

Abstract
Melt electrowriting (MEW) is an emerging high‐resolution additive manufacturing technique based on the electrohydrodynamic processing of polymers. MEW is predominantly used to fabricate scaffolds for biomedical applications, where the microscale fiber positioning has substantial implications in its macroscopic mechanical properties. This review gives an update on the increasing number of polymers processed via MEW and different commercial sources of the gold standard poly(ε‐caprolactone) (PCL). A description of MEW‐processed polymers beyond PCL is introduced, including blends and coated fibers to provide specific advantages in biomedical applications. Furthermore, a perspective on printer designs and developments is highlighted, to keep expanding the variety of processable polymers for MEW.
Keywords: 3D printing, additive manufacturing, biomedical materials, electrohydrodynamic materials, melt electrospinning writing
Melt electrowriting is a 3D‐printing technology that is increasingly investigated for biomedical applications. This review focuses on the different polymers used with this technology so far and provides a future perspective for additional requirements needed for melt electrowriting printers to help expand the possible processable materials and designs.
1. Introduction
Additive manufacturing (AM) is increasingly used for applications in biomaterials, tissue engineering, and biofabrication partly due to the potential fabrication of patient‐specific designs.[ 1 ] In general, every fabrication technology has an ideal printing resolution range and for many techniques this is ≈50–500 µm.[ 2 , 3 ] For biomedical applications that aim to replicate the tissue microarchitecture, it is necessary to be able to print even smaller structural features in the low micrometer or nanometer scale. Such small fiber diameters can be facilitated with electrohydrodynamic (EHD) techniques such as solution electrospinning (SES), melt electrospinning (MES), and melt electrowriting (MEW).[ 4 ] This review is directed at the latter AM technology, highlighting the different materials that have been used and provide a forward perspective on the material options for this technique.
1.1. What Is MEW?
MEW is a high‐resolution AM method with the capability to produce fibers in the micro‐ and nanometer range using a similar configuration to MES, except using different parameters and with a moving collector that facilitates direct‐writing.[ 5 ] A MEW device consists of a printing head equipped with a heating system to melt the material within a syringe, which is delivered to a metal nozzle. The MEW head is configured with a computer‐aided translating system so that direct writing onto a collector is established. A delivery system (typically air pressure) forces the extrusion of the polymer melt through the nozzle and an applied potential difference between the nozzle to the collector leads to an electrical field. While this configuration has similarities to electrospinning, there are important distinctions between MEW and MES with respect to ideal parameters, particularly the collector distance and the required electric field.[ 6 ]
Applying an electric field concentrates charges in the fluid drop at the closest point to the collector. In SES, these charges overcome the surface tension forces and a conical‐shaped jet is ejected toward the collector. The beginning of this jet and the remaining fluid at the tip of the nozzle is termed as the Taylor cone, named after Sir Geoffrey Taylor, a 20th century physicist who performed substantial fluid mechanics research including EHD phenomenon.[ 7 ] For SES, this is a rapid event with a Taylor cone generated within seconds, as soon as the applied electrical field overcomes the surface tension of the droplet at the tip of the nozzle.[ 8 , 9 ] As the jet approaches the collector for SES and MES, the surface charges increase in density to a level that secondary electrical instabilities, often called whipping, occur.[ 10 ] There are even further levels of electrical instability that stretch the jet into the nanoscale dimensions typically observed for SES.[ 8 ]
Figure 1A shows how jet initiation for MEW occurs. Similar to SES, applying an electric field results in charges aggregating at the part of the drop closest to the electric field. Polymer melts used in effective MEW, however, are typically more viscous and have less molecular species capable of charging compared to solvent‐based systems. As shown in Figure 1A, this charge concentration does not overcome surface tension and no jet is formed for over a minute. As the Taylor cone is filled with more molten polymer, the weight of the melt begins to neck from the initial droplet forming a thin column. This proceeds until the forces (gravitational and charges) draw the solidified polymer to be in contact with the collector (Figure 1A). While variations on this jet initiation can occur, such as the solidification of the polymer drop before reaching the collector, only when the polymer touches the collector can direct‐writing be commenced. The initiation of a jet within MEW depends on the mass flow rate to the nozzle, applied voltage, and collector distance, and can take in the order of several or even tens of minutes, depending on the flow rate and/or melt viscosity. The molten jet is stabilized with the voltage and the whipping that is associated with electrospinning does not occur.
Figure 1.
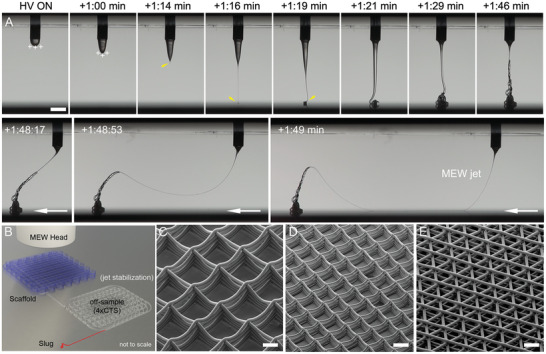
MEW jet initiation of poly(ɛ‐caprolactone) and examples of scaffolds. A) Photograph sequence with time indicated after high voltage (HV) application. When an electric field is applied to a PCL melt drop, charges aggregate at the surface of the liquid closest to the collector. As the drop increases in volume, it is distorted after +1:00 min, leading to a fluid column and jet at +1:16 min (yellow arrow). Another 30 s is required for the excess volume in the Taylor cone to be drawn to the collector, at which time (+1:48 min), the MEW jet is ready to direct‐write. B) Schematic showing how this jet initiation can be followed by a stabilization pattern off‐sample, with the slug being the collected material in the jet initiation step shown in (A). SEM images of MEW scaffolds in a box morphology using different diameter fibers of C) 20 µm and D) 10 µm and fiber spacings of 250 µm and 125 µm, respectively. E) The hexagonal laydown pattern results in a triangular morphology. A) Snapshots from a previously unpublished video kindly provided by Mr. Thomas Robinson. B) Adapted with permission under the terms of the Creative Commons Attribution‐NonCommercial License.[ 11 ] Copyright 2017 The Authors. Published by Wiley‐VCH GmbH. C–E) Reproduced with permission.[ 12 ] Copyright 2019, Mary Ann Liebert, Inc. Scale bars: (A) = 1 mm; (C)–(E) = 100 µm.
This EHD effect that prevents Plateau–Rayleigh instabilities allows the nozzle flow rate to be substantially reduced for MEW.[ 5 , 13 ] Additionally, the comparable high viscosity and low conductivity of melt fluids support the control and stability of the jet.[ 5 , 13 , 14 , 15 ] This in combination with the controllable collector plate, allows the defined deposition of the jet, which starts to solidify on the way to the collector enabling the layer‐by‐layer process.[ 5 , 13 ] Depending on the printing parameters, the resulting fiber diameter can be controlled, adjusted, and direct‐written in different configurations (Figure 1B–E).[ 11 , 16 ] The controlled fiber placement of small diameter fibers allows one to print porous structures, called scaffolds, that are used in biomedical sciences.[ 5 , 17 , 18 ]
Depending on the collector speed, the jet can be deposited as straight lines as soon as the collector speed is higher than the critical translation speed (CTS) of the polymer melt.[ 11 ] The CTS is defined as the speed at which the jet results in straight lines. This phenomenon is analogous to the liquid rope coiling of a viscous thread solution.[ 5 , 19 ]
2. Poly(ε‐caprolactone) (PCL)
Without question, the current gold standard polymer used for MEW is PCL.[ 11 , 16 ] In the literature, studies are performed with different designs including the typical box‐structured scaffolds[ 16 ] or scaffolds for cell experiments[ 20 , 21 , 22 ] to more complicated designs like sinusoid structures with horizontal layer stacking,[ 23 ] tubes,[ 24 , 25 ] or a fiber‐hydrogel composite with mechanical properties similar to that of a heart valve.[ 26 ]
2.1. Why PCL?
PCL is a semicrystalline, biodegradable polyester that is readily processable via MEW due to its low melting temperature[ 5 , 27 ] and rapid solidification.[ 28 ] It is favorable from the biomedical perspective as it is approved by the U.S. Food and Drug Administration (FDA) for certain clinical purposes[ 29 ] and offers a slow degradation rate up to several years.[ 18 , 27 , 30 ] PCL is also well‐studied in biomedical engineering as part of many other manufacturing technologies, such as SES, phase separation, and dip‐coating. Its interaction with different cell types has been widely used for tissue engineering and in the biomedical field[ 27 , 28 , 30 ] due to the availability in different molecular weights.
For MEW, the most widely studied polymer is medical‐grade PCL (PC12; Corbion, NL), which is made using good‐manufacturing practices (GMP) (Table 1 ). Medical‐grade PCL has improved purity compared to technical‐grade PCL and, in our experience, produces the highest printing quality. Table 1 demonstrates an interesting perspective for MEW, that the gold standard polymer with the most processing knowledge is one made under GMP conditions. As PCL is highlighted in several reviews on clinical translation of tissue engineering products, the transition from university‐led research to application in the clinic is often negatively impacted by the need to repeat experiments with medical‐grade polymers. In this instance, our understanding of processing via MEW is predominantly obtained with GMP‐manufactured PCL. Using medical‐grade PCL for MEW should therefore be the preferable material compared to technical‐grade PCL as the processed structures and designs have excellent control and accuracy (Figure 1).
Table 1.
Sources of linear PCL used for MEW
| Description | Properties | M w [g mol−1] (flow properties) | References |
|---|---|---|---|
| PURASORB PC12 | Medical grade |
Viscosity 1.2 dL g−1 Measured as M w = 80 kDa[ 31 ] |
[ 11 , 12 , 16 , 20 , 22 , 23 , 24 , 26 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 ] |
| Sigma Aldrich | Product number 440744[ 46 ] |
M n = 80 × 103[ 5 , 21 , 56 , 57 ] M n = 35 × 103 M w = 83 × 103[ 13 ] M n = 45 × 103[ 61 ] |
[ 5 , 13 , 21 , 56 , 57 , 58 , 59 , 60 , 61 ] |
| Capa 6400 |
M w ≈ 37 × 103 a) MFI = 40 b) |
[ 62 , 63 ] | |
| Capa 6430 |
M w ≈ 43 × 103 a) MFI = 40 b) M n = 83 kDa[ 14 ] M w = 37 kDa[ 64 ] |
[ 14 , 25 , 64 , 65 , 66 ] | |
| Capa 6500 |
M w ≈ 50 × 103 a) MFI = 7 b) M w = 50 kDa[ 64 ] |
[ 5 , 15 , 64 , 67 , 68 ] | |
| Capa 6500C | High clarity grade when molten |
MFI = 7 b) |
[ 69 , 70 ] |
| Capa 6506 | Medical grade | M w = 50 kDa[ 29 ] | [ 29 ] |
| Capa 6800 |
M w ≈ 80 × 103 a) MFI = 3 b) |
[ 71 ] | |
| CELLINK | M n = 5000[ 72 ] | [ 72 ] |
Note: Weight average molecular weight (M w); number average molecular weight (M n).
Approximate molecular weight
Typical values, melt flow index (MFI) measured with 1 in. PVC die, 2.16 kg weight, g/10 min at indicated temperature; for 6200 s at 80 °C, 6500 and 6800 s at 160 °C. Values/information provided by Perstorp CAPA brochure.
There are many studies investigating the printing parameters or phenomena using PCL[ 11 , 13 , 16 , 67 ] so as to make different scaffold designs for biomedical applications.[ 3 ] This includes the standard box pore morphology scaffolds (Figure 1C,D) or triangular pore scaffolds (Figure 1E) that are followed by understanding how this influences cell morphology and cell adhesion,[ 20 , 21 , 22 , 31 , 46 ] including mathematical modeling of pore bridging dynamics.[ 66 ] However, more recently, complex designs or further improvements for specific applications are gaining more and more interest. Some designs approached are discussed next, while an in‐depth review on MEW designs can be found elsewhere.
Importantly, MEW can be used to fabricate microstructures with tailored mechanical behavior and response. Depending on the direct‐written fibers, the structures can result in ultrastretchable scaffolds.[ 36 ] Those hexagonal scaffolds showed up to 40 times more elastic energy compared to rectangular box structures and exhibited large biaxial deformations. Furthermore, the hexagonal microstructures were used to engineer native‐like myocardial muscle.[ 36 ]
Medical‐grade PCL was used to fabricate constructs with different pattern designs similar to the wavy architecture of native collagen fibers to mimic the anisotropy and viscoelastic properties of native heart valve leaflets.[ 26 ] By changing design parameters like the radial fiber spacing, curvature degree, and number of printed layers, the fabricated constructs displayed tunable tensile properties. Furthermore, the scaffolds supported growth of human vascular smooth muscle cells and demonstrated excellent acute hydrodynamic performance under aortic physiological conditions in a custom‐made flow loop offering a promising design approach for heart valve tissue engineering.[ 26 ]
Complex designs can not only be used to improve approaches regarding their specific applications but also can change the mechanical properties or result in auxetics, which expand in transverse directions when applying tensile load (Figure 2A). Normally, constructs with a positive Poisson´s ratio shrink under tensile loading.[ 25 , 73 ] A similar approach is shown by Paxton et al.[ 25 ] fabricated MEW tubes with a negative Poisson´s ratio enabled expansion under tensile load based on a reentrant honeycomb unit cell design (Figure 2B). With this approach, the tubular scaffolds enabled a diameter increase up to 80.8%, depending on the applied tensile load, until the honeycomb design aligned with the axis of the tube.[ 25 ]
Figure 2.
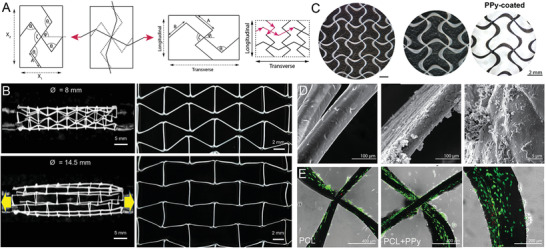
A) Schematic principle of auxetic designs with transverse expansion under tensile load and B) implemented in a tubular MEW scaffold. C) Polypyrrole (PPy) coating of auxetic patches enhancing electroconductive properties. D) Scanning electron microscope (SEM) images of neat PCL, PPy‐coated MEW fibers and E) showing Live/dead staining of neonatal cardiac fibroblasts after 48 h on PCL and PPy‐coated PCL fibers. A,C–E) Reproduced with permission.[ 58 ] Copyright 2020, Wiley‐VCH GmbH. B) Adapted under the terms of the Creative Commons CC‐BY license.[ 25 ] Copyright 2020 The Authors. Published by Elsevier Ltd.
2.2. Surface Coatings of PCL
PCL is a hydrophobic polymer and lacks bioactive cues, even though it supports cell attachment and proliferation through protein adsorption.[ 34 , 74 ] Due to the hydrophobicity, air bubbles trapped between the PCL fibers can cause handling issues especially within cell culture media. To overcome this limitation, several studies have focused on hydrophilizing the PCL fiber surface and improving properties for cells,[ 32 , 33 , 34 ] adding electroactive cues to aid cell alignment[ 60 ] or to introduce specific properties by coating with calcium phosphate (CaP)[ 31 , 51 ] or conductive materials.[ 58 , 61 ]
Olvera et al.[ 58 ] fabricated an auxetic PCL patch with fiber diameters of 80–120 µm and a fiber spacing set to 2 mm, which is designed to adapt the strains and stresses by the human myocardium and was treated with an electroconductive coating (Figure 2C,D). These patches were designed to offer the possibility of longitudinal and transverse deformations by the fabrication of missing‐rib geometry for anisotropic behavior compared to the typical box‐structured MEW scaffolds.[ 58 ] The PCL scaffolds were coated with polypyrrole (PPy) to impart the electroconductive properties for stimulating the human myocardium and those patches demonstrated cytocompatibility (Figure 2E).[ 58 ] The auxetic patches showed average values of electroconductivity of 2.51 ± 0.7 S m−1 in transverse direction and 2.07 ± 0.4 S m−1 in the longitudinal direction for a coating thickness of 0.77 ± 0.15 µm.[ 58 ]
Wang and colleagues sputter‐coated box‐structured PCL scaffolds with resulting fiber diameters of around 15 µm and fiber spacings of 200 µm with a nanolayer of gold (Au) ranging from 10 to 80 nm to enhance conductivity for the application in nerve regeneration (Figure 3A,C).[ 61 ] Due to the high flexibility and recoverability of the coated samples, the scaffolds were injectable through a needle. Furthermore, the Au coating enabled conductivity of 0.12 S cm−1 (Au thickness of 10 nm) to 1.39 S cm−1 (Au thickness of 80 nm) as demonstrated in Figure 3D and significantly increased the neurite number per cell and the neurite length, especially under electrical stimulation.[ 61 ]
Figure 3.
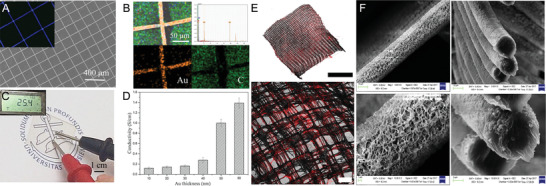
Various coating approaches for MEW scaffolds. A) SEM images of a gold (Au) coated PCL and B) EDS elemental analysis (Au in yellow and carbon in green). C) The resistance of the PCL scaffold coated with 50 nm Au and D) quantitative conductivity of the different thicknesses of Au coating. E) Confocal microscopy images of collagen‐functionalized PCL scaffolds highlighted through red staining. F) SEM images of the inorganic CaP coating deposited on PCL fibers showing the surface morphology and cross sections of the fibers. A–D) Reproduced with permission.[ 61 ] Copyright 2020, Elsevier B.V. All rights reserved. E) Reproduced with permission.[ 33 ] Copyright 2019, Wiley‐VCH GmbH. F) Reproduced with permission.[ 31 ] Copyright 2019, American Chemical Society. Scale bars: (B) = 5 mm and 200 µm and (F) = 10, 10, 1, and 3 µm, respectively.
As previously mentioned, coatings can reduce the hydrophobicity of PCL scaffolds resulting in improved handling during cell culture. Extracellular matrix (ECM) coatings on PCL are commonplace[ 75 ] and are also effective with MEW scaffolds. For example, an ECM suspension made from human decellularized adipose tissue provided an adipo‐inductive microenvironment for human bone‐marrow mesenchymal stromal cells (bmMSCs).[ 34 ] To both hydrophilize the PCL fibers and functionalize them, a hydrogel coating based on a six‐arm star‐shaped NCO‐poly(ethylene oxide‐stat‐propylene oxide) (sP(EO‐stat‐PO)) was used to prevent unspecific interactions with proteins and cells.[ 33 ] Additionally, this approach offers the possibility to covalently attach bioactive molecules resulting in a photoactivatable scaffold that enables the binding of sterically demanding molecules via postmodification irrespective of time and pH.[ 33 ] This leads to a successful collagen coating (Figure 3E) with the collagen fibrils along the PCL fibers and biofunctionalization was proven by postseeding of human MSCs showing a high cell viability after 8 d.[ 33 ]
CaP coatings are known to be hydrophilic,[ 31 , 76 ] offer higher surface areas supporting cell adhesion and subsequent cellular behavior,[ 31 , 77 ] and therefore offering promising results for bone regeneration using medical‐grade PCL scaffolds.[ 31 , 51 ] PCL fibers with diameters of 6 to 10 µm were coated with inorganic CaP and are shown in Figure 3F. The coating had no significant effect on the resulting pore size of the scaffolds or the fiber diameter but increased the surface area on the fiber.[ 31 ] Depending on the fiber spacing (250, 500, and 750 µm) within the scaffolds, as well as the fiber lay‐down pattern with offsets (30/70 and 50/50) or gradient, the scaffolds resulted in different mechanical properties with Young's modulus ranging from 0.91 ± 0.25 kPa for the 750 µm spacing to 4.34 ± 0.32 kPa for the 250 µm fiber spacing.[ 31 ]
In a different study, a cell‐accumulation and scaffold layering technique increased the thickness of medical‐grade PCL scaffolds resulting in oriented capillary‐like networks.[ 78 ] The PCL scaffolds were fibronectin‐coated and then seeded with human umbilical vein endothelial cells and, afterward, with human dermal fibroblasts incorporated in a gelatin nanofilm.[ 78 ] With this technique, the orientation of capillary‐like structures and the guidance of neovascular structures were successfully influenced into the center of the pores within the scaffolds. Furthermore, this approach should be easily applicable to other MEW‐processable thermoresponsive polymers with the advantage of being dissolvable resulting in microchannel networks.[ 78 ]
2.3. Dissolvable Channels
The embedding of particulates/fibers to later dissolve them away is one technique to introduce porosity/channels within a hydrogel[ 69 , 79 ] or polymer[ 80 ] as shown in Figure 4 . Haigh et al.[ 79 ] submersed PCL scaffolds within poly(2‐ethyl‐2‐oxazoline‐co‐2‐(3‐butenyl)‐2‐oxazoline) (PEtOx‐ButenOx) copolymer hydrogels (Figure 4A). Afterward, the PCL was dissolved with an acetone:water solution resulting in porous hydrogel structures. The dissolved channels were then back‐filled with a fluorescent dye.[ 79 ]
Figure 4.
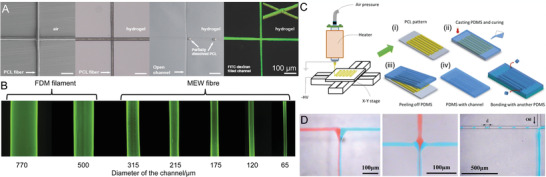
Dissolving MEW fibers to make channels. A) A series of images showing the PCL fibers embedded in a hydrogel that are then dissolved away and backfilled with fluorescent dye. B) Fluorescence images of FDM and MEW processed fibers resulting in channels with varying diameters. C) Schematic of inverse molding by curing the polymer PDMS around MEW fibers followed by delamination, for use as D) microfluidic systems. A) Reproduced with permission.[ 79 ] Copyright 2015, Wiley‐VCH GmbH. B) Reproduced under the terms of the Creative Commons CC‐BY license.[ 69 ] Copyright 2020 The Authors. Published by Wiley‐VCH GmbH. C–D) Reprinted by permission.[ 80 ] Copyright 2020, Springer Nature.
Wang and colleagues further built on this using both fused deposition modeling (FDM) and MEW to fabricate a PCL template, which was later embedded in a PEG hydrogel.[ 69 ] The swollen PEG enabled the simple removal of the PCL fibers and scaffolds (Figure 4B), then the resulting hollow structures post‐treated with cell‐adhesion peptides that allowed cell guidance throughout the channels without active perfusion.[ 69 ]
Another MEW fiber dissolution approach also demonstrates microfluidic potential (Figure 4C,D). Once PCL fibers are printed, they are embedded in poly(dimethyl siloxane) (PDMS) and dissolved away afterward.[ 80 ] Laminar flow and controlled mixing is demonstrated in this study.[ 80 ] MEW‐processed PCL was used as a dissolvable material embedded in an amorphous silica nanocomposite that is ultraviolet (UV)‐crosslinked and subsequently heated to 1300 °C.[ 39 ] Thermal debinding and sintering of the silica nanocomposite resulted in suspended hollow microstructures in fused silica glass for applications in optics and photonics, microfluidics, and lab‐on‐a‐chip.[ 39 ]
3. Materials beyond PCL
There is a general lack of well‐studied processable materials for MEW, contrasting with the many different polymers investigated and possible for SES.[ 18 , 81 ] This is an indication of the early stage of development for the process, as the electrohydrodynamic effect of jet stabilization is generally applicable to fluids. Polymer solutions can similarly be direct‐written with EHD principles,[ 82 , 83 ] and there are differences with respect to electric field and fiber placement.[ 83 ] For biomedical applications, melt processing provides an opportunity to process polymers without toxic solvents within the fabrication process. However, this comes with limitations on the types of polymers that can be processed, including those that are thermally stable. Nonetheless, several important polymers (Table 2 ) such as poly(propylene) (PP), poly(vinylidene fluoride) (PVDF), and poly(2‐oxazoline)s (POx) can be readily processed to provide different properties than MEW‐processed PCL.
Table 2.
List of MEW‐processed polymers beyond PCL showing the resulting fiber diameters and the printing parameters used
| Polymer class | Fiber diameter [µm] | Layers | Nozzle diameter [Gauge] | Temperature [°C] | Voltage [kV] | Pressure [bar] | Speed [mm min−1] | Distance [mm] | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Polyesters |
pHMGCL/PCL 40:60 |
3–7 |
25–30 200 × 200 µm 150 × 300 µm |
27 | 84 |
5–8.5 5 |
1–4 2 |
600–2400 300 |
3–5 3 |
[ 84 ] |
| Poly(urea‐siloxane)s; thermoplastic elastomer | 10.6 ± 0.6 to 19.5 ± 1.5 |
100 50 in x/y |
24 |
80–100 90 |
8–12 10 |
1–3 2 |
1500–4000 2500 |
8.5 | [ 85 ] | |
| Water‐soluble polymers | PEtOx | 8–183 | 180 | 30, 27, 25, and 23 |
200–220 210 |
3–7 4 |
1–3 2 |
200–400 |
3–7 5 |
[ 86 ] |
| Hydrogel | PEtOzi |
45 ± 5 (dry) 89 ± 12 (wet) |
10 500 µm spacing |
N/A |
130 Spinneret 150 |
2–4 | 1.5–2 | 1300–1800 | 3.3 | [ 87 ] |
| Photocurable polymers | Poly(LLA‐ε‐CL‐AC) | 24.6 ± 2.7 |
20 100 µm |
30 |
130 Spinneret 145 |
7 | 3 | 420 | 4.5 | [ 88 ] |
| Poly(ε‐CL‐AC) |
≈15–75 26.8 ± 1.9 |
10 5 in x/y 200 µm |
30 |
90 Spinneret 105 |
7 |
0.3–4 1 |
100–810 300 |
4.5 | [ 89 ] | |
| Nondegradable polymers | PP |
20–105 16.4 ± 0.2 |
10 on top (wall) 5 in x/y 200 µm |
25 |
215 Collector 70–90 80 |
6.2 |
0.5–1.0 0.5 |
25–150 750 |
3.3 ± 0.5 | [ 90 ] |
| Electroactive polymers | PVDF | 25–50 | 5 | 26 | 190 ± 2 | 2.70 ± 0.08 | 1.5–3.0 | 1800–5000 | 4.0 ± 0.5 | [ 91 ] |
| Compounding particulates | 3 and 7 wt% HAp in PCL |
16.84 ± 2.41 20.46 ± 1.09 |
190 µm | 21 | 80 | 7 | 20 mL h−1 | N/A | 10 | [ 47 ] |
| Bioactive milk proteins in PCL | ≈50 µm |
5 300 µm |
25 µm diameter | 85 | 20 | 1 | 720 | [ 29 ] | ||
| 5 wt% 45S5 BG in PLA‐PEG‐PLA/PLA 10 wt% | 31 ± 2 |
10 5 in x/y |
23 | 142 | 4 | 2 | 5000 | 3.5 | [ 92 ] | |
| SrBG in PCL | ≈100 µm |
≈8–10 ≈4–5 in x/y 1000 µm |
N/A | 55 | −6 |
3.8 0.69 0.36 |
N/A | 8 | [ 64 ] | |
| PEG and ROX in PCL | 3–10 |
0°–90° pattern 15 100 µm pore size |
45 | 95 | 2.3–2.6 kV | 0.015–0.03 | 600–1200 | 1.5 | [ 70 ] | |
3.1. Polyesters beyond PCL
While PCL is a polyester, this specific polymer has been described separately due to the substantial research already performed. Beyond pure PCL, Castilho and colleagues[ 84 ] blended a less hydrophobic material poly(hydroxymethylglycolide‐co‐ε‐caprolactone) (pHMGCL) with PCL for cardiac tissue engineering processed using MEW. The hydroxyl‐functionalized polyester pHMGCL has increased hydrophilicity compared to pure PCL,[ 93 ] and offers the possibility for further functionalization and tunable degradation rate.[ 84 , 94 ] Furthermore, the blend showed enhanced biocompatibility and improved fiber architecture stimulating cell retention and guiding cardiac cell growth.[ 84 ] The printability of pHMGCL compared to PURAC PC‐12 PCL, however, was reduced (Figure 5A). Since PCL is ductile, more elastic materials for MEW have been synthesized to overcome this limitation. For example, the MEW processability of a thermoplastic elastomer (TPE) based on an (AB) n ‐type segmented copolymer was studied.[ 85 ] This urea‐siloxane copolymer consists of hard urea units in combination with soft PDMS segments to provide a combination of typical thermoplastic polymer properties and elastomers. Materials with thermoreversible physical crosslinks were processable with MEW resulting in scaffolds and fibers with elastomeric properties. Additionally, this material could be continuously processed for 14 h at a moderate processing temperature of 90 °C. The resulting poly(urea‐siloxane) scaffolds printed with were especially well‐stacked without fiber sagging at intersecting points at 50 layers (Figure 5B) resulting in stacking ability superior to the benchmark medical‐grade PCL.[ 85 ]
Figure 5.

A–I) Scanning electron microscope (SEM) and microscope images of MEW‐processed materials beyond PCL and J–M) compounded particulates within polymers to improve the polymer properties. A) MEW‐processed polyester and B) printed thermoplastic elastomer with high printing accuracy similar to the gold‐standard material PCL. C) A printed scaffold of water‐soluble PEtOx and stereomicroscopic images of D) dry and E) swollen hydrogel PEtOzi scaffolds. SEM images of photocurable F) p(LLA‐CL‐AC) with smooth and uniform fiber surface and G) p(CL‐AC) with rougher fiber surface morphology. Typical box‐structure scaffold made of H) nondegradable PP and I) electroactive PVDF. PCL fibers including J) HAp particles and K) the fiber morphology of PLA‐PEG‐PLA blends including bioactive glass (BG) particles. L) PCL fibers with incorporated 33 wt% strontium‐substituted BG particles dissolved in chloroform and M) PCL‐PEG scaffold loaded with roxithromycin enabling antibacterial and drug‐release properties. A) Reprinted with permission.[ 84 ] Copyright 2017, Wiley‐VCH GmbH. B) Reproduced with permission.[ 85 ] Copyright 2018, Wiley‐VCH GmbH. C) Reproduced with permission.[ 86 ] Copyright 2014, the Authors. Published by Elsevier Ltd. D,E) Reproduced with permission.[ 87 ] Copyright 2020, The Royal Society of Chemistry. F) Reproduced with permission.[ 88 ] Copyright 2015, American Chemical Society. G) Reproduced with permission.[ 89 ] Copyright 2018, Elsevier Ltd. H) Reproduced with permission.[ 90 ] Copyright 2017, Elsevier B.V. I) Reproduced with permission.[ 91 ] Copyright 2018, Society of Chemical Industry. J) Reproduced with permission.[ 47 ] Copyright 2018, Elsevier Ltd. All rights reserved. K) Reproduced with permission.[ 92 ] Copyright 2017, the Authors. Published by Elsevier B.V. L) Reproduced with permission.[ 64 ] Copyright 2019, Wiley‐VCH GmbH. M) Reproduced with permission.[ 70 ] Copyright 2019, Elsevier B.V. All rights reserved. Scale bar: (A) = 300 and 100 µm, respectively.
3.2. Water‐Soluble Polymers
Another class of materials are hydrophilic polymers, often used in medical and pharmaceutical applications. One class of these polymers is POx with adjustable macromolecular structure known to be biologically compatible and for the ability as a drug delivery system. Poly(2‐ethyl‐2‐oxazoline) (PEtOx) is one type of POx with a high melting temperature between 200 and 220 °C demonstrated to be an excellent candidate for MEW as demonstrated in Figure 5C. Additionally, the sharp transition temperature for POx opens the possibility to design and adjust material properties during the polymer synthesis for thermoreversible behavior. This provides a tool for postprinting control through swelling,[ 87 ] as POx‐based polymers can be synthesized to control water solubility in temperature ranges for cell viability or to control drug release.[ 86 ]
3.3. Hydrogel MEW Fibers
Another hydrophilic polymer synthesized for MEW is poly(2‐ethyl‐2‐oxazine) (PEtOzi). This novel biomaterial ink spontaneously crosslinks via dynamic Diels–Alder click chemistry after processing as shown by Nahm and colleagues.[ 87 ] Therefore, the hydrogel can be MEW‐processed into scaffolds, which are both soft and robust with thermoreversible hydrogel characteristics in dry and swollen state (Figure 5D,E). The successful fabricated hydrogel scaffolds can be inserted into aqueous solutions and show temperature‐dependent deswelling from 2 to 80 °C by 50% of the relative area. The robust mechanical properties allowed the scaffold to be repeatedly aspirated and ejected through a canula without any visible structural damage. These properties are highly desirable for injectable scaffolds, which can be direct‐written into any essential microstructured design.[ 87 ]
3.4. Photocurable Polymers
Due to creep and the plasticizing effect of absorbed water when incorporating the fabricated constructs in an aqueous solution, the modulus of a swollen polymer can decrease.[ 95 , 96 ] Therefore, synthesizing and processing a polymer, which can be post‐crosslinked and thereby resisting the plasticizing effect in aqueous media could overcome this issue. On the basis of an already electrospun polymer poly(l‐lactide‐co‐acryloyl carbonate) (poly(LLA‐AC)),[ 95 ] which provided a high modulus in the hydrated scaffold after post‐photocrosslinking, Chen and colleagues synthesized a poly(l‐lactide‐co‐ε‐caprolactone‐co‐acryloyl carbonate) (poly(LLA‐ε‐CL‐AC)) polymer in an actual molar ratio of 60/31/9, which did slowly undergo premature crosslinking while melting.[ 88 ] However, the premature crosslinking was slow enough to enable MEW‐processing of the poly(LLA‐ε‐CL‐AC) polymer as shown in Figure 5F. The new synthesized polymer was prepared by mixing l‐lactide (LLA), a trimethylene carbonate‐based monomer (5‐methyl‐2‐oxo‐1,3‐dioxan‐5‐yl acrylate, or acryloyl carbonate (AC)) and ε‐caprolactone (ε‐CL). The ε‐CL was added to adjust the melting point required for MEW resulting in a melting onset temperature of 87 °C. Irgacure 651, a thermally stable crosslinking agent to temperatures up to 160 °C, was added to enable post‐crosslinking of the MEW‐processed scaffolds. The hydrated constructs showed an increased modulus and good stability after dynamic loading due to the successful post UV‐irradiation at room temperature.[ 88 ]
Hochleitner and colleagues[ 89 ] investigated another photopolymerizable polymer, poly(ε‐CL‐AC). Compared to the previous investigated polymer poly(LLA‐ε‐CL‐AC), poly(ε‐CL‐AC) showed considerably longer printing lifetime of up to 13 ± 7 h compared to 1.4 ± 0.7 h due to a remarkable slower crosslinking within the melt reservoir during MEW.[ 88 , 89 ] Additionally, the melting onset temperature of the polymer was reduced to 55 °C due to the absence of lactide in the polymer. With this, poly(ε‐CL‐AC) resulted in a stable and controllable printing process with homogeneous and accurate fiber placement (Figure 5G). The postprinting UV‐curing process improved the tensile strength and the Young´s modulus, as well as the elasticity and the creep resistance.[ 89 ] Therefore, the polymer was used to fabricate crimped patterns using sub‐CTS deposition of sinusoidal fibers to mimic tendon and ligament tissue as it is already shown that such morphologies are beneficial in generating fibroblast extracellular.[ 89 , 97 ]
3.5. Nondegradable Polymers
Compared to the elastic properties given by the aforementioned polymers, some applications require mechanical strength and long‐term stability of the scaffolds or implants. Therefore, another well‐known material, PP, with a long‐term stability in medical devices was tested for MEW to fabricate microscale fibers by Haigh and colleagues.[ 90 ] Using MEW to process PP offers the potential to fabricate small diameter fibers and constructs in an ordered manner (Figure 5H). However, compared to PCL and other MEW‐processed materials, PP requires a high set temperature of 215 °C to be extrudable using MEW. Together with the high viscosity, a melt flow rate of 16 g min−1 (ISO1 133 230 °C/2.16 kg), the extruded PP does not adhere to the collector, which inhibits a stable jet. Using a heated collector, as shown by other extrusion‐based techniques like FDM, enabled the fabrication of linear PP fibers and porous scaffolds.[ 90 ]
3.6. Electroactive Polymers
Another material family with remarkable properties, gaining more and more attention within the last years, are electroactive polymers (EAP). EAPs can generate electrical activity in response to deformations and are widely used in various electronic and biomedical applications.[ 98 , 99 ] PVDF‐based polymers are fluorinated electroactive materials, which are known for their remarkable ferroelectric and piezoelectric properties.[ 98 ] Compared to other piezoelectric materials such as lead zirconate titanate[ 100 ] or aluminum nitrite,[ 101 ] PVDF‐based polymers are mechanically flexible[ 98 ] and mainly processed using SES.[ 102 ]
Florczak et al.[ 91 ] processed PVDF using MEW for the first time. A processing temperature of around 170 °C was needed and a voltage of around 2.70 kV was applied.[ 91 ] The fibers were uniform in shape, had diameters in the range from 17 to 55 µm, and showed sufficient coherence resulting in stable constructs. Scaffolds were fabricated with up to five alternating layers in 0° and 90° (Figure 5I), however, higher amounts of printed layers were not possible due to lifting of the scaffold edges while printing.[ 91 ] The MEW‐processed polymer showed an increase in β‐phase content to 79% compared to unprocessed material (49%) resulting in good piezoresponse with d 33 values from 15.4 to 23.7 pm V−1.[ 91 ] The fabrication of electroactive PVDF fibers with high control over the fiber placement and design offers promising potential for the use as sensors or actuators.[ 91 ]
3.7. Compounding Particulates
For in vivo applications of PCL it is advantageous that the biodegradability can be tailored, however, due to its hydrophobic properties, there is a limitation regarding the attachment of cells.[ 64 , 103 ] Therefore, PCL can be modified by adding substitutes to design a PCL‐based material with the desired properties.
Therefore, hydroxyapatite (HAp) particles were added to medical‐grade PCL to enhance the bioactivity of MEW‐fabricated PCL scaffolds for the application in mineralized tissue reconstruction.[ 47 ] The PCL/HAp composites showed good control over the fiber placement and high stacking ability (Figure 5J). The influence of the incorporated particles on the fiber diameter was studied and showed an increasing fiber diameter with increasing amount of HAp particles. The fiber morphology of the PCL/HAp fibers was slightly rougher compared to plain medical‐grade PCL fibers and the HAp nanoparticles were uniformly distributed throughout the fibers. The resulting composite scaffolds induced higher cell growth of human osteoblasts cultured over 7 d and a significant acceleration in degradation rate compared to plain medical‐grade scaffolds.[ 47 ] In general, the PCL/HAp composites offer a promising material for MEW‐fabricated scaffolds for bone regeneration applications.[ 47 ]
Another approach to increase the bioactivity of MEW PCL scaffolds is demonstrated by Hewitt et al.[ 29 ] combining PCL with bioactive milk proteins, lactoferrin, and whey protein for skin regeneration. Printed scaffolds showed smooth and homogenous fibers.
Mixing and blending materials can easily improve limited or nonexisting material properties, as well as broaden up the range of MEW‐processable polymers. Therefore, Hochleitner and colleagues[ 92 ] used a poly(lactide‐block‐ethylene glycol‐block‐lactide) (PLA‐PEG‐PLA) triblock copolymer and blended it with low molecular weight PLA to lower the melting temperature of the degradable polymer. The PLA‐PEG‐PLA blend with 10 wt% PLA showed promising printing results regarding the shape fidelity of the scaffolds and additionally, incorporating 5 wt% solid 45S5 bioactive glass (BG) particles did not negatively influence the printability and processability (Figure 5K). This approach represents a promising novel material combination for bone tissue engineering and cell culture research.[ 92 ]
By adding strontium‐substituted bioactive glass (SrBG) to PCL, Paxton and colleagues[ 64 ] were able to combine the beneficial properties of the bioactive SrBG with the good mechanical properties and the simple processability of PCL.[ 103 , 104 ] With this approach, it was the first time that a PCL/SrBG composite was extruded and direct‐written with a SrBG concentration of 33 wt% demonstrated in Figure 5L. However, this was only possible by dissolving the PCL/SrBG 33 wt% composite in chloroform, an evaporable solvent, which reduced the viscosity of around 100 times and enabled MEW processability.[ 64 ] This study showed the advantage of combining a well‐studied MEW‐processable material with an additive resulting in the fabrication of direct‐written scaffolds using a high‐ceramic‐content polymer.[ 64 ]
Another approach to combine different materials for the application in bone repair and regeneration is shown by adding PEG, as well as roxithromycin (ROX) to PCL resulting in smooth and homogenous fibers (Figure 5M).[ 70 ] Therefore, PEG and ROX increased the hydrophilicity of the processed material and enabled effective antibacterial properties while retaining cell viability and cell growth on the scaffolds. Additionally, the fabricated scaffolds featured an initial short‐term burst release and subsequent long‐term sustained antibiotic release behavior due to the added ROX.[ 70 ]
4. Hybrid Fabrication
One general approach to improve deficiencies in scaffold fabrication is to combine different manufacturing processes overcoming limitations in any single technology. Hybrid fabrication[ 105 ] and a detailed summary of hybrid developments using MEW are given by Afghah et al.[ 106 ] Using composite materials and techniques offers promising strategies to combine advantageous properties of different materials or techniques to one design approach (Figure 6 ).
Figure 6.

Various hybrid fabrication approaches of soft network composites embedding poly(caprolactone) (PCL) fibers in A) PHEMA (core) and compressed collagen (skirt) as artificial cornea, and B) GelMA including amorphous magnesium phosphate (AMP). C) Combining different fabrication techniques like electrospinning and MEW resulting in bilayered tubular scaffolds and D) combining MEW and SES to design tight filters as E) on‐site substrate of circulating tumor cells stained for cytokeratin (magenta), DNA (blue), and CD45 (green). F) A bone–cartilage interface design using MEW of GelMA reinforced PCL and extrusion‐based printing of printable calcium phosphate (CaP). A) Reproduced with permission.[ 56 ] Copyright 2020, Elsevier Ltd. All rights reserved. B) Reproduced with permission.[ 72 ] Copyright 2020, Elsevier Ltd. All rights reserved. C) Reproduced with permission.[ 49 ] Copyright 2019, IOP Publishing Ltd. D,E) Reproduced with permission.[ 59 ] Copyright 2020, The Author(s). Published by Elsevier Ltd. F) Reproduced with permission.[ 52 ] Copyright 2020, The Author(s). Published by IOP Publishing Ltd. Scale bars: (B) = 100 µm and (F) = 50 µm.
4.1. Soft Network Composites
Soft network composites are the combination of MEW fibers and cast/bioprinted hydrogels. The synergistic mechanical reinforcement of MEW scaffolds with weak, cell‐friendly hydrogels offers the possibility to mitigate limitations regarding biomechanical properties. This is one approach to shift the biofabrication window to softer hydrogels and matrices while retaining handling or biomechanical requirements.[ 35 , 38 , 42 , 57 ]
Such composite approaches, reinforcing MEW‐fabricated PCL scaffolds into hydrogels that include gelatin methacryloyl (GelMA), show promising results regarding the stiffness increasing by up to 54‐fold.[ 42 ] In this fiber‐reinforced composite study, embedded human chondrocytes showed good viability and retained their roundish morphology, showing compatibility in terms of stiffness and biological cues for cartilage applications.[ 42 ]
To further improve and investigate the reinforcement mechanism behind the composite PCL/GelMA structures, the composites were tested under axial compression to obtain more in detail insights into the deformation process, which can help further planning for designs of the embedded MEW scaffolds.[ 35 ] Agarwal et al.[ 57 ] also embedded PCL scaffolds within a polyvinyl alcohol (PVA) matrix resulting in hybrid films with improved mechanical stability. Finally, to provide 3D neuronal cell culture models with a weak matrix microenvironment closer to the native tissue, MEW fabricated scaffolds were embedded into Matrigel.[ 38 , 41 ] Those reinforced scaffolds within the Matrigel improved the mechanical properties and therefore the handling of the delicate constructs.[ 38 , 41 ]
Another approach to mimic native tissue and its composition was demonstrated by using orthogonal microfiber scaffolds embedded into compressed collagen to design an artificial cornea.[ 56 ] Furthermore, the PCL scaffold served as an interface between the compressed collagen and a PHEMA hydrogel resulting in a core‐skirt design (Figure 6A).
To even further increase the stability of the hydrogel‐fiber composites, out‐of‐plane fibers stabilized an existing MEW wall‐like structure.[ 37 ] Those fibers were printed in sinusoids above the existing wall structure to specifically increase the shear modulus. Those composite structures were designed to better withstand mechanical load that the matrix is subjected to once implanted in the body.[ 37 ]
Similar to the previous studies, the combination of a porous MEW mesh with a soft material allows flexibility regarding the mechanical properties and additionally, for further modification by doping the hydrogels with bioactive cues. Dubey et al.[ 72 ] upgraded a GelMA hydrogel with amorphous magnesium phosphate to improve the osteogenic ability of the reinforced constructs for guided bone regeneration, shown in Figure 6B.
4.2. Combining Electrospinning with MEW
Combining different fiber orientations, diameters or even different fabrication techniques can improve the outcome of the design regarding the mechanical properties or the hierarchical design. Especially SES or MES with MEW is more frequently combined for different approaches.
Therefore, Zhang et al.[ 60 ] used SES to fabricate aligned PCL nanofibrous meshes, which were coated with gold in different thicknesses to introduce electroactive cues to the nanoscale PCL meshes. MEW was used to direct‐write microgrooves, also made of PCL on top of the coated meshes. Those hierarchically organized scaffolds were fabricated to investigate the influence of the nano‐ and microfibers in combination with the electroactive cues on myotube guidance for skeletal muscle regeneration.[ 60 ] Furthermore, the hierarchically hybrid scaffolds were rolled up to form fascicle mimicking the native bundles of parallel aligned myofibers within skeletal muscles.[ 60 ]
A hierarchically bilayered tubular scaffold was fabricated by combining the two techniques SES and MEW.[ 48 ] The inner SES layer enabled a dense fiber mesh with randomly oriented nanofibers and the outer layer was fabricated using MEW to control the microfiber placement and orientation. The tubular scaffold showed promising results directing the cell morphology and differentiation, as well as mimicking the intimal and medial layers of native vessels.[ 48 ] To further improve this approach, Pennings et al.[ 49 ] developed a bioreactor system for the coculture of endothelial colony forming cells and multipotent mesenchymal stromal cells within the hybrid tubular scaffolds described previously (Figure 6C).[ 48 ] The bioreactor induced simultaneous layer‐specific cell differentiation under shear stress. These studies showed promising results for bioengineered vascular grafts, particularly, mimicking the tunica intima and media.[ 48 , 49 ]
Hybrid scaffolds fabricated using the combination of SES and MEW can also be used for applications in cancer treatment.[ 59 ] The scaffolds made of non‐medical‐grade PCL were designed as a filter to catch circulating tumor cells within the blood, as well as the culture substrate directly on site (Figure 6D,E).[ 59 ] For the filters, different solvents were used to fabricate coiled fiber arrays by SES and on top of those tight membranes, MEW‐processed fibers were printed as a frame in the shape of typical box structures to stabilize the constructs.[ 59 ]
One strength of MEW is its solvent‐free perspective, and therefore combining with SES diminishes this standpoint. Recently, MES nanofibers with a diameter as small as 275 nm were generated by inserting an acupuncture needle in a nozzle.[ 107 ] In this way, medical‐grade PCL could be used to produce nanofibers that are of such interest for biomedical engineers. This study additionally combined this MES nozzle with MEW using a dual‐head printer, where electrospinning formed a thin barrier of nonwoven fibers and could be subsequently direct‐written upon. Therefore, a nanoscale/microscale product could be generated using GMP‐manufactured polymer on a single printer without the use of solvents.[ 107 ]
4.3. Combining Extrusion‐Based 3D Bioprinting with MEW
Soft network composites showed promising results regarding the adaptable mechanical properties and the handling of the samples. Additionally, the hydrogel can provide an improved biological environment for cells.[ 35 , 37 , 38 , 41 , 42 ] However, embedding the MEW scaffold into a gel prevents an accurate and controlled placement of the cells within the constructs. Therefore, de Ruijter et al.[ 43 ] used a simultaneous micropatterning approach by combining extrusion‐based bioprinting and MEW. 3D bioprinting enabled the accurate and controlled placement of the cell‐laden hydrogel and allowed the fabrication of more complex organized cellular structures by converging this technique with MEW.[ 43 ] GelMA including fluorescent dyes labeled equine‐derived mesenchymal stromal cells was printed into the pores of the medical‐grade PCL scaffold and the biological tolerance was demonstrated. This converged one‐step printing approach offers improved freedom in design and a defined control over cell deposition within the MEW scaffold.[ 43 ]
Diloksumpan et al.[ 52 ] used an extrusion‐based printer in combination with MEW to engineer a bone–cartilage interface. The hierarchically organized construct consisted of a bioceramic part of printable CaP‐based ink, which was deposited onto a MEW scaffold made of medical‐grade PCL to directly anchor the two components. To further mimic the chondral component, the MEW scaffold was embedded in GelMA,[ 52 ] as shown in Figure 6F.
5. Future Perspectives for MEW Printers
While the future of MEW may partly be an essential additional head for a bioprinter, there are several reasons to pursue research‐line, standalone devices. Both commercial and custom‐built MEW printers are available, with over 95% of MEW publications coming from the latter. Custom‐built systems allow specialized MEW‐processing research and provide indicators of required properties for any future MEW head. There are several different concept MEW printers described, for research perspectives such as Printomics [ 108 ] as well as scale‐up systems.
5.1. Collectors
Beyond a flat collector substrate, a cylindrical collector has been demonstrated for MEW.[ 24 , 25 , 48 , 49 , 53 , 109 , 110 ] When calculating the printing path, a current limitation of MEW is not being readily able to stop/start the jet. Therefore, a tubular shape such as shown in Figure 7A requires some mathematical calculations to generate the appropriate G‐codes for winding/translation.[ 24 , 109 ] An open‐access web‐based application “MEWTubes” allows such design of the tubes with different pore size, winding angle, and the lengths of translation, described in detail by McColl et al.[ 24 ] Similar to when performing MEW on flat collectors, the laydown pattern has a clear influence on the mechanical properties of the fabricated structures[ 24 , 109 ] and can result in auxetic tubular scaffolds.[ 25 ]
Figure 7.

SEM images of melt electrowritten scaffold designs using medical‐grade PCL: Different complex architectures include A) tubular constructs, including B) anatomically relevant tubes such as the aortic root model, C–F) microscale layer shifting and tilting due to increasing amplitudes resulting in fiber walls, triangle‐shaped pores, circular pores, and horizontal fibers. A) Reproduced with permission.[ 24 ] Copyright 2018 The Authors. Published by Elsevier Ltd. B) Reproduced under the terms of the Creative Common CC‐BY license.[ 54 ] Copyright 2020, the Authors. Published by Frontiers Media S.A. C–F) Reproduced under the terms of the Creative Common CC‐BY license.[ 23 ] Copyright 2020, the Authors. Published by Wiley‐VCH GmbH.
As previously discussed, the collector and its properties have a significant role for MEW to enable polymer processability and to fabricate constructs with accurate fiber placement even on tubular collectors. Since MEW is based on the EHD principle stabilizing the polymer jet[ 6 ] and therefore enabling the fabrication of fibers in the micrometer range due to a constant electrical field,[ 50 ] it also limits the fabrication of uniform fibers onto more complex, nonuniform collectors.[ 54 , 55 ] This technical boundary is due to the interaction of the collector with the EHD working principle leading to jet instabilities and limiting the accurate fiber placement.[ 54 , 55 ] One approach to overcome these limitations is shown by using a hybrid collector consisting of an aluminum (Al) mandrel in combination with a 3D‐printed PLA tube.[ 54 ] Compared to a full Al mandrel and a full conductive combination of Al and titanium, the Al‐PLA setup enabled a consistent electrical field strength. Using the double‐component collector consisting of a conductive and a nonconductive material combination, the PLA part collected the deposited fibers without causing electrical field disruptions and furthermore, enabled to possibility to fabricate patient‐specific designs due to the advantage of being 3D printed (Figure 7B).[ 54 ] Using this collector approach, the fabrication of an aortic wall including three aortic sinuses was possible and proved that MEW can be used to fabricate nonuniform complex designs.[ 54 ]
Another approach to print medical‐grade PCL in anatomical relevant structures with nonflat (wedge‐ and curved‐shaped) geometries was demonstrated with different collector substrates for nonconductive materials like magnesium phosphate (MgP) and PCL, as well as on conductive materials like GelMA and Al.[ 55 ] Comparing the different collector substrates, the conductive materials did result in bigger fiber diameters and higher scaffolds, as well as, for the first layer, a more flattened (ellipsoidal) fiber shape. Using computational stimulations, it was possible to relate these findings to a higher electrical field strength for conductive collecting materials compared to the nonconductive materials.[ 55 ] Furthermore, fiber deposition with high accuracy onto nonflat collectors was possible with a z‐correction in the printhead trajectory keeping the distance between the collector and the printhead, as well as the electrical field strength constant.[ 55 ]
The accuracy of MEW is also dependent on the stage quality, especially the axis precision.[ 90 ] Additionally, remaining charges as well as electrostatic attraction of the fibers is restricting the design and the result of MEW‐processed constructs. Therefore, understanding the principle of the jet formation, the influences on it and by controlling the jet lag for each layer while printing, inaccuracies can be corrected enabling control over fiber placement and horizontal layer stacking.[ 23 ] Medical‐grade PCL has been processed into complex structures with overhangs, wall texturing, and branching by introducing vertical layer shifting (Figure 7C,F).[ 23 ] Using those approaches, mechanical properties can be tailored by changing the scaffold design introduced by microscale layer shifting and furthermore, opening a new possibility to even more complex constructs.[ 23 ] Such shifts in printing path are as low as 6 µm, therefore stages are required that can deliver such precision.
5.2. Delivery of Melt to the Nozzle
Some of the first MEW printers used a piston attached to a syringe pump to control the melt flow to the nozzle.[ 5 , 14 ] More recently, air (or nitrogen) pressure has been used for pressurizing the syringe for melt flow with improved startup times. Furthermore, the use of digitally controlled pressure valves allows controlled alteration of air pressure, resulting in the capability to change the fiber diameter “on the fly” during a print. It is notable that there are instances when a higher pressure (than 5 bar) is required to achieve extrusion to the nozzle,[ 64 ] or lower pressures are needed to obtain small diameter fibers for MEW.[ 11 ]
Using the pressure to control fiber diameter, Hrynevich et al.[ 11 ] showed how variable diameters can be generated in a single print, through controlling the collector speed and applied pressure. The fabricated PCL scaffolds were adapted for spheroid seeding to develop a sheet‐like tissue scaffold including multicellular spheroids. The design was based on the typical box‐structured scaffolds, but included two 7 µm diameter catching fibers close to the bottom of the scaffold, to prevent spheroids passing through during seeding and cultivation.[ 11 , 40 ] This tissue‐sheet construct enabled simultaneous high spheroid seeding efficiency with ease of handling, high cell viability, and successful differentiation of the spheroids.[ 40 ]
5.3. Thermal Requirements
When considering polymers beyond PCL, an increased operation temperature is one significant area that is a prerequisite for many of the polymers highlighted in Table 2 and Figure 5. Melt processing temperatures above 100 °C are commonplace in other technologies and there are important engineering polymers that require processing at 200 °C and higher. There are three general approaches for heating the MEW/MES jet: 1) electrical, 2) circulating fluids/air (Figure 8A), and 3) infrared lasers. Both circulating air and infrared lasers have advantages in both upper temperature limits achievable and speed of jet initiation.[ 111 ] The most common approach, however, is the electrical heater (Figure 8B), designed to be isolated from the high voltage supply. Alternatively, a high voltage can be applied to the collector and the heating nozzle grounded. The implications of such voltage configurations on the fiber diameter, stacking, pulsing, and the CTS are still not fully elucidated.
Figure 8.
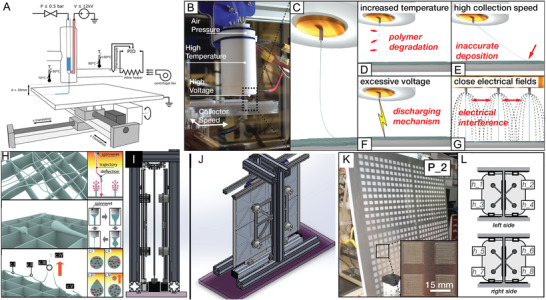
Overview of MEW printers. A) Schematic illustration of a MEW printer using an industrial heat gun as hot air stream supply, heating the polymer melt while the jet remains in ambient temperature conditions. B) Images of typical single material MEW head and C) the resulting polymer jet. There are limitations during fiber generation resulting from D) polymer degradation due to high temperatures, E) high printing speeds resulting in inaccurate fiber deposition, F) excessive voltage causing arcing, and G) electrical interference between close electrical fields. H) Schematic of fiber printing defects as increased build height due to electrostatic repulsion and/or attraction of the polymer melt, fiber pulsing, or fiber attraction toward the nozzle. I–J) CAD models showing different views of the high‐throughput MEW printer using eight simultaneous printheads extruding onto a vertical collector. K) Images of one side of the vertical collector with 576 scaffolds and L) the labeling of the eight printheads on both, the left and right side. A) Reproduced with permission.[ 21 ] Copyright 2018, Elsevier Ltd. All rights reserved. B–G) Reproduced with permission.[ 44 ] Copyright 2019, Mary Ann Liebert, Inc., publishers. H) Reproduced with permission.[ 50 ] Copyright 2018, Wiley‐VCH GmbH. I–L) Reproduced with permission.[ 44 ] Copyright 2019, Mary Ann Liebert, Inc.
Similar to FDM, MEW extrudes a polymer melt through a small nozzle generating layer‐by‐layer constructs with partially fiber fusion. However, the cooling down of the polymer within a specific shape can cause internal stresses leading to deformations (called warping) especially in corners.[ 112 ] Furthermore, when a substantial temperature difference between the nozzle and the ambient air is used, build plate adhesion issues are commonplace for FDM printing and can result in poor fiber adhesion to the collector. To overcome this issue, a heated collector has been shown to be necessary to achieve fiber collection. When processing PP with a spinneret temperature of 215 °C, for example, a heated collector of 70 °C is needed for adhesion of the fibers.[ 90 ] Ideally, controlling all aspects of the heating profile including nozzle, syringe, ambient temperatures, and heated collector would ensure control over jet processing, fiber adhesion, and fiber–fiber adhesion with the latter, having a substantial influence on the mechanical properties of the printed material. While an ideal ambient humidity of 25–45% r.h. at room temperature allows sample consistency, this level can be affected by seasonal changes.
5.4. Dynamic Electric Fields
MEW‐processed scaffolds are quite limited in thickness and volume when static collector distances and voltages are used, due to residual charges within the printed fibers that remain with increasing build height similar to the principles previously discussed for nonuniform collectors. Wunner and colleagues[ 50 ] investigated the underlying physical principles to overcome the current limitation regarding the thickness of the scaffolds, and identified a solution to attain thick (7 mm) MEW scaffolds. Since MEW requires an electrical field to form a stable jet, using a fixed voltage and collector distance between the printhead and the translating collector results in several complications. The resulting scaffolds are limited in the accurate fiber placement with increasing height due to the accumulation of excess charge (Figure 8H). These charges remaining in the deposited fibers result in attraction of the fibers back to the heated head and distorting of the scaffold architectures.[ 50 ] Maintaining the electrostatic force at a constant level during the print by an adjustable working distance between collector and printhead, and an increase of the voltage, a scaffold with a height of 7 mm was fabricated.[ 50 ] MEW printers therefore require the ability to digitally adjust both the collector distance and applied voltage to obtain thicker scaffolds.
5.5. Imaging and Visualization: Toward Digitization
While different AM technologies have their strengths and weaknesses, MEW has extremely good advantageous visual access to important monitoring information. While real‐time visual information from electron beam melting, stereolithography, or even FDM is of limited value, the MEW nozzle raised several millimeters above the collector provides an excellent view of the electrified jet. This information including jet angle provides process stability information and is relatively simple to collect with digital cameras (Figure 1A).[ 108 ] Such camera information and its subsequent digital processing was used to describe Printomics, the analysis of MEW printing parameters, through digital control and processing.[ 108 ] In this concept MEW printer, an inexpensive USB camera combined with a simple objective microscope was used to provide the jet angle and fiber diameter information, respectively. The lag in the jet is especially important for predictive fiber placement. Jin et al.[ 71 ] further established a model based on a reversed speed planning process to compensate the lag effect, which limits the accurate fiber placement depending on the printing speed using a high molecular weight PCL.
5.6. Scale‐Up Systems
An increasing number of AM technologies are used to fabricate 3D scaffolds for cell culture experiments especially for the application in tissue engineering and regenerative medicine. MEW, therefore, was used to produce scaffolds for researchers consisting of ordered fibers with micrometer size in diameter. However, scale‐up productions, which are necessary to effectively translate to industrial applications, are only commencing[ 44 ] and require uniform jet formation across different nozzles. Figure 8I–L shows a MEW system including eight simultaneously extruding printheads, which enabled to fabricate large‐scale structures based on printing parameters given from a single‐head system.[ 44 ] Scaffolds with identical morphologies and fiber diameters were successfully fabricated in dimensions up to 78 × 78 cm.[ 44 ] To establish a MEW printer ecosystem, which can be easily upscaled, the collector of the system was transferred to the vertical mounting.[ 44 ] Previous research by Wunner et al.[ 45 ] demonstrated the possibility to perform MEW printing from all directions (horizontal, lateral, and upside down) without affecting the fiber jet.[ 44 , 45 ]
5.7. Future Perspectives for Processable Materials
Exploring the compatibility of new polymers with MEW has only recently commenced, and improvements in printer designs and expansion into applications beyond biomedical will enhance this perspective. The capability of MEW printers is inextricably linked to exploring new polymers, however, based on our fundamental understanding of jet instabilities, it is possible to look forward and identify the properties of fluids that will be compatible with the process.
Fundamentally, fluid columns exist under many circumstances; a simple example is when water comes out of a faucet.[ 113 ] With unlimited space to fall, such a contiguous fluid column eventually breaks due to Plateau–Rayleigh instabilities (Figure 9A), where perturbations in the fluid column result in the formation of a discontinuous phase (Figure 9B).[ 114 ] Important and well‐known considerations that influence when this fluid break up include the flow rate, the shape of the outlet, and the viscosity of the fluid used.[ 113 , 115 ] When these fluids land on a solid collector, they buckle and form patterns depending on various parameters (Figure 9C–E).[ 115 , 116 ]
Figure 9.

Fluid jets and their breakup or stabilization. A) Photograph and schematic of a water jet that undergoes perturbations (red arrow) as part of Plateau–Rayleigh instabilities and prior to jet breakup. B) Photograph of 49 jets at different stages of Plateau–Rayleigh instabilities. Photographs of “liquid rope” coiling of C) honey and D) molten glass landing on a static collector with E) showing a render of how such fluid columns buckle and land when direct‐written. Photograph of F) a floating water bridge, where deionized water is suspended between two beakers using the application of a voltage, and G) from an infrared camera of a PCL MEW jet landing on a stationary collector and H) a photograph of a MEW jet, showing how the applied voltage can prevent this jet from undergoing Plateau–Rayleigh instabilities, even at low flow rates. A,B) Reproduced with permission.[ 114 ] Copyright 2010, AIP Publishing. C) Reproduced under the terms of the Creative Commons Agreement 4.0 (CC BY).[ 117 ] Copyright 2018, the Authors. Published by MDPI. D) Reproduced with permission.[ 116 ] Copyright 2017, The Royal Society. E) Reprinted figure with permission.[ 115 ] Copyright 2015, American Physical Society. F) Reproduced with permission.[ 118 ] Copyright 2010, AIP Publishing. G) Previously unpublished image provided by Mr. Andrei Hrynevich. H) Reproduced with permission.[ 108 ] Copyright 2019, IOP Publishing.
It is lesser known that applying a potential difference across this fluid column can suppress the Plateau–Rayleigh instabilities. Figure 9D shows how a column of deionized water can be horizontally suspended in mid‐air through the application of a voltage. This floating water bridge shows that charged species at the surface of the fluid column prevents local perturbations and therefore suppress fluid breakup.[ 118 ] This EHD effect was also shown by Sir Geoffrey Taylor in the 1960s[ 7 ] and is described in detail elsewhere in the context of MEW.[ 6 ] This is distinct to electrospraying water where the voltage is increased to such a high level that surface charges overcome surface tension.[ 119 ] It is these two EHD effects that resulted in the distinction between MES (where electrical instabilities in the jet are deliberately initiated) and MEW (Figure 9G). Once a fluid column is stabilized by a voltage, it can persist at low flow rates (Figure 9H) depending on the fluid properties.
When selecting a fluid to use for direct‐writing jetting in general, much can be learned from our understanding how noncharged fluid columns behave (Figure 9C,D). As previously mentioned, the flow rate to the nozzle is a significant indicator of whether a contiguous column is formed, as is the height of release. Higher flow rates can establish a fluid column, however of greater importance to the additive manufacturing community are smaller diameter fibers that result from low flow rates. Research on Plateau–Rayleigh instabilities is often performed with water/glycerol blends, where the viscosity can be substantially increased with minimal changes in surface tension.[ 113 ]
Certain fluids form contiguous columns at low flow rates, for example, honey, treacle, and certain oils.[ 120 ] Central to all of these materials are long polymer chains that result in macromolecular entanglement, which is also a commonality in all MEW‐processed polymers to date. The localized increased viscosity on the jet (which also increases during cooling) minimizes jet perturbations, especially at low flow rates. For MEW to achieve a small fiber diameter, an especially low flow rate is required—measured at 2.6 µL h−1 for 2–3 µm diameter fibers.[ 107 ] Therefore, low viscosity fluids are difficult to MEW process unless increased flow rates (and therefore diameters) are adopted.
The use of heat for MEW processing can also affect the fluid properties, both physical and chemical. The residency of a polymer at elevated temperatures is amplified by the need for low flow rates to achieve small diameter fibers. The heat applied to the melt can have 1) no effect on the melt, 2) initiate crosslinking, or 3) cause thermal degradation, depending on the chemical nature of the polymer used, and the level of heat required to achieve molten flow through the nozzle. For biologically derived polymers, denaturation[ 121 ] is an issue that needs to be overcome or avoided in future printing iterations.
The quantity of charged species within the polymer melt will also affect the printing properties of the polymer. As certain classes such as conductive polymers are MEW‐processed, the printer configurations are expected to change accordingly. Also observed with noncharged fluid columns, the diameter of the nozzle affects the resulting thinning of the jet—this effect was also observed in a previous MES paper.[ 122 ]
Understanding Plateau–Raleigh instabilities will enhance the selection of new polymers for MEW and excellent research studies and reviews on this phenomenon are found elsewhere. It is important to appreciate that, unlike electrospinning, MEW uses the applied voltage to stabilize the jet and to prevent it from breaking up droplets as known from noncharged falling fluids.[ 6 , 7 ] In combination with the significant lower flow rates and the smaller distance between collector and nozzle, it is essential that Plateau–Raleigh instabilities are prevented so that controlled direct‐writing can be achieved.[ 6 ]
Since the EHD effect of preventing fluid column breakup is general to fluids, it is expected that many fluids will work in this manner, although the lower dimensions achieved will be dependent on their macromolecular interactions. The minimum operating temperature is also crucial, as this affects the degradation profile (if any) and is one reason why the development of new polymers needs to go hand‐in‐hand with improvements to MEW printers. The temperature is important when delivering highly viscous fluids through small diameter nozzles can therefore pose problems in achieving certain flow rates. It is expected that over the next five to ten years, however, researchers will dramatically expand the number of polymers processable and enhance their specific properties with some of the design possibilities also outlined here.
6. Conclusion
While the vast majority of publications on MEW research use PCL, there is a growing number of studies demonstrating that many other polymers can be processed with this technique. Keeping in line with a biomedical application perspective, most other studies are performed with various chemical permutations of hydrolytically degradable polyesters. Furthermore, surface coatings of fabricated fibers and compounding additives into the melt phase are two strategies used to impart further function into the final printed structure. Beyond biomedical applications, other fields such as microfluidics,[ 80 ] optic guides,[ 123 ] or pressure sensors[ 124 ] are taking advantage of this high‐resolution AM technology. Expanding the number of processable polymers for MEW will also require improvements to current printers, which can lack the added features that aid in this type of processing. However, based on the current trends and the general interest in EHD‐assisted AM, we expect many more polymers will be processed via MEW.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
Proofreading by Dr. Biranche Tandon is greatly appreciated. This work was supported by funding from the Volkswagen Stiftung (Grant No. 93417). Open access funding enabled and organized by Projekt DEAL.
Biographies
Juliane C. Kade received her M.Sc. in biofabrication at the University of Würzburg, Germany, as part of the international double Master's degree with University of Wollongong, Australia. She has developed 3D printing approaches and bioinks for cartilage regeneration and is a Ph.D. candidate at the University Clinic Würzburg working on establishing new polymers for melt electrowriting in the field of biomedical materials.

Paul D. Dalton is a pioneer of melt electrowriting and an early adopter of melt electrospinning, for uses within the biomedical sciences. His career research philosophy is to develop biomaterial processing technologies within the university setting, which improve opportunities for translation to the clinic. A professor at the University of Würzburg, Germany, he has over 25 years’ experience working in biomaterials, tissue engineering, and biofabrication.

Kade J. C., Dalton P. D., Polymers for Melt Electrowriting. Adv. Healthcare Mater. 2020, 10, 2001232. 10.1002/adhm.202001232
References
- 1. Henkel J., Schantz J. T., Hutmacher D. W., Osteologie 2013, 22, 180. [Google Scholar]
- 2. Mao M., He J., Li X., Zhang B., Lei Q., Liu Y., Li D., Micromachines 2017, 8, 113. [Google Scholar]
- 3. Hutmacher D. W., Woodfield T. B. F., Dalton P. D., in Tissue Engineering (Eds: van Blitterswijk C., De Boer J.), Elsevier, Amsterdam: 2014, p. 311. [Google Scholar]
- 4. Brown T. D., Edin F., Detta N., Skelton A. D., Hutmacher D. W., Dalton P. D., Mater. Sci. Eng., C 2014, 45, 698. [DOI] [PubMed] [Google Scholar]
- 5. Brown T. D., Dalton P. D., Hutmacher D. W., Adv. Mater. 2011, 23, 5651. [DOI] [PubMed] [Google Scholar]
- 6. Dalton P. D., Curr. Opin. Biomed. Eng. 2017, 2, 49. [Google Scholar]
- 7. Taylor G. I., Van Dyke M. D., Proc. R. Soc. London, Ser. A 1969, 313, 453. [Google Scholar]
- 8. Reneker D. H., Yarin A. L., Polymer 2008, 49, 2387. [Google Scholar]
- 9.a) Yarin A. L., Koombhongse S., Reneker D. H., J. Appl. Phys. 2001, 90, 4836; [Google Scholar]; b) Vaseashta A., Appl. Phys. Lett. 2007, 90, 093115. [Google Scholar]
- 10.a) Han T., Reneker D. H., Yarin A. L., Polymer 2007, 48, 6064; [Google Scholar]; b) Shin Y. M., Hohman M. M., Brenner M. P., Rutledge G. C., Appl. Phys. Lett. 2001, 78, 1149; [Google Scholar]; c) Fridrikh S. V., Yu J. H., Brenner M. P., Rutledge G. C., Phys. Rev. Lett. 2003, 90, 144502. [DOI] [PubMed] [Google Scholar]
- 11. Hrynevich A., Elçi B. Ş., Haigh J. N., McMaster R., Youssef A., Blum C., Blunk T., Hochleitner G., Groll J., Dalton P. D., Small 2018, 14, 1800232. [DOI] [PubMed] [Google Scholar]
- 12. Youssef A., Hrynevich A., Fladeland L., Balles A., Groll J., Dalton P. D., Zabler S., Tissue Eng., Part C 2019, 25, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hochleitner G., Jüngst T., Brown T. D., Hahn K., Moseke C., Jakob F., Dalton P. D., Groll J., Biofabrication 2015, 7, 035002. [DOI] [PubMed] [Google Scholar]
- 14. Farrugia B. L., Brown T. D., Upton Z., Hutmacher D. W., Dalton P. D., Dargaville T. R., Biofabrication 2013, 5, 025001. [DOI] [PubMed] [Google Scholar]
- 15. Mota C., Puppi D., Gazzarri M., Bártolo P., Chiellini F., Polym. Int. 2013, 62, 893. [Google Scholar]
- 16. Hochleitner G., Youssef A., Hrynevich A., Haigh J. N., Jungst T., Groll J., Dalton P. D., BioNanoMaterials 2016, 17, 159. [Google Scholar]
- 17. Paxton N. C., Lanaro M., Bo A., Crooks N., Ross M. T., Green N., Tetsworth K., Allenby M. C., Gu Y., Wong C. S., Powell S. K., Woodruff M. A., J. Mech. Behav. Biomed. Mater. 2020, 105, 103695. [DOI] [PubMed] [Google Scholar]
- 18. Robinson T. M., Hutmacher D. W., Dalton P. D., Adv. Funct. Mater. 2019, 29, 1904664. [Google Scholar]
- 19. Blount M. J., Lister J. R., J. Fluid Mech. 2011, 674, 489. [Google Scholar]
- 20. Fuchs A., Youssef A., Seher A., Hochleitner G., Dalton P. D., Hartmann S., Brands R. C., Müller‐Richter U. D. A., Linz C., BMC Oral Health 2019, 19, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eichholz K. F., Hoey D. A., Acta Biomater. 2018, 75, 140. [DOI] [PubMed] [Google Scholar]
- 22. Tylek T., Blum C., Hrynevich A., Schlegelmilch K., Schilling T., Dalton P. D., Groll J., Biofabrication 2020, 12, 025007. [DOI] [PubMed] [Google Scholar]
- 23. Liashenko I., Hrynevich A., Dalton P. D., Adv. Mater. 2020, 32, 2001874. [DOI] [PubMed] [Google Scholar]
- 24. McColl E., Groll J., Jungst T., Dalton P. D., Mater. Des. 2018, 155, 46. [Google Scholar]
- 25. Paxton N. C., Daley R., Forrestal D. P., Allenby M. C., Woodruff M. A., Mater. Des. 2020, 193, 108787. [Google Scholar]
- 26. Saidy N. T., Wolf F., Bas O., Keijdener H., Hutmacher D. W., Mela P., De‐Juan‐Pardo E. M., Small 2019, 15, 1900873. [DOI] [PubMed] [Google Scholar]
- 27. Charuchinda A., Molloy R., Siripitayananon J., Molloy N., Sriyai M., Polym. Int. 2003, 52, 1175. [Google Scholar]
- 28. Mondal D., Griffith M., Venkatraman S. S., Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 255. [Google Scholar]
- 29. Hewitt E., Mros S., McConnell M., Cabral J., Ali A., Biomed. Mater. 2019, 14, 055013. [DOI] [PubMed] [Google Scholar]
- 30. Bartnikowski M., Dargaville T. R., Ivanovski S., Hutmacher D. W., Prog. Polym. Sci. 2019, 96, 1. [Google Scholar]
- 31. Abbasi N., Abdal‐hay A., Hamlet S., Graham E., Ivanovski S., ACS Biomater. Sci. Eng. 2019, 5, 3448. [DOI] [PubMed] [Google Scholar]
- 32. Bas O., D'Angella D., Baldwin J. G., Castro N. J., Wunner F. M., Saidy N. T., Kollmannsberger S., Reali A., Rank E., De‐Juan‐Pardo E. M., Hutmacher D. W., ACS Appl. Mater. Interfaces 2017, 9, 29430. [DOI] [PubMed] [Google Scholar]
- 33. Bertlein S., Hochleitner G., Schmitz M., Tessmar J., Raghunath M., Dalton P. D., Groll J., Adv. Healthcare Mater. 2019, 8, 1801544. [DOI] [PubMed] [Google Scholar]
- 34. Blum C., Schlegelmilch K., Schilling T., Shridhar A., Rudert M., Jakob F., Dalton P. D., Blunk T., Flynn L. E., Groll J., ACS Biomater. Sci. Eng. 2019, 5, 6655. [DOI] [PubMed] [Google Scholar]
- 35. Castilho M., Hochleitner G., Wilson W., van Rietbergen B., Dalton P. D., Groll J., Malda J., Ito K., Sci. Rep. 2018, 8, 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castilho M., van Mil A., Maher M., Metz C. H. G., Hochleitner G., Groll J., Doevendans P. A., Ito K., Sluijter J. P. G., Malda J., Adv. Funct. Mater. 2018, 28, 1803151. [Google Scholar]
- 37. de Ruijter M., Hrynevich A., Haigh J. N., Hochleitner G., Castilho M., Groll J., Malda J., Dalton P. D., Small 2018, 14, 1702773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Janzen D., Bakirci E., Wieland A., Martin C., Dalton P. D., Villmann C., Adv. Healthcare Mater. 2020, 9, 1901630. [DOI] [PubMed] [Google Scholar]
- 39. Kotz F., Risch P., Arnold K., Sevim S., Puigmartí‐Luis J., Quick A., Thiel M., Hrynevich A., Dalton P. D., Helmer D., Rapp B. E., Nat. Commun. 2019, 10, 1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMaster R., Hoefner C., Hrynevich A., Blum C., Wiesner M., Wittmann K., Dargaville T. R., Bauer‐Kreisel P., Groll J., Dalton P. D., Blunk T., Adv. Healthcare Mater. 2019, 8, 1801326. [DOI] [PubMed] [Google Scholar]
- 41. Schaefer N., Janzen D., Bakirci E., Hrynevich A., Dalton P. D., Villmann C., Adv. Healthcare Mater. 2019, 8, 1801226. [DOI] [PubMed] [Google Scholar]
- 42. Visser J., Melchels F. P. W., Jeon J. E., van Bussel E. M., Kimpton L. S., Byrne H. M., Dhert W. J. A., Dalton P. D., Hutmacher D. W., Malda J., Nat. Commun. 2015, 6, 6933. [DOI] [PubMed] [Google Scholar]
- 43. de Ruijter M., Ribeiro A., Dokter I., Castilho M., Malda J., Adv. Healthcare Mater. 2019, 8, 1800418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wunner F. M., Eggert S., Maartens J., Bas O., Dalton P. D., De‐Juan‐Pardo E. M., Hutmacher D. W., 3D Print. Addit. Manuf. 2019, 6, 82. [Google Scholar]
- 45. Wunner F. M., Maartens J., Bas O., Gottschalk K., De‐Juan‐Pardo E. M., Hutmacher D. W., Mater. Lett. 2018, 216, 114. [Google Scholar]
- 46. Gwiazda M., Kumar S., Świeszkowski W., Ivanovski S., Vaquette C., J. Mech. Behav. Biomed. Mater. 2020, 104, 103631. [DOI] [PubMed] [Google Scholar]
- 47. Abdal‐hay A., Abbasi N., Gwiazda M., Hamlet S., Ivanovski S., Eur. Polym. J. 2018, 105, 257. [Google Scholar]
- 48. Jungst T., Pennings I., Schmitz M., Rosenberg A. J. W. P., Groll J., Gawlitta D., Adv. Funct. Mater. 2019, 29, 1905987. [Google Scholar]
- 49. Pennings I., van Haaften E. E., Jungst T., Bulsink J. A., Rosenberg A. J. W. P., Groll J., Bouten C. V. C., Kurniawan N. A., Smits A. I. P. M., Gawlitta D., Biofabrication 2019, 12, 015009. [DOI] [PubMed] [Google Scholar]
- 50. Wunner F. M., Wille M.‐L., Noonan T. G., Bas O., Dalton P. D., De‐Juan‐Pardo E. M., Hutmacher D. W., Adv. Mater. 2018, 30, 1706570. [DOI] [PubMed] [Google Scholar]
- 51. Dondossola E., Alexander S., Holzapfel B. M., Filippini S., Starbuck M. W., Hoffman R. M., Navone N., De‐Juan‐Pardo E. M., Logothetis C. J., Hutmacher D. W., Friedl P., Sci. Transl. Med. 2018, 10, eaao5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Diloksumpan P., de Ruijter M., Castilho M., Gbureck U., Vermonden T., van Weeren P. R., Malda J., Levato R., Biofabrication 2020, 12, 025014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Black C., Kanczler J. M., de Andrés M. C., White L. J., Savi F. M., Bas O., Saifzadeh S., Henkel J., Zannettino A., Gronthos S., Woodruff M. A., Hutmacher D. W., Oreffo R. O. C., Biomaterials 2020, 247, 119998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saidy N. T., Shabab T., Bas O., Rojas‐González D. M., Menne M., Henry T., Hutmacher D. W., Mela P., De‐Juan‐Pardo E. M., Front. Bioeng. Biotechnol. 2020, 8, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peiffer Q. C., de Ruijter M., van Duijn J., Crottet D., Dominic E., Malda J., Castilho M., Mater. Des. 2020, 195, 109025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J., Chen Y., Bai Y., Quan D., Wang Z., Xiong L., Shao Z., Sun W., Mi S., Exp. Eye Res. 2020, 195, 108037. [DOI] [PubMed] [Google Scholar]
- 57. Agarwal K., Sahay R., Baji A., Polymers 2020, 12, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Olvera D., Molina M. S., Hendy G., Monaghan M. G., Adv. Funct. Mater. 2020, 1909880. [Google Scholar]
- 59. Jørgensen M. L., Müller C., Sikkersoq M., Nadzieja M., Zhang Z., Su Y., Just J., Spindler K.‐L. G., Chen M., Mater. Today Bio 2020, 6, 100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y., Zhang Z., Wang Y., Su Y., Chen M., Mater. Sci. Eng., C 2020, 116, 111070. [DOI] [PubMed] [Google Scholar]
- 61. Wang Y., Zhang Y., Zhang Z., Su Y., Wang Z., Dong M., Chen M., Colloids Surf., B 2020, 195, 111210. [DOI] [PubMed] [Google Scholar]
- 62. Powell S. K., Ristovski N., Liao S., Blackwood K. A., Woodruff M. A., Momot K. I., 3D Print. Addit. Manuf. 2014, 1, 95. [Google Scholar]
- 63. Dayan C. B., Afghah F., Okan B. S., Yıldız M., Menceloglu Y., Culha M., Koc B., Mater. Des. 2018, 148, 87. [Google Scholar]
- 64. Paxton N. C., Ren J., Ainsworth M. J., Solanki A. K., Jones J. R., Allenby M. C., Stevens M. M., Woodruff M. A., Macromol. Rapid Commun. 2019, 40, 1900019. [DOI] [PubMed] [Google Scholar]
- 65. Ristovski N., Bock N., Liao S., Powell S. K., Ren J., Kirby G. T. S., Blackwood K. A., Woodruff M. A., Biointerphases 2015, 10, 011006. [DOI] [PubMed] [Google Scholar]
- 66. Buenzli P. R., Lanaro M., Wong C. S., McLaughlin M. P., Allenby M. C., Woodruff M. A., Simpson M. J., Acta Biomater. 2020, 114, 285. [DOI] [PubMed] [Google Scholar]
- 67. Ding H., Cao K., Zhang F., Chang R. C., Mater. Des. 2019, 178, 107857. [Google Scholar]
- 68. Tourlomousis F., Babakhanov A., Ding H., Chang R. C., in Proc. 2015 Manufacturing Science and Engineering, ASME, New York: 2015, pp. 1–11. [Google Scholar]
- 69. Wang S., Sarwat M., Wang P., Surrao D. C., Harkin D. G., St John J. A., Bolle E. C. L., Forget A., Dalton P. D., Dargaville T. R., Macromol. Rapid Commun. 2020, 41, 2000295. [DOI] [PubMed] [Google Scholar]
- 70. Bai J., Wang H., Gao W., Liang F., Wang Z., Zhou Y., Lan X., Chen X., Cai N., Huang W., Tang Y., Int. J. Pharm. 2020, 576, 118941. [DOI] [PubMed] [Google Scholar]
- 71. Jin Y., Gao Q., Xie C., Li G., Du J., Fu J., He Y., Mater. Des. 2020, 185, 108274. [Google Scholar]
- 72. Dubey N., Ferreira J. A., Daghrery A., Aytac Z., Malda J., Bhaduri S. B., Bottino M. C., Acta Biomater. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lakes R. S., Annu. Rev. Mater. Res. 2017, 47, 63. [Google Scholar]
- 74.a) Ma Z., He W., Yong T., Ramakrishna S., Tissue Eng. 2005, 11, 1149; [DOI] [PubMed] [Google Scholar]; b) Yang F., Wolke J. G. C., Jansen J. A., Chem. Eng. J. 2008, 137, 154. [Google Scholar]
- 75. Woodruff M. A., Hutmacher D. W., Prog. Polym. Sci. 2010, 35, 1217. [Google Scholar]
- 76. Vaquette C., Ivanovski S., Hamlet S. M., Hutmacher D. W., Biomaterials 2013, 34, 5538. [DOI] [PubMed] [Google Scholar]
- 77. Escada A., Machado J., Alves Claro A. P., Mater. Res. 2015, 18, 3. [Google Scholar]
- 78. Bertlein S., Hikimoto D., Hochleitner G., Hümmer J., Jungst T., Matsusaki M., Akashi M., Groll J., Small 2018, 14, 1701521. [DOI] [PubMed] [Google Scholar]
- 79. Haigh J. N., Chuang Y.‐m., Farrugia B., Hoogenboom R., Dalton P. D., Dargaville T. R., Macromol. Rapid Commun. 2016, 37, 93. [DOI] [PubMed] [Google Scholar]
- 80. Zeng J., Wang H., Lin Y., Zhang J., Liang F., Fang F., Yang F., Wang P., Zhu Z., Chen X., Chen X., Wang Z., Cai N., Tang Y., Wu P., Microfluid. Nanofluid. 2018, 22, 23. [Google Scholar]
- 81. Brown T. D., Dalton P. D., Hutmacher D. W., Prog. Polym. Sci. 2016, 56, 116. [Google Scholar]
- 82. Jordahl J. H., Solorio L., Sun H., Ramcharan S., Teeple C. B., Haley H. R., Lee K. J., Eyster T. W., Luker G. D., Krebsbach P. H., Lahann J., Adv. Mater. 2018, 30, 1707196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim K.‐W., Oh H., Bae J. H., Kim H., Moon H. C., Kim S. H., ACS Appl. Mater. Interfaces 2017, 9, 18994. [DOI] [PubMed] [Google Scholar]
- 84. Castilho M., Feyen D., Flandes‐Iparraguirre M., Hochleitner G., Groll J., Doevendans P. A. F., Vermonden T., Ito K., Sluijter J. P. G., Malda J., Adv. Healthcare Mater. 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hochleitner G., Fürsattel E., Giesa R., Groll J., Schmidt H.‐W., Dalton P. D., Macromol. Rapid Commun. 2018, 39, 1800055. [DOI] [PubMed] [Google Scholar]
- 86. Hochleitner G., Hümmer J. F., Luxenhofer R., Groll J., Polymer 2014, 55, 5017. [Google Scholar]
- 87. Nahm D., Weigl F., Schaefer N., Sancho A., Frank A., Groll J., Villmann C., Schmidt H.‐W., Dalton P. D., Luxenhofer R., Mater. Horiz. 2020, 7, 928. [Google Scholar]
- 88. Chen F., Hochleitner G., Woodfield T., Groll J., Dalton P. D., Amsden B. G., Biomacromolecules 2016, 17, 208. [DOI] [PubMed] [Google Scholar]
- 89. Hochleitner G., Chen F., Blum C., Dalton P. D., Amsden B., Groll J., Acta Biomater. 2018, 72, 110. [DOI] [PubMed] [Google Scholar]
- 90. Haigh J. N., Dargaville T. R., Dalton P. D., Mater. Sci. Eng., C 2017, 77, 883. [DOI] [PubMed] [Google Scholar]
- 91. Florczak S., Lorson T., Zheng T., Mrlik M., Hutmacher D. W., Higgins M. J., Luxenhofer R., Dalton P. D., Polym. Int. 2018, 68, 735. [Google Scholar]
- 92. Hochleitner G., Kessler M., Schmitz M., Boccaccini A. R., Teßmar J., Groll J., Mater. Lett. 2017, 205, 257. [Google Scholar]
- 93.a) Seyednejad H., Ji W., Yang F., van Nostrum C. F., Vermonden T., van den Beucken J. J. J. P., Dhert W. J. A., Hennink W. E., Jansen J. A., Biomacromolecules 2012, 13, 3650; [DOI] [PubMed] [Google Scholar]; b) Seyednejad H., Vermonden T., Fedorovich N. E., van Eijk R., van Steenbergen M. J., Dhert W. J. A., van Nostrum C. F., Hennink W. E., Biomacromolecules 2009, 10, 3048. [DOI] [PubMed] [Google Scholar]
- 94.a) Boere K. W. M., Blokzijl M. M., Visser J., Linssen J. E. A., Malda J., Hennink W. E., Vermonden T., J. Mater. Chem. B 2015, 3, 9067; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Boere K. W. M., Visser J., Seyednejad H., Rahimian S., Gawlitta D., van Steenbergen M. J., Dhert W. J. A., Hennink W. E., Vermonden T., Malda J., Acta Biomater. 2014, 10, 2602. [DOI] [PubMed] [Google Scholar]
- 95. Chen F., Hayami J. W. S., Amsden B. G., Biomacromolecules 2014, 15, 1593. [DOI] [PubMed] [Google Scholar]
- 96.a) Hiljanen‐Vainio M. P., Orava P. A., Seppälä J. V., J. Biomed. Mater. Res. 1997, 34, 39; [DOI] [PubMed] [Google Scholar]; b) Surrao D. C., Hayami J. W. S., Waldman S. D., Amsden B. G., Biomacromolecules 2010, 11, 3624; [DOI] [PubMed] [Google Scholar]; c) Azevedo M. C., Reis R. L., Claase M. B., Grijpma D. W., Feijen J., J. Mater. Sci.: Mater. Med. 2003, 14, 103; [DOI] [PubMed] [Google Scholar]; d) Puppi D., Detta N., Piras A. M., Chiellini F., Clarke D. A., Reilly G. C., Chiellini E., Macromol. Biosci. 2010, 10, 887. [DOI] [PubMed] [Google Scholar]
- 97. Surrao D. C., Fan J. C. Y., Waldman S. D., Amsden B. G., Acta Biomater. 2012, 8, 3704. [DOI] [PubMed] [Google Scholar]
- 98. Hafner J., Teuschel M., Schneider M., Schmid U., Polymer 2019, 170, 1. [Google Scholar]
- 99. Rajabi A. H., Jaffe M., Arinzeh T. L., Acta Biomater. 2015, 24, 12. [DOI] [PubMed] [Google Scholar]
- 100. Kanno I., Kotera H., Wasa K., Sens. Actuators, A 2003, 107, 68. [Google Scholar]
- 101.a) Ababneh A., Schmid U., Hernando J., Sánchez‐Rojas J. L., Seidel H., Mater. Sci. Eng., B 2010, 172, 253; [Google Scholar]; b) Schneider M., Bittner A., Schmid U., J. Phys. D: Appl. Phys. 2015, 48, 405301. [Google Scholar]
- 102.a) Damaraju S. M., Shen Y., Elele E., Khusid B., Eshghinejad A., Li J., Jaffe M., Arinzeh T. L., Biomaterials 2017, 149, 51; [DOI] [PubMed] [Google Scholar]; b) Damaraju S. M., Wu S., Jaffe M., Arinzeh T. L., Biomed. Mater. 2013, 8, 045007; [DOI] [PubMed] [Google Scholar]; c) Lee Y.‐S., Arinzeh T. L., Tissue Eng., Part A 2012, 18, 2063; [DOI] [PubMed] [Google Scholar]; d) Lee Y.‐S., Collins G., Livingston Arinzeh T., Acta Biomater. 2011, 7, 3877. [DOI] [PubMed] [Google Scholar]
- 103. Lee H.‐H., Yu H.‐S., Jang J.‐H., Kim H.‐W., Acta Biomater. 2008, 4, 622. [DOI] [PubMed] [Google Scholar]
- 104.a) Hench L. L., J. Am. Ceram. Soc. 1991, 74, 1487; [Google Scholar]; b) Rahaman M. N., Day D. E., Bal B. S., Fu Q., Jung S. B., Bonewald L. F., Tomsia A. P., Acta Biomater. 2011, 7, 2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dalton P. D., Woodfield T. B. F., Mironov V., Groll J., Adv. Sci. 2020, 7, 1902953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Afghah S. F., Dikyol C., Altunbek M., Koc B., Appl. Sci. 2019, 9, 3540. [Google Scholar]
- 107. Großhaus C., Bakirci E., Berthel M., Hrynevich A., Kade J. C., Hochleitner G., Groll J., Dalton P. D., Small, unpublished. 10.1002/smll.202003471. [DOI] [PubMed] [Google Scholar]
- 108. Wunner F. M., Mieszczanek P., Bas O., Eggert S., Maartens J., Dalton P. D., De‐Juan‐Pardo E. M., Hutmacher D. W., Biofabrication 2019, 11, 025004. [DOI] [PubMed] [Google Scholar]
- 109. Brown T. D., Slotosch A., Thibaudeau L., Taubenberger A., Loessner D., Vaquette C., Dalton P. D., Hutmacher D. W., Biointerphases 2012, 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jungst T., Muerza‐Cascante M. L., Brown T. D., Standfest M., Hutmacher D. W., Groll J., Dalton P. D., Polym. Int. 2015, 64, 1086. [Google Scholar]
- 111. Xu H., Yamamoto M., Yamane H., Polymer 2017, 132, 206. [Google Scholar]
- 112.a) Alsoufi M., El‐Sayed A., Int. J. Mech. Mechatron. Eng. 2017, 17, 7; [Google Scholar]; b) Guerrero de Mier A., Espinosa M. M., Domínguez M., Proc. Eng. 2015, 132, 126. [Google Scholar]
- 113. Shi X. D., Brenner M. P., Nagel S. R., Science 1994, 265, 219. [DOI] [PubMed] [Google Scholar]
- 114. v. Hoeve W., Gekle S., Snoeijer J. H., Versluis M., Brenner M. P., Lohse D., Phys. Fluids 2010, 22, 122003. [Google Scholar]
- 115. Brun P. T., Audoly B., Ribe N. M., Eaves T. S., Lister J. R., Phys. Rev. Lett. 2015, 114, 174501. [DOI] [PubMed] [Google Scholar]
- 116. Brun P.‐T., Inamura C., Lizardo D., Franchin G., Stern M., Houk P., Oxman N., Phys. Eng. Sci. 2017, 375, 20160156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mier F. A., Bhakta R., Castano N., Garcia J., Hargather M. J., Fluids 2018, 3, 107. [Google Scholar]
- 118. Marin A. G., Lohse D., Phys. Fluids 2010, 22, 122104. [Google Scholar]
- 119. Bock N., Woodruff M. A., Hutmacher D. W., Dargaville T. R., Polymers 2011, 3, 131. [Google Scholar]
- 120. Chiu‐Webster S., Lister J. R., J. Fluid Mech. 2006, 569, 89. [Google Scholar]
- 121. Miles C. A., Burjanadze T. V., Bailey A. J., J. Mol. Biol. 1995, 245, 437. [DOI] [PubMed] [Google Scholar]
- 122. Zhmayev E., Cho D., Joo Y. L., Phys. Fluids 2011, 23, 073102. [Google Scholar]
- 123. Yu X., Feng M., Zhang R., Feng Y., You H., Guo F., Chen S., Zhang D., Org. Electron. 2017, 51, 442. [Google Scholar]
- 124. Lee G. Y., Lee H. T., Ryu W., Ahn S. H., Yang J., Smart Mater. Struct. 2018, 27, 11LT01. [Google Scholar]