

Abstract
Extracellular vesicles (EVs) are emerging as promising diagnostic and therapeutic tools for a variety of diseases. The characterization of EVs requires a series of orthogonal techniques that are overall time‐ and material‐consuming. Here, a microfluidic device is presented that exploits the combination of diffusion sizing and multiwavelength fluorescence detection to simultaneously provide information on EV size, concentration, and composition. The latter is achieved with the nonspecific staining of lipids and proteins combined with the specific staining of EV markers such as EV‐associated tetraspanins via antibodies. The device can be operated as a single‐step immunoassay thanks to the integrated separation and quantification of free and EV‐bound fluorophores. This microfluidic technique is capable of detecting and quantifying components associated to EV subtypes and impurities and thus to measure EV purity in a time scale of minutes, requiring less than 5 µL of sample and minimal sample handling before the analysis. Moreover, the analysis is performed directly in solution without immobilization steps. Therefore, this method can accelerate screening of EV samples and aid the evaluation of sample reproducibility, representing an important complementary tool to the current array of biophysical methods for EV characterization, particularly valuable for instance for bioprocess development.
Keywords: characterization, extracellular vesicles, fluorescence, immunoassay, microfluidics, purity, quantification
A crucial step to optimize large‐scale production of extracellular vesicles (EVs) is the development of characterization techniques capable of simultaneously monitoring physical and biochemical properties of EVs with high throughput. Here, a microfluidic technique is presented that can detect particle subpopulations, quantify their biomarkers, and measure their purity in a time scale of minutes and with no sample pretreatments.
1. Introduction
The discovery of the key role of extracellular vesicles (EVs) in mediating cell‐to‐cell communication motivated an increasing number of studies on their use for the diagnosis and treatment of numerous diseases.[ 1 , 2 ] EVs are a mixture of small (below 200 nm) and large (above 200 nm) membrane‐bound vesicles which can transfer messages from one cell to another through surface‐exposed or encapsulated biomolecules.[ 1 ] Recently, EVs have been increasingly investigated in clinical trials, mostly as early diagnostic tools in body fluids and less frequently as therapeutic agents.[ 3 , 4 , 5 , 6 ]
The relatively slow development of EVs in the biomedical field is partially related to the difficulties in producing the required large amounts of EVs in a reproducible manner.[ 7 , 8 , 9 ] Indeed, several challenges such as the heterogeneity of EV mixtures, the limited knowledge on functional EV subtypes and the presence of impurities with similar physicochemical properties to EVs (including protein and lipid aggregates), severely complicate their production as well as their characterization protocols.[ 10 , 11 ]
As described in details in the guidelines reported in the “Minimal Information for Studies of Extracellular Vesicles (MISEV),” the current protocol to characterize EV samples has to rely on a combination of multiple complementary techniques which include, among others, immunoblotting, single‐particle tracking techniques, electron microscopy, and quantification assays for lipids, proteins, and RNAs (Figure 1a).[ 12 , 13 , 14 , 15 ] Altogether these techniques provide comprehensive information on the concentration, morphology, and purity of EVs, but are considerably time‐ and sample‐consuming (Figure 1b).[ 16 , 17 , 18 , 19 ]
Figure 1.
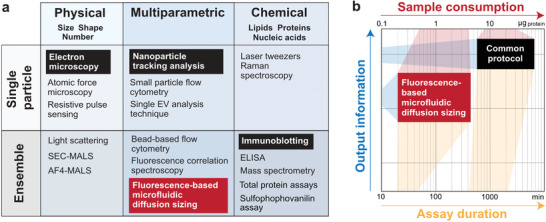
Examples of conventional methods for EV characterization. a) Different techniques provide information on physical or chemical properties, or a combination of both, and can be further classified based on single particle or ensemble analysis. The fluorescence‐based microfluidic diffusion sizing described in this work is an ensemble multiparametric technique. b) Fluorescence‐based microfluidic diffusion sizing (red box) provides a considerable amount of information on physical and chemical properties of the EV samples while requiring less material and time with respect to the common characterization protocol, i.e., the combination of immunoblotting, nanoparticle tracking analysis, and electron microscopy (black box). Although the microfluidic assay cannot replace the comprehensive characterization with multiple orthogonal techniques, it represents a convenient complementary tool, in particular for fast screening applications and bioprocess development.
Several applications, however, including the optimization of upstream and downstream operations for the large‐scale production of EVs, require intensive screening of EV samples.[ 9 , 20 ] For these applications, there is a need for a multiparametric analytical method able to quantitatively measure different EV‐specific properties and sample purity with higher throughput and lower sample amounts.
In this context, microfluidic technology offers a great opportunity to address this challenge. In microfluidic platforms, fluid volumes in the range from picolitres to microliters are manipulated in channels in the micrometer size scale. This miniaturization of sample volumes intrinsically reduces the amount of sample and analysis time. Currently, microfluidic devices have found several applications as diagnostic tools, in particular for EV fractionation or rare markers detection in EVs.[ 13 , 21 , 22 , 23 ] However, the potential of microfluidic technology for the rapid multiparametric characterization of EV batches during their production has remained essentially unexplored.
Here, we present a microfluidic technique that integrates particle separation, sizing, biomarker detection, and quantification on one single microfluidic chip, typically requiring only few microliters of nontreated sample and operating in a time scale of minutes. The core of the strategy relies on combining a diffusion‐sizing technique with multiwavelength fluorescence detection. To this aim, we developed a range of staining protocols that include dyes for high‐resolution imaging as well as common immunolabeling reagents, to achieve high signal‐to‐noise ratios without the need to remove nonreacted dye and individually track different EV biochemical components, such as lipids, primary amines, and EV‐specific proteins. We show that our fluorescence‐based microfluidic diffusion sizing technique (which is referred to in the following as fluoMDS) can robustly measure the average size of EV subpopulations. In addition, using labeled antibodies for EV staining, fluoMDS can quantify the amount of free and bound antibodies, without requiring secondary antibodies or washing steps, thereby working as a single‐step immunoassay for EV‐marker quantification. We further show that the combination of all these features allows to measure sample purity and overall obtain significant information with low sample consumption and high analytical throughput (Figure 1b). We demonstrate the power of this approach with the multiparametric characterization of EVs from human bone marrow stromal cell (MSC), human embryonic kidney (HEK) 293F cell lines and microalgae.[ 24 , 25 ]
Although the technique cannot replace the comprehensive characterization with multiple orthogonal techniques nor assess the full‐size distribution and the heterogeneity of EVs via single‐particle detection,[ 26 ] it holds great promise for applications that require rapid screening, quantification of biomolecules, and assessment of their relative variations in both pure and impure samples. The microfluidic platform is also amenable to multiplexing and parallelization, which is important to guarantee sufficient throughput.
This method has therefore important implications for instance for the development of bioprocesses for the reproducible preparation of high amounts of pure EVs.[ 9 ]
2. Results
2.1. fluoMDS for Multiparametric Characterization of Lipid Vesicles
A schematic illustration of the fluoMDS technique and its main components is shown in Figure 2 . In this device, which builds on previous diffusion‐sizing assays,[ 27 , 28 , 29 ] a fluorescent sample is hydrodynamically focused in the nozzle region between two sandwich buffer streams (Figure 2a). The fluorescent particles diffuse from the central region of the channel towards the lateral directions while flowing along the channel in laminar flow regime. Twelve images are acquired at different points along the channel using epifluorescence detection and converted into the corresponding lateral diffusion profiles at different diffusion times (Figure 2b). The diffusion profiles are fitted with a linear combination of simulated basis functions to provide the distribution of diffusion coefficients, which is translated into the size distribution of the sample via the Stokes–Einstein relationship (see also Experimental Section) (Figure 2c). The acquisition of an increasing number of diffusion profiles improves the robustness of the fitting, with a minimum of four diffusion profiles recommended for the assay.[ 27 ] The technique can detect species over a wide size range, from few Angstroms to hundreds of nanometers.[ 28 ] Moreover, since the fluorescence signal is linearly proportional to the amount of the different species present in the sample, the technique also enables the measurement of their relative concentration. Given the large number of components typically present in EVs, the fitting has been constrained to the sum of two Gaussian distributions to be able to capture different subpopulations in the sample while avoiding overfitting.[ 29 ]
Figure 2.
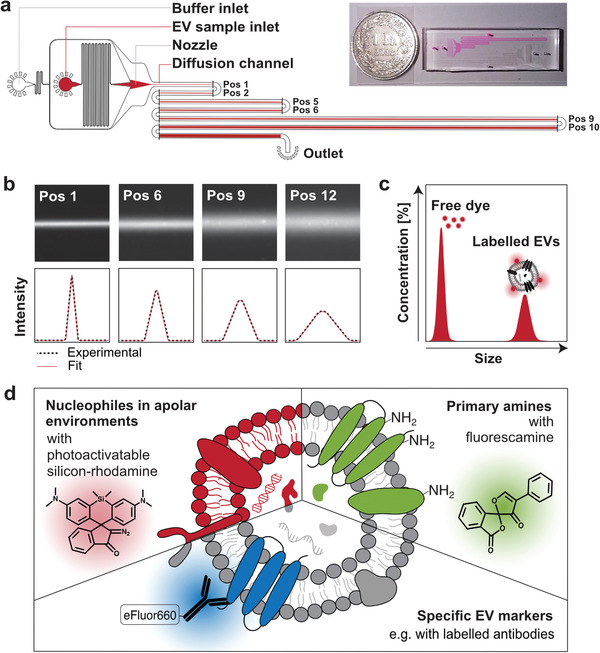
Fluorescence‐based microfluidic diffusion sizing for the multiparametric characterization and quantification of EVs. a) Schematic illustration and images of the microfluidic device used for EV analysis. The EV sample is hydrodynamically focused into a narrow stream in the middle of the channel by an auxiliary buffer. The sample diffuses laterally while flowing along the channel under steady‐state flow. Diffusion profiles in the direction orthogonal to the flow are measured by acquiring fluorescence images at 12 different positions (black marks), corresponding to 12 different diffusion times. This strategy avoids issues of photo‐bleaching of the fluorophores. b) Representative fluorescence images at different positions and corresponding extracted diffusion profiles (black dotted line). c) The size distribution is obtained by fitting the experimental diffusion profiles with a combination of simulated standard profiles (red line in panel (b)). The difference in size between the dye and the EVs allows their separation by diffusion in the channel without pretreatment of the sample. The two populations of free dye and EVs can be deconvoluted, allowing the estimation of the concentration of bound and unbound dye. d) Three different staining strategies were designed to target different EV components: lipids, primary amine groups and specific EV‐biomarkers.
fluoMDS measurements can be performed on a regular light microscope with a fluorescence detection system using few microliters of sample and applying multiple specific staining protocols on the same sample. In this way, different EV components can be simultaneously identified, therefore coupling biochemical analyses with sizing and quantification (Figure 2d). We note that this setup is also compatible with dyes susceptible to photobleaching such as fluorescamine. In fact, the device works under steady‐state conditions, and diffusion profiles along lateral directions are acquired in the presence of a constant flow along the longitudinal direction. Thus, fluorophores at different positions are continuously renewed by flow, hence avoiding photobleaching of the dyes.
All together, these features make fluoMDS a suitable technique for the characterization of EV subpopulations within heterogeneous mixtures. We first demonstrated this concept with a mixture of liposomes, which are structurally similar to EVs. Large (LUV) and small unilamellar labeled lipid vesicles (SUV) were synthetized by extruding a hydrated film of 14:0 Liss Rhod‐PE and individually sized by dynamic light scattering (DLS) analysis. LUVs and SUVs had number z‐average diameters of 197 ± 88 and 58 ± 15 nm, respectively. The lipid vesicles were then mixed to obtain SUV:LUV mass ratios of 1:2 and 1:0.9 and analyzed by fluoMDS. The size distributions of both mixtures exhibited two peaks corresponding to the average diameters of the two vesicle subpopulations (Figure S1, Supporting Information). These values were consistent with the DLS values of the individual homogenous SUV and LUV samples (Figure 3a). In particular, the SUV and LUV mean diameters values obtained by fluoMDS were respectively 52 ± 9 and 219 ± 3 nm for the 1:2 mixture, and 52 ± 9 and 201 ± 27 nm for the 1:0.9 mixture.
Figure 3.
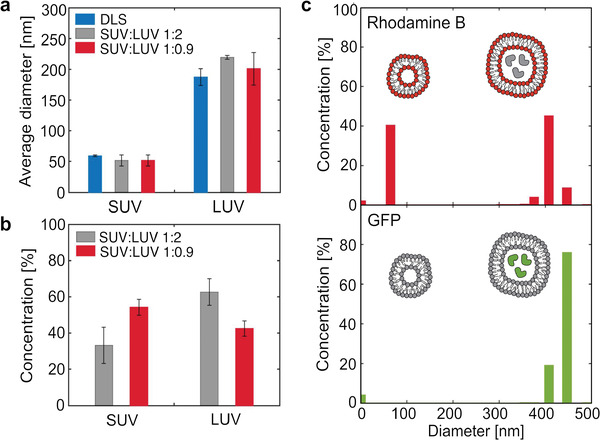
Size, concentration, and biochemical analysis of mixtures of lipid vesicles by fluoMDS. a) Size of the small (SUV) and large (LUV) unilamellar lipid vesicles populations in polydisperse mixtures corresponding to 1:2 (grey) and 1:0.9 (red) SUV:LUV mass ratios evaluated by fluoMDS. In both mixtures the measured average diameters are consistent with the values measured in individual homogeneous populations by DLS (blue). b) fluoMDS correctly detects changes in the mass fractions of SUV and LUV in the two mixtures. c) fluoMDS can detect different components of a mixture in one single analysis by using complementary staining methods and leveraging multiwavelengths fluorescence detection. fluoMDS detects rhodamine‐labeled SUVs and LUVs when membrane Rhod‐PE is excited (red), while only the LUV subpopulation with encapsulated GFP when GFP is excited (green). Experiments (a)–(c) were performed in triplicates and a representative plot is shown for (c). Data (a),(b) represent means ± standard deviations.
Moreover, the fluoMDS quantitatively detected the composition of the vesicle mixtures. As expected, the measured relative concentrations of SUVs and LUVs were, respectively, 33% and 63% in the 1:2 mixture, and 54% and 43% in the 1:0.9 mixture (Figure 3b). We note that this approach could also allow the measurement of absolute concentrations when coupled to a suitable calibration curve.
We finally exploited epifluorescence detection to couple the analysis of size and concentration with a multiplex assay for the detection of vesicle subpopulations characterized by specific biocomponents. For this purpose, we encapsulated green fluorescent protein (GFP) in rhodamine labeled LUV by freezing and thawing ten times a sample of fluorescent liposomes with GFP in the buffer. The number‐based diameter value of the obtained vesicles measured by DLS was 369 ± 197 nm. The GFP‐containing vesicles were mixed with rhodamine labeled SUVs to a 1:1 volume ratio and the resulting mixture was analyzed with the microfluidic device. As expected, we could detect both populations of rhodamine labeled lipid vesicles upon excitation at 540 nm, while only the peak corresponding to the LUV subpopulation was identified when exciting at 470 nm (Figure 3c; Figure S2, Supporting Information). Although in this case the measurement did not require any signal compensation, this aspect should be taken into account in the choice of the dyes.
Overall, these results demonstrate that the microfluidic technique allows us to identify the size, concentration, and presence of a biochemical component in a sub‐population of vesicles within a heterogeneous mixture.
2.2. Fluorescent Labeling and Multiparametric Analysis of EVs
After validating the fluoMDS technique with model vesicles, we next applied the assay to characterize EVs. A key prerequisite for this application is the development of fluorescent staining protocols to detect specific components of EVs and to allow their identification even in mixtures containing impurities of similar size. We selected three dyes to fulfill the following features: (i) target both unspecific and specific phenotypical properties of EVs, i.e., lipids, primary amines and tetraspanin‐CD63; (ii) avoid formation of dye aggregates in the size range of EVs, which can bias the results,[ 30 ] (iii) give a low background fluorescence also in presence of the unreacted dye, therefore avoiding the need for a free dye removal step and simplifying the assay (Figure 2d).
To stain the EV lipids, we selected a photoactivatable silicon‐rhodamine recently developed for super‐resolution imaging of lipid droplets (Figure 4a).[ 31 ] The dye precursor becomes fluorescent only after photoactivation with UV light in an apolar environment and upon reaction with a nucleophilic group. After photoactivation the dye migrates towards a polar environment and reacts with the closest nucleophiles.
Figure 4.
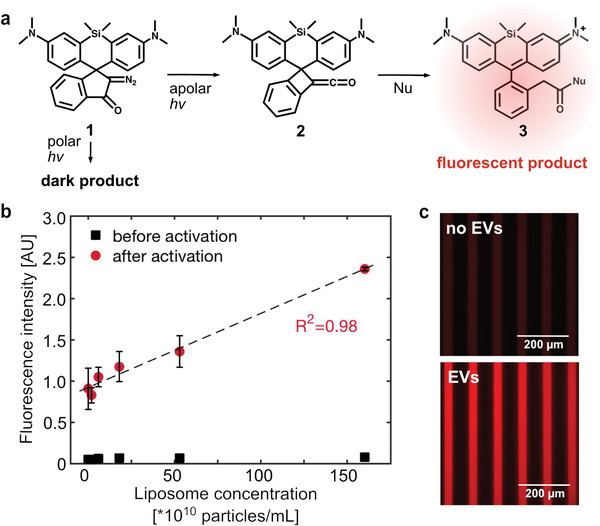
Photoactivatable fluorophore for high signal‐to‐noise unspecific staining of EVs. a) After photoactivation in apolar environment, the silicon‐rhodamine dye precursor (1) converts into a polar intermediate (2), which in turn reacts with a nucleophile and forms the fluorescent product (3). In contrast, photoactivation of the precursor in a polar environment generates a non‐fluorescent product. b) The increase in fluorescence intensity after photoactivation in liposome solutions is linearly proportional to the amount of vesicles. The experiment was performed in triplicates and the data is shown as mean ± standard deviation. c) The presence of EVs triggers the photoactivation of the fluorophore. Fluorescence signal in microfluidic channels after photoactivation of a sample without (top) and with (bottom) EVs.
Since hydrophobic regions and nucleophiles coexist in EVs, we exploited this dye as a novel method for EV labeling. First, we mixed the dye with different amounts of liposomes and showed that the dye could photoactivate in their lipid bilayers and the obtained fluorescence intensity was linearly proportional to the liposome concentration (Figure 4b). Second, we tested the dye with EVs in the microfluidic device and observed a strong fluorescence signal upon photoactivation also in this case (Figure 4c). Thanks to the combination of environment sensitive photoactivation and anchorage to nucleophilic groups, the detection with this fluorophore can yield significantly less background signal compared for instance with common small‐molecule probes such as BODIPY 493/503. Moreover, the dye does not form fluorescent aggregates that are typically observed with other lipophilic dyes (e.g., DiI, PKH26, and BODIPY‐PC) and that interfere with the measurement.[ 30 , 32 , 33 ] This dye is therefore very promising for the unspecific labeling of all hydrophobic components in EV mixtures, which can include lipoproteins and lipid particles in addition to EVs.
For the unspecific staining of proteins we employed fluorescamine, a commercial dye commonly used for protein quantification.[ 34 ] Similar to the silicon‐rhodamine dye, the precursor becomes fluorescent after reaction and binding with primary amine groups. As a result, the activation and the attachment of the fluorophore on the proteins minimize the amount of free fluorescent dye and make this dye convenient for unspecific protein labeling.
Lastly, specific staining of EV‐protein makers was achieved by immunolabeling, in particular with a labeled anti‐CD63 antibody. Following the standard procedures applied in western blotting and flow cytometry methods, we first blocked the unspecific binding sites with BSA and then added the antibody into the solution. A low antibody concentration of 0.1 ng µL−1 was employed in order to keep the background fluorescence intensity low, and detect EVs without performing any washing step to remove free antibodies.
All labeling strategies were individually tested on a sample of MSC‐derived EVs characterized in a previous work,[ 35 ] at concentrations between 1010 and 1014 particles mL−1. The fluoMDS analysis detected the presence of particles with an average diameter of ≈100 nm for all staining protocols, both unspecific and EV‐specific (Figure 5a–c; Figure S3, Supporting Information). Moreover, when the same sample was measured with the isotype control antibody, no peak in the size range of EVs was detected, confirming that the anti‐CD63 antibody specifically bound to EVs (Figure S4, Supporting Information).
Figure 5.
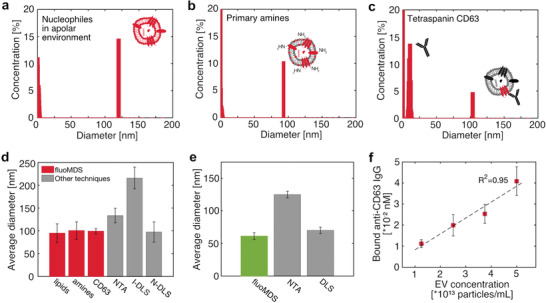
Single‐step immunoassay and fluoMDS multiparametric characterization of EVs extracted from human bone marrow stromal cells and microalgae and stained with unspecific and specific dyes. a–c) Particle populations detected by fluoMDS using different fluorophores: a) photoactivatable silicon‐rhodamine, which detects colocalized lipids and nucleophiles b) fluorescamine, which stains primary amines and c) anti‐CD63 immunoglobulin (IgG), which labels the EV surface‐marker tetraspanin CD63. In each panel the peaks in the low size ranges correspond to the free dye. d) Comparison between the particle average diameters measured by fluoMDS in (a)–(c) and the values measured by NTA in light‐scattering mode and from the DLS intensity (I‐DLS) and number (N‐DLS) based distributions (grey). e) Comparison between the average diameter of nanoalgosomes measured by fluoMDS (green) and the EV modes measured by NTA and DLS values reported in literature (grey).[ 24 ] For these measurements the nanoalgosomes were labelled with photoactivatable silicon‐rhodamine. f) Quantification of bound anti‐CD63 IgG in samples with different EV concentrations. The measured concentration of bound antibody scales linearly with the relative dilution, demonstrating that the device can accurately quantify the amount of CD63 in untreated samples. Experiments were performed in triplicates and data represent means ± standard deviations.
We note that these measurements can robustly provide only the average size of the broad size distribution of EV population. Our analysis provides a size distribution which exhibits only one narrow peak corresponding to the average size of the EV population, and cannot report the real broad distribution of EVs present in solution.
The average diameters estimated with fluoMDS were in agreement with the mean diameter of the DLS number based distribution and with the values measured by nanoparticle tracking analysis (NTA), which currently represents one of the most common EV sizing assays (Figure 5d).[ 12 , 18 , 26 ] These values are significantly smaller than the average diameter of the intensity‐based size distribution measured by DLS, which can be strongly biased by the presence of large particles in the sample.
We next further demonstrated the ability of the technique to size EV samples with different physico‐chemical properties by analyzing EVs derived by microalgae (nanoalgosomes),[ 24 , 25 ] which were stained with the apolar photoactivatable dye. The average diameter obtained by fluoMDS was in well agreement with the mode measured by DLS which we have reported previously,[ 24 ] and smaller than the mode measured by NTA,[ 24 ] possibly due to the limitations of NTA in detecting small particles (Figure 5e).[ 26 , 36 ]
Overall, these results demonstrated that even for very polydisperse samples such as EVs the fluoMDS assay robustly measures the average size of the particle population. This result is also due to the detection based on epifluorescence, which in contrast with scattering methods avoids biases towards larger size particles and makes the measurement less sensitive to sample impurities. We stress, however, that the assay is not a single‐particle method and cannot reconstruct the full‐size distribution without prior assumptions about the shape of the size distribution.
2.3. Semiquantification of Tetraspanin CD63 in EV Samples
Among the labeling strategies discussed in the previous paragraph, the immunolabeling is particularly attractive to quantify and size EV subpopulations even in the complex mixtures encountered not only during bioprocessing, but also in diagnostic applications.
The fluoMDS platform does not require the removal of excess primary antibodies, the addition of a secondary antibody or any washing step. The unbound antibody and the complex formed by the EV and the antibody differ in size almost by one order of magnitude, and are therefore characterized by different diffusion coefficients. The flow rate of the assay was optimized to 60 µL h−1 to capture the differences in diffusion profiles of the two species. At the end of the channel the unbound antibody had almost uniformly diffused along the width of the channel, while the complexes formed by the EVs and the antibodies remained more localized in the middle of the channel. These differences in diffusion profiles were captured by the deconvolution based on Gaussian distributions, which reported on both the average sizes and the relative concentrations of the anti‐CD63 antibody and the vesicle‐antibody complex (Figure 5c).
Under these conditions, the fluoMDS can therefore be used as single‐step immunoassay that not only identifies the presence of EVs but also quantifies the amount of their biomarkers using the primary antibodies in excess. To demonstrate this point, we stained samples of MSC‐derived EVs at different concentrations in the range from 1.25 × 1013 to 5*1013 particles mL−1 with 0.1 ng µL−1 labeled anti‐CD63 antibody. As expected, we observed that the relative concentrations of CD63‐labeled particles in the 100 nm size range increased linearly with the known concentration of EVs (Figure 5f), demonstrating that fluoMDS can quantitatively measure the fraction of bound antibody. From this fraction, the total amount of tetraspanin CD63 in the sample can be evaluated either semiquantitatively by assuming that each antibody binds to one CD63 molecule or quantitatively with a calibration curve. The analysis can be easily repeated with multiple antibodies targeting biomarkers of both EVs and contaminants, thus being a cheap and quick alternative to other immune‐based quantification methods, such as western blot, ELISA, and bead‐based flow cytometry. When the number of biomarker molecules per particle is known, the method could also be used to estimate the number of particles.
2.4. Analysis of EV Samples During Bioprocessing
Finally, we demonstrated the potential of fluoMDS to probe the purity of EV samples against contaminants that are typically coisolated with EVs. Currently, the presence of protein and lipid aggregates which are the most common EV contaminants in large scale preparations, is mostly estimated by measuring ratios of total protein, lipid, and particle amounts.[ 14 ] However, these approaches have low specificity and there is a severe need for more accurate techniques capable of including specific features of EVs and contaminants in the measurement.[ 37 ] When western blotting or proteomic analysis are included, contaminants can be specifically detected, but the analysis time and the sample consumption significantly increase and make the approach inconvenient both at large scales and in clinical settings. In this context, fluoMDS is attractive for its ability to detect different particle subpopulations and quantify specific markers on particles in a broad size range.
We analyzed a sample of 293F‐derived EVs which were only partially pre‐isolated by tangential flow filtration (TFF) (Figure S5, Supporting Information), and compared the results with an MSC‐derived EV sample isolated by ultracentrifugation (UC). We first verified that the 293F‐EVs were characterized by a lower particle to protein ratio with respect to the MSC‐EVs, indicative of the presence of a larger number of contaminating proteins in the sample (Figure 6a). This result was further confirmed by transmission electron microscopy (TEM), which showed the presence of more impurities in the 293F‐derived EV samples compared to the purified MSC‐derived EVs (Figure 6b).
Figure 6.
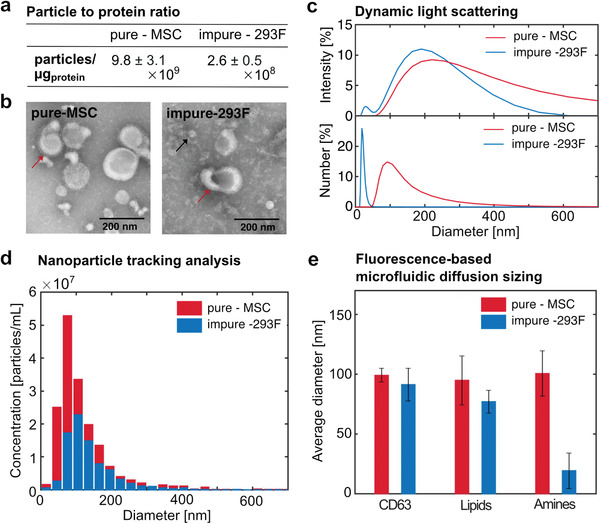
fluoMDS for the characterization of EV purity in bioprocessing. We analyzed human bone marrow stromal cell (MSC) EVs isolated by ultracentrifugation (pure, red) and 293F EVs isolated by tangential flow filtration (impure, blue). a) The pure‐MSC EVs have a higher particle to protein ratio than the impure‐293F EVs. b) TEM images of the pure‐MSC and impure‐293F samples show the presence of cup‐shaped vesicles (red arrows) in both samples, while large amounts of small contaminants were observed only in the impure sample (black arrow). c) DLS intensity‐weighted size distribution shows one single peak for pure‐MSC EVs and two peaks for the impure‐293F EVs, corresponding to EVs and impurities. The peak corresponsing to smaller impurities is even higher in the DLS number‐weighted size distribution. d) NTA measurements with light scattering detection are consistent with DLS analysis for the pure sample but do not detect the small contaminants in the impure sample. e) fluoMDS analysis with specific and unspecific orthogonal dyes shows that the two samples contain similar amounts of CD63‐positive and lipid particles, but drastically differ in primary amine‐rich particles. In particular, the particles containing primary amines in the impure‐293F EVs are characterized by an average diameter of 19 nm, in agreement with the a) particle to protein ratio, b) TEM, and c) DLS measurements. Experiments (a),(c),(d),(e) were performed in triplicates. For experiments (c),(d) representative plots are shown. Experiment (e) is shown as means ± standard deviations.
We further characterized the samples by DLS and NTA. Due to the bias of light scattering detection towards larger sizes, intensity‐, and number‐based size distributions can largely differ in DLS measurements, and the difference increases with the polydispersity of the sample. As expected, the intensity‐ and number‐based size distributions were more consistent for the purified MSC‐derived EVs than for the 293F‐derived EVs (Figure 6c). Specifically, for the MSC EVs one single main peak was observed at 215.8 ± 24.0 and 97.4 ± 22.2 nm in the intensity‐ and number‐based size distribution, respectively, demonstrating that the sample mainly consisted of particles in the EV size range. In contrast, the intensity‐weighed distribution of the 293F‐derived EVs exhibited two peaks at 33.8 ± 10.3 and 217.3 ± 104.1 nm, showing that the sample contained an additional population of smaller particles. The number‐weighed distribution of the 293F EVs showed a single peak at 23.2 ± 6.6 nm, further demonstrating that the impurities were much more abundant in number than the EV‐sized particles, in agreement with the particle to protein ratio and TEM analysis. Interestingly, the distributions obtained by number‐based DLS and NTA measurements were consistent only for the pure MSC sample. These results indicate that NTA provides a better representation of sample polydispersity than DLS, but in some cases it may have lower sensitivity towards smaller particles (Figure 6d).[ 36 ]
We next characterized the purity of the two different samples with our fluoMDS technique (Figure S6, Supporting Information). We first confirmed that both samples contained a population of CD63‐positive particles with an average size consistent with EVs (Figure 6e). The average size of these particles as well as of lipid‐containing particles was similar between the two samples. However, the average size of primary amine‐positive particles was significantly lower for the 293F sample. This result indicated the abundance of small impurities that are rich in primary amines and was consistent with the particle to protein ratio, the TEM and the DLS analysis. Therefore, the fluoMDS setup can assess EV purity by comparing the size values obtained for particles characterized by different components, such as lipids, proteins, and specific EVs biomarkers.
Alternatively, in analogy with the results shown in this work with the EV‐marker CD63, the technique can be further complemented in the future with specific staining protocols to detect contaminant‐specific markers and detect impurities with a similar size to EVs. This approach would allow one to quantify key impurities such as contaminating protein aggregates and lipoproteins and quantify their relative amounts with respect to the target EVs.
3. Discussion
In EV production, the analysis of the sample composition and of the abundance of impurities such as protein aggregates, lipid droplets, lipoproteins, and different EV subpopulations, is crucial to monitor sample reproducibility and product quality.[ 14 ] Currently, there is a lack of high‐throughput screening techniques for the quick and comprehensive analysis of EV samples.[ 9 , 20 , 26 ] In this paper, we tackled this gap and demonstrated that fluoMDS is a promising platform for the rapid characterization of the composition and the purity of EV samples. In particular, the ability to simultaneously size and quantify populations of particles with specific biochemical properties on a microfluidic scale provides important information in short time and with minimal sample consumption.
A crucial step to apply the fluoMDS technique to EV characterization was the development of orthogonal and reliable fluorescent labeling methods that did not bias results and targeted different particle subgroups. We addressed these challenges with fluorescent sensors, i.e., molecules which become fluorescent in response to a specific stimulus. We employed fluorescamine to label primary amine groups and a silicon‐rhodamine dye recently developed for high resolution imaging of lipid particles. These dyes prevented the formation of strong background fluorescence by simultaneously becoming fluorescent and being covalently retained on the particles, consequently making the removal of the free dye unnecessary. In particular, the working principle of the silicon‐rhodamine dye makes it suitable for labelling any lipid‐enclosed particle with nucleophiles on its surface (i.e., EVs, lipoproteins, lipid droplets). Moreover, the absence of long hydrophobic chains in the lipid probe avoids the formation of aggregates, which can bias results.[ 30 , 38 ] We envision that the resolution of the microfluidic sizing assay will largely benefit from developments of novel fluorescent sensors in the context of single molecule detection in high‐resolution imaging.[ 31 , 39 , 40 ]
In addition to unspecific labeling, we introduced specific EV staining by targeting EV surface markers with labeled antibodies conventionally applied in flow cytometry. In this modality, the technique works as a single‐step immunoassay for the semi‐quantitative or quantitative analysis of EV samples. The background fluorescence of the free dye can be reduced by working with low concentrations of dye, i.e., in the order of 0.7 × 10−9 m of labeled antibody. In this work, this feature was applied to quantify the CD63 EV‐marker, and in the future it can be exploited to measure the amount of other EV surface markers (e.g., CD81, CD9), other components (e.g., glycans) as well as contaminants, such as apolipoproteins.
The combination in one platform of these orthogonal labeling methods with sizing and quantification of particles makes fluoMDS also a unique high‐throughput tool for the analysis of sample purity. Currently, purity is assessed by measuring the amount of specific non‐EV biomarkers in the sample, for example with Western‐blotting and proteomic analysis, or by measuring the amount of lipids and proteins with well‐established methods such as sulfophosphovanilin assay and μBCA, and relating these values to other sample properties, such as particle number and RNA amount.[ 14 ] However, the need for more accurate methods which relate specific properties of EVs and contaminants was emphasized.[ 37 ] In this work, we showed that in fluoMDS the shifts in the average sizes of the unspecifically labeled particles with respect to the CD63‐positive ones reflect sample purity and can indicate the presence of small and large contaminants. Importantly, ensemble‐based measurements enable the detection of particles in a broad range of sizes, including small contaminants, which are below the resolution limit of conventional single particle techniques. Parallelly, since fluoMDS can quantify specific biomarkers on particles, the technique can also track the amount of impurities that are similar in size to EVs. Therefore, fluoMDS holds great promise to quickly analyze sample purity of EVs for therapeutic applications and thus to accelerate bioprocess optimization, and can be a useful tool in the diagnostic field where the detection and quantification of lipoproteins is still a major issue.[ 41 , 42 ]
We note that in the current configuration the fluoMDS technique cannot provide the full‐size distribution of the polydisperse EV populations and can measure an estimation of the average size. This information represents a convenient compromise to detect and quantify EVs in important applications, although single particle techniques remain crucial in the situations where the full‐size distribution of EV samples is required. In this context, the measurement of the EV average size for instance by fluoMDS, can complement the analysis of EVs with single particle techniques.[ 26 ]
In contrast with common multiparametric techniques such as NTA and flow cytometry, the fluoMDS analysis requires only few microliters of untreated EV samples, without any sample pretreatments to remove the free dye, therefore simplifying handling steps and reducing the risk of changing the sample composition. The technique is amenable to parallelization and multiplexing by using multiple fluorescence channels and a careful selection of dyes. The whole analysis can be performed with conventional epifluorescence microscopy tools without the need for high resolution instruments which are typically necessary for single particle analysis.[ 13 , 26 , 43 ] Moreover, unlike many quantification tools for EVs,[ 13 , 23 ] the measurement is performed directly in solution, without the requirement of functionalized surfaces for instance with antibodies or aptamers, thereby increasing the flexibility of the technique. In addition to the several microfluidic applications developed for EV fractionation and EV marker detection,[ 13 , 22 , 23 ] this study demonstrates the potential of microfluidic technology also for multiparametric EV characterization in large‐scale screening, which is particularly useful for instance in bioprocess development and analysis based on machine learning methods.[ 44 ] Indeed, the flexibility of the technique that can be used with specific and unspecific dyes for measurements of size, quantity, and purity of EVs, distinguishes fluoMDS from the microfluidic platforms available for the characterization of EVs.
4. Conclusion
In summary, we have shown that fluoMDS combined with three orthogonal staining methods can simultaneously size particle subpopulations, quantify particle biomarkers, and measure sample purity in short time, with limited amount of material and without time‐consuming handling steps before the analysis (Figure 7 ). Thus, fluoMDS has great potential to accelerate EV characterization and support the optimization of EV production and the preparation of reproducible EV samples. We envision that simplification of the device design and implementation of parallelization will lead to a scalable analytical platform for intensive sample screenings, which are required for instance for bioprocess development.
Figure 7.
fluoMDS is a multiparametric analytical assay that enables the measurement of average size, composition, and concentration of EV biomarker. Moreover, fluoMDS can also measure EV purity by comparing the relative amounts of different particle subpopulations characterized by different components. The microfluidic platform minimizes the sample and time required by the analysis, making fluoMDS a convenient technique for rapid screening of EV samples, with applications for instance for bioprocess development.
5. Experimental Section
Preparation of Liposomes
1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS, Avanti Polar Lipids) and 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine B sulfonyl) ammonium salt (14:0 Liss Rhod PE, Avanti Polar Lipids) were dissolved in chloroform and mixed to a DOPS/14:0 Liss Rhod PE molar ratio of 200:1. The chloroform was removed by rotary evaporation to yield a thin lipid film, which was further dried overnight under vacuum. The film was hydrated with phosphate buffer saline (PBS) and gently agitated at room temperature, before freezing the lipid suspension in liquid nitrogen and thawing it in a water bath for five times. The lipid suspension was then extruded through polycarbonate membranes with pore sizes of 1 µm and 50 nm (Sterlitech Corporation) for ten cycles per membrane to yield large and small fluorescent unilamellar lipid vesicles, respectively. Green fluorescent protein (GFP), which was produced as previously described,[ 45 ] was added to a suspension of small fluorescent unilamellar lipid vesicles extruded through the membrane with 50 nm pores and encapsulated in the lipid vesicles by freezing and thawing the mixture for ten times. The GFP‐containing lipid vesicles were then directly purified with PBS by centrifugation in PES centrifugal concentrators (Sartorius) at 1500 x g and room temperature. Polydisperse mixtures of lipid vesicles were produced by mixing vesicles of different sizes at different molar ratios. To prepare nonfluorescent liposomes, 100 mg pronanosome LIPO‐N formulation (Nanovex) was hydrated in PBS (2 mL) and gently shaken for 20 min at 65 °C. The solution was then extruded through polycarbonate membranes with pore size of 50 nm (Sterlitech Corporation). All lipid vesicles suspensions were stored at 4 °C.
Fabrication and Operation of fluoMDS Devices
Microfluidic devices were fabricated through soft lithography techniques. Master wafers produced by spin‐coating SU‐8 photoresists (Microchem) and exposing selected regions to UV light were used as device mold.[ 28 ] To replicate the channels, polydimethylsulfate (PDMS, Silicone elastomer 184, Dow Corning) mixed with carbon powder (Sigma‐Aldrich) was casted on the master wafers and cured for 4 h at 70 °C. After peeling off the PDMS from the master wafer, the PDMS channel and a glass slide were treated by plasma activation (ZEPTO plasma cleaner, Diener Electronics) and bonded together to make the microfluidic devices. Each device has a buffer inlet, a sample inlet, an outlet, and a nozzle where the sample is focused in between two buffer streams (Figure 2a). The channel height was 28.7 µm, the channel width was 40 µm in the hydrodynamic resistors before the nozzle, 3000 µm at the nozzle and 300 µm in the detection region. The channel length in the detection region was 10 cm. When specified, the channel was coated with poly(vinyl alcohol) (PVA, Sigma‐Aldrich) by incubating a PVA solution (1% w/v in ultrapure water) in the channel for 15 min before drying it with a nitrogen stream.
2–4 µL sample was loaded in the device with a gel loading tip after filling the channel and the buffer reservoir with buffer. PBS supplemented with 0.1% w/v bovine serum albumin (BSA, Sigma Aldrich) was used as auxiliary buffer for all samples except for the fluorescamine‐stained samples, whose analysis was run in PVA‐functionalized chips with PBS as auxiliary buffer. The fluid inside the channels was controlled by applying a constant negative pressure at the outlet of the device via an external syringe pump (Cetoni neMESYS, Cetoni GmbH) using a glass syringe (Hamilton). Flow rates were applied in the range from 20 to 60 µL h−1, corresponding to residence times between 120 and 40 s, respectively. Images of the analyte concentration over the channel width were taken at 12 different positions in the channel by moving the stage during the measurement (Figure 2b). The 12 positions corresponded to distances from the nozzle between 10 and 100 mm. Images were acquired on a Ti2‐U inverted microscope (Nikon) equipped with a LED light source (Omicron Laserage Laserprodukte GmbH) and a camera (Zyla sCMOS 4.2P‐CL10, Andor). The used filter cubes were DAPI HC BP Filter set F36‐500, CFP ET Filter set F46‐001, EGFP ET Filter set F46‐002, and Cy5 ET Filter set F46‐009 (AHF analysentechnik AG). Multiplexed measurements did not require signal compensation.
Analysis of Experimental Concentration Profiles
The experimental concentration profiles were analyzed following the approach previously described.[ 27 , 28 , 29 , 46 , 47 ] Briefly, a library of concentration profiles was created for a range of particles of known sizes ri by solving the diffusion advection equation within the channel boundaries.[ 46 ] The experimental concentration profiles P(t) integrated over the channel height were extracted from the acquired images at the 12 positions (Figure 2a,b). The distribution of diffusion coefficients was measured by fitting the concentration profiles with a linear combination of simulated profiles from the library of standard particles of different sizes. The linear combination that best describes the experimental profiles was obtained by minimizing the following function
| (1) |
where t is the diffusion time, y the lateral channel dimension, and ci the weight coefficient of each simulated profile . The relative weights of the linear combination were determined by a basin‐hopping algorithm implemented in Python with 2500 random displacements. The coefficients ci were constrained to a sum of two Gaussian distributions[ 28 , 29 ]
| (2) |
where δ i,0 is the Kronecker delta function which accounts for the diffusion occurring in the nozzle; r 1, σ 1, and r 2, σ 2 are the average radius and standard deviations of the two Gaussian distributions, and a and 1 − c0 − a are the factors that correct the relative fraction of each Gaussian population in the sample. For the quantification analyses, the relative fractions of the two distributions were computed neglecting coefficient c 0 to eliminate the background noise from the measured values.
The quality of the fit was measured from the reduced χ2 coefficient[ 28 ]
| (3) |
Where xi, data and xi, fit are the values of the experimental and simulated diffusion profile at pixel i of the channel width, σ noise is the signal standard deviation, n is the total number of pixels in one diffusion profile, and l the number of fit parameters. The distribution of diffusion coefficients was therefore converted into a distribution of hydrodynamic radii Ri thanks to the Stokes–Einstein relationship where kB is the Boltzmann constant, T the temperature and η the viscosity of the buffer in which the sample diffuses.
Production of Extracellular Vesicles
MSC‐derived EVs were produced and characterized in a previous work.[ 35 ] Briefly, human bone marrow stromal cell line HS‐5 (MSC, ATCC) were cultured for 48 h in Dulbecco's Modified Eagle's Medium supplemented with 100 units mL−1 penicillin, 100 µg mL−1 streptomycin, 2 × 10−3 m GlutaMAX and 1 × 10−3 m sodium pyruvate (Thermo Fisher Scientific) under humidified conditions at 37 °C and a 5% CO2. Fourty‐eight hours‐conditioned medium (200 mL) was harvested from ≈108 cells, clarified by low‐speed differential centrifugation and 0.22‐µm filtration. EVs were then isolated by ultracentrifugation (UC) at 100 000 x g for 70 min using an Optima XE‐90 equipped with a Type 45 Ti Fixed‐Angle Titanium Rotor (Beckman Coulter Life Sciences), resuspended in PBS (100 µL) and stored at −80 °C.
Nanoalgosomes were produced and characterized as described in a previous work.[ 24 ]
HEK293‐F cells (Gibco) were cultured at 37 °C, 8% CO2, and 125 rpm in CD 293 medium (Gibco) supplemented with 4 × 10−3 m GlutaMAX and 250 mg L−1 Pluronic F‐68 for 96 h. Conditioned media (200 mL) was harvested from ≈2.2 × 108 cells with 97% viability by alternating tangential flow filtration with an XCell ATF System (Repligen) and a 0.45 µm cut‐off hollow fiber membrane (N02‐P50U‐10‐N, Repligen). The permeate was clarified by 0.22‐µm filtration and processed by tangential flow filtration using a 500‐kDa cut‐off hollow fiber membrane (D02‐E500‐05‐S, Repligen) at 60 mL min−1 with no backpressure applied. The retentate was circulated until its volume was reduced to 20 mL, diluted in 300 mL PBS, and circulated again until reduction to 10 mL. The 10‐mL retentate was then concentrated 50–200 times with centrifugal concentrators (Sartorius) at 1500 x g, aliquoted and stored at −80 °C. The protein concentration was measured with the MicroBCA Protein Assay Kit (Thermo Fisher Scientific) following manufacturer's instructions.
Labeling of Liposomes and Extracellular Vesicles
Nonfluorescent liposomes were diluted in PBS to particle concentrations between 1010 and 2 × 1012 particles mL−1 and labeled with a photoactivatable silicon rhodamine probe produced as previously described.[ 31 ] The probe was used at a concentration of 20 × 10−6 m. Each solution was irradiated with a UV laser (Omicron Laserage Laserprodukte GmbH) at 377 nm and 0.76 V for 5 s. Pictures before and after irradiation were acquired and the overall intensity of each image was analyzed.
EVs were used at concentrations between 1010 and 1014 particles mL−1 as measured by NTA (ZetaView, Particle Metrix). EVs were first mixed with a dye in a microcentrifuge tube, and then loaded with a gel loading tip at the sample inlet of the microfluidic device. For the staining of lipophilic regions in the sample, EVs were mixed with 10 × 10−6 m photoactivatable silicon rhodamine. After loading the mixture in the microfluidic device and before applying flow in the channel, the sample in the tip at the inlet was photoactivated with UV light at 377 nm and 0.76 V for 2 min. For the staining of primary amine groups, EVs (4 μgprotein) were incubated with 1 × 10−3 m Fluram (Sigma‐Aldrich) for 15 min at room temperature. For immunolabeling staining and control, EVs were incubated in 0.1% BSA (Sigma‐Aldrich) in PBS for 1 h at room temperature in a microcentrifuge tube to block secondary interactions. Then, without washing the sample from the blocking buffer, solutions of 0.2 ng µL−1 CD63 monoclonal antibody (H5C6) – eFluor660 (Invitrogen) and Mouse IgG1 kappa isotype control – eFluor660 (Invitrogen) in 0.1% BSA in PBS were added in the tube in a 1:1 ratio to the blocked EVs to obtain a final antibody concentration of 0.1 ng µL−1. The mixtures were incubated for 1 h at room temperature and then directly loaded at the sample inlet in the device.
DLS Measurements
The size distributions of fluorescent and non‐fluorescent liposomes, MSC‐derived and 293F‐derived EVs were measured using a Zetasizer Nano ZSP DLS system (Malvern) working in backscattering mode at 173° at 20 °C. PBS was used as dilution buffer.
NTA Measurements
NTA measurements were performed on a ZetaView instrument equipped with a CMOS camera and a 405 nm laser (Particle Metrix). The chamber was calibrated daily with polystyrene nanoparticle standards according to manufacturer's recommendation. Samples were diluted in clean PBS to a particle concentration between 107 and 109 particles mL−1 and injected into the sample chamber using a 1 mL syringe until the chamber was filled. Video acquisition was performed for all samples at 11 positions applying an 80% scattering intensity, 150 shutter in light scattering mode, with a trace length of 15 frames and a framerate of 30 s−1. Data were analyzed by the ZetaView analysis software (ZetaView 8.04.02 SP2).
Transmission Electron Microscopy
293F‐EVs (3 µL) were placed on glow discharged (negatively at 25 mA for 30 s in an Emitech K100X glow discharge system, Quorum Technologies Ltd.) carbon‐coated grids (Quantifoil) and were allowed to adsorb for 60 s. Subsequently, the excess liquid was drained with a filter paper and the samples were subjected to negative staining with 2% phosphotungstic acid (PTA, pH 7.2) by two successive incubations of 1 and 15 s, respectively. The grids were air‐dried and imaged in a TEM FEI Morgagni 268 microscope (Thermo Fisher Scientific) in bright field mode operated at 100 kV.
Western Blot
293F‐EVs obtained concentrating the TFF retentate of 200 times by centrifugal filtration were lysed in lysis buffer (20 × 10−3 m Tris, 150 × 10−3 m NaCl, 5 × 10−3 m EDTA, 1% Triton‐X, 25 × 10−3 m NaF, 1 × 10−3 m PMSF, 1 × 10−3 m Na3VO4 and protease inhibitors) for 30 min at 4 °C. 293F cells were pelleted at 200 x g for 5 min and rinsed with PBS. The cells were then lysed in 1× lysis buffer for 30 min on ice and the cell debris were pelleted by centrifugation at 15 400 x g for 10 min. Cell lysate (35 µg) and EV lysate (12 µL) were loaded on 12% sodium dodecyl sulfate polyacrylamide gels and separated at 100 V for 2 h in a Mini‐PROTEAN Tetra Cell (Biorad). Proteins were transferred on a PVDF membrane (Biorad) with a Mini Trans‐Blot module (Biorad). The membrane was blocked with blocking buffer (5% BSA in TBS with 0.1% Tween‐20, TBS‐T) for 2 h at room temperature and incubated overnight at 4 °C under agitation with 1:1000 primary antibody dilutions (CD63 antibody Mx‐49.129.5, CD81 antibody 5A6, Alix antibody 1A12, TSG101 antibody C‐2, Calnexin antibody AF18, GAPDH antibody G‐9, Santa Cruz Biotechnology Inc.) in blocking buffer. The membrane was washed four times with TBS‐T and incubated for 2 h at room temperature with a 1:1000 secondary anti‐mouse HRP conjugated antibody dilution (m‐IgGκ BP‐HRP, Santa Cruz Biotechnology). Blots were detected using enhanced chemiluminescence solution (Western Blotting Luminol Reagent, Santa Cruz Biotechnology) and Chemidoc imaging system (Biorad).
EV Track
All relevant data of the experiments are submitted to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV200122).[ 48 ]
Statistical Analysis
Data analysis and graphs were performed using Matlab 2019a (Mathworks) and Python Spyder IDE (Anaconda). All data with error bars were presented as mean ± standard deviation (SD).
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was mainly supported by the VES4US project funded by the H2020‐EU.1.2.1‐FET Open programme via the Grant Agreement 801338. The work was additionally funded by the ETH Research Grants ETH‐33 17‐2 and ETH‐10 16.1. The electron microscopy work was performed by Stephan Handschin at the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zurich.
Open Access Funding provided by Eidgenossische Technische Hochschule Zurich.
Paganini C., Hettich B., Kopp M. R., Eördögh A., Capasso U. Palmiero, Adamo G., Touzet N., Manno M., Bongiovanni A., Rivera‐Fuentes P., Leroux J.‐C., Arosio P., Rapid Characterization and Quantification of Extracellular Vesicles by Fluorescence‐Based Microfluidic Diffusion Sizing. Adv. Healthcare Mater. 2022, 11, 2100021. 10.1002/adhm.202100021
Data Availability Statement
Research data are not shared.
References
- 1. Maas S. L. N., Breakefield X. O., Weaver A. M., Trends Cell Biol. 2017, 27, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kalluri R., LeBleu V. S., Science 2020, 367, eaau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mendt M., Kamerkar S., Sugimoto H., Mcandrews K. M., Wu C.‐C., Gagea M., Yang S., Rodriges Blanko E. V., Peng Q., Ma X., Marszalek J. R., Maitra A., Yee C., Rezvani K., Shpall E., Lebleu V. S., Kalluri R., JCI Insight 2018, 3, 99263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lener T., Gimona M., Aigner L., Börger V., Buzas E., Camussi G., Chaput N., Chatterjee D., Court F. A., del Portillo H. A., O'Driscoll L., Fais S., Falcon‐Perez J. M., Felderhoff‐Mueser U., Fraile L., Gho Y. S., Görgens A., Gupta R. C., Hendrix A., Hermann D. M., Hill A. F., Hochberg F., Horn P. A., de Kleijn D., Kordelas L., Kramer B. W., Krämer‐Albers E.‐M., Laner‐Plamberger S., Laitinen S., Leonardi T., Lorenowicz M. J., Lim S. K., Lötvall J., Maguire C. A., Marcilla A., Nazarenko I., Ochiya T., Patel T., Pedersen S., Pocsfalvi G., Pluchino S., Quesenberry P., Reischl I. G., Rivera F. J., Sanzenbacher R., Schallmoser K., Slaper‐Cortenbach I., Strunk D., Tonn T., Vader P., van Balkom B. W. M., Wauben M., El Andaloussi S., Théry C., Rohde E., Giebel B., J. Extracell. Vesicles 2015, 4, 30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ayers L., Pink R., Carter D. R. F., Nieuwland R., J. Extracell. Vesicles 2019, 8, 1593755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rohde E., Pachler K., Gimona M., Cytotherapy 2019, 21, 581. [DOI] [PubMed] [Google Scholar]
- 7. Ingato D., Lee J. U., Sim S. J., Kwon Y. J., J. Controlled Release 2016, 241, 174. [DOI] [PubMed] [Google Scholar]
- 8. Mendt M., Rezvani K., Shpall E., Bone Marrow Transplant. 2019, 54, 789. [DOI] [PubMed] [Google Scholar]
- 9. Paganini C., Capasso Palmiero U., Pocsfalvi G., Touzet N., Bongiovanni A., Arosio P., Biotechnol. J. 2019, 14, 1800528. [DOI] [PubMed] [Google Scholar]
- 10. Willms E., Cabañas C., Mäger I., Wood M. J. A., Vader P., Front. Immunol. 2018, 9, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang H., Freitas D., Kim H. S., Fabijanic K., Li Z., Chen H., Mark M. T., Molina H., Martin A. B., Bojmar L., Fang J., Rampersaud S., Hoshino A., Matei I., Kenific C. M., Nakajima M., Mutvei A. P., Sansone P., Buehring W., Wang H., Jimenez J. P., Cohen‐Gould L., Paknejad N., Brendel M., Manova‐Todorova K., Magalhães A., Ferreira J. A., Osório H., Silva A. M., Massey A., Cubillos‐Ruiz J. R., Galletti G., Giannakakou P., Cuervo A. M., Blenis J., Schwartz R., Brady M. S., Peinado H., Bromberg J., Matsui H., Reis C. A., Lyden D., Nat. Cell Biol. 2018, 20, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gardiner C., Di Vizio D., Sahoo S., Théry C., Witwer K. W., Wauben M., Hill A. F., J. Extracell. Vesicles 2016, 5, 32945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shao H., Im H., Castro C. M., Breakefield X., Weissleder R., Lee H., Chem. Rev. 2018, 118, 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Théry C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin‐smith G. K., Ayre D. C., Bach M., Bachurski D., Baharvand H., Balaj L., Bauer N. N., Baxter A. A., Bebawy M., Beckham C., Zavec A. B., Benmoussa A., Berardi A. C., Bielska E., Blenkiron C., Bobis‐wozowicz S., Boilard E., Boireau W., Bongiovanni A., Borràs F. E., Bosch S., Boulanger C. M., Breakefield X., Breglio A. M., Meadhbh Á., Brigstock D. R., Brisson A., Broekman M. L. D., Bromberg F., Bryl‐górecka P., Buch S., Buck A. H., Burger D., Busatto S., Buschmann D., Bussolati B., Buzás E. I., Byrd B., Camussi G., Carter D. R. F., Caruso S., Lawrence W., Chang Y., Chen C., Chen S., Cheng L., Chin R., Clayton A., Clerici S. P., Cocks A., Cocucci E., Coffey J., Cordeiro‐da‐silva A., Couch Y., Coumans F. A. W., Junior F. D. S., De Wever O., Portillo H. A., Deville S., Devitt A., Dhondt B., Di Vizio D., Dieterich L. C., Dolo V., Paula A., Rubio D., Dourado M. R., Driedonks T. A. P., Duarte F. V., Duncan M., Eichenberger R. M., Ekström K., Andaloussi S. E. L., Elie‐caille C., Erdbrügger U., Falcón‐pérez J. M., Fatima F., Fish J. E., Flores‐bellver M., Försönits A., Frelet‐barrand A., Gilbert C., Gimona M., Giusti I., Goberdhan D. C. I., Hochberg H., Hoffmann K. F., Holder B., Holthofer H., Ibrahim A. G., Ikezu T., Inal J. M., Isin M., Jenster G., Jiang L., Johnson S. M., Kusuma G. D., Kuypers S., Laitinen S., Langevin S. M., Lázaro‐ibáñez E., Le Lay S., Lee M., Xin Y., Lee F., Libregts S. F., Ligeti E., Lim R., Lim S. K., Linē A., Lorenowicz J., Lörincz Á. M., Lötvall J., Lovett J., Lowry M. C., J. Extracell. Vesicles 2018, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Görgens A., Bremer M., Ferrer‐Tur R., Murke F., Tertel T., Horn P. A., Thalmann S., Welsh J. A., Probst C., Guerin C., Boulanger C. M., Jones J. C., Hanenberg H., Erdbrügger U., Lannigan J., Ricklefs F. L., El‐Andaloussi S., Giebel B., J. Extracell. Vesicles 2019, 8, 1587567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu C., Zhao J., Tian F., Chang J., Zhang W., Sun J., J. Am. Chem. Soc. 2019, 141, 3817. [DOI] [PubMed] [Google Scholar]
- 17. Ramirez M. I., Amorim M. G., Gadelha C., Milic I., Welsh J. A., Freitas V. M., Nawaz M., Akbar N., Couch Y., Makin L., Cooke F., Vettore A. L., Batista P. X., Freezor R., Pezuk J. A., Rosa‐Fernandes L., Claudia A., Carreira O., Devitt A., Jacobs L., Silva I. T., Coakley G., Nunes D. N., Carter D., Palmisano G., Dias‐Neto E., Nanoscale 2018, 10, 881. [DOI] [PubMed] [Google Scholar]
- 18. Hartjes T. A., Mytnyk S., Jenster G. W., van Steijn V., van Royen M. E., Bioengineering 2019, 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Daaboul G. G., Gagni P., Benussi L., Bettotti P., Ciani M., Cretich M., Freedman D. S., Ghidoni R., Ozkumur A. Y., Piotto C., Prosperi D., Santini B., Ünlü M. S., Chiari M., Sci. Rep. 2016, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thane K. E., Davis A. M., Hoffman A. M., Sci. Rep. 2019, 9, 12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Contreras‐Naranjo J. C., Wu H.‐J., Ugaz V. M., Lab Chip 2017, 17, 3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo S. C., Tao S. C., Dawn H., J. Extracell. Vesicles 2018, 7, 1508271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J., Ma P., Kim D. H., Liu B.‐F., Demirci U., Nano Today 2021, 37, 101066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adamo G., Barone M. E., Fierli D., Aranyos A., Romancino D. P., Picciotto S., Gai M., Carrotta R., Morsbach S., Raccosta S., Stanly C., Paganini C., Cusimano A., Martorana V., Noto R., Librizzi F., Randazzo L., Parkes R., Rao E., Paterna A., Santonicola P., Kisslinger A., Kralj‐igliˇ V., Palmiero U. C., Corcuera L., Di Schiavi E., Liguori G. L., Landfester K., Arosio P., Pocsfalvi G., Touzet N., Bongiovanni A., J. Extracell. Vesicles 2021, 10, e12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Picciotto S., Barone M. E., Fierli D., Aranyos A., Adamo G., Božič D., Romancino D. P., Stanly C., Parkes R., Morsbach S., Raccosta S., Paganini C., Cusimano A., Martorana V., Noto R., Carrotta R., Librizzi F., Capasso Palmiero U., Santonicola P., Iglič A., Gai M., Corcuera L., Kisslinger A., Di Schiavi E., Landfester K., Liguori G. L., Kralj‐Iglič V., Arosio P., Pocsfalvi G., Manno M., Touzet N., Bongiovanni A., Biomater. Sci. 2021, 9, 2917. [DOI] [PubMed] [Google Scholar]
- 26. Arab T., Mallick E. R., Huang Y., Dong L., Liao Z., Zhao Z., Gololobova O., Smith B., Haughey N. J., Pienta K. J., Slusher B. S., Tarwater P. M., Tosar J. P., Zivkovic A. M., Vreeland W. N., Paulaitis M. E., Witwer K. W., J. Extracell. Vesicles 2021, 10, e12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arosio P., Müller T., Rajah L., Yates E. V., Aprile F. A., Zhang Y., Cohen S. I. A., White D. A., Herling T. W., De Genst E. J., Linse S., Vendruscolo M., Dobson C. M., Knowles T. P. J., ACS Nano 2016, 10, 333. [DOI] [PubMed] [Google Scholar]
- 28. Kopp M. R. G., Villois A., Palmiero U. C., Arosio P., Ind. Eng. Chem. Res. 2018, 57, 7112. [Google Scholar]
- 29. Gang H., Galvagnion C., Meisl G., Müller T., Pfammatter M., Buell A. K., Levin A., Dobson C. M., Mu B., Knowles T. P. J., Anal. Chem. 2018, 90, 3284. [DOI] [PubMed] [Google Scholar]
- 30. Morales‐Kastresana A., Telford B., Musich T. A., Mckinnon K., Clayborne C., Braig Z., Rosner A., Demberg T., Watson D. C., Karpova T. S., Freeman G. J., Dekruyff R. H., Pavlakis G. N., Terabe M., Robert‐Guroff M., Berzofsky J. A., Jones J. C., Sci. Rep. 2017, 7, 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eördögh A., Paganini C., Pinotsi D., Arosio P., Rivera‐Fuentes P., ACS Chem. Biol. 2020, 15, 2597. [DOI] [PubMed] [Google Scholar]
- 32. Takov K., Yellon D. M., Davidson S. M., J. Extracell. Vesicles 2017, 6, 1388731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simonsen J. B., J. Extracell. Vesicles 2019, 8, 1582237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Udenfriend S., Stein S., Böhlen P., Dairman W., Leimgruber W., Weigele M., Science (80‐.) 1972, 178, 871. [DOI] [PubMed] [Google Scholar]
- 35. Hettich B., Ben‐Yehuda M., Werner S., Leroux J.‐C., Adv. Sci. 2020, 7, 2002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Filipe V., Hawe A., Jiskoot W., Pharm. Res. 2010, 27, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tian Y., Gong M., Hu Y., Liu H., Zhang W., Zhang M., Hu X., Aubert D., Zhu S., Wu L., Yan X., J. Extracell. Vesicles 2020, 9, 1697028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gardiner C., Ferreira Y. J., Dragovic R. A., Redman C. W. G., Sargent I. L., J. Extracell. Vesicles 2013, 2, 19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Halabi E. A., Thiel Z., Trapp N., Pinotsi D., Rivera‐Fuentes P., J. Am. Chem. Soc. 2017, 139, 13200. [DOI] [PubMed] [Google Scholar]
- 40. Panagopoulou M. S., Wark A. W., Birch D. J. S., Gregory C. D., J. Extracell. Vesicles 2020, 9, 1710020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takov K., Yellon D. M., Davidson S. M., J. Extracell. Vesicles 2019, 8, 1560809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuana Y., Levels J., Grootemaat A., Sturk A., Nieuwland R., J. Extracell. Vesicles 2014, 3, 23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tian Y., Ma L., Gong M., Su G., Zhu S., Zhang W., Wang S., Li Z., Chen C., Li L., Wu L., Yan X., ACS Nano 2018, 12, 671. [DOI] [PubMed] [Google Scholar]
- 44. Narayanan H., Dingfelder F., Butté A., Lorenzen N., Sokolov M., Arosio P., Trends Pharmacol. Sci. 2021, 42, 151. [DOI] [PubMed] [Google Scholar]
- 45. Faltova L., Kü A. M., Hondele M., Weis K., Arosio P., ACS Nano 2018, 12, 9991. [DOI] [PubMed] [Google Scholar]
- 46. Müller T., Arosio P., Rajah L., Cohen S. I. A., Yates E. V., Vendruscolo M., Dobson C. M., Knowles T. P. J., Int. J. Nonlinear Sci. Numer. Simul. 2016, 17, 175. [Google Scholar]
- 47. Arosio P., Hu K., Aprile F. A., Müller T., Knowles T. P. J., Anal. Chem. 2016, 88, 3488. [DOI] [PubMed] [Google Scholar]
- 48. Van Deun J., Mestdagh P., Agostinis P., Akay Ö., Anand S., Anckaert J., Martinez Z. A., Baetens T., Beghein E., Bertier L., Berx G., Boere J., Boukouris S., Bremer M., Buschmann D., Byrd J. B., Casert C., Cheng L., Cmoch A., Daveloose D., De Smedt E., Demirsoy S., Depoorter V., Dhondt B., Driedonks T. A. P., Dudek A., Elsharawy A., Floris I., Foers A. D., Gärtner K., Garg A. D., Geeurickx E., Gettemans J., Ghazavi F., Giebel B., Kormelink T. G., Hancock G., Helsmoortel H., Hill A. F., Hyenne V., Kalra H., Kim D., Kowal J., Kraemer S., Leidinger P., Leonelli C., Liang Y., Lippens L., Liu S., Lo Cicero A., Martin S., Mathivanan S., Mathiyalagan P., Matusek T., Milani G., Monguió‐Tortajada M., Mus L. M., Muth D. C., Németh A., Nolte‐’T Hoen E. N. M., O'Driscoll L., Palmulli R., Pfaffl M. W., Primdal‐Bengtson B., Romano E., Rousseau Q., Sahoo S., Sampaio N., Samuel M., Scicluna B., Soen B., Steels A., Swinnen J. V., Takatalo M., Thaminy S., Théry C., Tulkens J., Van Audenhove I., Van Der Grein S., Van Goethem A., Van Herwijnen M. J., Van Niel G., Van Roy N., Van Vliet A. R., Vandamme N., Vanhauwaert S., Vergauwen G., Verweij F., Wallaert A., Wauben M., Witwer K. W., Zonneveld M. I., De Wever O., Vandesompele J., Hendrix A., Nat. Methods 2017, 14, 228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.