

Abstract
Messenger RNA (mRNA)‐based therapies offer great promise for the treatment of a variety of diseases. In 2020, two FDA approvals of mRNA‐based vaccines have elevated mRNA vaccines to global recognition. However, the therapeutic capabilities of mRNA extend far beyond vaccines against infectious diseases. They hold potential for cancer vaccines, protein replacement therapies, gene editing therapies, and immunotherapies. For realizing such advanced therapies, it is crucial to develop effective carrier systems. Recent advances in materials science have led to the development of promising nonviral mRNA delivery systems. In comparison to other carriers like lipid nanoparticles, polymer‐based delivery systems often receive less attention, despite their unique ability to carefully tune their chemical features to promote mRNA protection, their favorable pharmacokinetics, and their potential for targeting delivery. In this review, the central features of polymer‐based systems for mRNA delivery highlighting the molecular design criteria, stability, and biodistribution are discussed. Finally, the role of targeting ligands for the future of RNA therapies is analyzed.
Keywords: ligand‐mediated targeting, messenger RNA, polymer‐based delivery systems
Messenger RNA (mRNA) has emerged as a powerful and versatile platform for treating different diseases. Polymer‐based nanovectors exhibit distinct advantages for mRNA delivery, though they still present several challenges for breaking into the mainstream. This review provides insights on polymeric carriers for constructing efficient mRNA delivery platforms toward future therapies.
1. Introduction
The delivering of messenger RNA (mRNA) is a versatile and powerful approach for next‐generation therapies.[ 1 ] The recent approval of two mRNA‐based vaccines for COVID‐19 is a testament of the potential of this strategy holds.[ 2 ] For successful mRNA‐based treatments, mRNA must reach the cytosol of cells and engage with the translation machinery to produce therapeutic proteins they encode. However, mRNA is rapidly degraded by nucleases, has limited cellular uptake, and inherent immunogenicity on its own.[ 1 , 3 ] For that reason, the development of safe and effective vehicles is essential for the application of mRNA as a therapeutic agent.
Various types of vectors are being developed for mRNA delivery.[ 1 , 4 ] Common challenges of these vectors relate to increasing translation efficiency, promoting advantageous pharmacokinetic profiles with high mRNA stability, achieving tissue and cellular selectivity, and employing simple manufacturing and storage. Viral vectors have shown outstanding properties for delivering mRNA, such as in vivo application and high translation rates.[ 5 ] However, they are limited by their maximum packing size, cytotoxicity, immunogenicity and intricate manufacturing.[ 6 ] Nonviral vectors based on biocompatible materials have potential for overcoming the drawbacks of viruses through precise engineering of their nanostructure. These nonviral carriers are mainly prepared from lipids, polymers, and their combinations.[ 4 , 7 ] Lipid nanoparticles (LNPs) are at the forefront of clinical translation, showing high delivery efficiency. mRNA formulated in LNPs can be delivered to several organs and cell types, leading to a variety of therapeutic applications, for example, vaccines for cancer and infectious diseases.[ 1 , 2 , 8 ] These advanced studies have demonstrated the safety and high translation ability of lipid carriers, but also identified limitations, such as thermostability issues that correspond with expensive cold chains and logistics,[ 9 ] and in many cases tropism to the liver.[ 10 ] While polymer‐based mRNA delivery systems are less advanced in the clinic than lipids and show certain challenges, such as relatively low transfection efficiency and potential toxicities,[ 11 ] they have potential for providing unique features, such as the assembly of various nanostructures in aqueous conditions, ability of lyophilization and long‐term storage,[ 12 ] and distinct pharmacokinetics that could be useful for developing advanced mRNA therapies. However, it is important to note that precise engineering of the polymers, controlled chemical structure, and high batch‐to‐batch reproducibility are key features for the translation of polymer‐based mRNA delivery systems into therapeutics. In this regard, not all polymer‐based systems may satisfy the abovementioned properties, and thus, may not be suitable for clinical application.[ 13 ] In our work, we discussed the unique strengths, advantages, and disadvantages of polymer systems. In every system, when the data were available, we discussed the polymer structure, in vitro and in vivo stability, biodistribution, and outcomes of the in vivo studies. We also reviewed current ligand strategies capable of translation efficacy, and cellular and tissue targeting by improving selectivity and intracellular delivery.
2. Current Designs
A variety of polymeric structures have been suggested for delivering nucleic acids. Major approaches include polyplexes, sterically stabilized polyplexes, and polymeric micelles. Particularly, polymer‐based approaches have shown capability for enhancing the delivery of different nucleic acids by protecting them from degradation and promoting cellular uptake and endosomal escape. Along with the development of new synthetic polymers, routes of administration have been expanded besides parenteral (intravascular, intramuscular, subcutaneous, and inhalation administration) administration, and several studies have even demonstrated oral delivery of nucleic acids using polymeric nanoparticles.[ 14 ] In this section, we will introduce each design and highlight recent strategies for delivering mRNA toward in vivo application.
2.1. Polyplexes
A simple form of polymeric cargo‐carrier complex consists of a usually spherical particle made up from polymer molecules engaging in electrostatic interactions with negatively charged nucleic acids. These complexes, termed polyplexes, have been extensively studied and reviewed as delivery systems for DNA and RNA‐based drugs. In this section, we give an overview of polymeric carriers used to form polyplexes for mRNA delivery. Moreover, we examined the strategies to adapt them to suit the specific needs for this class of nucleic acid.
2.1.1. PEI
Poly(ethyleneimine) (PEI) (Figure 1 ) is one of the most widely‐used cationic polymers for nucleic acid delivery.[ 15 ] PEI is known for its strong cationic properties contributing to efficient endosomal disruption.[ 16 ] More specifically, PEI has been reported to form relatively large pores in biological membranes at endosomal pH conditions, promoting the escape of PEI‐based carriers from endosomes.[ 17 ] However, this is paired with concerns of poor biocompatibility due to excess cationic charge and lack of biodegradable bonds[ 18 ] (Table 1 ). PEI is toxic, causing necrosis, apoptosis, and inflammation.[ 18 , 19 ] For most mRNA‐based therapeutic strategies, such as vaccination and protein replacement, the cellular damage and the reduction of the cell viability are critical issues. Thus, major efforts have been dedicated to reduce the toxicity of PEI, such as tuning the dose and the molecular weight, introducing degradable disulfide bonds in the PEI structure in the PEI structure[ 18b ] and modifying the surface of PEI‐based polyplexes.[ 20 ] Appropriate in vitro models of toxicity are essential for anticipating the in vivo safety of the engineered polyplexes. In this regard, it is important that the in vitro models ponder the biodistribution of polyplexes and the area under the concentration–time curve (AUC) of the polycations in the tissues, as the toxicities can depend on the cell type, and the dose and time span that the tissues are exposed to the polyplexes, respectively. Additionally, excessive stability of PEI‐RNA complexes has been reported to impair translational efficiency due to limited release from the complex.[ 21 ] For the delivery of RNA, PEI has been used to form polyplexes as‐is[ 22 ] or with modifications to optimize physical characteristics, tolerability, and delivery efficiency.[ 20 , 23 ] Specifically, modulating the hydrophobicity or molecular weight of PEI has resulted in more favorable RNA delivery performance.[ 24 ] Incorporating stearic acid blocks into PEI showed improved transfection in antigen‐presenting cells (APCs) and reduced toxicity, resulting in enhanced anti‐HIV1 gag protein immune responses with high antigen‐specific antibody secretion and pro‐inflammatory cytokine production.[ 25 ] Similarly, deoxycholic acid‐conjugated PEI was used to deliver mRNA into brain. The deoxycholic acid‐PEI/mRNA polyplex could protect mRNAs from fourfold excess heparin attack and RNase attack up to 60 min, resulting in higher mRNA transfection after local injection in the brain compared to larger PEI variants and lipofectamine.[ 26 ] While the applicability of PEI for systemic delivery remains limited, with reports of lung and liver embolisms when polyplexes were formed with linear PEI and DNA.[ 27 ] However, PEI‐based nanoparticles modified with various PEG terminal groups enable targeted delivery to the lungs, despite showing decreased stability against heparin than PEI polyplexes. Especially, PEI polyplexes modified with PEG chains having amino or glycine terminal groups showed the highest gene expression level in the lungs[ 28 ] (Figure 2a). These results indicate the important role of the PEG coating on the performance of polyplexes, as well as spur further work for fully understanding how the terminal group of PEG influences mRNA delivery. Still, most reports of mRNA delivery using PEI focus on applications with local injections, such as vaccines (Table 2 ).[ 23 , 24 , 29 ]
Figure 1.
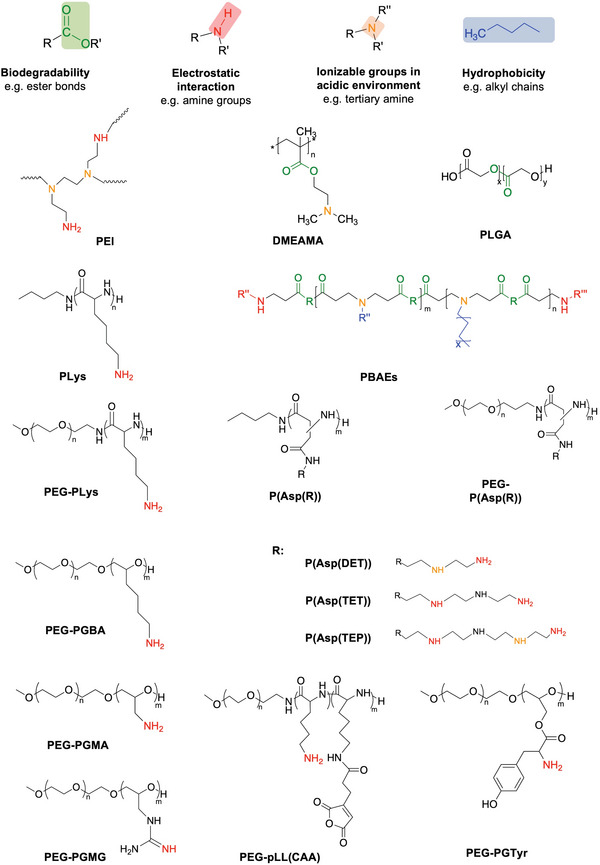
Overview of design elements of polymeric mRNA carriers to improve delivery efficiency (The full name of polymers can be found in the main text.).
Table 1.
Summary of the advantages of disadvantages of representative types of polyplexes for mRNA delivery
| Type of polymer (Examples) a) | Advantages | Disadvantages |
|---|---|---|
| PEI | Efficient endosome escape | Charge‐dependent toxicity; poor biocompatibility; lack of biodegradable bonds |
| Poly(amino acids) (P(Lys), P(Orn), P(Asp)) | Biodegradability; endosomal escape capacity | Charge‐dependent toxicity |
| Polyesters (PLGA, PBAE, PACE) | Biodegradability; facile synthesis monomer availability | No cationic charge; inefficient loading |
| Natural polymers (Chitosan, Protamine) | Biodegradability; Biocompatibility | Low batch‐to‐batch variability |
| RAFT polymer (DMAEMA) | Easy production; well‐controlled side chain moieties | Charge‐dependent toxicity |
| Dendrimers (PAMAM) | Good water solubility; stability against hydrolysis; tunable degradation | Charge‐dependent toxicity especially in high generation of PAMAM; difficulty in synthesizing high molecular weight |
The full name of polymers can be found in the main text.
Figure 2.
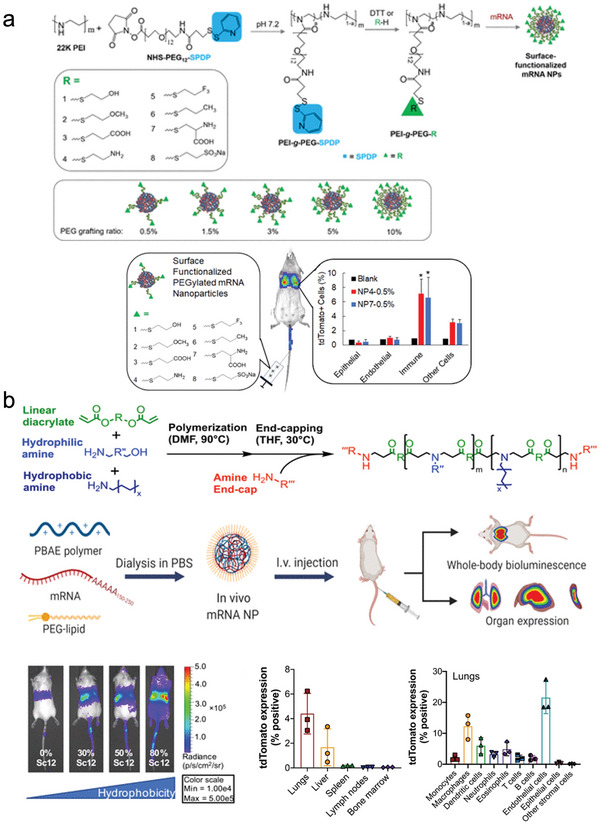
a) PEI‐g‐PEG/mRNA nanoparticles with various PEG terminal groups and PEG grafting ratios deliver mRNA to pulmonary immune cells. Adapted with permission.[ 28 ] Copyright 2020, American Chemical Society. b) Fine‐tuning of PBAE‐derived polymers with hydrophobic side chain and end‐cap allows for systemic mRNA delivery to the lung and the liver. Adapted with permission.[ 48a ] Copyright 2022, American Association for the Advancement of Science (AAAS).
Table 2.
Polyplex systems to deliver mRNA
| Polymer component a) | Molecular weight | Preparation Method | Average size [nm] |
Zeta potential [mV] |
Cation to RNA ratio | Design elements | Experimental application | Outcome b) | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| PEI | 25 kDa | Mixing in HBG (20 × 10‐3 m of HEPES, 5% glucose; pH 7.4) | 78–93 | 10–18 | N/P = 12 |
Fatty acids Disulfide linker |
Intratracheal | Local expression | [23a] |
| bPEI | 2 kDa | Mixing | 234 | 47.3 ± 3.6 | N/P = 16 | Cyclodextrin | I.m. vaccine | Local expression | [23b] |
| PEI | 1.8 kDa | Mixing in RNase‐free water | 145 | 64.2 ± 0.9 | N/P = 32 | Vitamin E succinate | I.m. vaccine | Local expression | [24b] |
| PEI | 25 kDa | Mixing | 118 ± 4 | Not reported | 4:1 w/w | Stearic acid | Subcutaneous | Antigen‐specific immune response | [25] |
| PEI | 2 kDa | Mixing | 75 ± 11 | 27.5 ± 3.7 | 3:1 w/w | Deoxycholic acid | Intracerebral | Reduced Ischemic stroke | [26] |
| PEI | 2 kDa | Mixing in RNase‐free water | 80 | 26 | N/P = 16 | Cyclodextrin | I.m. vaccine | Local expression | [29] |
| PEI + PEG | 22 kDa | Mixing | 60 | 10 | N/P = 8 | Amino group | i.v. | Delivery to lungs | [28] |
| pAsp | Not reported | Mixing in 10 × 10‐3 m HEPES (pH 7.3) | ≈110 | 15–5 | N/P = 5 | Cyclohexylethyl | i.v. | Lungs, Spleen | [38] |
| PLGA + chitosan | 4–15 kDa | Emulsion‐diffusion‐evaporation | 157 ± 1 | 30.8 ± 0.115 | 25:1 w/w | Chitosan | Intratracheal | Lungs | [43] |
| PLGA + chitosan | 4–15 kDa | Not reported | 142 ± 4 | 21.9 ± 2.7 | Not reported | Chitosan | Intratracheal and i.v. | Lungs | [44] |
| PBAE+PEG lipid | 2.2 kDa | Mixing in 25 × 10‐3 m NaOAc buffer | 164 | 14.6 ± 5.6 | Not reported | Caprolactone | i.v. | Spleen | [47] |
| PBAE+PEG lipid | 4–10 kDa | Mixing in 25 × 10‐3 m magnesium acetate buffer (MgAc2, pH 5) | 100 | 42 | 30:1 w/w | Hydrophobicity and amine groups | i.v. | Lungs, Liver | [48a] |
| PBAE+PEG lipid | 2.7 kDa | Mixing in acidic buffer (pH 5.3) | 187 ± 44 | 39.3 | N/P = 57 | Amine groups and PEG lipid | i.v. | Lungs, Liver | [48b] |
| PBAE+PGA | Not reported | Mixing in NaOAc buffer | 107 | 4 ± 2 | 60:1 w/w | Ab for targeting | i.v. | T cells | [49] |
| Hyperbranched PBAE | 20 kDa | Mixing | 160 | 45 | 50:1 w/w | Amine groups | Nebulized intratracheal | Lungs | [51] |
| Polyesters+surfactant | 4.2–7 kDa | Mixing in 10 × 10‐3 m citric acid/trisodium citrate buffer (pH 4.2) | 100 | −17–1 | 30:1 w/w |
Alkyl length Amine groups |
i.v. | Lungs, Spleen | [52] |
| PACE+PACE‐PEG | PEG: 5k Da | Mixing in 25 × 10‐3 m sodium acetate buffer (pH 6) | 200 | −22–‐3.1 | 100:1 w/w | PEG density optimized | i.v. | Lungs, Spleen | [53] |
| Chitosan | Not reported | Mixing in 4% acetic acid solution | 100 | Not reported | Not reported | Protein coating | Intranasal | Local expression | [57] |
| Protamine | Not reported | Not reported | Not reported | 1:2 w/w | Protamine MW | i.d. | Local expression | [60b] | |
| PAMAM+PEG lipid | Not reported | Microfluidic preparation | 120 | 5:1 w/w | Alkyl‐modified | I.m. vaccine | Local expression | [79] | |
| pABOL | 8 kDa | Titrating saRNA solutions into polymer solutions | ≈70 | + 23 | 45:1 w/w |
Bioreducible MW optimized |
I.m. vaccine | Local expression | [80] |
| p(BAC‐TETIm/AD), p(BAC‐TET‐Im/β‐CD)), PEG‐p(BAC‐TET‐Im/AD)–PEG | 8.5–19.5 kDa | Mixing in 25 × 10‐3 m NaOAc buffer (pH 5.5) | 168‐179 | −2.5–7.9 | Not reported | Redox‐responsive; Host–guest interaction between β‐CD and AD | Intracellular | HEK 293, RAW 264.7, HCT116, and NHDF cells | [91] |
The full name of polymers can be found in the main text
HEK293: human embryonic kidney 293 cell line; RAW 264.7: murine macrophage cell line; HCT116: human colorectal carcinoma cell line; NHDF: human dermal fibroblast cell line.
2.1.2. Poly(amino acid)s
Poly(amino acid)s such as poly(lysine) have long been used for delivering various nucleic acids (Figure 1).[ 30 ] More recently, poly(L‐lysine) (P(Lys)) has also been modified with weak basic groups such as histidine to improve endosomal escape[ 31 ] (Table 1). Dirisala et al. employed P(Lys) and poly(L‐ornithine) (P(Orn)) to form a polyplex with mRNA, using an anionic derivative of poly(aspartic acid) as a charge conversion polymer, protecting mRNA in 50% FBS and facilitating endosomal escape.[ 32 ]
Cationic polymers consisting of N‐substituted poly(aspartamide)s (P(Asp)) with varying numbers of side chain amines represent a safer alternative to simple polycations like PEI, due to their biodegradability and endosomal escape capacity[ 33 ] (Table 1). The variation in the protonation status of side chain amines under different pH conditions[ 34 ] is a key factor that affects their buffering capacity and endosomal escape.[ 34 , 35 ] Optimizing the number of amines in the side‐chain has resulted in efficient mRNA delivery, with odd‐numbered repeats outperforming even‐numbered side‐chain amines.[ 34 ] The effect of the side‐chain structure on translation efficiency was also studied, revealing that binding efficiency with translation initiation factor eIF4E was affected by the number of aminoethylene repeats.[ 36 ] In a similar trend to PEI, modulations of the biodegradability and hydrophobicity of the polymeric carrier have recently been carried out (Figure 1).[ 37 ] The R‐modified PAsp(DET/R)s polyplexes with logD7.3 > − 2.4 were more stable in 10% FBS, allowing higher in vitro transfection. The R‐polyplexes with logD7.3 between −1.8 and −1.3 enable in vivo mRNA delivery to the lungs after i.v. injection.[ 38 ] Poly(amino acid)s thus represent a promising class of polymeric delivery systems for mRNA capable of both local and systemic mRNA delivery (Table 2).
2.1.3. Polyesters
Poly(lactic‐co‐glycolic acid) (PLGA) is an FDA‐approved polyester widely used for drug delivery.[ 39 ] It is an attractive candidate for gene delivery formulation because of its biodegradability through hydrolysis of ester linkages, its noncytotoxic degradation products, small size, structural integrity, stability, facile preparation, and adaptability of PLGA nanoparticles (Figure 1) (Table 1).[ 40 ] However, several issues have been identified for formulating PLGA‐based carriers for nucleic acids. For example, formation and loading of particles can be inefficient and damaging to the DNA and hydrolysis can lower pH inside the particle to 1.5, potentially resulting in DNA hydrolysis.[ 41 ] Moreover, as PLGA alone does not possess the necessary cationic charge to complex nucleic acids at neutral pH, it has been modified with cationic groups, such as chitosan, PEI, or cetyltrimethylammonium bromide (CTAB).[ 42 ] The approach has been successfully used in vivo for delivering nuclease‐encoding mRNA, resulting in site‐specific genome editing.[ 43 ] In another study, the intratracheal and intravenous delivery of chitosan‐coated PLGA nanoparticles was compared in a mouse model of cystic fibrosis.[ 44 ] The nanoparticles loaded a chemically modified mRNA encoding the transmembrane conductance regulator (CFTR). The i.v. injection increased the nanoparticle accumulation in the lungs compared to the intratracheal injection, allowing significant improvement of the lungs function.[ 44 ] Such differences of nanoparticle delivery after intratracheal and intravenous delivery may be related to the size and charge of the nanoparticles, which would affect the accessibility of the carriers to tissues. Probably, intratracheally injected nanoparticles may have not been able to reach the cells in the airways of mice due to retention in the severe mucus barrier of cystic fibrosis. On the other hand, after intravenous injection, the nanoparticles with positively charge surface would allow for accumulating in the lung capillaries.
Because of their biodegradability, poly(𝛽‐amino ester)s (PBAEs) are considered a safe alternative for nucleic acid delivery (Figure 1).[ 45 ] Another advantage is the facile synthesis and monomer availability, enabling the generation of a library of distinct polymers (Table 1). Initially, PBAEs were screened for DNA transfection ability, revealing a trend of hydrophobic polymers performing best.[ 46 ] Further side‐chain optimization for use with mRNA resulted in significantly enhanced delivery to the spleen than Jet PEI after systemic administration,[ 47 ] which offer advantages for cancer immunotherapy with antigen‐encoding mRNAs. Combining the PBAE polyplex with PEG–lipid to form polymer–lipid nanoparticles further permits tunable targeting of other organs such as the lungs or the liver[ 48 ] (Figure 2b). A formulation without lipid consisting of a PBAE/mRNA polyplex coated with antibody‐conjugated polyglutamic acid for cell targeting enabled the in situ generation of CAR‐T cells after intravenous infusion,[ 49 ] which supports the potential for working in vivo without the need of having lipids in the formulation. Hyperbranched PBAEs/mRNA polyplexes also achieved uniform mRNA distribution in the lungs.[ 50 ] Based on this platform, nebulized formulations of hyperbranched PBAE loading Cas13a mRNA and guide RNA showed high translation and gene editing efficiency in the respiratory tract to treat SARS‐CoV‐2 and influenza infections.[ 51 ] Even though there are several studies on the PBAE system with encouraging results, little is known about how these nanoparticles protect mRNA in physiological conditions. Such information would be useful for predicting the potential for other applications.
Polyester‐based carriers were also used for a structure optimization study focused on mRNA delivery.[ 52 ] Results indicated that adapting molar ratio and alkyl chain length allows for selective delivery to either lungs or spleen after intravenous injection. Moreover, mRNA delivery to the lungs or spleen in mice could be tuned by optimizing the surface‐modification of polyplexes based on poly(amine‐co‐ester) (PACE) with PEG.[ 53 ] From dynamic light scattering (DLS) measurements, the size of the mRNA polyplex was found to be stable in 10% FBS with a PACE‐PEG content as low as 0.25%. On the other hand, in vivo studies used formulations with higher PEG content (5%). Besides studying the size stability of the polyplexes, elucidating the effect of PACE‐PEG content on the stability of the mRNA payload would allow further optimization of the nanocarriers for in vivo application.[ 53 ] Evidently, polyester‐based polymeric carriers are suitable for mRNA delivery both in vitro and in vivo, with similar trends to other polymer‐based systems in terms of optimization of polymer hydrophobicity and targeting of lungs and spleen after systemic administration (Table 2).
2.1.4. Natural Polymers
Other than synthetic carriers, polymers found in nature have also attracted attention as nucleic acid vehicles due to their inherent biocompatibility (Table 1). Chitosan is a polysaccharide derived from chitin which is able to interact electrostatically with nucleic acids. Due to these properties, it has been investigated for its potential in the delivery of genetic material.[ 54 ] For mRNA delivery, chitosan is generally combined with other materials to improve stability and delivery such as with PLGA to deliver nuclease‐encoding mRNA.[ 43 ] The combination with hyaluronic acid is used for its ability to ameliorate cellular uptake and stability, leading to favorable transgene expression in vitro, especially at slightly acidic pH (pH 6.4–6.5).[ 55 ] The results also showed that an increase in molecular weight and deacetylation degree of the chitosan was beneficial for protecting mRNA against heparin attack.[ 55a ] Similarly, addition of poly(2‐propylacrylic acid) to increase endosomolytic capacity resulted in transfection efficiency similar to lipid control.[ 56 ] In vivo, protein‐coated polyplexes of chitosan and mRNA have been successfully used as a vaccine against avian influenza viruses.[ 57 ]
The use of protamine, a category of naturally occurring arginine‐rich polypeptides, for RNA delivery goes back as far as 1961.[ 58 ] Its natural ability to condense nucleic acids make it an attractive candidate as an RNA vehicle. It has thus been applied as the basis of many drug delivery systems.[ 59 ] Particularly, the ability of mRNA complexes with protamine to stimulate the immune system has led to the development of protamine‐based mRNA vaccines.[ 60 ] The Tuebingen‐based company CureVac has developed a proprietary mRNA platform using a mixture of 50% non‐chemically modified mRNA and 50% mRNA complexed with protamine.[ 61 ] This technology platform is a lyophilized, temperature‐stable formulation that is stable at 5–25 °C for 36 months and at 40 °C for 6 months. This formulation with mRNAs encoding tumor‐associated antigens has shown promise in the context of therapeutic mRNA cancer vaccines of melanoma,[ 62 ] non‐small cell lung cancer (NSCLC) (CV9201 and CV9202)[ 63 ] and prostate cancer (CV9104)[ 64 ] in preclinical and clinical studies. After intradermal injection of CV9201 vaccines, 63% of the patients with NSCLC reacted against the administered mRNA antigen and 27% had an antigen‐specific T‐cell response.[ 63a ] Nevertheless, clinical trials for CV9104 and CV9201 eventually failed to improve overall survival compared to historical controls,[ 63 , 65 ] which may be a result of tumor‐induced immunosuppression. Based on these results, the company further combined CV9202 vaccines with immune checkpoint inhibitors,[ 63b ] which is undergoing phase 1/2 studies, with results yet to be published. Further results have demonstrated the proof‐of‐concept of mRNA for a rabies vaccines using the cationic protamine formulation (CV7201).[ 66 ] Preclinical studies in mice and pigs have established that this vaccine could induce specific, long‐lived, and protective adaptive immunity by intradermal and intramuscular injection.[ 67 ] Further clinical research demonstrated that it was able to induce rabies antibodies in humans.[ 66 ] However, the company has since shifted focus to the use of lipid‐based carriers (CV7202) since the induction of adequate immune responses by the protamine‐based platform was dependent upon the mode of administration and required specialized devices.[ 68 ]
While the ability to create a consistent pharmaceutical drug substance is imperative for successful clinical application, both chitosan and protamine, as well as other products derived from natural materials, may present limitations in clinical translation due to batch‐to‐batch variability. Indeed, small differences in molecular weight can significantly impact the delivery efficiency of RNA for both chitosan and protamine.[ 13 ] Overall, applications of natural polymers for mRNA delivery have hitherto been limited to local administrations such as vaccines, but with optimizations in stability may be amenable to systemic delivery as well (Table 2).
2.1.5. Other Polymers and Dendrimers
Besides the abovementioned polymers, other polyplex examples can be found in the literature. Among them, polymers prepared from reversible addition‐fragmentation chain‐transfer (RAFT) polymerization are particularly useful for easily producing copolymers with controlled side chain moieties (Table 1). For example, research on polymeric delivery systems based on dimethylaminoethyl methacrylate (DMAEMA) has shown promising features for nucleic acid delivery.[ 69 ] While being originally conceived for siRNA and other oligonucleotide drugs (Figure 1),[ 69 ] DMAEMA copolymers can be applied to mRNA delivery. Notably, DMAEMA copolymers forming polyplexes with mRNA were optimized for molecular weight and hydrophobicity to attain a balance between cytotoxicity and in vitro transfection efficiency, outperforming the standard PEI.[ 70 ] Moreover, mRNA polyplexes could be modified with ligands that can promote intracellular delivery and translation. For example, polyplexes formed with poly((carbonic acid 2‐dimethylamino‐ethyl ester 1‐methyl‐2‐(2‐methacryloylamino)‐ethyl ester)‐N‐[2‐(2‐pyridyldithio)]ethyl methacrylamide‐azidoethylmethyacryl) p(HPMA‐DMAE‐co‐PDTEMA‐co‐AzEMAm) copolymers and decorated with membrane‐disruptive GALA peptides augmented cell uptake and endosomal escape in dendritic cells.[ 71 ] The effect of introducing ligands on the efficiency of the delivery is covered in Section 2.4.
Dendrimers, which are a class of polymeric molecules branching out to form a spherical structure, are also good candidates for nucleic acid delivery thanks to their excellent stability, solubility, and inherent multivalency of functional groups (Table 1). Poly(amidoamine) (PAMAM) is one of the most studied dendrimers.[ 72 ] As with PEI and PLL, PAMAM dendrimers exert in vitro cytotoxicity due to a relatively open network with cationic groups.[ 73 ] 2.0 generation PAMAM dendrimers without modification were found to be cytotoxic at concentrations above 700 × 10‐6 m. Higher‐generation dendrimers may have hemolytic toxicity due to the greater overall cationic charge.[ 74 ] Nevertheless, some general trends are clear. PAMAM can be functionalized easily by engineering surface functional groups, modifying core and branching, which might help to balance efficiency and toxicity.[ 75 ] While PAMAM has been used for the delivery of siRNA[ 76 ] and pDNA,[ 77 ] it has yet to be successfully used by itself for mRNA delivery.[ 78 ] However, by combining a modified PAMAM dendrimer with PEG‐lipid, a replicon RNA‐based vaccine was developed that provided protective immunity against several pathogens.[ 79 ] Bioreducible poly(cystamine bisacrylamide‐co‐4‐amino‐1‐butanol) (pABOL) was also used to formulate self‐replicating RNA, a protein‐encoding RNA about 10‐fold larger than a typical mRNA. The polymer was effectively applied in vivo, and the results showed that an increase in molecular weight of the polymer was beneficial for promoting transfection efficiency at the injected site after intramuscular and intradermal injection.[ 80 ] Moreover, a study using a library of copolymers consisting of poly(2‐ethyl‐2‐oxazoline) and PEI showed that the polymer molecular weights must be optimized depending on the carried nucleic acid, with large RNAs, such as self‐amplifying RNAs, requiring longer polymer chains.[ 81 ] However, stability tests of nanoparticles to protect mRNA under physiological condition are not available in these studies. Polyplex stability in physiological condition should be taken into consideration in optimizing molecular weight for efficient polymer‐based mRNA therapies.
2.2. Sterically Stabilized Polyplexes
Since charge‐neutral polyplexes are likely to aggregate and precipitate, polyplexes are usually stabilized under excess cationic polymers ([amine groups in polymer (N)]/[phosphate groups in RNA (P)] (N/P ratio) >1). The positively charged polyplexes have shown potential to improve cellular uptake through interaction with negatively charged cell membranes.[ 82 ] Meanwhile, high‐density surface positive charges are also associated with the observed toxicity[ 17 , 83 ] and poor colloidal stability under physiological conditions due to interaction with negatively charged serum proteins such as albumin.[ 84 ] Thus, they are largely limited to local applications in vivo. Therefore, the surface of the polyplexes is typically modified with hydrophilic and biocompatible polymers for in vivo application because such modifications can limit the ion exchange with anionic proteoglycans on the cell membranes and also reduce interaction with serum proteins.[ 85 ] A very potent and commonly used hydrophilic polymer to protect the payload from the exterior environment is PEG. PEG is an FDA‐approved biocompatible material, which has been widely used in the pharmaceutical field to prolong the blood circulation time of proteins and reduce their immunogenicity.[ 86 ] However, PEG shielding can reduce the cellular internalization of nanoparticles; the so‐called “PEG dilemma.”[ 87 ] Nevertheless, in the case of negatively charged nucleic acid, the cellular uptake is even lower due to the electrostatic repulsion with the negatively charged cellular surface.[ 88 ] Moreover, electrostatically mediated nanoparticles may undergo ion exchange with anionic glycosaminoglycan components of the cell membrane surface, leading to nanoparticle dissociation and impaired uptake.[ 89 ] Sterically stabilizing polyplexes by PEGylation provides improved resistance against polyanion attack in biological settings, particularly in vivo. For example, poly(amine‐co‐ester) PACE polyplexes showed different biological activity and biodistribution after being modified with different PEGs. Thus, high concentration of PEG (>1%) decreased mRNA transfection efficiency of PACE polyplexes in vitro, but improved local delivery of mRNA to the lung[ 53 ] (Table 2). These results are consistent with previous studies of PEGylation on DNA transfection,[ 90 ] which showed that in vitro screens of optimal PEG content correlate poorly with in vivo results. Moreover, another study using a PEG‐modified PEI system showed that PEI polyplexes with PEG grafting ratios of 0.5% achieved high gene expression levels in the lung after systemic administration[ 28 ] (Table 2). More recently, a study using poly(N,N′‐bis‐(acryloyl)cystamine‐poly(aminoalkyl)) (PBAP)‐based polymers for delivering a variety of negatively charged payloads, including mRNA,[ 91 ] demonstrated that the mRNA polyplexes modified by host‐guest chemistry with a PEG‐PBAP‐PEG‐bearing adamantane achieved high stability in 10% FBS‐containing cell culture media and 4% BSA solution[ 91 ] (Table 2). Importantly, the modification of polyplexes with PEG–lipids has also shown that it is possible to tune mRNA delivery to the lungs or liver[ 48 ] (Table 2).
2.3. Block Copolymer‐Based Delivery Systems
Block copolymer systems allow self‐assembling mRNA‐loaded micelles by engineering the shielding and core‐forming blocks. The most common systems are based on catiomers having a hydrophilic neutral segment and a cationic block for complexing with mRNA. When catiomers are mixed with mRNA in aqueous conditions, they spontaneously self‐assemble into polymeric micelles having mRNA incorporated in the core.[ 88 , 92 ] As discussed in Section 2.2, the addition of a PEG shell decreases cellular uptake, endosomal escape and transfection efficiency. These issues could be solved by developing systems with PEG‐sheddable function. For example, PEG‐sheddable systems with stimuli‐responsive linkages, such as pH[ 93 ] and enzyme‐sensitive linkers,[ 93b ] have been shown to improve intracellular delivery. In the following sections, we describe the key parameters for constructing effective shell and mRNA‐loading components.
2.3.1. Engineering Shielding Domains in Block Copolymers
Shielding domains of block copolymers have been mainly based on PEG. PEG blocks can yield strong protection of the mRNA payload from nuclease digestion and avoid toll‐like‐receptor recognition in host immune cells.[ 94 ] Since PEG can be synthesized with a wide range of molecular weights (MW), optimizing the MW of PEG is vital for designing efficient formulations. It was found that the PEG length range varies among different categories of delivery systems. In lipid‐based or polymer–lipid hybrid mRNA nanoparticles, the molecular weight of the PEG can vary from 350 to 3000 Da,[ 95 ] and PEG‐lipids with a MW of around 2000 Da are usually incorporated into the formulations together with cationic or ionizable lipids and cholesterol to achieve high colloidal stability and improve in vivo mRNA delivery efficiency.[ 4 , 48 , 79 , 96 ] Of note, this length of PEG may not be enough for assembling effective mRNA‐loaded polymeric micelle systems. The effect of the PEG shell of polymeric micelles on pDNA and siRNA delivery has been investigated.[ 97 ] The effect of the PEG crowding on the plasmid DNA transcription in a cell‐free system was studied in PEG‐PLys block copolymers.[ 97b ] PEG‐PLys having a fixed P(Lys) degree of polymerization (DP) of around 70 and PEGs of 12 and 21 kDa showed appreciable efficiency compared to polymers with PEGs of 30 and 42 kDa, which is in good agreement with the reduction of protein adsorption mediated by PEG. The 30 and 42 kDa PEGs present an adequate level of PEG crowding to effectively reduce protein adsorption (⟨L⟩/2R g below 0.47; ⟨L⟩ means the closest distance between tethering PEG sites, and R g is the radius of gyration of PEG).[ 98 ] The effect of the PEG shell on micelles for preventing adsorption of biological compounds in blood was evaluated in systems based on PEG‐PLys using intravital real‐time confocal laser scanning microscopy (IVRTCLSM). The results showed that better blood circulation was observed for pDNA PEG‐PLys micelles with PEG in a squeezed conformation.[ 97c ] Another study exploring the effect of the PEG shell on siRNA delivery efficiency indicated that increasing the PEG MW incorporated into diblock polymer poly[dimethylaminoethyl methacrylate‐b‐(dimethylaminoethyl methacrylate‐copropylacrylic acid‐co‐butyl methacrylate)] (poly[DMAEMA‐b‐(DMAEMA‐co‐PAA‐co‐BMA)]) from 5 to 20 kDa could prevent aggregation and adsorption to blood components, leading to increased circulation time for systemic siRNA delivery.[ 97a ]
As discussed in the previous section, N‐substituted polyaspartamides (P(Asp)) with varying number of aminoethane repeats offers a significant endosome escape capacity[ 34 ] and safe profile due to its self‐degradation property in physiological conditions.[ 99 ] Incorporating PEG blocks into (P(Asp))‐based systems has proven beneficial for in vivo delivery of mRNA. Compared with P(Asp(DET)) polyplexes, PEG‐P(Asp(DET))‐based (Figure 1) polymeric micelles (MWPEG: 12–23 kDa) suppressed the immune responses, resulting in high transfection in central nervous system,[ 94a ] nasal cavity,[ 100 ] liver[ 101 ] and knee joint[ 102 ] (Table 3 ). More recently, the same system further demonstrated the potential to co‐encapsulate Cas9 mRNA and sgRNA for gene editing in the murine brain[ 103 ] (Table 3). Moreover, micelles from PEG‐P(Asp(DET)) (MWPEG: 42 kDa) exhibited a prolonged gene expression (around 7 d) after intraspinal injection, demonstrating the feasibility of mRNA‐loaded micelles to treat spinal cord injury[ 104 ] (Table 3).
Table 3.
Block copolymer‐based systems to deliver mRNA
| Polymer component a) | Molecular weight | Preparation method | Average size [nm] | Zeta potential [mV] | Cation to RNA ratio | Design elements | Experimental application | Outcome b) | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| PEG‐P(Asp(DET)) | 22 kDa | Self‐assembly in 10 × 10‐3 m HEPES buffer | 50 | Not reported | N/P = 8 | Endosomal escape | Intrathecal | Expression in central nervous system | [94a] |
|
PEG‐P(Asp(DET)) P(DMAEMA‐co‐OEGMA) |
22 kDa 54 kDa |
Self‐assembly in 10 × 10‐3 m HEPES buffer Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.3) |
Not reported |
Not reported 0.071 ± 0.159 |
N/P = 3 | N/P = 3 | Intranasal | Local expression | [100] |
| 50 | N/P = 3 | N/P = 3 | Hydrodynamic injection | Liver | [101] | ||||
|
Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.3) Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.4) |
50 65 ± 5 |
N/P = 8 | N/P = 8 | Intra‐articular | Local expression | [102] | |||
| Not reported | N/P = 3 | N/P = 3 | Intracerebral | Genome editing in brain | [103] | ||||
|
Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.4) Self‐assembly in HEPES buffered saline (HBS, 150 × 10‐3 m NaCl, 10 × 10‐3 m HEPES, pH 7.4) |
Not reported |
Not reported 37 ± 15 |
N/P = 3 | N/P = 3 | Intraspinal injections | Spinal cord injury treatment | [104] | ||
| 20–22 kDa | 37 | N/P 10–40 | PEGylation | Intracellular | BEAS‐2B cells | [105] | |||
| PEGMA‐p(DMAEMA‐co‐BMA)) | 61.7 kDa | Self‐assembly in nuclease‐free water | 86 ± 16 | 12.4 ± 0.7 | N/P = 3 | Endosomal escape | Intracellular | T cells | [106] |
| P(MEO3MA)‐b‐P(H2N‐Cys‐MA) | 69.3 kDa | Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.4) | 87 | Not reported | N/P = 5 | Redox‐responsive by disulfide bonds | Intracellular | RAW macrophages and 3T3 fibroblasts | [107] |
| P(EtOx)‐P(EI) | 45 kDa for mRNA, and 72 kDa | Self‐assembly in DMEM | 80–450 | −5–2 | 7000 (molar ratio) | Molar mass and charge density optimized | Intracellular | HEK 293T cells | [81] |
| PEG‐P(Asp(TEP))‐Chol | 34 kDa | Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.3) | 55 | Not reported | N/P = 8 | Hydrophobicity and endosomal escape | i.v. | BxPC‐3 pancreatic tumor | [110] |
| PEG‐P(Asp(DET))‐Chol | 25 kDa | Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.4) | 60–65 | 1.1 | N/P = 3 | Chol (+)‐OligoRNA | Intratracheal | Local expression | [112a] |
| PEG‐PLys | 19 kDa |
Self‐assembly in 10 × 10‐3 m HEPES buffer (pH 7.4) Mixing polymer solution (10 × 10‐3 m HEPES buffer, pH 3.5) with mRNA solution (10 × 10‐3 m HEPES buffer, pH 7.4) |
100 | Not reported | N/P = 2 | mRNA bundling | Intracerebral |
Improved blood circulation Local expression |
[112b] |
| PEG‐PGBA | 22 kDa | 56 | 1.1 | N/P = 3 | Flexible polycation | Intranasal |
Improved blood circulation; Local expression |
[115] | |
| PEG‐PGMG | 18 kDa | 62 | 0.90 | N/P = 3 | Multivalent interactions from guanidine | Intracellular | HuH‐7 cells | [116] | |
| PEG‐PGTyr | 26 kDa | 75 | Not reported | N/P = 3 | π–π stacking | i.v. and i.m. |
Improved blood circulation Local expression |
[117] | |
| cRGD‐PEG/PNIPAM‐PLys(SH) | 12 kDa/5 kDa | 59 ± 2 | 1.89 ± 0.28 | N/P = 1.5 | Redox‐responsive crosslinking; thermo‐responsive | i.v. | U‐87 glioma tumor | [119] | |
| PEG‐PLys(AMP) | 23 kDa | 53 ± 2 | −1.23 ± 0.4 | N/P = 2 | Redox‐responsive crosslinking | Intracellular | HuH‐7 cells | [120] | |
| PEG‐PAsp(DET/FPBA)‐Chol, PEG‐PAsp(DET/GlcAm)‐Chol | 22 kDa | 70–90 | −2.5 ± 0.9 | N/P = 1.5 | ATP‐responsive crosslinking; Chol (+)‐OligoRNA | Intracellular | Improved blood circulation | [112c] | |
| PEG‐pLL(CAA) | 20 kDa | 94 ± 1 | Not reported | Not reported | pH‐responsive crosslinking | Intratumoral | Improved expression in CT 26 tumors | [123] | |
| PEG‐PAsp(DET/FPBA) | 24 kDa | 59–72 | Not reported | N/P = 1.5 | ATP‐responsive crosslinking | Intratracheal | Local expression | [112d] |
The full name of polymers can be found in the main text;
BEAS‐2B: human non‐tumorigenic lung epithelial cell line; 3T3: mouse embryonic fibroblast cell line, HEK 293T: a stable clone derivative of the human embryonic kidney (HEK) 293 cell line; HuH‐7: hepatocyte‐derived carcinoma cell line.
Other hydrophilic polymers are also being evaluated as alternatives to PEG as shielding material for mRNA delivery. For example, a series of block copolymers based on poly‐N,N dimethylaminoethylmethacrylate‐oligo(ethylene glycol) methyl ether methacrylate (P(DMAEMA‐co‐OEGMA)) have been developed for mRNA delivery. Among these copolymers, OEGMA(9)10 influenced mRNA binding with p(DMAEMA)110, and improved transfection efficiency[ 105 ] (Table 3). Cheng et al. extended the p(DMAEMA) based systems and further developed a variety of triblock polymers with PEG containing poly(ethylene glycol) methyl ether methacrylate (PEGMA) blocks to provide hydrophilic stabilization. They found that the position and length of PEGMA block in the triblock polymer will influence mRNA binding and in vitro transfection efficiency[ 106 ] (Table 3). Recently, other hydrophilic polymers like poly‐tri(ethylene glycol) methyl ether methacrylate (PMEO3MA)[ 107 ] and poly(2‐ethyl‐2‐oxazoline) (P(EtOx))[ 81 ] were synthesized and evaluated for their ability to provide stabilization for mRNA[ 81 , 107 ] and RepRNA[ 81 ] transfection in vitro. However, the ability of these hydrophilic polymers for protecting mRNA as compared to PEG is unknown so far.
2.3.2. Engineering Core Forming Domains
While polymeric micelles have improved stability under physiological salt conditions, the stability of polymeric micelles in blood and other biological fluids presents another critical issue, especially for the systemic delivery of mRNA. The interaction of polymeric micelles with the widely distributed small ribonucleases (size of RNase A: 3.8*2.8*2.2 nm),[ 108 ] polyelectrolytes, proteins, or cellular surfaces could result in partial or complete dissociation of nanoparticles.[ 109 ] Therefore, more stable packaging of mRNA inside the core is required in addition to shielding with the hydrophilic blocks.
Efforts to Enhance Core Condensation by Noncovalent Interactions
A feasible strategy to increase micelle condensation within the core can be achieved by introducing hydrophobic moieties in polycation segments to improve polymer mRNA interaction. For example, installation of a hydrophobic cholesteryl group at the end of block copolymer PEG‐P(Asp(TEP)) (Figure 1) demonstrated significantly improved blood circulation after intravenous injections, achieving three orders of magnitude higher concentration in blood than that of naked mRNA. Eventually, this system exhibited significant antitumor efficiency against pancreatic cancer by systemic delivery of cholesterol‐modified polymeric micelles with mRNAs encoded antiangiogenic sFlt‐1 gene.[ 110 ]
Another direction for enhancing core packing has been focusing on engineering the mRNA. Functionalized RNA oligonucleotides (OligoRNAs) have been introduced to hybridize mRNA, resulting in highly structured mRNA nano‐assemblies with around 100‐fold improved stability compared with unhybridized mRNA.[ 4 , 11 ] Hybridized mRNA can be further assembled with classical block copolymers.[ 112 ] OligoRNAs modified with a hydrophobic cholesterol (Chol) moiety (Chol‐OligoRNAs) were hybridized to mRNA and then mixed with PEG‐P(Asp(DET))‐Chol (+) block copolymers to prepare polymeric micelles. Hybridizing mRNA (783 nt) with even one single Chol (+)‐OligoRNA (17 nt) drastically improved the tolerability against the nuclease of the polymeric micelle. Eventually, this system exhibited efficient mRNA introduction into mouse lungs via intratracheal administration[ 112a ] (Table 3).
The effect of RNA rigidity on polyion complex micelle formation has been recently noted. Rigid double‐strand RNA (dsRNA) stays in the primary assembly state whereas flexible single‐stranded RNA could promote secondary association into micelles, probably because the steric repulsion derived from PEG chains could be compensated by the increase in entropy from the enhanced conformational freedom within the micelle core.[ 113 ] Thus, we designed a block catiomer termed PEG‐poly(glycidyl butyl amine) (PEG‐PGBA) (Figure 1) (Table 3) with flexible polycation backbone comprising ether bonds to prepare micelles and compared them to micelles prepared from the relatively rigid PEG‐PLys bearing peptide bonds. Molecular dynamic simulations allowed to visualize the effect of the flexibility of the backbone on the binding to RNA molecules. In the case of double‐stranded RNA (dsRNA), the PEG‐PGBA/dsRNA complexes presented a lower R g than PEG‐PLys/dsRNA.[ 114 ] The average distance analysis also showed that the amines in the PEG‐PGBA were closer to the phosphates of dsRNA than the amines of PEG‐PLys (Figure 3a). Thus, the improved contact surface area between PEG‐PGBA and dsRNA (Figure 3b) allows for more efficient protection of siRNA from polyanion attack and improves the in vitro delivery efficiency of siRNA.[ 114 ] Based on this model, herein, we simulated the interaction of the polymers with single‐stranded RNA (ssRNA) using the same protocol as we used in our previous work.[ 114 ] Again, the PEG‐PGBA with flexible polycations was much closer to ssRNA than the relatively more rigid PEG‐PLys (Figure 3c), resulting in a larger contact surface area (Figure 3d). The increased binding of the PEG‐PGBA to mRNA was confirmed experimentally, where PEG‐PGBA allowed more than 50‐fold stronger binding to mRNA than PEG‐PLys, as determined by isothermal calorimetry analysis (Figure 3e). Eventually, mRNA‐loaded PEG‐PGBA micelles exhibited increased protein translation and prolonged blood circulation (Figure 3f), indicating the significance of polycation flexibility on the assembly of polyion complexes with mRNA[ 115 ] (Table 3). We also found that further applying multivalent interactions by introducing guanidine moieties (Figure 1) into the flexible polycation segments, which increase the binding by ionic pairing and hydrogen bonding with the phosphate groups in mRNA, can also contribute to increased stability and enhanced delivery[ 116 ] (Table 3). More recently, we designed mRNA‐loaded micelles composed of biocompatible block copolymers having functional amino acid moieties for tunable interaction with mRNA for enhanced in vivo delivery (Figure 3g). The block copolymer was based on poly(ethylene glycol)‐poly(glycerol) (PEG‐PG) with flexible polyether backbone, and modified with glycine (Gly), leucine (Leu) or tyrosine (Tyr) via ester bonds. PEG‐PGTyr (Figure 1), which formed micelles and π–π stacking with mRNA (Figure 3g,h), displayed excellent stability against polyanions, 50% FBS and biological settings (Figure 3i). Thus, PEG‐PGTyr/m improved the efficacy of mRNA delivery in vivo after intramuscular injection.[ 117 ]
Figure 3.
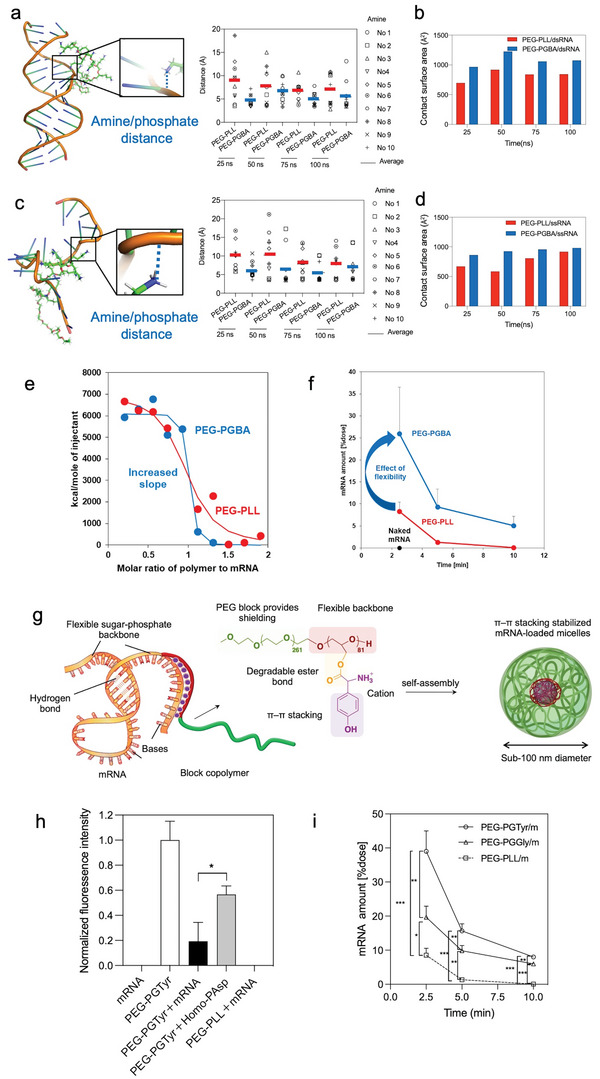
Control of the core forming domains enhances the micelle performance. a) Amine/phosphate distance in PEG‐PLys/dsRNA and PEG‐PGBA/dsRNA, each amine is identified by residue numbers (No.). Reproduced with permission.[ 114 ] Copyright 2021, Taylor & Francis. b) Contact surface area between PEG‐PLys/dsRNA and PEG‐PGBA/dsRNA. c) Amine/phosphate distance in PEG‐PLys/ssRNA and PEG‐PGBA/ssRNA, each amine is identified by residue numbers (No.). d) Contact surface area between PEG‐PLys/ssRNA and PEG‐PGBA/ssRNA. e) Isothermal titration calorimetry shows that the flexibility of the polycation segment affects the binding affinity to mRNA, as more flexible PEG‐PGBA binds tighter than PEG‐PLL. Reproduced with permission.[ 115 ] Copyright 2020, John Wiley and Sons. f) The remaining mRNA amount after systemic mRNA delivery by PEG‐PLys and PEG‐PGBA‐based micelles. Reproduced with permission.[ 115 ] Copyright 2020, John Wiley and Sons. g) PEG‐PGTyr enhanced mRNA delivery via π–π stacking‐assisted micellar assembly. h) π–π stacking assessment by Tyr fluorescence quenching. i) Remaining mRNA amount in the mice bloodstream after intravenous injection of mRNA‐loaded micelles. g–i) Reproduced with permission.[ 117 ] Copyright 2023, Taylor & Francis.
Efforts to Enhance Core Stabilization by Stimuli‐Responsive Crosslinking
As noted in the previous sections, the success of mRNA therapy is critically dependent on the carriers to protect mRNA and deliver them to the cytoplasm in the desired tissues. Modifying polymeric systems with stimuli‐responsive crosslinking systems that can release active mRNA in response to a desired stimulus could provide a smart strategy for enhancing mRNA stability, intracellular delivery, and specificity. A variety of endogenous and exogenous stimuli can be exploited for designing these systems, such as changes in redox potential, pH or metabolites, and light or ultrasound, respectively. Redox‐sensitive crosslinking systems by disulfide bridges are among the most investigated stimuli‐responsive strategies since they are reversible and easily reduced due to the presence of a higher concentration of glutathione (GSH) in the intracellular environment (≈2–10 × 10‐3 m) than in the extracellular environments (≈2–10 × 10‐6 m).[ 118 ] Redox‐sensitive core crosslinked micelles have been implemented in self‐assembled mRNA particles with block copolymers like PEG/PNIPAM‐PLys(SH),[ 119 ] PEG‐PLys(AMP),[ 120 ] PEG‐P(Asp‐AED‐ICA),[ 121 ] and P(MEO3MA)‐b‐P(H2N‐Cys‐MA)[ 107 ] containing disulfide bonds in the side chain of the polycation (Table 3). Another example of effective mRNA protection by micelle crosslinking was demonstrated with an ATP‐responsive crosslinking of micelles based on the ester formation from 4‐carboxy‐3‐fluorophenylboronic acid (FPBA) and polyol moieties[ 112c ] (Table 3). These ATP‐sensitive micelles achieved effective intracellular delivery and release of mRNA since the phenylboronate ester linkages are cleaved selectively in the intracellular environment due to the higher ATP concentration.[ 122 ] The optimized formulation was further stabilized by introducing cholesterol moieties into both the mRNA and the block copolymer, exhibiting ATP‐responsive mRNA release and a 10‐200‐fold increase of the amount of intact mRNA amount in the blood compared with that of non‐crosslinked micelles[ 112c ] (Table 3). Recently, we have developed a pH‐sensitive micelle system for mRNA delivery with cross‐linked core formed by cis‐aconitic anhydride‐modified poly(ethylene glycol)‐poly(l‐lysine) (PEG‐pLL(CAA)) block copolymers (Figure 1, Figure 4a). The resulting cross‐linked micelles maintained their size at physiological pH 7.4, as well as at intratumoral pH 6.5. However, they disassociated at pH lower than pH 6.5, which corresponds to the endosomal pH range (Figure 4b). The core cross‐linked micelles effectively protected the mRNA against counter polyanion exchange and FBS attack. Thus, the cross‐linked micelles showed improved fluc expression in vivo in CT 26 tumor‐bearing mice compared to non‐cross‐linked micelles and even to standard PEI‐based polyplexes (Figure 4c,d) (Table 3).[ 123 ] Besides cross‐linking of polymers, polymer/mRNA crosslinking may provide a prominent stabilizing effect on mRNA. Thus, an ATP‐responsive polymer/mRNA cross‐linked system was proposed for further core stabilization[ 112d ] (Table 3). First, mRNA was hybridized with a 17 nt engineered OligoRNA with a GlcAm moiety at the 5′ end and ribose at the 3′ end. The ATP‐responsive core crosslinked micelles are formed via phenylboronate ester linkages between FPBA in the polycations and GlcAm at the 5′end of and the diol moiety at 3′ end ribose (Figure 4e). Eventually, these micelles exhibited excellent stability against polyion exchange reaction and ribonuclease attack, resulting in improved mRNA transfection in the lungs of mice over noncrosslinked micelles after intratracheal administration[ 112d ] (Table 3). Therefore, stabilization of the polymeric micelle core with elements responsive to intracellular stimuli, such as redox and ATP concentration, became a strategy to regulate mRNA release in vitro and in vivo.
Figure 4.
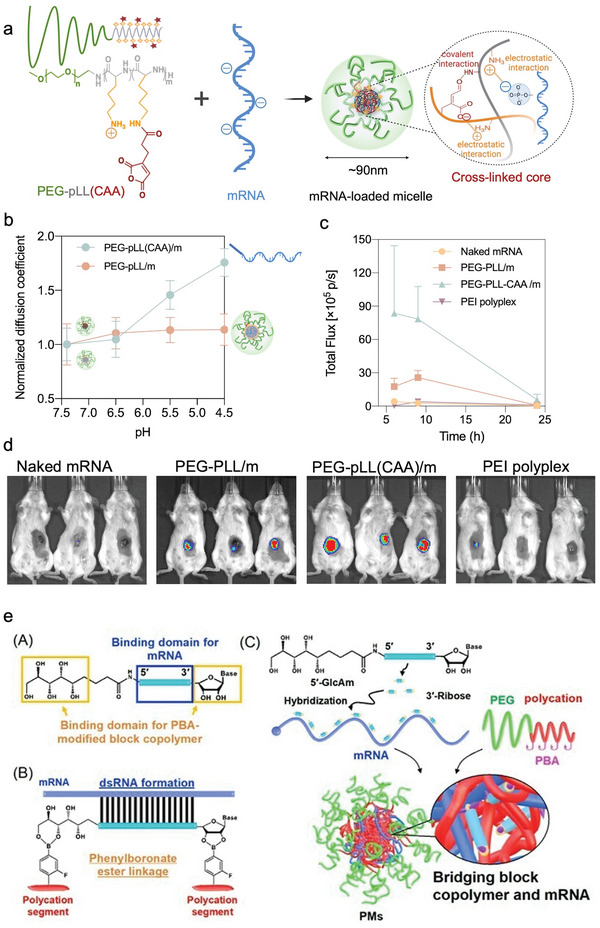
Micelle core stabilization by stimuli‐responsive crosslinking. a) pH‐sensitive micelle system with cross‐linked core formed by cis‐aconitic anhydride‐modified PEG‐pLL(CAA). b) Diffusion coefficient of PEG‐pLL/m and PEG‐pLL(CAA)/m versus buffer having different pHs. c) Quantification analysis luminescent signals at indicated time points. d) Bioluminescence images 9 h postinjection following in vivo delivery of naked mRNA, PEG‐pLL/m, PEG‐pLL(CAA)/m in CT26 tumor‐bearing mice. (a–d) Reproduced with permission.[ 123 ] Copyright 2022, MDPI. e) ATP‐responsive bridging between block copolymers and mRNA. Reproduced with permission.[ 112d ] Copyright 2021, Wiley‐VCH GmbH.
2.4. Enhancing mRNA Delivery through Ligand‐Mediated Targeting
The addition of targeting ligands has great potential to overcome urgent problems of current mRNA delivery strategies using polymeric nanocarriers. On the one hand, hydrophilic, polymeric shells like PEG on nanoparticles are often essential as they reduce the exposure of positive charges within the core, protect the mRNA from nucleases in the blood and increase the circulation time significantly. On the other hand, they also impair cellular uptake and reduce the internalization of the nanocarrier into targeted cells before degradation. This is why more precise and efficient mRNA therapies using actively targeted delivery systems with ligands are highly desirable.[ 88 , 124 ]
Through ligand‐receptor interactions, the system can deliver mRNA to certain types of cells or tissues,[ 125 ] and promote cellular uptake by receptor‐mediated endocytosis[ 126 ] (Figure 5 ). Ligands can improve extravasation at targeting sites by interacting with specific ligands on endothelial cells for example within tumor vasculatures[ 127 ] or in the brain[ 128 ] and they can modulate signaling pathways.[ 129 ] Certain ligands can even improve intracellular delivery after cell uptake by facilitating endosomal escape, a critical step for successful translation of mRNA after endocytosis.[ 124 , 130 ]
Figure 5.
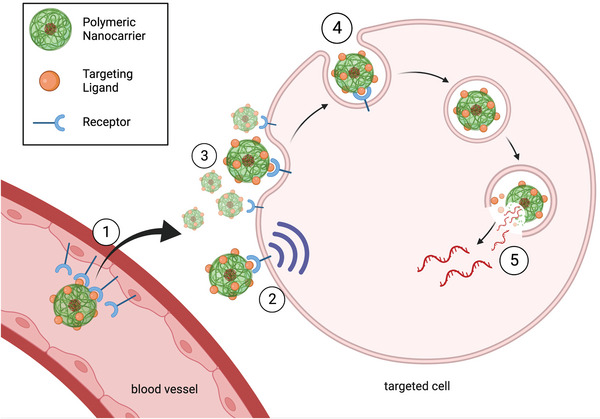
Targeting ligand interactions. 1) Active ligand‐mediated transport through blood vessels. 2) Ligand‐mediated cell signaling 3) Ligand‐mediated retention on cell surface. 4) Ligand‐mediated endocytosis. 5) Endosome escape through ligand interactions. Figure was created with Biorender (Biorender.com).
Up to this point, research on improving polymeric nanocarrier formulations for mRNA delivery mainly focused on prolonging circulation time and on shielding the cargo from the harsh biological environment in vivo (see Section 2.3. Block Copolymer‐Based Delivery Systems). Especially in cancer nanomedicine development, the enhanced permeability and retention (EPR) effect has been often described to be responsible for increased accumulation in tumors and has been used as a basis for this “stealthy” nanocarrier design.[ 131 ] However, the significance of this targeting effect in human patients has been heavily debated in recent years.[ 132 ] Clinical observations of the EPR effect show that it fluctuates greatly between different cancer types, patients and even within the same tumor mass.[ 133 ] On top of that, circulation times required for meaningful passive accumulation may be hard to achieve when degradation‐prone mRNA is delivered. For those reasons, passive targeting strategies might not exhaust the full potential of polymeric nanocarriers, and a fast and direct delivery of mRNA should be worked toward instead.
First and foremost, it is of great importance to choose a suitable ligand for the type of cell or tissue to be targeted. However, it can be difficult to find targets solely expressed by the targeted cells or tissues. In that case, off‐targeting side effects should be assessed. For example, CD3 antibodies can direct nanoparticles towards T‐cells expressing CD3 on their surfaces, but may also trigger cellular responses leading to T‐cell anergy and immunosuppression.[ 134 ] Similarly, CD138 targeting ligands on nanoparticles to target melanoma cells in vivo led to decreased tumor uptake compared to the non‐targeting nanoparticles due to the rapid internalization by healthy lymphocytes upon injection.[ 135 ] High‐affinity antibodies can create a barrier between the delivery vehicle and the targeted cell reducing cellular uptake.[ 124 , 132 ] Furthermore, ligands involved in receptor‐mediated vascular translocation,[ 127 ] endocytosis[ 126 ] and endosomal escape[ 130 ] can help to transfect cells that are difficult to reach or transfect otherwise, like T cells, tumor cells, and cells across the blood brain barrier (BBB).
A higher density of targeting ligands on the surface of a nanocarrier does not always translate into a higher targeting ability of the nanocarrier. In fact, the opposite can often be the case and high ligand density often decreases the targeting effect in vivo. One reason for this is because targeting ligands can significantly change the circulation time of nanocarriers and promote rapid clearance by the RES.[ 136 ] Targeting ligands also promote the formation of a protein corona with proteins in the blood, which shield targeting moieties and mitigate their efficacy. A protein corona around nanocarriers further promotes fast clearance and can lead to off‐targeting effects that can be hard to predict.[ 137 ] The impact of the protein corona on targeting nanomedicines is summarized in a review paper by Farshbaf et al.[ 138 ] and may be an essential part to consider when designing carriers for mRNA delivery in vivo.
2.4.1. Targeting Tumors
Passive tumor targeting might be insufficient for effective mRNA delivery to tumor sites.[ 132c ] This is especially relevant due to the fast degradation of mRNA in vivo. Direct and fast delivery of mRNA encoding therapeutic peptides, cytotoxic proteins or antibodies for cancer therapy directly to the tumor site can reduce side effects and circumvent costs associated with protein synthesis.[ 139 ] Tumors and tumor environments often overexpress specific antigens which can act as a targeting receptor for ligand‐based nanocarrier designs. Those include CD105 on tumor‐associated vascular endothelium,[ 140 ] α V β 3 integrins overexpressed in tumors[ 141 ] or folate receptors[ 142 ] among many others. Furthermore, a majority of cancers also overexpress glucose transporters (GLUTs), likely due to the increased energy consumption cancers require to facilitate their uncontrolled growth.[ 143 ]
Ligands promoting fast and direct delivery to tumor‐specific antigens can be implemented into the polymer design for mRNA delivery to tumor sites before mRNA is degraded in vivo. Chen et al. decorated crosslinked polymeric micelles loading mRNA with a cyclic Arg‐Gly‐Asp peptide (cRGD) to successfully deliver mRNA to tumors in vivo.[ 119 ] The cRGD is targeting α V β 3 and α V β 5 integrins that are overexpressed on the surface of certain tumor cells and endothelial cells in tumor vasculature.[ 144 ] The cRGD‐containing nanocarriers in this study increased tumor accumulation and GFP protein expression in the tumor 10‐fold compared to those without the cRGD targeting ligand.[ 119 ] Glucose‐decorated nanocarriers likewise have shown to increase tumor uptake[ 145 ] and the excess of GLUTs on the tumor vascular endothelium promoted translocation and accumulation in cancers.[ 127 ]
2.4.2. Targeting Macrophages
Macrophages are immune cells that play an essential role in the context of inflammation and cancer progression. Furthermore, they are antigen‐presenting cells which makes them promising targets for immunotherapy and for treating inflammatory diseases and cancer. Generally, two types of macrophages exist, classically activated M1 macrophages and alternatively activated M2 macrophages. Among others, M1 macrophages express CD86, tumor necrosis factor (TNF)‐α and nitric oxide synthase as possible targeting sites. Reprogramming M1 macrophages by targeted mRNA delivery could be explored to treat inflammatory diseases like rheumatoid arthritis, multiple sclerosis, or lupus.[ 146 ]
Tumor‐associated macrophages (TAMs) are immunosuppressing and cancer‐promoting M2 macrophages that form in the TME. Unlike classically activated M1 macrophages, protumoral TAMs are involved in tumor progression, metastasis and tumor resistances.[ 147 ] Among others, M2 macrophages express CD163 and CD200R and release IL‐10. Reprogramming those TAMs can enhance antitumor treatments and reverse immunosuppressing effects around the tumor.[ 146 ] For example, targeted delivery of two mRNAs (IRF5/IKKβ) using PBAE based nanocarriers with di‐mannose ligands could reprogram TAMs to regular, proinflammatory M1 macrophages. Around 28% more TAMs could be reprogrammed in vitro compared to the same nanocarrier without the targeting ligand. The delivery of mRNA to macrophages was also demonstrated in vivo treating three different cancer models as well as in vitro using human macrophages. In this study, i.v. injections of nanoparticles at 30 µg mRNA per dose resulted mainly in accumulation in spleen, liver, and lungs, which is why side effects, e.g., on liver functions after repeated administration on these organs should be assessed. Furthermore, in vivo results in this study were achieved using relatively high amounts of mRNA per dose (50 µg mRNA per dose i.v. and 100 µg mRNA per dose i.p. bi‐weekly).[ 148 ]
2.4.3. Targeting Dendritic Cells
Sufficient antigen‐presentation on APCs is essential for immunotherapy. Dendritic cells (DCs) are known to be professional antigen presenters and are heavily involved in initiating and maintaining an immune response.[ 149 ] Several receptors on DCs can provide targeting points for delivery systems to effectively deliver sufficient amounts of mRNA to DCs and generate an immune response against a variety of diseases. Extensive lists of DC receptors and ligands can be found in other reviews.[ 150 ]
A promising example is CD11c, which is mainly expressed on DCs, which makes it an excellent target for CD11c antibody fragments, fibrinogen[ 151 ] or heparin[ 152 ] ligands. Specifically, Castro et al. demonstrated superior immune responses when CD11c was targeted with its corresponding antibody fragment compared to CD205, MHC‐II, TLR2, or CD40, which are also commonly expressed on DCs.[ 153 ] Another strategy to target DCs is through their mannose receptors: Mannose‐decorated PEI nanoparticles have been shown to increase antigen delivery to dendritic cells[ 154 ] and preclinical trials with mannose‐coated liposomes have been successful in delivering mRNA to dendritic cells.[ 155 ]
2.4.4. Targeting T Cells
T‐cell therapy aims to reprogram cytotoxic T cells in our immune system to recognize and kill diseased cells. But T cells are known to be hard to transfect and besides physical transfection methods, targeted delivery is usually necessary to accomplish meaningful transfection efficiencies. CD8 and CD3, which are expressed on the surface of T cells, are commonly targeted to direct and internalize nanoparticles into T cells., Moffett et al. demonstrated that programing T‐cells ex vivo through a CD3‐targeted PBAE‐based mRNA polyplex can express genome‐editing agent in anti‐cancer T‐cells[ 156 ] (Figure 6a). Further, Parayath et al. electrochemically adsorbed CD8 antibody fragments on PBAE polymer loading mRNA to initiate rapid receptor‐induced endocytosis and deliver mRNA to T‐cells in vivo. By switching the mRNA in the core, the same delivery system was able to produce cytotoxic T cells targeting lymphoma, prostate cancer, and hepatitis B‐ induced hepatocellular carcinoma[ 49 ] (Figure 6b). Similarly, Future targeting agents might selectively deliver mRNA to activated lymphocytes expressing CD25, 4‐1BB, or CD40L by exchanging the targeting ligand to their respective antibodies on a similar polymeric delivery system.
Figure 6.
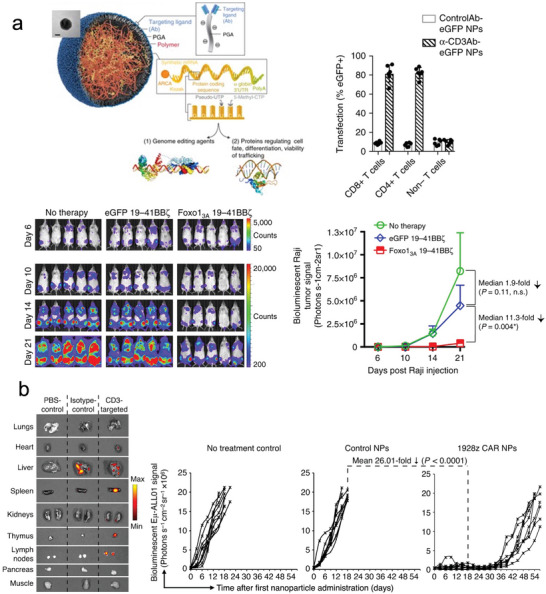
Creating nanoparticles with CD8/CD3 antibody to program therapeutic T‐cells. a) T cells were programmed ex vivo to express genome‐editing proteins using a CD3‐targeted PBAE‐based mRNA polyplex. Reproduced with permission.[ 156 ] Copyright 2017, Springer Nature. b) The same system can target CD3+ cells to generate CAR‐T cells in situ, resulting in strong antitumor activity. Reproduced with permission.[ 49 ] Copyright 2020, Springer Nature.
3. Conclusion and Future Perspectives
Since the first clinical trial of a naked mRNA cancer vaccine in 2008,[ 157 ] there has been a surge in technology platforms to overcome the instability and the inherent immunogenicity of IVT mRNA and exploit effective mRNA delivery systems. Many mRNA structural optimization methods, like modifying nucleosides, caps, poly(A) tails, and engineering the untranslated regions, have been applied to improve stability and reduce the innate immunogenicity of synthetic mRNA. Regarding delivery, polymer‐based nanocarriers have shown considerable potential in mRNA therapeutics. The studies described in this review assessed the recent progress in polymer design and selection of targeting ligands for mRNA therapeutics, which will help to pave the way for advanced therapies.
Although polyplexes are simple in structure compared to nanoparticulate systems with more elaborated architectures, the opportunities for modification are myriad. The creation of an efficient delivery system for nucleic acids demands fine‐tuning of different aspects of the carrier. The molecular weight of a cationic polymer and the carrier to RNA ratio can strongly influence the cytotoxicity and needs to match the type of nucleic acid being encapsulated. Some systems for mRNA delivery directly employ formulations that were optimized for a different type of nucleic acid, missing crucial steps in carrier optimization.[ 26 ] Another factor is the toxicity of the polymer, where labile bonds, such as ester or disulfide groups, can improve biocompatibility. Endosomal escape is a major barrier towards cytosolic delivery of mRNA and incorporating ionizable groups with a pKa around 6.0–6.5 or membrane‐active peptides can ameliorate transfection efficiency. The addition of surface ligands can contribute to effective targeting of cells and increase cellular uptake. Surface modification with a hydrophilic moiety, such as PEG, can work to improve colloidal stability of particulate mRNA formulations. In fact, most of the systems reviewed for systemic administration relied on PEG or some other surface coating to stabilize the particle (Table 2). Whereas most polymer‐based platforms are geared towards local applications, such as vaccines, the recent trend in optimizing for side‐chain hydrophobicity has further improved biological stability enabling systemic targeting of mRNA cargo to the lungs, which was previously unfeasible. This enables further exploitation of mRNA‐based advanced therapies, such as protein replacement in cystic fibrosis or immunotherapy in cancer.
Polymeric micelle delivery vectors have some natural advantages due to their unique core–shell structure and significantly improved shielding. The improved understanding of the structure–function relationships has led to more sophisticated polymer structures. Recently, it is clear that polycation properties such as hydrophobicity, flexibility, and hydrogen bonding can be tuned to improve polycation/mRNA binding. Hybridizing mRNA with functional oligoRNAs could be applied as an effective method to condense and functionalize the micelle core. Besides physical stabilization, integrating stimuli‐responsive crosslinking moieties in the polycation can offer excellent in vivo and in vitro stability and also selective release of the mRNA payload inside the target cell. Considerable recent advances have allowed therapeutic applications of mRNA polymeric micelles including genome editing therapy and cancer therapy after local or systemic administration (Table 3). Micelles have the potential for tissue selective delivery of mRNA after systemic administration. Further improvements in the ability of micelles to protect mRNA during circulation in the bloodstream are likely to enhance the selectivity of the delivery, as well as increase the levels of protein translation in target tissues. Moreover, the micelles could integrate stimuli‐responsive functions for triggering mRNA translation in specific cells, which might be optimized according to the therapeutic demands. For example, we have recently used the increased endo/lysosomal acidification of some cancer cells compared to noncancerous cells for designing polymeric micelles with tunable pH‐activated intracellular delivery.[ 158 ] These micelles allowed selective protein delivery inside the cytosol of the cancer cells.[ 158 ] Thus, it would also be possible to control the specificity of mRNA delivery by regulating the access of micelles into the cytosol, which would provide an additional targeting approach to the conventional targeting strategies of pharmacokinetic control and ligand‐mediated tissue accumulation and cell uptake.
The proteins adsorbed on the nanocarriers, i.e., the protein corona, can greatly affect their physicochemical characteristics and delivery efficiency. For example, the adsorption of bovine serum albumin on the surface of PEI/mRNA complexes altered the translation.[ 159 ] Moreover, as the protein corona is dynamic and depends on the biological environment surrounding the nanocarriers,[ 160 ] it may be possible that the proteins adsorbing on the surface of polymeric nanocarriers may change depending on the tissues where the nanocarriers are locating, which could influence their interaction with cells and eventually their activity. Moreover, the protein corona may also determine the biodistribution of the polymeric nanocarriers in vivo. In the case of mRNA‐loaded LNPs, the proteins adsorbed on their surface were found to play an important role in their tropism to liver, spleen, or lungs.[ 161 ] Thus, further understanding of the effects of the protein corona on the performance of polymeric delivery systems of mRNA may allow designing innovative strategies with high efficacy. Notably, polymeric micelles have shown negligible adsorption of proteins on their surface probably due to their high PEG density,[ 162 ] which could be a useful feature for controlling the biodistribution and cellular targeting based on their physicochemical properties and the installation of ligands.
Targeting ligands could provide polymeric nanocarriers an extra boost to increase target accumulation, uptake, and release of mRNA in targeted cells. With their inclusion, limitations due to the insufficient ability to transport mRNA into the cytoplasm of specific cells before their degradation could be improved in various examples presented in this review. We believe that fast and specific delivery can also make up for the low passive accumulation and cellular uptake of mRNA delivery systems that come with the low circulation times. Polymeric nanocarriers can easily be decorated with targeting ligands on their surfaces which makes them suitable candidates for targeted mRNA delivery through ligand‐receptor interactions. However, despite the plethora of benefits, one must also keep in mind the risks of unpredicted off‐targeting effects due to changes in particle size, surface charge, or the formation of a protein corona in blood. For now, the amount of literature using targeting ligands in polymeric formulations to deliver mRNA is still limited. For example, despite great interest to deliver therapeutic agents to the brain and various studies describing receptor mediated strategies to improve drug uptake,[ 128 , 163 ] no studies of systemic mRNA delivery using this approach have been reported to this date. Ligand requirements, like density or binding affinity, need to be optimized specifically according to different situations. Thus, the development of relevant in vitro models that allow predicting the in vivo performance of ligand‐installed mRNA delivery systems are highly desired. In this regard, organ‐on‐a‐chip and body‐on‐a‐chip systems are emerging as new predictive platforms for toxicity and efficacy assessment of nanoparticles, and could have a central role in the optimization of ligand‐installed mRNA delivery systems. This is likely to change within the near future due to the rising interest in mRNA‐based therapies and the potential that targeting ligands and nanocarriers have demonstrated across the fields of various delivery systems.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) (Project No. 16 cm0106202h0001; H.C.) from the Japan Agency for Medical Research and Development (AMED) and GAP Fund from the University of Tokyo (H.C.). The study was also partially supported by Grants‐in‐Aid for Scientific Research B (20H04524; H.C.). This work was supported by fellowship of the Chugai Foundation for Innovative Drug Discovery Science:C‐FINDs (W.Y.).
Biographies
Wenqian Yang received her M.Sc. in pharmaceutics from Shenyang Pharmaceutical University, Shenyang, China, in 2018. Then, she received her Ph.D. in bioengineering from the University of Tokyo in 2021. She was granted a Japan Society for the Promotion of Science (JSPS) fellowship for her research on developing novel delivery system for messenger RNA (mRNA) using polymers. Since 2021, she is a postdoctoral fellow at Cabral's lab based on the prestigious fellowship of the Chugai Foundation for Innovative Drug Discovery Science. Her work focuses on the analysis and optimization of interactions between polymeric carriers and RNA cargo.

Lucas Mixich is a Ph.D. candidate at the Department of Bioengineering, Graduate School of Engineering, the University of Tokyo. He holds an M.Sc. in technical chemistry from the Graz University of Technology and was awarded a Monbukagakusho (MEXT) scholarship in 2020 to pursue his Ph.D. in Japan under the supervision of associate professor Horacio Cabral. His research is centered on developing polymeric mRNA delivery systems for cancer treatment.

Eger Boonstra received his M.Sc. degree from Utrecht University in 2018 under the supervision of Prof. Hennink. He was granted the Monbukagakusho (MEXT) scholarship from the Japanese government and joined Dr. Cabral's lab as a Ph.D. student in 2019. He received his Ph.D. in bioengineering from the University of Tokyo in 2022. He has a broad background in the mRNA delivery field, with specific expertise in endosomal escape and non‐viral RNA delivery for genetic vaccines.

Horacio Cabral is an associate professor at the Department of Bioengineering, Graduate School of Engineering, the University of Tokyo. He received his Ph.D. in materials engineering from the University of Tokyo, in 2007 under the supervision of Prof. Kazunori Kataoka. His research interests focus on nanomedicine for diagnosis and treatment of intractable diseases. His findings have provided breakthroughs in understanding the design of nanomedicines for tumor‐targeted therapy. Moreover, his work has led to several clinical studies of nanotherapeutics. In 2021, he co‐founded Red Arrow Therapeutics aiming to develop next‐generation therapeutics through targeted delivery of biologics.

Yang W., Mixich L., Boonstra E., Cabral H., Polymer‐Based mRNA Delivery Strategies for Advanced Therapies. Adv. Healthcare Mater. 2023, 12, 2202688. 10.1002/adhm.202202688
References
- 1. Sahin U., Kariko K., Tureci O., Nat. Rev. Drug Discovery 2014, 13, 759. [DOI] [PubMed] [Google Scholar]
- 2.a) Polack F. P., Thomas S. J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J. L., Perez Marc G., Moreira E. D., Zerbini C., Bailey R., Swanson K. A., Roychoudhury S., Koury K., Li P., Kalina W. V., Cooper D., Frenck R. W. Jr., Hammitt L. L., Tureci O., Nell H., Schaefer A., Unal S., Tresnan D. B., Mather S., Dormitzer P. R., Sahin U., Jansen K. U., Gruber W. C., Group C. C. T., N. Engl. J. Med. 2020, 383, 2603; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shroff R. T., Chalasani P., Wei R., Pennington D., Quirk G., Schoenle M. V., Peyton K. L., Uhrlaub J. L., Ripperger T. J., Jergovic M., Dalgai S., Wolf A., Whitmer R., Hammad H., Carrier A., Scott A. J., Nikolich‐Zugich J., Worobey M., Sprissler R., Dake M., LaFleur B. J., Bhattacharya D., Nat. Med. 2021, 27, 2002; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jackson L. A., Anderson E. J., Rouphael N. G., Roberts P. C., Makhene M., Coler R. N., McCullough M. P., Chappell J. D., Denison M. R., Stevens L. J., Pruijssers A. J., McDermott A., Flach B., Doria‐Rose N. A., Corbett K. S., Morabito K. M., O'Dell S., Schmidt S. D., Swanson P. A. 2nd, Padilla M., Mascola J. R., Neuzil K. M., Bennett H., Sun W., Peters E., Makowski M., Albert J., Cross K., Buchanan W., Pikaart‐Tautges R., et al, N. Engl. J. Med. 2020, 383, 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morse D. E., Yanofsky C., Nature 1969, 224, 329. [DOI] [PubMed] [Google Scholar]
- 4.a) Uchida S., Perche F., Pichon C., Cabral H., Mol. Pharm. 2020, 17, 3654; [DOI] [PubMed] [Google Scholar]; b) Hou X., Zaks T., Langer R., Dong Y., Nat. Rev. Mater. 2021, 6, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naldini L., Nature 2015, 526, 351. [DOI] [PubMed] [Google Scholar]
- 6. Bessis N., GarciaCozar F. J., Boissier M. C., Gene Ther. 2004, 11, S10. [DOI] [PubMed] [Google Scholar]
- 7. Islam M. A., Reesor E. K., Xu Y., Zope H. R., Zetter B. R., Shi J., Biomater. Sci. 2015, 3, 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chaudhary N., Weissman D., Whitehead K. A., Nat. Rev. Drug Discovery 2021, 20, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whitley J., Zwolinski C., Denis C., Maughan M., Hayles L., Clarke D., Snare M., Liao H., Chiou S., Marmura T., Zoeller H., Hudson B., Peart J., Johnson M., Karlsson A., Wang Y., Nagle C., Harris C., Tonkin D., Fraser S., Capiz L., Zeno C. L., Meli Y., Martik D., Ozaki D. A., Caparoni A., Dickens J. E., Weissman D., Saunders K. O., Haynes B. F., et al, Transl. Res. 2022, 242, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Cheng Q., Wei T., Jia Y., Farbiak L., Zhou K., Zhang S., Wei Y., Zhu H., Siegwart D. J., Adv. Mater. 2018, 30, 1805308; [DOI] [PubMed] [Google Scholar]; b) DeRosa F., Guild B., Karve S., Smith L., Love K., Dorkin J. R., Kauffman K. J., Zhang J., Yahalom B., Anderson D. G., Heartlein M. W., Gene Ther. 2016, 23, 699; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang X., Zhao W., Nguyen G. N., Zhang C., Zeng C., Yan J., Du S., Hou X., Li W., Jiang J., Deng B., McComb D. W., Dorkin R., Shah A., Barrera L., Gregoire F., Singh M., Chen D., Sabatino D. E., Dong Y., Sci. Adv. 2020, 6, eabc2315; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu J., Chang J., Jiang Y., Meng X., Sun T., Mao L., Xu Q., Wang M., Adv. Mater. 2019, 31, 1902575; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Qiu M., Glass Z., Chen J., Haas M., Jin X., Zhao X., Rui X., Ye Z., Li Y., Zhang F., Xu Q., Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fischer D., Li Y., Ahlemeyer B., Krieglstein J., Kissel T., Biomaterials 2003, 24, 1121. [DOI] [PubMed] [Google Scholar]
- 12.a) Guerrero‐Cazares H., Tzeng S. Y., Young N. P., Abutaleb A. O., Quinones‐Hinojosa A., Green J. J., ACS Nano 2014, 8, 5141; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fornaguera C., Castells‐Sala C., Lazaro M. A., Cascante A., Borros S., Int. J. Pharm. 2019, 569, 118612. [DOI] [PubMed] [Google Scholar]
- 13.a) Alameh M., Lavertu M., Tran‐Khanh N., Chang C. Y., Lesage F., Bail M., Darras V., Chevrier A., Buschmann M. D., Biomacromolecules 2018, 19, 112; [DOI] [PubMed] [Google Scholar]; b) Jarzebska N. T., Lauchli S., Iselin C., French L. E., Johansen P., Guenova E., Kundig T. M., Pascolo S., Drug Delivery 2020, 27, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Wilson D. S., Dalmasso G., Wang L., Sitaraman S. V., Merlin D., Murthy N., Nat. Mater. 2010, 9, 923; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ballarin‐Gonzalez B., Dagnaes‐Hansen F., Fenton R. A., Gao S., Hein S., Dong M., Kjems J., Howard K. A., Mol. Ther.– Nucleic Acids 2013, 2, e76; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Han L., Tang C., Yin C., Biomaterials 2014, 35, 4589. [DOI] [PubMed] [Google Scholar]
- 15.a) Demeneix B., Behr J. P., Adv. Genet. 2005, 53PA, 215; [DOI] [PubMed] [Google Scholar]; b) Shim B. S., Park S. M., Quan J. S., Jere D., Chu H., Song M. K., Kim D. W., Jang Y. S., Yang M. S., Han S. H., Park Y. H., Cho C. S., Yun C. H., BMC Immunol. 2010, 11, 65; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Boussif O., Lezoualc'h F., Zanta M. A., Mergny M. D., Scherman D., Demeneix B., Behr J. P., Proc. Natl. Acad. Sci. USA 1995, 92, 7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wojnilowicz M., Glab A., Bertucci A., Caruso F., Cavalieri F., ACS Nano 2019, 13, 187. [DOI] [PubMed] [Google Scholar]
- 17. Boonstra E., Hatano H., Miyahara Y., Uchida S., Goda T., Cabral H., J. Mater. Chem. B 2021, 9, 4298. [DOI] [PubMed] [Google Scholar]
- 18.a) Lv H., Zhang S., Wang B., Cui S., Yan J., J. Controlled Release 2006, 114, 100; [DOI] [PubMed] [Google Scholar]; b) Breunig M., Lungwitz U., Liebl R., Goepferich A., Proc. Natl. Acad. Sci. USA 2007, 104, 14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Thomas M., Lu J. J., Ge Q., Zhang C. C., Chen J. Z., Klibanov A. M., Proc. Natl. Acad. Sci. USA 2005, 102, 5679; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Choi Y. J., Kang S. J., Kim Y. J., Lim Y. B., Chung H. W., Drug Chem. Toxicol. 2010, 33, 357; [DOI] [PubMed] [Google Scholar]; c) Beyerle A., Irmler M., Beckers J., Kissel T., Stoeger T., Mol. Pharm. 2010, 7, 727. [DOI] [PubMed] [Google Scholar]
- 20.a) Peng Q., Zhong Z., Zhuo R., Bioconjugate Chem. 2008, 19, 499; [DOI] [PubMed] [Google Scholar]; b) Taranejoo S., Liu J., Verma P., Hourigan K., J. Appl. Polym. Sci. 2015, 132, 1.25866416 [Google Scholar]
- 21. Bettinger T., Carlisle R. C., Read M. L., Ogris M., Seymour L. W., Nucleic Acids Res. 2001, 29, 3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Rejman J., Tavernier G., Bavarsad N., Demeester J., De Smedt S. C., J. Controlled Release 2010, 147, 385; [DOI] [PubMed] [Google Scholar]; b) Demoulins T., Ebensen T., Schulze K., Englezou P. C., Pelliccia M., Guzman C. A., Ruggli N., McCullough K. C., J. Controlled Release 2017, 266, 256; [DOI] [PubMed] [Google Scholar]; c) Johler S. M., Rejman J., Guan S., Rosenecker J., PLoS One 2015, 10, e0137504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Krhac Levacic A., Berger S., Muller J., Wegner A., Lachelt U., Dohmen C., Rudolph C., Wagner E., J. Controlled Release 2021, 339, 27; [DOI] [PubMed] [Google Scholar]; b) Tan L., Zheng T., Li M., Zhong X., Tang Y., Qin M., Sun X., Drug Delivery Transl. Res. 2020, 10, 678. [DOI] [PubMed] [Google Scholar]
- 24.a) Chiper M., Tounsi N., Kole R., Kichler A., Zuber G., J. Controlled Release 2017, 246, 60; [DOI] [PubMed] [Google Scholar]; b) Ren J., Cao Y., Li L., Wang X., Lu H., Yang J., Wang S., J. Controlled Release 2021, 338, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao M., Li M., Zhang Z., Gong T., Sun X., Drug Delivery 2016, 23, 2596. [DOI] [PubMed] [Google Scholar]
- 26. Oh J., Kim S. M., Lee E. H., Kim M., Lee Y., Ko S. H., Jeong J. H., Park C. H., Lee M., Biomater. Sci. 2020, 8, 3063. [DOI] [PubMed] [Google Scholar]
- 27. Chollet P., Favrot M. C., Hurbin A., Coll J. L., J. Gene Med. 2002, 4, 84. [DOI] [PubMed] [Google Scholar]
- 28. Ke X., Shelton L., Hu Y., Zhu Y., Chow E., Tang H., Santos J. L., Mao H. Q., ACS Appl. Mater. Interfaces 2020, 12, 35835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li M., Li Y., Peng K., Wang Y., Gong T., Zhang Z., He Q., Sun X., Acta Biomater. 2017, 64, 237. [DOI] [PubMed] [Google Scholar]
- 30. Laemmli U. K., Proc. Natl. Acad. Sci. USA 1975, 72, 4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.a) Bello Roufaï M., Midoux P., Bioconjugate Chem. 2001, 12, 92; [DOI] [PubMed] [Google Scholar]; b) Alazzo A., Gumus N., Gurnani P., Stolnik S., Rahman R., Spriggs K., Alexander C., J. Mater. Chem. B 2022, 10, 236. [DOI] [PubMed] [Google Scholar]
- 32. Dirisala A., Uchida S., Li J., Van Guyse J. F. R., Hayashi K., Vummaleti S. V. C., Kaur S., Mochida Y., Fukushima S., Kataoka K., Macromol. Rapid Commun. 2022, 43, 2100754. [DOI] [PubMed] [Google Scholar]
- 33. Itaka K., Ishii T., Hasegawa Y., Kataoka K., Biomaterials 2010, 31, 3707. [DOI] [PubMed] [Google Scholar]
- 34. Uchida H., Itaka K., Nomoto T., Ishii T., Suma T., Ikegami M., Miyata K., Oba M., Nishiyama N., Kataoka K., J. Am. Chem. Soc. 2014, 136, 12396. [DOI] [PubMed] [Google Scholar]
- 35. Uchida H., Miyata K., Oba M., Ishii T., Suma T., Itaka K., Nishiyama N., Kataoka K., J. Am. Chem. Soc. 2011, 133, 15524. [DOI] [PubMed] [Google Scholar]
- 36. Uchida H., Itaka K., Uchida S., Ishii T., Suma T., Miyata K., Oba M., Nishiyama N., Kataoka K., J. Am. Chem. Soc. 2016, 138, 1478. [DOI] [PubMed] [Google Scholar]
- 37. Naito M., Otsu Y., Kamegawa R., Hayashi K., Uchida S., Kim H. J., Miyata K., Sci. Technol. Adv. Mater. 2019, 20, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yum J., Kim B. S., Ogura S., Kamegawa R., Naito M., Yamasaki Y., Kim H. J., Miyata K., J. Controlled Release 2022, 342, 148. [DOI] [PubMed] [Google Scholar]
- 39. Shive M. S., Anderson J. M., Adv. Drug Delivery Rev. 1997, 28, 5. [DOI] [PubMed] [Google Scholar]
- 40. Holvold L. B., Fredriksen B. N., Bogwald J., Dalmo R. A., Fish Shellfish Immunol. 2013, 35, 890. [DOI] [PubMed] [Google Scholar]
- 41. Fu K., Pack D. W., Klibanov A. M., Langer R., Pharm. Res. 2000, 17, 100. [DOI] [PubMed] [Google Scholar]
- 42.a) Xiao B., Zhang Z., Viennois E., Kang Y., Zhang M., Han M. K., Chen J., Merlin D., Theranostics 2016, 6, 2250; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pai Kasturi S., Qin H., Thomson K. S., El‐Bereir S., Cha S. C., Neelapu S., Kwak L. W., Roy K., J. Controlled Release 2006, 113, 261; [DOI] [PubMed] [Google Scholar]; c) Singh M., Ugozzoli M., Briones M., Kazzaz J., Soenawan E., O'Hagan D. T., Pharm. Res. 2003, 20, 247. [DOI] [PubMed] [Google Scholar]
- 43. Mahiny A. J., Dewerth A., Mays L. E., Alkhaled M., Mothes B., Malaeksefat E., Loretz B., Rottenberger J., Brosch D. M., Reautschnig P., Surapolchai P., Zeyer F., Schams A., Carevic M., Bakele M., Griese M., Schwab M., Nürnberg B., Beer‐Hammer S., Handgretinger R., Hartl D., Lehr C.‐M., Kormann M. S. D., Nat. Biotechnol. 2015, 33, 584. [DOI] [PubMed] [Google Scholar]
- 44. Haque A., Dewerth A., Antony J. S., Riethmuller J., Schweizer G. R., Weinmann P., Latifi N., Yasar H., Pedemonte N., Sondo E., Weidensee B., Ralhan A., Laval J., Schlegel P., Seitz C., Loretz B., Lehr C. M., Handgretinger R., Kormann M. S. D., Sci. Rep. 2018, 8, 16776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.a) Yin H., Kanasty R. L., Eltoukhy A. A., Vegas A. J., Dorkin J. R., Anderson D. G., Nat. Rev. Genet. 2014, 15, 541; [DOI] [PubMed] [Google Scholar]; b) Gao Y., Huang J. Y., O'Keeffe Ahern J., Cutlar L., Zhou D., Lin F. H., Wang W., Biomacromolecules 2016, 17, 3640. [DOI] [PubMed] [Google Scholar]
- 46. Anderson D. G., Lynn D. M., Langer R., Angew. Chem., Int. Ed. Engl. 2003, 42, 3153. [DOI] [PubMed] [Google Scholar]
- 47. Capasso Palmiero U., Kaczmarek J. C., Fenton O. S., Anderson D. G., Adv. Healthcare Mater. 2018, 7, 1800249. [DOI] [PubMed] [Google Scholar]
- 48.a) Rui Y., Wilson D. R., Tzeng S. Y., Yamagata H. M., Sudhakar D., Conge M., Berlinicke C. A., Zack D. J., Tuesca A., Green J. J., Sci. Adv. 2022, 8, eabk2855; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kaczmarek J. C., Patel A. K., Kauffman K. J., Fenton O. S., Webber M. J., Heartlein M. W., DeRosa F., Anderson D. G., Angew. Chem., Int. Ed. Engl. 2016, 55, 13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parayath N. N., Stephan S. B., Koehne A. L., Nelson P. S., Stephan M. T., Nat. Commun. 2020, 11, 6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patel A. K., Kaczmarek J. C., Bose S., Kauffman K. J., Mir F., Heartlein M. W., DeRosa F., Langer R., Anderson D. G., Adv. Mater. 2019, 31, 1805116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blanchard E. L., Vanover D., Bawage S. S., Tiwari P. M., Rotolo L., Beyersdorf J., Peck H. E., Bruno N. C., Hincapie R., Michel F., Murray J., Sadhwani H., Vanderheyden B., Finn M. G., Brinton M. A., Lafontaine E. R., Hogan R. J., Zurla C., Santangelo P. J., Nat. Biotechnol. 2021, 39, 717. [DOI] [PubMed] [Google Scholar]
- 52. Yu X., Liu S., Cheng Q., Lee S. M., Wei T., Zhang D., Farbiak L., Johnson L. T., Wang X., Siegwart D. J., Pharmaceutics 2021, 13,1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grun M. K., Suberi A., Shin K., Lee T., Gomerdinger V., Moscato Z. M., Piotrowski‐Daspit A. S., Saltzman W. M., Biomaterials 2021, 272, 120780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chuan D., Jin T., Fan R., Zhou L., Guo G., Adv. Colloid Interface Sci. 2019, 268, 25. [DOI] [PubMed] [Google Scholar]
- 55.a) Lallana E., Rios de la Rosa J. M., Tirella A., Pelliccia M., Gennari A., Stratford I. J., Puri S., Ashford M., Tirelli N., Mol. Pharm. 2017, 14, 2422; [DOI] [PubMed] [Google Scholar]; b) Soliman O. Y., Alameh M. G., De Cresenzo G., Buschmann M. D., Lavertu M., J. Pharm. Sci. 2020, 109, 1581. [DOI] [PubMed] [Google Scholar]
- 56. Jeandupeux E., Alameh M. G., Ghattas M., De Crescenzo G., Lavertu M., J. Pharm. Sci. 2021, 110, 3439. [DOI] [PubMed] [Google Scholar]
- 57. Hajam I. A., Senevirathne A., Hewawaduge C., Kim J., Lee J. H., Vet. Res. 2020, 51, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Amos H., Biochem. Biophys. Res. Commun. 1961, 5, 1. [Google Scholar]
- 59.a) Jarzebska N. T., Mellett M., Frei J., Kundig T. M., Pascolo S., Pharmaceutics 2021, 13, 877; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ruseska I., Fresacher K., Petschacher C., Zimmer A., Nanomaterials 2021, 11, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.a) Scheel B., Teufel R., Probst J., Carralot J.‐P., Geginat J., Radsak M., Jarrossay D., Wagner H., Jung G., Rammensee H.‐G., Hoerr I., Pascolo S., Eur. J. Immunol. 2005, 35, 1557; [DOI] [PubMed] [Google Scholar]; b) Petsch B., Schnee M., Vogel A. B., Lange E., Hoffmann B., Voss D., Schlake T., Thess A., Kallen K. J., Stitz L., Kramps T., Nat. Biotechnol. 2012, 30, 1210. [DOI] [PubMed] [Google Scholar]
- 61. Kallen K. J., Heidenreich R., Schnee M., Petsch B., Schlake T., Thess A., Baumhof P., Scheel B., Koch S. D., Fotin‐Mleczek M., Hum. Vaccines Immunother. 2013, 9, 2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weide B., Pascolo S., Scheel B., Derhovanessian E., Pflugfelder A., Eigentler T. K., Pawelec G., Hoerr I., Rammensee H. G., Garbe C., J. Immunother. 2009, 32, 498. [DOI] [PubMed] [Google Scholar]
- 63.a) Sebastian M., Schroder A., Scheel B., Hong H. S., Muth A., von Boehmer L., Zippelius A., Mayer F., Reck M., Atanackovic D., Thomas M., Schneller F., Stohlmacher J., Bernhard H., Groschel A., Lander T., Probst J., Strack T., Wiegand V., Gnad‐Vogt U., Kallen K. J., Hoerr I., von der Muelbe F., Fotin‐Mleczek M., Knuth A., Koch S. D., Cancer Immunol. Immunother. 2019, 68, 799; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Papachristofilou A., Hipp M. M., Klinkhardt U., Fruh M., Sebastian M., Weiss C., Pless M., Cathomas R., Hilbe W., Pall G., Wehler T., Alt J., Bischoff H., Geissler M., Griesinger F., Kallen K. J., Fotin‐Mleczek M., Schroder A., Scheel B., Muth A., Seibel T., Stosnach C., Doener F., Hong H. S., Koch S. D., Gnad‐Vogt U., Zippelius A., J. Immunother. Cancer 2019, 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kubler H., Scheel B., Gnad‐Vogt U., Miller K., Schultze‐Seemann W., Vom Dorp F., Parmiani G., Hampel C., Wedel S., Trojan L., Jocham D., Maurer T., Rippin G., Fotin‐Mleczek M., von der Mulbe F., Probst J., Hoerr I., Kallen K. J., Lander T., Stenzl A., J. Immunother. Cancer 2015, 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Stenzl A., Feyerabend S., Syndikus I., Sarosiek T., Kübler H., Heidenreich A., Cathomas R., Grüllich C., Loriot Y., Perez Gracia S. L., Gillessen S., Klinkhardt U., Schröder A., Schönborn‐Kellenberger O., Reus V., Koch S. D., Hong H. S., Seibel T., Fizazi K., Gnad‐Vogt U., Ann. Oncol. 2017, 28, v408. [Google Scholar]
- 66. Alberer M., Gnad‐Vogt U., Hong H. S., Mehr K. T., Backert L., Finak G., Gottardo R., Bica M. A., Garofano A., Koch S. D., Fotin‐Mleczek M., Hoerr I., Clemens R., von Sonnenburg F., Lancet 2017, 390, 1511. [DOI] [PubMed] [Google Scholar]
- 67. Schnee M., Vogel A. B., Voss D., Petsch B., Baumhof P., Kramps T., Stitz L., PLoS Neglected Trop. Dis. 2016, 10, e0004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aldrich C., Leroux‐Roels I., Huang K. B., Bica M. A., Loeliger E., Schoenborn‐Kellenberger O., Walz L., Leroux‐Roels G., von Sonnenburg F., Oostvogels L., Vaccine 2021, 39, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.a) Convertine A. J., Diab C., Prieve M., Paschal A., Hoffman A. S., Johnson P. H., Stayton P. S., Biomacromolecules 2010, 11, 2904; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fliervoet L. A. L., Zhang H., van Groesen E., Fortuin K., Duin N. J. C. B., Remaut K., Schiffelers R. M., Hennink W. E., Vermonden T., Nanoscale 2020, 12, 10347; [DOI] [PubMed] [Google Scholar]; c) Jacobson M. E., Wang‐Bishop L., Becker K. W., Wilson J. T., Biomater. Sci. 2019, 7, 547. [DOI] [PubMed] [Google Scholar]
- 70. Ulkoski D., Munson M. J., Jacobson M. E., Palmer C. R., Carson C. S., Sabirsh A., Wilson J. T., Krishnamurthy V. R., ACS Appl. Bio Mater. 2021, 4, 1640. [DOI] [PubMed] [Google Scholar]
- 71. Lou B., De Koker S., Lau C. Y. J., Hennink W. E., Mastrobattista E., Bioconjugate Chem. 2019, 30, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kheraldine H., Rachid O., Habib A. M., Al Moustafa A.‐E., Benter I. F., Akhtar S., Adv. Drug Delivery Rev. 2021, 178, 113908. [DOI] [PubMed] [Google Scholar]
- 73. Kolhatkar R. B., Kitchens K. M., Swaan P. W., Ghandehari H., Bioconjugate Chem. 2007, 18, 2054. [DOI] [PubMed] [Google Scholar]
- 74.a) Jevprasesphant R., Penny J., Jalal R., Attwood D., McKeown N. B., D'Emanuele A., Int. J. Pharm. 2003, 252, 263; [DOI] [PubMed] [Google Scholar]; b) Jain K., Kesharwani P., Gupta U., Jain N. K., Int. J. Pharm. 2010, 394, 122. [DOI] [PubMed] [Google Scholar]
- 75. Shetty A., Chikhaliwala P., Suryawanshi J., Chandra S., Curr. Pathobiol. Rep. 2021, 9, 57. [Google Scholar]
- 76. Biswas S., Deshpande P. P., Navarro G., Dodwadkar N. S., Torchilin V. P., Biomaterials 2013, 34, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.a) Verminnen K., Beeckman D. S. A., Sanders N. N., De Smedt S., Vanrompay D. C. G., Vaccine 2010, 28, 3095; [DOI] [PubMed] [Google Scholar]; b) Choi Y., Thomas T., Kotlyar A., Islam M. T., Baker J. R. Jr., Chem. Biol. 2005, 12, 35. [DOI] [PubMed] [Google Scholar]
- 78.a) Bøe S. L., Jørgensen J. A. L., Longva A. S., Lavelle T., Sæbøe‐Larssen S., Hovig E., Nucleic Acid Ther. 2013, 23, 160; [DOI] [PubMed] [Google Scholar]; b) Abedi‐Gaballu F., Dehghan G., Ghaffari M., Yekta R., Abbaspour‐Ravasjani S., Baradaran B., Ezzati Nazhad Dolatabadi J., Hamblin M. R., Appl. Mater. Today 2018, 12, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chahal J. S., Khan O. F., Cooper C. L., McPartlan J. S., Tsosie J. K., Tilley L. D., Sidik S. M., Lourido S., Langer R., Bavari S., Ploegh H. L., Anderson D. G., Proc. Natl. Acad. Sci. USA 2016, 113, E4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Blakney A. K., Zhu Y., McKay P. F., Bouton C. R., Yeow J., Tang J., Hu K., Samnuan K., Grigsby C. L., Shattock R. J., Stevens M. M., ACS Nano 2020, 14, 5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Blakney A. K., Yilmaz G., McKay P. F., Becer C. R., Shattock R. J., Biomacromolecules 2018, 19, 2870. [DOI] [PubMed] [Google Scholar]
- 82. Frohlich E., Int. J. Nanomed. 2012, 7, 5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Monnery B. D., Wright M., Cavill R., Hoogenboom R., Shaunak S., Steinke J. H. G., Thanou M., Int. J. Pharm. 2017, 521, 249. [DOI] [PubMed] [Google Scholar]
- 84. Nishikawa M., Huang L., Hum. Gene Ther. 2001, 12, 861. [DOI] [PubMed] [Google Scholar]
- 85.a) Laga R., Carlisle R., Tangney M., Ulbrich K., Seymour L. W., J. Controlled Release 2012, 161, 537; [DOI] [PubMed] [Google Scholar]; b) Poon G. M. K., Gariépy J., Biochem. Soc. Trans. 2007, 35, 788. [DOI] [PubMed] [Google Scholar]
- 86.a) Abuchowski A., McCoy J. R., Palczuk N. C., van Es T., Davis F. F., J. Biol. Chem. 1977, 252, 3582; [PubMed] [Google Scholar]; b) Knop K., Hoogenboom R., Fischer D., Schubert U. S., Angew. Chem., Int. Ed. 2010, 49, 6288. [DOI] [PubMed] [Google Scholar]
- 87.a) Hatakeyama H., Akita H., Harashima H., Biol. Pharm. Bull. 2013, 36, 892; [DOI] [PubMed] [Google Scholar]; b) Yu M., Huang S., Yu K. J., Clyne A. M., Int. J. Mol. Sci. 2012, 13, 5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cabral H., Miyata K., Osada K., Kataoka K., Chem. Rev. 2018, 118, 6844. [DOI] [PubMed] [Google Scholar]
- 89. Zuckerman J. E., Choi C. H., Han H., Davis M. E., Proc. Natl. Acad. Sci. USA 2012, 109, 3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.a) Mastorakos P., Zhang C., Berry S., Oh Y., Lee S., Eberhart C. G., Woodworth G. F., Suk J. S., Hanes J., Adv. Healthcare Mater. 2015, 4, 1023; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Williford J. M., Archang M. M., Minn I., Ren Y., Wo M., Vandermark J., Fisher P. B., Pomper M. G., Mao H. Q., ACS Biomater. Sci. Eng. 2016, 2, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wang Y., Ma B., Abdeen A. A., Chen G., Xie R., Saha K., Gong S., ACS Appl. Mater. Interfaces 2018, 10, 31915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.a) Kataoka K., Harada A., Nagasaki Y., Adv. Drug Delivery Rev. 2001, 47, 113; [DOI] [PubMed] [Google Scholar]; b) Jarak I., Pereira‐Silva M., Santos A. C., Veiga F., Cabral H., Figueiras A., Appl. Mater. Today 2021, 25, 101217. [Google Scholar]
- 93.a) Trichet V., Layrolle P., Escriou V., Nanomedicine 2012, 7, 181; [DOI] [PubMed] [Google Scholar]; b) Li J., Ge Z., Liu S., Chem. Commun. 2013, 49, 6974. [DOI] [PubMed] [Google Scholar]
- 94.a) Uchida S., Itaka K., Uchida H., Hayakawa K., Ogata T., Ishii T., Fukushima S., Osada K., Kataoka K., PLoS One 2013, 8, e56220. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Uchida S., Kataoka K., J. Biomed. Mater. Res., Part A 2019, 107, 978. [DOI] [PubMed] [Google Scholar]
- 95. Oberli M. A., Reichmuth A. M., Dorkin J. R., Mitchell M. J., Fenton O. S., Jaklenec A., Anderson D. G., Langer R., Blankschtein D., Nano Lett. 2017, 17, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.a) Chahal J. S., Fang T., Woodham A. W., Khan O. F., Ling J., Anderson D. G., Ploegh H. L., Sci. Rep. 2017, 7, 252; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Su X. F., Fricke J., Kavanagh D. G., Irvine D. J., Mol. Pharmaceutics 2011, 8, 774; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Eygeris Y., Gupta M., Kim J., Sahay G., Acc. Chem. Res. 2022, 55, 2. [DOI] [PubMed] [Google Scholar]
- 97.a) Miteva M., Kirkbride K. C., Kilchrist K. V., Werfel T. A., Li H., Nelson C. E., Gupta M. K., Giorgio T. D., Duvall C. L., Biomaterials 2015, 38, 97; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Takeda K. M., Osada K., Tockary T. A., Dirisala A., Chen Q., Kataoka K., Biomacromolecules 2017, 18, 36; [DOI] [PubMed] [Google Scholar]; c) Tockary T. A., Osada K., Chen Q. X., Machitani K., Dirisala A., Uchida S., Nomoto T., Toh K., Matsumoto Y., Itaka K., Nitta K., Nagayama K., Kataoka K., Macromolecules 2013, 46, 6585. [Google Scholar]
- 98.a) Kenausis G. L., Vörös J., Elbert D. L., Huang N., Hofer R., Ruiz‐Taylor L., Textor M., Hubbell J. A., Spencer N. D., J. Phys. Chem. B 2000, 104, 3298; [Google Scholar]; b) Harada‐Shiba M., Yamauchi K., Harada A., Takamisawa I., Shimokado K., Kataoka K., Gene Ther. 2002, 9, 407. [DOI] [PubMed] [Google Scholar]
- 99. Murthy N., Campbell J., Fausto N., Hoffman A. S., Stayton P. S., Bioconjugate Chem. 2003, 14, 412. [DOI] [PubMed] [Google Scholar]
- 100. Baba M., Itaka K., Kondo K., Yamasoba T., Kataoka K., J. Controlled Release 2015, 201, 41. [DOI] [PubMed] [Google Scholar]
- 101. Matsui A., Uchida S., Ishii T., Itaka K., Kataoka K., Sci. Rep. 2015, 5, 15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Aini H., Itaka K., Fujisawa A., Uchida H., Uchida S., Fukushima S., Kataoka K., Saito T., Chung U. I., Ohba S., Sci. Rep. 2016, 6, 18743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Abbasi S., Uchida S., Toh K., Tockary T. A., Dirisala A., Hayashi K., Fukushima S., Kataoka K., J. Controlled Release 2021, 332, 260. [DOI] [PubMed] [Google Scholar]
- 104. Crowley S. T., Fukushima Y., Uchida S., Kataoka K., Itaka K., Mol. Ther.–Nucleic Acids 2019, 17, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Uzgun S., Nica G., Pfeifer C., Bosinco M., Michaelis K., Lutz J. F., Schneider M., Rosenecker J., Rudolph C., Pharm. Res. 2011, 28, 2223. [DOI] [PubMed] [Google Scholar]
- 106. Cheng C., Convertine A. J., Stayton P. S., Bryers J. D., Biomaterials 2012, 33, 6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Nuhn L., Kaps L., Diken M., Schuppan D., Zentel R., Macromol. Rapid Commun. 2016, 37, 924. [DOI] [PubMed] [Google Scholar]
- 108. Kartha G., Bello J., Harker D., Nature 1967, 213, 862. [DOI] [PubMed] [Google Scholar]
- 109. Burke R. S., Pun S. H., Bioconjugate Chem. 2008, 19, 693. [DOI] [PubMed] [Google Scholar]
- 110. Uchida S., Kinoh H., Ishii T., Matsui A., Tockary T. A., Takeda K. M., Uchida H., Osada K., Itaka K., Kataoka K., Biomaterials 2016, 82, 221. [DOI] [PubMed] [Google Scholar]
- 111. Yoshinaga N., Cho E., Koji K., Mochida Y., Naito M., Osada K., Kataoka K., Cabral H., Uchida S., Angew. Chem., Int. Ed. Engl. 2019, 58, 11360. [DOI] [PubMed] [Google Scholar]
- 112.a) Yoshinaga N., Uchida S., Naito M., Osada K., Cabral H., Kataoka K., Biomaterials 2019, 197, 255; [DOI] [PubMed] [Google Scholar]; b) Koji K., Yoshinaga N., Mochida Y., Hong T., Miyazaki T., Kataoka K., Osada K., Cabral H., Uchida S., Biomaterials 2020, 261, 120332; [DOI] [PubMed] [Google Scholar]; c) Yoshinaga N., Uchida S., Dirisala A., Naito M., Osada K., Cabral H., Kataoka K., J. Controlled Release 2021, 330, 317; [DOI] [PubMed] [Google Scholar]; d) Yoshinaga N., Uchida S., Dirisala A., Naito M., Koji K., Osada K., Cabral H., Kataoka K., Adv. Healthcare Mater. 2022, 11, 2102016. [DOI] [PubMed] [Google Scholar]
- 113. Hayashi K., Chaya H., Fukushima S., Watanabe S., Takemoto H., Osada K., Nishiyama N., Miyata K., Kataoka K., Macromol. Rapid Commun. 2016, 37, 486. [DOI] [PubMed] [Google Scholar]
- 114. Yang W., Miyazaki T., Chen P., Hong T., Naito M., Miyahara Y., Matsumoto A., Kataoka K., Miyata K., Cabral H., Sci. Technol. Adv. Mater. 2021, 22, 850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Miyazaki T., Uchida S., Nagatoishi S., Koji K., Hong T., Fukushima S., Tsumoto K., Ishihara K., Kataoka K., Cabral H., Adv. Healthcare Mater. 2020, 9, 2000538. [DOI] [PubMed] [Google Scholar]
- 116. Miyazaki T., Uchida S., Hatano H., Miyahara Y., Matsumoto A., Cabral H., Eur. Polym. J. 2020, 140, 110028. [Google Scholar]
- 117. Yang W., Miyazaki T., Nakagawa Y., Boonstra E., Masuda K., Nakashima Y., Chen P., Mixich L., Barthelmes K., Matsumoto A., Mi P., Uchida S., Cabral H., Sci. Technol. Adv. Mater. 2023, just‐accepted, 2170164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.a) Mura S., Nicolas J., Couvreur P., Nat. Mater. 2013, 12, 991; [DOI] [PubMed] [Google Scholar]; b) Manickam D. S., Li J., Putt D. A., Zhou Q. H., Wu C., Lash L. H., Oupicky D., J. Controlled Release 2010, 141, 77; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng F., Hennink W. E., Zhong Z., Biomaterials 2009, 30, 2180. [DOI] [PubMed] [Google Scholar]
- 119. Chen Q., Qi R., Chen X., Yang X., Wu S., Xiao H., Dong W., Mol. Ther. 2017, 25, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dirisala A., Uchida S., Tockary T. A., Yoshinaga N., Li J., Osawa S., Gorantla L., Fukushima S., Osada K., Kataoka K., J. Drug Targeting 2019, 27, 670. [DOI] [PubMed] [Google Scholar]
- 121. Chen G., Ma B., Wang Y., Gong S., ACS Appl. Mater. Interfaces 2018, 10, 18515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.a) Traut T. W., Mol. Cell. Biochem. 1994, 140, 1; [DOI] [PubMed] [Google Scholar]; b) Gorman M. W., Feigl E. O., Buffington C. W., Clin. Chem. 2007, 53, 318. [DOI] [PubMed] [Google Scholar]
- 123. Yang W., Chen P., Boonstra E., Hong T., Cabral H., Pharmaceutics 2022, 14, 2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.a) Houseley J., Tollervey D., Cell 2009, 136, 763; [DOI] [PubMed] [Google Scholar]; b) Tsui N. B., Ng E. K., Lo Y. M., Clin. Chem. 2002, 48, 1647; [PubMed] [Google Scholar]; c) Mi P., Cabral H., Kataoka K., Adv. Mater. 2020, 32, 1902604. [DOI] [PubMed] [Google Scholar]
- 125.a) Deshayes S., Cabral H., Ishii T., Miura Y., Kobayashi S., Yamashita T., Matsumoto A., Miyahara Y., Nishiyama N., Kataoka K., J. Am. Chem. Soc. 2013, 135, 15501; [DOI] [PubMed] [Google Scholar]; b) Oyewumi M. O., Yokel R. A., Jay M., Coakley T., Mumper R. J., J. Controlled Release 2004, 95, 613. [DOI] [PubMed] [Google Scholar]
- 126.a) Xiong X. B., Lavasanifar A., ACS Nano 2011, 5, 5202; [DOI] [PubMed] [Google Scholar]; b) Xue B., Kozlovskaya V., Sherwani M. A., Ratnayaka S., Habib S., Anderson T., Manuvakhova M., Klampfer L., Yusuf N., Kharlampieva E., Biomacromolecules 2018, 19, 4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Suzuki K., Miura Y., Mochida Y., Miyazaki T., Toh K., Anraku Y., Melo V., Liu X., Ishii T., Nagano O., Saya H., Cabral H., Kataoka K., J. Controlled Release 2019, 301, 28. [DOI] [PubMed] [Google Scholar]
- 128. Anraku Y., Kuwahara H., Fukusato Y., Mizoguchi A., Ishii T., Nitta K., Matsumoto Y., Toh K., Miyata K., Uchida S., Nishina K., Osada K., Itaka K., Nishiyama N., Mizusawa H., Yamasoba T., Yokota T., Kataoka K., Nat. Commun. 2017, 8, 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.a) Allen T. M., Nat. Rev. Cancer 2002, 2, 750; [DOI] [PubMed] [Google Scholar]; b) Kanapathipillai M., Brock A., Ingber D. E., Adv. Drug Delivery Rev. 2014, 79‐80, 107. [DOI] [PubMed] [Google Scholar]
- 130.a) Gu F., Zhang L., Teply B. A., Mann N., Wang A., Radovic‐Moreno A. F., Langer R., Farokhzad O. C., Proc. Natl. Acad. Sci. USA 2008, 105, 2586; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Oe Y., Christie R. J., Naito M., Low S. A., Fukushima S., Toh K., Miura Y., Matsumoto Y., Nishiyama N., Miyata K., Kataoka K., Biomaterials 2014, 35, 7887. [DOI] [PubMed] [Google Scholar]
- 131.a) Matsumura Y., Maeda H., Cancer Res. 1986, 46, 6387; [PubMed] [Google Scholar]; b) Maeda H., Wu J., Sawa T., Matsumura Y., Hori K., J. Controlled Release 2000, 65, 271. [DOI] [PubMed] [Google Scholar]
- 132.a) Lammers T., J. Controlled Release 2019, 294, 372; [DOI] [PubMed] [Google Scholar]; b) Shi Y., van der Meel R., Chen X., Lammers T., Theranostics 2020, 10, 7921; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ashford M. B., England R. M., Akhtar N., Adv. Ther. 2021, 4, 2000285. [Google Scholar]
- 133. Prabhakar U., Maeda H., Jain R. K., Sevick‐Muraca E. M., Zamboni W., Farokhzad O. C., Barry S. T., Gabizon A., Grodzinski P., Blakey D. C., Cancer Res. 2013, 73, 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wolf H., Muller Y., Salmen S., Wilmanns W., Jung G., Eur. J. Immunol. 1994, 24, 1410. [DOI] [PubMed] [Google Scholar]
- 135. Omstead D. T., Mejia F., Sjoerdsma J., Kim B., Shin J., Khan S., Wu J., Kiziltepe T., Littlepage L. E., Bilgicer B., J. Hematol. Oncol. 2020, 13, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.a) Miura Y., Takenaka T., Toh K., Wu S., Nishihara H., Kano M. R., Ino Y., Nomoto T., Matsumoto Y., Koyama H., Cabral H., Nishiyama N., Kataoka K., ACS Nano 2013, 7, 8583; [DOI] [PubMed] [Google Scholar]; b) Min H. S., Kim H. J., Ahn J., Naito M., Hayashi K., Toh K., Kim B. S., Matsumura Y., Kwon I. C., Miyata K., Kataoka K., Biomacromolecules 2018, 19, 2320; [DOI] [PubMed] [Google Scholar]; c) Kawamura W., Miura Y., Kokuryo D., Toh K., Yamada N., Nomoto T., Matsumoto Y., Sueyoshi D., Liu X., Aoki I., Kano M. R., Nishiyama N., Saga T., Kishimura A., Kataoka K., Sci. Technol. Adv. Mater. 2015, 16, 035004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Salvati A., Pitek A. S., Monopoli M. P., Prapainop K., Bombelli F. B., Hristov D. R., Kelly P. M., Åberg C., Mahon E., Dawson K. A., Nat. Nanotechnol. 2013, 8, 137. [DOI] [PubMed] [Google Scholar]
- 138. Farshbaf M., Valizadeh H., Panahi Y., Fatahi Y., Chen M., Zarebkohan A., Gao H., Int. J. Pharm. 2022, 614, 121458. [DOI] [PubMed] [Google Scholar]
- 139.a) Rybakova Y., Kowalski P. S., Huang Y., Gonzalez J. T., Heartlein M. W., DeRosa F., Delcassian D., Anderson D. G., Mol. Ther. 2019, 27, 1415; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hoppenz P., Els‐Heindl S., Beck‐Sickinger A. G., Front. Chem. 2020, 8, 571; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Asrorov A. M., Gu Z., Min K. A., Shin M. C., Huang Y., ACS Pharmacol. Transl. Sci. 2020, 3, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Fonsatti E., Altomonte M., Nicotra M. R., Natali P. G., Maio M., Oncogene 2003, 22, 6557. [DOI] [PubMed] [Google Scholar]
- 141. Liu Z., Wang F., Chen X., Drug Dev. Res. 2008, 69, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zwicke G. L., Mansoori G. A., Jeffery C. J., Nano Rev. 2012, 3, 18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.a) Medina R. A., Owen G. I., Biol. Res. 2002, 35, 9; [DOI] [PubMed] [Google Scholar]; b) Gillies R. J., Robey I., Gatenby R. A., J. Nucl. Med. 2008, 49, 24S. [DOI] [PubMed] [Google Scholar]
- 144.a) Vellon L., Menendez J. A., Liu H., Lupu R., Differentiation 2007, 75, 819; [DOI] [PubMed] [Google Scholar]; b) Takayama S., Ishii S., Ikeda T., Masamura S., Doi M., Kitajima M., Anticancer Res. 2005, 25, 79. [PubMed] [Google Scholar]
- 145. Yi Y., Kim H. J., Zheng M., Mi P., Naito M., Kim B. S., Min H. S., Hayashi K., Perche F., Toh K., Liu X., Mochida Y., Kinoh H., Cabral H., Miyata K., Kataoka K., J. Controlled Release 2019, 295, 268. [DOI] [PubMed] [Google Scholar]
- 146. Hu G., Guo M., Xu J., Wu F., Fan J., Huang Q., Yang G., Lv Z., Wang X., Jin Y., Front. Immunol. 2019, 10, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.a) Caux C., Ramos R. N., Prendergast G. C., Bendriss‐Vermare N., Ménétrier‐Caux C., Cancer Res. 2016, 76, 6439; [DOI] [PubMed] [Google Scholar]; b) Chen Y., Zhang S., Wang Q., Zhang X., J. Hematol. Oncol. 2017, 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhang F., Parayath N. N., Ene C. I., Stephan S. B., Koehne A. L., Coon M. E., Holland E. C., Stephan M. T., Nat. Commun. 2019, 10, 3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.a) Sabado R. L., Balan S., Bhardwaj N., Cell Res. 2017, 27, 74; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang W., Zhu G., Wang S., Yu G., Yang Z., Lin L., Zhou Z., Liu Y., Dai Y., Zhang F., Shen Z., Liu Y., He Z., Lau J., Niu G., Kiesewetter D. O., Hu S., Chen X., ACS Nano 2019, 13, 3083. [DOI] [PubMed] [Google Scholar]
- 150.a) Tacken P. J., de Vries I. J., Torensma R., Figdor C. G., Nat. Rev. Immunol. 2007, 7, 790; [DOI] [PubMed] [Google Scholar]; b) Wang F., Ullah A., Fan X., Xu Z., Zong R., Wang X., Chen G., J. Controlled Release 2021, 333, 107. [DOI] [PubMed] [Google Scholar]
- 151. Loike J. D., Sodeik B., Cao L., Leucona S., Weitz J. I., Detmers P. A., Wright S. D., Silverstein S. C., Proc. Natl. Acad. Sci. USA 1991, 88, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Vorup‐Jensen T., Chi L., Gjelstrup L. C., Jensen U. B., Jewett C. A., Xie C., Shimaoka M., Linhardt R. J., Springer T. A., J. Biol. Chem. 2007, 282, 30869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Castro F. V. V., Tutt A. L., White A. L., Teeling J. L., James S., French R. R., Glennie M. J., Eur. J. Immunol. 2008, 38, 2263. [DOI] [PubMed] [Google Scholar]
- 154. Pei M., Xu R., Zhang C., Wang X., Li C., Hu Y., Colloids Surf., B 2021, 197, 111378. [DOI] [PubMed] [Google Scholar]
- 155. Moignic A. L.e, Malard V., Benvegnu T., Lemiègre L., Berchel M., Jaffrès P. A., Baillou C., Delost M., Macedo R., Rochefort J., Lescaille G., Pichon C., Lemoine F. M., Midoux P., Mateo V., J. Controlled Release 2018, 278, 110. [DOI] [PubMed] [Google Scholar]
- 156. Moffett H. F., Coon M. E., Radtke S., Stephan S. B., McKnight L., Lambert A., Stoddard B. L., Kiem H. P., Stephan M. T., Nat. Commun. 2017, 8, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Weide B., Carralot J. P., Reese A., Scheel B., Eigentler T. K., Hoerr I., Rammensee H. G., Garbe C., Pascolo S., J. Immunother. 2008, 31, 180. [DOI] [PubMed] [Google Scholar]
- 158. Chen P., Yang W., Hong T., Miyazaki T., Dirisala A., Kataoka K., Cabral H., Biomaterials 2022, 288, 121748. [DOI] [PubMed] [Google Scholar]
- 159. Zhu D., Yan H., Zhou Z., Tang J., Liu X., Hartmann R., Parak W. J., Shen Y., Feliu N., Adv. Healthcare Mater. 2021, 10, 2100125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Nguyen V. H., Lee B. J., Int. J. Nanomed. 2017, 12, 3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Dilliard S. A., Cheng Q., Siegwart D. J., Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Alberg I., Kramer S., Schinnerer M., Hu Q., Seidl C., Leps C., Drude N., Mockel D., Rijcken C., Lammers T., Diken M., Maskos M., Morsbach S., Landfester K., Tenzer S., Barz M., Zentel R., Small 2020, 16, e1907574. [DOI] [PubMed] [Google Scholar]
- 163.a) Teleanu D. M., Negut I., Grumezescu V., Grumezescu A. M., Teleanu R. I., Nanomaterials 2019, 9, 371>; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Uchida Y., Ito K., Ohtsuki S., Kubo Y., Suzuki T., Terasaki T., J. Neurochem. 2015, 134, 97; [DOI] [PubMed] [Google Scholar]; c) Englert C., Trutzschler A. K., Raasch M., Bus T., Borchers P., Mosig A. S., Traeger A., Schubert U. S., J. Controlled Release 2016, 241, 1; [DOI] [PubMed] [Google Scholar]; d) Kreuter J., Adv. Drug Delivery Rev. 2014, 71, 2. [DOI] [PubMed] [Google Scholar]