

Abstract
Advances in DNA nanotechnology allow the design and fabrication of highly complex DNA structures, uisng specific programmable interactions between smaller nucleic acid building blocks. To convey this concept to the fabrication of metallic nanoparticles, an assembly platform is developed based on a few basic DNA structures that can serve as molds. Programming specific interactions between these elements allows the assembly of mold superstructures with a range of different geometries. Subsequent seeded growth of gold within the mold cavities enables the synthesis of complex metal structures including tightly DNA‐caged particles, rolling‐pin‐ and dumbbell‐shaped particles, as well as T‐shaped and loop particles with high continuity. The method further supports the formation of higher‐order assemblies of the obtained metal geometries. Based on electrical and optical characterizations, it is expected that the developed platform is a valuable tool for a self‐assembly‐based fabrication of nanoelectronic and nanooptic devices.
Keywords: DNA origami, DNA templating, gold nanoparticles, seeded growth, shape programming
A versatile construction kit for the bottom‐up synthesis of complex metal nanostructures with programmable shapes is presented. It uses different DNA elements that can be docked together to produce hollow mold superstructures for metal filling. Electrical and optical characterizations of the obtained metallic nanostructures explore their potential for the self‐assembly of nanoelectronic and nanooptic devices.
1. Introduction
DNA nanotechnology[ 1 , 2 , 3 , 4 ] has become a rapidly advancing field for assembling complex nucleic acid structures. A range of highly efficient techniques has been developed that allows fabrication of 2D and 3D nano‐ and micrometer‐sized DNA structures with programmable shape and nanometer feature sizes.[ 5 , 6 , 7 , 8 , 9 ] For many applications, for example, in nanoelectronics[ 10 , 11 , 12 ] and nanophotonics,[ 13 , 14 ] it would be highly desirable if the shape of the DNA structure could be directly transferred to other materials of choice, for example, metals or semiconductors. This idea has inspired different approaches,[ 15 ] such as the usage of 2D DNA shapes as masks to pattern thin metal[ 16 , 17 ] and graphene films[ 18 ] as well as the employment of rigid DNA structures as scaffolds for the positioning of inorganic nanoparticles.[ 19 , 20 , 21 ] The most versatile and direct way toward a desired shape transfer is currently DNA‐templated electroless deposition of thin metal layers. It has been realized both on linear DNA molecules[ 22 , 23 , 24 , 25 ] as well as on rigid DNA nanostructures.[ 11 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 ] This method employs metal seeds which are attached on the DNA template where they serve as nucleation centers for the subsequent material deposition. Inhomogeneities in seed distribution or material growth as well as a lack of control of the material deposition often provide rather grainy and coarse structures when using electroless deposition. A recent approach could overcome part of these challenges by using DNA origami nanostructures[ 34 , 35 ] as nanoscale molds.[ 36 , 37 , 38 ] The DNA mold walls allowed the metal growth to be confined and guided. This way metallic cuboids of different sizes could be fabricated. By coupling individual origami molds to form long linear chains, micrometer‐sized metallic wires of homogeneous diameter with metal‐like conductivity were obtained.[ 36 ] By introducing a set of specific interfaces between individual monomers, more recently, linear mold superstructures of defined length could be fabricated in which each monomer remained addressable.[ 39 ] This supported the fabrication of linear gold nanostructures of defined length and allowed a controlled site‐specific metallization.
So far, the mold‐based fabrication scheme was limited to quasi‐1D mold superstructures, that is, rods and wires. Using mold elements with different geometries should, however, enable the fabrication of much more complex inorganic nanostructures that extend in more than one dimension and that possess well‐controlled local shapes and sizes. Designing specific interfaces with high affinity for the mold‐to‐mold docking should allow the placement of different structural elements at pre‐determined positions. This way a range of differently shaped mold superstructures would be obtained based on the same set of monomers, similar to stacking different types of LEGO bricks together. Following metallization, the geometry of the resulting metal structures would, in this case, be flexibly programmed by the DNA sequences encoding the interface specificities between particular monomers.
Here, we demonstrate the establishment of such a versatile fabrication platform for the self‐assembly of metallic nanostructures with complex shapes. Our approach is based on four different structural elements that are used as building blocks, such as molds with different diameters and additional docking sites as well as junctions. The interfaces on each of the elements are designed to support specific docking to any of the other element types. This way we can produce metal nanostructures that are tightly caged by DNA, that possess controlled spacings between metal interfaces as well as constrictions, and most importantly that expand in two dimensions, such as branched structures or closed loops. We note that our platform also fully supports the integration of other functional materials, such as semiconductor nanoparticles.[ 40 ] Overall, our method provides a significantly improved control and flexibility for metal nanostructure assembly and establishes a useful basis for a self‐assembly‐based fabrication of nanoelectronic and nanooptical devices with complex geometries. For example, structure caging prevents inter‐structure aggregation allowing for a highly parallelized bulk solution fabrication. Defined DNA gaps between particles can, in a length‐dependent manner, be employed for electrical insulation between devices or for harnessing spin‐selective charge transport in DNA (see below). An increased constriction of metal growth can facilitate true nanoscopic contacts to other nanomaterials. Most importantly, the integration of branches paves the way toward 2D or 3D circuit structures.
2. Results and Discussion
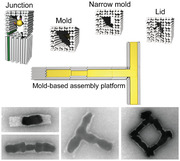
We based our modular assembly platform on a set of four different structural elements. These elements were assembled by the origami method[ 34 ] and supported the desired structural diversity and complexity while maintaining simplicity. As elements, we used (see Figure 1a): i) a previously employed linear mold structure possessing an inner cavity with quadratic cross‐section and 17 nm diameter whose walls were formed by a double layer of DNA helices;[ 36 ] ii) a linear mold termed 3‐wall structure whose walls were formed by a triple layer of DNA helices providing a narrow cavity diameter of only 11 nm; iii) a lid structure used to block the gold growth at mold ends; and iv) a junction structure used to introduce 3‐way branch points. These elements were designed to support docking to each other in a fully flexible and programmable fashion. This should allow the formation of extended mold superstructures (Figure 1b). Preloading the individual elements with gold nanoparticle (AuNP) seeds before the docking should enable a site‐specific “casting” of gold inside the extended cavities of the superstructures, resulting in gold nanostructures with predesigned complex shapes (Figure 1c).
Figure 1.
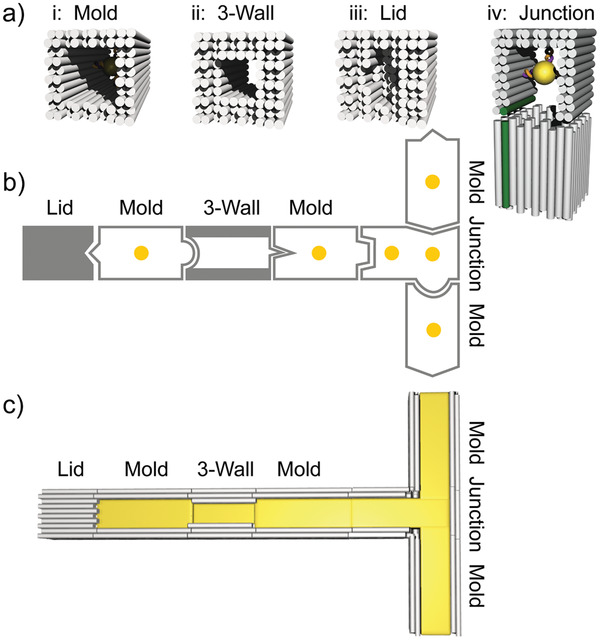
Mold elements of the assembly platform. a) 3D sketches of the four elements of the assembly platform: two tubular structures with inner diameters of 17 nm (mold) and 11 nm (3‐wall), a lid structure without a central channel, and a junction element that can bind three other elements. b) Example of an assembly of elements into a mold superstructure using different specific interfaces between neighboring elements (indicated by different geometric forms). The yellow dots indicate bound gold nanoparticle seeds. c) 3D scheme of the mold superstructure after gold “casting”.
To support the integration of the different structural elements into a single superstructure, all elements shared the same interface geometry based on a 10 × 10 lattice of DNA helix ends (see Note S1 and Figure S1, Supporting Information). The pattern of recessed and exposed helix ends in the outer two DNA layers corresponded for all structures to the design of the standard mold element. All DNA helices that formed the interface had the same length (for design details, see Figure S1, Supporting Information) providing that the two ends of an element (called left end L and right end R) were asymmetric.[ 39 ] Interacting helix ends of the interface docked only gap‐free onto each other, if a left mold end docked onto a right mold end (Note S1, Supporting Information). Elements inside a mold superstructure were therefore always oriented in a specific polarity. To allow specific docking between the different elements, the helix ends of an interface were chosen to either interact in an attractive or a repulsive manner as accomplished by specifically extended or recessed staple overhangs (Note S1, Supporting Information). Specific interfaces between different elements were established by using different overhang types, patterns of attractive versus repulsive helix ends as well as overhang sequences[ 36 , 39 ] (see below). Simple changes of the terminal staples that defined the interactions between the different elements should thus allow a flexible programming of the composition of the mold superstructure that determines the shape of the final gold nanostructure.
To demonstrate that different structural elements can be integrated into our assembly platform, we first tested combinations of the linear mold with the lid element. As seen previously, gold growth inside linear molds often resulted in an outgrowth of gold at the ends of the mold structures[ 39 ] leading to aggregation. Capping the mold ends with lids would prevent the undesired outgrowth of gold. Furthermore, integration of a lid between mold elements would establish well‐defined gaps between gold nanoparticles, for example, for the use in nanooptical applications.[ 41 ] Compared to the standard mold structure, the designed lid had the same cross‐sectional dimension but its cavity was filled with additional DNA helices (Figure 2a). To differentiate between the “left” and the “right” end of the lid, a niche was designed at its left end. Transmission‐mode scanning electron microscopy (tSEM) imaging confirmed that the lid structure was correctly formed including the niche (Figure 2a).
Figure 2.
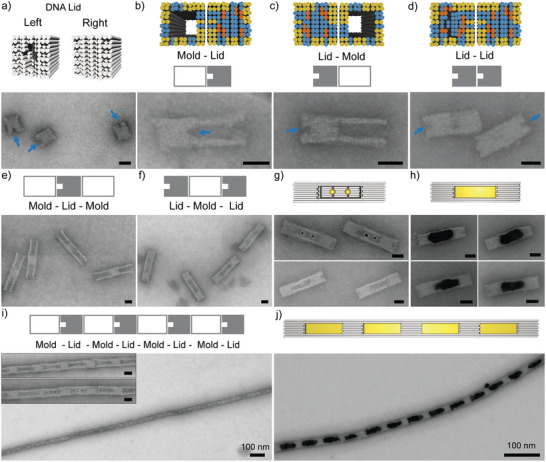
Fabrication of DNA cages using lid structures. Design schemes are shown on top and corresponding tSEM images at the bottom of each subfigure. Colors at the helix ends indicate attractive (in yellow), repulsive (in blue), and “neutral” ends (in orange). a) DNA origami lid structure. A niche of ≈5 nm in depth on the left end of the structure allows the two ends of the lid to be distinguished (see blue arrows in the tSEM image). b,c) Formation of lid–mold dimers with the mold being specifically attached on either the left or the right end of the lid (as sketched). d) Formation of lid–lid dimers. e) Assembly of mold–lid–mold trimers. f) Assembly of trimeric lid–mold–lid DNA nanocages. g) Cage structures encapsulating either two (top) or one (bottom) AuNPs. h) Formation of cage filling gold nanoparticles by seeded Au growth inside the cage. i) Formation of micrometer‐long chains with an alternating lid–mold pattern. The inset shows an enlarged view onto a chain in absence (top) and presence (bottom) of two AuNP seeds. j) Chain with a periodic mold–lid pattern (one AuNP) after seeded gold growth. All scale bars are 20 nm unless indicated.
In order to integrate the lid structure into the mold system, specific interfacing of a lid to a mold or another lid required 40 attractive helix ends in the outer two DNA layers of lid and mold (marked in yellow in Figure 2b–d). The remaining helix ends were repulsive (marked in blue in Figure 2b–d) except of 12 “neutral” non‐interacting ends” at which the end staples were entirely omitted (marked orange in Figure 2b–d, see Note S1, Supporting Information, for details). Compared to mold–mold interactions that use only 24 attractive ends,[ 39 ] the increased number of attractive ends required for lid interactions was most likely due to interfacial tension. The DNA‐filled cavity of the lid caused an increased electrostatic repulsion and thus a larger transversal expansion of the lid element compared to the mold.[ 42 ] We next demonstrated that the designed interfaces supported stable and also specific interactions to both ends of the lid. For this, lids with one interacting and one non‐interacting interface (in which all helix ends were repulsive) were prepared and mixed with a corresponding mold structure carrying a complementary interface part. Specific mold docking was possible at high yields of 96 ± 3% on either side of the lid structure, as also indicated by the correct position of the niche (Figure 2b,c and Figure S2a,b, Supporting Information). Similarly, specific lid–lid dimers could be formed at high yields of 94 ± 1% supporting the versatility of the approach (Figure 2d; Figure S2c, Supporting Information).
Equipping the lid with two interfaces for mold binding allowed mold–lid–mold trimers to be formed (Figure 2e; Figure S3a, Supporting Information). Correspondingly, two lid‐binding interfaces on the mold supported the formation of lid–mold–lid trimers (Figure 2f; Figure S3b, Supporting Information). For the latter, a closed cage was created. Preloading the mold before cage assembly with one or two 5 nm AuNP seeds allowed their encapsulation (Figure 2g). Applying a seeded gold growth procedure (see Experimental Section) allowed growth of DNA‐encapsulated elongated gold nanoparticles inside the cage (Figure 2h; Figure S3c, Supporting Information). Equipping both the mold as well as the lid with two interfaces for binding the other element supported the formation of long chains with an alternating mold–lid pattern (Figure 2i). Preloading the mold elements with one (Figure S4a, Supporting Information) or two AuNP seeds (inset in Figure 2i) following a seeded gold growth allowed a chain of encapsulated gold blocks with well‐defined DNA spacers in between to be fabricated (Figure 2g) in contrast to previously assembled particle chains without tight lid spacers.[ 39 ] Using just a single AuNP seed per mold, we studied the dependence of the gold deposition on the gold precursor. When increasing the gold precursor concentration, the initially small roundish particles filled the cage space more and more until leakage of the gold growth through the cage side‐walls walls occurred (Figure S4b, Supporting Information). Noticeably, leakage was preferred over rupture of the chain.
Overall these data show that lid elements can be successfully integrated into the mold‐based assembly platform allowing the flexible formation of different DNA structures and the confined internal deposition of metal.
After demonstrating the preparation of Au nanoparticles inside DNA cages, we tested whether DNA molds can be used to produce particles with specifically designed constrictions. To this end, we employed the 3‐wall structure (Figure 3a). It possessed the same outer cross‐sectional dimensions as the standard mold. Its walls comprised, however, three DNA layers such that the cavity diameter was only ≈11 nm. While the nominal diameter of the standard mold is ≈17 nm in solution,[ 37 ] grown Au nanoparticles had typical diameters of ≈25 nm.[ 36 ] This indicated that DNA double‐layer walls were too soft, to allow a full confinement. The increased stiffness of the mold structure with a triple DNA layer constituting the walls might better control the particle dimensions during the metal growth.[ 38 ]
Figure 3.
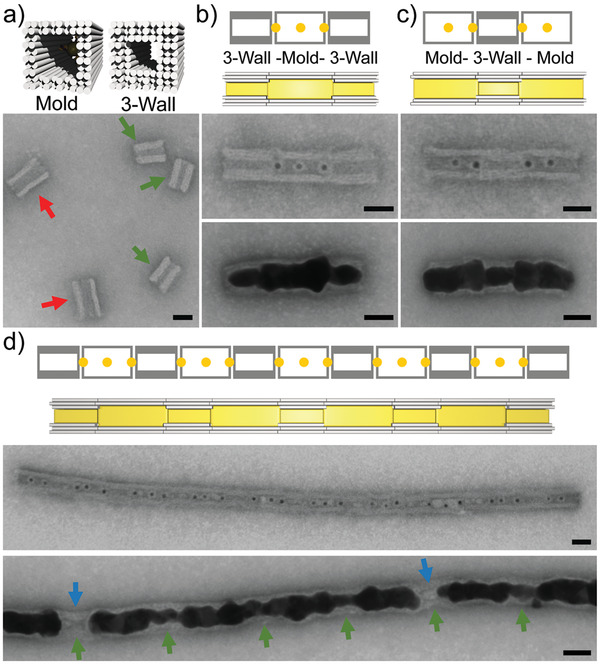
Synthesis of gold nanoparticles with constrictions using 3‐wall structures. Design schemes are shown on top and corresponding tSEM images at the bottom of each subfigure. a) Mold structure based on DNA double layers (indicated by the red arrows) compared to the 3‐wall structure based on a DNA triple layer (indicated by the green arrows). b) 3‐wall–mold–3‐wall trimer. AuNP seeds were loaded into the mold centers and onto the ends of each 3‐wall structure (center). Seeded gold growth provided rolling‐pin‐shaped gold particles (bottom). c) Mold–3‐wall–mold trimer. AuNP seeds were loaded into the mold centers and onto both ends of the 3‐wall structure (center). Seeded gold growth provided dumbbell‐shaped gold particles (bottom). d) Chains with an alternating mold–3‐wall pattern (center) supported the formation of gold nanowires with periodically changing diameter (bottom). The positions of the 3‐wall elements are indicated by green arrows. Occasional empty 3‐wall elements are marked by the blue arrows. The scale bars are 20 nm.
tSEM imaging confirmed that the designed 3‐wall structure formed correctly (Figure 3a). Since we used the same scaffold (p8064) for all DNA origami elements, the 3‐wall structure had the same width but was shorter (32 nm × 25 nm) compared to the mold structure (40 nm × 25 nm).[ 43 ] To allow specific docking between 3‐wall and mold elements, we applied the same concept as before using 40 attractive helices in the outer two DNA layers. Incorporating two attractive interfaces either on the 3‐wall or the mold structure allowed the formation of mold–3‐wall–mold and 3‐wall‐mold‐3‐wall trimer structures (Figure 3b,c and Figure S5a,b, Supporting Information). To allow gold casting, we pre‐loaded 5 nm AuNP seeds to the element monomers before the trimer formation. For the mold structure, one AuNP was loaded in the cavity center. Since the cavity of the 3‐wall structure was too narrow, AuNP seeds could only be placed at the cavity ends (see Figure 3b,c top). After the seeded gold growth, cylindrical particles with non‐uniform diameters were obtained as programmed by the non‐uniform diameter of the mold cavity. The 3‐wall–mold–3‐wall trimer gave “rolling‐pin” like particles with two external constrictions while the mold–3‐wall–mold trimer provided dumbbell‐shaped particle with one internal constriction. When using two attractive interfaces on both the mold and the 3‐wall structure, long chains with an alternating mold–3‐wall pattern could be obtained which supported the formation of gold nanowires with periodically changing diameter (Figure 3d; Figure S5c, Supporting Information). The average particle diameters inside the mold and inside the 3‐wall structure were 21 ± 3 nm and 13 ± 2 nm, respectively. This indicated a tighter control of the particle diameter for the 3‐wall compared to the mold element. Gold growth inside the constriction originated from seeds at the cavity ends of the 3‐wall structure. The success rate of this process was 77 ± 6%. In the remaining cases, the constriction remained either just partially filled or it remained empty as if the “surface tension” of the growing particle was too high to enter the cavity (see Figure S5a,b, Supporting Information).
So far, the produced mold superstructures were exclusively linear, that is, the cavity axes of the individual elements aligned onto each other. To expand the mold system into two dimensions, we thought to introduce T‐shaped branches. To this end, we designed a junction structure using a single scaffold strand. Its bottom part was a shortened version of the standard mold (Figure 4a). Its top part had a U‐shaped form, corresponding to the standard mold geometry in which one sidewall was removed. A scaffold loop connected the upper and the bottom part with six unpaired spacer nucleotides at the connection sites to decrease tension. To further connect the bottom part and the top part, six additional end staple overhangs were added on each side of the bottom part that connected to the U‐structure. These overhangs were connected to the top part either by direct hybridization to the scaffold loop (single staple geometry, see Figure 4b) or by hybridization to staple overhangs from the top part (zipper geometry, see Figure S6, Supporting Information, for sequences). CanDo simulations[ 43 ] of the junction structure confirmed the feasibility of the design (Figure S6, Supporting Information) and tSEM imaging confirmed the correct assembly of the structure (Figure 4a). The two different attachment geometries provided similar results for all the following experiments. In the following, only results for the zipper geometry are shown.
Figure 4.

Synthesis of branched nanostructures using junction elements. a) Design scheme of the junction element including two AuNP seeds in the top and bottom part (right) as well as tSEM images (left) of the two possible orientations of the junction with the cavity axis of the top part being perpendicular (top) and parallel (bottom) to the surface. The helices marked in green correspond to helix 0 in the standard mold design. b) Two different connector types between the two junction parts. Either staples from the bottom and top part hybridizing to each other in a zipper geometry or end staples from the bottom part with a 6 nt spacer hybridizing with the scaffold of the top part were used (see enlarged views on the top). The locations of the connections between the two junction parts mapped onto the helix ends of the bottom part are shown at the bottom. Scaffold crossings are shown in blue. Connector positions are shown in pink. c) tSEM image and design (inset) of a T‐shaped mold superstructure formed from a junction element and three standard molds. d) tSEM image of the T‐shaped mold after seed loading. e) tSEM image of the T‐shaped mold after gold growth. f) tSEM image and design (inset) of a chain of T‐shaped elements. An extra mold element (orange) interconnected the individual T‐structures. g) tSEM image of the branched chain structure after gold growth. h) Design of a square‐loop mold superstructure based on junction and mold elements that were connected by alternating Y and Z interfaces. i) tSEM image of a square‐loop mold superstructure. j) tSEM image of the square loop with loaded AuNP seeds. k) tSEM image of the metallized square loop. Scale bars without labels correspond to 20 nm.
The junction element possessed three different sites for mold docking for which we designed corresponding interfaces. The top part had a left (L) end, being part of interface “X”, and a right (R) end, being part of interface “Y”, for mold docking (Figure S7a, Supporting Information). For both interfaces, a total of 34 attractive helices were taken. The bottom part had an L end, being part of interface “Z”, for which 40 attractive helices were used (Figure S7b, Supporting Information). When applied in mold docking, the X and Y interfaces of the top part of the junction supported the formation of L‐shaped dimer elements (Figure S7a, Supporting Information), while the bottom part supported the formation of linear dimers (Figure S7b, Supporting Information). When incorporating attractive interfaces at all sides of the junction, three mold monomers could be bound on the junction element to form a T‐shaped structure (Figure 4c; Figure S8, Supporting Information). The assembly yield of the full T‐shaped structure reached 72 ± 11%. Preloading the mold and junction monomers with AuNPs (Figure 4a) allowed an efficient decoration of the T‐structure with Au seeds (Figure 4d). Following the seeded growth procedure, this supported the formation of T‐shaped gold structures (Figure 4e) in which a perpendicular arrangement of the two T‐sections was typically maintained (see also Figure S9, Supporting Information). The yield for metallized T‐shaped structures was 61 ± 11%.
Individual T‐structures could be further assembled into higher‐order superstructures. To this end, we introduced additional attractive A and B interfaces at the mold elements[ 39 ] that were docked to the top part of the junction (i.e., by using YL‐AR and BL‐XR mold monomers, see Figure 4f). When mixed with mold monomers that carried complementary AL‐BR interfaces, the T‐shaped mold superstructures could be connected to each other to form comb‐like structures, that is, long chains with periodically occurring branches of equal length (Figure 4f). Preloading of the monomeric elements with AuNP seeds supported again a seeded gold growth and thus the formation of branched gold chains (Figure 4g).
The results above demonstrated that gold nanostructures with complex shapes (e.g., including constrictions and branches) can be obtained by combining standard molds with additional elements. Next, we showed that with a different selection of interfaces between linear mold and junction elements other shapes of gold nanostructures can be programmed. To this end, we formed a square‐loop mold superstructure. It consisted of four L‐shaped mold–junction dimers. The junction elements carried a YR interface and a repulsive interface at the top part as well as a ZL interface at the bottom part. The mold elements carried corresponding YL and ZR interfaces. This would provide an alternating junction–mold chain which would self‐terminate upon loop‐closure (Figure 4h). The junction and mold monomers were mixed at equal stoichiometry. tSEM imaging revealed the formation of closed square loops consisting of eight individual elements (Figure 4i). In addition to closed square‐loops, pentagon‐shaped as well as incomplete open structures were found (Figure S10, Supporting Information). The latter often exhibited some aggregation. The yield of correctly assembled square loops was 65 ± 15%, which could not be increased using a second lower‐affinity interface Z´ (Figure S11, Supporting Information). Next, seed‐loaded square loops were prepared from monomers that were correspondingly pre‐loaded with AuNPs. The mold monomers were either loaded with one (Figure 4j) or with two AuNPs (Figure S12a, Supporting Information). After gold growth, small square gold loops were realized (Figure 4k). The metal film became continuous once enough gold precursor was supplied (Figure S12b, Supporting Information). Defect mold structures were also metallized incl. partial squares, pentagons and mold squares with missing seeds (Figure S12d, Supporting Information). The yield of square loop metallization was 29 ± 10%.
The square loop geometry was further employed to realize larger network structures, where individual loops were mutually connected using an additional linear element. This allowed the formation of even larger superstructures as a proof of principle (see Note S2 and Figures S13–S15, Supporting Information). Due to the accumulation of assembly effects, the periodicity of these structures was however limited.
To experimentally demonstrate that the fabricated metallic nanostructures can be used in nanooptical applications, we measured and modeled the scattering cross‐section of single T‐ and L‐shaped structures by single particle spectroscopy (see Note S3, Supporting Information). To spectrally resolve the scattered light from selected structures, we employed dark‐field (DF) spectroscopy correlated with the scanning electron microscopy (SEM) (see Figures S16 and S17, Supporting Information). The combination of the two techniques ensured that the optical response originated only from the anticipated structure. We observed for both types of structures a characteristic spectral shape in qualitative agreement with numerical simulations. The experimentally measured unpolarized response of both structures was characterized by a main plasmonic mode (>800 nm for T‐shaped and ≈650 nm for L‐shaped structure). Using simulations with linear polarized excitation, these modes could be assigned to one geometrical axis of the nanostructure. The optical response was dominated by a longitudinal mode being excited by the electric field component of the incident light parallel to the longer geometric axis.[ 44 ] Furthermore, a transversal mode, representing surface charge oscillations along the shorter axis, could be recognized. With the single particle spectroscopy, the basic plasmonic modes could be quantified, which is a prerequisite for nano‐optical applications.
Recently, the electronic properties of DNA have gained increasing interest due to the discovered chiral induced spin selectivity (CISS effect) of charge transport through helical biomolecules.[ 45 ] Based on the spin‐selective charge transport, first thin‐film spin torque devices[ 46 ] as well as memristor elements[ 47 ] could be fabricated. To demonstrate that our assembly scheme readily supports the integration and electric contacting of well‐oriented DNA nanolayers at nanoscale dimensions, we constructed a nanolid structure comprising a ≈10 nm (31 bp) thin DNA layer through which charge transport was probed. The design of the nanolid element was based on the standard mold by densely filling the cavity center with additional DNA helices. In contrast to the lid element, the thickness of the additional DNA layer on the nanolid element was only ≈10 nm (Figure 5a) to allow efficient charge transport as well as spin coherence.[ 48 ] The DNA nanolayer comprised a total of 36 DNA helices that were oriented parallel to the mold axis. We designed suitable interfaces (E and F) to enable docking of standard mold elements (see Figure S18, Supporting Information). This allowed the formation of mold–nanolid and nanolid–mold dimers as well as mold–nanolid–mold trimers at efficiencies of 97±2% (N = 211), 98±2% (N = 209) and 90±5% (N = 256), respectively (Figure S18, Supporting Information). To establish nanoscale contacts to the DNA layer, we docked onto each side of the nanolid mold trimers, in which the two molds that were proximal to the nanolid were each preloaded with two AuNP seeds (Figures 5a,b). This structure consisting of seven mold elements formed at 68 ± 8% (N = 252) efficiency (see Figure S19a, Supporting Information). Nanoscale contacts were finally fabricated by seeded gold growth. TEM imaging confirmed the successful contacting of the DNA nanolayer by two ≈100 nm long Au nanoelectrodes of ≈25 nm diameter (Figure 5b and Figure S19b, Supporting Information).
Figure 5.
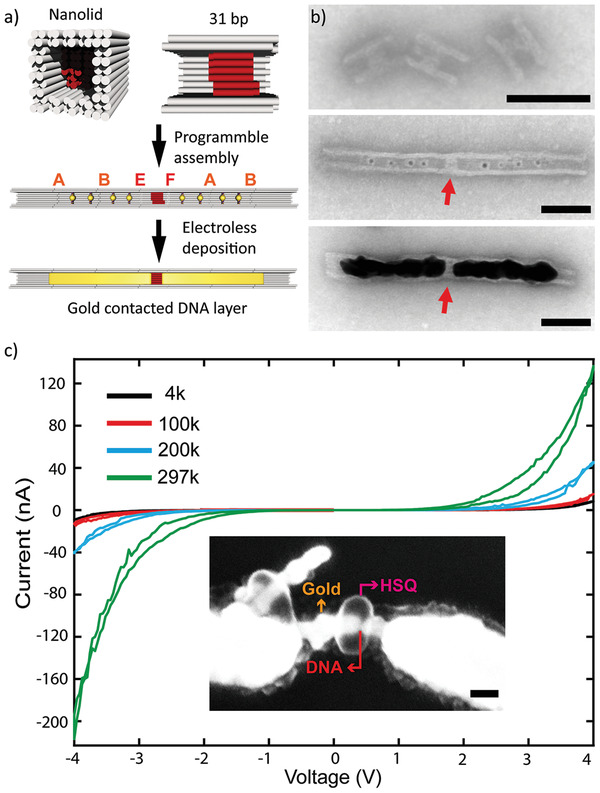
Contacting and conductance of a 10 nm DNA nanolayer. a) Top: design scheme of a nanolid element consisting of a standard mold element containing a central 31 bp (≈10 nm) DNA nanolayer (shown in red) in which the DNA helices align with the mold axis. Middle: Integrating the nanolid element with three AuNP‐loaded mold elements on either side. The letters indicate the employed interfaces. Bottom: Electric contacting of the DNA nanolayer by the growth of Au nanoelectrodes inside the mold elements b) tSEM images of nanolid elements before (top) and after integration (middle) with AuNP‐loaded mold elements and Au nanoelectrode growth (bottom). The red arrows point to the DNA nanolayer. c) Current–voltage characteristics of the DNA nanolayer measured at different temperatures (4, 100, 200, and 297 K). Inset: SEM image of the contacted DNA nanolayer–nanoelectrode assembly. The arrows mark the DNA nanolayer (red), the protective HSQ resist layer (magenta), and the gold nanoelectrodes (orange). The scale bars are 50 nm.
To probe the conductance of the contacted DNA nanolayer, the nanoelectrode assemblies were deposited on SiO2 substrates. Using electron beam lithography (EBL), the DNA nanolayers were protected by a small patch of hydrogen silsesquioxane (HSQ) resist (see inset in Figure 5c). Subsequently, microelectrodes integrating the nanoelectrode assembly into a microchip were fabricated by EBL (see Note S4 and Figure S20, Supporting Information, for a more detailed description of the site‐specific EBL procedure). Contacted and integrated DNA nanolayers were electrically characterized at different temperatures ranging from room temperature to 4 k. The I–V characteristics were strongly non‐linear. The samples showed low currents below a voltage of 1 V but exhibited a strong increase in current above 2 V (Figure 5c; Figure S21, Supporting Information). Furthermore, the conductance appeared to be thermally activated. This resembled semiconductive behavior, which has been previously observed for single DNA molecules trapped in break junctions[ 49 ] and self‐assembled monolayers of thiol‐functionalized DNA. The temperature‐dependence of the conductance suggested hopping as charge transport mechanism along the DNA, as expected for DNA molecules, which are in contact with a substrate.[ 50 ] The overall resistance of the DNA nanolayer was on the order of 20 MΩ at room temperature, which was comparable to previous measurements.[ 51 ] Control measurements on a continuous gold nanowire or on the bare HSQ resist layer provided conductance values that were several orders of magnitude higher or lower, respectively (Figure S21a, Supporting Information,). This suggests that effects from the metallic nanoelectrodes as well as the protecting resist can be neglected in the obtained I–V characteristics of the DNA nanolayers.
3. Conclusion
We successfully established a versatile platform that allowed the fabrication of gold nanostructures in a modular and programmable manner. Our approach was based on four DNA mold elements. Programming of the interactions between the different elements enabled the assembly of mold superstructures with a range of different predetermined geometries. Superstructure assembly relied on a total of 16 different specific interfaces between the element monomers compared to four interfaces introduced previously.[ 39 ] “Casting” of gold into the mold cavities enabled the subsequent synthesis of complex metal nanoparticles as predetermined by the designed DNA template. So far, similar nanoparticle geometries could not or only with difficulties be obtained by other “non‐mold" methods. This includes tightly DNA‐caged particles using the lid elements, rolling‐pin‐ and dumbbell‐shaped particles, as well as L‐shaped, T‐shaped, and loop particles. Furthermore, the individual superstructures could be employed in a next level of hierarchical self‐assembly to form larger 1D or even 2D periodic structures.
Generally, our mold‐based procedure supported rather high yields, homogeneity, and continuity of the obtained gold nanostructures (see Table S1, Supporting Information, summarizing yields of mold superstructure assembly and metallization). The most critical point was the assembly efficiencies of the mold superstructures, particularly for the most complex structures. Generally, linear mold assemblies formed at high efficiencies (up to 90%), demonstrating that the interface specificity was not the limiting factor. Involvement of the junction element provided however reduced yields. While assembly of the T‐shaped mold exhibited a smaller yield reduction (72 ± 11%), the yield for the closed loop structure was more markedly reduced (65 ± 15%). We attribute these problems in part to the flexibility of the junction element as well as to unsuccessful self‐termination events. Using an improved and more rigid junction design as well as specific rather than periodic interfaces should allow these difficulties to be overcome, which currently provide a bottle neck for forming higher order networks (see Note S2, Supporting Information). The yield of the mold superstructure metallization (Table S1, Supporting Information) was also high given the complexity of the produced structures. Main difficulties were aggregate formation due to open mold ends at which uncapped metal surfaces could grow out as well as a somewhat grainy morphology of the nanoparticles in particular for the finest mold features (Figure 3d). The latter arises mainly from the fact that the seeded growth involves the formation of multiple nanocrystallites. Their expansion is not fully controlled by the soft mold walls given the bulk and surface energies of the crystal lattice. Nonetheless, the obtained control over the nanoparticle morphology within molds is significantly improved compared to an external metal deposition on DNA templates.
A further advantage of the developed method is that all elements with unique interfaces remain individually addressable.[ 39 ] We anticipate that this will support the formation of complex heterostructures from different metallic,[ 32 ] semiconducting materials,[ 40 ] and organic materials.[ 52 ] Different seeds in specific molds should support the electroless growth of different metals at the desired location. As shown previously, our mold‐based approach allows a direct integration and contacting of semiconducting nanorods.[ 40 ] In this work, we furthermore showed that our approach allowed the electric contacting and charge injection into a heterostructure comprising gold nanocontacts and DNA as an organic conductive material possessing spin‐selectivity. Using the established toolset of our assembly platform, including the assembly of heterostructures, controlled gaps, and junctions, we anticipate a greatly facilitated fabrication of locally addressable switchable electric devices. Upon further optimization of the assembly efficiencies, even 2D and 3D arrangement of several devices could be established. We also demonstrated that the obtained structures possess specific geometry‐dependent plasmonic properties and identified the fundamental plasmonic modes using single‐particle spectroscopy as a prerequisite for their further applications in nanooptics. If assembled in a periodic manner, the produced structures could generate collective resonance. In the simplest case, the Bragg mode interacts coherently with the fundamental plasmonic modes, resulting in a surface lattice resonance with significantly improved plasmonic mode quality. Such modes with low radiation losses are particularly well suited as optical sensors and nonlinear optical effects.[ 53 ] For advanced applications, such as wavefront and beam shaping, one can take the advantage of changing the relative orientation of the asymmetric nanostructures within the lattice.[ 54 ] Such assemblies can be achieved through patterning the substrate to immobilize the DNA‐origami nanostructures.[ 55 , 56 , 57 ]
We therefore think that our mold‐based platform for the fabrication of metal nanoparticles with programmable shape will be a promising tool to realize different applications in nanoelectronics and nanooptics.
4. Experimental Section
Preparation of Seed‐Loaded Origami Monomers
The different DNA origami elements were designed with CaDNAno[ 58 ] using a square lattice for the helix arrangement[ 42 ] (see Figures S22–28, Supporting Information, for the design schemes). The origami structures were assembled in folding buffer (FB, Eurofins) containing 5 × 10−3 m Tris‐HCl, 1 × 10−3 m EDTA, and 11 × 10−3 m MgCl2 (pH 8.0) following a one‐pot assembly protocol.[ 39 ] Subsequently, the mold monomers were purified using precipitation with poly(ethylene glycol) to remove excess staples.[ 59 ] 5 nm AuNPs (Sigma‐Aldrich) were densely coated with 15 nt polythymidine oligonucleotides carrying a 5′‐thiol modification using the method of salt aging.[ 60 ] The particle concentration was estimated from the absorbance at 520 nm using an extinction coefficient of 1.1 × 107 m −1 cm−1. For preloading the elements with seeds, the DNA‐functionalized gold nanoparticles were mixed with the purified DNA origami elements in the presence of 350 mm NaCl at a molar ratio of 3:1 for each seed binding site. The mixture was slowly heated to 40 °C and afterward cooled down to 23 °C over a duration of 5 h to allow hybridization of the AuNPs with the complementary capture strands on the elements. The seed‐loaded elements were purified by an additional precipitation step with PEG to remove excess seeds before using them in superstructure assembly.
Formation of Mold Superstructures
Mold monomers and different elements of a particular superstructure carrying corresponding end staples to allow specific docking to their left or right ends (see Figures S29–31, Supporting Information, for the design details of all interfaces) were mixed at equal stoichiometry in FB supplemented with 350 × 10−3 m NaCl and incubated overnight. For the assembly of branched chain structures, individual T‐shaped structures were formed first. At the next day, the additional mold elements with A‐B interfaces were added to the mixture following an additional overnight incubation. For the network assembly, loop structures were assembled first, followed by the addition of connector‐dimers at the next day. Typically, mold superstructures that were internally decorated with AuNPs were subjected to PEG precipitation after the superstructures were formed in order to remove excess AuNPs. Loop and network structures were not subjected to a final PEG precipitation step. Rather, PEG precipitation was performed already after preloading the origami monomers with AuNP seeds. For tSEM imaging, 5 µL of a 2–5 × 10−9 m solution of origami structures were placed onto glow‐discharged carbon‐coated TEM grids for 5 min. The samples were subsequently stained using a filtered 2% solution of uranyl formate in 5 × 10−3 m NaOH for 1–2 min, followed by two washing steps with 5 µL ultrapure water for 10 s. tSEM imaging was performed on a Gemini SEM500 scanning electron microscope (Zeiss) operated in transmission mode at 25 kV. TEM imaging was performed using a Jeol JEM2100Plus transmission electron microscope.
Seeded‐Growth of Gold within Mold Superstructures
For seed‐mediated deposition of gold inside the mold superstructures,[ 61 ] the mold concentration was adjusted, such that the solution contained a total of 0.5 × 10−9 m AuNPs in folding buffer (100 µL final volume). Hydroxylamine (NH2OH) used as a reducing agent was premixed with H[AuCl4] gold precursor in a 1 to 1 molar ratio at concentrations of 450 or 675 × 10−6 m unless indicated otherwise. The gold deposition was initiated by injecting the corresponding amount of mold superstructures into the reaction mixture and was allowed to proceed for 1 min.
Dark‐Field Microscopy and Spectroscopy
Single‐particle scattering spectroscopy was performed with a Nikon Ti‐U inverted microscope in transmission mode. The data were recorded with IsoPlane‐160 spectrometer and a PIXIS 256 charge‐coupled device camera (Princeton Instruments). The measurements were performed with a dark‐field condenser (air, NA 0.8–0.95) and a 60× air objective (CFI S Plan Fluor ELWD, NA 0.7, Nikon, Japan) under the illumination by an Energetic EQ‐99 laser‐driven light source. The measured spectra were corrected by subtracting the dark current at the detector and normalizing against the white light scattering spectrum of a roughened glass surface. The optical measurements were correlated with SEM imaging to verify the particles shape. A NEON 40 FIB‐SEM workstation (Carl Zeiss Microscopy GmbH, Oberkochen, Germany) operated at accelerating voltage of 1 kV, with the standard aperture size of 30 µm was used in a secondary‐electron‐detection mode to obtain scanning electron microscopy images.
Electromagnetic Simulations
A finite‐difference time domain (FDTD) method was used (FDTD: 3D Electromagnetic Simulator) to perform the numerical calculations (Lumerical Inc.).[ 62 ] The structures were represented by the rectangular shapes with the dimensions, matching the ones, determined by the SEM imaging (Figure S16, Supporting Information). To simulate the optical response, a total‐field scattered‐field source (λ = 450–800 nm) was used, illuminating the structure at a normal incidence along the z‐axis, as demonstrated in the Figure S16, Supporting Information. To simulate the unpolarized light, two separate simulations with orthogonally polarized beams were performed.
Integration and Electrical Measurements of DNA Nanolayers
The DNA nanolayer were electrically contacted using EBL similarly as described before.[ 36 ] To protect the DNA nanolayer from damage during the contacting procedure, a patch of HSQ resist above each nanolayer was defined (see Note S4, Supporting Information, for details). The electrical characterization was performed in a cryogenic probestation (LakeShore) using an Agilent parameter analyzer (4156C). In this setup, various devices could be tested within one cooling cycle, thus minimizing the time necessary for analyzing multiple configurations.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors gratefully acknowledge Markus Löffler and the Dresden Center for Nanoanalysis for access, training, and support for the tSEM imaging. The authors furthermore acknowledge Jonathan Doye, Dominik Kauert, and Fatih N. Gür for helpful discussions and comments. The authors also thank Ulrich Kemper for TEM imaging support. This work was supported by the Deutsche Forschungsgemeinschaft within the Cluster of Excellence Center for Advancing Electronics Dresden (cfaed/TU Dresden) as well as grant SE 1646/8‐1 to R.S. This project was financially supported by the Volkswagen Foundation through a Freigeist Fellowship to T.A.F.K. as well as the Deutsche Forschungsgemeinschaft (404818834, O.A. and T.A.F.K.). J.Y. and T.B. acknowledge support by the Helmholtz Association through IHRS for Nanoelectronic Networks NanoNet (VH‐KO‐606).
Open access funding enabled and organized by Projekt DEAL.
Ye J., Aftenieva O., Bayrak T., Jain A., König T. A. F., Erbe A., Seidel R., Complex Metal Nanostructures with Programmable Shapes from Simple DNA Building Blocks. Adv. Mater. 2021, 33, 2100381. 10.1002/adma.202100381
Data Availability Statement
Research data are not shared.
References
- 1. Seeman N. C., Sleiman H. F., Nat. Rev. Mater. 2018, 3, 17068. [Google Scholar]
- 2. Bathe M., Rothemund P. W. K., MRS Bull. 2017, 42, 882. [Google Scholar]
- 3. Zhang F., Nangreave J., Liu Y., Yan H., J. Am. Chem. Soc. 2014, 136, 11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pinheiro A. V., Han D., Shih W. M., Yan H., Nat. Nanotechnol. 2011, 6, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seeman N. C., Annu. Rev. Biochem. 2010, 79, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hong F., Zhang F., Liu Y., Yan H., Chem. Rev. 2017, 117, 12584. [DOI] [PubMed] [Google Scholar]
- 7. Wang W., Chen S., An B., Huang K., Bai T., Xu M., Bellot G., Ke Y., Xiang Y., Wei B., Nat. Commun. 2019, 10, 1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin C., Liu Y., Rinker S., Yan H., ChemPhysChem 2006, 7, 1641. [DOI] [PubMed] [Google Scholar]
- 9. Tikhomirov G., Petersen P., Qian L., Nature 2017, 552, 67. [DOI] [PubMed] [Google Scholar]
- 10. Gates E. P., Dearden A. M., Woolley A. T., Crit. Rev. Anal. Chem. 2014, 44, 354. [DOI] [PubMed] [Google Scholar]
- 11. Liu J., Geng Y., Pound E., Gyawali S., Ashton J. R., Hickey J., Woolley A. T., Harb J. N., ACS Nano 2011, 5, 2240. [DOI] [PubMed] [Google Scholar]
- 12. Zhao M., Chen Y., Wang K., Zhang Z., Streit J. K., Fagan J. A., Tang J., Zheng M., Yang C., Zhu Z., Sun W., Science 2020, 368, 878. [DOI] [PubMed] [Google Scholar]
- 13. Samanta A., Banerjee S., Liu Y., Nanoscale 2015, 7, 2210. [DOI] [PubMed] [Google Scholar]
- 14. Shen B., Kostiainen M. A., Linko V., Langmuir 2018, 34, 14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Z., Liu C., Cao F., Ren J., Qu X., Chem. Soc. Rev. 2018, 47, 4017. [DOI] [PubMed] [Google Scholar]
- 16. Shen B., Linko V., Tapio K., Kostiainen M. A., Toppari J. J., Nanoscale 2015, 7, 11267. [DOI] [PubMed] [Google Scholar]
- 17. Shen B., Linko V., Tapio K., Pikker S., Lemma T., Gopinath A., Gothelf K. V., Kostiainen M. A., Toppari J. J., Sci. Adv. 2018, 4, eaap8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jin Z., Sun W., Ke Y., Shih C.‐J., Paulus G. L. C., Hua Wang Q., Mu B., Yin P., Strano M. S., Nat. Commun. 2013, 4, 1663. [DOI] [PubMed] [Google Scholar]
- 19. Liu N., Liedl T., Chem. Rev. 2018, 118, 3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gür F. N., Schwarz F. W., Ye J., Diez S., Schmidt T. L., ACS Nano 2016, 10, 5374. [DOI] [PubMed] [Google Scholar]
- 21. Kuzyk A., Schreiber R., Fan Z., Pardatscher G., Roller E.‐M., Högele A., Simmel F. C., Govorov A. O., Liedl T., Nature 2012, 483, 311. [DOI] [PubMed] [Google Scholar]
- 22. Braun E., Eichen Y., Sivan U., Ben‐Yoseph G., Nature 1998, 391, 775. [DOI] [PubMed] [Google Scholar]
- 23. Richter J., Seidel R., Kirsch R., Mertig M., Pompe W., Plaschke J., Schackert H. K., Adv. Mater. 2000, 12, 507. [Google Scholar]
- 24. Keren K., Krueger M., Gilad R., Ben‐Yoseph G., Sivan U., Braun E., Science 2002, 297, 72. [DOI] [PubMed] [Google Scholar]
- 25. Mertig M., Colombi Ciacchi L., Seidel R., Pompe W., De Vita A., Nano Lett. 2002, 2, 841. [Google Scholar]
- 26. Teschome B., Facsko S., Schönherr T., Kerbusch J., Keller A., Erbe A., Langmuir 2016, 32, 10159. [DOI] [PubMed] [Google Scholar]
- 27. Schreiber R., Kempter S., Holler S., Schüller V., Schiffels D., Simmel S. S., Nickels P. C., Liedl T., Small 2011, 7, 1795. [DOI] [PubMed] [Google Scholar]
- 28. Schreiber R., Do J., Roller E.‐M., Zhang T., Schüller V. J., Nickels P. C., Feldmann J., Liedl T., Nat. Nanotechnol. 2014, 9, 74. [DOI] [PubMed] [Google Scholar]
- 29. Uprety B., Gates E. P., Geng Y., Woolley A. T., Harb J. N., Langmuir 2014, 30, 1134. [DOI] [PubMed] [Google Scholar]
- 30. Yan H., Park S. H., Finkelstein G., Reif J. H., LaBean T. H., Science 2003, 301, 1882. [DOI] [PubMed] [Google Scholar]
- 31. Pilo‐Pais M., Goldberg S., Samano E., LaBean T. H., Finkelstein G., Nano Lett. 2011, 11, 3489. [DOI] [PubMed] [Google Scholar]
- 32. Geng Y., Pearson A. C., Gates E. P., Uprety B., Davis R. C., Harb J. N., Woolley A. T., Langmuir 2013, 29, 3482. [DOI] [PubMed] [Google Scholar]
- 33. Pearson A. C., Liu J., Pound E., Uprety B., Woolley A. T., Davis R. C., Harb J. N., J. Phys. Chem. B 2012, 116, 10551. [DOI] [PubMed] [Google Scholar]
- 34. Rothemund P. W. K., Nature 2006, 440, 297. [DOI] [PubMed] [Google Scholar]
- 35. Douglas S. M., Dietz H., Liedl T., Högberg B., Graf F., Shih W. M., Nature 2009, 459, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bayrak T., Helmi S., Ye J., Kauert D., Kelling J., Schönherr T., Weichelt R., Erbe A., Seidel R., Nano Lett. 2018, 18, 2116. [DOI] [PubMed] [Google Scholar]
- 37. Helmi S., Ziegler C., Kauert D. J., Seidel R., Nano Lett. 2014, 14, 6693. [DOI] [PubMed] [Google Scholar]
- 38. Sun W., Boulais E., Hakobyan Y., Wang W. L., Guan A., Bathe M., Yin P., Science 2014, 346, 1258361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ye J., Helmi S., Teske J., Seidel R., Nano Lett. 2019, 19, 2707. [DOI] [PubMed] [Google Scholar]
- 40. Weichelt R., Ye J., Banin U., Eychmüller A., Seidel R., Chem. A Eur. J. 2019, 25, 9012. [DOI] [PubMed] [Google Scholar]
- 41. Gür F. N., McPolin C. P. T., Raza S., Mayer M., Roth D. J., Steiner A. M., Löffler M., Fery A., Brongersma M. L., Zayats A. V., König T. A. F., Schmidt T. L., Nano Lett. 2018, 18, 7323. [DOI] [PubMed] [Google Scholar]
- 42. Ke Y., Douglas S. M., Liu M., Sharma J., Cheng A., Leung A., Liu Y., Shih W. M., Yan H., J. Am. Chem. Soc. 2009, 131, 15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim D.‐N., Kilchherr F., Dietz H., Bathe M., Nucleic Acids Res. 2012, 40, 2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prescott S. W., Mulvaney P., J. Appl. Phys. 2006, 99, 123504. [Google Scholar]
- 45. Gohler B., Hamelbeck V., Markus T. Z., Kettner M., Hanne G. F., Vager Z., Naaman R., Zacharias H., Science 2011, 331, 894. [DOI] [PubMed] [Google Scholar]
- 46. Ben Dor O., Morali N., Yochelis S., Baczewski L. T., Paltiel Y., Nano Lett. 2014, 14, 6042. [DOI] [PubMed] [Google Scholar]
- 47. Al‐Bustami H., Koplovitz G., Primc D., Yochelis S., Capua E., Porath D., Naaman R., Paltiel Y., Small 2018, 14, 1801249. [DOI] [PubMed] [Google Scholar]
- 48. Kumar A., Capua E., Fontanesi C., Carmieli R., Naaman R., ACS Nano 2018, 12, 3892. [DOI] [PubMed] [Google Scholar]
- 49. Liu S. P., Artois J., Schmid D., Wieser M., Bornemann B., Weisbrod S., Marx A., Scheer E., Erbe A., Phys. Status Solidi B 2013, 250, 2342. [Google Scholar]
- 50. Livshits G. I., Stern A., Rotem D., Borovok N., Eidelshtein G., Migliore A., Penzo E., Wind S. J., Di Felice R., Skourtis S. S., Cuevas J.‐C., Gurevich L., Kotlyar A. B., Porath D., Nat. Nanotechnol. 2014, 9, 1040. [DOI] [PubMed] [Google Scholar]
- 51. Cohen H., Nogues C., Ullien D., Daube S., Naaman R., Porath D., Faraday Discuss. 2006, 131, 367. [DOI] [PubMed] [Google Scholar]
- 52. Zessin J., Fischer F., Heerwig A., Kick A., Boye S., Stamm M., Kiriy A., Mertig M., Nano Lett. 2017, 17, 5163. [DOI] [PubMed] [Google Scholar]
- 53. Kravets V. G., Kabashin A. V., Barnes W. L., Grigorenko A. N., Chem. Rev. 2018, 118, 5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen H.‐T., Taylor A. J., Yu N., Rep. Prog. Phys. 2016, 79, 076401. [DOI] [PubMed] [Google Scholar]
- 55. Gopinath A., Rothemund P. W. K., ACS Nano 2014, 8, 12030. [DOI] [PubMed] [Google Scholar]
- 56. Woo S., Rothemund P. W. K., Nat. Commun. 2014, 5, 4889. [DOI] [PubMed] [Google Scholar]
- 57. Shaali M., Woller J. G., Johansson P. G., Hannestad J. K., de Battice L., Aissaoui N., Brown T., El‐Sagheer A. H., Kubatkin S., Lara‐Avila S., Albinsson B., Jesorka A., J. Mater. Chem. C 2017, 5, 7637. [Google Scholar]
- 58. Douglas S. M., Marblestone A. H., Teerapittayanon S., Vazquez A., Church G. M., Shih W. M., Nucleic Acids Res. 2009, 37, 5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stahl E., Martin T. G., Praetorius F., Dietz H., Angew. Chem. Int. Ed. 2014, 53, 12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hurst S. J., Lytton‐Jean A. K. R., Mirkin C. A., Anal. Chem. 2006, 78, 8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ye J., Weichelt R., Kemper U., Gupta V., König T. A. F., Eychmüller A., Seidel R., Small 2020, 16, 2003662. [DOI] [PubMed] [Google Scholar]
- 62.Nanophotonic FDTD Simulation Software – Lumerical FDTD, https://www.lumerical.com/products/fdtd/ (accessed: September 2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.