

Abstract
Collagen mineralization is a biological process in many skeletal elements in the animal kingdom. Examples of these collagen‐based skeletons are the siliceous spicules of glass sponges or the intrafibrillar hydroxyapatite platelets in vertebrates. The mineralization of collagen in vitro has gained interest for two reasons: understanding the processes behind bone formation and the synthesis of scaffolds for tissue engineering. In this paper, the efforts toward collagen mineralization in vitro are reviewed. First, general introduction toward collagen type I, the main component of the extracellular matrix in animals, is provided, followed by a brief overview of collagenous skeletons. Then, the in vitro mineralization of collagen is critically reviewed. Due to their biological abundance, hydroxyapatite and silica are the focus of this review. To a much lesser extent, also some efforts with other minerals are outlined. Combining all minerals and the suggested mechanisms for each mineral, a general mechanism for the intrafibrillar mineralization of collagen is proposed. This review concludes with an outlook for further improvement of collagen‐based tissue engineering scaffolds.
Keywords: bioinspired materials, collagen, hydroxyapatite, mineralization, silica
The in vitro mineralization of collagen deals with the mechanisms of bone formation and the synthesis of tissue engineering scaffolds. Due to their biological relevance, the extra‐ and intrafibrillar mineralization of collagen with hydroxyapatite and silica are the focus of this review. A general mechanism for the intrafibrillar mineralization of collagen is proposed.
1. Introduction
Biomineralization in general has been an inspiration for scientists for decades. Often, the interplay between an organic matrix or template and an inorganic mineral phase in biomineralization gives rise to extraordinary properties. Nacre, for example, exhibits characteristic iridescent optical properties, arising from calcium carbonate disks stacked in a chitin matrix.[ 1 ] Another example is bone, which derives its load‐bearing properties from hydroxyapatite (HAp) platelets hierarchically deposited in a collagen matrix.[ 2 ]
Collagen is the most ubiquitous protein found in animals (from sponges to humans). Together with chitin and chitin‐like molecules, collagen is the most common template for the formation of skeletal elements.[ 3 ] In vertebrates, collagen templates the formation of HAp platelets in bone and teeth. Also, more recently, it has been found that collagen‐like molecules might template the formation of amorphous silica in the spicules of certain sponges.[ 4 ] In this respect the most important minerals are probably those related to calcium ions (HAp) and silicon ions (amorphous silica). Although many other biominerals are known, such as calcium carbonate and magnetite (iron), these are not encountered with collagen in biological systems. Moreover, even though there is no record of collagen‐based calcium carbonate skeletons, the first successful experiments for intrafibrillar mineralization of collagen were achieved for this mineral.[ 5 ] The mechanism of formation of calcium carbonate inside collagen, however, may have illuminated that of HAp and silica. The formation of minerals, whether crystalline or amorphous, in or around a collagen template is generally termed collagen mineralization (a misnomer as another compound crystallizes within collagen).
The reasoning behind collagen mineralization in vitro is two‐fold: 1) understanding the processes behind bone mineralization and 2) mimicking bone tissue via synthetic procedures. Although these are two very different aspects, these often go hand‐in‐hand. Hence, numerous studies, both in living and synthetic systems, were performed to investigate the processes involved in collagen mineralization.[ 6 ]
In this paper, we deal with the mineralization of collagen with minerals of calcium and silicon and, to a much lesser extent, other minerals, like yttria‐stabilized zirconia and combinations of silica and HAp. In Section 2, we first provide a detailed summary of the molecular structure of collagen type I, the collagen type of interest for mineralization purposes. We provide an overview of collagen‐based hybrid materials in nature in Section 3. In Section 4, we first cover the in vitro efforts toward mineralization with calcium carbonate and calcium phosphate (Section 4.1), followed by in vitro silicification (Section 4.2). This section is concluded with mineralization with multiphase minerals and other minerals (Section 4.3). In Section 5, the work reviewed here is placed into perspective. We provide some comments on the mechanisms involved in collagen mineralization and end this section with some concluding remarks and prospects for further research.
2. Collagen Type I
2.1. General Introduction about Collagen
Collagen is a ubiquitous protein in animals. Only among the vertebrates, already 28 different types of collagen can be found.[ 7 ] All collagen‐containing supramolecular structures in an organism are formed by different types of collagen and also noncollagenous components. The combination of collagen types,[ 8 ] postsynthesis modifications,[ 9 ] and the interaction with noncollagenous components is specific for each tissue and its function.[ 10 ]
Collagen type I is the most abundant fibrillar protein of the extracellular matrix (ECM), being present in bone, skin, tendons, and ligaments.[ 11 ] It is a heterotrimeric molecule composed of two α1‐chains and one α2‐chain. These left‐handed chains adopt a right‐handed triple helix as tertiary structure. Each α‐chain has a characteristic [Gly‐Xaa‐Yaa] repeating unit, with Xaa and Yaa being majorly proline (Pro) and 4‐hydroxyproline (4‐Hyp), respectively. The Gly‐Pro‐Hyp sequence represents the 12% of the amino acids.[ 12 ] Glycine residues showed to be essential for the formation of the triple helix and mutations in its position are related to diseases, such as osteogenesis imperfecta (also known as the brittle bone disease).[ 13 ]
2.2. Structure of Collagen Microfibrils and Fibrils
The collagen molecules (Figure 1a) have a length of ≈300 nm and a width of ≈1.5 nm. They spontaneously assemble into fibrils and fibers that can be up to 1 cm in length and 1 µm in diameter, depending on the tissue.
Figure 1.
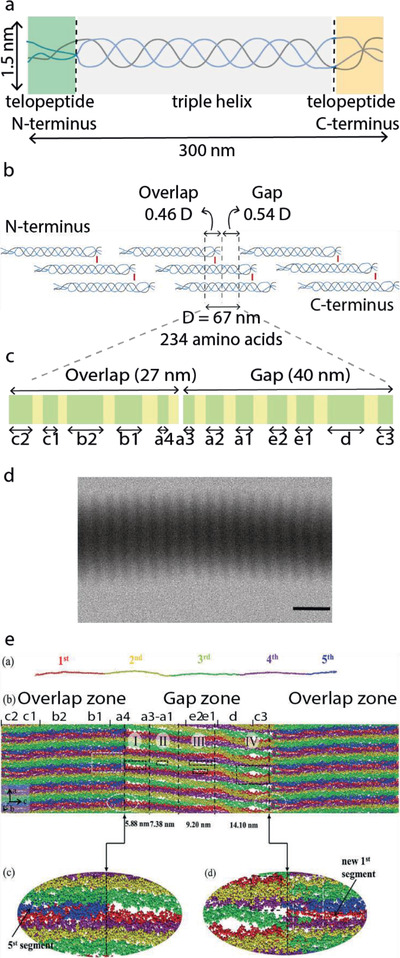
Collagen structure. a) Tropocollagen molecule b) Collagen fibril formed by staggered tropocollagen molecules. c) This leads to bands with different high (green) and low (yellow) electron density observed inside the overlap and gap regions. d) TEM micrograph of a collagen fibril, unstained, showing the characteristic D banding pattern (scale bar: 200 nm). e) Full‐atom structure of one and a half unit cells within a microfibril obtained by MD simulations shows the presence of voids in the a‐ and e‐bands of the gap region. e) Reproduced with permission.[ 19 ] Copyright 2018, Royal Society of Chemistry.
Fibrils are composed of smaller rope‐like building blocks called microfibrils. These are formed of five tropocollagen molecules arranged with an offset of 67 nm between neighboring molecules (Figure 1b). Their organization in a quasihexagonal unit cell with an axial length of 67 nm[ 14 ] was confirmed by high‐resolution mapping combined with crystallographic information and modeling.[ 15 ] Orgel et al. were able to fit the amino sequence of collagen to the experimental electron density map of collagen molecules obtained in situ.[ 15 ] The quasihexagonal structure is maintained throughout the microfibril and each unit cell contains the molecular segments of four tropocollagen molecules and a gap region. Being able to visualize the full path covered by a single molecule through successive unit cells allowed to determine that neighboring microfibers are interdigitated by crosslinks, which explains why it is not possible to isolate microfibrils.[ 7d ] The interpretation of the electron density map also provides insight into the collagen regions that interact with other biomolecules.[ 16 ] Moreover, collagen fibrils can bundle into fibers. The orientation in which the fibrils align within the fibers largely contributes to the mechanical properties of different tissues.
The staggered arrangement of collagen molecules within the microfibrils into gap and overlap regions leads to a characteristic banding pattern that can be easily observed by electron microscopy as zones of low and high contrast, as shown in Figure 1d. This banding pattern is termed D‐periodicity, D‐spacing, or D‐band pattern, with D‐periodicity the most commonly used term.
With chemical staining, several sub‐bands can be identified. Positive staining using phosphotungstate (PTA) reveals the position of positively charged amino acid residues in the collagen sequence.[ 17 ] PTA are large anions that react with the positively charged sidechains of Lys, Hyl, and Arg. These amino acids are nonuniformly distributed in groups along the axial direction of the microfibril. When collagen fibrils are positively stained with PTA and imaged with transmission electron microscopy (TEM), up to 12 peaks of different intensities are observed. These peaks, schematically shown in Figure 1c can be assigned to the positively charged groups in the gap (a3, a2, a1, e2, e1, d, c3), overlap (c2, c1, b2, b1) and both regions (a4).[ 17 ]
The precise location and orientation of charged amino acids along the collagen molecule can give insight on the potential sites for intrafibrillar infiltration of minerals as well as the interaction sites for non‐collageneous proteins (NCPs).[ 18 ] Recently, Xu et al.[ 18 , 19 ] performed molecular dynamics simulations on a collagen microfibril to understand how the relative position of the collagen molecules changes along the c‐axis leading to the formation of possible infiltration paths within the fibrils (Figure 1e). They managed to describe with atomic level detail the shape and size of the void space between molecules in a microfibril and confirm the presence of channels across adjacent microfibrils. Their findings indicate that early mineralization would be favored in the channels formed in the a‐ and e‐bands, which agrees with experimental results.[ 20 ] The calculated dimensions for the a‐band are 1.5 nm in height (a‐axis) × 5.8 nm in width (c‐axis) and for the “e” band are 1.5 × 5.6 nm2.[ 19 ] The length of the channels (b‐axis) would correspond to the diameter of a fibril, that is between 100 and 200 nm2. While there are also voids formed in the d‐band, these channels appear tortuous for infiltration. Moreover, Xu et al. showed that the density of water within the gap region is 0.7 g cm−3, less than in bulk (1 g cm−3). This reduction in water density promotes mineralization by reducing the enthalpic penalty for desolvation of mineralization precursors.
2.3. Sources, Extraction Methods, and Formulations
Collagen type I used for biomedical, commercial, and basic research applications is mainly extracted from animal tissues, mostly from skin and tendon of porcine, bovine, and ovine origin.[ 21 ] Less common are collagens from fish[ 22 ] and sponges.[ 23 ] Due to the relatively easy and mild extraction method, it is also common to use collagen extracted from rat tail tendon for research purposes.
The methods used to extract collagen from different sources depend on the source (animal and tissue) of the collagen. Tissues where collagen fibrils present a low degree of crosslinking, such as tendon, require mild methods like acidic solutions, while tissues with high degree of crosslinking require harsher methods, usually including proteolytic digestion. The source and extraction method will affect the purity and integrity of the collagen: cross‐links and telopeptides regions might be lost during the process.[ 24 ] This can affect the kinetics of polymerization and final properties of the collagenous material. Moreover, it is important to note that the lack of standardization among collagen sources makes the comparison between studies difficult (see also Section 5).
The use of collagen from animal sources for biomedical applications has the disadvantage of being potentially immunogenic (i.e., provoke an immune response in the host) and of being susceptible to batch‐to‐batch variabilities. The use of recombinant collagen, produced by genetically modified organisms, could overcome both issues but has low yields and the resulting collagen lacks of some post‐translational modifications.[ 25 ] Small synthetic peptides with collagen‐like sequences have been used to gain insight on the molecular structure, biochemistry, stability, and self‐assembly of collagen.[ 26 ] While their use for biomedical applications is still very limited, they have been used for studies of mineralization.[ 27 ]
Research on collagen mineralization, which will be discussed in Section 4, has been done in reconstituted collagen in different formulations, such as, soft hydrogels, assembled fibrils, freeze‐dried sponges, and demineralized tissues. The election of the collagen formulation depends on the research goal and the desired application.
2.3.1. Collagen Hydrogels
Collagen hydrogels are water‐swollen collagen networks typically formed by neutralization of an acidic collagen solution and incubation at 37 °C. The kinetics of self‐assembly, the morphological and structural characteristics depend on various parameters, such as the source and extraction method of collagen, the collagen concentration, the pH, ionic strength, and temperature.[ 28 ] For a given application, it can be necessary to induce chemical, physical, or biological crosslinks[ 12 ] to strength the reconstituted hydrogels and avoid their disintegration during its handling and use.
2.3.2. Assembled Fibrils
Collagen fibrils can be assembled on 2D substrates such as glass cover slides[ 29 ] or TEM grids,[ 30 ] following neutralization methods similar to the preparation of 3D collagen hydrogels. This facilitates the use of characterization techniques that require thin specimens. Collagen mineralization has been studied in self‐assembled grids in TEM grids,[ 20 ] allowing TEM imaging and electron tomography measurements without further treatments to the specimen.
2.3.3. Freeze‐Dried Sponges
Typically, collagen suspensions are immersed and freeze in a cryogenic bath at temperatures between −20 and −45 °C. The composition of the suspension and the freezing parameters can be tuned to regulate the pore size of the sponge.[ 31 ]
2.3.4. Demineralized Tissues
The minerals present in mineralized tissues such as bovine bones or fish scales can be removed by chemical treatments leading to a collagenous structure.[ 32 ] Following this process, the hierarchical levels of the tissues are preserved and hence, the templates can be tougher and stronger than other forms of reconstituted collagen.
3. Collagen‐Based Biological Materials
The mineralized tissues present in animals are composed of mineralized biomacromolecules.[ 3a ] The most common organic templates for mineralization are collagen and chitin[ 3b ] and the inorganic components intimately associated with these biomolecules are limited to a small variety of minerals: amorphous silica,[ 4 , 33 ] polymorphs of calcium carbonate (aragonite and calcite),[ 1 , 34 ] and carbonated apatite.[ 6 , 35 ] In this section, we will focus on the examples in nature where collagen acts as a template for mineral formation: HAp in tissues of vertebrate animals[ 6a ] and silica in glass sponges.[ 4 ] These examples are summarized in Table 1 .
Table 1.
Collagen mineralization in collagen‐based natural materials
| Bone (vertebrates) | Tooth (vertebrates) | Spicules (sponges) | |
|---|---|---|---|
| Mineral | HAp | HAp | Amorphous Silica |
| Ratio organic:inorganic | 35:65 wt% | 35:65 wt% (cementum) 30:70 wt% (dentin) | n.a. a) |
| Ratio extra‐:intrafibrillar (inorganic content) | 75:25 vol% | 75:25 vol% (cementum) 65:35 vol% (dentin) | n.a. a) |
| Mineral size b) | (2–7) nm × (15–200) nm × (10–80) nm c) | (2–7) nm × (15–200) nm × (10–80) nm d) | n.a. a) |
To the best of our knowledge, collagen has been found to template silica formation only in the hexactinellida sponges Hyalonema sieboldii.[ 4 ] The slow‐etched method developed by Ehrlich et al. allowed the authors to obtain and characterize the collagen‐like molecule that templates silica deposition but no details regarding organic–inorganic ration or the location of the minerals are given. In other hexactinellida and demosponges, fibrillar collagen is found as a net that surrounding the spicules
Mineral size is given in (thickness) × (length) × (width)
The size of HAp crystals in bone varies among species, age, and different regions of bone. Size of extrafibrillar crystals is comparable to the size of intrafibrillar crystals
The HAp crystals in dentin are platelet‐shaped in dentin and are more needle‐shaped toward the pulp. Size of extrafibrillar crystals is comparable to the size of intrafibrillar crystals.
3.1. Bone
Bone is a dense connective tissue found in vertebrates and presents a complex hierarchical structure (Figure 2a), providing, e.g., structural support, protection for soft organs, and storage of ions. The bone marrow, which produces red and white blood cells, is found within the bone in the medullary cavity (Figure 2b).[ 36 ] The structure of bone depends on sex, age, and location in the body.[ 37 ]
Figure 2.
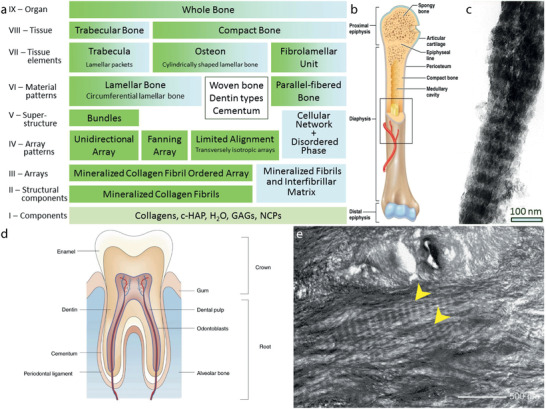
Bone and tooth. a) The 9 hierarchical levels of bone as determined by Reznikov et al.[ 40 ] b) Schematic overview of human bone. c) TEM image of a mineralized collagen fibril in bone. Scale bar is 100 nm. d) Schematic overview of the human tooth. e) TEM image showing the microstructure of undamaged elephant dentin in a tusk, which is very similar to human dentin.[ 64b ] The mineralized collagen fibrils are indicated with an arrow. Scale bar is 500 nm. a,c) Reproduced with permission.[ 40 ] Copyright 2014, Elsevier. b) Reproduced with permission.[ 36 ] Copyright 2019, Taylor & Francis. d) Reproduced with permission.[ 61 ] Copyright 2019, Springer Nature. e) Reproduced with permission.[ 64b ] Copyright 2005, Elsevier.
The hierarchical arrangement within bone leads to extraordinary mechanical properties.[ 38 ] Weiner and Wagner[ 39 ] assessed the complex structure of bone and identified seven levels of hierarchy. More recently, their model was updated with two additional hierarchical levels for the ordered bone structures by Reznikov et al.[ 40 ] (Figure 2a). In the next paragraphs, we will present a concise overview of important features of bone on the first two levels of hierarchy: the (chemical) components and the structural components.
The organic matrix of bone (≈25 wt%) consists mostly of collagen fibrils, predominantly collagen type I.[ 41 ] The rest of the organic matrix includes proteoglycans and NCPs.[ 6a ] NCPs make up 10–15% of the organic matrix of bone.[ 42 ] Although most NCPs are not bone specific, many were found to play important roles in the formation and mineralization of bone.[ 43 ] NCPs are generally highly negatively charged, due to the abundance of acidic amino acids or phosphorylation of threonine and/or serine residues.[ 44 ]
Following secretion of the organic collagen matrix by the bone‐forming cells (osteoblasts), inorganic carbonated HAp is deposited inside and around the collagen fibrils (intrafibrillar and extrafibrillar, respectively) (≈65 wt%).[ 41 , 45 ]
Carbonated HAp differs from synthetic (stoichiometric) HAp in chemical composition. The hydroxyl content of biological apatite is lower compared to synthetic HAp.[ 46 ] Of bone apatite, 4–6 wt% consists of carbonate ions, which replace either the hydroxyl groups (A‐type) or phosphate ions (B‐type) in the crystal lattice.[ 47 ] Other substitutions, for example with citrate, chloride, and fluoride, are encountered as well.[ 48 ] Substitution with HPO4 2− is unique to biological apatite.[ 49 ] Cationic substitutions are also possible, for example with magnesium or sodium.[ 50 ] Ion substitutions influence the biological and physiochemical properties of the HAp crystals.[ 50 , 51 ] Furthermore, the theoretical calcium to phosphorus (Ca/P) ratio is 1.67,[ 48 ] while for synthetic and biological HAp both lower (1.51) as well as higher (up to 1.86) values are reported.[ 51 , 52 ]
The larger part of the bone mineral is present as extrafibrillar mineral (≈75 vol%), the rest is intrafibrillar mineral.[ 53 ] The crystals form through an amorphous precursor. Although earlier observations suggested a needle‐like appearance, the HAp crystals are platelet‐shaped, for both the intra‐ and extrafibrillar minerals.[ 54 ] The intrafibrillar crystals are arranged in periodical arrays with a 67 nm repeat (Figure 2c),[ 55 ] due to templating effects of the collagen matrix,[ 54b ] and are oriented with the long axis of the crystals (c‐axis) parallel to the long axis of the collagen.[ 39 ] The majority of the extrafibrillar crystals are also aligned with the collagen fibrils,[ 56 ] though local differences in alignment were observed for the disordered regions of bone.[ 57 ] Typically, the crystals, both intra‐ and extrafibrillar, are between 2 and 7 nm thick, between 10 and 80 nm wide and between 15 and 200 nm long,[ 54 , 58 ] though it should be noted that the size of the crystals varies between different regions of bone, species, and age.[ 52e ]
The presence of water in bone should not be neglected. Water is present throughout bone (≈10 wt%), in both the organic and inorganic components.[ 41 ] Water can be adsorbed on crystal surfaces, for example, unbound water is found between the collagen triple helices[ 53 , 59 ] and it is thought to play an important role during bone mineralization.[ 60 ]
3.2. Teeth
In most vertebrates, teeth consist of four main components: enamel, cementum, pulp, and dentin (Figure 2d).[ 61 ] Enamel is the hard, visible layer covering the tooth. It is highly mineralized with HAp but does not contain collagen. Cementum is the bone‐like layer surrounding the roots of teeth and molars and connecting the tooth to the bone. In the pulp, the inner part of the tooth, nerves, and blood vessels are located. At this site, the formation of dentin takes place. Dentin is the most abundant mineralized tissue in the tooth and serves as a support for the outer enamel layer.[ 62 ]
Both cementum and dentin are a collagenous structure, mainly collagen type I, mineralized intra‐, and extrafibrillarly with carbonated HAp (Figure 2e). The crystals are needle‐like near the pulp region and become more plate‐shaped in regions close to the enamel.[ 63 ] The intrafibrillar crystals are arranged in periodic arrays along the collagen fibrils and the crystal dimensions are comparable to those of bone.[ 64 ] As in bone, NCPs are involved in the mineralization of the organic matrix.[ 65 ]
The extent of mineralization and organization of cementum is comparable to that of bone, while dentin has a slightly higher mineral content (≈70 wt%), of which approximately 65 vol% is extrafibrillar mineral, and does not have the same extent of hierarchical organization as the ordered bone structures.[ 64 , 66 ] Both cementum and dentin contain a significant amount of water (≈10 wt%). Like in bone, water is present in both the organic and inorganic components.[ 66 ]
3.3. Sponges
The sponges from the classes Demospongiae and Hexactinellida present skeletons whose structural elements, the spicules, are formed by amorphous silica and a collagen‐containing matrix (Figure 3 ).[ 67 ]
Figure 3.
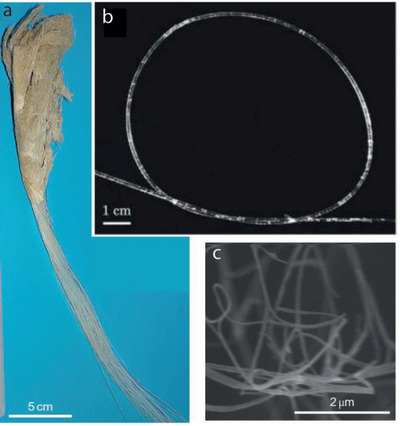
Collagen‐silica biocomposites. a) The anchoring spicules of the glass sponge Hyalonema sieboldii has a multilayered structure. b) the meter‐long spicule presents great flexibility probably due to the plywood organization of collagen nanofibrils. c) SEM of fibrillar collagen extracted by a slow etching method. Reproduced with permission.[ 4 ] Copyright 2010, Springer Nature.
In demosponges the spicules are produced by the enzymatic activity of silicateins that induce intracellular silica condensation inside of specialized cells called sclerocytes that uptake ortho silicic acid from the environment.[ 68 ] Once they are extruded to the extracellular space, the spicules keep growing both in length and in diameter and are embedded into a fibrous organic network that contains collagen. Although the process is not well understood yet, it is believed that the proteinaceous coat around the spicule plays a role in its final morphology.
In hexactinellida collagen is not only involved in shaping the final morphology of the spicule but it also acts as a template for silicification. Ehrlich et al. developed a slow etching method that allowed them to isolate the protein content from the glass spicules of the hexactinellida sponge Hyalonema sieboldii, and showed the double role of collagen molecules during the formation of the spicules.[ 4 ] Biochemical characterization of the isolated proteins in H. sieboldii revealed that the collagen molecules have hydroxylated prolines in the repetitive sequence of amino acids [Gly‐Xaa‐Yaa] n that can interact with silicic acid through hydrogen bonds and promote silica formation.[ 4 ] The authors propose that the incorporation of collagen as silicification templates enabled the formation of meter‐long anchoring glass spicules present in hexactinellida sponges, in comparison with the silicatein‐based spicules in demosponges that are ten orders of magnitude shorter.
4. In Vitro Mineralization of Collagen
In this section, in vitro approaches toward the mineralization of collagen with different minerals are reviewed. Section 4.1 focuses on HAp mineralization. Section 4.2 continues with silicification of collagen, while Section 4.3 briefly describes some efforts with combinations of HAp and silica and other inorganic components. Extrafibrillar mineralization of collagen scaffolds is generally more straight‐forward compared to intrafibrillar mineralization. Hence, we start each mineral section with the synthesis of collagen scaffolds with extrafibrillar mineral, after which we describe the efforts toward intrafibrillar mineralization.
4.1. Mineralization of Collagen with Hydroxyapatite and Calcium Carbonate
4.1.1. Extrafibrillar Mineralization of Collagen with Hydroxyapatite
In bone tissue engineering, both pure collagen and pure HAp scaffolds were investigated. Although collagen scaffolds have a high biodegradability and excellent biological performance, the scaffold is generally too soft to serve as bone implant material.[ 69 ] HAp scaffolds, on the other hand, have a higher elastic modulus, but their brittleness and poor biodegradability are limiting their use as bone replacements.[ 70 ] Thus, combining the advantages of collagen with those of HAp, collagen‐HAp scaffolds were designed as bone tissue engineering candidates. In general, two methods are used to synthesize HAp‐collagen scaffolds: 1) deposition of HAp on top of an existing collagen scaffold and 2) coprecipitation of HAp and collagen. Both methods are briefly covered in this section.
The first method first involves the synthesis of a collagen scaffold with the desired porosity. The porosity of the scaffold greatly influences cell adhesion, cell growth and matrix deposition.[ 71 ] Generally, a porous collagen scaffold is synthesized by increasing the pH of a collagen solution, leading to self‐assembly of the fibrils. Chemical crosslinking is sometimes performed to avoid break‐up of the self‐assembled structure and the scaffold is finally freeze‐dried to maintain a porous structure.[ 69 , 72 ] By immersing this collagen scaffold in a solution supersaturated with respect to HAp, like simulated body fluid (SBF), HAp particles can be deposited on top of the collagen scaffold.[ 73 ] The presence of the soluble extracellular domain of discoidin domain receptor 2 (ECD‐DDR2), a promotor of HAp nucleation, enhances the formation of extrafibrillar mineral in SBF (Figure 4 ).[ 74 ] However, ECD‐DDR2 does not influence the amount of extrafibrillar in polymer‐stabilized amorphous calcium phosphate (ACP) solutions, nor the amount of intrafibrillar mineral. The presence of other additives in the mineralization medium generally results in the formation of intrafibrillar mineral, as described in Section 4.1.2.
Figure 4.

Extrafibrillar mineralization of collagen with HAp. a) Mineralization in mSBF. b) Mineralization in mSBF in the presence of ECD‐DDR2, enhancing the amount of extrafibrillar mineral. Reproduced with permission.[ 74 ] Copyright 2019, Elsevier.
In the second method, the self‐assembly of collagen is combined with the precipitation of HAp. A solution of collagen is mixed with SBF. Upon increasing the pH, the collagen self‐assembles, and HAp particles are deposited around the collagen fibrils.[ 75 ] Freeze‐drying of the scaffold maintains its structure. By tuning the freezing rate and direction, the pore size of unidirectionally aligned macropores can be tuned.[ 76 ]
The scaffolds obtained with the methods described above, as well as commercially available collagen‐HAp scaffolds, were tested in vitro and in vivo. In vitro, the mechanical properties as well as cell adhesion and cell proliferation were tested. The HAp coating increases the stiffness of the collagen scaffold four times compared to pure collagen scaffolds. By depositing other calcium phosphate phases instead of or in combination with HAp, the stiffness could be increased even further, depending on the method used.[ 77 ] The porosity of the scaffolds decreases upon coating with HAp or calcium phosphate, but porosities over 90% could still easily be obtained.[ 73 , 77 ]
In vivo, the biological performance of collagen‐HAp scaffolds as bone implant material was investigated. It was shown that there is no significant difference in performance between laboratory‐made scaffolds and commercially available scaffolds.[ 75 ] Both are well accepted by the host tissue but lack sufficient adhesion between scaffold and native bone to be suitable as bone implants for load‐bearing bones.[ 75 ] However, in nonload bearing bones or with additional mechanical fixation of the scaffold, these collagen‐HAp scaffolds could be promising as bone‐graft materials.
4.1.2. Intrafibrillar Mineralization of Collagen with Hydroxyapatite and Calcium Carbonate
Although HAp‐collagen scaffolds are promising for bone tissue engineering applications, the mechanical properties (stiffness, strength, resistance to crack propagation, etc.) of the bone‐graft need some optimization, especially for load‐bearing bones. As bone consists largely of intrafibrillar HAp crystals, the formation of intrafibrillar HAp in a collagen matrix was investigated. The goal is not only to develop new bone‐like materials in a laboratory environment, but also to understand the processes involved in bone formation. A review by Nudelman et al.[ 78 ] summarizes a great part of the work done on collagen mineralization with HAp in vitro. Here, we summarize first the most important results including some of the more recent additions to the research on HAp mineralization in collagen. Thereafter, combining all available knowledge at this stage, we speculate on the mechanisms involved in intrafibrillar HAp nucleation and growth as presented at the end of this section.
Surprisingly, the first successful attempt toward intrafibrillar mineralization of collagen in vitro was not with the native mineral HAp, but with calcium carbonate. Knowing that many biominerals are formed via an amorphous precursor in the presence of highly acidic biomacromolecules,[ 79 ] Gower and Odom used poly(aspartic acid) (pAsp) to influence CaCO3 growth.[ 80 ] Even in the presence of pAsp at concentrations as low as 20 µg mL−1, the polypeptide binds ions and inhibits nucleation, thereby inducing a liquid–liquid phase separation between the crystallization medium and the precursor phase.[ 81 ] As the polymer plays an essential role in the formation of the precursor, the authors termed this new phase “polymer‐induced liquid‐phase precursor” (PILP). This PILP‐phase has a highly liquid‐like character, and as such, the amorphous calcium carbonate can seep into the cracks of a substrate that is present in the mineralizing solution. Upon solidification of the PILP phase, the crystals maintain their shape and can thus be “molded” into different shapes, even at room temperature.[ 81 ] At the same time, poly(acrylic acid) (pAA) was found to exhibit a similar influence on the CaCO3 crystallization process,[ 82 ] although the term PILP was only introduced later for this polymer.[ 83 ]
Not long after this discovery, Olszta et al. showed that the PILP phase could be used to mineralize reconstituted collagen tapes with calcium carbonate.[ 83 ] After exposure of collagen fibrils to the PILP phase, the presence of calcite in a periodic structure along the collagen was observed in scanning electron microscopy (SEM) analysis, while in the absence of the acidic polymer only extrafibrillar mineralization was observed.[ 5 ] With selective etching of the mineral or the collagen with acid or bleach, respectively, intrafibrillar mineralization was further confirmed. Upon treatment with a dilute acid solution, the mineral phase surrounding the collagen was removed, leaving a remnant fibrous structure, supporting that the mineral had indeed infiltrated the collagen fibrils. Upon bleach etching, the collagen was removed. Instead of the typical platelet shape that was found for HAp crystals in bone, disk‐like calcite crystals were found, spanning the full diameter of the collagen fibrils. The absence of visible voids in the mineral phase indicated that the minerals had formed inside, rather than around the collagen fibrils.[ 84 ]
After mineralization of collagen with calcium carbonate via a PILP‐phase, Olszta et al. suggested that a similar process might be suitable to mineralize collagen with HAp.[ 85 ] Indeed, when using pAsp, collagen was successfully mineralized with HAp, while in its absence, only extrafibrillar minerals were observed. They proposed that the mechanism behind the intrafibrillar mineralization depends on capillary forces, dragging the liquid‐like precursor into the collagen, where it subsequently solidifies to form crystalline HAp.
Deshpande and Beniash studied the use of pAsp as a mimic for NCPs using a calcium concentration closer to that in biological situations.[ 86 ] Instead of reconstituted collagen tapes, they used a self‐assembled layer of collagen on a TEM grid as mineralization templates. The observations, however, are similar: collagen could be mineralized with HAp in the presence of pAsp, while without polymer only extrafibrillar crystals were observed. They proposed a different mechanism, in which the polymer creates a local supersaturation around the collagen fibrils, driving the formation of intrafibrillar minerals.
The results above suggest that there are three essential factors in collagen mineralization: 1) the collagen fibril itself, 2) the mineral, and 3) the NCP‐mimic or (polymer) additive. The role of each component will be addressed in the following sections. The work of Olszta et al. and Deshpande et al. also sparked the discussion on the mineralization mechanism. Most of the follow‐up works choose either one of the two mechanisms, until some more recent contributions proposed other mechanisms. This section concludes with a general discussion on the mineralization mechanism.
The Role of Collagen in Intrafibrillar Collagen Mineralization
Even though Zhang et al.[ 87 ] did not achieve intrafibrillar mineralization, these authors were one of the first to indicate that the collagen itself might play an essential role in calcium phosphate nucleation. In their experiments they coassembled collagen with calcium phosphate without the use of pAsp or another NCP mimic. They observed that a HAp phase was formed around the self‐assembled collagen, with a crystallographic orientation in which the c‐axis of the HAp crystals was aligned along the collagen fibrils. From these experiments, they inferred that the collagen should have an important function in the nucleation of HAp.
Another early study on demineralized fish bones showed that the collagen matrix plays an important role as structural template for the HAp crystals. Chen et al.[ 88 ] first demineralized fish bone with either HCl or ethylenediaminetetraacetic acid (EDTA). Both are popular decalcification agents in histology, but with different results. Whereas demineralization with HCl is generally much faster, EDTA presents a much gentler way of decalcifying.[ 89 ] More recently, EDTA conjugated to glycol‐chitosan was shown to remove extrafibrillar mineral exclusively.[ 90 ]
After demineralization, Chen et al. remineralized the collagen scaffold and investigated the result of the demineralization agent on the HAp crystals obtained after remineralization of the acid‐treated scaffold. In the case of demineralization with EDTA, the collagen matrix could be remineralized and an oriented mineral deposit was obtained. Demineralization with HCl, on the other hand, led to hydrolysis of the collagen and hence to a break‐up of the macromolecular organization, and no oriented minerals were obtained after remineralization. These results show that the organization of the collagen molecules templates the formation of oriented HAp platelets, whereas without such organization, the collagen is not able to induce HAp formation in oriented arrays.
In a time‐resolved cryo‐TEM study, Nudelman et al. studied the influence of collagen on the mineralization process, in the presence of pAsp.[ 20 ] Using uranyl acetate staining, the banding pattern throughout the reconstituted collagen fibrils was visualized. By correlating the stained banding pattern with the amorphous calcium carbonate phases in and around the collagen, it could be observed that ACP preferentially enters the collagen around the a‐bands, a region full of charged amino acids (Figure 5 ).
Figure 5.
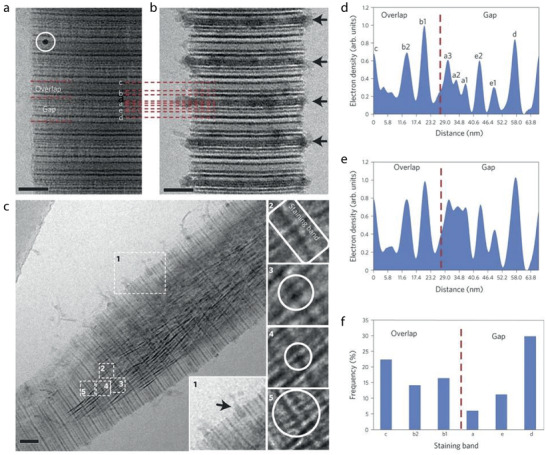
Collagen mineralization preferentially occurs around the staining bands. a) CryoTEM image of stained, nonmineralized collagen. Staining bands are labeled according to ref. [ 17 ] White circle: 10 nm gold marker for electron tomography. b) CryoTEM image of stained collagen, mineralized for 24 h. Calcium phosphate is associated with the fibril in a regular pattern, following the staining bands (black arrows). Peaks are labeled corresponding to their respective staining band. c) CryoTEM image of a stained fibril mineralized for 48 h. Apatite crystals are found within an ACP bed, which can still be seen infiltrating into the fibril through the a‐band (inset 1, black arrow). Insets 2–5 show crystals nucleating on the staining bands. d) Intensity profile of a, nonmineralized collagen. Dashed line: border between overlap and gap zones (C‐terminus). e) Intensity profile of b, collagen mineralized for 24 h. Dashed line: border between overlap and gap zones (C‐terminus). f) Histogram of the distribution of the number of nucleating crystals per staining band. Dashed line: border between overlap and gap zones. Scale bars: 50 nm. a–f) Reproduced with permission.[ 20 ] Copyright 2010, Springer Nature.
Uranyl acetate staining was also used to identify nucleation sites for the calcium phosphate within the collagen. It was observed that the early‐stage calcium phosphate crystals are always located at a staining band, with a slight preference for the d‐band. Surprisingly, the final crystals are not concentrated at one band specifically but are randomly located in the fibrils. This is also observed for other infiltration studies and will be further addressed in the Section “The role of additives in intrafibrillar collagen mineralization.” It was hypothesized that pAsp binds to highly charged regions in the collagen and thereby induces nucleation. To investigate this, collagen was also mineralized in the presence of fetuin, a protein that cannot enter the collagen fibrils. Another set of experiments was performed with the C‐terminal domain of DMP‐1 (dentin matrix protein 1), which is a mineralization promotor in the presence of collagen. The results from these experiments are very similar: early‐stage crystals are only observed on staining bands, suggesting that collagen has a distinct spatial arrangement of charged amino acid groups, providing a structural template for oriented calcium phosphate mineralization.
Mimicking fibrillogenesis after secretion of tropocollagen by collagen‐producing cells, Wang et al.[ 91 ] designed a model system to recreate the dynamics of this process. An acidic collagen solution was continuously injected into a permeable dialysis membrane. Via reverse dialysis against a high concentration of polyethylene glycol (PEG) polymer, a high concentration of the collagen matrix could be achieved, whereas this is not possible using other strategies. Both solutions contained calcium, phosphate, and carbonate ions as the mineral precursors. Coprecipitation of collagen and calcium phosphate occurred via neutralization with ammonia vapor. The mineral content in the collagen fibrils was enhanced by continuing the mineralization reaction in SBF. Comparing the synthetic product with natural bone, the two are indistinguishable in both X‐ray diffraction (XRD) and nuclear magnetic resonance (NMR). In contrast to the earlier experiments, no additive was needed to induce intrafibrillar mineralization, indicating that the formation of apatite is directed by the collagen fibrils, even at atomic scale.
Although collagen has been suggested to provide a 3D‐environment which coordinates calcium and phosphate ions to nucleate HAp, most in vitro efforts show that nucleation can happen throughout the collagen fibril, and not only in the gap region. This indicates that, although collagen is capable of directing its own mineralization, other mechanisms are involved in directing nucleation to the gap region. Some NCPs are shown to bind to the gap region, facilitating nucleation at specific sites.[ 92 ] The role of protein additives and polymer NCP mimics is discussed in more detail in Section “The role of additives in intrafibrillar collagen mineralization.”
The Role of the Mineral Phase in Intrafibrillar Collagen Mineralization
The existence of amorphous precursors is well‐known for biomineralization processes. Also for bone mineralization, it was hypothesized that an amorphous phase would be present before crystallization into the more stable crystalline HAp would occur. It is, however, difficult to distinguish amorphous phases in bulk material, due to its transient nature and the small size of precursor particles. ACP was identified in the newly formed bone of continuously growing zebrafish fin bones.[ 93 ] ACP is present in vesicles in the bone‐making cells (osteoblasts) of zebrafish fine bone,[ 94 ] but also in mouse bones.[ 95 ]
In bone, the ACP‐containing vesicles were found to fuse to the preformed collagen matrix. At this mineralization front, ACP particles become platelet‐shaped crystalline carbonated HAp, but the mechanism behind the amorphous‐to‐crystalline transition is still poorly understood.[ 94 ] An octa‐calcium phosphate (OCP) intermediate has been postulated.[ 93 ] So far, OCP has only been found in association with the NCP osteocalcin,[ 96 ] but it could not be identified as intermediate for the transition from ACP to crystalline HAp inside the collagen fibrils.
The amorphous phase was also identified in in vitro procedures before crystalline HAp is observed.[ 85 , 86 ] In the presence of Cu+, a well‐known stabilizer of ACP,[ 97 ] it was shown that the collagen fibrils could be fully infiltrated with ACP, which appeared to be stable for weeks.[ 98 ] This indicates that, although it is generally assumed that the ACP phase should be stabilized to facilitate infiltration into the collagen,[ 78 ] it should not be too stable to ensure conversion to crystalline HAp. A polymer additive (see Section “The role of additives in intrafibrillar collagen mineralization”) might aid in stabilization of the ACP precursor phase. How and when the amorphous‐to‐crystalline transition occurs, is still topic of debate, but it is generally accepted today that the mineral infiltrates as an amorphous phase.
The Role of Additives in Intrafibrillar Collagen Mineralization
As discussed earlier, the results from Nudelman et al.[ 20 ] and Wang et al.[ 91 ] strongly suggested that collagen is directing its own nucleation and templates the formation of HAp crystals. It is, however, difficult to fully mineralize reconstituted collagen matrices without additives. NCPs, comprising 10–15% of the organic matrix of bone,[ 42 ] were found to be essential in the mineralization processes in vivo. Gene knock‐out studies support this finding. Some NCPs act as nucleation promotors in solution, others as nucleation inhibitors, but almost all NCPs promote intrafibrillar mineralization of collagen. A detailed overview of the use of NCPs to promote intrafibrillar mineralization in vitro was presented by Nudelman et al.[ 78 ]
Generally, NCPs are highly acidic, due to the abundance of aspartic and/or glutamic acid residues. Post‐translational phosphorylation of serine and threonine residues leads to high net negative charges at neutral pH, which are ideal in the sequestration of Ca2+ ions. It is, however, not always straightforward to obtain NCPs from the ECM.[ 99 ] Dilute acid treatment of bone could lead to dephosphorylation of proteins, altering their molecular structure. In vivo, the extent of phosphorylation of recombinant NCPs is generally higher than phosphorylation obtained in vitro. Recombinant approaches to express certain NCPs in bacteria are generally time‐consuming and have a high cost. As such, researchers have searched for simpler mimics of the NCPs present in bone and teeth. A brief overview of the NCP‐mimics used in in vitro collagen mineralization experiments is presented below and is summarized in Table 2 .
Table 2.
In vitro mineralization of collagen with HAp. A selection of past work is made to show the wide range of conditions that can be used to mineralize collagen templates with HAp. We are not aiming to provide a recipe for intrafibrillar collagen mineralization with HAp. We refer the reader to the original paper for the exact experimental details
| Reference | Additive | M w [kDa] | Additive [µg mL−1] a) | Mineralization Conditions b) | Concentrations [× 10−3 m] c) | Collagen Matrix d) | Collagen Source | Periodicity e) | Mineral Content f) |
|---|---|---|---|---|---|---|---|---|---|
| Nudelman et al.[ 20 ] | pAsp | 2–11 | 10 | HEPES (pH 7.4) | [Ca2+] = 2.7 [HPO4 2−] = 1.35 | Self‐assembled fibrils on TEM grid | Horse | No | n.q. |
| Olszta et al.[ 85 ] | pAsp | 6.2 | 15 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Collagen sponge | Cow | No | n.q. |
| Li et al.[ 168 ] | pAsp | 10.5 | 50 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Collagen film 0% crosslinking | Cow | No | 48 wt% (14 d) |
| pAsp | 10.5 | 50 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Collagen film 26% crosslinking | Cow | No | 48 wt% (14 d) | |
| pAsp | 10.5 | 50 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Collagen film 59% crosslinking | Cow | No | 53 wt% (14 d) | |
| pAsp | 10.5 | 50 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Collagen film 82% crosslinking | Cow | No | 64 wt% (14 d) | |
| Jee et al.[ 105 ] | pAsp | 5.5 | 50 | Deionized water | [Ca2+] = 3.0 [HPO4 2−] =? g) | Collagen sponge | Cow | No | n.q. |
| Jee et al. [ 103 ] | pAsp | 10.3 | 50 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Collagen sponge | Cow | No | 34 wt% (8 d); 60 wt% (16 d) |
| pAsp | 32.2 | 50 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Collagen sponge | Cow | No | 58 wt% (8 d); 74 wt% (16 d) | |
| Deshpande et al.[ 86 ] | pAsp | 5–15 | 62.5 | PBS (pH 7.7) h) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid | Rat | No | n.q. |
| Niu et al.[ 111a ] | pAsp | 27 | 75 | HEPES (pH 7.4) i) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Self‐assembled fibrils on TEM grid | Rat | No | n.q. |
| Jee et al.[ 104 ] | pAsp | 10.3 | 100 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.7 | Tendon | Turkey | No | n.q. |
| Thula et al.[ 169 ] | pAsp | 10.5 | 100 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Demineralized bone | Manatee | No | 30 wt% (7 d) |
| pAsp | 27 | 100 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Demineralized bone | Manatee | No | 45 wt% (7 d) | |
| Olszta et al.[ 85 ] | pAsp | 6.2 | 100 | TBS (pH 7.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Tendon | Turkey | No | n.q. |
| Lausch et al.[ 106 ] | pAsp | 5–15 | 125 | PBS h) , j) | [Ca2+] = 1.7 [HPO4 2−] = 9.1 | Cryo‐sectioned demineralized molars | Mouse | No | n.q. |
| Shao et al.[ 126 ] | pAsp | 27 | 10 | m‐SBF + HEPES (10 × 10−3 m) j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid | Not reported | No | 0.14 k) (6 h) |
| pAsp | 27 | 10 | m‐SBF + HEPES (10 × 10−3 m) j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid (citrate coated) | Not reported | No | 1.0 k) (6 h) | |
| pAsp | 27 | 15 | m‐SBF + HEPES (10 × 10−3 m) j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid | Not reported | No | 0.13 k) (6 h) | |
| pAsp | 27 | 15 | m‐SBF + HEPES (10 × 10−3 m) j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid (citrate coated) | Not reported | No | 1.0 k) (6 h) | |
| pAsp | 27 | 50 | m‐SBF j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid | Not reported | No | 0.11 k) (6 h) | |
| pAsp | 27 | 50 | m‐SBF j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid (citrate coated) | Not reported | No | 0.65 k) (6 h) | |
| pAsp | 27 | 120 | m‐SBF j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid | Not reported | No | 0.48 k) (6 h) | |
| pAsp | 27 | 120 | m‐SBF j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid (citrate coated) | Not reported | No | 1.0 k) (6 h) | |
| pAsp | 27 | 240 | m‐SBF j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid | Not reported | No | 0.54 k) (6 h) | |
| pAsp | 27 | 240 | m‐SBF j) | [Ca2+] = 1.67 [HPO4 2−] = 9.5 | Self‐assembled fibrils on TEM grid (citrate coated) | Not reported | No | 1.0 k) (6 h) | |
| Qi et al.[ 107 ] | pAA | 2 | 10 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 57 wt% (7 d) |
| pAA | 50 | 10 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 62 wt% (7 d) | |
| pAA | 450 | 10 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 53 wt% (7 d) | |
| pAA | 2 | 25 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 53 wt% (7 d) | |
| pAA | 50 | 25 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 63 wt% (7 d) | |
| pAA | 450 | 25 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 63 wt% (7 d) | |
| pAA | 2 | 50 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 4 wt% (7 d) | |
| pAA | 50 | 50 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 64 wt% (7 d) | |
| pAA | 450 | 50 | TBS (pH 6.4) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Cross‐linked collagen matrices | Cow | No | 65 wt% (7 d) | |
| Song et al.[ 108 ] | pAA | 450 | 50 | HEPES (pH 7.4) l) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Self‐assembled fibrils on TEM grid | Rat | No | n.q. m) |
| pAA | 450 | Covalently bound n) | HEPES (pH 7.4) l) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Self‐assembled fibrils on TEM grid | Rat | No | n.q. m) | |
| pAA | 2 | Covalently bound n) | HEPES (pH 7.4) l) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Self‐assembled fibrils on TEM grid | Rat | No | inhibition | |
| Ping et al.[ 29 ] | pAA | Not reported | 100 | HEPES (pH 7.4) | [Ca2+] = 2.7 [HPO4 2−] = 1.35 | Self‐assembled fibrils on glass coverslip | Cow | No | n.q. |
| pAA BSP‐HApBP | Not reported | 100 2 | HEPES (pH 7.4) | [Ca2+] = 2.7 [HPO4 2−] = 1.35 | Self‐assembled fibrils on glass coverslip | Cow | Yes | n.q. | |
| Hu et al.[ 116 ] | pAA TPP | 2 | 0.8–5.6 mg mL−1 1.2 wt% | m‐SBF + HEPES (pH 7.2) | [Ca2+] = 31.7 [HPO4 2−] = 12.5 | Fibrils in solution | Rat | Yes | n.q. o) |
| Liu et al.[ 114 ] | pAA | 1.8 | 500 | SBF (pH 7.4) | [Ca2+] = 2.5 p) [HPO4 2−] = 1 | Partially demineralized dentin (STMP coated) | Human | Yes | 41 vol% q) (4 m) |
| Dai et al.[ 170 ] | pAA | 1.8 | 0.28 × 10−3 m | SBF h) , i) (pH 7.4) | [Ca2+] = 3.75 p) [HPO4 2−] = 1.5 | Self‐assembled fibrils on TEM grid (TPP coated) | Cow | Yes | n.q. |
| Niu et al.[ 111a ] | pAH | 15 | 200 | HEPES (pH 7.4) i) | [Ca2+] = 4.5 [HPO4 2−] = 2.1 | Self‐assembled fibrils on TEM grid | Rat | No | n.q. |
| Liang et al.[ 101b ] | G3‐PAMAM‐NH2 | 6.9 | Preloaded on collagen | Artificial saliva (pH 7.0) | [Ca2+] = 1.5 [HPO4 2−] = 0.9 | Self‐assembled fibrils on TEM grid | Cow | No | n.q. |
| Li et al.[ 101a ] | G4‐PAMAM‐COOH | 10.1 | Preloaded on collagen | Artificial saliva (pH 7.0) | [Ca2+] = 1.5 [HPO4 2−] = 0.9 | Self‐assembled fibrils on TEM grid | Cow | No | n.q. r) |
| G3‐PAMAM‐COOH | 4.9 | Preloaded on collagen | Artificial saliva (pH 7.0) | [Ca2+] = 1.5 [HPO4 2−] = 0.9 | Demineralized dentin | Human | No | n.q. r) | |
| G4‐PAMAM‐COOH | 10.1 | Preloaded on collagen | Artificial saliva (pH 7.0) | [Ca2+] = 1.5 [HPO4 2−] = 0.9 | Demineralized dentin | Human | No | n.q. r) | |
| Yu et al.[ 109 ] | PEG‐COOH TPP s) | 42 | 0.8 mg mL−1 1.2 wt% | m‐SBF + HEPES (pH 7.2) t) | [Ca2+] = 2.5 [HPO4 2−] = 1.0 | Self‐assembled fibrils on TEM grid | Rat | Yes | 62 wt% (24 h) |
| PEG‐pAA TPP s) | 44 | 0.8 mg mL−1 1.2 wt% | m‐SBF + HEPES (pH 7.2) t) | [Ca2+] = 2.5 [HPO4 2−] = 1.0 | Self‐assembled fibrils on TEM grid | Rat | Yes | 64 wt% (24 h) | |
| pAA TPP | 2 | 0.8 mg mL−1 1.2 wt% | m‐SBF + HEPES (pH 7.2) t) | [Ca2+] = 2.5 [HPO4 2−] = 1.0 | Self‐assembled fibrils on TEM grid | Rat | Yes | 58 wt% (24 h) |
Results are sorted based on additive concentration to facilitate easy comparison between similar experiments
Mineralization conditions refer to mineralization medium, pH, and temperature. Buffers were generally made via standardized protocols. Temperature is 37 °C, unless indicated otherwise. Experiments were performed in lab atmosphere, unless indicated otherwise
Starting concentrations are given
With collagen sponge we refer to commercially available collagen sponges or tapes. With tendon we refer to extracted tendon with the protective sheath removed and used as is
With periodicity we mean intrafibrillar mineralization in the gap region exclusively. Periodic mineralization is determined via author statements (or, occasionally, estimated from their TEM images)
n.q. = not quantified; only experiments that led to intrafibrillar mineralization with HAp are included in this table. Mineral content was determined via TGA unless stated otherwise. Mineralization time is given in brackets, with the following abbreviations: h = hours, d = days, m = months
Phosphate ions were delivered via enzymatic cleavage of rac‐glycerol 1‐phosphate with alkaline phosphatase in solution. Hence the concentration is uncertain
100% humidity atmosphere
Room temperature
pH not specified
Mineral content was determined using the method of Smeets et al.[ 171 ] This method uses the TEM images to quantify the mineral content. A mineral content of 0 means that there is no mineral present in the sample, a mineral content of 1 means that the sample is fully mineralized.
Mineralization temperature not reported
Not quantified, but it was noted that only prenucleation clusters were formed when pAA was used in solution. When using pAA crosslinked to collagen, mineralization occurred
Chemically crosslinked to collagen via a carbodiimide and N‐hydroxysulfosuccinimide reaction
Not quantified, but it was established that the D‐banding becomes more apparent with decreasing pAA concentration.
Portland cement was used as a continuous source of calcium ions during the mineralization reaction
Vol% was determined via µ‐CT scans
Not quantified, but the authors state that G4‐PAMAM dendrimers are more effective in promoting intrafibrillar mineralization compared to G3‐PAMAM dendrimers (for the same mineralization time)
PEG‐COOH and PEG‐pAA as listed here are brush‐like polymers
Two temperature process: 1 h at 25 °C, 23 h at 37 °C.
Often referred to as artificial proteins, dendrimers are interesting candidates to mimic NCPs.[ 100 ] Poly(amidoamine) (PAMAM) dendrimers, both carboxyl‐ and amino‐terminated, were shown to be capable of remineralizing human dentine and reconstituted collagen fibrils, respectively.[ 101 ]
Even simpler additives were explored for their function in the intrafibrillar mineralization of collagen. NCPs often contain large acidic domains, rich in aspartic acid and/or glutamic residues with little secondary structure and very flexible in nature. As such, acidic linear polymers, like pAsp were proposed as model system for NCP mimics.[ 42 ]
Bradt et al.[ 102 ] were the first to indicate that pAsp might be important in facilitating intrafibrillar mineralization. Mimicking the multistep process of bone formation, they combined self‐assembly of a collagen template with calcium phosphate formation.[ 102 ] By accurately tuning the experimental conditions, collagen fibrils were formed alongside an ACP phase. This amorphous phase subsequently mineralizes to form crystalline calcium phosphate in clusters around the collagen. Upon the addition of pAsp, separate needle‐like calcium phosphate crystals were found covering the collagen. Although the exact role of pAsp was not yet fully understood, Bradt et al. already noted that pAsp interacts with the ACP phase, thereby delaying the amorphous to crystalline transition. Additionally, it was found that pAsp improves the connection between collagen and calcium phosphate.
Both Olszta et al.[ 85 ] and Deshpande et al.[ 86 ] found that, in absence of pAsp, randomly oriented extrafibrillar HAp crystals were formed. When using pAsp at a concentration at which the formation of crystalline HAp would be inhibited due to formation of pAsp‐stabilized ACP, HAp crystals formed inside the collagen fibrils. The crystals, unlike those found in mineralized collagen in bone, were randomly distributed throughout the collagen fibril. From thermogravimetric analysis (TGA), it was found that the mineral content increased with increasing molecular weight of the pAsp chains and that higher molecular weight pAsp leads to faster mineralization kinetics.[ 103 ] The use of pAsp was also proven to lead to intrafibrillar mineralization of denser collagen substrates, such as turkey tendon.[ 104 ] Similarly, pAsp was found to be essential to achieve intrafibrillar collagen mineralization in experiments where the phosphate ions were provided via enzyme cleavage of organophosphate esters with alkaline phosphatase.[ 105 ]
In a set of experiments in which the role of the ECM was investigated, demineralized cryo‐sectioned mouse molars were exposed to solutions of SBF to remineralize the collagen matrices.[ 106 ] From these experiments, it was found that the ECM itself can control the rate of mineral deposition, but only led to the formation of ACP in the sectioned molars. In contrast, in a pAsp stabilized solution, intrafibrillar HAp crystals formed within the cryo‐sections, again highlighting the importance of pAsp or a similar additive.
As demonstrated for the PILP phase of CaCO3, pAsp could be easily replaced with the cheaper pAA. The effect of molecular weight of the pAA chains in combination with the concentration of the polymer on intrafibrillar mineralization of reconstituted collagen fibrils was studied in detail.[ 107 ] It was found that low molecular weight pAA at high concentrations results in intrafibrillar mineralization, while higher molecular weights at lower concentrations lead to the formation of extrafibrillar crystals only. Experiments by Song et al. demonstrate that the polymer does not necessarily need to be in solution. These authors used a high‐molecular weight pAA (450 kDa) to mineralize collagen. When the polymer was in solution, only prenucleation clusters were observed, while crosslinking this high‐molecular weight pAA to collagen leads to intrafibrillar mineralization.[ 108 ] Crosslinking a low‐molecular weight (2 kDa) pAA to collagen leads to full inhibition of HAp mineralization.
In an alternative approach, following a coassembly strategy, brush‐like polymers were used as sequestration analogues to induce intrafibrillar mineralization of collagen fibrils.[ 109 ] Tripolyphosphate (TPP) was added as a templating analogue (vide infra). TGA showed that the intrafibrillar mineral content was enhanced with brush‐like polymers, compared to its linear counterparts. This was ascribed to the higher back‐bone rigidity due to steric hindrance of the grafted side chains, which leads to less entanglements compared to the linear pAsp or pAA, making the carboxyl groups more accessible to stabilize ACP clusters.
Polyanionic polymers were investigated for their resemblance to the acidic NCPs. Nevertheless, polycations, like poly(allylamine hydrochloride) (PAH), were also known to induce a PILP phase for calcium carbonate.[ 110 ] Moreover, PAH is often exploited in intrafibrillar mineralization of collagen with silica (see Section 4.2.3). Previously, it was thought that negatively charged polymers form a negatively charged ACP complex which can be drawn to or into the collagen via electrostatic attraction. However, the work with PAH demonstrates that also positively charged polymers can promote intrafibrillar mineralization, rejecting the hypothesis that only negatively charged complexes can interact with the collagen.[ 111 ]
Although the use of acidic polymers as sequestration analogues led to intrafibrillar mineralization, the periodic distribution of mineral as observed in native bone was not achieved. Instead, randomly distributed HAp crystals formed inside the collagen matrix. Therefore, it was suggested that a templating analogue was necessary, to guide the formation of mineral into the gap zone of collagen only.[ 112 ] The phosphorylated moieties of NCPs were suggested to act as templating additives in collagen mineralization and induce the formation of HAp in the gap regions of the collagen fibril only. As such, phosphorylated polymers and recombinant proteins were suggested as mimics for phosphorylated NCPs to act as templating analogues in collagen mineralization. Ping et al. designed a multifunctional protein with a domain based on bone sialoprotein (BSP) and a HAp binding peptide (HApBP).[ 29 ] When using this BSP‐HApBP protein, randomly distributed HAp platelets form on the surface of the collagen fibril, just as in the control without additives. When adding PAA, however, the formation of intrafibrillar crystals in a periodic array is observed. This suggests that the BSP‐HApBP protein acts as a templating analogue in the mineralization of collagen (Figure 6 ), similar to the role of phosphorylated NCPs in bone.
Figure 6.

Comparison between collagen mineralization in the absence a) and presence b) of BSP‐HApBP as templating analogue and pAA as a sequestration analogue. a) TEM image of collagen mineralized for 72 h in the presence of pAA. HAp crystals form randomly inside the collagen fibril. Scale bar is 50 nm. b) TEM image of collagen mineralized for 72 h in the presence of BSP‐HApBP and pAA. HAp forms in periodic arrays along the collagen fibril. Scale bar is 100 nm. Insets: corresponding SAED pattern of the TEM image. Reproduced with permission.[ 29 ] Copyright 2015, Royal Society of Chemistry.
The periodic intrafibrillar arrangement of HAp platelets could also be obtained by using phosphorylated collagen as the mineralization template. Collagen could be phosphorylated via a pre‐treatment of the collagen matrix with TPP,[ 113 ] sodium trimetaphosphate (STMP),[ 114 ] or via immobilization of poly(vinylphosphonic acid) (PVPA).[ 115 ] Similarly, TPP can be added to the mineralizing solution to obtain periodic intrafibrillar mineral arrays.[ 116 ] It is, however, necessary to add a polymer additive like pAsp or pAA, as phosphorylation of the collagen alone is not sufficient to achieve intrafibrillar mineralization.[ 117 ]
The influence of small molecules on intrafibrillar collagen mineralization has been sparingly investigated in literature. It was established that amino acids influence nucleation and growth of HAp, but less studies investigated the influence of amino acids on collagen mineralization.[ 118 ] In a very recent contribution l‐Glutamic acid (Glu) was investigated in the mineralization of collagen gels with HAp. It was shown that at low Glu concentrations (between 50 and 200 × 10−3 m), the mineral content in the gels increased with increasing Glu concentration, but unfortunately, the authors did not investigate whether the minerals were formed intra‐ or extrafibrillarly.[ 119 ]
Serine was used to stabilize ACP by Shen et al., albeit in combination with other molecules.[ 120 ] These authors aimed to mimic matrix vesicles that are thought to be important in collagen mineralization in vivo[ 121 ] by first mixing serine‐stabilized ACP with PEG and incorporating the PEG‐serine‐stabilized ACP into polysorbate micelles. Using these micelles for mineralization of collagen resulted in extrafibrillar crystals, but when the micelles were broken up with isopropyl alcohol, however, serine‐stabilized ACP was released from the micelles, which could infiltrate the collagen and intrafibrillar HAp crystals were obtained. This again highlights the importance of a stabilizing agent, even when other molecules are used to deliver the ACP to the collagen fibril.
Another small molecule that was investigated in collagen mineralization is the citrate ion. Citrate is known to be present in bone, in levels up to 2 wt%.[ 122 ] With advanced solid state NMR techniques, Hu et al.[ 123 ] looked into the connections between citrate and HAp in natural bone. They found that citrate inhibits the formation of additional phosphate layers, leading to the formation of very thin HAp platelets. This is in close agreement with earlier observations, where thinner HAp crystals were formed in the presence of citrate.[ 124 ] Combining solid state NMR with powder XRD, Davies et al.[ 125 ] further investigated the relation between citrate and HAp in bone. Their results indicate that citrate is present in a hydrated layer around the HAp, separating the thin crystals from one another.
Shao et al. explored the role of citrate in collagen mineralization in vitro.[ 126 ] They exposed collagen to a citrate solution to induce adsorption of citrate on the collagen matrix. The high affinity between collagen and citrate leads to stable and efficient adsorption, close to values found in natural bone.[ 122 ] Citrate alone is not sufficient to induce intrafibrillar mineralization. In contrast to when using pAsp, the intrafibrillar mineralization degree increases. Citrate enhances intrafibrillar mineralization in thin self‐assembled collagen fibrils on TEM grids, as well as in demineralized dentin substrates. The authors argued that the most important role of citrate is not to stabilize the ACP precursor, as experiments with Mg2+, a well‐known ACP stabilizer,[ 97 ] do not lead to improved intrafibrillar mineralization. Instead, the results indicate that the contact angle between collagen and ACP is reduced upon coating collagen with citrate, lowering the energy barrier for ACP to deposit on the collagen matrix. Thus, citrate was found to improve the interface wetting between collagen and the ACP precursor, thereby enhancing infiltration of the ACP into the collagen matrix and subsequent intrafibrillar mineralization (Figure 7 ).
Figure 7.

Schematic of the effect of citrate on intrafibrillar collagen mineralization. Citrate reduces the contact angle between ACP (blue) and collagen (gray rods), promoting intrafibrillar HAp (yellow) mineralization. In the presence of citrate, the contact angle between the ACP droplets and the collagen fibril decreases, leading to spreading of the ACP droplet, which enhances infiltration of the ACP precursor into the collagen, as demonstrated in the middle column. The last column shows the relative amount of intrafibrillar HAp mineral for different concentrations of citrate. Reproduced with permission.[ 126 ] Copyright 2018, Wiley‐VCH.
The results reviewed above strongly suggest that additives, being NCPs or charged polymers acting as NCP‐mimics, are necessary to induce intrafibrillar mineralization. The presence of citrate might enhance the intrafibrillar mineral content. In many cases, however, the mineral is distributed randomly in the collagen fibril. The use of phosphorylated peptides or phosphorylation of the collagen aids in obtaining periodic mineral arrays, similar to as observed in bone.
The Mechanism Behind Intrafibrillar Collagen Mineralization
Since the first successful in vitro attempts to mineralize collagen fibrils with HAp, the mechanisms behind the intrafibrillar mineralization have been—and still are—under debate. In this section, we will briefly summarize some of the results for intrafibrillar HAp mineralization, and then speculate about possible mechanisms.
A great deal of the discussion on the mechanisms behind intrafibrillar collagen mineralization has focused on the role of the polymer or protein. First of all, the charge of the polymeric additive has been subject of the mechanistic debates. After the in vitro experiments with polyanions like pAsp and pAA were done, it was hypothesized that the polymer is either drawn to the collagen on itself or as a negatively charged polymer‐ACP complex. It was shown, however, that also polycationic polymers, previously thought to be repelled by the collagen fibrils, could induce intrafibrillar mineralization.[ 111 ]
Olszta et al.[ 85 ] hypothesized that for both calcium carbonate and calcium phosphate the pAsp‐induced liquid‐like precursor phase was drawn into the collagen via capillary action. To support their hypothesis, they conducted confocal microscopy experiments using fluorescently labeled pAsp. From these experiments, they confirmed that the polymer penetrates the collagen matrix well beyond 200 µm. It is, however, not clear whether the polymer infiltrates as a PILP‐phase or as a pure polymer. Thus, they performed control experiments, in which formation of the PILP‐phase was inhibited. In these experiments, no such penetration depth was observed for the polymer. Although these experiments indicate that capillary forces indeed might play a role, due to the limited resolution of confocal microscopy, it could not be confirmed whether indeed the PILP phase was diffusing into the collagen matrix.
Deshpande and Beniash[ 86 ] argued that pAsp does not enter the collagen, but instead forms a complex with the collagen, creating a local supersaturation around the fibrils. This then leads to intrafibrillar mineralization, which starts as an amorphous phase within the fibrils, only later transforming to crystalline apatite crystals.
The discussion above raises the question whether the polymer enters the collagen matrix. Moreover, does the polymer need to enter the collagen to fulfill its function as intrafibrillar mineralization promotor? In the experiments performed by Price et al.[ 127 ] and Nudelman et al.,[ 20 ] it was shown that in the presence of fetuin, a 48 kDa protein too large to enter the collagen fibril, intrafibrillar mineralization was enhanced.[ 128 ] This demonstrates that the additive does not necessarily need to enter the collagen to promote intrafibrillar mineralization. It should be mentioned here, that although smaller polymers might enter the collagen fibrils, it is not clear whether this actually happens during mineralization. For smaller molecules that prevent transition of the ACP phase to the crystalline phase, like Cu+ ions, it is known that they fully suppress the formation of HAp crystals, also inside the collagen. For the polymer or protein additives, it is generally accepted nowadays that these simply inhibit HAp crystallization in solution, thereby driving the formation of intrafibrillar crystals.
Indeed, a more recent study demonstrates that the driving force for intrafibrillar mineralization with HAp in the presence of polymer additives is very straight‐forward. Using classical nucleation theory in combination with in situ X‐ray experiments, Kim et al.[ 129 ] examined the nucleation rates for extra‐ and intrafibrillar mineralization. From these nucleation rates, the nucleation energies could be deduced.
They found that without pAsp, extrafibrillar mineralization has the lowest nucleation barrier and is thus preferred over intrafibrillar mineralization. In the presence of pAsp, however, the interfacial energy between the nuclei and the mineralizing solution increases, thereby increasing the nucleation barrier for extrafibrillar mineralization. The gap region is a confined space for nucleation, which minimizes the surface area of the nuclei and thereby the Gibbs (surface) energy. As the gap region now has a lower nucleation energy compared to the bulk solution with pAsp, nucleation inside the gap region is favored over extrafibrillar mineralization.
A very recent contribution to the field, however, demonstrates that polymer additives are not always necessary to obtain intrafibrillar mineralization. Du et al.[ 130 ] tried to mimic the microvariations in bone structure that arise during bone development under applied loads. To imitate the applied loads during vertebrate movement, Du et al. applied fluid shear stress to collagen. They found that the applied fluid shear stress promotes the transition from ACP to HAP and leads to highly mineralized intrafibrillar mineralized collagen. Under a specific set of periodic loading conditions and in the presence of TPP, hierarchical collagen‐HAp composites were formed even in the absence of pAA. This work demonstrates a novel approach to mineralizing hierarchical collagen scaffolds. This work also indicates that polymer additives are not always required to obtain intrafibrillar mineralization, as long as a different driving force is present to induce the formation of intrafibrillar crystals.
The role of the collagen and the mineral itself in intrafibrillar collagen mineralization should not be excluded. Collagen has been shown to direct nucleation, as well as crystal size, shape, orientation, and alignment of the HAp crystals, serving as a template for HAp formation. It is generally accepted that the mineral infiltrates the collagen as an amorphous precursor, though the mechanism behind the amorphous to crystalline conversion from ACP to HAp is still under debate.
Furthermore, the role of water in collagen mineralization cannot be neglected. It has been shown that residual water is present at the surface of the HAp platelets.[ 131 ] Moreover, water in the gap region is argued to be of a lower density (≈0.7 g cm−3) compared to water in the bulk.[ 18 ] If true, this would imply that the enthalpic penalty for ion desolvation is lower, thus lowering the energy barrier for intrafibrillar HAp nucleation.
Despite our knowledge on collagen mineralization is expanding rapidly, many other questions remain unanswered so far. At this point, it is neither clear how nucleation is regulated inside the gap region, nor why the presence of a second additive like TPP is necessary to induce periodic mineral arrangements inside the collagen fibril. Moreover, the research described in this section only focused on HAp due to its biological presence in bone. However, the infiltration and mineralization of other minerals have been studied using similar approaches. Section 4.2 will address silicification, while Section 4.3 briefly describes efforts with other—nonbiological—minerals.
4.2. In Vitro Mineralization of Collagen with Silica
Silicon is the most abundant element on Earth and quartz, a crystalline form of silicon oxide, the most abundant mineral. Silicon is important for animal growth, as it proved to be an essential nutrient.[ 132 ] It participates in several biological processes such as the formation of bone, cartilage, and connective tissue.[ 132 ] While quartz can be found in nature as a mineral, amorphous silica can be found in the skeleton of sponges[ 133 ] and the frustules of diatoms.[ 134 ]
Most of the work done to create collagen‐silica hybrids, either by extra or intrafibrillar mineralization, has been done with the goal of contributing to the field of tissue engineering. Even though silicified collagen is not found in higher animals but only in sponges, the biochemical role of silicon in the development of tissues makes it a great candidate to be combined with collagen, the main component of the ECM, to prepare scaffolds for tissue engineering. These materials should unite a series of properties before they can be considered as (possibly) suitable scaffolds for biomedical applications. First, they should provide macroscopic porous networks where cells can attach, proliferate and differentiate and nutrients and metabolism products can be transported through. Moreover, the scaffolds should be stiff enough to resist matrix contraction and biodegradable in a rate compatible with its use. The ability of cells to adhere to a scaffold depends on its surface topography and their ability to proliferate is linked to the mechanical stability.[ 135 ] Finally, in vitro and in vivo studies need to be performed in order to study the biocompatibility as well as the response of the organisms to the scaffolds during the wound healing process. Heinemann et al. reviewed the efforts on making silica‐collagen hybrids for biomedical applications until 2013.[ 136 ] In this section, we aim to provide an overview of the past and most recent work and a summary in Table 3 .
Table 3.
In vitro mineralization of collagen with silica. A selection of past work is made to show the wide range of conditions that can be used to silicify collagen templates. We are not aiming to provide a recipe for collagen silicification. We refer the reader to the original paper for the exact experimental details
| Reference | Silica precursor | Silica precursor Concentration [× 10−3 m] a) | Additive | Additive Concentration [× 10−3 m] a) | Mineralization Conditions b) | Collagen Matrix c) | Collagen [mg mL−1] | Collagen Source | Extra/intrafibrillar infiltration |
|---|---|---|---|---|---|---|---|---|---|
| Eglin et al.[ 138 ] | Sodium silicate | 0.166–10 | — | — | Tris‐HCl (pH 7.4) 21 °C | Soluble | 2 | 30 days old rat tail tendon | Extra |
| Silica particles | 0.166–10 | — | — | Tris‐HCl (pH 7.4) 21 °C | Soluble | 2 | 30 days old rat tail tendon | Extra | |
| Shen et al.[ 139 ] | Sodium silicate | 2–8 | — | — | PBS (pH 7.4) 4 °C | Soluble | 0.8 | Calf skin | Extra |
| Niu et al.[ 151 ] | Silbond | 1.5 wt% | Choline chloride | 72 | pH 5.5 37 °C | Sponge | — | — | Intra |
| Silbond | 1.5 wt% | Choline chloride | 72 | pH 5.5 37 °C | Tendon fascicles (EDS/NHS cross‐linked and PAH enriched) | — | 70 days old rat tail tendon fascicles | Intra | |
| Niu et al.[ 160 ] | Silbond | 5 wt% | PAH | 667 | pH 5.5 37 °C 2 d | Sponge | — | Intra | |
| Silbond | 5 wt% | PAH | 667 | pH 5.5 37 °C 10 d | Demineralized scale | — | Fish (C. carpio) | Intra | |
| Silbond | 5 wt% | PAH | 667 | pH 5.5 37 °C 7 7 d | Demineralized trabecular bone | — | Cow rib bone | Intra | |
| Hu et al.[ 154 ] | Silbond | 3 wt% | — | — | HEPES (pH 6.4) 37 °C 1 d | Soluble | 0.3 | Rat tail | Extra |
| Silbond | 3 wt% | PAH | 267 | HEPES (pH 6.4) 37 °C 1 d | Soluble | 0.3 | Rat tail | Intra | |
| Silbond | 3 wt% | PAH TPP | 267 1.2 wt% | HEPES (pH 6.4) 37°C 1 d | Soluble | 0.3 | Rat tail | Intra | |
| Silbond | 3 wt% | — | — | HEPES (pH 6.4) 37 °C 1 d | Self‐assembled hydrogel | 0.3 | Rat tail | Extra | |
| Silbond | 3 wt% | PAH | 267 | HEPES (pH 6.4) 37 °C 1 d | Self‐assembled hydrogel | 0.3 | Rat tail | Intra | |
| Silbond | 3 wt% | PAH TPP | 267 1.2 wt% | HEPES (pH 6.4) 37 °C 1 d | Self‐assembled hydrogel | 0.3 | Rat tail | Intra | |
| Desimone et al.[ 141 ] | Silica particles | 5–10 | — | — | PBS, NaOH (pH 7) | Soluble | 0.6 | Rat tail | Extra |
| Chen et al.[ 145 ] | GPTMS | — | — | R.T. 1 d | Soluble | 10 | Pig | Extra | |
| Wang et al.[ 150 ] | APTMS | 100 | — | — | PBS (pH 7.4) 2 h | Demineralized cancellous bone grafted with DMAEMA | — | Pig femur and tibia | Extra |
| MPTMS | 100 | — | — | PBS (pH 7.4) 2 h | Demineralized cancellous bone grafted with DMAEMA | — | Pig femur and tibia | Extra | |
| TMS | 100 | — | — | PBS (pH 7.4) 2 h | Demineralized cancellous bone grafted with DMAEMA | — | Pig femur and tibia | Extra | |
| Foglia et al.[ 148 ] | APTES | 50–200 | — | — | 4 °C 1 h | Self‐assembled hydrogel | 1.6 | Rat tail | Extra |
| Quignard et al.[ 140 ] | Sodium silicate | 25 | — | — | Tris‐HCl (pH 7.5) RT 1 h | Soluble | 5 | Rat tail | Extra |
| Silica particles | 25 | — | — | Tris‐HCl (pH 7.5) RT 1 h | Soluble | 5 | Rat tail | Extra | |
| Desimone et al. 2011[ 142 ] | Sodium silicate | 10 | — | — | NaOH pH 7 | Soluble | 0.8–3 | Rat tail | Extra |
| Silica particles | 10 | — | — | NaOH pH 7 | Soluble | 0.8–3 | Rat tail | Extra |
Silica precursor and additive concentrations are given in × 10−3 m unless indicated otherwise
Mineralization conditions refer to the nature of neutralization agent used (if any), pH, temperature, and time with the following abbreviations h = hours, d = days
Soluble collagen refers to tropocollagen molecules dissolved in acidic conditions. With collagen sponge we refer to commercially available collagen sponges or tapes.
It was discussed in Section 2.3 that collagen can be used for research in variety of forms: soluble collagen, fibrils, hydrogels, or demineralized tissues. The interaction of silica with all these different collagen formulations has been studied in the last 20 years (Figure 8 ). The use of soluble collagen gave insight in the influence of silicon species in the self‐assembly of collagen and the interaction between the organic and inorganic components, while silicification of hydrogels or demineralized tissues was used as a method to stiffen the organic template for biomedical applications.
Figure 8.
Overview of mineralization of collagen with silica. Collagen‐silica composites can be formed by combining collagen in different forms (triple helixes, fibrils or 3D scaffolds, such as hydrogels, sponges, and demineralized tissues) using silica as molecular precursor (R = —H, —Me, —Et) or silica particles that can be functionalized or loaded with biomolecules of interest.
4.2.1. Effect of Silicon Species in the Fibrillogenesis Process
Ono et al. were the first to study the interaction of a silica precursor with soluble collagen.[ 137 ] By assembling soluble collagen in the presence of tetraethyl orthosilicate (TEOS) at neutral pH given by a saline phosphate buffer at 37 °C, they obtained tubular silica structures with an inner diameter of 25–50 nm (corresponding to the diameter of assembled fibrils in solution) and silica coatings 50 and 100 nm thick. The core–shell structure presented silica protrusions with a periodicity of 60–80 nm. The collagen fibrils act as a template for silica condensation due to the electrostatic interaction between the negatively charged silicic acid and the positively charged gap zones of collagen. This preferred localization for silica condensation explains the formation of protrusions with a spacing of ≈67 nm.
Eglin et al. performed an in vitro study on the effect of three different biomimetic silicon species (sodium silicate, sodium catecholate and 12 nm silica particles) on the kinetics of the self‐assembly process of collagen type I extracted from rat tail tendon.[ 138 ] Because collagen molecules scatter more light upon aggregation, they followed the fibrillogenesis process via spectrometric turbidity measurements using a wavelength of 400 nm, and measured the amount of silicon species in solution. When molecular silica precursors are added, they break down into ortho‐silicic acid (Si(OH)4). At concentrations larger than 2 × 10−3 m, Si(OH)4 undergoes poly‐condensation and oligomeric species are formed. Therefore, it is not surprising to see that the fibrillogenesis process below and above this concentration is affected in different ways. While below 2 × 10−3 m fibrillogenesis is enhanced, the opposite occurs for higher concentrations. Moreover, the process seems to be totally inhibited above 10 × 10−3 m. A similar concentration‐dependent inhibition was observed with silica nanoparticles. At high concentrations, the adsorption of poly‐silicic acid and particles to collagen hinder the fibrillogenesis until it is completely inhibited. This is explained by the fact that fibrillogenesis can still occur if most of the triple helices are free to interact with each other, and therefore the amount of silicon species that can be added is limited. As long as the amount of oligomeric species or colloid content is low, tropocollagen molecules are still able to interact and assemble. Unfortunately, the authors did not perform any analysis on the morphology of the aggregates.
Shen et al. also studied the effect of sodium silicate on the self‐assembly of collagen type I extracted from calf skin.[ 139 ] The turbidity measurements show the same trend as those observed for the results obtained by Eglin et al.[ 138 ] for rat tail tendon: sodium silicate enhances fibrillogenesis below 2 × 10−3 m and inhibits it above that concentration. These results are confirmed by morphological analysis made by atomic force microscopy (AFM) and SEM. Collagen fibrils with the characteristic D‐periodicity were obtained for sodium silica concentrations up to 4 × 10−3 m while for larger concentrations only clustered collagen aggregates were formed.
It is worth noting that fibrillogenesis is sensitive to a wide range of parameters, from collagen source and concentration to the nature of the buffer used. Moreover, the self‐assembly process is not only affected by those individual parameters but also by their cumulative effects. The results obtained by different groups must be carefully compared and cannot necessarily be translated to other collagen systems. While the above‐mentioned studies show that there is an apparent upper limit concentration for silicon species where fibrillogenesis can still take place, this concentration depends on the specific conditions used for the experiments.
4.2.2. Extrafibrillar Silicification of Collagen
The main interest in studying the interaction between collagen and silica species lies in the fact that they present potential for biomedical applications as scaffolds for tissue engineering. A general look at the work done in this field indicates that there is a synergistic effect between collagen and silica, but that there is a need to find a good compromise between structural and mechanical properties of the scaffold and its biocompatibility.[ 136 ]
The strategies to produce collagen‐silica hybrids can be divided into two groups. On the one hand, mineralization can be achieved in a one‐step approach by the simultaneous self‐assembly of collagen and silica poly‐condensation and, on the other hand, by a two‐step approach where the formation of collagen matrixes is followed by silicification. In both cases, silicification of collagen has been achieved both extra‐ and intrafibrillary.
For the one‐step approach, tropocollagen molecules or collagen oligomers in acidic solution are mixed with silica precursors followed by neutralization of the system to trigger fibrillogenesis simultaneously with silica condensation. If the chosen conditions allow it, cell cultures can be directly added prior the process, which, however, limits further processing. When cells are added after the formation of the hybrid silica‐collagen material, this hybrid material can undergo different drying treatments according to its final use, such as freeze‐drying, critical point drying, or controlled solvent evaporation. While freeze‐drying can disrupt the 3D‐structure of the scaffold, critical point drying is a better method to conserve this structure, but the resulting aerogel might have poor mechanical properties due to its high porosity and vulnerability to moisture. Solvent evaporation, or controlled air‐drying, leads to xerogels with a stiffer structure.
The work by Eglin et al.[ 138 ] indicates that the amount of silica precursors that can be added during fibrillogenesis is limited, which can present a problem if larger amounts of silica are needed to achieve desired properties. In order to increase the amount of silica without inhibiting collagen self‐assembly, higher concentrations of collagen are needed. However, concentrated solutions of collagen become rather viscous and can be hard to work with.[ 140 ] Desimone et al.[ 141 ] managed to prepare collagen‐silica materials from a 3 mg mL−1 collagen solution with both silicate and silica particles as silica sources in concentrations that would inhibit the fibrillogenesis if lower collagen concentrations were used.[ 138 ] Quignard et al.[ 140 ] overcame this problem and successfully used 5 mg mL−1 collagen solutions by using a Tris‐HCl buffer as neutralization agent instead of NaOH solutions or phosphate buffer saline (PBS). This illustrates the sensitivity of the process for the precise experimental conditions (see Table 3).
Depending on whether silicate solutions or silica particles are used as inorganic source, the final scaffolds might have different structural and biological properties. Quignard et al.[ 140 ] prepared hybrid materials by mixing a rat tail collagen solution with either sodium silicate or 12, 80, and 390 nm silica particles followed by neutralization at room temperature (Figure 9a). While using silicate leads to a material with a tenfold increase in elastic modulus G′ compared to bare collagen hydrogels, little to no increase in G′ is observed when nanoparticles are used. This indicates that the interaction of nanoparticles and collagen is weaker than the interaction of the silicate and its poly‐condensation product with the collagen. Biocompatibility assays with human dermal fibroblast showed that silicates and silica particles of 12 and 80 nm enhance cell growth compared to collagen hydrogels, while 390 nm particles are detrimental to cell growth. Even though gel stiffness is key for cell growth, an extensive silica coverage can be detrimental for the adhesion of cells. A compromise between the material stiffness and material roughness, a controlling factor for cell adhesion, needs to be found for each application.[ 141 , 142 ] Moreover, loading of silica nanoparticles with biomolecules, such as antibiotics[ 143 ] or genes,[ 144 ] and incubating them with collagen leads to a formation of functional materials with potential applications in drug delivery for tissue engineering.
Figure 9.

Extrafibrillar silicification of collagen type I. a) Collagen‐silica hydrogels obtained by a one‐step approach. The SEM images show the gel surface of the hydrogel after 1 day (cell‐free, on the left) and 21 days (with Human Dermal Fibroblasts culture) for 1,2) bare collagen, 3,4) collagen and 80 nm silica particles and 5,6) collagen and 390 nm silica particles. Scale bar: 5 µm. b) Collagen‐silica hydrogels obtained by a two‐step approach. Collagen hydrogels (1) were functionalized with increasing concentration of APTES (2,3,4). Scale bar: 1 µm. a) Reproduced with permission.[ 140 ] Copyright 2012, Wiley‐VCH. b) Reproduced with permission.[ 148 ] Copyright 2013, Royal Society of Chemistry.
Chen et al.[ 145 ] prepared dense 2D hybrid silica‐collagen membranes by coating polydimethylsiloxane (PDMS) chips with a microgrooved pattern. The authors mixed an acidic mixture of porcine collagen type I (probably from skin) and 3‐glycidoxeypropyltrimethoxysilane (GPTMS) as silica source. The epoxy groups of GPTMS react with the free amino groups of the collagen, while the Si—OCH3 groups are hydrolyzed and condensed into Si—O—Si bonds forming a silica network. These hybrid membranes have adequate stiffness, biodegradability, and biocompatibility to enhance the growth of skeletal myoblasts. Moreover, the use of microgrooved surfaces strongly directed the myoblast alignment and elongation. This material presents potential applications for regeneration of tissues with cells aligned in a certain direction, such as musculoskeletal tissue.
The two‐step method, where a collagen matrix is formed before the addition of a silica source, can be used to avoid incompatibilities between the desired silica concentration and the self‐assembly process. In the same way that the collagen hydrogel stiffness is enhanced with the use of fixatives as glutaraldehyde (commonly used for fixation prior preparation for image analysis),[ 146 ] molecules with silicon moieties, such as GPTMS[ 147 ] and 3‐aminopropyl triethoxysilane (APTES)[ 148 ] can act as cross‐linker and silica source at the same time.
Foglia et al.[ 148 ] fixed collagen fibers with glutaraldehyde molecules which, instead of acting as cross‐linkers, remain immobilized and pendant, and remain available to react with the aminosilane forming a colored Schiff base. By increasing the amount of glutaraldehyde while leaving the amount of APTES constant, the authors obtained gels with homogeneous silica coverage, reaching average fibril diameters of 100 nm (double in size compared to control collagen gels). The amount of APTES, that can bind covalently the aldehyde or polymerize via sol–gel with another APTES molecule, establishes the silica content of the hybrid material (Figure 9b). Hydrogels prepared with APTES and glutaraldehyde were chosen to pursue in vitro and in vivo experiments,[ 149 ] as these matrices showed a 60‐fold increment in rheological properties and lower rates of degradation by collagenase type I, compared to collagen gels.
Wang et al.[ 150 ] prepared porous nanosilica‐collagen scaffolds derived from porcine demineralized cancellous bone with a porous hierarchical structure and a nanoroughened surface. In order to ensure that silicification occurs homogeneously throughout the scaffold, the surface of the demineralized bone was first coated with three bilayers of poly(diallyldimethylammonium chloride)/poly(acrylic acid‐g‐alkyl bromide) (PDADMAC/PAA‐g‐B), followed by polymerized dimethylaminoethyl methacrylate (PDMAEMA). Three different silica precursors were used: (3‐aminopropyl)trimethoxysilane (APTMS), (3‐mercaptopropyl)trimethoxysilane (MPTMS) and tetramethoxysilane (TMS). Energy dispersive spectroscopy (EDS) elemental mapping performed on cross‐sections of the scaffold showed that the amounts of silicon were similar throughout the gel. The extent of silicon deposition was directly related to the surface roughness and the scaffold sturdiness. Scaffolds made with TMS showed larger silica deposition due to the negative charge induced by the hydrolysis of the precursors. The amino groups of APTMS and MPTMS have a positive charge that is repelled by the PDMAEMA. In vitro and in vivo studies indicates that these scaffolds could be appropriate scaffolds for in situ bone tissue engineering. After implantation, the scaffolds can recruit and host mesenchymal stem cells.
4.2.3. Intrafibrillar Silicification of Collagen
Niu et al. were the first to show that intrafibrillar mineralization of collagen with silica was possible.[ 151 ] They proposed a biomimetic method to infiltrate a polyamine‐enriched collagen sponge with a fluidic precursor phase of stabilized polysilicic acid. Silicic acid is obtained from the hydrolysis of TEOS and is stabilized by choline chloride to prevent the formation of a silica gel during the experiment. PAH acts as a catalyst for silica condensation, but whether it is forming macromolecular aggregates inside the fibrils or only coating the collagen fibers surface is not clear. TEM micrographs (Figure 10a) of sponge sections show that the moldable fluidic silica phase infiltrates the collagen fibrils already after 1 day of silicification, replicating the cross‐banding patterns and microfibrillar architecture of the collagen fibrils. The authors suggest that the infiltration occurs via a PILP‐like mechanism (discussed in Section 4.1.2), but they dwell into details. The intrafibrillar silicified collagen sponges have been used in in vitro and in vivo studies and showed their potential for biomedical applications and tissue engineering. They promoted the proliferation and osteogenic differentiation of bone marrow‐derived mesenchymal stem cells[ 152 ] and human dental pulp stem cells.[ 153 ] In the same work, the authors used this method to infiltrate the collagen present in tendon fascicles of mature rats (Figure 10b).[ 151 ] Similar to the sponges, the fascicles became stiffer after silica treatment. The intrafibrillar silicification was confirmed by electron tomography.
Figure 10.

Intrafibrillar silicification of different collagen type I. a) TEM micrograph showing that the mineral infiltrates the collagen fibers of a commercial collagen sponge replicates the typical banding pattern. Scale bar: 100 nm. b) Intrafibrillar mineralization of rat tendon fascicles. The white arrows indicate the continuity of the silica microfibrillar strands. Scale bar: 50 nm. Reproduced with permission.[ 151 ] Copyright 2011, Wiley‐VCH.
Slightly modified, this approach was applied to infiltrate demineralized tissues.[ 32a ] After removing the carbonated apatite present in fish scales and bovine rib bones, the authors obtained a dense collagen structure that retained the hierarchical structure of the tissues. Although the collagen tapes could be successfully infiltrated by a two‐step method,[ 151 ] the demineralized matrices are much denser making infiltration of the silica precursors more challenging. They modified the previous method by directly stabilizing silicic acid with PAH, instead of using choline chloride. PAH prevents condensation of silica in solution and keeps the silicic acid in a fluidic state, either by the formation of hydrogen bonds between silanol and amine groups or by masking the silanol groups with the long polymer chain. This fluidic phase was transported via transversal canals and perpendicular sheet‐like structures in the fish scale and through Haversian canal and its branches in the osteon to reach and infiltrate the collagen fibrils.
Recently, Hu et al.[ 154 ] presented a one‐step approach to infiltrate silica into collagen fibrils via the simultaneous self‐assembly of collagen molecules (extracted from rat tail tendon) and silica infiltration and condensation. Inspired by the proteins responsible for silica formation in diatoms, the authors use PAH and TPP as biomimetic replacement for positively and negatively charges proteins, respectively, to ensure the stabilization of the silica precursor and its infiltration to the intrafibrillar space. The presence of PAH as a silicic acid stabilizer is a requirement for intrafibrillar silicification. In its absence, the silicification occurs only extrafibrillar. While the presence of TPP in the system is not necessary for intrafibrillar silicification, its presence has a significant effect on the packing of the collagen molecules. When the self‐assembly was triggered in the absence of TPP, twisted silicified collagen fibrils with a periodicity of ≈145 nm were obtained, while in the presence of TPP a clear banded structure with a 67 nm periodicity was observed. The authors explained this difference as a consequence of the interaction between PAH and the carboxyl end groups of the collagen that puts the PAH as a spacer between molecules. TPP molecules interact strongly with the positively charged groups of the gap zone while PAH stabilized silicic acid infiltrates de collagen fibrils via capillary action.
Hu et al. also managed to infiltrate preassembled fibers with silica using PAH and TPP. Their results seem to conflict with the previous work of Niu et al.[ 151 ] The latter showed that the presence of (poly)phosphate groups was not necessary, even counter‐productive, for the intrafibrillar mineralization of a preformed collagen template, while Hu et al. do not observe intrafibrillar silicification in the two‐step approach, unless they add TPP to the system. The difference might be due to the difference in the collagen templates used (collagen sponges obtained by freeze‐drying in contrast with self‐assembled collagen) and provides another illustration of the sensitivity to the precise experimental conditions.
4.3. Multiphase In Vitro Mineralization of Collagen and Mineralization with Other Inorganic Components
Most of the work done combining collagen with non‐natural occurring minerals or incorporating more than one mineral phase leads to the formation of composites, with the inorganic moieties interacting extrafibrillarly with the collagen. For example, homogeneous, machinable, and biocompatible HAp/silica/collagen xerogels can be formed with a composition of at least 40 wt% silica, at least 10 wt% collagen, and a maximum of 20 wt% HAp.[ 155 ] Moreover, cross‐linking collagen with nanoparticles can replace the use of commonly used toxic chemical cross‐linkers and improve physicochemical and biological properties of the collagen matrices and add different functionalities to the material.[ 156 ] For example, oleic acid‐capped magnetite particles can act as cross‐linker for collagen and lead to materials with potential for oil removal applications and leather making.[ 157 ] Collagen crosslinked with bi‐metallic Fe—Zn[ 158 ] and ZnO,[ 159 ] nanoparticles have potential for biomedical applications.
The intrafibrillar infiltration of collagen with inorganic compounds other than silica, calcium carbonate, and HAp remains mostly unexplored. In this section, the few examples of intrafibrillar mineralization of collagen with two minerals as well as non‐naturally occurring minerals will be dealt with. The mineralization strategies discussed in Sections 4.1 and 4.2.3 can be adapted to infiltrate collagen fibrils with both silica and HAp and with yttria‐stabilized zirconia (YSZ).
The methods used to achieve intrafibrillar mineralization with both silica and apatite were combined in a two‐step bottom‐up approach to obtain collagen‐silica‐apatite hybrids.[ 160 ] The concept was proven using commercial collagen sponges and later translated to rat tail collagen as a model material for soft tissue and trabecular bone as model material for hard tissue. In a first step, collagen sponges were infiltrated with PAH‐stabilized silicic acid solution. After 2 days, the silicified sponges were washed and soaked in calcifying medium, a solution prepared by mixing CaCl2, K2HPO4, and pAsp, for up to 7 days. TEM characterization performed after 4 days of apatite deposition showed that plate‐like apatite and silica could be distinguished within the intrafibrillar milieu. After 7 days, the crystallites occupied the entire volume masking the band characteristics of silicified collagen. It is proposed that, after silicification, active silanol groups attracts calcium from the pAsp‐ACP solution and calcium phosphate gets deposited over the silica phase. To confirm that both silica and apatite coexist inside the fibrils, the authors performed a selective dissolution in alkaline or acidic solutions, respectively. When the same approach was used on rat‐tail collagen, the mechanical properties of this multiphase material fell in between the ones with only one mineral phase. The addition of apatite to silicified sponges increases the crystallinity of the material making it stiffer but less resilient when compared with collagen that is silicified only. Homogeneous mineralization was achieved in trabecular bone. Moreover, in vitro studies performed with biphasic mineralized collagen sponges show that this material is biocompatible.[ 161 ] Silica and HAp seem to have not only an effect in the mechanical properties of the scaffold but also on aspects of bone metabolism.
The intrafibrillar infiltration strategies were also tested for only one non‐naturally occurring mineral. Zhou et al. achieved the infiltration of YSZ into the fibrils of collagen sponges, rat‐tail tendon, and demineralized porcine trabecular bone.[ 162 ] Like for other materials, the precursors need to be stabilized in solution to decrease the rate of condensation or inhibit it. In this case, PAH‐coated and glutaraldehyde‐crosslinked collagen matrices were infiltrated with acetylacetone‐inhibited zirconia precursor nanodroplets. TEM studies indicated that YSZ amorphous aggregates infiltrate the fibrils of collagen sponges after 4 days, although the D‐band pattern becomes noticeable after 8 days, when the infiltration is denser. With longer infiltration times, the pattern is masked due to the high mineral density. These precursor aggregates were also observed along the surface of the fibrils.
Although researchers mainly focused on the mineralization of collagen with HAp and silica, the use of other minerals inside or around the collagen matrix could potentially lead to novel hybrid materials with extraordinary properties and maybe even further elucidate the mechanisms involved in collagen mineralization.
5. Conclusions and Perspective
In Section 1, we have reviewed synthetic efforts toward collagen mineralization in vitro with HAp, silica, combinations of HAp and silica and some other inorganic components. The reasoning behind collagen mineralization in vitro is two‐fold: 1) gaining a fundamental knowledge of the processes involved in bone formation and 2) creating functional scaffolds with potential applications in tissue engineering.
Regarding the first, gaining a fundamental understanding of the processes involved in bone formation, many in vitro collagen mineralization experiments were performed as model systems for the biological process. Even though in vitro experiments can give insight on the molecular mechanisms of bone formation, it is important to note that they are a simplified version of what happens in vivo. Nevertheless, well designed in vitro models can be useful materials for understanding biological processes.[ 163 ]
The experiments reviewed here have focused on collagen type I, as type I is the most abundant protein in animals and is mineralized in the bones of vertebrates. A critical remark we would like to make regarding those in vitro experiments is that the collagen substrates used throughout the literature reviewed here, differ greatly in collagen source, network density, and amount of crosslinking. The choice of collagen substrate depends on the experimental needs and application of the material. For studying the fundamentals behind collagen mineralization, thin self‐assembled fibrils that can easily be investigated in electron microscopes are desired. These smaller substrates could be mineralized with HAp in mineral contents close to natural bone (Table 2). Denser collagen substrates, on the other hand, are desired for investigating the macroscopic properties of the mineralized material, but intrafibrillar mineralization of such denser collagen substrates has been challenging due to diffusion limitations in dense networks. Due to the differences in collagen substrates, direct comparison between these studies might not be straightforward.
Moreover, each group seems to have developed its own mineralization protocol, making it difficult to reproduce experimental results and hampering comparison of results between different papers. Compare, for example, the experiments of Qi et al.[ 107 ] and Song et al.[ 108 ] (Table 2). Both use 450 kDa pAA, at a concentration of 50 µg mL−1 in solution, but whereas Qi et al. observed intrafibrillar mineralization, Song et al. only observed the formation of prenucleation clusters. The differences between their results might be due to variations in their experimental procedures, such as pH (6.4 vs 7.4, respectively), collagen source (cow vs rat), and density of the collagen network (cross‐linked matrices vs self‐assembled fibrils). Similarly, for intrafibrillar silicification, Niu et al.[ 164 ] and Hu et al.[ 154 ] used comparable procedures, obtaining different results (Table 3). Whereas Niu et al. found that the addition of TPP leads to limited intrafibrillar silicification of collagen, Hu et al. found that in absence of TPP silicified collagen with a banding pattern larger than 67 nm was obtained. Again, differences in pH (5.5 vs 6.4), collagen source (cow and fish vs rat), and density of the collagen network (demineralized bone, demineralized fish scales and commercial sponges vs self‐assembling matrices) could be the reason for different results.
Even though it is often difficult to compare experiments, some general remarks can be made concerning the mechanism behind intrafibrillar mineralization of collagen. This is summarized in Figure 11 . Intrafibrillar mineralization of collagen with HAp, silica, and other minerals is achieved almost exclusively in the presence of a polymer additive. Phosphate additives like TPP can be used in HAp mineralization to direct nucleation to the gap region of the collagen only, while the results for phosphate additives in silicification are contradictory.
Figure 11.
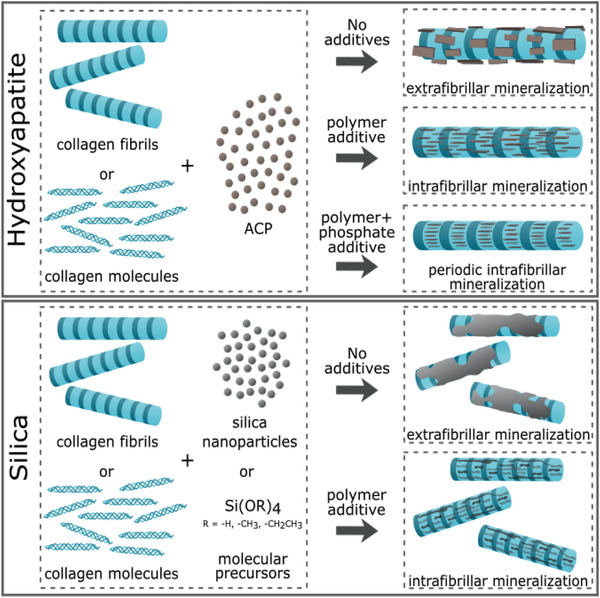
Overview of collagen mineralization mechanisms. Collagen fibrils represent self‐assembled fibrils, freeze dried sponges or demineralized tissues.
The exact role of additives in collagen mineralization has been under debate since the early stages of the mineralization work. Two mechanisms were proposed early on for intrafibrillar HAp mineralization: capillary action of the PILP‐phase[ 85 ] or a local supersaturation around the collagen.[ 86 ] Both mechanisms rely on electrostatic interactions between the polymer or the polymer‐precursor complex and the collagen. In collagen mineralization with HAp, typically acidic polymers are used to promote intrafibrillar mineralization, due to their resemblance of NCPs. For silica, on the other hand, the positively charged PAH is often used to achieve intrafibrillar silicification, as PAH is a well‐known stabilizer of silicic acid. Connecting these two minerals, Niu et al.[ 111a ] tested the use of PAH in HAp mineralization. It was shown that PAH also promotes intrafibrillar mineralization with HAp, indicating that the charge of the polymer‐precursor complex is of lesser importance in obtaining intrafibrillar crystals.
In later stages of the collagen mineralization work, it was proposed that the polymer additive is simply stabilizing the (amorphous) precursor in solution, thereby inhibiting nucleation in bulk. Recently, it was shown that the polymer indeed is increasing the nucleation barrier in solution, thereby favoring nucleation inside the collagen.[ 129 ] For intrafibrillar silicification, the polymer might play a similar role by stabilizing silicic acid in solution and thereby driving the formation of intrafibrillar silica particles. Despite the continuously growing knowledge about the mechanism behind intrafibrillar mineralization, some questions remain unanswered. At this point, though it is known that the polymer does not need to enter the collagen fibril to promote intrafibrillar mineralization, it is still unclear where the polymer additive resides during the mineralization process. Furthermore, the role of phosphate additives is still poorly understood.
Using the existing knowledge of bone formation in vivo and collagen mineralization in vitro, the goal is, ultimately, to create functional materials that can serve as scaffolds for tissue engineering. Ideally, these scaffolds are biocompatible and bioresorbable, promote cell growth and match the mechanical properties of the host tissue. In practice, it is important to keep in mind that a scaffold for tissue engineering should not necessarily match all the characteristics of the native tissue, but should provide a temporary and suitable template to promote tissue regeneration.
Designing a successful scaffold implies a tradeoff between mechanical strength and porosity that is needed for cell‐adhesion and mass transport.[ 165 ] From the wide range of materials that have been studied for tissue engineering, in this review we only focused on the collagen‐based ones. While collagen alone will be too soft for most applications and minerals alone can be too brittle, combining the two can lead to scaffolds with synergistic properties.
Mineralized collagen matrices were tested for in vivo applications, showing their potential to be used in tissue engineering. Collagen‐silica scaffolds, though not as strong as natural bone, with either extra‐ or intrafibrillar silica particles, were shown to be promising as bone implants.[ 149 , 152 ] Moreover, the release of silicic acid from collagen‐silica scaffolds can promote bone growth.[ 132 ]
In most HAp‐collagen scaffolds tested in vivo as scaffolds for bone tissue engineering the mineral is present as extrafibrillar mineral.[ 166 ] These scaffolds were shown to be well accepted by the host, however, the adhesion between the scaffold and the native bone can be improved.[ 75 ] In vitro mechanical testing of intrafibrillar collagen‐HAp scaffolds shows their potential as bone grafts, but collagen scaffolds with intrafibrillar HAp were barely tested in vivo.[ 76 ] Simultaneous incorporation of HAp and silica into collagen leads to scaffolds which mechanical properties are in between those of HAp‐collagen and silica‐collagen. In vitro testing of multiphase scaffolds showed their potential to stimulate bone formation.[ 161 ]
In conclusion, the efforts made in the last decades to study the mineralization of collagen with HAp and other minerals improved our understanding of the processes involved in bone formation. 3D mineralized collagen scaffolds potentially suitable for tissue engineering were already designed and in vivo studies could give more insight in the advantages and limitations of these laboratory‐made mineralized collagen scaffolds. The vast knowledge about collagen mineralization we have now, should be translated to novel techniques like 3D‐bioprinting to extend the possibilities of the tissue engineering scaffolds.[ 167 ]
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
B.M.O. and M.P.V. contributed equally to this work. The work of B.M.O. is funded by a TopPunt Grant (“Bi‐Hy”, 718.016.003) of the Netherlands Organisation for Scientific Research (NWO). The work of M.P.V. is partially funded by EU H2020 Marie Sklodowska‐Curie Action project “MULTIMAT” (676045). The authors thank Dr. Sandra Hofmann for critically reviewing the manuscript.
Biographies
Bernette Oosterlaken obtained her B.Sc. degree in Chemical Engineering and Chemistry at the Eindhoven University of Technology (TU/e), followed by an M.Sc. degree in Molecular Systems and Materials Chemistry at the same university. After obtaining her M.Sc. degree in 2017, she joined the “Bi‐Hy” project as a Ph.D. student. Using a combination of advanced electron microscopy and spectroscopic techniques, her research focuses on the mechanisms involved in the bio‐inspired synthesis of iron oxides in biological and synthetic templates.

Paula Vena graduated in Chemical Sciences from the University of Buenos Aires, Argentina, in 2013. After working as a research assistant at the Instituto de Química Física de los Materiales, Medio Ambiente y Energía (INQUIMAE), in 2016 she moved to the Netherlands to pursue her Ph.D. at the Eindhoven University of Technology. She joined the Innovative Training Network MULTIMAT. Her research comprises the study of the self‐assembly of synthetic and natural polymers using advanced electron microscopy techniques and the formation of hybrid silica‐polymer structures for biomedical applications.

Gijsbertus de With is a professor of materials science at Eindhoven University of Technology (TU/e). After graduating (Utrecht University) and receiving his Ph.D. (1977, Twente University), he joined Philips Research Laboratories, Eindhoven. In 1985, he was appointed as a part‐time professor and became a full professor at TU/e in 1995. His research interests include structure and interfacial phenomena related to the chemical and thermomechanical behavior of multiphase materials.

Oosterlaken B. M., Vena M. P., de G. With, In Vitro Mineralization of Collagen. Adv. Mater. 2021, 33, 2004418. 10.1002/adma.202004418
References
- 1. Nassif N., Pinna N., Gehrke N., Antonietti M., Jäger C., Cölfen H., Proc. Natl. Acad. Sci. USA 2005, 102, 12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weiner S., Traub W., FASEB J. 1992, 6, 879. [PubMed] [Google Scholar]
- 3.a) Weiner S., Addadi L., J. Mater. Chem. 1997, 7, 689; [Google Scholar]; b) Ehrlich H., Int. Geol. Rev. 2010, 52, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ehrlich H., Deutzmann R., Brunner E., Cappellini E., Koon H., Solazzo C., Yang Y., Ashford D., Thomas‐Oates J., Lubeck M., Baessmann C., Langrock T., Hoffmann R., Wörheide G., Reitner J., Simon P., Tsurkan M., Ereskovsky A. V., Kurek D., Bazhenov V. V., Hunoldt S., Mertig M., Vyalikh D. V., Molodtsov S. L., Kummer K., Worch H., Smetacek V., Collins M. J., Nat. Chem. 2010, 2, 1084. [DOI] [PubMed] [Google Scholar]
- 5. Olszta M. J., Odom D. J., Douglas E. P., Gower L. B., Connect. Tissue Res. 2003, 44, 326. [PubMed] [Google Scholar]
- 6.a) Lowenstam H. A., Weiner S., On Biomineralization, Oxford Univeristy Press, New York: 1989; [Google Scholar]; b) Christoffersen J., Landis W. J., Anat. Rec. 1991, 230, 435. [DOI] [PubMed] [Google Scholar]
- 7.a) Gordon M. K., Hahn R. A., Cell Tissue Res. 2009, 339, 247; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brodsky B., Persikov A. V., Adv. Protein Chem. 2005, 70, 301; [DOI] [PubMed] [Google Scholar]; c) Brinckmann J., Collagen, Springer, Berlin, Heidelberg: 2005, p. 1; [Google Scholar]; d) Hulmes D. J., J. Struct. Biol. 2002, 137, 2; [DOI] [PubMed] [Google Scholar]; e) Shoulders M. D., Raines R. T., Annu. Rev. Biochem. 2009, 78, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Meek K. M., Biophys. Rev. 2009, 1, 83; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Banos C. C., Thomas A. H., Kuo C. K., Birth Defects Res., Part C. 2008, 84, 228. [DOI] [PubMed] [Google Scholar]
- 9. Gjaltema R. A., Bank R. A., Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74. [DOI] [PubMed] [Google Scholar]
- 10. Wess T. J., Adv. Protein Chem. 2005, 70, 341. [DOI] [PubMed] [Google Scholar]
- 11. Bou‐Gharios G., Abraham D., de Crombrugghe B., in Principles of Bone Biology, Academic Press, Cambridge, Massachusetts, USA: 2020, p. 295. [Google Scholar]
- 12. Sorushanova A., Delgado L. M., Wu Z., Shologu N., Kshirsagar A., Raghunath R., Mullen A. M., Bayon Y., Pandit A., Raghunath M., Adv. Mater. 2019, 31, 1801651. [DOI] [PubMed] [Google Scholar]
- 13. Bodian D. L., Madhan B., Brodsky B., Klein T. E., Biochemistry 2008, 47, 5424. [DOI] [PubMed] [Google Scholar]
- 14.a) Hodge A. J., in Aspects of Protein Structure, (Ed: Ramachandran G. N.), Academic Press, New York: 1963, p. 289; [Google Scholar]; b) Hulmes D. J., Miller A., Nature 1979, 282, 878. [DOI] [PubMed] [Google Scholar]
- 15. Orgel J. P. R. O., Irving T. C., Miller A., Wess T. J., Proc. Natl. Acad. Sci. USA 2006, 103, 9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Orgel J., San Antonio J., Antipova O., Connect. Tissue Res. 2011, 52, 2. [DOI] [PubMed] [Google Scholar]
- 17. Chapman J. A., Tzaphlidou M., Meek K. M., Kadler K. E., Electron Microsc. Rev. 1990, 3, 143. [DOI] [PubMed] [Google Scholar]
- 18. Xu Z., Yang Y., Zhao W., Wang Z., Landis W. J., Cui Q., Sahai N., Biomaterials 2015, 39, 59. [DOI] [PubMed] [Google Scholar]
- 19. Xu Z., Zhao W., Wang Z., Yang Y., Sahai N., Phys. Chem. Chem. Phys. 2018, 20, 1513. [DOI] [PubMed] [Google Scholar]
- 20. Nudelman F., Pieterse K., George A., Bomans P. H. H., Friedrich H., Brylka L. J., Hilbers P. A., de With G., Sommerdijk N. A. J. M., Nat. Mater. 2010, 9, 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Antoine E. E., Vlachos P. P., Rylander M. N., Tissue Eng., Part B 2014, 20, 683; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu D., Nikoo M., Boran G., Zhou P., Regenstein J. M., Annu. Rev. Food Sci. Technol. 2015, 6, 527. [DOI] [PubMed] [Google Scholar]
- 22. Liu D., Liang L., Regenstein J. M., Zhou P., Food Chem. 2012, 133, 1441. [Google Scholar]
- 23. Rahman M. A., Mar. Drugs 2019, 17, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kreger S., Bell B., Bailey J., Stites E., Kuske J., Waisner B., Voytik‐Harbin S., Biopolymers 2010, 93, 690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Ruggiero F., Exposito J.‐Y., Bournat P., Gruber V., Perret S., Comte J., Olagnier B., Garrone R., Theisen M., FEBS Lett. 2000, 469, 132; [DOI] [PubMed] [Google Scholar]; b) Olsen D., Yang C., Bodo M., Chang R., Leigh S., Baez J., Carmichael D., Perälä M., Hämäläinen E.‐R., Jarvinen M., Adv. Drug Del. Rev. 2003, 55, 1547; [DOI] [PubMed] [Google Scholar]; c) Yang C., Hillas P. J., Báez J. A., Nokelainen M., Balan J., Tang J., Spiro R., Polarek J. W., BioDrugs 2004, 18, 103. [DOI] [PubMed] [Google Scholar]
- 26. Fallas J. A., O'Leary L. E., Hartgerink J. D., Chem. Soc. Rev. 2010, 39, 3510. [DOI] [PubMed] [Google Scholar]
- 27. Weiher F., Schatz M., Steinem C., Geyer A., Biomacromolecules 2013, 14, 683. [DOI] [PubMed] [Google Scholar]
- 28. Zhu S., Yuan Q., Yin T., You J., Gu Z., Xiong S., Hu Y., J. Mater. Chem. B 2018, 6, 2650. [DOI] [PubMed] [Google Scholar]
- 29. Ping H., Xie H., Su B.‐L., Cheng Y.‐b., Wang W., Wang H., Wang Y., Zhang J., Zhang F., Fu Z., J. Mater. Chem. B 2015, 3, 4496. [DOI] [PubMed] [Google Scholar]
- 30. Bancelin S., Aimé C., Gusachenko I., Kowalczuk L., Latour G., Coradin T., Schanne‐Klein M.‐C., Nat. Commun. 2014, 5, 1. [DOI] [PubMed] [Google Scholar]
- 31.a) Schoof H., Apel J., Heschel I., Rau G., J. Biomed. Mater. Res. 2001, 58, 352; [DOI] [PubMed] [Google Scholar]; b) Haugh M. G., Murphy C. M., O'Brien F. J., Tissue Eng., Part C 2010, 16, 887. [DOI] [PubMed] [Google Scholar]
- 32.a) Niu L.‐n., Jiao K., Ryou H., Diogenes A., Yiu C. K., Mazzoni A., Chen J.‐h., Arola D. D., Hargreaves K. M., Pashley D. H., Biomacromolecules 2013, 14, 1661; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nikiforuk G., Sreebny L., J. Dent. Res. 1953, 32, 859. [DOI] [PubMed] [Google Scholar]
- 33. Ehrlich H., Krautter M., Hanke T., Simon P., Heinemann C., Heinemann S., Worch H., J. Exp. Zool., Part B 2007, 308, 473. [DOI] [PubMed] [Google Scholar]
- 34. Shechter A., Berman A., Singer A., Freiman A., Grinstein M., Erez J., Aflalo E. D., Sagi A., Biol. Bull. 2008, 214, 122. [DOI] [PubMed] [Google Scholar]
- 35. Iijima M., Moriwaki Y., Calcif. Tissue Int. 1990, 47, 237. [DOI] [PubMed] [Google Scholar]
- 36. Chahal S., Kumar A., Hussian F. S. J., J. Biomater. Sci., Polym. Ed. 2019, 30, 1308. [DOI] [PubMed] [Google Scholar]
- 37.a) Keaveny T. M., Yeh O. C., J. Musculoskelet. Neuron. Interact. 2002, 2, 205; [PubMed] [Google Scholar]; b) Martin R. B., Atkinson P. J., J. Biomech. 1977, 10, 223; [DOI] [PubMed] [Google Scholar]; c) Atkinson P. J., Calcif. Tissue Res. 1967, 1, 24. [DOI] [PubMed] [Google Scholar]
- 38.a) Currey J. D., Science 2005, 309, 253;16002605 [Google Scholar]; b) Currey J. D., Zioupos P., Peter D., Casinos A., Proc. R. Soc. London, Ser. B 2001, 268, 107; [Google Scholar]; c) Currey J. D., Osteoporosis Int. 2003, 14, 29; [Google Scholar]; d) Launey M. E., Buehler M. J., Ritchie R. O., Annu. Rev. Mater. Res. 2010, 40, 25; [Google Scholar]; e) Natali A. N., Meroi E. A., J. Biomed. Eng. 1989, 11, 266; [DOI] [PubMed] [Google Scholar]; f) Saha S., Pal S., J. Biomed. Mater. Res. 1984, 18, 435; [DOI] [PubMed] [Google Scholar]; g) Wang X., Puram S., Ann. Biomed. Eng. 2004, 32, 123; [DOI] [PubMed] [Google Scholar]; h) Weiner S., Traub W., Wagner H. D., J. Struct. Biol. 1999, 126, 241. [DOI] [PubMed] [Google Scholar]
- 39. Weiner S., Wagner H. D., Annu. Rev. Mater. Sci. 1998, 28, 271. [Google Scholar]
- 40. Reznikov N., Shahar R., Weiner S., Acta Biomater. 2014, 10, 3815. [DOI] [PubMed] [Google Scholar]
- 41. Currey J. D., J. Exp. Biol. 1999, 202, 2495. [DOI] [PubMed] [Google Scholar]
- 42. Boskey A. L., Connect. Tissue Res. 1996, 35, 357. [DOI] [PubMed] [Google Scholar]
- 43.a) Al‐Qtaitat A. I., Am. J. Life Sci. 2014, 2; [Google Scholar]; b) Roach H. I., Cell Biol. Int. 1994, 18, 617. [DOI] [PubMed] [Google Scholar]
- 44. George A., Veis A., Chem. Rev. 2008, 108, 4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fratzl P., Gupta H. S., Paschalis E. P., Roschger P., J. Mater. Chem. 2004, 14, 2115. [Google Scholar]
- 46. Pasteris J. D., Wopenka B., Freeman J. J., Rogers K., Valsami‐Jones E., van der Houwen J. A. M., Silva M. J., Biomaterials 2004, 25, 229. [DOI] [PubMed] [Google Scholar]
- 47.a) Landi E., Celotti G., Logroscino G., Tampieri A., J. Eur. Ceram. Soc. 2003, 23, 2931; [Google Scholar]; b) Leventouri T., Biomaterials 2006, 27, 3339. [DOI] [PubMed] [Google Scholar]
- 48. Elliott J. C., Structure and Chemisty of the Apatites and Other Calcium Orthophosphates, Elsevier, Amsterdam: 1994. [Google Scholar]
- 49. Rey C., Combes C., Drouet C., Glimcher M. J., Osteoporosis Int. 2009, 20, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhuofan C., Baoxin H., Haobo P., Darvell B. W., Cryst. Growth Des. 2009, 9, 2816. [Google Scholar]
- 51.a) Samavedi S., Whittington A. R., Goldstein A. S., Acta Biomater. 2013, 9, 8037; [DOI] [PubMed] [Google Scholar]; b) Huang J., Best S. M., Brooks R. A., Rushton N., Bonfield W., J. Biomed. Mater. Res. A 2008, 87A, 598; [DOI] [PubMed] [Google Scholar]; c) Chen Z. F., Darvell B. W., Leung V. W. H., Arch. Oral Biol. 2004, 49, 359; [DOI] [PubMed] [Google Scholar]; d) Liu Q., Huang S., Matinlinna J. P., Chen Z., Pan H., BioMed. Res. Int. 2013, 2013, 929748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.a) Kuhn L. T., Grynpas M. D., Rey C. C., Wu Y., Ackerman J. L., Glimcher M. J., Calcif. Tissue Int. 2008, 83, 146; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barakat N. A. M., Khalil K. A., Sheikh F. A., Omran A. M., Gaihre B., Khil S. M., Kim H. Y., Mater. Sci. Eng. C 2008, 28, 1381; [Google Scholar]; c) Janus A. M., Faryna M., Haberko K., Rakowska A., Panz T., Microchim. Acta 2008, 161, 349; [Google Scholar]; d) Joschek S., Nies B., Krotz R., Göpferich A., Biomaterials 2000, 21, 1645; [DOI] [PubMed] [Google Scholar]; e) Eliaz N., Metoki N., Materials 2017, 10, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bonar L. C., Lees S., Mook H. A., J. Mol. Biol. 1985, 181, 265. [DOI] [PubMed] [Google Scholar]
- 54.a) McNally E. A., Schwarcz H. P., Botton G. A., Arsenault A. L., PLoS One 2012, 7, e29258; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Landis W. J., Song M. J., Leith A., Mcewen L., Mcewen B. F., J. Struct. Biol. 1993, 110, 39. [DOI] [PubMed] [Google Scholar]
- 55.a) Boskey A. L., J. Dent. Res. 1997, 76, 1433; [DOI] [PubMed] [Google Scholar]; b) Hassenkam T., Fantner G. E., Cutroni J. A., Weaver J. C., Morse D. E., Hansma P. K., Bone 2004, 35, 4. [DOI] [PubMed] [Google Scholar]
- 56. McNally E., Nan F., Botton G. A., Schwarcz H. P., Micron 2013, 49, 46. [DOI] [PubMed] [Google Scholar]
- 57. Grünewald T. A., Liebi M., Wittig N. K., Johannes A., Sikjaer T., Rejnmark L., Gao Z., Rosenthal M., Guizar‐Sicairos M., Birkedal H., Burghammer M., Sci. Adv. 2020, 6, eaba4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rubin M. A., Jasiuk I., Taylor J., Rubin J., Ganey T., Apkarian R. P., Bone 2003, 33, 270. [DOI] [PubMed] [Google Scholar]
- 59. Wilson E. E., Awonusi A., Morris M. D., Kohn D. H., Tecklenburg M. M. J., Beck L. W., J. Bone Miner. Res. 2005, 20, 625. [DOI] [PubMed] [Google Scholar]
- 60. Dorvee J. R., Veis A., J. Struct. Biol. 2013, 183, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Millar S. E., Nat. Mater. 2019, 18, 530. [DOI] [PubMed] [Google Scholar]
- 62.a) Marshall G. W., Marshall S. J., Kinney J. H., Balooch M., J. Dent. 1997, 25, 441; [DOI] [PubMed] [Google Scholar]; b) Kinney J. H., Marshall S. J., Marshall G. W., Crit. Rev. Oral Biol. Med. 2003, 14, 13. [DOI] [PubMed] [Google Scholar]
- 63. Kinney J. H., Pople J. A., Marshall G. W., Marshall S. J., Calcif. Tissue Int. 2001, 69, 31. [DOI] [PubMed] [Google Scholar]
- 64.a) Goldberg M., Kulkarni A. B., Young M., Boskey A., Front. Biosci. 2011, 3, 711; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nalla R. K., Porter A. E., Daraio C., Minor A. M., Radmilovic V., Stach E. A., Tomsia A. P., Ritchie R. O., Micron 2005, 36, 672; [DOI] [PubMed] [Google Scholar]; c) Bertassoni L. E., Dent. Mater. 2017, 33, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.a) Orsini G., Ruggeri A., Mazzoni A., Nato F., Manzoli L., Putignano A., Di Lenarda R., Tjäderhane L., Breschi L., Endod. Topics 2012, 21, 1; [Google Scholar]; b) Mazzoni A., Breschi L., Carrilho M., Nascimento F. D., Orsini G., Ruggeri A. Jr., Gobbi P., Manzoli L., Tay F. R., Pashley D. H., Tjäderhane L., Endod. Topics 2009, 21, 19. [Google Scholar]
- 66. Linde A., Goldberg M., Crit. Rev. Oral Biol. Med. 1993, 4, 679. [DOI] [PubMed] [Google Scholar]
- 67. Ehrlich H., Wysokowski M., Żółtowska‐Aksamitowska S., Petrenko I., Jesionowski T., Mar. Drugs 2018, 16, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Müller W. E. G., Wang X., Cui F.‐Z., Jochum K. P., Tremel W., Bill J., Schröder H. C., Natalio F., Schlossmacher U., Wiens M., Appl. Microbiol. Biotechnol. 2009, 83, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.a) Harley B. A., Leung J. H., Silva E. C., Gibson L. J., Acta Biomater. 2007, 3, 463; [DOI] [PubMed] [Google Scholar]; b) Chamberlain L. J., Yannas I. V., Hsu H. P., Strichartz G., Spector M., Exp. Neurol. 1998, 154, 315; [DOI] [PubMed] [Google Scholar]; c) Yannas I. V., Lee E., Orgill D. P., Skrabut E. M., Murphy G. F., Proc. Natl. Acad. Sci. USA 1989, 86, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.a) Rezwan K., Chen Q. Z., Blaker J. J., Boccaccini A. R., Biomaterials 2006, 27, 3413; [DOI] [PubMed] [Google Scholar]; b) Marcacci M., Kon E., Moukhachev V., Lavroukov A., Kutepov S., Quarto R., Mastrogiacomo M., Cancedda R., Tissue Eng. 2007, 13, 947. [DOI] [PubMed] [Google Scholar]
- 71.a) Chvapil M., J. Biomed. Mater. Res. 1977, 11, 721; [DOI] [PubMed] [Google Scholar]; b) Tsuruga E., Takita H., Itoh H., Wakisaka Y., Kuboki Y., J. Biochem. 1997, 121, 317; [DOI] [PubMed] [Google Scholar]; c) Zeltinger J., Sherwood J. K., Graham D. A., Müeller R., Griffith L. G., Tissue Eng. 2001, 7, 557. [DOI] [PubMed] [Google Scholar]
- 72.a) O'Brien F. J., Harley B. A., Yannas I. V., Gibson L., Biomaterials 2004, 25, 1077; [DOI] [PubMed] [Google Scholar]; b) O'Brien F. J., Harley B. A., Yannas I. V., Gibson L. J., Biomaterials 2005, 26, 433; [DOI] [PubMed] [Google Scholar]; c) O'Brien F. J., Harley B. A., Waller M. A., Yannas I. V., Gibson L. J., Prendergast P. J., Technol. Health Care 2007, 15, 3. [PubMed] [Google Scholar]
- 73. Al‐Munajjed A. A., Plunkett N. A., Gleeson J. P., Weber T., Jungreuthmayer C., Levingstone T., Hammer J., O'Brien F. J., J. Biomed. Mater. Res., Part B 2009, 90, 584. [DOI] [PubMed] [Google Scholar]
- 74. Farzadi A., Renner T., Calomeni E. P., Presley K. F., Karn N., Lannutti J., Dasi L. P., Agarwal G., Mater. Sci. Eng., C 2019, 104, 109905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Villa M. M., Wang L., Huang J., Rowe D. W., Wei M., J. Biomed. Mater. Res., Part B 2015, 103, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xia Z., Villa M. M., Wei M., J. Mater. Chem. B 2014, 2, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Al‐Munajjed A. A., O'Brien F. J., J. Mech. Behav. Biomed. Mater. 2009, 2, 138. [DOI] [PubMed] [Google Scholar]
- 78. Nudelman F., Lausch A. J., Sommerdijk N. A., Sone E. D., J. Struct. Biol. 2013, 183, 258. [DOI] [PubMed] [Google Scholar]
- 79. Aizenberg J., Addadi L., Weiner S., Lambert G., Adv. Mater. 1996, 8, 222. [Google Scholar]
- 80. Gower L. A., Tirrell D. A., J. Cryst. Growth 1998, 191, 153. [Google Scholar]
- 81. Gower L. B., Odom D. J., J. Cryst. Growth 2000, 210, 719. [Google Scholar]
- 82. Kato T., Suzuki T., Amamiya T., Irie T., Komiyama M., Yui H., Supramol. Sci. 1998, 5, 411. [Google Scholar]
- 83. Olszta M. J., Douglas E. P., Gower L. B., Calcif. Tissue Int. 2003, 72, 583. [DOI] [PubMed] [Google Scholar]
- 84. Olszta M. J., Douglas E. P., Gower L. B., Mater. Res. Soc. Symp. Proc. 2003, 774, 127. [Google Scholar]
- 85. Olszta M. J., Cheng X. G., Jee S. S., Kumar R., Kim Y. Y., Kaufman M. J., Douglas E. P., Gower L. B., Mater. Sci. Eng. R 2007, 58, 77. [Google Scholar]
- 86. Deshpande A. S., Beniash E., Cryst. Growth Des. 2008, 8, 3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang W., Liao S. S., Cui F. Z., Chem. Mater. 2003, 15, 3221. [Google Scholar]
- 88. Chen J. L., Burger C., Krishnan C. V., Chu B., Hsiao B. S., Glimcher M. J., Macromol. Chem. Phys. 2005, 206, 43. [Google Scholar]
- 89. Rolls G., An Introduction to Decalcification, https://www.leicabiosystems.com/knowledge-pathway/an-introduction-to-decalcification/ (accessed: June 2020).
- 90. Guo J. M., Makvandi P., Wei C. C., Chen J. H., Xu H. K., Breschi L., Pashley D. H., Huang C., Niu L. N., Tay F. R., Acta Biomater. 2019, 90, 424. [DOI] [PubMed] [Google Scholar]
- 91. Wang Y., Azais T., Robin M., Vallee A., Catania C., Legriel P., Pehau‐Arnaudet G., Babonneau F., Giraud‐Guille M. M., Nassif N., Nat. Mater. 2012, 11, 724. [DOI] [PubMed] [Google Scholar]
- 92.a) Traub W., Jodaikin A., Arad T., Veis A., Sabsay B., Matrix 1992, 12, 197; [DOI] [PubMed] [Google Scholar]; b) He G., George A., J. Biol. Chem. 2004, 279, 11649. [DOI] [PubMed] [Google Scholar]
- 93. Mahamid J., Sharir A., Addadi L., Weiner S., Proc. Natl. Acad. Sci. USA 2008, 105, 12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mahamid J., Aichmayer B., Shimoni E., Ziblat R., Li C., Siegel S., Paris O., Fratzl P., Weiner S., Addadi L., Proc. Natl. Acad. Sci. USA 2010, 107, 6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mahamid J., Sharir A., Gur D., Zelzer E., Addadi L., Weiner S., J. Struct. Biol. 2011, 174, 527. [DOI] [PubMed] [Google Scholar]
- 96. Simon P., Gruner D., Worch H., Pompe W., Lichte H., El Khassawna T., Heiss C., Wenisch S., Kniep R., Sci. Rep. 2018, 8, 13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Okamoto Y., Hidaka S., J. Biomed. Mater. Res. 1994, 28, 1403. [DOI] [PubMed] [Google Scholar]
- 98. Nudelman F., Bomans P. H. H., George A., de With G., Sommerdijk N. A. J. M., Faraday Discuss. 2012, 159, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gerstenfeld L. C., Feng M., Gotoh Y., Glimcher M. J., Calcif. Tissue Int. 1994, 55, 230. [DOI] [PubMed] [Google Scholar]
- 100. Esfand R., Tomalia D. A., Drug Discov. Today 2001, 6, 427. [DOI] [PubMed] [Google Scholar]
- 101.a) Li J., Yang J., Li J., Chen L., Liang K., Wu W., Chen X., Li J., Biomaterials 2013, 34, 6738; [DOI] [PubMed] [Google Scholar]; b) Liang K., Gao Y., Li J., Liao Y., Xiao S., Zhou X., Li J., J. Biomater. Sci., Polym. Ed. 2015, 26, 963. [DOI] [PubMed] [Google Scholar]
- 102. Bradt J.‐H., Mertig M., Teresiak A., Pompe W., Chem. Mater. 1999, 11, 2694. [Google Scholar]
- 103. Jee S.‐S., Thula T. T., Gower L. B., Acta Biomater. 2010, 6, 3676. [DOI] [PubMed] [Google Scholar]
- 104. Jee S. S., Kasinath R. K., DiMasi E., Kim Y.‐Y., Gower L., CrystEngComm 2011, 13, 2077. [Google Scholar]
- 105. Jee S. S., Culver L., Li Y., Douglas E. P., Gower L. B., J. Cryst. Growth 2010, 312, 1249. [Google Scholar]
- 106. Lausch A. J., Quan B. D., Miklas J. W., Sone E. D., Adv. Funct. Mater. 2013, 23, 4906. [Google Scholar]
- 107. Qi Y., Ye Z., Fok A., Holmes B. N., Espanol M., Ginebra M.‐P., Aparicio C., ACS Biomater. Sci. Eng. 2018, 4, 2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Song Q., Jiao K., Tonggu L., Wang L. G., Zhang S. L., Yang Y. D., Zhang L., Bian J. H., Hao D. X., Wang C. Y., Ma Y. X., Arola D. D., Breschi L., Chen J. H., Tay F. R., Niu L. N., Sci. Adv. 2019, 5, eaav9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yu L., Martin I. J., Kasi R. M., Wei M., ACS Appl. Mater. Interfaces 2018, 10, 28440. [DOI] [PubMed] [Google Scholar]
- 110. Cantaert B., Kim Y.‐Y., Ludwig H., Nudelman F., Sommerdijk N. A. J. M., Meldrum F. C., Adv. Funct. Mater. 2012, 22, 907. [Google Scholar]
- 111.a) Niu L. N., Jee S. E., Jiao K., Tonggu L., Li M., Wang L., Yang Y. D., Bian J. H., Breschi L., Jang S. S., Chen J. H., Pashley D. H., Tay F. R., Nat. Mater. 2017, 16, 370; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiao K., Niu L. N., Ma C. F., Huang X. Q., Pei D. D., Luo T., Huang Q., Chen J. H., Tay F. R., Adv. Funct. Mater. 2016, 26, 6858. [Google Scholar]
- 112.a) Tay F. R., Pashley D. H., Biomaterials 2008, 29, 1127; [DOI] [PubMed] [Google Scholar]; b) Kim Y. K., Gu L.‐s., Bryan T. E., Kim J. R., Chen L., Liu Y., Yoon J. C., Breschi L., Pashley D. H., Tay F. R., Biomaterials 2010, 31, 6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Liu Y., Kim Y.‐K., Dai L., Li N., Khan S. O., Pashley D. H., Tay F. R., Biomaterials 2011, 32, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu Y., Li N., Qi Y., Niu L.‐n., Elshafiy S., Mao J., Breschi L., Pashley D. H., Tay F. R., Dent. Mater. 2011, 27, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu Y., Li N., Qi Y.‐p., Dai L., Bryan T. E., Mao J., Pashley D. H., Tay F. R., Adv. Mater. 2011, 23, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hu C., Zhang L., Wei M., ACS Biomater. Sci. Eng. 2015, 1, 669. [DOI] [PubMed] [Google Scholar]
- 117. Li X., Chang J., J. Biomed. Mater. Res. A 2008, 85A, 293. [DOI] [PubMed] [Google Scholar]
- 118. Tavafoghi M., Cerruti M., J. R. Soc. Interface 2016, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bu H., Yang H., Shen L., Liu W., Li G., Colloids Surf., B 2020, 190, 110892. [DOI] [PubMed] [Google Scholar]
- 120. Shen M., Lin M., Zhu M., Zhang W., Lu D., Liu H., Deng J., Que K., Zhang X., Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 167. [DOI] [PubMed] [Google Scholar]
- 121. Hasegawa T., Histochem. Cell Biol. 2018, 149, 289. [DOI] [PubMed] [Google Scholar]
- 122. Dickens F., Biochem. J. 1941, 35, 1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hu Y.‐Y., Rawal A., Schmidt‐Rohr K., Proc. Natl. Acad. Sci. USA 2010, 107, 22425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.a) López‐Macipe A., Gómez‐Morales J., Rodríguez‐Clemente R., Adv. Mater. 1998, 10, 49; [Google Scholar]; b) Tenhuisen K. S., Brown P. W., J. Mater. Sci. Mater. Med. 1994, 5, 291. [Google Scholar]
- 125. Davies E., Müller K. H., Wong W. C., Pickard C. J., Reid D. G., Skepper J. N., Duer M. J., Proc. Natl. Acad. Sci. USA 2014, 111, E1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shao C., Zhao R., Jiang S., Yao S., Wu Z., Jin B., Yang Y., Pan H., Tang R., Adv. Mater. 2018, 30, 1704876. [DOI] [PubMed] [Google Scholar]
- 127. Price P. A., Toroian D., Lim J. E., J. Biol. Chem. 2009, 284, 17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Toroian D., Lim J. E., Price P. A., J. Biol. Chem. 2007, 282, 22437. [DOI] [PubMed] [Google Scholar]
- 129. Kim D., Lee B., Thomopoulos S., Jun Y. S., Nat. Commun. 2018, 9, 962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Du T., Niu X., Hou S., Xu M., Li Z., Li P., Fan Y., J. Mater. Chem. B. 2020, 8, 2562. [DOI] [PubMed] [Google Scholar]
- 131. Von Euw S., Chan‐Chang T. H. C., Paquis C., Haye B., Pehau‐Arnaudet G., Babonneau F., Azais T., Nassif N., Geosciences 2018, 8, 466. [Google Scholar]
- 132. Carlisle E. M., Sci. Total Environ. 1988, 73, 95. [DOI] [PubMed] [Google Scholar]
- 133. Müller W. E., Wang X., Cui F.‐Z., Jochum K. P., Tremel W., Bill J., Schröder H. C., Natalio F., Schloßmacher U., Wiens M., Appl. Microbiol. Biotechnol. 2009, 83, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kröger N., Poulsen N., Annu. Rev. Genet. 2008, 42, 83. [DOI] [PubMed] [Google Scholar]
- 135. Discher D. E., Janmey P., Wang Y.‐l., Science 2005, 310, 1139. [DOI] [PubMed] [Google Scholar]
- 136. Heinemann S., Coradin T., Desimone M. F., Biomater. Sci. 2013, 1, 688. [DOI] [PubMed] [Google Scholar]
- 137. Ono Y., Kanekiyo Y., Inoue K., Hojo J., Nango M., Shinkai S., Chem. Lett. 1999, 28, 475. [Google Scholar]
- 138. Eglin D., Shafran K. L., Livage J., Coradin T., Perry C. C., J. Mater. Chem. 2006, 16, 4220. [Google Scholar]
- 139. Shen L., Bu H., Yang H., Liu W., Li G., Int. J. Biol. Macromol. 2018, 115, 635. [DOI] [PubMed] [Google Scholar]
- 140. Quignard S., Copello G. J., Aimé C., Bataille I., Hélary C., Desimone M. F., Coradin T., Adv. Eng. Mater. 2012, 14, B51. [Google Scholar]
- 141. Desimone M. F., Hélary C., Rietveld I. B., Bataille I., Mosser G., Giraud‐Guille M.‐M., Livage J., Coradin T., Acta Biomater. 2010, 6, 3998. [DOI] [PubMed] [Google Scholar]
- 142. Desimone M. F., Hélary C., Quignard S., Rietveld I. B., Bataille I., Copello G. J., Mosser G., Giraud‐Guille M.‐M., Livage J., Meddahi‐Pellé A., Coradin T., ACS Appl. Mater. Interfaces 2011, 3, 3831. [DOI] [PubMed] [Google Scholar]
- 143. Alvarez G. S., Hélary C., Mebert A. M., Wang X., Coradin T., Desimone M. F., J. Mater. Chem. B 2014, 2, 4660. [DOI] [PubMed] [Google Scholar]
- 144. Wang X., Hélary C., Coradin T., ACS Appl. Mater. Interfaces 2015, 7, 2503. [DOI] [PubMed] [Google Scholar]
- 145. Chen S., Chinnathambi S., Shi X., Osaka A., Zhu Y., Hanagata N., J. Mater. Chem. 2012, 22, 21885. [Google Scholar]
- 146. Damink L. O., Dijkstra P., Van Luyn M., Van Wachem P., Nieuwenhuis P., Feijen J., J. Mater. Sci. Mater. Med. 1995, 6, 460. [Google Scholar]
- 147. Chen S., Osaka A., Ikoma T., Morita H., Li J., Takeguchi M., Hanagata N., J. Mater. Chem. 2011, 21, 10942. [Google Scholar]
- 148. Foglia M. L., Camporotondi D. E., Alvarez G. S., Heinemann S., Hanke T., Perez C. J., Diaz L. E., Desimone M. F., J. Mater. Chem. B 2013, 1, 6283. [DOI] [PubMed] [Google Scholar]
- 149. Foglia M. L., Mitarotonda R., De Marzi M. C., Desimone M. F., Mater. Sci. Eng. C 2019, 99, 47. [DOI] [PubMed] [Google Scholar]
- 150. Wang S. J., Jiang D., Zhang Z. Z., Chen Y. R., Yang Z. D., Zhang J. Y., Shi J., Wang X., Yu J. K., Adv. Mater. 2019, 31, 1904341. [DOI] [PubMed] [Google Scholar]
- 151. Niu L. n., Jiao K., Qi Y. p., Yiu C. K., Ryou H., Arola D. D., Chen J. h., Breschi L., Pashley D. H., Tay F. R., Angew. Chem., Int. Ed. 2011, 50, 11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.a) Sun J. L., Jiao K., Niu L. N., Jiao Y., Song Q., Shen L. J., Tay F. R., Chen J. H., Biomaterials 2017, 113, 203; [DOI] [PubMed] [Google Scholar]; b) Niu L.‐N., Jiao K., Qi Y.‐P., Nikonov S., Yiu C. K., Arola D. D., Gong S.‐Q., El‐Marakby A., Carrilho M. R., Hamrick M. W., FASEB J. 2012, 26, 4517; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sun J.‐l., Jiao K., Song Q., Ma C.‐f., Ma C., Tay F. R., Niu L.‐n., Chen J.‐h., Acta Biomater. 2018, 67, 354. [DOI] [PubMed] [Google Scholar]
- 153. Niu L.‐n., Sun J.‐q., Li Q.‐h., Jiao K., Shen L.‐j., Wu D., Tay F., Chen J.‐h., J. Dent. 2014, 42, 839. [DOI] [PubMed] [Google Scholar]
- 154. Hu C., Yu L., Wei M., RSC Adv. 2017, 7, 34624. [Google Scholar]
- 155.a) Heinemann S., Coradin T., Worch H., Wiesmann H., Hanke T., Compos. Sci. Technol. 2011, 71, 1873; [Google Scholar]; b) Heinemann S., Heinemann C., Jager M., Neunzehn J., Wiesmann H. P., Hanke T., ACS Appl. Mater. Interfaces 2011, 3, 4323. [DOI] [PubMed] [Google Scholar]
- 156. Gu L., Shan T., Ma Y.‐x., Tay F. R., Niu L., Trends Biotechnol. 2019, 37, 464. [DOI] [PubMed] [Google Scholar]
- 157. Alliraja C., Rao J. R., Thanikaivelan P., RSC Adv. 2015, 5, 20939. [Google Scholar]
- 158. Srivatsan K., Lakra R., Sai K. P., Kiran M., J. Mater. Chem. B 2016, 4, 1437. [DOI] [PubMed] [Google Scholar]
- 159. Agban Y., Lian J., Prabakar S., Seyfoddin A., Rupenthal I. D., Int. J. Pharm. 2016, 501, 96. [DOI] [PubMed] [Google Scholar]
- 160. Niu L. n., Jiao K., Ryou H., Yiu C. K., Chen J. h., Breschi L., Arola D. D., Pashley D. H., Tay F. R., Angew. Chem., Int. Ed. 2013, 52, 5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Jiao K., Niu L.‐n., Li Q.‐h., Chen F.‐m., Zhao W., Li J.‐j., Chen J.‐h., Cutler C. W., Pashley D. H., Tay F. R., Acta Biomater. 2015, 19, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zhou B., Niu L. n., Shi W., Zhang W., Arola D. D., Breschi L., Mao J., Chen J. h., Pashley D. H., Tay F. R., Adv. Funct. Mater. 2014, 24, 1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. de Wildt B. W. M., Ansari S., Sommerdijk N. A. J. M., Ito K., Akiva A., Hofmann S., Curr. Opin. Biomed. Eng. 2019, 10, 107. [Google Scholar]
- 164. Niu L.‐n., Angew. Chem., Int. Ed. 2011, 50, 11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Hollister S. J., Nat. Mater. 2005, 4, 518. [DOI] [PubMed] [Google Scholar]
- 166. Murphy C. M., Schindeler A., Gleeson J. P., Yu N. Y., Cantrill L. C., Mikulec K., Peacock L., O'Brien F. J., Little D. G., Acta Biomater. 2014, 10, 2250. [DOI] [PubMed] [Google Scholar]
- 167.a) An J., Teoh J. E. M., Suntornnond R., Chua C. K., Engineering 2015, 1, 261; [Google Scholar]; b) Liu C. Z., Xia Z. D., Han Z. W., Hulley P. A., Triffitt J. T., Czernuszka J. T., J. Biomed. Mater. Res., Part B 2008, 85B, 519; [DOI] [PubMed] [Google Scholar]; c) Yeong W. Y., Chua C. K., Leong K. F., Chandrasekaran M., Lee M. W., Rapid Prototyp. J. 2006, 12, 229; [Google Scholar]; d) Inzana J. A., Olvera D., Fuller S. M., Kelly J. P., Graeve O. A., Schwarz E. M., Kates S. L., Awad H. A., Biomaterials 2014, 35, 4026; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Murphy S. V., Atala A., Nat. Biotechnol. 2014, 32, 773. [DOI] [PubMed] [Google Scholar]
- 168. Li Y., Thula T. T., Jee S., Perkins S. L., Aparicio C., Douglas E. P., Gower L. B., Biomacromolecules 2011, 13, 49. [DOI] [PubMed] [Google Scholar]
- 169. Thula T. T., Rodriguez D. E., Lee M. H., Pendi L., Podschun J., Gower L. B., Acta Biomater. 2011, 7, 3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Dai L., Qi Y.‐P., Niu L.‐N., Liu Y., Pucci C. R., Looney S. W., Ling J.‐Q., Pashley D. H., Tay F. R., Cryst. Growth Des. 2011, 11, 3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Smeets P. J. M., Finney A. R., Habraken W. J. E. M., Nudelman F., Friedrich H., Laven J., De Yoreo J. J., Rodger P. M., Sommerdijk N. A. J. M., Proc. Natl. Acad. Sci. USA 2017, 114, E7882. [DOI] [PMC free article] [PubMed] [Google Scholar]