

Abstract
Atherosclerotic arteries are commonly treated using drug‐eluting stents (DES). However, it remains unclear whether and how the properties of atherosclerotic plaque affect drug transport in the arterial wall. A limitation of the currently used atherosclerotic animal models to study arterial drug distribution is the unpredictability of plaque size, composition, and location. In the present study, the aim is to create an artificial atherosclerotic plaque—of reproducible and controllable complexity and implantable at specific locations—to enable systematic studies on transport phenomena of drugs in stented atherosclerosis‐mimicking arteries. For this purpose, mixtures of relevant lipids at concentrations mimicking atherosclerotic plaque are incorporated in gelatin/alginate hydrogels. Lipid‐free (control) and lipid‐rich hydrogels (artificial plaque) are created, mounted on DES and successfully implanted in porcine coronary arteries ex‐vivo. Matrix‐assisted laser desorption ionization mass spectrometry imaging (MALDI‐MSI) is used to measure local drug distribution in the arterial wall behind the prepared hydrogels, showing that the lipid‐rich hydrogel significantly hampers drug transport as compared to the lipid‐free hydrogel. This observation confirms the importance of studying drug transport phenomena in the presence of lipids and of having an experimental model in which lipids and other plaque constituents can be precisely controlled and systematically studied.
Keywords: arterial drug transport, atherosclerosis, drug‐eluting stents, hydrogels, MALDI imaging
The aim of this work is to create an implantable artificial atherosclerotic plaque of reproducible and controllable composition. Mixtures of relevant lipids are incorporated in gelatin/alginate hydrogels, which are mounted on DES and implanted in swine coronary arteries ex‐vivo. Drug concentrations in the arterial wall behind the plaque show that the lipid‐rich hydrogel hampers drug transport as compared to the lipid‐free hydrogel.
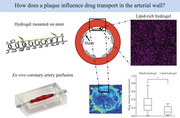
1. Introduction
Heart disease is the main cause of death worldwide, with coronary artery disease (CAD) accounting for nearly half of the victims.[ 1 ] The dominant cause of CAD is atherosclerosis, i.e., the narrowing of coronary arteries due to the build‐up of fatty deposits in the arterial wall, known as plaque. Atherosclerotic plaque consists primarily of cholesterol, fatty acids, cellular waste products, dead cells, calcifications, and fibrin.[ 2 ] Progression, destabilization, and rupture of plaque may ultimately cause acute coronary events and sudden death.[ 3 , 4 ] The primary choice for acute treatment of symptomatic atherosclerosis is the implantation of a stent, with drug‐eluting stents (DES) generally being favored over bare metal stents (BMS) to reduce the formation of excessive scar tissue in the healing process following implantation.[ 5 ]
Despite successful clinical results, the efficacy of DES is limited in some groups of patients and plaque types.[ 6 , 7 , 8 ] Furthermore, delayed healing, local hypersensitivity reactions and neoatherosclerosis remain a concern.[ 9 , 10 ] The efficacy of DES depends on drug uptake by, and drug distribution within, the arterial wall. However, precise mechanisms of drug release from DES, and drug transport in the atherosclerotic arterial wall remain unclear. Such mechanisms are dependent on many factors including the type of stent, the physiochemical properties of the drug, and the composition of the atherosclerotic plaque.[ 11 ]
Preclinical research on coronary DES is focused on vascular healing and the majority of these studies are based on porcine coronary arteries due to their close resemblance to human arteries.[ 12 ] While most of these studies are performed in healthy young animals, the field is increasingly aware that drug transport and vascular healing is different in animals with atherosclerosis.[ 13 , 14 ] While atherosclerotic plaque was found to resemble human‐like lesions[ 15 ] in some atherosclerotic animal models and the vascular response to DES was similar to that in patients in terms of endothelialization,[ 16 , 17 ] a major drawback of such disease animal models is that atherosclerotic plaque development, composition, and location in the arterial tree are highly variable. Additionally, study costs are considerable due to the time associated with natural plaque development. These issues limit the utility of atherosclerotic animal models to systematically study how plaque properties affect arterial drug distribution and vascular healing following DES placement. The lack of low‐cost, well‐controlled, and readily available experimental models for atherosclerotic plaque with a predictable and reproducible composition and location hampers a systematic and cost‐effective study of treatment options for specific types of atherosclerotic plaque.
The potential and relevance of such experimental models could significantly impact vascular research.[ 18 , 19 , 20 ] For an excellent overview of advances in DESanalysis, design, and performance, we refer to the special issue published in the Annals of Biomedical Engineering.[ 21 ] In this issue, McKittrick et al. reviewed in‐vitro, ex‐vivo, in‐vivo and mathematical models to incorporate the impact of atherosclerosis on drug uptake and distribution in the arterial wall, pointing out the different pros and cons of each model and their relevance to human atherosclerotic plaques. McKittrick also underlines the lack of knowledge regarding the influence of atherosclerotic plaque on drug uptake and distribution in the arterial wall.[ 22 ]
To date, only a few studies have investigated drug distribution in an artificial in‐vitro model for atherosclerotic plaques. Several studies have focused on flow‐through cell models, which are based on a hydrogel with a tubular structure, into which a stent can be implanted in order to study the release of drugs from DES and the distribution in the hydrogel.[ 23 , 24 , 25 ] While Seidlitz et al. studied the transport in a homogeneous hydrogel,[ 24 ] other studies incorporated hydrophobic (lipid‐rich) domains to mimic differential tissue composition seen in atherosclerotic arteries.[ 23 , 25 , 26 ] These studies illustrate that hydrophobic/lipophilic drugs like triamterene and fluvastatin partition over the hydrophilic and hydrophobic regions in the hydrogel, with partition coefficients in the hydrophobic regions several‐fold higher than in the hydrophilic regions, and increasing with increasing lipid concentration.[ 25 , 26 ] On top of affinity to lipids, Tzafriri et al. showed that binding of drugs to intra‐ and extracellular proteins plays an important role in the distribution of drugs in human excised vessels containing atherosclerotic plaque, underscoring the potential of artificial atherosclerotic artery models for DES testing.[ 27 ] While the artificial plaque models described above aimed to mimic selected in‐vivo aspects that influence drug distribution in a standardized and reproducible manner, it was not the explicit intention of any of these studies to simulate atherosclerotic arterial tissue. Furthermore, these plaque models were not designed for ex‐vivo and in‐vivo implantation, hence drug transport studies in animal models were not possible.
In light of these considerations, we hypothesized that a novel artificial atherosclerotic plaque that can be co‐implanted with DES to obtain a stented atherosclerosis‐mimicking artery can serve as a way to investigate—in the preclinical setting—the influence of plaque composition on drug transport in the arterial wall. This approach opens the door to pharmacokinetic and pharmacodynamic studies of coronary DES on precisely engineered atherosclerotic arteries. Consequently, the aim of the present study was two‐fold. First, we created an implantable artificial atherosclerotic plaque, allowing for a designable plaque composition. For this purpose, we developed gelatin/alginate hydrogels that carry the most common plaque lipids in concentrations reported for atherosclerotic plaques. Second, we mounted the artificial plaques on a DES, implanted it in a porcine coronary artery ex‐vivo, and studied drug distribution into the arterial wall for lipid‐free and lipid‐rich artificial plaques, using MALDI‐MSI, to provide a proof‐of‐principle of the potential use of the herein introduced approach for drug transport studies.
2. Results and Discussion
2.1. Artificial Plaque Development
The method we developed to fabricate artificial atherosclerotic plaque by incorporation of lipids in a gelatin/alginate hydrogel is illustrated in Figure 1 . Lipid type and composition were selected to create a type IV artificial atherosclerotic plaque, which is defined as a lesion presenting a lipid‐rich core with distinct solid deposits.[ 28 ] We determined the composition by combining data from literature,[ 29 , 30 ] see Table 1 . The table describes the only available data in literature concerning the mass contribution of the different lipid components, which are for carotid and femoral atherosclerotic plaques. As our main goal is to understand the influence of atherosclerosis on drug distribution in the diseased arterial wall, we used this data for our work on coronary atherosclerosis.
Figure 1.
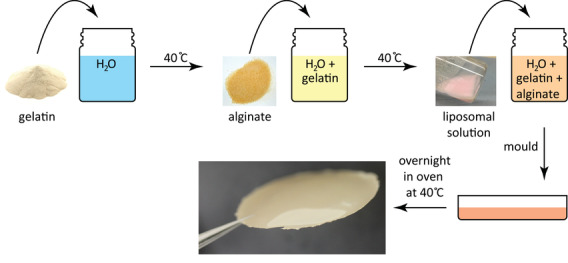
Artificial plaque is created by making a gelatin/alginate/liposome solution that is molded and crosslinked by CaCl2. The photograph shows the final appearance of the artificial plaque.
Table 1.
Atherosclerotic plaque lipid composition as based on endarterectomy samples (excluding healthy arterial wall) taken from patients’ carotid and femoral artery plaques and the percentage of the most abundant cholesterol esters. Dry weight is calculated from mg/g wet weight with the assumption that ≈79% of tissue is water. Each cholesterol ester composition is given as the percentage of the total cholesterol ester amount. This composition is used for incorporation into the artificial plaque
| Lipids | Wet weight | Dry weight | ||
|---|---|---|---|---|
| [mg g−1] | [wt%] | [mg g−1] | [wt%] | |
| Cholesterol[ 29 ] | 13.1 | 1.30 | 62.4 | 6.24 |
| Cholesterol ester[ 29 ] | 9.50 | 0.95 | 45.2 | 4.52 |
| Cholesterol palmitate (C16:0)[ 30 ] | 16.7% | 0.16 | 16.7% | 0.75 |
| Cholesterol oleate (C18:1)[ 30 ] | 33.3% | 0.32 | 33.3% | 1.51 |
| Cholesterol linoleate (C18:2)[ 30 ] | 50.0% | 0.48 | 50.0% | 2.26 |
| Phospholipid[ 29 ] | 9.70 | 0.97 | 46.2 | 4.62 |
The resulting hydrogel appearance is shown in the photograph in Figure 1. Hydrogels made from biopolymers such as gelatin and alginate and combinations thereof, have proven their use as implantable (cardiac) tissue.[ 31 ] Furthermore, sodium alginate has proven to be a great addition to gelatin providing good stability and strength to the hydrogel network.[ 32 ] Thus, based on previous work, we hypothesized that gelatin/alginate gels form a good basis for making an artificial lipid‐rich plaque. Two important criteria in this study were the ability to stably incorporate physiologically relevant lipid levels in the hydrogels and the ability to handle the gels such that they could be implanted. Therefore, the stability of films in the HEPES buffer mix at 37 °C and the mechanical resistance towards manual manipulation were also taken into consideration. Incubation in the HEPES buffer mix did not show any sign of degradation after 5 h, and the hydrogels could be easily handled, pulled, and stretched with tweezers. The fabricated hydrogels hence met the above‐mentioned requirements and they were considered suitable for implantation.
2.2. Lipid Dispersion and Retention within the Artificial Plaque
Lipid dispersion within the artificial plaque was assessed using confocal microscopy. DOPE‐Rhodamine B was added to the lipid solution during liposome preparation. Similar to the distribution of lipids in atherosclerotic plaque (Figure 2A–C), lipids in the artificial plaque were homogeneously dispersed (Figure 2D–E). In addition, visual inspection of the confocal micrographs indicated that the dispersion of lipids in the hydrogel remained unchanged over a period of 17 days (Figure 2F). Following the 17 days incubation, the hydrogels did not show any signs of visual damage. Their ability to be manually handled with tweezers and to be slightly stretched was comparable to the hydrogels that did not undergo water incubation. While the current study pursued the creation of a type IV atherosclerotic plaque according to the modified AHA classification,[ 28 ] our approach of an implantable artificial plaque also allows the incorporation of more complex materials such as cholesterol crystals, calcifications, and necrotic core, typically observed in advanced atherosclerosis. This allows the creation of plaques of specific, well documented, and reproducible complexity to study different strategies geared towards plaque specific pharmacotherapy, with plaque regression as an interesting area of application. We illustrate the ability to create complex plaques resembling coronary arteries with advanced plaque components (Figure 3A) with the example of an artificial plaque with crystalline material in the core (Figure 3B). However, we will focus on the type IV lesions as a model system for the work in the remainder of the paper.
Figure 2.
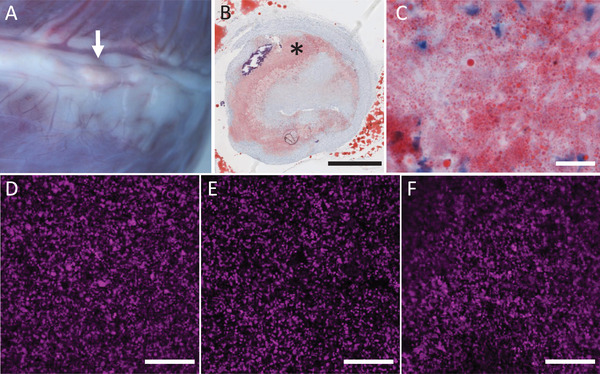
Examples of macroscopy A) and microscopy (B,C; Oil‐Red‐O staining) of coronary atherosclerotic plaque, and of labeled liposome (DOPE‐Rhodamine B) distribution in the hydrogels (D–E). The presence of plaque is shown from the epicardial view (A, arrow). Histology of that plaque in B (detail in C) shows lipid droplets as a light red area (*). Confocal micrographs show the distribution of labelled liposomes throughout artificial plaque before crosslinking D), after crosslinking E), and after 17 days of incubation in water F) and shows no visible difference in distribution. Scale bars B: 1 mm; C: 20 µm; D–F: 50 µm.
Figure 3.
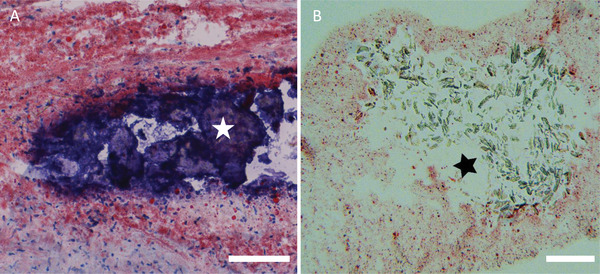
Example of coronary artery calcification A,*) surrounded by a lipid‐rich plaque (Oil‐Red‐O staining). Complex artificial plaques can be fabricated by including components typical of complex plaque such as cholesterol crystals, calcifications and necrotic core. Illustration of a gelatin/alginate hydrogel filled with lipids (B, stained red) and crystalline material in the core of the gel B,*). Scale bars: 100 µm.
2.3. Mounting the Artificial Plaque on DES and Implanting it in a Coronary Artery
Two consecutive thin strips of hydrogel, one lipid‐rich (artificial plaque) and the other lipid‐free (blank) were successfully mounted on an Everolimus‐eluting stent using polypropylene sutures, as illustrated for one of the two strips in the schematic in Figure 4A. Part of the stent is in direct contact with the arterial wall, while another part is separated from the (healthy) arterial wall by the artificial plaque (Figure 4B). The hydrogel‐loaded stent was implanted in a coronary artery mounted inside a perfusion chamber (Figure 4C), with the lipid‐rich hydrogel at the distal part of the artery and the lipid‐free hydrogel at the proximal part. This strategy enabled comparing drug levels in the arterial wall behind the lipid‐rich and lipid‐free hydrogels in the same artery in the same perfusion experiment. Mounting the strips of hydrogel onto the stent and implanting the assembly inside a coronary artery mounted in the perfusion chamber was feasible as judged by the two strips of hydrogel remaining firmly attached to the stent after passing the guiding catheter, placement in the coronary artery and following inflation and deflation of the stent delivery balloon.
Figure 4.
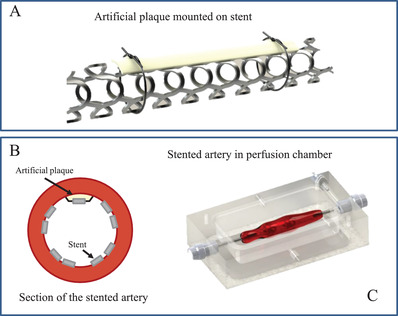
A) Ex‐vivo study on drug transport from stent to an arterial wall through lipid‐free and lipid‐rich hydrogels. Artist impression of hydrogel mounted on a stent using sutures. B) Cross‐section of a stented artery, showing that part of the stent is in direct contact with the arterial wall and another part through the hydrogel. C) Artist impression of a perfusion chamber mounted with a stented porcine coronary artery.
2.4. Drug Distribution in Stented Porcine Coronary Arteries
The left anterior descending (LAD) coronary arteries (n = 4) were dissected from slaughterhouse porcine hearts. The LAD was mounted in a perfusion chamber (Figure 4C) to be perfused with the HEPES buffer mix. The plaque‐mounted stent‐balloon system, containing both a lipid‐free (control) and a lipid‐rich hydrogel (artificial plaque), was inserted into the dissected coronary artery and inflated. The arteries were perfused between 30 min and 5 h. The effect of lipids on drug transport was investigated by quantitatively analyzing cross‐sections of the artery using MALDI MS imaging. Examples of MALDI‐MS images of arterial wall regions with lipid‐free and lipid‐rich hydrogels for 30‐ and 5 h perfusions are shown in Figure 5A, with the rectangles indicating where the hydrogels were mounted during the perfusions. A comparison of normalized Everolimus intensities behind the lipid‐rich and lipid‐free hydrogels (data combined for perfusions between 30 min and 5 h) is illustrated in Figure 5B. Everolimus intensity in the artery wall behind the blank hydrogel was 0.26 [0.01–0.75] (n = 80) while behind the lipid‐rich hydrogel it was 0.09 [0.01–0.31] (n = 80). Data show that Everolimus in the arterial wall behind the artificial plaque was significantly lower than behind the lipid‐free hydrogel (p < 0.05), showing that the lipids in the artificial atherosclerotic plaque present a significant barrier for drug transport from the DES into the arterial wall.
Figure 5.
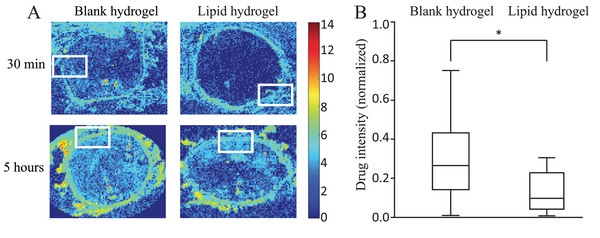
MALDI‐MS images show the Everolimus distribution in the arterial wall behind the lipid‐free and lipid‐rich hydrogels after 30 min and 5 h of perfusion. The rectangles indicate the location of the hydrogels as obtained from images with HE staining (not shown) (A). The Rainbow scale shows an Ln scale of the total ion count for Everolimus. Analysis shows a difference between normalized Everolimus intensities in arterial wall regions behind the lipid‐free and lipid‐rich hydrogel. Intensities in the arterial wall behind the lipid‐rich and lipid‐free hydrogel were normalized against the maximum intensity in the corresponding MALDI images, which was found in the tunica media (excluding stent struts) not covered by the hydrogel. The variations in normalized intensity behind the blank hydrogel run between 0.01 and 0.75. The absolute intensities behind the lipid‐rich hydrogel are lower, so are the variations, which run from 0.01 to 0.31. Data are given as a boxplot illustrating the median and interquartile range, with the ends of the whiskers indicating the minimum and maximum observed values. Data are combined for all perfusions between 30 min and 5 h (n = 80 ROIs for blank and n = 80 ROIs for lipid‐rich) (B). * = p < 0.05
Our finding that the drug concentration in the arterial wall behind the lipid‐rich hydrogel is lower than behind the lipid‐free hydrogel implies that everolimus binds to the lipids and thereby diffuses slower through the lipid‐rich hydrogel, in line with the findings by Guo et al.[ 26 ] While important for the artificial plaque demonstrated in this work, we note that there may be other factors even more important for drug transport than the binding of the drug to lipid‐rich environments as demonstrated by Tzafriri et al.[ 27 ] They measured the partition coefficient of paclitaxel, Everolimus, and sirolimus in atherosclerotic plaques of human aortas and found the partition coefficients in high cholesterol regions to be lower than in low cholesterol regions. This points to the importance of drug affinity to extra‐ and intracellular proteins. The artificial plaque presented in this work allows a systematic study of the different roles and mechanisms.
The here presented proof‐of‐concept on the engineering of an implantable artificial plaque with controlled composition and co‐implantation with a DES to obtain stented atherosclerosis‐mimicking arteries is an encouraging step towards systematic ex‐vivo drug transport studies. Before concluding, we sketch several exciting avenues for further research. A first avenue is to gain mechanistic insights on drug transport by performing longitudinal studies. Such insights are expected to be instrumental in the further development and validation of predictive mathematical models, that will increase our understanding of the behavior of drug and drug release in the context of complex plaques.[ 33 , 34 , 35 , 36 ] While the chosen time window in the present study was relatively short in comparison to the release of drugs from the stent, this work already showed the influence of lipids in the first hours after implantation in which the burst release takes place. Current efforts focus on addressing the challenges in keeping arteries viable in ex‐vivo cultures by employing a cultured artery model,[ 37 ] enabling studies over several weeks. This opens the opportunity to study dynamical effects, for example, the importance of drug binding/unbinding to plaque constituents, which makes the plaque act as a reservoir.[ 33 ] A second avenue is to unravel the role of each of the constituents of atherosclerotic plaques on drug transport, which has remained difficult to study and hence is still poorly understood.[ 18 , 25 , 26 , 38 ] Kakizaki and Nomoto investigated the relation between plaque composition and vascular healing responses after DES implantation and reported that human coronary arteries showed thicker neointima growth in the presence of lipid‐rich plaques, as compared to stent struts laying on more fibrous plaques.[ 39 , 40 ] Additionally, there is evidence of limited efficacy of DES in different plaques (e.g., complex lesions) and in patient groups presenting comorbidities such as diabetes,[ 6 , 7 , 41 ] which might be explained by a differential drug uptake and retention in different tissue types. This clinical evidence underlines the necessity to incorporate plaque components in in‐vitro, ex‐vivo and in‐vivo models to understand the relation between plaque components and DES efficacy. The creation of an artificial plaque allows the controlled addition of each of these constituents, the feasibility of which is shown in Figure 3, and allows systematic study of their effect on the transport of drugs, both in terms of stent‐based and systemic drug delivery. A third avenue is to implant the artificial plaque in‐vivo, both in the presence and in absence of co‐morbidities such as hypertension and diabetes, to unravel co‐morbidity effects. The here proposed approach may therefore lead to a significant refinement of atherosclerotic animal models in cardiovascular research. Additionally, the ability to implant an artificial plaque, and allowing the body to incorporate it,[ 42 ] opens the door to studies on plaque regression and its treatment. We hence also foresee a potential impact of the artificial plaque and the here presented approach beyond preclinical DES research.
3. Conclusion
The present study is the first to fabricate an artificial atherosclerotic plaque with the aim to co‐implant it with DES in coronary arteries to obtain stented atherosclerosis‐mimicking arteries that allow systematic studies on the role of plaque properties on drug transport and DES treatment. In this work, we provided a proof‐of‐concept, demonstrating that lipids in the artificial atherosclerotic plaque present a significant barrier for drug transport from DES. The here presented approach, using (healthy) porcine coronary arteries from the slaughterhouse in ex‐vivo perfusion studies, also provides an important means towards reducing, refining and replacing animal models in cardiovascular research.
4. Experimental Section
Hydrogel Preparation
Gelatin/alginate hydrogels (mass ratio of 1:3) were prepared as follows: gelatin (111 mg, Type B, Sigma‐Aldrich, St. Louis, MO) was added to water (5 ml) at 40 °C at constant stirring. Once dissolved, sodium alginate (333 mg, viscosity: 1% in water at 25 o C is 5.0–40.0 cps, M/G ratio = 1.56, Sigma‐Aldrich, St. Louis, MO, product number: W201502) was added to the solution and stirred until dissolved.
Liposome Solution Preparation
Liposome solutions were prepared as follows: powdered lipids (cholesterol, cholesterol palmitate, cholesterol oleate, cholesterol linoleate (all from Sigma‐Aldrich, St. Louis, MO) and 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphocholine (DPPC) (Tokyo Chemical Industry Co., Tokyo, Japan)) were first dissolved in chloroform (VWR International, The Netherlands) in a round‐bottom glass flask in a 1:10 mass ratio for full lipid dissolution. The flask was placed on a rotary evaporator (Buchi Rotovapor R‐215, Sigma‐Aldrich, St. Louis, MO) until the chloroform had fully evaporated. This resulted in a thin lipid film on the wall of the flask. This film was then soaked in water (MilliQ grade) (also in 1:10 mass ratio) and the flask was left in a sonication bath (Bransonic 2510E‐MTH, Branson Ultrasonics, Brookfield, CT) for 30 min at 35 °C to create a liposome solution. Lipid concentrations used are shown in Table 1. Given the low mass fraction of lipids (around 4%), we do not expect that the incorporation of lipids affects the bulk properties of the hydrogel such as the porosity.
Artificial Atherosclerotic Plaque Preparation
The prepared liposome solutions were subsequently mixed with gelatin/alginate solutions under constant stirring at 40 °C until homogenization. The resulting uniform mixture was poured into plastic Petri dishes (inner diameter of 5 cm) and left overnight at 40 °C to form thin, dry gel films. Films were then soaked in an aqueous CaCl2 (100 mM, Sigma‐Aldrich, St. Louis, MO) solution for 24 h at room temperature for crosslinking. The procedure to prepare the artificial atherosclerotic plaque is shown in Figure 1.
Lipid Dispersion within the Artificial Plaque
Confocal microscopy (Zeiss LSM 710) was used to examine lipid dispersion within hydrogels. For this, 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine B sulfonyl) (DOPE‐Rhodamine B) (supplied in chloroform, Avanti Lipids, Inc., Alabama, USA) was added to the lipid solution in chloroform during liposome preparation (final DOPE‐Rhodamine B concentration in water not exceeding 4 µM for imaging).
Lipid Retention in Artificial Atherosclerotic Plaque
Lipid retention was investigated by incubating hydrogels for 17 days in water at room temperature. Crosslinked hydrogels were left in water (3 ml, MilliQ grade) in tightly sealed Petri dishes. Hydrogels were imaged with confocal microscopy to investigate changes during incubation. To assess stability in water following water incubation for 17 days, the hydrogels were pulled and stretched with tweezers.
Ex‐Vivo Artificial Atherosclerotic Plaque Implantation and Perfusion in Porcine Coronary Arteries
Porcine hearts (n = 4, local slaughterhouse) were transported to Erasmus MC in ice cold KREBS buffer (1.26 M NaCl, 25 mM KCl, 250 mM NaHCO3, 12 mM NaH2PO4, 12 mM MgCl2, 25 mM CaCl2, 25 mM glucose) within two hours post‐excision. The left anterior descending (LAD) coronary artery together with approximately 1 cm of myocardial tissue was dissected from the hearts in KREBS buffer immersed in ice, mounted in a perfusion chamber, with large side branches tied off to maintain flow through the LAD.
The perfusion chamber was placed on a heating plate (37 °C). The chamber was perfused with HEPES buffer mix (325ml 1x HEPES saline buffer, 40 ml phosphate buffer containing 62 mg ml−1 sucrose and 8.2 mg ml−1 mannitol). Prior to perfusion experiments, hydrogels were incubated at 37 °C in HEPES buffer mix up to 5 h in order to evaluate their stability in this medium. After incubation, the hydrogels were manually pulled and stretched with tweezers, and they were sutured around a test catheter to check if it could be firmly secured around the stent. Two thin strips of hydrogel, one with lipids (artificial plaque) and one without (blank), were cut to size and mounted onto Everolimus‐eluting stents (Xience Prime, Abbott Vascular, Illinois, USA) using polypropylene suture (8–0 Prolene Blue 2 × 18" BV130‐5 Double Armed, Ethicon, USA). The plaque‐mounted stent‐balloon system, containing both a lipid‐free hydrogel (proximal) and a lipid‐rich hydrogel (distal), was inserted into the artery and inflated for at least 20 s to ensure full stent expansion. To avoid oversizing and vascular damage while ascertaining stent apposition, balloons were inflated until slight but visible expansion of the vessel from the outside. Inflation pressures (13 to 20 atm.) were dependent on artery and stent diameter (2.5–3.5 mm). After deflation, the catheter was removed. To avoid drug loss prior to expansion, we did not deploy the stent under flow. After removing the catheter, the perfusion chamber was connected to a heart‐lung machine, with a pressure of approximately 80 mmHg and a flow of 100–120 ml min−1. The pump used to generate the flow was a roller pump (Stockert Shiley). We did not measure the flow pattern, but we expect it to be similar to flow in in‐vivo implantation. The perfusion was stopped between 30 min and 5 h following DES implantation. After experiments, arteries were removed from the chamber, flushed with embedding medium (polyvinyl alcohol (PVA)), quickly frozen on dry ice and stored at −80 °C until cryosectioning in preparation for MALDI‐MS imaging for further analysis.
Cryosectioning of Stented Arteries
Arteries (n = 4) were cut into multiple 3 mm blocks for cryosectioning (Microm HM560, Germany). Sections (12 µm in thickness) were mounted on glass slides and stored at −80 °C until further analysis. Blocks containing lipid‐free or lipid‐rich plaque were selected for sectioning.
Matrix Assisted Laser Desorption/Ionization Mass Spectrometry Imaging (MALDI‐MSI)
Matrix was prepared by dissolving 2,5‐dihydroxybenzoic acid (DHB) (5 mg ml−1 , Sigma‐Aldrich, St. Louis, MO) in acetone. 10 ml was pipetted on a sublimation device (designed and manufactured in‐house, Erasmus MC) and heated to 50 °C until acetone was evaporated. Glass slides containing sections of the artery were mounted onto the sample holder and subjected to vacuum. At 5 mbar, the matrix table was heated to 125 °C (10 min) to sublimate DHB. Following sublimation, slides were subjected to methanol vapor to recrystallize the DHB matrix to enhance Everolimus detection, and immediately analyzed by MALDI‐MS imaging (MALDI Synapt G2‐Si; Waters, Wilmslow UK) with a step size of 75 µm and laser energy of 150 (arbitrary units). Polarity was positive with target enhancement at 980 Da for Everolimus.
Drug Distribution Following Ex‐Vivo Artificial Plaque Implantation
Raw data was processed with HD Imaging and Masslynx (version 4.1; Waters, Wilmslow UK) for m/z values of 980.5 or 980.6 for Everolimus. Data were analyzed using the regions of interest (ROI) tool to measure Everolimus intensity in the arterial wall behind the hydrogels. In one MALDI MS image, ROIs were analyzed behind hydrogels with (n = 20) or without lipids (n = 20) for all the conditions (n = 4 perfusions between 30 min and 5 h). Intensities in each image were normalized with the highest intensity in the arterial wall (specifically excluding the stent struts themselves) in that same image, in order to allow comparing normalized intensities obtained in different MALDI‐MS images.
Statistical Analysis
Prior to analysis, outliers from data sets were removed by analyzing MALDI MS images to justify the removal of an outlier (e.g., a piece of stent strut). Everolimus intensities measured in the arterial wall behind the hydrogels were normalized with respect to the highest intensity in the same MALDI‐MS image, without taking into account the stent struts themselves, always occurring in the area of the arterial wall without hydrogel. Results are presented as a median [min, max]. Data sets were tested for normality taking into account the Shapiro‐Wilk test as well as Q–Q plots. Since the data were not normally distributed, a non‐parametric Mann‐Whitney U test was performed to compare arterial wall regions behind lipid‐free (n = 80) and lipid‐rich (n = 80) hydrogels. We considered p values < 0.05 statistically significant. The analysis was performed using SPSS 24 (IBM SPSS statistics).
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
H.M.M.v.B. and V.v.S. contributed equally to this work. This study was supported by Grants ZonMw (91113020) to H.M.M.v.B., ZonMW (114021510) to H.M.M.v.B. and V.v.S., VENI NWO‐STW (13137) to V.v.S., FP7 Marie Curie ETN “Smartnet” (316656) to M.L. and J.v.E. G. Springeling is thanked for his help in designing the perfusion chamber and preparing the artist's illustrations. P.C. Burgers is thanked for his indispensable help with MALDI‐MS imaging.
Razzi F., Lovrak M., Gruzdyte D., Den Y. Hartog, Duncker D. J., van J. H. Esch, van V. Steijn, van H. M. M. Beusekom, An Implantable Artificial Atherosclerotic Plaque as a Novel Approach for Drug Transport Studies on Drug‐Eluting Stents. Adv. Healthcare Mater. 2022, 11, 2101570. 10.1002/adhm.202101570
Contributor Information
Volkert van Steijn, Email: V.vanSteijn@tudelft.nl.
Heleen M. M. van Beusekom, Email: h.vanbeusekom@erasmusmc.nl.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Vähätalo J., Holmström L., Pakanen L., Kaikkonen K., Perkiömäki J., Huikuri H., Junttila J., Am. J. Cardiol. 2021,147, 33. [DOI] [PubMed] [Google Scholar]
- 2. Ross R., N. Engl. J. Med. 1999, 340, 115. [DOI] [PubMed] [Google Scholar]
- 3. Farb A., Tang A. L., Burke A. P., Sessums L., Liang Y., Virmani R., Circulation 1995, 92,1701. [DOI] [PubMed] [Google Scholar]
- 4. Kolodgie F. D., Burke A. P., Farb A., Gold H. K., Yuan J., Narula J., Finn A. V., Virmani R., Curr. Opin. Cardiol. 2001,16, 285. [DOI] [PubMed] [Google Scholar]
- 5. Kassimis G., Banning A. P., Eur. Heart J. 2016, 37, 3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiarito M., Mehran R., Catheter. Cardiovasc. Interventions 2020, 96, 253. [DOI] [PubMed] [Google Scholar]
- 7. Torii S., Jinnouchi H., Sakamoto A., Mori H., Park J., Amoa F. C., Sawan M., Sato Y., Cornelissen A., Kuntz S. H., Kutyna M., Paek K. H., Fernandez R., Braumann R., Mont E. K., Surve D., Romero M. E., Kolodgie F. D., Virmani R., Finn A. V., Eur. Heart J. 2020, 41, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernelli C., Chan J., Chieffo A., Expert Rev. Cardiovasc. Ther. 2014, 12, 95. [DOI] [PubMed] [Google Scholar]
- 9. Torii S., Jinnouchi H., Sakamoto A., Kutyna M., Cornelissen A., Kuntz S., Guo L., Mori H., Harari E., Paek K. H., Fernandez R., Chahal D., Romero M. E., Kolodgie F. D., Gupta A., Virmani R., Finn A. V., Nat. Rev. Cardiol. 2020, 17, 37. [DOI] [PubMed] [Google Scholar]
- 10. Joner M., Finn A. V., Farb A., Mont E. K., Kolodgie F. D., Ladich E., Kutys R., Skorija K., Gold H. K., Virmani R., J. Am. Coll. Cardiol. 2006, 48, 193. [DOI] [PubMed] [Google Scholar]
- 11. Borhani S., Hassanajili S., Tafti S. H. A., Rabbani S., Prog. Biomater. 2018, 7, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwartz R. S., Edelman E. R., Carter A., Chronos N., Rogers C., Robinson K. A., Waksman R., Weinberger J., Wilensky R. L., Jensen D. N., Zuckerman B. D., Virmani R., Circulation 2002, 106, 1867. [DOI] [PubMed] [Google Scholar]
- 13. Iqbal J., Chamberlain J., Francis S. E., Gunn J., Ann. Biomed. Eng. 2016, 44, 453. [DOI] [PubMed] [Google Scholar]
- 14. van Ditzhuijzen N. S., van den Heuvel M., Sorop O., van Duin R. W., Krabbendam‐Peters I., van Haeren R., Ligthart J. M., Witberg K. T., Duncker D. J., Regar E., van Beusekom H. M., van der Giessen W. J., Neth. Heart J. 2011, 19, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoogendoorn A., den Hoedt S., Hartman E. M. J., Krabbendam‐Peters I., Te Lintel Hekkert M., van der Zee L., van Gaalen K., Witberg K. T., Dorst K., Ligthart J. M. R., Drouet L., Van der Heiden K., van Lennep J. R., van der Steen A. F. W., Duncker D. J., Mulder M. T., Wentzel J. J., Arterioscler., Thromb., Vasc. Biol. 2019, 39, 2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Llano R., Winsor‐Hines D., Patel D. B., Seifert P. S., Hamamdzic D., Wilson G. J., Wang H., Keane M. G., Huibregtse B. A., Wilensky R. L., Circ.: Cardiovasc. Interventions 2011, 4, 438. [DOI] [PubMed] [Google Scholar]
- 17. Tellez A., Seifert P. S., Donskoy E., Sushkova N., Pennington D. E., Milewski K., Krueger C. G., Kaluza G. L., Eppihimer M. J., Huibregtse B. A., Dawkins K. D., Granada J. F., Coron. Artery Dis. 2014, 25, 198. [DOI] [PubMed] [Google Scholar]
- 18. Levin A. D., Vukmirovic N., Hwang C. W., Edelman E. R., Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mongrain R., Faik I., Leask R. L., Rodés‐Cabau J., Larose E., Bertrand O. F., J. Biomech. Eng. 2007, 129, 733. [DOI] [PubMed] [Google Scholar]
- 20. Cunnane E. M., Walsh M. T., Drug Discovery Today 2016, 21, 1512. [DOI] [PubMed] [Google Scholar]
- 21. McHugh P., Barakat A., McGinty S., Ann. Biomed. Eng. 2016, 44, 274. [DOI] [PubMed] [Google Scholar]
- 22. McKittrick C. M., Kennedy S., Oldroyd K. G., McGinty S., McCormick C., Ann. Biomed. Eng. 2016, 44, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neubert A., Sternberg K., Nagel S., Harder C., Schmitz K. P., Kroemer H. K., Weitschies W., J. Controlled Release 2, 2008, 130, 2. [DOI] [PubMed] [Google Scholar]
- 24. Seidlitz A., Nagel S., Semmling B., Grabow N., Martin H., Senz V., Harder C., Sternberg K., Schmitz K. P., Kroemer H. K., Weitschies W., Eur. J. Pharm. Biopharm. 2011, 78, 36. [DOI] [PubMed] [Google Scholar]
- 25. Semmling B., Nagel S., Sternberg K., Weitschies W., Seidlitz A., AAPS PharmSciTech 2013, 14, 1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo J., Saylor D. M., Glaser E. P., Patwardhan D. V., J. Pharm. Sci. 2013, 102, 1905. [DOI] [PubMed] [Google Scholar]
- 27. Tzafriri A. R., Vukmirovic N., Kolachalama V. B., Astafieva I., Edelman E. R., J. Controlled Release 2010, 142, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stary H. C., Chandler A. B., Dinsmore R. E., Fuster V., Glagov S., Insull Jr W., Rosenfeld M. E., Schwartz C. J., Wagner W. D., Wissler R. W., Circulation 1995, 92, 1355. [DOI] [PubMed] [Google Scholar]
- 29. Marinello E., Setacci C., Giubbolini M., Cinci G., Frosi B., Porcelli B., Terzuoli L., Life Sci. 2003, 72, 2689. [DOI] [PubMed] [Google Scholar]
- 30. Rapp J. H., Connor W. E., Lin D. S., Inahara T., Porter J. M., J. Lipid Res. 1983, 24, 1329. [PubMed] [Google Scholar]
- 31. Lee K. Y., Mooney D. J., Prog. Polym. Sci. 2012, 37, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosellini E., Cristallini C., Barbani N., Vozzi G., Giusti P., J. Biomed. Mater. Res., A 2009, 91, 447. [DOI] [PubMed] [Google Scholar]
- 33. Escuer J., Aznar I., McCormick C., Pena E., McGinty S., Martinez M. A., Biomech. Model. Mechanobiol 2020, 20, 767. [DOI] [PubMed] [Google Scholar]
- 34. Bozsak F., Chomaz J. M., Barakat A. I., Biomech. Model. Mechanobiol 2014, 13, 327. [DOI] [PubMed] [Google Scholar]
- 35. Mcginty S., McKee S., Wadsworth R. M., McCormick C., SIAM J. Appl. Math. 2013, 73, 2004. [Google Scholar]
- 36. Karanasiou G. S., Papafaklis M. I., Conway C., Michalis L. K., Tzafriri R., Edelman E. R., Fotiadis D. I., Ann. Biomed. Eng. 2017, 45, 853. [DOI] [PubMed] [Google Scholar]
- 37. Vanerio N., Stijnen M., de Mol B. A. J. M., Kock L. M., J. Med. Devices Trans. ASME 2019, 2, 041003. [Google Scholar]
- 38. Sirianni R. W., Kremer J., Guler I., Chen Y. L., Keeley F. W., Saltzman W. M., Biomacromolecules 2008, 9, 2792. [DOI] [PubMed] [Google Scholar]
- 39. Kakizaki R., Minami Y., Hashikata T., Nemoto T., Hashimoto T., Fujiyoshi K., Meguro K., Shimohama T., Tojo T., Ako J., Coron. Artery Dis. 2018, 29, 624. [DOI] [PubMed] [Google Scholar]
- 40. Nomoto Y., Nakagawa M., Shirai N., Kajio K., Mizutani K., Yamazaki T., Sugioka K., Kamimori K., Ueda M., Izumiya Y., Yoshiyama M., Medicine 2019, 98, e17097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baber U., Kini A. S., Sharma S. K., Nat. Rev. Cardiol. 2010, 7, 485. [DOI] [PubMed] [Google Scholar]
- 42. van der Giessen W. J., Lincoff A. M., Schwartz R. S., van Beusekom H. M., Serruys P. W., Holmes Jr D. R., Ellis S. G., Topol E. J., Circulation 1996, 94, 1690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.