

Abstract
Amyloid fibrils may serve as building blocks for the preparation of novel hydrogel materials from abundant, low‐cost, and biocompatible polypeptides. This work presents the formation of physically cross‐linked, self‐healing hydrogels based on bovine serum albumin at room temperature through a straightforward disulfide reduction step induced by tris (2‐carboxyethyl) phosphine hydrochloride. The structure and surface charge of the amyloid‐like fibrils is determined by the pH of the solution during self‐assembly, giving rise to hydrogels with distinct physicochemical properties. The hydrogel surface can be readily functionalized with the extracellular matrix protein fibronectin and supports cell adhesion, spreading, and long‐term culture. This study offers a simple, versatile, and inexpensive method to prepare amyloid‐based albumin hydrogels with potential applications in the biomedical field.
Keywords: antifouling, cell mechanosensing, fibrillar hydrogels, self‐assembly, sustainable materials
A facile and straightforward fabrication method for viscoelastic albumin hydrogels via the assembly and physical interactions of amyloid fibrils is presented. The resulting hydrogels exhibit self‐healing behavior and tunable mechanical properties and support cell adhesion and growth post‐functionalization with the extracellular matrix protein fibronectin.
![]()
1. Introduction
Hydrogels are a class of materials with wide‐ranging applications in the biomedical field as coatings, implants, or scaffolds for organoid development.[ 1 , 2 , 3 ] Their success is owing to a number of attractive properties such as their high water‐content, malleable structure, and tunable mechanical properties, which match those of the various soft tissues.[ 4 ] These properties are beneficial for cell adaptation/differentiation and/or integration with cells/living tissue. In general, hydrogels consist of a cross‐linked macromolecular network, which entraps a large volume of water depending on the cross‐linking density as well as the hydrophilicity of the material used.[ 5 ] The materials are typically based on synthetic or natural (bio)polymers, while the network is formed through physical or chemical cross‐links among the building blocks.
Albumin is the most abundant plasma protein and an attractive building block for constructing hydrogels. Albumin is approved by the US Food and Drug Administration (FDA) for use in clinical practice; it is readily available, inexpensive, and can potentially be harvested from patients to create personalized, autogenic materials, minimizing risks of immunogenicity. Hence unsurprisingly, it has been previously considered by many as the building block for creating albumin‐based hydrogels.[ 6 , 7 , 8 , 9 , 10 , 11 ] In most cases however, the use of chemical reagents to either chemically modify the albumin, or induce cross‐linking of side chains, introduces concerns related to cytotoxicity, reproducibility, and undesired side products. An alternative approach that we explored in this study, is to denature albumin and drive its assembly into amyloid‐like fibrils, which would then physically interact to create a free‐standing hydrogel material.
The assembly of amyloid fibrils and associated plaques has been extensively studied, mostly in the context of pathological amyloid plaques forming in the brains of patients suffering from neurodegenerative diseases.[ 12 ] However, functional and beneficial amyloid fibrils have also been reported in living organisms.[ 13 , 14 ] It is now accepted that most proteins may form amyloid fibrils under certain conditions, albeit with variable morphologies and intermolecular interactions.[ 12 , 14 , 15 ] Consequently, a wide variety of proteins has been considered to fabricate biomaterials based on the amyloid state,[ 16 , 17 , 18 , 19 , 20 , 21 ] in an emerging field that has been previously reviewed.[ 22 , 23 ] Despite the distinction between amyloid and amyloid‐like fibrils still being debated, a consensus exists for the presence of intermolecular beta sheet structures along their main axis, with the beta strands aligned perpendicular to that axis, and their identification through the use of specific dyes, such as Congo red and thioflavin T (ThT).[ 12 , 16 , 24 ]
In the case of serum albumin, assembly into amyloid‐like fibrils was reported following denaturation induced by changes in pH,[ 25 ] temperature,[ 26 , 27 , 28 , 29 , 30 ] reducing agents,[ 15 , 31 ] or combination of these parameters.[ 10 , 15 , 25 , 32 , 33 ] Each of the above parameters have distinct effects on albumin folding and denaturation. Therefore, differences in the morphology and structure of the resulting amyloid fibrils are expected, although this has not yet been systematically studied. For example, in the case of pH, a transition from the so‐called N‐form at physiological pH 7.4 to the F‐form occurs at pH 4,[ 25 , 27 ] below the isoelectric point of 4.7. At pH values above 8, bovine serum albumin (BSA) adopts the B (basic) form. While the aggregation of BSA as a function of the combination of temperature and pH has been previously reported,[ 25 , 27 , 32 , 34 ] the effect of pH on BSA structure upon disulfide reduction remains unknown.
In this study we present the fabrication of albumin hydrogels through physical cross‐linking of amyloid‐like fibrils, assembled following a straightforward disulfide reduction step at room temperature. We have opted for utilizing tris(2‐carboxyethyl) phosphine hydrochloride (TCEP) as the reducing agent due to favorable kinetics and building on previous work that exploited this agent to assemble amyloid‐based films. In particular, the group of Yang pioneered the above strategy to fabricate functional and/or antifouling surface coatings.[ 19 , 35 , 36 , 37 , 38 ] Here instead, we have explored the possibility of preparing physically crosslinked hydrogels through albumin fibril assembly. Interestingly, the pH at which protein denaturation had occurred following disulfide reduction had a pronounced effect on amyloid fibril structure and resulting hydrogel properties. The surface of the formed hydrogels could be functionalized in a straightforward manner with the extracellular matrix (ECM) protein fibronectin (Fn), supporting cell adhesion, cell growth and demonstrating cytocompatibility. The biomaterial presented here is therefore a promising candidate for further development. Comparisons with similar materials and potential applications are discussed.
2. Results
2.1. Hydrogel Formation Induced by Disulfide Reduction of Albumin
The native structure of bovine serum albumin (BSA) is stabilized through 17 disulfide bonds. We first investigated whether reduction of these disulfides leads to protein denaturation and amyloid‐like fibril assembly as previously reported.[ 15 ] When a 2% w/v BSA solution was heated at 90 °C in presence of the reducing agent TCEP, amyloid‐like fibrils were formed within 90 min as evidenced by atomic force microscopy (AFM) imaging (Figure S1, Supporting Information). Instead, a 2% w/v BSA solution heated at 90 °C without TCEP did not assemble into amyloid‐like fibrils even after 24 h (Figure S1, Supporting Information). We hypothesized that increasing the concentration of BSA could result in fibril association and/or entanglements that would drive the formation of an amyloid‐based hydrogel. Indeed, 5 and 10% BSA solutions incubated with 40 mm TCEP and heated under magnetic stirring at 90 °C gelled within minutes. We next examined whether gelation would also occur at room temperature (RT), solely through addition of TCEP, within a time period of 24 h. A lower temperature would minimize protein degradation and render the process more facile, convenient, and economic. Gelation was observed for aqueous BSA solutions treated with 40 µm TCEP with a concentration larger than 3% (Figure S2, Supporting Information), indicating that the addition of TCEP, without thermal denaturation, is sufficient for gelation. The presence of the reducing agent was necessary for gelation; simply lowering the pH to 3–3.5 in a 5% BSA solution was not sufficient for gel formation within 24 h (Figure S2, Supporting Information). Another deciding factor during protein denaturation and amyloid fibril assembly is the presence of salts.[ 39 , 40 ] In preliminary experiments, we tested the effect of increasing ionic strength, by adding 150 mm NaCl in the precursor mixtures, and observed quick aggregation as evidenced by clouding of the solution and precipitation. Therefore, we prepared all subsequent samples in water. Nevertheless, we note that a small amount of salt was present since TCEP was provided as the HCl salt and 1 n NaOH was used to adjust the pH.
We next examined the combined effect of pH, BSA and TCEP concentrations on gelation. Since addition of TCEP reduces the solution pH, the pH was adjusted using 1 n NaOH for 5 and 10% BSA aqueous solutions. Of note, NaOH and TCEP were added rapidly one after the other, since prolonged incubation at basic pH after addition of NaOH led to inconsistent results, most likely due to pH‐dependent BSA denaturation.[ 41 ] After 24 h, samples were examined visually and a gel inversion method was performed, to assess their transparency and gel formation, respectively (Figure 1A–C). The ability for gelation and the optical properties of the resulting gels varied in a non‐trivial manner as a function of TCEP, pH, and BSA concentration. The main observations can be summarized in the following points: 1) In the pH range from 4.5 to 5, close to the isoelectric point of BSA (pIBSA = 4.7–4.8), the solution turned quickly opaque, indicating aggregation; 2) at very low (<2) or high (>10) pH values, there was no gelation observed; 3) at pH values below the pIBSA the gels were transparent when formed; 4) at pH values above the pIBSA the optical transparency of the gels was lower and depended on both pH and TCEP concentration: gels shifted from opaque to semi‐transparent as the pH increased up to a maximal value, above which no gelation was observed. Increasing the TCEP concentration shifted this transition to higher pH values. However, above a certain TCEP concentration, gels were always opaque. These observations indicated that the protein network responsible for gel formation differed depending on both pH and TCEP concentration in a non‐trivial manner.
Figure 1.
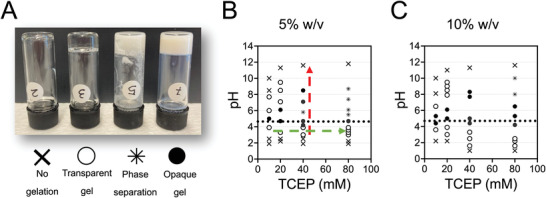
TCEP‐pH phase diagrams for BSA gelation at room temperature. A) Photographs of BSA solutions exhibiting four different conditions. From left to right: no gelation of solution, transparent gel, phase separation, and opaque gel. The distinction between opaque and transparent gels was made based on the ability to clearly see an object through the gel. B,C) Phase diagrams for (B) 5% and 10% (C) BSA solutions with pH on the y‐axis and TCEP concentration on the x‐axis constructed on the base of conditions presented in (A). Dotted line at pH 4.7 corresponds to the isoelectric point of BSA. Red and green dashed arrows are a guide to the eye to aid the discussion in the text.
In order to gain more detailed understanding of the process, we focused on a single BSA concentration (5%) and first studied the effect of pH for a fixed TCEP concentration of 40 mm TCEP (red dashed line in Figure 1B). We performed rheology to determine the gelation kinetics and estimate the mechanical properties of the hydrogel. As shown for a typical kinetic curve after the addition of TCEP (t = 0), the elastic (storage) modulus of the BSA solution started to rise following a lag phase (Figure 2A). Preliminary experiments showed that a plateau value for the elastic modulus was reached after 24 h. Near the isoelectric point, the lag time was the shortest (Figure 2B), but the resulting gels were opaque, suggesting the presence of large aggregates. Furthermore, the hydrogels had significantly lower shear elastic moduli compared to gels prepared at more acidic or more basic conditions (Figure 2C). At acidic conditions (pH 3–4), the lag phase was the longest (25 min; average of three measurements), and the resulting gels were stiffer (Figure 2B,C). At basic pH (7.4), the lag phase decreased to 14 min (average of three measurements) and the shear modulus was the highest (Figure 2B,C). These results indicate that the gelation kinetics and final structure of the protein network responsible for the free‐standing gel is largely dependent on pH and suggest that quick aggregation near pIBSA prevents the formation of a well‐formed three‐dimensional (3D) protein network.
Figure 2.
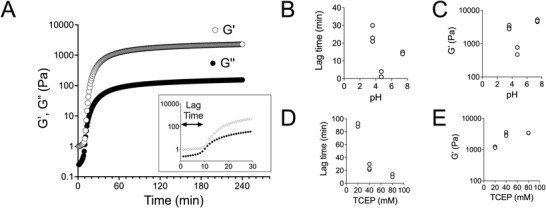
Rheological characterization of 5% BSA hydrogels as a function of pH and TCEP. A) Gelation kinetics were monitored using oscillatory rheology at 1% strain and angular frequency of 1 Hz. The storage (Gʹ) and loss (Gʺ) moduli were measured every minute following mixing of TCEP with BSA (t = 0). The storage modulus started to rapidly increase following a lag phase (inset). B,C) Lag phase and plateau storage modulus, determined 24 h after mixing TCEP (40 mm) with BSA (5%) as a function of solution pH. At pH values close to the isoelectric point, gelation occurred rapidly, but the resulting gels were not as stiff, compared to gels prepared at acidic pH (3.6) or physiological pH (7.4). D,E). Lag phase and plateau storage modulus as a function of TCEP concentration used for 5% BSA gels at pH 3.6. Increasing the TCEP concentration led to faster gelation. The plateau storage modulus increased when increasing TCEP concentration from 20 to 40 mm, but remained similar for 80 mm TCEP. Each data point in (B–E) corresponds to an independent experiment (N = 2 or 3).
Using rheology, we also studied the effect of TCEP concentration for 5% BSA gels prepared at a constant pH of 3.6 ± 0.2 (green dashed line in Figure 1B). We postulate that the main effect of varying TCEP concentration at a constant pH is the change in extent of disulfide bond reduction and ensuing denaturation of the protein. Accordingly, we expected that with increasing TCEP concentration, the hydrophobic domains of BSA would be exposed more and faster, thus accelerating gelation. Indeed, the lag time for gelation decreased with TCEP concentration (Figure 2D). However, doubling the TCEP concentration from 40 to 80 mm did not noticeably affect hydrogel stiffness (Figure 2E), suggesting that a molar ratio of TCEP to BSA of 53:1 is sufficient for optimal network formation. Our results so far are in line with our hypothesis that TCEP acts by denaturing BSA, leading to amyloid fibril formation and gelation above a concentration threshold.
2.2. Amyloid‐like Fibril Formation of BSA upon TCEP Treatment
In order to establish the amyloid character of the assembled structures and follow the kinetics of their formation we monitored the fluorescence of the ThT dye. ThT increases its fluorescence upon binding planar fibrillar assemblies formed via beta‐sheets and is an accepted marker of amyloid fibrils.[ 42 ] Since ThT is cationic and thus its binding is sensitive to the pH of the solution,[ 42 ] samples were analyzed in buffered solutions at different time points. Low concentration of BSA was used in this assay in order to avoid gelation and thus allow pipetting. Initially we examined whether the kinetics and absolute ThT intensity differed at acidic versus alkaline conditions. Upon TCEP addition to a 2% BSA solution at room temperature, ThT fluorescence rapidly increased, without any lag phase and reached a plateau within 1 day (Figure 3A). In absence of TCEP, there was no increase in ThT fluorescence within 24 h as expected. These results indicated that amyloid‐like fibril assembly was occurring as a result of TCEP‐mediated disulfide reduction. The increase of ThT fluorescence was higher for pH 3.6 compared to 7.4, hinting at a more efficient amyloid‐like fibril assembly at the acidic pH (Figure 3A). However, the intensity of ThT fluorescence is also influenced by fibril surface charge[ 42 ] and therefore, it cannot be used as a quantitative measure of amyloid fibril assembly.
Figure 3.
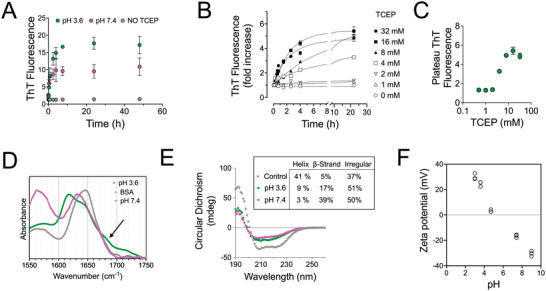
TCEP‐induced denaturation of BSA leads to beta‐sheet forming amyloid‐like structures. A) Kinetics of ThT fluorescence increase after addition of 20 mm TCEP to a 2% BSA solution (t = 0) at different pH values, and negative control (2% BSA without ThT). The mean values and standard deviation from three independent experiments are presented. B) ThT fluorescence increase as a function of time after mixing 2% BSA with TCEP to reach indicated TCEP concentrations at pH 3.6. Data were normalized and presented as fold‐increase compared to the initial measurement immediately before TCEP addition. The lines correspond to best fits for the equation y = y M − y 0 e −kx. C) The values of ThT fluorescence 24 h after mixing TCEP with a 2% BSA solution at pH 3.6, as a function of the final TCEP concentration. Data from one out of three independent experiments are presented. D) Normalized FTIR spectra in the amide I′ region of BSA solutions in D2O, in absence or presence of TCEP at pD 3.6 or 7.4. The shift of the maximum band from 1648 to 1620 cm−1 for pD 3.6 and 1630 cm−1 for pD 7.4 indicated the shift toward a higher beta‐sheet content. The arrow points to a shoulder peak at 1680 cm−1, indicative of antiparallel β‐sheets. E) Circular dichroism spectra of a control aqueous BSA solution and BSA solutions incubated with TCEP at different pH values. The BSA control exhibits two peaks at 208 and 225 nm, characteristic of a protein rich in alpha helical structure, while TCEP‐induced denaturation of BSA results in the attenuation of these two peaks and the emergence of one peak centered ≈ 215 nm, typical of beta‐sheet structures. The secondary structure prediction of helical, β‐strand, and irregular elements shown in the inset was calculated using the web‐based server tool CAPITO. F) Zeta potential measurements of 2% BSA solutions treated with 16 mm TCEP for 24 h at room temperature as a function of the solution pH. Each data point corresponds to an independent experiment (N = 3 experiments).
Next, ThT fluorescence was monitored as a function of TCEP concentration for a 2% BSA solution at acidic pH. Both the rate of ThT fluorescence intensity increase and the plateau value reached after 24 h depended on TCEP concentration (Figure 3B). A threshold TCEP concentration of 2 mm, which corresponds to a TCEP:cysteine molar ratio of 0.19, was required for a noticeable increase in ThT fluorescence intensity (Figure 3B,C). A plateau intensity value for ThT fluorescence was reached above a TCEP concentration of 16 mm, which corresponds to a TCEP:cysteine molar ratio of 1.5 (Figure 3B,C). Additionally, the rate of increase of the ThT fluorescence was not increased above the 1.5 TCEP:cysteine ratio. This ratio was the same as the one determined from the rheology study (Figure 2E), indicating that it represents an optimal value for the formation of fibrillar beta‐sheets structures. Therefore, the corresponding TCEP:BSA molar ratio of 53:1 was used for all subsequent experiments, unless otherwise stated.
The shift in the secondary structure of the protein was additionally monitored using Fourier Transform Infrared (FTIR) and circular dichroism (CD) spectroscopy. Both methods confirmed the shift from a structure with predominantly alpha‐helical content to one with increased beta‐sheet content (Figure 3D,E). The amide I′ band monitored by FTIR spectroscopy for 10% BSA solutions showed a clear shift from a maximum at wavelength number of 1650 cm−1, indicative of alpha helices, to 1619 cm−1, demonstrating the formation of beta‐sheets, characteristic of amyloid‐like fibrils (Figure 3D).[ 43 ] The presence of a shoulder peak at 1685 cm−1 additionally indicated the presence of antiparallel intermolecular beta sheets (Figure 3D).[ 44 ] Consistent with the FTIR data, the CD spectrum of untreated BSA showed twin peaks at 208 and 220 nm, characteristic for proteins with high alpha‐helical content, whereas TCEP‐treated BSA at both pHs, showed a shift toward a shallower peak centered at 215 nm, characteristic of beta‐sheets (Figure 3E). In sum, the above data confirm the amyloid‐like structure of the gel network.
In order to visualize the structure of the amyloid‐like fibrils, we performed AFM imaging of TCEP‐treated BSA solutions deposited on mica. First, we imaged BSA fibrils prepared at basic conditions (pH 7.4), which readily adsorbed at the mica surface. The surface of the mica was not homogeneously covered with structures; instead, regions of interconnected short fibrils, surrounded by empty mica areas were observed (Figure 4A). This surface distribution, coupled with the lack of isolated fibrils on the surface, suggested that upon BSA denaturation and fibril assembly, the fibrils rapidly associated with each other and formed a network. When we attempted to use freshly‐cleaved mica to image the sample prepared under acidic conditions (pH 3.6), the protein formed a multi‐layered aggregate on the surface. The surface charge of mica is negative, and we assumed that the interaction of the amyloid‐like fibrils prepared at pH 3.6, which were below the isoelectric point, were the reason for this behavior. Hence, the mica was coated with (3‐aminopropyl)triethoxysilane (APTES) prior to fibril deposition. Under these conditions, short fibrils homogeneously covered the mica surface (Figure 4B). Interestingly, the average height of the fibrils prepared at different pH was different: at acidic conditions, fibrils were roughly twice as thick compared to the ones prepared at basic conditions (Figure 4A,B). The observation that amyloid fibrils prepared at pH values below and above the pIBSA interacted differently with the mica surface also suggests that their surface charge changes depending on the pH at which they are assembled. Zeta potential measurements confirmed this prediction and showed a reversal from positive values when assembly occurred at pH values below the pIBSA to negative charges above (Figure 3F). Overall, our combined results demonstrated that while TCEP treatment induced amyloid‐like fibrils at both pH values, the structure of these fibrils was dependent on the pH in which denaturation and self‐assembly occurred.
Figure 4.
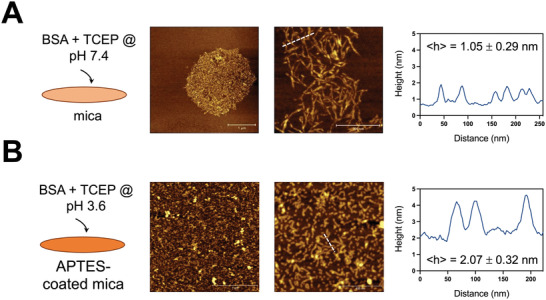
BSA amyloid‐like fibril structure differs depending on the pH used for their preparation. A) Tapping mode AFM images under ambient conditions of BSA amyloid‐like fibrils deposited on freshly‐cleaved mica. A 2% BSA solution prepared with 20 mm TCEP at pH 7.4 was pipetted on the mica and then rinsed with water. Fibrils were observed clustered at regions of the mica surface. A height profile corresponding to the dashed white line is shown in the left, and the average height noted (mean and standard deviation from 50 measurements from two independent experiments). B) Tapping mode AFM images under ambient conditions of BSA amyloid‐like fibrils deposited on APTES‐treated mica. In this case, the BSA/TCEP mixture was adjusted to pH 3.6. The surface was homogeneously covered with short fibrils that were roughly twice as high compared to the ones prepared at basic conditions.
2.3. The Dynamic Mechanical Properties of BSA Hydrogels Depend on the pH Used during their Formation
Our results so far have demonstrated that the physical association of denatured BSA fibrils resulted in self‐standing hydrogels under both acidic and basic conditions, albeit with differences in mechanical properties (Figure 2) and fibril structure (Figure 4). We next characterized in more detail the dynamic mechanical properties of these hydrogels using rheology. The storage (elastic, Gʹ) and loss (viscous, Gʺ) moduli showed a low dependence on the angular frequency, with the storage modulus being more than one order of magnitude greater, validating that BSA hydrogels are viscoelastic solids (Figure 5A). The value of the loss tangent, tan(δ) = Gʺ/Gʹ, was consistently higher for gels prepared at pH 7.4 compare to pH 3.6 (Figure 5B). The angular frequency dependence of the storage modulus was also slightly higher for hydrogels prepared at pH 7.4, with an average slope of 0.080 ± 0.02 on a log–log scale (mean ± standard deviation (SD) from 3 independent measurements), compared to a slope of 0.050 ± 0.01 for those prepared at pH 3.6 (Figure 5A). These results indicated a more elastic character for the hydrogels prepared at the lower pH, despite their lower storage moduli.
Figure 5.
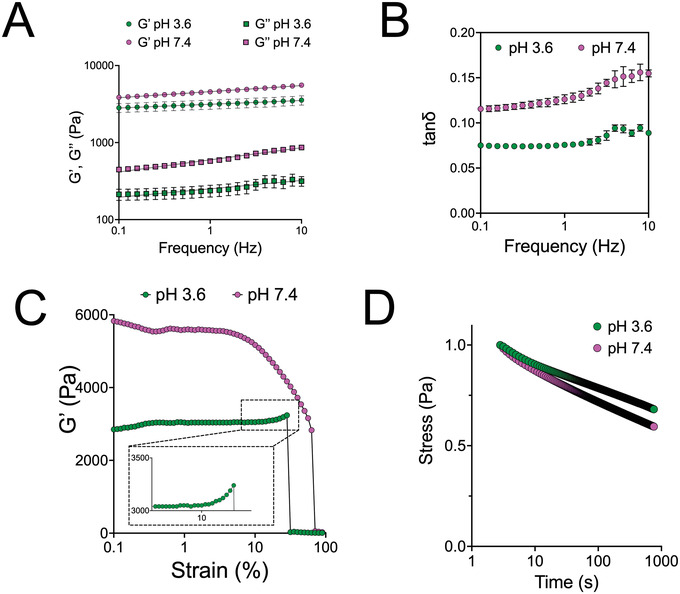
Rheological properties of BSA amyloid‐based hydrogels depend on the pH used during gelation. A) Storage (Gʹ) and loss (Gʺ) moduli for 5% hydrogels prepared with 40 mm TCEP as a function of angular frequency. The frequency dependence of Gʹ for gels prepared at pH 7.4 was slightly larger for those prepared at pH 3.6. Data from three different batches and standard deviation are presented. Red lines represent log(Y) = A + B × log(X). B) Loss tangent (tanδ = Gʺ/Gʹ) as a function of angular frequency, calculated from data presented in (A). C) Storage modulus for 5% hydrogels prepared with 40 mm TCEP as a function of strain. Gels prepared at pH 7.4 showed strain softening above a strain of ≈ 5%, whereas gels prepared at pH 3.6 strain stiffening, before breaking at 30%. One of three independent experiments is shown. Inset shows magnified the region where strain stiffening is apparent. D) Stress relaxation (normalized to maximum stress) after rapid application of 10% strain for 5% hydrogels prepared with 40 mm TCEP. The mean of three independent experiments is presented; error bars (SD) are too small to be visible. Hydrogels prepared at pH 7.4 exhibited faster stress relaxation compared to those prepared at pH 3.6.
The linear viscoelastic regime was found to extend to strains above 10% for gels prepared at pH 3.6, but was lower for gels prepared at pH 7.4 (Figure 5C). Interestingly, the hydrogels showed divergent behavior in response to increasing strain. Hydrogels prepared at pH 7.4 showed strain softening (thixotropy), before material failure (Figure 5C). On the other hand, gels prepared at pH 3.6 showed a small degree of strain stiffening prior to abrupt macroscopic damage over a threshold strain (Figure 5C). Finally, due to the viscoelastic nature of the hydrogels and the physical interactions holding the network together, stress relaxation was also observed (Figure 5D). Again, a difference was observed depending on the pH used: gels prepared at pH 7.4 exhibited faster stress relaxation. Combined, these results suggest that gels prepared at pH 7.4 are thixotropic, have a more viscous character, and more quickly rearrange upon stress application, compared to hydrogels prepared at pH 3.6.
2.4. BSA Hydrogel Swelling Depends on Preparation Conditions
When exposed to aqueous environments, hydrogels may exchange water with their surroundings to reach an energetically favorable equilibrium state. Whether hydrogels will take up or expel water depends on a combination of factors such as the initial cross‐linking density, the hydrophilicity of the hydrogel network, as well as entropic factors.[ 45 ] BSA hydrogels prepared at pH 3.6 or pH 7.4 were exposed to different aqueous solutions and their swelling behavior monitored over time. When BSA gels that were prepared at acidic conditions were exposed to pure water, the gels soaked up water over time, eventually losing their integrity after 1 day (Figure 6A). Intriguingly, the same gels exhibited shrinkage when exposed to 10 mm PBS, reaching equilibrium after a few hours, and almost no change in swelling when exposed to physiological salt concentration (Figure 6A). The shrinkage of gels in PBS, but not in 150 mm NaCl, indicated that the change in pH triggered tighter association between individual fibrils. We speculate that this is due to the neutralization of charges on the surface of fibrils when exposed to the buffer of pH 7.4.
Figure 6.
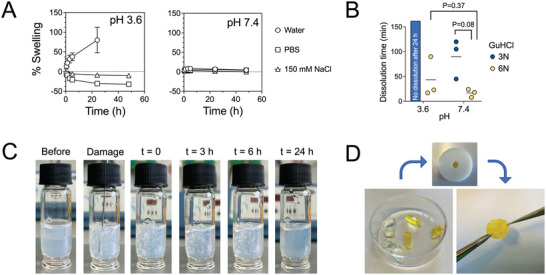
Swelling behavior of BSA hydrogels depends on the pH used during preparation. A) Swelling of 5% BSA hydrogels (250 µL), prepared with 40 mm TCEP at pH 3.6 or pH 7.4, in Milli‐Q water, PBS 10 or 150 mm NaCl over time. Mean and standard deviations from three independent experiments are presented. B) The dissolution time of 5% BSA hydrogels (250 µL), prepared with 40 mm TCEP at pH 3.6 or pH 7.4, in presence of 3 or 6 m GuHCl in PBS. Each data point corresponds to an independent experiment (N = 3). Data were compared using unpaired t‐tests with Welch's correction. C) Self‐healing demonstration for a 5% BSA hydrogel prepared at pH 3.6 with 40 mm TCEP. After 24 h, the hydrogel was damaged using a syringe needle and its recovery was monitored over time. D) Two 10% BSA hydrogels, prepared at pH 3.6 with 80 mm TCEP, were cut into pieces and then placed in a Teflon mold overnight. The hydrogel pieces were joined together and formed a hydrogel that could resist force application by tweezer pulling.
BSA gels prepared at physiological pH value exhibited different swelling behavior. There was low (<10%) swelling when gels were immersed in water, even lower swelling (<5%) when immersed in PBS and no change when immersed in 150 mm NaCl (Figure 6A). This finding is consistent with the tighter inter‐fibril association suggested by the AFM imaging experiments, where fibrils prepared at pH 7.4 assembled in clusters and did not disperse over the entire surface (Figure 4A).
Since amyloid self‐assembly is a result of intra‐protein beta sheet formation, we hypothesized that in presence of the chaotropic guanidinium chloride (GuHCl), the fibrils would dissolve and the hydrogel would break down. Indeed, addition of 6 m GuHCl in PBS led to quick dissolution of hydrogels prepared at both acidic and physiological pH, while 3 m GuHCl effectively dissolved only gels prepared at pH 7.4 (Figure 6B and Figure S3, Supporting Information). The kinetics of dissolution were faster for the gels prepared at pH 7.4, indicating less stable intramolecular bonds (Figure 6B). In both cases, the hydrogel appeared to erode, instead of first swelling and dissolving, supporting a diffusion rate‐limiting mechanism of individual fibril dissolution (Figure S3, Supporting Information). Another prediction arising from the physical nature of amyloid fibril assembly and non‐covalent cross‐linking of the fibrils was that hydrogels would exhibit self‐healing behavior.[ 46 , 47 , 48 ] The ability of the BSA amyloid gel to self‐heal was verified by performing two experiments. First, after damaging a transparent, 5% BSA gel using a syringe needle, the hydrogel recovered its original coherent structure over time, showing no residual damage after 24 h (Figure 6C). Second, a 10% BSA gel was prepared, cut into small pieces and then brought together into a mold. After 24 h, it was possible to obtain a recovered free‐standing hydrogel (Figure 6D).
Overall, our results suggest that the BSA hydrogel network is kept in place through physical cross‐links and differences in denatured BSA conformation adopted during amyloid fibril formation as a function of the pH, largely determine the swelling and degradation behavior of the resulting hydrogels.
2.5. The Surface of BSA Hydrogels can be Functionalized with ECM Proteins
During the swelling studies, we noticed that when water came into contact with hydrogels, it often formed droplets with significant contact angles. Measurements using water or PBS on 5% BSA hydrogels prepared at pH 3.6 revealed contact angles that were initially ≈ 50° and slightly decreased over time (Figure 7A and Figure S4, Supporting Information). In contrast, when the hydrogels were prepared at pH 7.4, the contact angles of hydrogels were >120° denoting a hydrophobic hydrogel surface. We hypothesized that the hydrophilicity of the surface is largely determined by the surface charge of the denatured protein. Indeed, increasing the pH to 9.0, which is expected to increase protein negative charges, led to a decrease in contact angles (Figure 7A).
Figure 7.
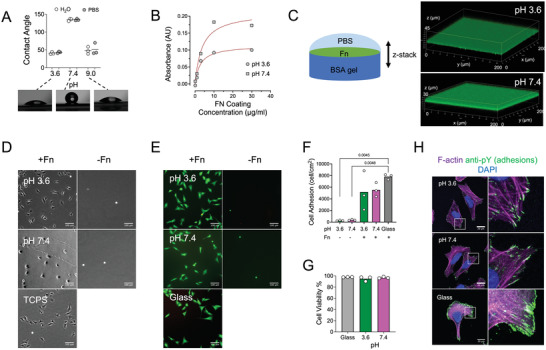
BSA hydrogels allow for fibronectin adsorption and subsequent cell culture. A) Water contact angle measurements for 5% BSA hydrogels prepared with 40 mm TCEP at pH 3.6, 7.4 or 9.0. There was no difference between using Milli‐Q water or PBS for preparing the droplet. The contact angles for 3 independent experiments are presented on top and images of aqueous droplets on the hydrogels from one of the experiments on the bottom. B) The coating efficiency of adsorbed Fn on 5% BSA hydrogels prepared at pH 3.6 or 7.4 was estimated using a modified ELISA assay and the monoclonal anti‐Fn antibody P1H11, which recognizes the Fn central cell binding domain. A higher amount of Fn was adsorbed on hydrogels prepared at pH 7.4. The average value from 2 independent experiments is presented. Lines correspond to fits of the equation y = A(1 − e −kx). C) Confocal z‐stack fluorescence microscopy images of fluorescent Fn adsorbed on 5% BSA hydrogels prepared at pH 3.6 or 7.4. Fn was adsorbed homogeneously over the hydrogel surface and did not penetrate into the hydrogel interior. D) Phase contrast images of live U2OS cells 24 h after seeding on 5% BSA hydrogels prepared with 40 mm TCEP at indicated pH values, and tissue culture polystyrene (TCPS) as control. Cells adhered and spread only when hydrogels were coated with Fn. Scale bars: 100 µm. E) Epifluorescence microscopy images of live U2OS cells stained with calcein (green) and dead U2OS cells stained with ethidium homodimer (red), 20 h after cell seeding. Cells were seeded on Fn‐coated or uncoated hydrogels prepared at indicated pH or Fn‐coated glass as a control. F) Surface concentration of live U2OS cells on different substrates 20 h post‐seeding. G) Viability of U2OS cells on different substrates 20 h post‐seeding was calculated as the ratio of live to total cells. H) Confocal microscopy images of fixed U2OS cells seeded for 5 h on Fn‐coated glass or Fn‐coated 5% BSA hydrogels prepared with 40 mm TCEP at pH 3.6 or 7.4. The cells were immunostained against pY, which stains adhesion clusters, and stained with Phalloidin‐TRITC (F‐actin) and DAPI (nucleus; DNA). Focal adhesions and stress fibers were evident inside cells on both hydrogels. Data in (F) and (G) were analyzed using a Brown‐Forsythe and Welch ANOVA statistical test, comparing all data sets with each other. Only p values from statistically significant comparisons (p < 0.05) are plotted. Scale bars: 20 µm.
The hydrophobic surface of BSA hydrogels prompted us to examine the possibility of adsorbing biologically‐active molecules on their surface. To this end, we incubated BSA hydrogels prepared at basic and acidic conditions with soluble fibronectin (Fn), an ECM protein that promotes cell adhesion.[ 49 ] An ELISA‐type assay using an antibody against the cell‐binding domain of Fn demonstrated a concentration‐dependent increase in adsorbed protein on the surface of hydrogels, reaching a plateau when a coating concentration of 10 µg mL−1 was used (Figure 7B). Interestingly, the amount of adsorbed Fn was higher for hydrogels prepared at pH 7.4, which exhibited higher contact angles. In order to examine the homogeneity of the protein coating, fluorescently‐labeled Fn was used to coat the hydrogel surface and confocal microscopy z‐stack reconstructions were acquired (Figure 7C). The results demonstrated that Fn covered homogeneously the hydrogel surface at the optical resolution examined, and did not penetrate into the interior of the hydrogel, presumably due to its large (440 kDa) size.
2.6. BSA Hydrogels as Substrates for Cell Culture
Finally, we examined the possibility of using amyloid‐based BSA hydrogels as cell culture substrates and initially tested their cytocompatibility and their ability to support cell adhesion and spreading. The osteosarcoma cell lines U2OS and NIH3T3, routinely used for adhesion and motility assays,[ 50 , 51 ] adhered on Fn‐coated hydrogels, but not on control uncoated hydrogels, irrespective of the pH used for their preparation (Figure 7D and Figure S5, Supporting Information). The few adherent cells were not able to spread, even after 24 h of incubation (Figure 7D). These findings indicated that pre‐coating hydrogels with Fn was necessary for cell adhesion, and showed that uncoated BSA hydrogels did not promote non‐specific cell adhesions. U2OS cells were able to spread, migrate over the Fn‐coated surface and divide after cell seeding (Movie S1, Supporting Information). Cells were viable over at least 1 week on the hydrogel confirming the cytocompatible character of the hydrogels (Figure S6, Supporting Information). In order to quantify cell adhesion and viability on BSA hydrogels as a function of pH and presence of an Fn coating, we counted the number of adherent living and number of dead cells 24 h after cell seeding. Living cells were identified by staining with calcein, which is rendered fluorescent by metabolically‐active cells, while the nuclei of dead cells were stained red with ethidium homodimer. U2OS (Figure 7E,F) and NIH3T3 (Figure S5, Supporting Information) adhesion was higher when hydrogels were coated with Fn prior to seeding and similar to positive controls (Fn‐coated glass), confirming visual observations. Moreover cell viability was not compromised on BSA amyloid‐based hydrogels, irrespective on the pH at which they were prepared (Figure 7E,G and Figure S5, Supporting Information). A closer look on adhesion cluster formation and the actin cytoskeleton revealed that cells assembled robust stress fibers originating from focal adhesions on hydrogels prepared at both tested pH values (Figure 7H). Overall, BSA hydrogels are a suitable substrate for cell culture studies.
3. Discussion
In this study we present the facile and straightforward fabrication of cytocompatible, amyloid‐based hydrogels at room temperature through the addition of the reducing agent TCEP to aqueous albumin solutions. Building on established work, which showed that reduction of native disulfide bonds of albumin leads to its denaturation and subsequent amyloid‐fibril assembly,[ 15 , 36 ] we here describe how the physical interactions of such fibrils, above a threshold concentration, result in hydrogel formation. The presented approach complements a large body of literature based on short, designed peptide sequences and similar self‐assembling principles.[ 16 , 52 ] Engineered peptides developed as hydrogelators typically rely on hydrogen‐bonding along their backbone to form amyloid‐like fibrillar structures that constitute the hydrogel network.[ 16 ] Alternatively, peptides have been modified with hydrophobic lipid tails to induce self‐assembly into fibrillar structures based on hydrophobic interactions.[ 53 , 54 , 55 ] Both strategies have been successful in creating hydrogels, but are more expensive, require peptide synthesis/modification and the resulting hydrogels are often less stable than the amyloid‐based hydrogels presented here. Further advantages of using albumin are linked to its availability and biocompatibility: being the most abundant plasma protein, it is available at low cost in large quantities and can potentially be harvested from the blood of patients for the fabrication of autogenous hydrogels. While our experiments were performed with albumin from bovine sources, the high sequence homology with human albumin and previous work comparing the behavior of different albumins suggests that similar hydrogels could be prepared by human serum albumin.[ 11 , 56 ]
Previous albumin‐based hydrogels were chemically modified to introduce reactive groups on the protein for its subsequent cross‐linking into 3D networks.[ 7 , 8 , 9 , 11 ] While such approaches result in more robust hydrogels due to covalent bond formation, they are associated with extra reaction and purification steps, concerns of side effects and extra costs. Amyloid‐like fibril formation of non‐modified albumin was previously considered, but usually entailed a heating step that most likely causes protein degradation, as shown for similar proteins undergoing amyloidogenesis.[ 57 ] In this work we have investigated the formation of BSA hydrogels through disulfide reduction at room temperature. TCEP acts as the reducing agent, breaking some of the 17 disulfide bonds present in BSA. The number of exposed or partially buried disulfides depends on the pH of the solution and only ≈ 5 disulfide bonds have been reported to be solvent‐accessible and cleaved by TCEP, without the use of other denaturants.[ 58 , 59 ] While the exact number of reduced disulfides was not determined in this study, the fact that above a certain TCEP:BSA ratio there was no change in ThT fluorescence of the amyloid fibrils (Figure 3), nor a shift in mechanical properties of the hydrogels (Figure 2), supports the idea that the limiting factor is disulfide accessibility. Following amyloid‐based hydrogel formation, TCEP was washed away; however, its use raised concerns over possible effects of its remnants on living matter. The performed cell experiments demonstrated no adverse effects on cells, even in long‐term culture. Moreover, the replacement with alternative, biocompatible reducing agents such as glutathione is a possibility that merits testing.
The assembly of high aspect ratio structures does not necessarily designate amyloid fibril formation,[ 57 , 60 , 61 , 62 ] nor is there a single type of amyloid fibril, as polymorphism was previously shown in various systems.[ 15 , 33 ] Secondary structure characterization of the fibrils prepared through TCEP‐treatment of albumin confirmed the presence of beta‐sheet formation, which was further corroborated by ThT fluorescence measurements, validating the presence of amyloid‐like fibrils. However, the structure of these amyloid‐like fibrils was not unique, but instead was determined by the pH of the solution during amyloid assembly. In particular, fibrils formed at pH 7.4 had a negative surface charge, were thinner and tended to interact more with each other as evidenced by AFM imaging (Figure 4). At acidic pH (3.6), below the isoelectric point of albumin, the surface charge of the fibrils was positive and fibrils were thicker. A more detailed structural analysis using X‐ray scattering[ 10 , 57 ] and/or electron microscopy techniques[ 60 ] could provide the atomic‐scale structure of the formed fibrils in the future.
The formation of the 3D network from amyloid‐like fibrils above a threshold concentration occurred through physical interactions. The hydrogels were stable following several washes with PBS and remained intact for days in PBS or cell culture medium, whereas incubation with 6 m of the chaotropic agent Gu‐HCl resulted in rapid (within minutes) gel dissolution. Moreover, the self‐healing behavior of the hydrogels (Figure 6) supports the conclusion that physical interactions are keeping the gel intact. The nature of these inter‐fibril interactions was not elucidated in this work, but our data point toward major differences between the nature of these interactions for the hydrogels prepared at different pH values. For hydrogels prepared at acidic pH, the swelling studies suggested electrostatic interactions being critical: hydrogels dissolved over time in presence of pure water, but not in 150 mm NaCl solution, which was expected to screen charges via the Debye–Hückel double layer of ions. This finding indicated that the electrostatic repulsion between fibrils eventually drove them apart from each other in an excess of solvent. This effect should not be confounded with the effect of salt during amyloid fibril formation: when NaCl was added in the precursor mixture, albumin precipitated rapidly, causing phase separation and an increase in turbidity, similar to what was previously reported in literature.[ 63 ] The use of different salts to distinguish the relative contributions of charge screening versus ion binding to charged elements of proteins could provide a more detailed understanding of the inter‐fibrillar interactions.[ 40 ] On the other hand, hydrogels prepared at pH 7.4 did not swell considerably in water or PBS, indicating that hydrophobic interactions are more likely to contribute in 3D network formation.
The differences in amyloid‐like fibril structure and inter‐fibril interactions for hydrogels prepared at the different pH values also translated into distinct mechanical properties of the resulting hydrogels. Hydrogels prepared at pH 7.4 exhibited shear thinning, making them interesting candidates for injectable formulations. When hydrogels were instead prepared at pH 3.6 the hydrogel stiffened with increasing strain, before losing integrity macroscopically. The recent appreciation that dynamic, viscoelastic properties of the substrate control cell physiology, beyond stiffness,[ 64 ] makes BSA amyloid‐based hydrogels a worthwhile system to investigate toward that direction as well.
The surface of amyloid‐based albumin hydrogels prepared at physiological pH was hydrophobic, and exhibited a contact angle of ≈ 50° when prepared at pH 3.6. Importantly, incubation with fibronectin for both conditions led to its physical adsorption on their surface, which enabled the adhesion and spreading of cells on the hydrogels. The Fn coating was necessary for cell adhesion, since cells cultured on uncoated hydrogels were unable to adhere. It is important to note that Fn was coated directly on prepared hydrogels, and was retained after washing. When hydrogels were washed prior to Fn addition, the amount of adsorbed Fn decreased dramatically. We speculated that washing might affect surface hydrophilicity; indeed, when the contact angles were measured a few minutes after washing with PBS, the surface was hydrophilic with a significant drop in contact angle (Figure S5, Supporting Information). The hydrophobicity of the surface recovered with time in air, reminiscent of the behavior of silicone elastomers upon surface oxidation.[ 65 ] This type of recover further points to the dynamic, physical character of the hydrogel network.
Previous studies using amyloid fibrils have reported contradictory findings in terms of cell adhesion. Some studies showed that the resulting coatings were antifouling,[ 36 , 56 ] while others claimed that amyloid fibrils are permissive to cell adhesion, even in absence of adhesive ligands.[ 66 , 67 ] Our findings using two different cell types agree with the former result, and clearly demonstrated that the hydrogels presented here do not support cell adhesion without prior functionalization with Fn. A number of reasons may be responsible for the conflicting reports. The thickness of the coating might influence the ability of cells to sense the underlying substrate. In studies that described cell adhesion on amyloid fibrils, the fibrils were deposited directly on stiff substrates, such as glass or mica[ 66 , 67 ]; here instead a viscoelastic hydrogel was formed, and in the case of the work from Hu et al.[ 36 ] a film of 30–130 nm was assembled. Different experimental protocols for amyloid assembly or adhesion evaluation could also influence the results. For example, in this work we removed non‐adherent cells 1 h after cell seeding; prolonged incubation of non‐adherent cells with the substrates might have enabled cells to eventually adhere to the fibrils or even interact with the surface in between these.
As a first step toward potential bio‐applications of the amyloid‐based albumin hydrogels presented here, we showed that they support cell culture over prolonged periods of time. Albumin hydrogels were so far considered for a number of applications to serve as drug delivery depots, implantable materials or coatings. Interestingly, albumin gels have also shown favorable antifouling and anti‐bacterial properties. In one study, albumin was used to prepare amyloid‐based films as versatile coating that exhibited protection against microorganisms and bacterial strains.[ 36 ] Overall, the hydrogels presented here merit further investigation as inexpensive, sustainable, versatile and tunable biomaterials.
4. Experimental Section
Materials
The reagents and antibodies used in this study are given in Tables 1 and 2 , respectively.
Table 1.
Reagents used in our study
| Reagent | Abbreviation | Supplier | Cat. No. |
|---|---|---|---|
| Bovine serum albumin | BSA | Sigma | A7030 |
| Fibronectin | Fn | Sigma | F1141‐1MG |
| Fibronectin HiLyte 488 | FFn | Cytoskeleton | FNR02‐A |
| Sodium hydroxide solution 1 N | NaOH | ||
| Tris(2‐carboxyethyl) phosphine Hydrochloride | TCEP | Sigma | C4706 |
| Thioflavin T | ThT | Sigma | T3516 |
| Hydrochloric acid | HCl | Merck | 109057 |
| Phosphate buffer saline | PBS | ||
| (3‐Aminopropyl)triethoxysilane | APTES | Sigma‐Aldrich | 440140 |
| 4′,6‐Diamidino‐2‐phenylindol | DAPI | Thermo Fisher | D1306 |
| Phalloidin‐tetramethylrhodamine B isothiocyanate | TRITC‐phalloidin | Sigma | P1951 |
| 4% Paraformaldehyde | PFA | Santa Cruz Biotechnologies | 30525‐89‐4 |
| Triton X‐100 | — | Thermo Fisher | HFH10 |
| Silicone oil AP 150 Wacker | — | Sigma | 50384 |
| Sodium deuteroxide solution | NaOD | Sigma | 372072 |
| Deuterium Oxide | D2O | Carl Roth | 6672.2 |
| Guanidinium chloride | Gu HCl | Sigma | G3272 |
| Calcein‐acetoxymethylester | Calcein‐AM | Sigma | 17783 |
| Ethidium homodimer | EthD | Sigma | 46043 |
Table 2.
Antibodies used in our study
| Antibody | Clone | Application/Dilution | Supplier | Cat. No. |
|---|---|---|---|---|
| Anti‐fibronectin | P1H11 | ELISA 1:5000 | Millipore | MAB1926 |
| Anti‐pY | PY99 | Immunofluorescence/1:100 | Santa Cruz | sc‐7020 |
| Anti‐mouse IgG AlexaFluor488 conjugate | Polyclonal | Immunofluorescence/1:150 | Thermo‐Fisher | A11001 |
| Anti‐mouse HRP conjugate | Polyclonal | ELISA/1:2000 | R&D | HAF007 |
Hydrogel Formation
BSA was dissolved in Milli‐Q water, filtered (0.22 µm filter) and mixed with predetermined amounts of 1 n NaOH or 1 n HCl. A fresh 1 m solution of TCEP was prepared in Milli‐Q water and mixed with the BSA solution to give the desired concentration of BSA and TCEP. In the case of heating, a glass vial was placed in an oil bath set at 90 °C under magnetic stirring.
ThT Assay
For the ThT assay, 4 µL aliquots from the different BSA samples (pH 3.6 and pH 7.4) were mixed with 196 µL of a 10 µm ThT solution in 10 mm PBS. The fluorescence measurements were carried out using a SPARK Multimode Microplate Reader (TECAN) at 25 °C in a flat, black 96‐well plate. The excitation wavelength was set at 440 nm and the emission was measured at 485 nm. ThT fluorescence was also monitored during BSA fibril formation by incubating ThT (5 µm) with a mixture of BSA and TCEP directly in the 96‐well plate. In this case, the pH of the solution was kept at 7.4 through addition of 1 n NaOH.
Rheology
All rheology measurements were performed on a Malvern Kinexus rheometer. The precursor solution was prepared, and TCEP was added last. The solution (500 µL) was then placed on the bottom plate and the top flat plate was used to compress the liquid so that the whole surface was covered and a positive meniscus was formed between the plates (gap between 1.3 and 1.4 mm). Immediately after, a kinetic measurement was performed by measuring every minute the shear elastic and viscous moduli in oscillatory mode, at a frequency of 1 Hz and 1% strain, for a total of 4 h. The temperature was set at 25 °C. The beginning of the measurement was initialized within 1 min from TCEP addition. Next, a frequency sweep (0.1–100 Hz) and strain sweep (0.1–10%) were performed. Silicone oil was then applied to prevent evaporation and the sample was left overnight. After 24 h, a frequency sweep (0.1–10 Hz) was performed.
Atomic Force Microscopy Imaging
For AFM imaging, samples were prepared by diluting the BSA/TCEP mixtures with Milli‐Q water to a final protein concentration of 0.01% w/v. Then, 20 µL aliquots were deposited on freshly cleaved mica for 1 min, rinsed with Milli‐Q water and gently dried with N2 flow. For the acidic BSA/TCEP samples, mica surfaces were coated with APTES. Therefore, freshly cleaved mica and 1 mL APTES in a small open glass vial were placed inside a desiccator. Vacuum was applied and the mica surfaces were left overnight inside the desiccator. AFM imaging was carried out with a JPK Nanowizard 3 operated in air tapping mode and with NSG01 (NT‐MDT) cantilevers having a resonance frequency of ≈ 150 kHz.
Circular Dichroism Spectroscopy
The protein samples were diluted to 0.02% (m/v) in water and CD spectra at 25 °C were collected in quartz cuvettes with 1 mm optical path length using a Jasco J‐810 Spectropolarimeter equipped with a Peltier temperature‐controlled cell holder. CD spectra were measured in continuous scanning mode between 190 and 260 nm at a rate of 50 nm min−1, with a step size of 1 nm, response time of 0.5 s, 1 nm bandwidth, and ten accumulations. The baseline scans were subtracted from the protein sample spectra, which were subsequently smoothed using the Means‐Movement method with a convolution width of 5. Secondary structure prediction from the measured spectra was performed using the web‐based server analysis tool CAPITO.[ 68 ]
FTIR Measurements
Attenuated Total Reflection (ATR) FTIR Spectroscopy was performed on a Shimadzu IR Tracer‐100 FT‐IR spectrometer equipped with a MCT detector. 4 µL of a BSA precursor solution prepared in D2O was pipetted onto the surface of the prism and the spectral recording was initiated. The TCEP solution was also prepared in D2O and pD was adjusted with NaOD The spectra were obtained with the following parameters: resolution 2 cm−1, range from 1800 to 1400 cm−1, 100–512 scans recorded, scanning velocity: 20 kHz, zero‐filling factor: 4, apodization function: a Blackman‐ Harris 3‐term. As a reference, D2O cell was used.
Zeta Potential Measurements
In order to measure the surface zeta potential of BSA amyloid‐like fibrils, 2% BSA solutions with 16 mm of TCEP at different pH conditions were prepared. The samples were kept at room temperature during 24 h. For the measurement, the protein samples were diluted to 1:20 in Milli‐Q water and collected into a DTS1070 capillary zeta cell from Malvern. The surface zeta potential was determined using a Zetasizer Nano ZS instrument.
Contact Angle Measurements
For contact angle measurements, 500 µL gels were formed for 24 h in a humidified atmosphere at room temperature on a 24 × 60 glass coverslip. Measurements were performed in the middle of gels, which presented a quasi‐flat surface. The sessile drop technique was used for optical measurement of the contact angle using a Drop Shape Analyzer DSA25 (Krüss, Germany). The volume of the droplets was 10 µL. The drop profile was fitted by an ellipse (tangent−1) function provided by the Krüss ADVANCE software in order to get the contact angle. The mean value from the left and right contact angles was used. For the contact angle determination, two independent experiments were performed.
Swelling Measurements
For swelling measurements, 250 µL of the BSA‐TCEP mixture was deposited on a 24 × 24 coverslip glass for 24 h. These glass‐supported gels were weighted and then, were placed on a 6‐well plate. The gels were completely covered with the different solutions. After certain time, the gels were removed from the wells, carefully dried and weighted again. Two independent experiments were performed.
Self‐Healing Measurements
In order to verify the self‐healing nature of the hydrogels, two experiments were performed. First, a 5% BSA/40 mm TCEP gel at pH 3,6 was polymerized into a glass vial for 24 h. Using a needle (StericanIntramuskular G18), the gel was damaged. The needle was removed and the hydrogel was left under static conditions during the next 24 h. Photographs were acquired at regular time intervals. For the second experiment, a 10% BSA/80 mm TCEP at pH 3.6 was polymerized for 24 h. The free‐standing BSA gel was then broken into small pieces. These pieces were brought together into a Teflon mold during another 24 h. Finally, the gel was removed from the mold and manipulated with tweezers in order to confirm the self‐healing ability of the gel.
Protein Adsorption
Bovine plasma fibronectin (Fn) was coated on BSA hydrogels prepared 16–24 h before. The relative coating efficiencies were calculated through a modified ELISA assay as previously described.[ 69 ] Of note, washing of hydrogels prior to coating dramatically reduced coating efficiency. Additionally, pipetting on top of the gel should be avoided as shear stress appeared to damage the hydrogel surface. For visualizing Fn adsorption, BSA hydrogels were inverted on a droplet of fluorescently‐labeled Fn solution (10 µg mL−1 in PBS) that was placed on parafilm. The hydrogels were incubated for 1 h at room temperature and washed four times for 5 min with an excess of PBS prior to imaging. Z‐stacks from the hydrogel interior to the PBS solution above were acquired on a Zeiss LSM 880 laser scanning confocal microscope (LSCM) using a 20×/0.8 NA objective (Figure 7C). For cell experiments, a 10 µg mL−1 solution of non‐labeled Fn in PBS was used, and gels were washed four times for 5 min with an excess of PBS prior to cell seeding.
Cell Culture
U2OS cells, a human osteosarcoma cell line, was bought from the DMSZ‐German collection of microorganisms and cell cultures GmbH and gifted by the laboratory of Kai Johnsson (Max Planck Institute for medical research, Heidelberg, Germany). U2OS were cultured in McCoy 5A medium supplemented with 10% fetal bovine serum (FBS) and 100 U mL−1 penicillin‐streptomycin at 37 °C and 5% CO2. NIH3T3 cells were purchased from ATCC and cultured in Dulbecco's modified eagle medium (DMEM; Gibco 41 965) supplemented with 10% fetal bovine serum (FBS) and 100 U mL−1 penicillin‐streptomycin.
Cell Experiments
Cells were seeded on hydrogels at a cell density of 5000 cells per cm2 in supplemented culture medium. After 1 h incubation, non‐adherent and loosely‐attached cells were removed by exchanging once the medium. For live‐cell, time‐lapse imaging, cells were transferred to a Leica DMi8 microscope, equipped with an environmental chamber and imaged using a 10×/0.32 NA objective. For immunofluorescence studies, cells were fixed with 4% PFA in PBS for 10 min, 4 h after cell seeding. Cells were then washed with PBS, permeabilized with 0.1% Triton X‐100 in PBS for 5 min, blocked with 1% BSA in PBS for 30 min and then washed three times for 5 min with PBS. Primary anti‐pY antibodies (diluted 1:100 in 1% BSA) were incubated for 1 h at room temperature or overnight at 4 °C. Cells were washed and incubated with secondary Alexa Fluor‐labeled antibodies (diluted 1:150 in 1% BSA) for 1 h at room temperature. DAPI and TRITC‐phalloidin were used to stain nuclei and filamentous actin (F‐actin). Images were acquired on a Zeiss LSM 880 laser scanning confocal microscope (LSCM) using a 63×/1.4 NA oil‐immersion objective (Zeiss).
Cell adhesion and viability on 5% BSA hydrogels prepared with 40 µm TCEP at pH 3.6 or pH 7.4 were quantified using the cell viability dyes calcein and ethidium homodimer. Hydrogels were prepared on 20 × 20 coverslips that were previously treated in O2 plasma (0.4 mBar, 150 W) for 5 min to improve wettability. After hydrogel formation overnight, the coverslips were transferred to 6‐wells and hydrogels were incubated with 10 µg mL−1 Fn in PBS or just with PBS as a control for 1 h. Then hydrogels were washed four times with an excess of PBS inside the wells and incubated with supplemented cell culture medium for 10 min prior to cell seeding. Cells (5 × 103 cells per cm2) were seeded on the functionalized and control hydrogels, as well as on Fn‐coated glass coverslips; after 1 h, non‐adherent cells were removed following aspiration and addition of fresh culture medium. Cells were incubated for 20 h. Then, cells were incubated with 1 µm calcein AM and 2 µm ethidium homodimer in PBS for 30 min, transferred to the Leica DMi8 microscope, equipped with an environmental chamber, and imaged using a 10×/0.32 NA objective. 4–6 random field of views were acquired for each sample. The number of live cells, identified by a strong calcein AM fluorescent signal, and the number of dead cells, identified by a strong ethidium homodimer signal were counted. Cell adhesion was expressed as live cells per cm2 and cell viability was expressed as the number of live cells/total (live + dead) cells. Three independent experiments were performed.
Statistical Analysis
Statistical analyses were performed using Prism (version 9; GraphPad Inc.). Experimental data were analyzed using a Brown‐Forsythe and Welch ANOVA statistical test or unpaired t‐tests with Welch's correction as indicated in figure legends. Exact p values are presented in graphs only when p < 0.05.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supplemental Movie 1
Acknowledgements
The authors thank Prof. Joachim P. Spatz for hosting the work in his department at the Max Planck Institute for Medical Research. The authors also thank Mr. Lukas Brenner for assistance with experiments, Dr. Miroslav Tarnawski for assistance with circular dichroism measurements, and Dr. Tamás Haraszti for comments on the manuscript. C.D. gratefully acknowledges the Alexander von Humboldt Foundation for the Georg Forster Fellowship for Experienced Researchers.
Open access funding enabled and organized by Projekt DEAL.
Diaz C., Missirlis D., Amyloid‐Based Albumin Hydrogels. Adv. Healthcare Mater. 2023, 12, 2201748. 10.1002/adhm.202201748
Contributor Information
Carolina Diaz, Email: carolina.diaz@mr.mpg.de.
Dimitris Missirlis, Email: dimitris.missirlis@mr.mpg.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Gaharwar A. K., Singh I., Khademhosseini A., Nat. Rev. Mater. 2020, 5, 686. [Google Scholar]
- 2. Kratochvil M. J., Seymour A. J., Li T. L., Paşca S. P., Kuo C. J., Heilshorn S. C., Nat. Rev. Mater. 2019, 4, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dimatteo R., Darling N. J., Segura T., Adv. Drug Delivery Rev. 2018, 127, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caliari S. R., Burdick J. A., Nat. Methods 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Slaughter B. V., Khurshid S. S., Fisher O. Z., Khademhosseini A., Peppas N. A., Adv. Mater. 2009, 21, 3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ong J., Zhao J., Justin A. W., Markaki A. E., Biotechnol. Bioeng. 2019, 116, 3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mao S.‐Y., Peng H.‐W., Wei S.‐Y., Chen C.‐S., Chen Y.‐C., ACS Biomater. Sci. Eng. 2021, 7, 3293. [DOI] [PubMed] [Google Scholar]
- 8. Xia T., Jiang X., Deng L., Yang M., Chen X., Colloids Surf., B 2021, 208, 112042. [DOI] [PubMed] [Google Scholar]
- 9. Hong L., Chen G., Cai Z., Liu H., Zhang C., Wang F., Xiao Z., Zhong J., Wang L., Wang Z., Cui W., Adv. Sci. (Weinh) 2022, 9, 2200281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amdursky N., Mazo M. M., Thomas M. R., Humphrey E. J., Puetzer J. L., St‐Pierre J.‐P., Skaalure S. C., Richardson R. M., Terracciano C. M., Stevens M. M., J. Mater. Chem. B 2018, 6, 5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharifi S., Saei A. A., Gharibi H., Mahmoud N. N., Harkins S., Dararatana N., Lisabeth E. M., Serpooshan V., VéGvári Á. K., Moore A., Mahmoudi M., ACS Appl. Bio. Mater. 2022, 5, 2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sawaya M. R., Hughes M. P., Rodriguez J. A., Riek R., Eisenberg D. S., Cell 2021, 184, 4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cereghetti G., Wilson‐Zbinden C., Kissling V. M., Diether M., Arm A., Yoo H., Piazza I., Saad S., Picotti P., Drummond D. A., Sauer U., Dechant R., Peter M., Nat. Cell Biol. 2021, 23, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maji S. K., Perrin M. H., Sawaya M. R., Jessberger S., Vadodaria K., Rissman R. A., Singru P. S., Nilsson K. P. R., Simon R., Schubert D., Eisenberg D., Rivier J., Sawchenko P., Vale W., Riek R., Science 2009, 325, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cao Y., Adamcik J., Diener M., Kumita J. R., Mezzenga R., J. Am. Chem. Soc. 2021, 143, 11473. [DOI] [PubMed] [Google Scholar]
- 16. Belwal V. K., Chaudhary N., Soft Matter 2020, 16, 10013. [DOI] [PubMed] [Google Scholar]
- 17. Yang L., Li H., Yao L., Yu Y., Ma G., ACS Omega 2019, 4, 8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou J., Li T., Peydayesh M., Usuelli M., Lutz‐Bueno V., Teng J., Wang L., Mezzenga R., Adv. Sci. 2021, 9, e2104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang D., Ha Y., Gu J., Li Q., Zhang L., Yang P., Adv. Mater. 2016, 28, 7414. [DOI] [PubMed] [Google Scholar]
- 20. Palika A., Armanious A., Rahimi A., Medaglia C., Gasbarri M., Handschin S., Rossi A., Pohl M. O., Busnadiego I., GüBeli C., Anjanappa R. B., Bolisetty S., Peydayesh M., Stertz S., Hale B. G., Tapparel C., Stellacci F., Mezzenga R., Nat. Nanotechnol. 2021, 16, 918. [DOI] [PubMed] [Google Scholar]
- 21. Ha Y., Kwon Y., Nam E.‐J., Park H., Paik S. R., Acta Biomater. 2022, 145, 52. [DOI] [PubMed] [Google Scholar]
- 22. Das S., Jacob R. S., Patel K., Singh N., Maji S. K., Biomacromolecules 2018, 19, 1826. [DOI] [PubMed] [Google Scholar]
- 23. Xuan Q., Wang Y., Chen C., Wang P., Front. Bioeng. Biotechnol. 2021, 9, 718883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lendel C., Solin N., RSC Adv. 2021, 11, 39188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baler K., Michael R., Szleifer I., Ameer G. A., Biomacromolecules 2014, 15, 3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borzova V. A., Markossian K. A., Chebotareva N. A., Kleymenov S. Y., Poliansky N. B., Muranov K. O., Stein‐Margolina V. A., Shubin V. V., Markov D. I., Kurganov B. I., PLoS One 2016, 11, e0153495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Juárez J., Taboada P., Mosquera V.C., Biophys. J. 2009, 96, 2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khanna S., Singh A. K., Behera S. P., Gupta S., Mater. Sci. Eng., C 2021, 119, 111590. [DOI] [PubMed] [Google Scholar]
- 29. Pearce F. G., Mackintosh S. H., Gerrard J. A., J. Agric. Food Chem. 2007, 55, 318. [DOI] [PubMed] [Google Scholar]
- 30. Murata M., Tani F., Higasa T., Kitabatake N., Doi E., Biosci. Biotechnol., Biochem. 1993, 57, 43. [DOI] [PubMed] [Google Scholar]
- 31. Zhang X., Jiang S., Yan T., Fan X., Li F., Yang X., Ren B.o, Xu J., Liu J., Soft Matter 2019, 15, 7583. [DOI] [PubMed] [Google Scholar]
- 32. Navarra G., Peres C., Contardi M., Picone P., San Biagio P. L., Di Carlo M., Giacomazza D., Militello V., Arch. Biochem. Biophys. 2016, 606, 134. [DOI] [PubMed] [Google Scholar]
- 33. Usov I., Adamcik J., Mezzenga R., Faraday Discuss. 2013, 166, 151. [DOI] [PubMed] [Google Scholar]
- 34. Arabi S. H., Aghelnejad B., Schwieger C., Meister A., Kerth A., Hinderberger D., Biomater. Sci. 2018, 6, 478. [DOI] [PubMed] [Google Scholar]
- 35. Liu R., Zhao J., Han Q., Hu X., Wang D., Zhang X.u, Yang P., Adv. Mater. 2018, 30, 1802851. [DOI] [PubMed] [Google Scholar]
- 36. Hu X., Tian J., Li C., Su H., Qin R., Wang Y., Cao X., Yang P., Adv. Mater. 2020, 32, 2000128. [DOI] [PubMed] [Google Scholar]
- 37. Wu Z., Yang P., Adv. Mater. Interfaces 2015, 2, 1400401. [Google Scholar]
- 38. Li C., Xu L., Zuo Y. Y., Yang P., Biomater. Sci. 2018, 6, 836. [DOI] [PubMed] [Google Scholar]
- 39. Xuan Q., He J., Zhang W., Zhang W., Zhang Q.i, Zhou Y., Wei A., Wang H., Li H., Chen C., Wang P., Biomacromolecules 2022, 23, 3318. [DOI] [PubMed] [Google Scholar]
- 40. Goto Y., Adachi M., Muta H., So M., Biophys. Rev. 2018, 10, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahmad B., Kamal M., Khan R., Protein Pept. Lett. 2004, 11, 307. [DOI] [PubMed] [Google Scholar]
- 42. Arad E., Green H., Jelinek R., Rapaport H., J. Colloid Interface Sci. 2020, 573, 87. [DOI] [PubMed] [Google Scholar]
- 43. Moran S. D., Zanni M. T., J. Phys. Chem. Lett. 2014, 5, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zou Y., Li Y., Hao W., Hu X., Ma G., J. Phys. Chem. B 2013, 117, 4003. [DOI] [PubMed] [Google Scholar]
- 45. Hoshino K.‐I., Nakajima T., Matsuda T., Sakai T., Gong J. P., Soft Matter 2018, 14, 9693. [DOI] [PubMed] [Google Scholar]
- 46. Zaguri D., Shaham‐Niv S., Chakraborty P., Arnon Z., Makam P., Bera S., Rencus‐Lazar S., Stoddart P. R., Gazit E., Reynolds N. P., ACS Appl. Mater. Interfaces 2020, 12, 21992. [DOI] [PubMed] [Google Scholar]
- 47. Bolisetty S., Harnau L., Jung J.‐M.i, Mezzenga R., Biomacromolecules 2012, 13, 3241. [DOI] [PubMed] [Google Scholar]
- 48. Jacob R. S., Ghosh D., Singh P. K., Basu S. K., Jha N. N., Das S., Sukul P. K., Patil S., Sathaye S., Kumar A., Chowdhury A., Malik S., Sen S., Maji S. K., Biomaterials 2015, 54, 97. [DOI] [PubMed] [Google Scholar]
- 49. Pankov R., Yamada K. M., J. Cell Sci. 2002, 115, 3861. [DOI] [PubMed] [Google Scholar]
- 50. Hotulainen P., Lappalainen P., J. Cell Biol. 2006, 173, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wong S., Guo W.‐H., Wang Y.‐L., Proc. Natl. Acad. Sci. USA 2014, 111, 17176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Levin A., Hakala T. A., Schnaider L., Bernardes G. J. L., Gazit E., Knowles T. P. J., Nat. Rev. Chem. 2020, 4, 615. [Google Scholar]
- 53. Aida T., Meijer E. W., Stupp S. I., Science 2012, 335, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Black M., Trent A., Kostenko Y., Lee J. S., Olive C., Tirrell M., Adv. Mater. Weinheim 2012, 24, 3845. [DOI] [PubMed] [Google Scholar]
- 55. Okesola B. O., Wu Y., Derkus B., Gani S., Wu D., Knani D., Smith D. K., Adams D. J., Mata A., Chem. Mater. 2019, 31, 7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ong J., Zhao J., Levy G. K., Macdonald J., Justin A. W., Markaki A. E., Sci. Rep. 2020, 10, 12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lara C., Gourdin‐Bertin S., Adamcik J., Bolisetty S., Mezzenga R., Biomacromolecules 2012, 13, 4213. [DOI] [PubMed] [Google Scholar]
- 58. Katchalski E., Benjamin G. S., Gross V., J. Am. Chem. Soc. 1957, 79, 4096. [Google Scholar]
- 59. Iyer K. S., Klee W. A., J. Biol. Chem. 1973, 248, 707. [PubMed] [Google Scholar]
- 60. Pohl C., Effantin G., Kandiah E., Meier S., Zeng G., Streicher W., Segura D. R., Mygind P. H., Sandvang D., Nielsen L. A., Peters G. H. J., Schoehn G., Mueller‐Dieckmann C., Noergaard A., Harris P., Nat. Commun. 2022, 13, 3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gosal W. S., Morten I. J., Hewitt E. W., Smith D. A., Thomson N. H., Radford S. E., J. Mol. Biol. 2005, 351, 850. [DOI] [PubMed] [Google Scholar]
- 62. Tayeb‐Fligelman E., Tabachnikov O., Moshe A., Goldshmidt‐Tran O., Sawaya M. R., Coquelle N., Colletier J.‐P., Landau M., Science 2017, 355, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hill S. E., Miti T., Richmond T., Muschol M., PLoS One 2011, 6, e18171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chaudhuri O., Cooper‐White J., Janmey P. A., Mooney D. J., Shenoy V. B., Nature 2020, 584, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Morra M., Occhiello E., Marola R., Garbassi F., Humphrey P., Johnson D., J. Colloid Interface Sci. 1990, 137, 11. [Google Scholar]
- 66. Reynolds N. P., Charnley M., Bongiovanni M. N., Hartley P. G., Gras S. L., Biomacromolecules 2015, 16, 1556. [DOI] [PubMed] [Google Scholar]
- 67. Jacob R. S., George E., Singh P. K., Salot S., Anoop A., Jha N. N., Sen S., Maji S. K., J. Biol. Chem. 2016, 291, 5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wiedemann C., Bellstedt P., Görlach M., Bioinformatics 2013, 29, 1750. [DOI] [PubMed] [Google Scholar]
- 69. Missirlis D., Haraszti T. S., Heckmann L., Spatz J. P., Biophys. J. 2020, 119, 2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Movie 1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.