared to two other technologies to confirm the superior maintained quality of the released anti‐PD‐1 antibody. Also, we confirmed that particles can be formed with partially oxidized alginate with enhanced degradability properties and the modified oxidized alginate does not negatively affect the protein activity or cause aggregation. Furthermore, using low MW mannuronic rich, with 1.5% uronic group partial oxidation, excellent biodegradation of the particles in simulated body fluids was achieved. Evaluating the crystalline mAb laden particles to achieve long‐term sustained release through hydrogel modifications to benefit from the stability of immobilized proteins can be next step for this research. Furthermore our in vitro and in vivo studies provide more insight for SC particle‐based administration of immunotherapy agents. The crystalline anti‐PD‐1 antibody‐laden alginate particles were administrated subcutaneously in Wistar Han rats, and the PK of the drug was evaluated. Crystalline mAb and mAb‐laden alginate hydrogel demonstrated similar PK parameters suggesting that absorption is not differentiated by the hydrogel dosing group upon SC administration. The results of this study can shape the future trend of therapeutic antibody delivery by allowing the development of stable concentrated formulations.
4. Experimental Section
Materials
All chemicals used were of analytical grade and were used without any further purification. Low endotoxin level, ultrapure NaALGs were purchased from DuPont NovaMatrix. Four different types of sodium alginates were used, and their properties (provided by the vendor) are summarized in Table 4 . CaCl2, caffeine, and 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) were purchased from Sigma. Poly(ethylene glycol) (PEG, 3350 Da) was from Hampton Research. Purified, humanized mAb was provided by Merck.
Table 4.
Molecular weight (MW), apparent viscosity, and guluronic to mannuronic ratio (G/M) of sodium alginates used in this work (provided by the vendor)
| Type | MW weight [kDa] | Apparent viscosity [mPa s−1] | G/M |
|---|---|---|---|
| VLVG | <75 | < 20 | ≥ 1.5 |
| VLVM | <75 | <20 | ≤1 |
| MVG | >200 | >200 | ≥1.5 |
| MVM | >200 | >200 | ≤1 |
For cell culture and in vitro assays, RAW264.7 (TIB‐71) cells were obtained from ATCC. HEK‐Blue mTLR4 reporter cells (hbk‐mtlr4) and required selection (1X HEK‐Blue Selection) antibiotics were obtained from InvivoGen. Cell growth media was prepared using DMEM (Corning 10‐013‐CV), fetal bovine serum (FBS; Gibco 10437‐028), and Penicillin/Streptomycin (Sigma P4333; 100 U mL−1, 100 µg mL−1), and was sterile‐filtered before use. For HEK‐Blue mTLR4 cells, the cell growth media was appropriately supplemented with required selection antibiotics. For HEK‐Blue mTLR4 assays, the assay media was prepared using heat‐inactivated FBS (by heating at 56 °C for 30 min) to prevent high background signal. Quanti‐Blue reagent and lipopolysaccharide (LPS‐EB from E. coli O111‐B4) were obtained from InvivoGen. Presto Blue reagent for quantifying cell viability was obtained from Life Technologies. Mouse TNFα DuoSet (DY410) and Mouse IL‐6 DuoSet (DY406) ELISA kits were obtained from R&D Systems. 1‐Step Ultra TMB‐ELISA substrate solution was obtained from ThermoFisher Scientific. 4N sulfuric acid was obtained from Sigma–Aldrich.
Crystalline mAb and mAb‐Laden Hydrogel Particles Preparation and Characterizations
For crystallization of anti‐PD‐1 antibody, a “PEG buffer” for crystallization buffer was prepared as a 10% w/v PEG solution in 50 × 10−3 m HEPES, pH 7.0. Caffeine solution (co‐solute for crystallization) was prepared as a 2.5% w/v caffeine in 20 × 10−3 m L‐His, pH 5.4 buffer. The mAb was kept frozen at 45 mg mL−1 in 20 mm l‐His, pH 5.4 buffer and thawed right before the crystallization process. Solutions were prepared with distilled water and were filtered with a 0.22‐micron filter. Anti‐PD‐1 antibody crystals were grown in batches at total volume of 2 mL, with each batch yielding ≈12 mg of the antibody in the crystalline form. For each batch, the antibody, PEG buffer, and caffeine solution were combined at a volume ratio of 3:6:1 respectively. Crystallization mixture was kept at room temperature for 4–16 h while rotating at 12 rpm on a tube revolver (Thermo Scientific, model 88 881 001). Antibody crystals were recovered from the batches by centrifugation at 1700 RCF for 2 h, transferred into fresh PEG buffer, resuspended. Crystals were used immediately or stored at 4 °C.
For preparation of the mAb ALG pre‐gel suspension, at first NaALG powder was dissolved in the 10% w/w PEG crystallization buffer overnight and later filtered using 0.2‐micron filter. ALG was dissolved in the PEG buffer to help maintain the antibody in crystalline form. The NaALG solution was added to the mAb crystal and mixed to generate a homogenous suspension. Later the suspension was concentrated by centrifugation. Small quantities of the suspension were removed and the mAb concentration was measured. Antibody concentration was measured using Nanodrop UV–Vis Spectrophotometer using the 280 nm absorbance method after 20‐fold dilution in PBS. Centrifugation was continued until the suspension reached the desired mAb concentration. This final suspension was used as the pre‐gel to form the particles.
For preparation of antibody‐laden particles, the pre‐gel suspension was filled inside a plastic syringe (with the plunger removed) connected to a 26G (ID = 260 µm, OD = 464 µm), 30G (ID = 159 µm, OD = 312 µm), 32G (ID = 108 µm, OD = 235 µm), or 34G (ID = 52 µm, OD = 159 µm) (1/4" in length) blunt‐tip dispenser attached to it. A cross‐linking solution consisting of 5 – 50 × 10−3 m CaCl2, 10% w/v PEG 3350 (for stabilization of anti‐PD‐1 antibody crystals), 50 × 10−3 m HEPES pH 7.0 and 0–0.06% v/v Tween 20 nonionic surfactant (for reducing the impact force) was prepared. The cross‐linking solution was filled inside a 15 mL centrifuge tube to form the CaCl2 bath. The distance from the tip of the dispenser and the CaCl2 bath was 2–10 mm. The device was centrifuged for 10–30 min at 200–2000 RCF. Upon completion the bath solution was replaced with 10% w/v PEG3350, 50 × 10−3 m HEPES and particles were resuspended.
For particle antibody loading and encapsulation efficiency measurement, mAb + ALG pre‐gel suspensions were prepared, and the antibody concentration was measured using the protein 280 nm absorbance method. Later particles were formed using the same suspensions. Hydrogel particles were redispersed, and the excess aqueous solution was wiped using a tissue paper and the mass of the mAb‐laden hydrogel was measured. The total amount of the encapsulated mAb was measured based on the amount of released antibody in PBS to calculate the particle loading. To evaluate the encapsulation efficiency the concentration of antibody in the CaCl2 bath and the PEG buffer for resuspension were measured and compared to the total amount of mAb.
Microscopy was used for particle size distribution, using a Zeiss Axiovert microscope. A minimum of 30 particles were measured (ImageJ) for each sample for each reported mean diameter. SONICC (Formulatrix) was utilized to collect micrographs of microsphere samples in the following modes: bright‐field, ultraviolet two‐photon excited fluorescence (UV‐TPEF), and second harmonic generation (SHG).
For preparation of partially oxidized NaALG, 2 g of NaALG was dissolved in DI water at 2% w/v. Sodium periodate was dissolved in DI water at 3 mg mL−1 (for 1.5 molar % uronic oxidation) and 6 mg mL−1 (for 3 molar % uronic oxidation). The sodium periodate solution (50 mL) was added to the NaALG solution to carry out the oxidation reaction. The reaction was continued for 24 h in dark conditions while mixing. Reactions byproducts and unreacted species were carefully removed. For this purpose, the mixture was purified by dialysis using a 3.5 KDa snakeskin dialysis tubes for 48 h. Furthermore, the product was concentrated using a 5 kDa centrifuge concentrator to ≈3% w/v NaALG and was later freeze dried. The final dried products were stored at 4 °C.
Stimulated body fluid (SBF) was prepared to mimic the amount of monovalent and divalent ions in the SC environment, based on the literature[ 41 ] at sodium chloride 7.996 mg mL−1, sodium bicarbonate 0.350 mg mL−1, potassium chloride 0.224 mg mL−1, potassium phosphate dibasic trihydrate 0.228 mg mL−1, magnesium chloride hexahydrate 0.305 mg mL, 40 mL of 1 m hydrochloric acid, CaCl2 0.278 mg mL−1, sodium sulfate 0.071 mg mL−1, and tris(hydroxymethyl) aminomethane 6.057 mg mL−1.
Biodegradation of ALG particles was evaluated at 37 °C in the simulated body fluid over the course of 45 days using a gravimetric assay (no agitation or stirring). Particles were formed using 10 mg mL−1 sodium alginate, using a 30‐gauge dispenser at 400 RCF in a 20 × 10−3 m CaCl2 bath. For this purpose, the biodegradation (dissolution and hydrolysis) of particles formed using six different ALG types were compared. Batches of particles were divided to equal aliquots (each ≈15 mg particles solid basis) and were immersed in 5 mL SBF. The SBF in the test tubes was replaced with fresh SBF daily or every 2 days to keep the sink conditions. At different time intervals three replicate tubes from each sample type were removed from storage temperature and swashed with DI water (to remove excess ions and dissolved polymers). The residual particles were dried at 75 °C and weighed using a microbalance with 0.01 mg precision. This mass was compared to the original ≈15 mg solid to calculate the retained mass %. To increase the accuracy, the cumulative mass of tube and the sample was measured, and the tube weight was later subtracted to obtain the particles mass. The intervals between each sampling were varied between 1 and 4 days.
To measure the ALG hydrogel's swelling ratio, particles formed using different NaALGs were immersed in SBF and were stored at 37 °C for 24 h (samples in triplicate). Later the excess buffer was removed from the hydrogel particles using a paper tissue and hydrogel particles were weighed. Samples were dried at 75 °C in vacuum and the dried samples were weighed again. Swelling ration was calculated as:
| (2) |
To evaluate the release of mAb from the hydrogel, ≈50 µL of the mAb‐laden hydrogel was dispersed in the PEG buffer and moved to a 2 mL tube. Later at time zero the excess buffer was replaced by the prewarmed (37 °C) SBF, and small volumes of the sample (10 µL) were removed at different time intervals to measure the mAb concentration in the SBF. Concentration of the mAb was evaluated by the 280 nm absorbance methods using a Thermo Scientific Nanodrop‐2000 UV–Vis Spectrophotometer (samples in triplicates).
Bioanalytical studies were carried out to evaluate the quality of the released anti‐PD‐1 antibody. SEC was used to determine the amount of the antibody monomer and aggregates. Multiangle light scattering (MALS) was utilized to determine the MWs. This method utilizes the Waters Acquity UPLC H‐Class Bio system (attached to a Wyatt UP LS laser and UP tRex detector) with a Waters Acquity BEH200 SEC column to separate molecules based on their hydrodynamic radius. Resolved peaks were detected by the absorbance at 280 nm, light scattering intensity, and the differential refractive index (dRI). SEC experiments were carried out under isocratic conditions at a flow rate of 0.5 mL min−1 in a buffer composed of 50 × 10−3 m phosphate, 450 × 10−3 m arginine at pH 7.0. IEX provides an ion exchange HPLC method to determine charge variants present in pembrolizumab samples. Analysis was performed using a Dionex ProPac WCX‐10, 10 µm 4 × 250 mm column and a mobile phase gradient from 24 × 10−3 m MES pH 6.1, 4% acetonitrile to 20 × 10−3 m NaPO4, 95 × 10−3 m NaCl pH 8, 4% acetonitrile. UV detection is performed at 280 nm The antibody samples were analyzed for mass determination by ESI time‐of‐flight (TOF) MS. Samples were separated without deglycosylation on an Acquity H‐class UPLC (Waters Corporation, MA) with a Supelco BioShell IgG 1000Ǻ C4 column (10 cm × 2.1 mm, 2.7 µm). Mobile phase A was 0.05% trifluoroacetic acid (TFA) in water; mobile phase B was 0.05% TFA in acetonitrile. The column was maintained at 80 °C and eluted with a gradient of 25%–30% B between 0–2 min, followed by 30%–45% B between 2–9 min, and stripped by 99% B. The flow rate was 0.3 mL min−1. ESI TOF mass signals were recorded online by Waters Xevo G2‐XS Q‐TOF mass spectrometer (Waters Corporation) operating in the positive ion mode. The mass spectrometer was externally calibrated with sodium iodide tuning mix (Waters Corporation) in the mass range of 500–4000 m/z. MS conditions were as follows: capillary voltage—3.2 kV, cone voltage—200 V, and desolvation temperature—500 °C. Mass data were acquired in the mass range of 600–4000 m/z. The mass spectra were integrated and deconvoluted using MassLynx analysis software (Waters Corporation).
ELISA bioassays were carried out to identify the anti‐PD‐1 antibody bioactivity using competitive binding assays. To evaluates the ability of the antibody to other ligand to bind with correct receptors, the reference material and test samples were serially diluted and mixed with an equal volume of the ligands before transfer to ELISA plates. The levels of bound ligand to the ELISA plate were detected by conjugation with streptavidin and chemiluminescence substrate. Luminescence is measured using a microplate reader and resulting inhibition response curves were analyzed with curve fitting software (e.g., SoftMax Pro). Biological activity is expressed as % relative activity of the reference sample.
In Vitro Assays
RAW 264.7 cells viability during treatment with ALG particles were evaluated. Cells were maintained in cell growth media (DMEM + 10% v/v FBS + 100 U mL−1 Penicillin + 0.1 mg mL−1 Streptomycin) at 37 °C and 5% v/v CO2. Cells were handled using standard aseptic techniques in a biosafety cabinet. NaALG solution, ALG particles and mAb‐laden Alg particles were prepared in either 1× PBS or diluent buffer at 10× the final required concentration for cell assay. To set up the assay, RAW 264.7 cell suspension was prepared in cell growth media. In a 96‐well tissue culture‐treated plate, 20 µL of samples were first dispensed followed by addition of 180 µL (25 000 cells) of cell the suspension. The treated cells were incubated at 37 °C for 20 h. At the end of the assay, the assay plate was centrifuged at 250 RCF for 2 min, and 110 µL of cell supernatant was aspirated and stored in a 96‐well nontreated plate (Greiner Bio‐One 237 105) at ‐20 °C until use. To quantify cell viability, 10 µL of Presto Blue was added to the assay plate and incubated at 37 °C for 30–60 min. Cell viability was measured using a plate reader (Tecan Infinite M200) at an excitation wavelength of 550 nm and emission wavelength of 600 nm. Cell viability is reported relative to untreated cells taken as 100% viable at the end of the assay.
To quantify TNFα and IL‐6 release from RAW 264.7 cells, the cell supernatant was assayed using ELISA kits following the kit protocol. Costar 3690 half‐area high bind plates were used for ELISA. Reagent volumes were reduced to one‐quarter (25 µL per well) of the suggested volumes in the protocol. The cytokine standards were prepared in the cell growth media. Similarly, the samples were appropriately diluted using the cell growth media. Cytokine concentration was measured off the standard curve prepared on the same ELISA plate.
To quantify endotoxin levels in the samples HEK‐Blue mTLR4 cells were maintained in cell growth media (DMEM + 10 v/v % FBS + 100 U mL−1 Penicillin + 0.1 mg mL−1 Streptomycin) supplemented with 100 µg mL−1 Normocin and 1× HEK‐Blue Selection antibiotics and at 37 °C and 5% v/v CO2. HEK‐Blue mTLR4 cell suspension was prepared in the assay media (DMEM + 10% heat‐inactivated FBS + 100 U mL−1 Penicillin + 1 mg mL−1 Streptomycin + 1 mg mL−1 Normocin). To quantify endotoxin levels in the samples, LPS standards were prepared in 1x PBS. In a 96‐well tissue culture‐treated plate, 20 µL of samples and LPS standards was first dispensed followed by addition of 180 µL (25 000 cells) of cell suspension. The treated cells were incubated at 37 °C for 20 h. At the end of the assay, the assay plate was centrifuged at 250 RCF for 2 min, and 110 µL of cell supernatant was aspirated. 20 µL of the aspirated supernatant was added to a 96‐well plate and 180 µL of Quanti‐Blue reagent was added to each well. The plate was incubated at 37 °C for 30 min or until the LPS standards developed dark blue color. The plate was read using a plate reader (Tecan Infinite M200) at 620 nm.
In Vivo Studies
The study described here was designed to evaluate the kinetics of SC administered hydrogel formulation of anti‐PD‐1 compared to SC administration of anti‐PD‐1 crystalline formulation in naïve male Wistar Rats.
Procedures involving the care and use of animals in the study were reviewed and approved by the Institutional Animal Care and Use Committee at Merck Research Laboratories, Palo Alto, CA. During the study, the care and use of animals were conducted in accordance with the principles outlined in the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), the Animal Welfare Act, the American Veterinary Medical Association (AVMA) Euthanasia Panel on Euthanasia, and the Institute for Laboratory Animal Research (ILAR) Guide to the Care and Use of Laboratory Animals.
Sample Preparation
The mAb‐laden hydrogel particles were prepared inside the biosafety safety cabinet under sterile conditions. Pre‐gel suspension of mAb + ALG was centrifuged to 200–240 mg mL−1. The hydrogel particles were produced at 1200 RCF centrifugal speed using a 30G dispenser. The bath solution contained 40 × 10−3 m CaCl2, 0.01w/v tween 20 and 10% w/v PEG 3350. Samples were tested for endotoxin levels (≈0.01–0.05 EU mL−1). Hydrogel particles were formulated at 6% w/v PEG 3350 and contained ≈1.5 mole caffeine/mole anti‐PD‐1, 50 × 10−3 m HEPES (at pH 7.0), NaALG: ≈1% w/v, CaCl2 <5 × 10−3 m, Tween 20: ≪ 0.01% w/v. The particles were filled inside the syringe and were packed using gravity. Material loaded in each syringe was injected into a test tube and the amount of the mAb was measured using spectrophotometry after mAb dissolution in SBF. After filling the syringes were stored at 4 °C.
Group Designation, Dose Level, and Dosing Schedule
Following an acclimation period, the animals were assigned to the study based on acceptable health as determined by a staff veterinarian. Overnight fasting was not required. Each animal received a single 100 µL SC dose in the shoulder blades, at target dose levels indicated in Table 5 .
Table 5.
Group designation, dose level, and dosing schedule for the pharmacokinetics (PK) study
| Group | Total number of animals/sex/group | Test article | Actual dose concentration [mg mL−1] | Formulation | ROA |
|---|---|---|---|---|---|
| 1 | 2/M | Anti‐PD‐1 | 157 | Crystalline: 6% PEG 3350, ≈1.5 mole caffeine/mole anti‐PD‐1, 50 × 10−3 m HEPES pH 7.0 | SC |
| 2 | 5/M | Anti‐PD‐1 | 93.4 | Alginate hydrogel: 6% PEG 3350, ≈1.5 mole caffeine/mole anti‐PD‐1, 50 × 10−3 m HEPES, pH 7.0 | SC |
Collection, Processing, and Analysis of Blood Samples
For study, whole blood samples (0.25 mL each) were collected from the tail vein. Blood samples were collected from all animals at 0.5, 1, 6, 24, 48, 72, 96, 168, 216, 336, 408, and 504 h after dosing. Blood samples were placed into 500 µL capillary blood collection tubes and placed on wet ice immediately after collection. Serum was collected by centrifuging the whole blood at 6000 rpm for 6 min. The resulting serum was transferred into tubes, and immediately placed on dry ice. Dosing retainer aliquots were put into respective assay plates as well. Serum samples were stored at ‐70 °C until analysis.
For analysis of the dosing solutions, aliquots of each formulation were centrifuged in microfuge for 3 min at 3500 RPM. The supernatants were removed the resulting pellets were resuspended in 900 µL ml PBS solution and rotated on a Labnet rotator or mixing table for 2 h at room temperature. The resulting solutions were centrifuged in microfuge for 3 min at 3500 RPM to remove alginate particles. The supernatants were measured for protein determination using the Nanodrop 260/280 ratio protocol. Samples were later studied for IEX, SEC, and ELISA analyses.
For anti‐PD‐1 analysis, serum samples and dosing solutions were analyzed at Merck Research Laboratories, CA. For determination of test article concentration, a mesoscale discovery (MSD) immunosorbent assay platform was used. The minimum reportable concentration or lower limit of quantitation (LLOQ) was 0.00002 mg mL−1 of test article in rat serum.
Statistical Analysis
The data of the experiments were presented as mean ± standard deviation (SD). All experimental samples were in triplicate unless otherwise notes. Data were compared using one‐way ANOVA with Tukey's multiple comparisons test. Adjusted p values were indicated as: **p < 0.01, *p < 0.05, ns > 0.05. Statistical analysis was carried out using GraphPad Prism.
Conflict of Interest
The authors have filed a patent application based on the results in this article. Patent application no. PCT/US2021/043916.
Supporting information
Supporting Information
Supplemental Video 1
Supplemental Video 2
Acknowledgements
The authors acknowledge Merck & Co. Inc. for the financial support of the work.
Erfani A., Schieferstein J. M., Reichert P., Narasimhan C. N., Pastuskovas C., Parab V., Simmons D., Yang X., Shanker A., Hammond P., Doyle P. S., Crystalline Antibody‐Laden Alginate Particles: A Platform for Enabling High Concentration Subcutaneous Delivery of Antibodies. Adv. Healthcare Mater. 2023, 12, 2202370. 10.1002/adhm.202202370
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Shanmugaraj B., Siriwattananon K., Wangkanont K., Phoolcharoen W., Asian Pac. J. Allergy Immunol. 2020, 38, 10. [DOI] [PubMed] [Google Scholar]
- 2. Levine M. M., Monoclonal Antibody Therapy for Ebola Virus Disease, Massachusetts Medical Society, MA, USA: 2019. [DOI] [PubMed] [Google Scholar]
- 3. Tsumoto K., Isozaki Y., Yagami H., Tomita M., Immunotherapy 2019, 11, 119. [DOI] [PubMed] [Google Scholar]
- 4. Sharma P., Allison J. P., Science 2015, 348, 56. [DOI] [PubMed] [Google Scholar]
- 5. Viola M., Sequeira J., Seiça R., Veiga F., Serra J., Santos A. C., Ribeiro A. J., J. Controlled Release 2018, 286, 301. [DOI] [PubMed] [Google Scholar]
- 6. Shpilberg O., Jackisch C., Br. J. Cancer 2013, 109, 1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y., Guo L., Dong S., Cui J., Hao J., Adv. Colloid Interface Sci. 2019, 266, 1. [DOI] [PubMed] [Google Scholar]
- 8. Rule S., Collins G. P., Samanta K., J. Med. Econ. 2014, 17, 459. [DOI] [PubMed] [Google Scholar]
- 9. Ghosh R., Calero‐Rubio C., Saluja A., Roberts C. J., J. Pharm. Sci. 2016, 105, 1086. [DOI] [PubMed] [Google Scholar]
- 10. Pawate A. S., Šrajer V., Schieferstein J., Guha S., Henning R., Kosheleva I., Schmidt M., Ren Z., Kenis P. J., Perry S. L., Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2015, 71, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang M. X., Shenoy B., Disttler M., Patel R., McGrath M., Pechenov S., Margolin A. L., Proc. Natl. Acad. Sci. USA 2003, 100, 6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schieferstein J. M., Pawate A. S., Varel M. J., Guha S., Astrauskaite I., Gennis R. B., Kenis P. J., Lab Chip 2018, 18, 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schieferstein J. M., Reichert P., Narasimhan C. N., Yang X., Doyle P. S., Adv. Ther. 2021, 4, 2000216. [Google Scholar]
- 14. Wechsler M. E., Stephenson R. E., Murphy A. C., Oldenkamp H. F., Singh A., Peppas N. A., Biomed. Microdevices 2019, 21, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J., Mooney D. J., Nat. Rev. Mater. 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bak A., Ashford M., Brayden D. J., Adv. Drug Delivery Rev. 2018, 136, 2. [DOI] [PubMed] [Google Scholar]
- 17. Liu M., Cao Z., Zhang R., Chen Y., Yang X., ACS Appl. Mater. Interfaces 2021, 13, 33874. [DOI] [PubMed] [Google Scholar]
- 18. Ren X., Wang N., Zhou Y., Song A., Jin G., Li Z., Luan Y., Acta Biomater. 2021, 124, 179. [DOI] [PubMed] [Google Scholar]
- 19. Zhang Z.‐Q., Kim Y.‐M., Song S.‐C., ACS Appl. Mater. Interfaces 2019, 11, 34634. [DOI] [PubMed] [Google Scholar]
- 20. Shewan H. M., Stokes J. R., J. Colloid Interface Sci. 2015, 442, 75. [DOI] [PubMed] [Google Scholar]
- 21. Caló E., Khutoryanskiy V. V., Eur. Polym. J. 2015, 65, 252. [Google Scholar]
- 22. Hamrang Z., Rattray N. J., Pluen A., Trends Biotechnol. 2013, 31, 448. [DOI] [PubMed] [Google Scholar]
- 23. Jiskoot W., Randolph T. W., Volkin D. B., Middaugh C. R., Schöneich C., Winter G., Friess W., Crommelin D. J., Carpenter J. F., J. Pharm. Sci. 2012, 101, 946. [DOI] [PubMed] [Google Scholar]
- 24. van de Weert M., Hennink W. E., Jiskoot W., Pharm. Res. 2000, 17, 1159. [DOI] [PubMed] [Google Scholar]
- 25. Egbu R., Brocchini S., Khaw P. T., Awwad S., Eur. J. Pharm. Biopharm. 2018, 124, 95. [DOI] [PubMed] [Google Scholar]
- 26. Alvarez‐Urena P., Davis E., Sonnet C., Henslee G., Gugala Z., Strecker E. V., Linscheid L. J., Cuchiara M., West J., Davis A., Tissue Eng., Part A 2017, 23, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruff L. E., Mahmoud E. A., Sankaranarayanan J., Morachis J. M., Katayama C. D., Corr M., Hedrick S. M., Almutairi A., Integr. Biol. 2013, 5, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abasalizadeh F., Moghaddam S. V., Alizadeh E., Kashani E., Fazljou S. M. B., Torbati M., Akbarzadeh A., J. Biol. Eng. 2020, 14, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matyash M., Despang F., Ikonomidou C., Gelinsky M., Tissue Eng., Part C 2014, 20, 401. [DOI] [PubMed] [Google Scholar]
- 30. Pescosolido L., Piro T., Vermonden T., Coviello T., Alhaique F., Hennink W. E., Matricardi P., Carbohydr. Polym. 2011, 86, 208. [Google Scholar]
- 31. Ghaffarian R., Pérez‐Herrero E., Oh H., Raghavan S. R., Muro S., Adv. Funct. Mater. 2016, 26, 3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhai P., Chen X., Schreyer D. J., Mater. Sci. Eng., C 2015, 56, 251. [DOI] [PubMed] [Google Scholar]
- 33. Bruchet M., Melman A., Carbohydr. Polym. 2015, 131, 57. [DOI] [PubMed] [Google Scholar]
- 34. Boontheekul T., Kong H.‐J., Mooney D. J., Biomaterials 2005, 26, 2455. [DOI] [PubMed] [Google Scholar]
- 35. Eral H. B., Safai E. R., Keshavarz B., Kim J. J., Lee J., Doyle P., Langmuir 2016, 32, 7198. [DOI] [PubMed] [Google Scholar]
- 36. Li M., Reichert P., Narasimhan C., Sorman B., Xu W., Cote A., Su Y., Mol. Pharmaceutics 2022, 19, 936. [DOI] [PubMed] [Google Scholar]
- 37. Wang S. S., Yan Y., Ho K., Antibody Ther. 2021, 4, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marquette S., Peerboom C., Yates A., Denis L., Goole J., Amighi K., Eur. J. Pharm. Biopharm. 2014, 86, 393. [DOI] [PubMed] [Google Scholar]
- 39. Johansen P., Moon L., Tamber H., Merkle H. P., Gander B., Sesardic D., Vaccine 1999, 18, 209. [DOI] [PubMed] [Google Scholar]
- 40. Li H., Mergel O., Jain P., Li X., Peng H., Rahimi K., Singh S., Plamper F. A., Pich A., Soft Matter 2019, 15, 8589. [DOI] [PubMed] [Google Scholar]
- 41. Marques M. R., Loebenberg R., Almukainzi M., Dissolution Technol. 2011, 18, 15. [Google Scholar]
- 42. Ryman J. T., Meibohm B., CPT: Pharmacometrics Syst. Pharmacol. 2017, 6, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao L., Ji P., Li Z., Roy P., Sahajwalla C. G., J. Clin. Pharmacol. 2013, 53, 314. [DOI] [PubMed] [Google Scholar]
- 44. Zhou R., Shi X., Gao Y., Cai N., Jiang Z., Xu X., J. Agric. Food Chem. 2015, 63, 160. [DOI] [PubMed] [Google Scholar]
- 45. Bilati U., Allémann E., Doelker E., Eur. J. Pharm. Biopharm. 2005, 59, 375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Video 1
Supplemental Video 2
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.