

Abstract
The initial contact with blood and its components, including plasma proteins and platelets, directs the body's response to foreign materials. Natural scaffolds of extracellular matrix or fibrin contain fibrils with nanoscale dimensions, but how platelets specifically respond to the topography and architecture of fibrous materials is still incompletely understood. Here, planar and nanofiber scaffolds are fabricated from native fibrinogen to characterize the morphology of adherent platelets and activation markers for phosphatidylserine exposure and α‐granule secretion by confocal fluorescence microscopy and scanning electron microscopy. Different fibrinogen topographies equally support the spreading and α‐granule secretion of washed platelets. In contrast, preincubation of the scaffolds with plasma diminishes platelet spreading on planar fibrinogen surfaces but not on nanofibers. The data show that the enhanced interactions of platelets with nanofibers result from a higher locally accessible surface area, effectively increasing the ligand density for integrin‐mediated responses. Overall, fibrinogen nanofibers direct platelets toward robust adhesion formation and α‐granule secretion while minimizing their procoagulant activity. Similar results on fibrinogen‐coated polydimethylsiloxane substrates with micrometer‐sized 3D features suggest that surface topography could be used more generally to steer blood‐materials interactions on different length scales for enhancing the initial wound healing steps.
Keywords: hemocompatibility, platelet‐rich plasma, self‐assembly, thrombogenicity, wound healing
Fibrinogen is a natural component of blood clots and a candidate material for wound dressings. Here, fibrinogen coatings of different topography are demonstrated to activate platelets to different degrees. It is uncovered that the increased surface area of 3D fibrinogen nanofibers favors platelet noncoagulant responses, while plasma components render the scaffolds less adhesive. Topography thus steers blood‐material interactions and platelet responses.

1. Introduction
The treatment of acute and chronic wounds has evolved into one of the greatest challenges of our rapidly aging population.[ 1 ] Recent wound healing approaches have benefited immensely from advances in nanotechnology,[ 2 ] where a wide range of engineered nanomaterials has been explored.[ 3 , 4 ] When synthetically prepared materials are used for wound treatment, they come into direct contact with blood, resulting in adsorption of various proteins and subsequent adhesion of blood cells.[ 5 , 6 ] These blood‐material interactions depend on the particular surface chemistry and topography of a scaffold material and have a direct effect on its hemocompatibility.[ 7 ] Although consensus on some general design principles for medical device coatings in contact with blood has been reached, the specific requirements for wound dressings have received much less attention, and the influence of nanoscale topography on protein adsorption and cell adhesion is incompletely understood.[ 6 , 7 ]
When a blood vessel gets injured, platelets (i.e., thrombocytes) adhere to the subendothelial matrix and form a thrombus through fibrinogen‐mediated platelet‐platelet aggregation.[ 8 ] The plasma protein fibrinogen then is enzymatically cleaved by thrombin at the wound site and a nanofibrous fibrin clot is formed during secondary hemostasis.[ 9 ] This fibrin mesh serves as a provisional extracellular matrix to support the migration of fibroblasts, endothelial cells, and immune cells involved in wound repair.[ 10 , 11 ] Platelets attach to fibrin(ogen) via the highly abundant platelet integrin α IIb β 3 whose activation is tightly regulated to avoid erroneous thrombosis.[ 12 ] Immobilized, but not soluble fibrinogen, is sufficient to induce platelet adhesion, spreading and contraction which is strictly dependent on functional α IIb β 3 integrins[ 13 ] and further modulated by ligand density,[ 14 ] substrate stiffness,[ 15 , 16 ] glycoprotein VI signaling,[ 17 ] as well as by other platelet agonists such as thrombin. In native 3D nanofibrous fibrin clots, platelets extend filopodia along fibrin fibers to pull on them to drive clot compaction.[ 18 ] Platelets can foster thrombin generation by exposing “procoagulant” phosphatidylserine (PS) on their surface[ 19 ] which serves as platform for the assembly of the intrinsic tenase and prothrombinase complexes. Platelet activation can also lead to the secretion of α‐granules which contain “prohemostatic” adhesion proteins and clotting factors, but also growth factors, cytokines, and antimicrobial peptides important for wound healing.[ 20 , 21 ] How local microenvironmental cues selectively tune platelet activation to differentially regulate these diverse platelet functions is incompletely understood.[ 8 , 20 ] A detailed characterization of a scaffold's ability to steer platelet responses is thus important to evaluate its suitability for wound healing applications.
So far, the role of surface topography on platelet‐material interactions has been investigated predominantly in the context of dental implants. Microtextured titanium disks with rough surfaces led to enhanced platelet adhesion from platelet rich plasma (PRP)[ 22 , 23 ] as well as to elevated P‐selectin expression[ 23 ] or growth factor release[ 24 ] as a consequence of α‐granule secretion. The coarser the microtexture, the higher was the platelet activation, independent of calcium or phosphate ions on the surface.[ 24 ] However, such synthetic microtextures were fabricated by different techniques and potential differences in surface chemistry[ 25 ] could affect the composition of the adsorbed protein layer[ 6 ] and therefore complicate the interpretation of platelet‐topography interactions. Polydimethylsiloxane (PDMS) substrates with approximately micrometer‐sized holes and well‐defined fibrinogen coating revealed two distinct modes of how platelets touch, sense, and bridge the holes,[ 26 ] leading to more irregular platelet shapes but similar spreading area compared to flat surfaces.[ 27 ] Despite these isolated insights obtained on different synthetically prepared surfaces, an overarching mechanistic understanding of how platelets sense and respond to different topographies is missing.
Fibrin gels can be readily fabricated from (autologous) plasma or from purified fibrinogen by the addition of thrombin and calcium. While fibrin gels offer a simple route to 3D nanofibrous scaffolds for tissue engineering applications,[ 28 ] human thrombin is costly, and its retention can cause thrombogenic complications[ 29 ] when not counter‐acted, e.g., by their modification with heparin.[ 30 ] As a better‐defined alternative to fibrin gels, synthetically prepared fibrinogen nanofibers have become of particular interest as scaffolds to support wound healing.[ 31 ] Techniques to prepare fibrinogen nanofibers include electrospinning,[ 32 ] template‐assisted extrusion[ 33 ] or various self‐assembly approaches.[ 34 ] While electrospun fibrinogen fibers have been shown to promote the growth of fibroblasts,[ 35 , 36 , 37 ] endothelial cells,[ 38 ] smooth muscle cells,[ 39 ] and mesenchymal stem cells,[ 40 , 41 ] their preparation involves organic solvents and high electric fields, which may affect their bioactivity.[ 42 ] For fibrinogen in particular, electrospinning requires very high protein concentrations up to 200 mg mL–1,[ 32 , 38 ] which renders the production of large wound healing scaffolds very resource intensive. To overcome these limitations, we have recently introduced salt‐induced self‐assembly as a physiological process to prepare fibrinogen nanofibers using fibrinogen concentrations of only 5 mg mL–1.[ 43 , 44 ] These self‐assembled fibrinogen nanofibers resemble the architecture of native fibrin clots, exhibit mechanical properties similar to fibrin upon rehydration, promote fibroblast adhesion, and prevent infiltration with Escherichia coli bacteria.[ 45 ] To further explore the potential of self‐assembled fibrinogen nanofibers as new scaffold material for the treatment of acute and chronic wounds, we here assessed the interaction of different fibrinogen topographies with human platelets in the absence or presence of plasma proteins. Unlike fibrin which offers only limited tuning of fibril dimensions,[ 46 , 47 ] the salt‐induced self‐assembly of fibrinogen can be controlled to yield planar 2D films or nanofibrous 3D scaffolds with nearly identical surface chemistries, as previously characterized by circular dichroism and Fourier‐Transform Infrared (FTIR) spectroscopy.[ 43 , 44 ] Our unique approach thus allowed us to specifically investigate the effect of (nano)topography on platelet activation in terms of adhesion and spreading, α‐granule secretion, and PS exposure.
2. Results
2.1. Nanofibers Promote Platelet Adhesion and Spreading on Fibrinogen Scaffolds in the Presence of Plasma
We first characterized the surface topography of nanofibrous versus planar fibrinogen scaffolds. As a reference to facilitate comparisons with previous platelet studies, we included coverslips that were coated with fibrinogen by physisorption from solution. Scanning electron microscopy (SEM) images and confocal microscopy images showed flat, almost featureless surfaces for the physisorbed glass and planar scaffolds, with an increased fibrinogen staining density on planar scaffolds (Figure 1a,b). Nanofibrous scaffolds had the strongest fibrinogen staining (Figure S1, Supporting Information) and exhibited fibrils with submicrometer diameters piled up into bundles which formed an ca. 6 µm high, mountainous landscape with undulated microtopography (Figure 1c), in agreement with previous studies.[ 43 , 44 , 45 ]
Figure 1.
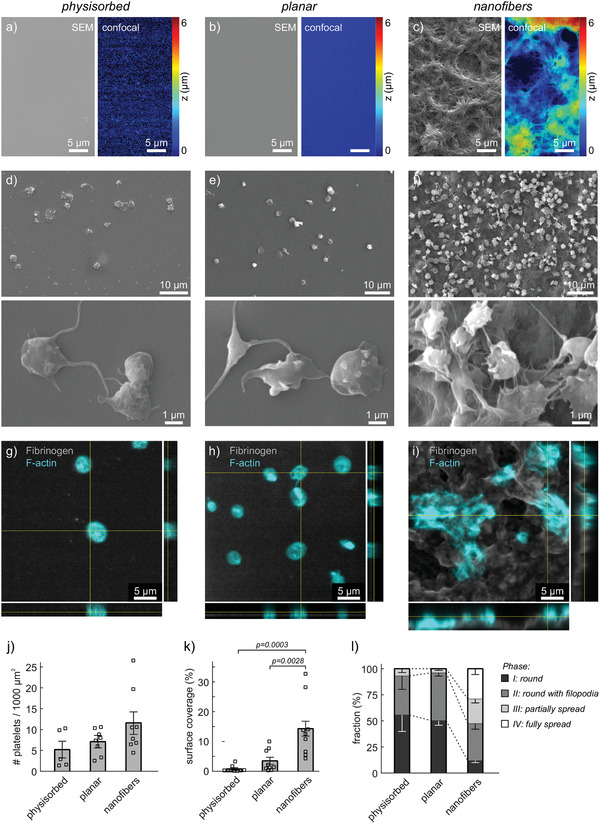
Interactions of human platelet‐rich plasma (PRP) with fibrinogen scaffolds of different topographies. Samples comprised: a,d,g) physisorbed fibrinogen on glass coverslips; b,e,h) planar or c,f,i) nanofibrous fibrinogen scaffolds prepared by salt‐induced self‐assembly. a–c) Characterization of surface topography by scanning electron microscopy (SEM; left) or by confocal z‐stacks (color‐coded; right). Note that SEM and confocal images show different sample regions. d–l) PRP from citrated blood was incubated for 1 h at 37 °C on the surfaces before fixation and sample processing for imaging. d–f) Representative SEM images of fixed, dehydrated, and gold‐coated adherent platelets on the three different scaffolds. g–i) Representative confocal images and orthogonal views of adhered platelets on the three different scaffolds. Gray: fibrinogen stain. Cyan: phalloidin stain. j–l) Quantitative comparison of platelet interactions with the three different scaffolds with respect to j) number of adhered platelets, k) surface coverage by platelets, and l) spreading phase of platelets. Each data point in (j,k) represents one image. Bars and error bars show mean and standard error of the mean (s.e.m.) of replicates, respectively. For (j,k), differences between conditions were assessed by one‐way ANOVA with Tukey post hoc multiple comparison. Only p‐values smaller than 0.05 are displayed.
Static incubation of PRP from healthy donors on the three different surfaces resulted in platelet adsorption and spreading to different degrees. SEM images showed mainly round platelets without or with filopodia on physisorbed and planar fibrinogen surfaces, whereas platelets appeared denser and more spread on fibrinogen nanofibers (Figure 1d–f and Figure S2, Supporting Information). Accordingly, platelets on physisorbed and planar scaffolds showed mainly cortical f‐actin staining with some filopodia protrusions, while on fibrinogen nanofibers, the actin cytoskeleton exhibited clear f‐actin bundles spanning across spread platelets or small platelet aggregates (Figure 1g–i). Counting of platelets revealed a trend toward more adhered platelets on nanofiber scaffolds which did not reach statistical significance (Figure 1j). In contrast, the surface area covered by platelets was highly significantly larger on nanofibers compared to both flat fibrinogen surfaces (Figure 1k). A visual evaluation of the adhesion morphology revealed that about 50% of adherent platelets were partially or fully spread on nanofibers, whereas more than 90% of platelets on flat surfaces remained round or were round with filopodia (Figure 1l). In summary, platelets from PRP adhered and spread much more strongly on nanofibrous scaffolds than on planar scaffolds or on physisorbed fibrinogen control surfaces.
2.2. Washed Platelets Adhere and Spread Normally on All Fibrinogen Surfaces
The failure of platelets from PRP to spread on flat fibrinogen surfaces was unexpected considering that fibrinogen is the primary ligand of the most abundant integrin on platelets, α IIb β 3. Since plasma proteins can affect cell‐material[ 7 ] and receptor‐ligand[ 48 ] interactions, we next tested how washed platelets interact with the three different fibrinogen surfaces in the absence of plasma proteins. Washed platelets adhered and spread on all three substrates (Figure 2a–c and Figure S3, Supporting Information). On flat surfaces, platelets formed extensive flat lamellipodia outward from a central, slightly elevated granulomere region (Figure 2a,b), resulting in the prototypic and commonly reported “fried egg” appearance.[ 49 ] On nanofibers, platelets spread over the rough surface and further extended finger‐like protrusions along fibrinogen fibers (Figure 2c, asterisks), similar to platelets in native fibrin meshes.[ 18 ] Platelets on all substrates showed pronounced bundled f‐actin filaments spanning the cell (Figure 2d–f). Vinculin, an adaptor protein re‐enforcing focal adhesions under mechanical tension,[ 50 ] localized to the ends of these f‐actin bundles, as well as to the outer rim of the lamellipodium on flat fibrinogen surfaces (Figure 2d,e), in agreement with previous findings.[ 49 ] On nanofibers (Figure 2f), focal adhesions and f‐actin bundles appeared more distinct than on flat surfaces; however, a systematic analysis of f‐actin cytoskeletal morphology[ 49 ] did not show significant differences in the self‐alignment of actin filaments within single platelets (Figure 2j). Interestingly, this pronounced formation of f‐actin bundles on nanofiber scaffolds contrasts with our previous study in which fibroblasts exhibited a more diffuse cytoskeleton on fibrinogen fibers compared with aligned actin filaments on planar fibrinogen.[ 45 ] An analysis of platelet outlines revealed a comparable spreading area and overall elliptical platelet shape on all surfaces (Figure 2g,h), whereas the circularity revealed significantly more irregular cell shapes on nanofibers (Figure 2i), as explained by the protrusions seen on SEM images (Figure 2c). Taken together, the interaction of platelets alone with fibrinogen surfaces of different topography resulted in comparable activation of platelet adhesion and spreading, indicating a diminished spreading of platelets on planar surfaces in the presence of plasma proteins.
Figure 2.
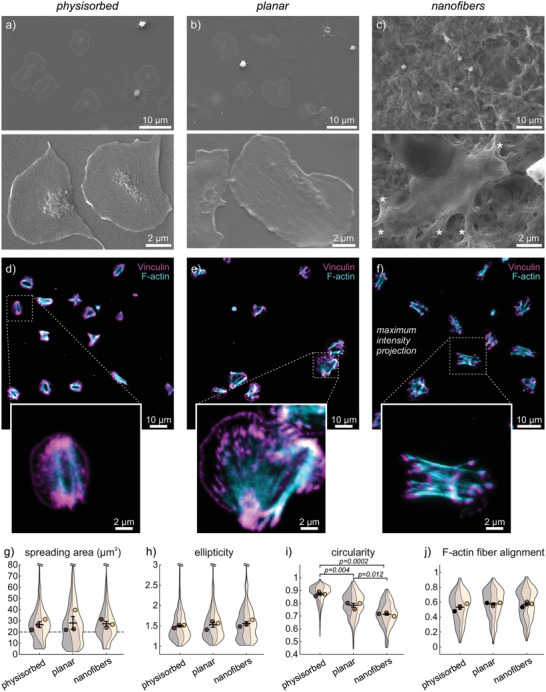
Interactions of washed human platelets with fibrinogen scaffolds of different topographies. Samples comprised: a,d) physisorbed fibrinogen on glass coverslips; b,e) planar or c,f) nanofibrous fibrinogen scaffolds prepared by salt‐induced self‐assembly. Washed platelets were incubated for 1 h at 37 °C on the surfaces before fixation and sample processing for imaging. a–c) Representative SEM images of adhered platelets on the three different scaffolds. Asterisks denote pseudopodia. d,e) Representative confocal slices or f) maximum intensity projections of z‐stacks of adhered platelets on the three different scaffolds. Cyan: phalloidin stain. Magenta: vinculin stain. g–j) Comparison of platelet interactions with the three different scaffolds. Shown are Violin SuperPlots[ 51 ] representing the (non‐normal) distribution of parameters within each biological replicate (band) of three independent experiments, overlaid with the replicate means (circles) and the means’ mean and s.e.m. (error bars). g) Spreading area of single platelets. The number n of platelets per replicate was between 56 and 288. k) Ellipticity, i) circularity, and j) f‐actin fiber alignment of platelets with a spreading area larger than 20 µm2 (dashed line in (g)). The number n of platelets per replicate was between 46 and 234. Differences between conditions were assessed by one‐way ANOVA with Tukey post hoc multiple comparison. Only p‐values smaller than 0.05 are displayed.
2.3. Nanofibers Steer Platelets toward a Noncoagulant Biomechanical Phenotype
To investigate how platelet functions beyond adhesion, spreading and aggregation were affected by fibrinogen scaffold topography, we assessed P‐selectin expression as a marker for α‐granule secretion, and Annexin‐V binding as a marker for PS exposure, in the presence and absence of plasma (Figure 3 ). An f‐actin counterstain showed adhesion morphologies similar to previous experiments (cf. Figures 1 and 2), with predominantly nonspread platelets from PRP on planar scaffolds and partially or fully spread platelets in other conditions (Figure 3a–d). About 50% of platelets expressed P‐selectin on their surface, irrespective of the fibrinogen topography (Figure 3e). PS exposure was considerable (>60%) on planar substrates in the presence of plasma but significantly reduced to <12% on nanofibrous scaffolds, with no significant difference for washed platelets (Figure 3f). Most pronounced in PRP on nanofibers was the reduction for PS+ only platelets (Figure 3g) which typically had a dissolved f‐actin cytoskeleton (cf. Figure 3a for a comparison to planar). Accordingly, the proportion of double negative platelets increased (Figure 3g) which was also seen in the images on nanofibers as platelets containing only the f‐actin stain (Figure 3b). We thus conclude that nanofiber topographies limited the amount of PS exposure on platelets by promoting their spreading, while equally allowing for the secretion of α‐granules.
Figure 3.
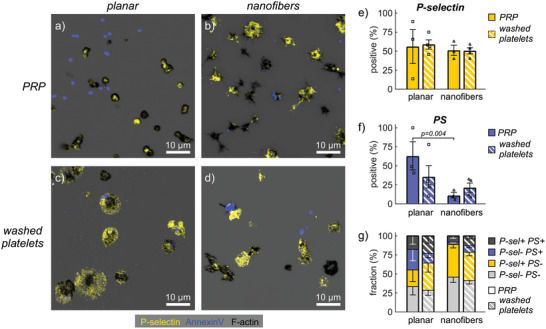
Platelet activation markers on fibrinogen scaffolds of different topographies. a,c) Representative confocal slices on planar scaffolds or b,d) maximum intensity projections of z‐stacks on nanofibrous scaffolds of human platelets stained for phalloidin (black), P‐selectin (yellow), and Annexin‐V (blue). a,b) PRP or c,d) washed platelets were incubated for 45 min at 37 °C on the surfaces and then for 30 min in the presence of fluorescently labeled Annexin‐V before fixation and sample processing for imaging. e–g) Comparison of e) P‐selectin expression, f) PS exposure, and g) combinations thereof by scaffold topography and by absence/presence of plasma. Bars and error bars show mean and s.e.m. of replicates, respectively. For (e,f), differences between topographies were assessed by a ratio paired t‐test. Only p‐values smaller than 0.05 are displayed.
2.4. Nanofibers Promote Platelet Spreading by Locally Increasing the Accessible Surface Area
We next investigated how different aspects of the nanofiber topography contributed to platelet responses. We first assessed whether the micrometer‐scale substrate curvature affected platelet adhesion and spreading. Platelets on nanofibers were found to spread in both, convex regions (“peaks”) or in concave regions (“valleys”) of the undulated 3D landscape (Figure 4a). While platelets on peaks were relatively flat (ca. 1 µm), platelets in valleys often spanned a larger z‐range (2–4 µm) or bridged gaps by forming pronounced focal adhesions on opposing mountain sides interconnected by f‐actin bundles (arrows). To distinguish between peaks and valleys, we obtained a height profile of the surface topography from confocal z‐stacks and used a Laplace filter with a scale of 2 µm to separate convex from concave regions (Figure 4b). A statistical analysis of the surface coverage of platelets in peak versus valley regions, relative to the total surface coverage in the whole image, revealed no preference for convex or concave regions for washed platelets, but a pronounced preference of platelets from PRP for peak regions (Figure 4c).
Figure 4.
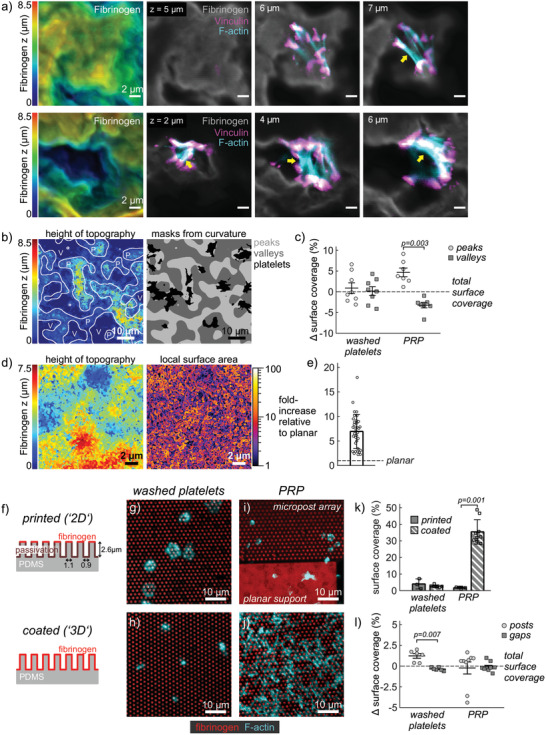
Influence of scaffold curvature and locally accessible surface area on platelet interactions. a) Representative confocal slices of single platelets on nanofiber scaffolds. Gray: fibrinogen stain. Cyan: phalloidin stain. Magenta: vinculin stain. b) Determination of masks for concave (valley) versus convex (peak) regions. c) Difference in platelet surface coverage of valley versus peak regions relative to the overall surface coverage in the respective image. The mean overall surface coverage was 18% for washed platelets and 28% for PRP. d) Detailed height profile of nanofiber scaffolds (left) and corresponding actual local surface area (right). Data analysis was based on high‐resolution deconvolved confocal z‐stacks. e) Fold‐increase of surface area averaged over 10 µm × 10 µm regions (n = 35) on four different scaffolds. f) Schematic and dimensions of microcontact printed (top) or homogeneously coated (bottom) micropost arrays as 2D and 3D topography models. g,h) Washed platelet and i,j) PRP interactions with g,i) printed or h,j) coated micropost arrays. Red: fluorescently labeled fibrinogen. Cyan: phalloidin stain. k) Comparison of surface coverage by platelets. l) Difference in surface coverage of platelets on posts or in gaps relative to the overall surface coverage. Bars and error bars in (c,k,l) show mean and s.e.m. of replicates, respectively, while bars and error bars in (e) show mean and standard deviation. For (k,l), differences between substrate topographies were assessed by a paired t‐test. Only p‐values smaller than 0.05 are displayed. Scale bars: a,d) 2 µm, b,g–j) 10 µm.
Apart from forming a mountainous landscape on the micrometer scale, fibrinogen nanofibers offer a substantially increased surface area due to their thin diameter and porous network architecture (cf. Figure 1c). To estimate the increase in surface area associated with this undulated submicrometer topography, we deconvolved high‐resolution confocal z‐stacks, determined a detailed height profile and calculated the fold‐increase of the local surface area (compared to a flat surface) following standard procedures.[ 52 ] The involved nanofiber architecture locally resulted in a more than 10x increased surface area (Figure 4d), with an average fold‐increase of 6.9 ± 3.5 (mean ± std) compared to a planar surface (Figure 4e). Note that this simplistic analysis underestimates the true surface area because finest structural details are not resolved by diffraction‐limited confocal imaging, and any overhanging regions are not accounted for. The surface area accessible to platelets for adhesion and spreading thus might well be >10x larger than on the planar scaffold, an estimate which is further supported by the measured increase in surface roughness of nanofiber scaffolds by 15‐fold compared to planar.[ 45 ]
Based on this analysis, we hypothesized that an increased locally accessible surface area due to the mountainous 3D topography directly modulated platelet spreading by offering more immobilized fibrinogen molecules that can be bound by platelet integrins. To test our hypothesis in a more generic setting, we turned toward PDMS micropost arrays as a geometrically well‐defined 3D topography model. We compared two cases: arrays in which only the tops of posts were microcontact‐printed with fibrinogen while the surfaces between posts were passivated, restricting platelet adhesion and spreading to the printed post tops, versus arrays that were uniformly coated with fibrinogen by physisorption, allowing platelets to adhere on posts or in the gaps between posts (Figure 4f). The uniformly coated arrays offer a ca. 13‐fold larger local surface area covered by fibrinogen than the printed arrays, which can be compared to the fold‐increase of the surface area on nanofiber versus planar fibrinogen scaffolds. Washed platelets adhered and spread on the tops of the fibrinogen‐printed posts (Figure 4g), as exploited commonly in traction force measurements,[ 53 , 54 ] while they engulfed single posts or adhered between posts to maximize their contact area with the homogeneously fibrinogen‐coated substrate (Figure 4h). Incubation of the top‐printed arrays with PRP resulted in very little platelet adsorption and rounded morphologies, similar to the printed flat support structures next to the micropost arrays (Figure 4i). In stark contrast, PRP incubation on homogeneously coated arrays resulted in an extended mesh of interconnected platelets covering posts and filling the gaps in‐between (Figure 4j). The surface coverage by platelets from PRP was highly significantly increased on homogeneously coated arrays versus printed arrays (Figure 4k), with only a slight preference of platelets for (convex) posts (Figure 4l). In summary, the 3D topography of homogeneously coated microposts led to strong interactions with platelets in PRP while the 2D printed post tops failed to do so, analogous to the interactions of 3D nanofiber and 2D planar fibrinogen scaffolds with PRP.
2.5. Priming of Scaffolds by Plasma Reduces Platelet Adhesion and Spreading
It is well established that the activation threshold of platelets in solution depends on plasma components,[ 55 ] but how plasma‐material interactions might affect the activation threshold of platelets is less well understood. To further investigate how plasma modulated platelet activation in response to different scaffold topographies, we pretreated planar and nanofiber scaffolds with platelet‐poor plasma (PPP) before thorough washing and seeding of washed platelets in plasma‐free spreading buffer. Comparable to the effect of plasma during seeding (Figure 5a, cf. Figure 1), preincubation of the scaffolds with PPP inhibited platelet spreading on planar scaffolds but much less so on nanofibers (Figure 5b). PPP‐pretreatment can thus be understood to result in a general reduction of the activating stimulus strength of scaffolds for platelet adhesion. Since the reduction of platelet spreading was much more pronounced on planar scaffolds, we conclude that the nanotopography of fibrous scaffolds provided an effectively stronger stimulus. This general line of reasoning could explain why the spreading of washed platelets was differentially affected by plasma pretreatment, without the necessity of postulating a topography‐specific interaction of scaffolds with plasma proteins.
Figure 5.
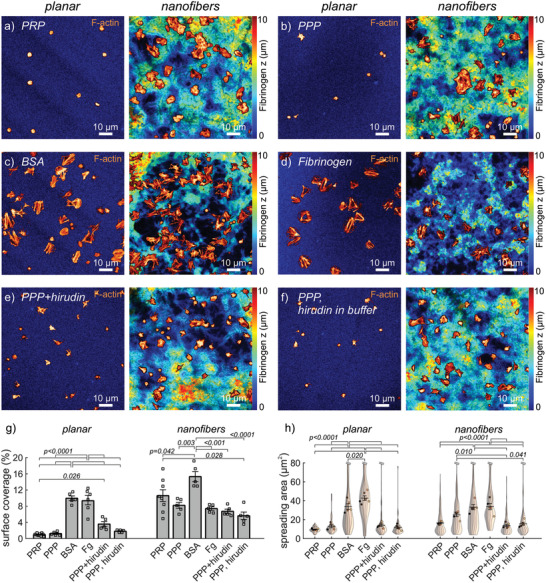
Modulation of platelet adhesion on fibrinogen scaffolds by plasma components. a–f) Representative confocal images of a) PRP on untreated scaffolds or b–f) washed platelets on scaffolds preincubated with b) platelet‐poor plasma (PPP), c) bovine serum albumin (BSA, 1 mg mL–1), d) fibrinogen (1.5 mg mL–1), e) PPP and hirudin (1 U mL–1), and f) PPP while hirudin (1 U mL–1) was added to the spreading buffer. Shown are overlays of the f‐actin stain (orange‐hot) with the fibrinogen stain (color‐coded from blue to red according to z‐position). g) Surface coverage for different treatments on planar or nanofiber fibrinogen scaffolds. h) Violin SuperPlots of platelet spreading area for different treatments by scaffold topography. The number n of platelets per replicate was between 5 and 32 on planar or between 12 and 129 on nanofiber scaffolds, respectively. Bars and error bars in (g,h) show mean and s.e.m. of replicates, respectively. Differences between treatments on the same topography were assessed by one‐way ANOVA with Tukey post hoc multiple comparison. Only adjusted p‐values smaller than 0.05 are displayed.
To test whether the reduction in platelet adhesion was due to a blocking effect, we preincubated the scaffolds with albumin, a major component of plasma commonly used to reduce unspecific protein adsorption or cell adhesion to artificial surfaces. Washed platelets spread normally on albumin‐blocked planar and nanofibrous scaffolds (Figure 5c), comparable to untreated scaffolds (cf. Figure 2), ruling out that albumin was responsible for the observed reduced platelet spreading. Fibrinogen is another major component of plasma which competes to bind activated α IIb β 3 integrins that are required for platelet spreading on immobilized fibrinogen, and soluble fibrinogen has been shown to prevent adsorption of platelets to fibrinogen coatings under flow.[ 56 ] However, pretreatment of scaffolds with soluble fibrinogen (Figure 5d) did not significantly reduce spreading of washed platelets (Figure 5h) while it mainly reduced the number of adhered platelets on nanofibers (Figure S4, Supporting Information). A potential preferential adsorption of fibrinogen from plasma to curved regions[ 57 ] that could enhance interactions on nanofiber scaffolds did thus not play a significant role. These results and the similar behavior of platelets on both physisorbed fibrinogen and planar scaffolds (Figures 1 and 2) further demonstrate that the formaldehyde vapor treatment of scaffolds did not affect plasma‐scaffold and platelet‐scaffold interactions.
The thrombogenicity of materials often is an indirect effect mediated by activation of coagulation,[ 6 , 7 ] potentially depending on surface topography.[ 58 ] Thrombin is a potent activator of platelets[ 59 ] that is found in plasma in its inactive form but can be activated via the contact‐mediated coagulation cascade initiated by binding of factor XII to negatively charged surfaces.[ 60 ] To investigate whether thrombin positively or negatively modulated the response of platelets to the fibrinogen scaffolds, we blocked thrombin activity using the selective inhibitor hirudin, which abolished thrombin‐induced aggregation of washed platelets in solution (Figure S5, Supporting Information). Adding hirudin to PPP during the preincubation of scaffolds before seeding (Figure 5e) or seeding washed platelets on PPP‐pretreated scaffolds in the presence of hirudin (Figure 5f) had similar effects, indicating that any action of thrombin was mediated by its interaction with the scaffold rather than its direct action on platelets. Thrombin inhibition by hirudin did not change the trend of more platelets being found on nanofibers than on planar fibrinogen, even though numbers on planar substrates were tendentially increased (Figure S4, Supporting Information). Thrombin inhibition slightly decreased platelet spreading, and thus surface coverage, on nanofiber scaffolds but did not affect these parameters on planar scaffolds (Figure 5g,h). These opposing trends only partially reduced the differences in platelet behavior between different topographies. Since thrombin inhibition only had a minor effect on platelet adhesion and spreading, a potential activation of the contact pathway on scaffolds, although not directly tested here, did not account for the main inhibitory effect seen with plasma.
2.6. Nanofiber Topographies Facilitate Platelet Adhesion under Shear Flow
Many blood‐biomaterial interactions take place under shear flow which is known to affect platelet‐surface interactions through adhesion receptor mechanotransduction.[ 16 ] We thus extended the investigation of platelet‐scaffold interactions to a previously described flow assay using whole blood.[ 61 ] At arterials shear rates (1500 s–1) where resting platelets do not bind fibrinogen[ 56 , 62 ] but depend on von Willebrand Factor (vWF) capture by the surface and vWF‐GPIb signaling to activate firm platelet adhesion,[ 63 ] no interactions of platelets with planar fibrinogen scaffolds occurred (Figure 6a,b), in stark contrast to platelet accumulation on vWF‐coated channels (Figure S6a, Supporting Information). Very short transient binding of single platelets on planar fibrinogen was observed at elevated venous shear rate (300 s–1), while transient small aggregates formed at 100 s–1 (Figure 6a). At low venous shear rate (50 s–1), platelet interactions with planar scaffolds remained sparse, and when microaggregates formed, they remained smaller than ca. 10 µm and persisted in the field of view for typically less than a minute before detaching or disassembling (Figure 6c, cf. also Figure S6b, Supporting Information). Platelets also adhered transiently and formed microaggregates on nanofiber scaffolds, with a tendency for more prolonged platelet‐surface interactions (see kymographs in Figure 6d and Figure S6c, Supporting Information), resulting in more platelets accumulating over time than on planar scaffolds (Figure 6e). The growth, sliding, and rupture of small aggregates occurred repeatedly at different locations on both planar and nanofiber surfaces and always locally aligned with the flow direction (Movie S1, Supporting Information), which is typical for platelet adhesion to vWF strings. These data indicate that the underlying nanofibrous fibrinogen topography might be more efficient than planar surfaces to capture vWF from blood which mediates the adhesion of platelets to fibrinogen scaffolds under flow. These findings match published results that showed increased platelet adhesion from whole blood at 300 s–1 shear to 3D polymer surfaces with dimensions similar to the peaks and valleys of our mountainous nanofiber topographies, compared to 2D polymer surfaces.[ 64 ]
Figure 6.
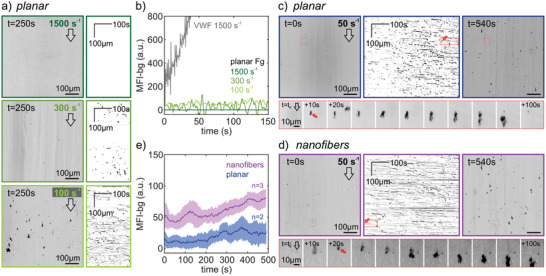
Interactions of whole blood with planar or nanofiber fibrinogen scaffolds under flow. Scaffolds were integrated into a flow channel and whole blood was flown over the scaffolds using a defined flow rate. Platelets were visualized by the fluorescent DiOC6 membrane stain in epifluorescence at two frames per second. a) End frame (left, inverted colormap) and kymographs (right) of time‐lapse acquisitions at shear rates of 1500 s–1 (top), 300 s–1 (middle), or 100 s–1 (bottom). Direction of flow is indicated by an arrow. Kymographs were obtained by performing a maximum intensity projection of each frame onto the direction of flow (y‐axis) and plotting this over time (x‐axis). b) Background‐subtracted mean fluorescence intensity of platelet accumulation over time. Shown are the different shear rates (color coded) on planar fibrinogen scaffolds (see (a)). For comparison, platelet accumulation on a vWF‐coated surface is shown at arterial shear (gray). c,d) Kymographs with start and end frames (top) at a shear rate of 50 s–1 on c) planar or d) nanofiber fibrinogen scaffolds. The exemplary interaction event series (bottom) shown corresponds to the red arrow and boxed regions in the movie frames and kymographs. e) Comparison of platelet accumulation over time on planar (blue) or nanofiber (magenta) fibrinogen scaffolds. Shown are the mean and s.e.m. (40 s moving average) of the background‐subtracted mean fluorescence intensity of platelet accumulation over time.
3. Discussion
The two major findings of our study are that a 3D nanofiber topography facilitates platelet interactions with fibrinogen scaffolds by increasing the local surface area (Figures 1, 2, and 4), and that plasma components interacting with the scaffolds differentially modulate platelet activation responses (Figures 3 and 5). Our approach complements previous works on platelet‐scaffold interactions using organic or inorganic materials with different microtextures[ 5 , 24 , 25 , 57 ] and exceeds these in several aspects. Fibrinogen scaffolds ensure well‐defined binding interactions through α IIb β 3 integrins alone, in contrast to arginine‐glycine‐aspartic acid (RGD) peptides which are bound by many different integrins, thereby ensuring physiological signaling responses of platelets and facilitating the investigation of topography effects independent of composition. Moreover, dissecting blood into its components allowed us to differentiate between platelet‐intrinsic responses versus responses which were mediated by plasma‐scaffold interactions.
Different platelet activation outcomes, most importantly proadhesive integrin activation (enabling attachment at wound sites and platelet aggregation), procoagulant PS exposure (contributing to thrombin‐driven coagulation), as well as prohemostatic and proangiogenic α‐granule secretion (providing a source of adhesion molecules and growth factors at the wound site) are regulated through numerous activation and inhibitory pathways[ 13 , 55 , 65 , 66 ] and contribute critically to the thrombotic risk and the healing response in blood‐material interactions.[ 7 ] The observed counter‐regulation of proadhesive and procoagulant platelet activation responses, as here induced by the nanofiber topography (Figure 3), agrees with previous findings which showed that PS exposing platelets are noncontractile,[ 67 , 68 ] and that PS exposure is upregulated upon inhibition of platelet contractility[ 69 ] or upon reduced platelet spreading on nonadhesive topographies.[ 70 ] Previous reports also observed the highest levels of platelet adhesion and α‐granule secretion on surfaces with a roughness of 105 nm[ 71 ] which is comparable to the ca. 120 nm roughness of fibrinogen nanofibers.[ 45 ] It has been proposed that the conformation of physisorbed fibrinogen on planar surfaces influences platelet binding and activation.[ 72 , 73 ] The preparation of fibrinogen scaffolds using different salt concentrations results in very similar protein conformations, with a slight increase in β‐sheets for nanofibers as compared to more α‐helical content in planar fibrinogen.[ 44 ] Although we thus cannot exclude that the conformation of fibrinogen molecules in our scaffolds differentially affects platelet binding, the qualitatively similar response of platelets to artificial 3D topographies (Figure 4f–l) that were coated with fibrinogen under physiological conditions strongly indicates that the observed enhanced interaction with 3D environments does not depend on fibrinogen conformation, but rather is an intrinsic property of 3D topographies.
Immobilized fibrinogen is a well‐known activating stimulus that induces platelet spreading.[ 13 ] The spreading of platelets depends on integrin clustering and downstream signaling, which are strongly influenced by ligand density of flat fibrinogen coatings.[ 14 ] A tight spacing of binding sites for α IIb β 3 integrins, and the concomitant avidity effect, has recently been proposed to enable fibrin to overcome an elevated activation threshold for platelet aggregation in the presence of integrin inhibitors.[ 74 ] Due to the high surface‐to‐volume ratio of nanofibers and their dense packing, a single platelet on fibers can bind many more fibrinogen molecules in its vicinity than on planar fibrinogen. Our data suggest that the 3D presentation of a larger number of binding sites, as present on nanofibers but also on coated micropost arrays (Figure 4f–l), can enhance platelet responses analogously to reduced ligand spacing. This concept of a locally accessible surface area[ 75 ] as the relevant parameter for platelet adhesion and spreading is also consistent with a reduced platelet adhesion on nanopost[ 57 ] or nanopore[ 76 ] substrates with gaps that are too narrow for platelets to squeeze into.
The interaction of plasma components with the fibrinogen scaffolds that happened before platelet adhesion, rather than their direct action on platelets, led to reduced platelet‐scaffold interactions (Figure 5). The observed reduced interactions could in principle be explained by a competitive inhibition of integrin binding to the fibrinogen scaffolds by a plasma component.[ 56 ] More studies are needed to identify which plasma component might bind to, or mask, integrin binding sites on the fibrinogen scaffolds, since we could exclude a role for soluble fibrinogen and albumin by our experiments. Based on the available data, we speculate that a reduced adhesiveness of plasma‐exposed scaffolds could not only explain the reduced spreading on planar scaffolds,[ 14 , 77 ] but also the preference of platelets in PRP, but not of washed platelets, for peak regions (Figure 4b,c). By engulfing substrate protrusions, less integrin‐mediated friction is needed to hold on to and spread over the surface, compared to platelets attaching to two opposing sides of a valley and pulling transversally at the substrate (cf. Figure 4a), since a similar mechanism has been shown to underlie the sensing of nanowire topographies by fibroblasts using fifilopodia.[ 78 ]
4. Conclusions
Altogether, our study clearly demonstrates the feasibility of employing self‐assembled biomimetic nanofibrous fibrinogen scaffolds to enhance platelet adhesion and spreading as compared to planar fibrinogen surfaces. Further studies are required to elucidate the interactions of plasma proteins with these scaffolds and their capacity to modulate platelet‐scaffold interactions. It is expected that these differential blood‐scaffold interactions based on topography also change the interactions of other cells with the material. This opens up the opportunity to employ fibrinogen scaffolds with defined topography‐function relationships for various tissue engineering applications, e.g., as novel wound dressing materials or implant coatings. Toward future applications in wound healing where platelets are known to recruit and instruct immune cells, it will be critical to assess the interactions of immune cells with nanostructured fibrinogen scaffolds, ideally taking PRP exposure into account. Targeted modulation of platelet responses by topography may have potential to enhance responses in patients who suffer from impaired wound healing. Other avenues for fibrous fibrinogen scaffolds are the cocultivation of skin cells to develop new biomaterials for skin tissue engineering or the tailoring of the topography of fibrinogen‐based biomaterials for stem cell differentiation or blood vessel replacement.
Our more general finding that surface topography on the nanometer to micrometer scale differentially affected certain platelet activation pathways suggests that topography could be exploited as a design parameter for new biomaterials to prevent excessive clotting while supporting the physiological function of platelets. In this respect, we speculate that (nano)topography might be usable as an alternative to heparin coatings of implants.
Generally, the comprehensive methodology used in this study could be applied to delineate the particular contributions of plasma proteins and platelets to define blood‐material interactions also for other (bio)materials. Improved understanding of how the interactions of plasma and/or PRP with tissue engineered scaffolds, either by design[ 79 ] or during implantation, direct the responses of immune cells, stem cells, and tissue forming cells could aid the successful translation of in vitro findings into better in vivo outcomes.
5. Experimental Section
Substrates, Buffers, and Protein Solution Preparation
Glass slides coated with (3‐aminopropyl)triethoxysilane (APTES) were used as substrate to prepare nanofibrous and planar fibrinogen scaffolds following a previously published routine.[ 45 ] Fifteen millimeter round glass coverslips (VWR, Darmstadt, Germany) were first cleaned with freshly prepared piranha solution for 5 min by immersion. For the piranha solution, sulfuric acid (95%; VWR) was mixed with hydrogen peroxide solution (30%; VWR) in a ratio of 3:1 v/v. After piranha treatment the glass slides were washed five times with deionized water from a water purification system (TKA MicroPure UV; Thermo Fisher Scientific, Schwerte, Germany). Cleaned glass coverslips were dried with nitrogen and modified with APTES by overnight immersion in ethanol (Honeywell, VWR) containing APTES (5% v/v; Sigma‐Aldrich, Steinheim, Germany) at room temperature. The modified APTES glasses were washed with ethanol and dried with N2 for subsequent modification with fibrinogen. Fibrinogen stock solutions (10 mg mL–1) were prepared by dissolving fibrinogen from human plasma (Merck KGaA, Darmstadt, Germany) in ammonium hydrogen carbonate solution (10 × 10−3 m; Roth, Karlsruhe, Germany). Subsequently, dialysis was carried out overnight to remove low molecular weight compounds with 14 kDa cutoff cellulose membrane dialysis tubing (Sigma‐Aldrich). Phosphate buffered saline (PBS, Thermo Fischer Scientific) was prepared by dissolving PBS tablets in deionized water.
Preparation of Fibrinogen Scaffolds
As previously introduced,[ 45 ] planar scaffolds were prepared by adding fibrinogen stock solution (100 µL) and deionized water (100 µL) onto APTES‐coated glass slides. For fibrous scaffolds instead of water, PBS (5x; 100 µL) was added to yield the final concentration of fibrinogen on the scaffolds (5 mg mL–1 in 2.5x PBS). All fibrinogen scaffolds were dried in a home‐built humidity chamber overnight at 24 °C and 30% relative humidity. After drying, scaffolds were crosslinked for 2 h by incubation in a sealed beaker containing formaldehyde solution (1 µL cm–3, 37%; AppliChem, Darmstadt, Germany). After crosslinking, all scaffolds were washed with deionized water four times for 15 min each. Washed samples were dried at room temperature for further analysis. To enable visualization of substrate topography by confocal microscopy, the substrates were labeled directly before use with ATTO647N‐NHS dye (1 × 10−6 m in 50 × 10−3 m NaHCO3; AttoTec, Germany) for 1 h at room temperature, washed with PBS.
Preparation of Micropost Arrays
Micropost arrays containing micropillars of 2.65 µm in height and 1 µm in diameter arranged in hexagonal arrays between solid supports were prepared as previously described.[ 54 ] Briefly, hard PDMS base and crosslinker (Gelest Inc, Germany) were mixed 1:1 w/w, degassed under vacuum for 5 min, placed on an air plasma‐treated (PDC‐32G‐2; Harrick, USA) 20 mm round coverslips (Hecht Assistant, Germany) and brought into contact with a negative mold. Samples were degassed under vacuum for 30 min, baked at 80 °C for 16 h, and then unmolded. Prior to surface functionalization with labeled fibrinogen, micropost arrays were treated by UV ozone for 7 min (Novascan PSD PRO‐UV4). Human fibrinogen (Sigma‐Aldrich) was labeled with Alexa Fluor 488 N‐hydroxysuccinimide (NHS) ester (Thermo Fisher Scientific). To restrict fibrinogen coating to the tops of posts and the support structures, a mixture of labeled and unlabeled fibrinogen (1:1 molar ratio, final concentration 100 µg mL–1) was physisorbed onto flat PDMS stamps (1:10 base:crosslinker w/w, Sylgard 184; Dow Corning Inc, USA) for 1 h at room temperature, stamps were dried under nitrogen, and protein was transferred by microcontact printing. Alternatively, to achieve homogeneous fibrinogen coverage of the micropost arrays, a droplet of the same fibrinogen mixture was placed directly onto the substrate for 1 h at room temperature. Unbound fibrinogen was removed by washing with PBS. Uncoated surfaces of the micropost arrays were blocked with bovine serum albumin (BSA, 0.5 mg mL–1 in PBS; 9647; Sigma‐Aldrich) conjugated to DyLight405 NHS ester (Thermo Fisher Scientific) for 1 h at room temperature. Pluronic F127 (0.5% in distilled water; Sigma‐Aldrich) was added to the substrates for a further 1 h at room temperature. Multiple PBS exchanges were performed to remove unbound BSA or Pluronic F127.
Preparation of Flow Chambers
Fibrinogen scaffolds were directly prepared on 24 mm × 50 mm rectangular glass coverslips. A small drop of Alexa Fluor 488 NHS ester (0.5 µL; 10 × 10−6 m in 10 × 10−3 m NaHCO3) was placed in the center and air dried in an oven at 80 °C for 1 min. Previously described flow chambers[ 61 ] (EFJ Engineering; Dublin, Ireland) with channel width 1.5 mm and height 50 µm were assembled onto these coverslips. As a control, plain coverslips were coated with von Willebrandt factor (vWF, Haemate‐P; CSL Behring) for 1 h at room temperature. All chambers were rinsed with PBS (2 mL), blocked with BSA (1 mL; 1% in PBS) for 1 h at room temperature, then rinsed with PBS.
Preparation of Platelets from Whole Blood
Whole blood samples were collected from healthy consenting volunteers in accordance with RCSI research ethics (REC1391 and REC1504) and the Declaration of Helsinki. A 20‐gauge butterfly needle was used to draw blood into a 2.7 mL ethylenediaminetetraacetic acid (EDTA) tube (S‐Monovette; Sarstedt) followed by 2–4 10 mL tubes containing sodium citrate (S‐Monovette; Sarstedt). Blood collected in EDTA was used to get a full blood count (Sysmex KX‐21N). 5 mL citrated whole blood was placed in 15 mL Falcon tubes and centrifuged at 170 relative centrifugal force (RCF) for 10 min at room temperature without deceleration. Platelet‐rich plasma (PRP) was transferred into a clean Falcon tube and recalcified with CaCl2 (1.8 × 10−3 m final concentration) for 10 min prior to seeding on substrates. PPP was prepared by centrifugation of PRP at 1000 RCF for 10 min. To prepare washed platelets, PRP was transferred into a clean Falcon tube, and acid‐citrate‐dextrose solution (0.4 mL; 124 × 10−3 m dextrose, 85 × 10−3 m sodium citrate tribasic, 38 × 10−3 m citric acid) and prostaglandin E1 (PGE1; 2 µL; 1 × 10−3 m in ethanol; Sigma‐Aldrich) were added. The time between PGE1 treatment and seeding on substrates was at least 1 h to ensure normal platelet responsiveness. The tubes were centrifuged at 900 RCF for 5 min without deceleration. A micropipette was used to remove as much supernatant as possible without disturbing the pellet. Platelets were resuspended in washing buffer (1 mL; 10 × 10−3 m sodium citrate, 150 × 10−3 m NaCl, 1 × 10−3 m EDTA, 1% (w/v) dextrose, pH 7.4), and two Falcon tubes of platelets were combined into a clean Falcon tube and centrifuged at 720 RCF without deceleration. After removal of supernatant, platelets were resuspended in Tyrode's buffer (134 × 10−3 m NaCl, 2.68 × 10−3 m KCl, 0.4 × 10−3 m Na2HPO4, 11.9 × 10−3 m NaHCO3, pH 6.5) containing dextrose (5 × 10−3 m; Sigma‐Aldrich) and apyrase (0.5 U mL–1; from potato; Sigma‐Aldrich). Platelet count was determined and adjusted to 3–3.5 × 105 µL–1 with Tyrode's buffer containing dextrose (5 × 10−3 m). Washed platelets were recalcified to 1.8 × 10−3 m CaCl2 at least 10 minutes before use in experiments.
Platelet‐Material Interaction Experiments
Five million washed platelets in spreading buffer (1 mL; Tyrode's buffer containing 1.8 × 10−3 m CaCl2 and 5 × 10−3 m adenosine diphosphate) were added to fibrinogen‐coated coverslips, planar or nanofibrous fibrinogen scaffolds, or micropost arrays in a 12‐well plate. Alternatively, undiluted recalcified PRP (0.8 mL) was added. These samples were incubated at 37 °C with 5% CO2 for 1 h, before washing with Tyrode's buffer, and fixing in paraformaldehyde (3% in PBS) at room temperature for 15 min. For Annexin‐V staining, platelets were spread for 45 min, and the spreading solution was replaced with Annexin‐V binding buffer (100 µL) containing Annexin‐V (Alexa Fluor 555 conjugate; Thermo Fisher Scientific). The samples were incubated at 37 °C with 5% CO2 for a further 30 min before washing and fixation.
Scanning Electron Microscopy
Fixed platelets on fibrinogen substrates were washed with MilliQ water to prevent salt crystals appearing as artifacts. Fixed samples in 12‐well plates were incubated with 50% ethanol:water v/v for 2.5 h at room temperature or at 4 °C overnight. The plates were sealed with parafilm at all stages to reduce evaporation. Samples were incubated with 75% ethanol overnight at 4 °C, followed by 15 min sequential incubations with 87.5%, 93.8%, 96.9%, and 98.4% ethanol. Samples were placed in plastic boxes and allowed to air dry in a fume hood. Before SEM analysis all samples were sputter coated with 7 nm of gold using an EM ACE600 high vacuum sputter coater (Leica Microsystems, Wetzlar, Germany). SEM analysis was conducted with a Desktop SEM (Phenom XL, Phenom‐World BV, Eindhoven, the Netherlands) with acceleration voltages of 10 kV.
Fluorescence Staining
Fixed samples containing fluorescently labeled Annexin‐V were blocked with BSA (3% in PBS) for 45 min at room temperature and incubated with mouse anti‐P‐selectin monoclonal antibody (1:100 in 3% PBS; Abcam Ltd, Cambridge, UK) overnight at 4 °C. Unbound antibody was washed off and samples were incubated with goat anti‐mouse Alexa Fluor 647 (1:100; Fisher Scientific) and Alexa Fluor 488 Phalloidin (1:40; F‐actin stain; Thermo Fisher Scientific) for 2 h at room temperature. Samples containing ATTO647N‐NHS pre‐stained scaffolds were permeabilized with 0.5% Triton X‐100 for 5 min, incubated with Alexa Fluor 488 Phalloidin (1:40; F‐actin stain; Thermo Fisher Scientific) for 2 h at room temperature, washed three times in PBS for 5 min, and mounted in Mowiol© 4‐88 containing 1,4‐Diazabicyclo 2.2.2 octane (DABCO; 4%; Sigma‐Aldrich) on glass object slides. Samples of washed platelets for morphological analysis were permeabilized with 0.5% Triton X‐100 for 15 min, blocked with 3% BSA in PBS for 45 min at room temperature and incubated with mouse anti‐vinculin monoclonal antibody (1:100; Sigma‐Aldrich) overnight at 4 °C. Unbound antibody was washed off and samples were incubated with goat anti‐mouse Alexa Fluor 546 (1:100; Fisher Scientific) and Alexa Fluor 488 Phalloidin (1:40) for 2 h at room temperature. All samples were washed three times and mounted as described above.
Confocal Microscopy
Samples were imaged on two confocal microscopes. Data for Figures 1, 2, 3, and 4a,g–j were imaged with a 40x Plan‐Apochromat oil objective lens on an Examiner Z1 confocal microscope (Zeiss, Oberkochen, Germany) with 70 nm pixel size in xy and 0.3 µm steps for z‐stacks, which was increased to 280 nm in xy and 1 µm in z for P‐selectin and PS stainings on nanofibrous scaffolds to avoid bleaching. The pinhole was set to 1 Airy unit and laser intensity was adjusted to exploit the full dynamic range. Data for Figures 4b,d and 5 were imaged on a Stellaris confocal microscope (Leica, Mannheim, Germany) using excitation lines at 488, 561, and 638 nm of a white light laser and a 100x oil immersion objective (HC PL APO CS2, 1.40 NA). Z‐stacks for Figures 4b and 5 were acquired with 178 nm pixel size and 1 µm step size in z. Z‐stacks for Figure 4d were acquired with 46 nm pixel size and 124 nm step size in z and further processed by adaptive deconvolution (Lightning; Leica).
Image Analysis
A morphometric analysis of the platelet actin cytoskeleton was performed as previously described.[ 49 ] Briefly, platelet actin images were used to segment individual platelets and single platelet parameters (spreading area, circularity, F‐actin alignment, and vinculin adhesion morphology) were calculated. Single platelet P‐selectin and Annexin‐V stainings were categorized in a blinded fashion as either negative or positive by three independent individuals to achieve an unbiased quantification of the data. For the analysis of effective surface area of the nanofiber scaffolds (Figure 4d,e), deconvolved stacks were upsampled 5x in the z‐direction and an argmax z‐projection was performed using the CLIJ2 plugin in Fiji.[ 80 ] From these digital height maps, the calculation of the surface area per pixel was performed according to a published procedure[ 52 ] and implemented in a custom MATLAB (Mathworks) script.
Flow Assay and Analysis
Whole blood samples were stained with 3,3'‐dihexyloxacarbocyanine iodide (1 × 10−3 m; DiOC6) for 5 min before connecting via a ca. 10 cm long tubing to the flow chamber. Flow was driven through the flow chamber by suction using a 10 mL glass syringe (Micromate) using a syringe pump (PHD 2000, Harvard apparatus). The flow rate was adapted to achieve the desired shear rate of 50 s–1 (or higher, as indicated) at the bottom of the rectangular channel. The chambers were mounted on the stage of on an inverted epifluorescence microscope (Nikon Eclipse Ti2) in an incubation chamber (Okolab) at 37 °C. Illumination around 470 nm (CoolLED) was used together with a green fluorescene protein (GFP) filterset and a 525/50 bandpass emission filter. The bottom of chambers was prefocused using the fluorescently marked spot on the scaffold before turning on the perfect focus system and moving ca. 3–5 mm upstream. This pre‐focussing allowed acquisitions to be started within 3–6 s after the blood entered the channel. Images were acquired on an sCMOS camera (Orca Fusion BT; Hamamatsu) at 2 frames s−1 at 15x magnification (pixel size 0.653 µm) for 9 min. Movies were processed using an anisotropic 3D median filter in ImageJ to remove fluorescence of fast‐moving labeled platelets in the blood stream. 2D kymographs were obtained from filtered movies by a maximum intensity projection in the transverse direction and postfiltering by an anisotropic 2D median filter before a background subtraction. To measure fluorescence intensity of platelet accumulation on scaffolds over time, the filtered movies were resized 0.25‐fold to speed up processing and then a 3D median‐filtered background movie was subtracted to remove the increasing background fluorescence which occurred due to DiOC6 binding to the scaffolds, before measuring the mean intensity of each frame. Mean and standard deviation of experimental replicates were determined over a 40 s moving window before rescaling the standard deviation by the square root of the replicate number to obtain mean ± s.e.m. (cf. Figure 6e).
Statistical Analysis
Continuous variables are expressed as mean ± s.e.m. Mean values from different replicates were treated as normally distributed with equal variances. Paired t‐tests were used to determine differences between two conditions. One‐way analysis of variance (ANOVA) was used to determine differences between three or more experimental conditions, followed by Tukey's post hoc test for multiple comparisons. The significance level was set at p ≤ 0.05. Results below this value were considered statistically significant. All analyses were conducted using GraphPad Prism (version 8.0.0). Violin SuperPlots were generated using the MATLAB implementation from the respective publication.[ 51 ]
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supplemental Movie 1
Acknowledgements
The authors would like to thank Thomas Lukasczyk (Fraunhofer IFAM) for access to the SEM device. The authors gratefully acknowledge the super‐resolution image consortium (SRIC) at the Royal College of Surgeons in Ireland for equipment access and technical expertise. D.B. gratefully acknowledges funding by the Emmy Noether program of the German Research Foundation (DFG) via grant number 267326782. St.S. was financially supported by the internal program Fraunhofer TALENTA start. This work was further supported by RCSI under a StAR lecturership and through the MPharm student‐selected projectP scheme (I.S.).
Open access funding provided by IReL.
Kenny M., Stamboroski S., Taher R., Brüggemann D., Schoen I., Nanofiber Topographies Enhance Platelet‐Fibrinogen Scaffold Interactions. Adv. Healthcare Mater. 2022, 11, 2200249. 10.1002/adhm.202200249
Contributor Information
Dorothea Brüggemann, Email: brueggemann@uni-bremen.de.
Ingmar Schoen, Email: ingmarschoen@rcsi.ie.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Dreifke M. B., Jayasuriya A. A., Jayasuriya A. C., Mater. Sci. Eng., C 2015, 48, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kalashnikova I., Das S., Seal S., Nanomedicine 2015, 10, 2593. [DOI] [PubMed] [Google Scholar]
- 3. Parani M., Lokhande G., Singh A., Gaharwar A. K., ACS Appl. Mater. Interfaces 2016, 8, 10049. [DOI] [PubMed] [Google Scholar]
- 4. Rajendran N. K., Kumar S. S. D., Houreld N. N., Abrahamse H., J. Drug Delivery Sci. Technol. 2018, 44, 421. [Google Scholar]
- 5. Weber M., Steinle H., Golombek S., Hann L., Schlensak C., Wendel H. P., Avci‐Adali M., Front. Bioeng. Biotechnol. 2018, 6, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brash J. L., Horbett T. A., Latour R. A., Tengvall P., Acta Biomater. 2019, 94, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gorbet M., Sperling C., Maitz M. F., Siedlecki C. A., Werner C., Sefton M. V., Acta Biomater. 2019, 94, 25. [DOI] [PubMed] [Google Scholar]
- 8. van der Meijden P. E. J., Heemskerk J. W. M., Nat. Rev. Cardiol. 2019, 16, 166. [DOI] [PubMed] [Google Scholar]
- 9. Mosesson M. W., J. Thromb. Haemostasis 2005, 3, 1896. [DOI] [PubMed] [Google Scholar]
- 10. Reinke J. M., Sorg H., Eur. Surg. Res. 2012, 49, 35. [DOI] [PubMed] [Google Scholar]
- 11. Rodrigues M., Kosaric N., Bonham C. A., Gurtner G. C., Physiol. Rev. 2019, 99, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Durrant T. N., Van Den Bosch M. T., Hers I., Blood 2017, 130, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Savage B., Shattil S. J., Ruggeri Z. M., J. Biol. Chem. 1992, 267, 11300. [PubMed] [Google Scholar]
- 14. Jirouskova M., Jaiswal J. K., Coller B. S., Blood 2007, 109, 5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qiu Y., Brown A. C., Myers D. R., Sakurai Y., Mannino R. G., Tran R., Ahn B., Hardy E. T., Kee M. F., Kumar S., Bao G., Barker T. H., Lam W., Proc. Natl. Acad. Sci. USA 2014, 111, 14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hansen C. E., Qiu Y., McCarty O. J. T., Lam W. A., Annu. Rev. Biomed. Eng. 2018, 20, 253. [DOI] [PubMed] [Google Scholar]
- 17. Mangin P. H., Onselaer M.‐B., Receveur N., Le Lay N., Hardy A. T., Wilson C., Sanchez X., Loyau S., Dupuis A., Babar A. K., Miller J. L. C., Philippou H., Craig E., Herr A. B., Ariëns R. A. S., Mezzano D., Haematologica 2018, 103, 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim O. V., Litvinov R. I., Alber M. S., Weisel J. W., Nat. Commun. 2017, 8, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoenwaelder S. M., Yuan Y., Josefsson E. C., White M. J., Yao Y., Mason K. D., O'Reilly L. A., Henley K. J., Ono A., Hsiao S., Willcox A., Roverts A. W., Huang D. C. S., Salem H. H., Kile B. T., Jackson S. P., Blood 2009, 114, 663. [DOI] [PubMed] [Google Scholar]
- 20. Golebiewska E. M., Poole A. W., Blood Rev. 2015, 29, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nurden A. T., Nurden P., Sanchez M., Andia I., Anitua E., Front. Biosci. 2008, 13, 3532. [DOI] [PubMed] [Google Scholar]
- 22. Kämmerer P. W., Gabriel M., Al‐Nawas B., Scholz T., Kirchmaier C. M., Klein M. O., Clin. Oral Implants Res. 2012, 23, 504. [DOI] [PubMed] [Google Scholar]
- 23. Park J. Y., Gemmell C. H., Davies J. E., Biomaterials 2001, 22, 2671. [DOI] [PubMed] [Google Scholar]
- 24. Kikuchi L., Park J. Y., Victor C., Davies J. E., Biomaterials 2005, 26, 5285. [DOI] [PubMed] [Google Scholar]
- 25. Milleret V., Tugulu S., Schlottig F., Hall H., Eur. Cells Mater. 2011, 21, 430. [DOI] [PubMed] [Google Scholar]
- 26. Sandmann R., Köster S., Sci. Rep. 2016, 6, 22357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sandmann R., Henriques S. S. G., Rehfeldt F., Köster S., Soft Matter 2014, 10, 2365. [DOI] [PubMed] [Google Scholar]
- 28. Shaikh F. M., Callanan A., Kavanagh E. G., Burke P. E., Grace P. A., McGloughlin T. M., Cells Tissues Organs 2008, 188, 333. [DOI] [PubMed] [Google Scholar]
- 29. Frost‐Arner L., Spotniz W. D., Rodeheaver G. T., Drake D. B., Plast. Reconstr. Surg. 2001, 108, 1655. [DOI] [PubMed] [Google Scholar]
- 30. Kaplan O., Hierlemann T., Krajewski S., Kurz J., Nevoralová M., Houska M., Riedel T., Riedelová Z., Zárubová J., Wendel H. P., et al., J. Biomed. Mater. Res., Part A 2017, 105, 2995. [DOI] [PubMed] [Google Scholar]
- 31. Rajangam T., An S. S. A., Int. J. Nanomed. 2013, 8, 3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wnek G. E., Carr M. E., Simpson D. G., Bowlin G. L., Nano Lett. 2003, 3, 213. [Google Scholar]
- 33. Raoufi M., Aslankoohi N., Mollenhauer C., Boehm H., Spatz J. P., Brüggemann D., Integr. Biol. 2016, 8, 1059. [DOI] [PubMed] [Google Scholar]
- 34. Stamboroski S., Joshi A., Noeske P.‐L. M., Köppen S., Brüggemann D., Macromol. Biosci. 2021, 21, 2000412. [DOI] [PubMed] [Google Scholar]
- 35. Yuan T. T., DiGeorge Foushee A. M., Johnson M. C, Jockheck‐Clark A. R, Stahl J. M, Nanoscale Res. Lett. 2018, 13, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McManus M. C., Boland E. D., Simpson D. G., Barnes C. P., Bowlin G. L., J. Biomed. Mater. Res., Part A 2007, 81A, 299. [DOI] [PubMed] [Google Scholar]
- 37. Sell S. A., Francis M. P., Garg K., McClure M. J., Simpson D. G., Bowlin G. L., Biomed. Mater. 2008, 3, 045001. [DOI] [PubMed] [Google Scholar]
- 38. Gugutkov D., Gustavsson J., Ginebra M. P., Altankov G., Biomater. Sci. 2013, 1, 1065. [DOI] [PubMed] [Google Scholar]
- 39. McManus M., Boland E., Sell S., Bowen W., Koo H., Simpson D., Bowlin G., Biomed. Mater. 2007, 2, 257. [DOI] [PubMed] [Google Scholar]
- 40. Forget J., Awaja F., Gugutkov D., Gustavsson J., Gallego Ferrer G., Coelho‐Sampaio T., Hochman‐Mendez C., Salmerón‐Sánchez M., Altankov G., Salmeron‐Sánchez M., Altankov G., Macromol. Biosci. 2016, 16, 1348. [DOI] [PubMed] [Google Scholar]
- 41. Nedjari S., Awaja F., Altankov G., Sci. Rep. 2017, 7, 15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palchesko R. N., Sun Y., Zhang L., Szymanski J. M., Jallerat Q., Feinberg A. W., in Springer Handbook of Nanomaterials, Springer, Berlin, Heidelberg: 2013. [Google Scholar]
- 43. Stapelfeldt K., Stamboroski S., Mednikova P., Brüggemann D., Biofabrication 2019, 11, 025010. [DOI] [PubMed] [Google Scholar]
- 44. Stapelfeldt K., Stamboroski S., Walter I., Suter N., Kowalik T., Michaelis M., Brüggemann D., Nano Lett. 2019, 19, 6554. [DOI] [PubMed] [Google Scholar]
- 45. Suter N., Joshi A., Wunsch T., Graupner N., Stapelfeldt K., Radmacher M., Müssig J., Brüggemann D., Mater. Sci. Eng., C 2021, 126, 112156. [DOI] [PubMed] [Google Scholar]
- 46. Herbert C. B., Nagaswami C., Bittner G. D., Hubbell J. A., Weisel J. W., J. Biomed. Mater. Res. 1998, 40, 551. [DOI] [PubMed] [Google Scholar]
- 47. Rowe S. L., Lee S. Y., Stegemann J. P., Acta Biomater. 2007, 3, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nicolai L., Schiefelbein K., Lipsky S., Leunig A., Hoffknecht M., Pekayvaz K., Raude B., Marx C., Ehrlich A., Pircher J., Zhang Z., Saleh I., Marel A. K., Löf A., Petzold T., Lorenz M., Stark K., Pick R., Rosenberger G., Weckbach L., Uhl B., Xia S., Reichel C. A., Walzog B., Schulz C., Zheden V., Bender M., Li R., Massberg S., Gaertner F., Nat. Commun. 2020, 11, 5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lickert S., Sorrentino S., Studt J. D., Medalia O., Vogel V., Schoen I., Sci. Rep. 2018, 8, 5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Atherton P., Stutchbury B., Jethwa D., Ballestrem C., Exp. Cell Res. 2016, 343, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kenny M., Schoen I., Mol. Biol. Cell 2021, 32, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jenness J. S., Wildl. Soc. Bull. 2004, 32, 829. [Google Scholar]
- 53. Feghhi S., Munday A. D., Tooley W. W., Rajsekar S., Fura A. M., Kulman J. D., López J. A., Sniadecki N. J., Biophys. J. 2016, 111, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lickert S., Selcuk K., Kenny M., Mehl J. L., Bender M., Früh S. M., Burkhardt M. A., Studt J. D., Nieswandt B., Schoen I., Vogel V., Sci. Adv. 2022, 8, eabj8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Veninga A., Baaten C. C. F. M. J., De Simone I., Tullemans B. M. E., Kuijpers M. J. E., Heemskerk J. W. M., van der Meijden P. E. J., Thromb. Haemostasis 2021, Online ahead of print, 10.1055/s-0041-1735972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Endenburg S. C., Lindeboom‐Blokzijl L., Zwaginga J. J., Sixma J. J., de Groot P. G., Arterioscler. Thromb. Vasc. Biol. 1996, 16, 633. [DOI] [PubMed] [Google Scholar]
- 57. Koh L. B., Rodriguez I., Venkatraman S. S., Biomaterials 2010, 31, 1533. [DOI] [PubMed] [Google Scholar]
- 58. Ferraz N., Nilsson B., Hong J., Ott M. K., J. Biomed. Mater. Res., Part A 2008, 87, 575. [DOI] [PubMed] [Google Scholar]
- 59. Sang Y., Roest M., de Laat B., de Groot P. G., Huskens D., Blood Rev. 2021, 46, 100733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Samuel M., Pixley R. A., Villanueva M. A., Colman R. W., Villanueva G. B., J. Biol. Chem. 1992, 267, 19691. [PubMed] [Google Scholar]
- 61. Kent N. J., Basabe‐Desmonts L., Maede G., MacCraith B. D., Corcoran B. G., Kenny D., Ricco A. J., Biomed. Microdevices 2010, 12, 987. [DOI] [PubMed] [Google Scholar]
- 62. Bonnefoy A., Liu Q., Legrand C., Frojmovic M. M., Biophys. J. 2000, 78, 2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Savage B., Saldívar E., Ruggeri Z. M., Cell 1996, 84, 289. [DOI] [PubMed] [Google Scholar]
- 64. Chen H., Song W., Zhou F., Wu Z., Huang H., Zhang J., Lin Q., Yang B., Colloids Surf., B 2009, 71, 275. [DOI] [PubMed] [Google Scholar]
- 65. Altschuler D., Lapetina E. G., J. Biol. Chem. 1993, 268, 7527. [PubMed] [Google Scholar]
- 66. Eigenthaler M., Ullrich H., Geiger J., Horstrup K., Honig‐Liedl P., Wiebecke D., Walter U., J. Biol. Chem. 1993, 268, 13526. [PubMed] [Google Scholar]
- 67. Nechipurenko D. Y., Receveur N., Yakimenko A. O., Shepelyuk T. O., Yakusheva A. A., Kerimov R. R., Obydennyy S. I., Eckly A., Léon C., Gachet C., Brishchuk E. L., Ataullakhanov F. I., Mangin P. H., Panteleev M. A., Arterioscler. Thromb. Vasc. Biol. 2019, 39, 37. [DOI] [PubMed] [Google Scholar]
- 68. Zhang Y., Qiu Y., Blanchard A. T., Chang Y., Brockman J. M., Ma V. P.‐Y., Lam W. A., Salaita K., Proc. Natl. Acad. Sci. USA 2018, 115, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Agbani E. O., Van Den Bosch M. T. J., Brown E., Williams C. M., Mattheij N. J. A., Cosemans J. M. E. M., Collins P. W., Heemskerk J. W. M., Hers I., Poole A. W., Circulation 2015, 132, 1414. [DOI] [PubMed] [Google Scholar]
- 70. Ferraz N., Hong J., Karlsson Ott M., J. Biomater. Appl. 2010, 24, 675. [DOI] [PubMed] [Google Scholar]
- 71. Ahmed M., Ghanbari H., Cousins B. G., Hamilton G., Seifalian A. M., Acta Biomater. 2011, 7, 3857. [DOI] [PubMed] [Google Scholar]
- 72. Firkowska‐Boden I., Jandt K. D., Helbing C., Dauben T. J., Pieper M., Langmuir 2020, 36, 11573. [DOI] [PubMed] [Google Scholar]
- 73. Zhang L., Casey B., Galanakis D. K., Marmorat C., Skoog S., Vorvolakos K., Simon M., Rafailovich M. H., Acta Biomater. 2017, 54, 164. [DOI] [PubMed] [Google Scholar]
- 74. Buitrago L., Lefkowitz S., Bentur O., Padovan J., Coller B., Blood Adv. 2021, 5, 3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen L., Han D., Jiang L., Colloids Surf., B 2011, 85, 2. [DOI] [PubMed] [Google Scholar]
- 76. Ferraz N., Carlsson J., Hong J., Ott M. K., J. Mater. Sci.: Mater. Med. 2008, 19, 3115. [DOI] [PubMed] [Google Scholar]
- 77. Elosegui‐Artola A., Oria R., Chen Y., Kosmalska A., Pérez‐González C., Castro N., Zhu C., Trepat X., Roca‐Cusachs P., Nat. Cell Biol. 2016, 18, 540. [DOI] [PubMed] [Google Scholar]
- 78. Albuschies J., Vogel V., Sci. Rep. 2013, 3, 1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. do Amaral R. J. F. C., Zayed N. M. A., Pascu E. I., Cavanagh B., Hobbs C., Santarella F., Simpson C. R., Murphy C. M., Sridharan R., González‐Vázquez A., et al., Front. Bioeng. Biotechnol. 2019, 7, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schindelin J., Arganda‐Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.‐Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Nat. Methods 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Movie 1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.