

Abstract
Their natural functions in intercellular communication render extracellular vesicles (EV) highly attractive for drug delivery applications. However, the loading efficiency of present methods to incorporate particularly hydrophilic low molecular weight drugs of biomedical interest is largely unexplored, as is the impact these methods may have on the intrinsic structural and biological vesicle properties. Here, different methods are exploited to incorporate hydrophilic non‐membrane permeable compounds into stem cell‐derived small EV, and to assess the vesicle characteristics after the different loading processes. When comparing several methods head‐to‐head, the loading capacity increases in the order saponin ≤ sonication < fusion < freeze‐thawing ≤ osmotic shock. Interestingly, the structural and biological functions of small EV are dependent on the applied encapsulation process, with the functional properties being altered at a greater extent. Therefore, the importance of including additional characterization parameters to probe alterations of the biological functionality of small EV is clearly demonstrated. Here, freeze‐thawing and particularly the osmotic shock have proven to be the most appropriate methods for EV loading, as they achieve a high drug encapsulation and yet preserve the investigated structural and biological vesicle characteristics.
Keywords: CD73 | wound healing, drug loading, extracellular vesicles, hydrophilic low molecular weight compounds
Small extracellular vesicles have inherent functions in cell‐to‐cell communication, which render them attractive for drug delivery applications. This study investigates the loading of hydrophilic compounds into stem cell‐derived small extracellular vesicles using different membrane permeabilization methods, and emphasizes the importance of a thorough characterization of the intrinsic structural and biological vesicle properties after the encapsulation process.
1. Introduction
Drug delivery platforms can substantially improve the therapeutic index of drugs by enhancing the pharmacological response within disease sites and minimizing off‐target effects.[ 1 , 2 ] Artificial nanocarriers (e.g., liposomes, drug‐polymer conjugates) have demonstrated some clinical success for this purpose, hence making their way into several approved drug formulations (e.g., Doxil, Adagen).[ 2 , 3 ] Nevertheless, synthetic systems often suffer from targeting inefficiency, low cargo transfer into the host cells, and tolerance issues.[ 1 , 4 , 5 ] Fueled by these limitations, small extracellular vesicles (EV) such as exosomes have been proposed as an appealing alternative.[ 1 , 6 ]
Small EV are cell‐secreted vesicles with diameters of no more than 200 nm.[ 7 , 8 ] As intercellular communication messengers, they have intrinsic biological functions and targeting capabilities that make them interesting from a biomedical perspective.[ 7 , 9 , 10 ] Particularly stem cell‐derived small EV possess a broad therapeutic activity spectrum, rendering them suitable for the management of chronic wounds and graft‐versus‐host disease, among other pathologies.[ 11 , 12 ] As demonstrated in rodent and a few human studies, stem cell‐derived EV are poorly immunogenic and not toxic.[ 12 , 13 , 14 ] Moreover, they are believed to be relatively degradation‐resistant and to efficiently traffic their payload to host cells, potentially outperforming artificial nanocarriers in these aspects.[ 4 , 6 , 15 ]
In terms of drug loading into EV, different strategies can be followed.[ 16 ] Hydrophobic and amphiphilic drugs (e.g., paclitaxel, doxorubicin) can be incorporated by simple coincubation with the vesicles, albeit the achieved loading levels are generally low.[ 16 , 17 , 18 ] More hydrophilic and macromolecular drugs (e.g., nucleic acids, enzymes) require a transient permeabilization of the vesicle membrane to be encapsulated, which has been achieved by electroporation, sonication, saponin treatment, freeze‐thawing, and osmotic shock approaches.[ 1 , 16 , 19 ] Electroporation has been applied mostly for nucleic acids,[ 16 ] but has proven inefficient for the incorporation of other types of compounds into EV, such as an intermediately hydrophilic analogue of the photosensitizer porphyrin.[ 20 , 21 , 22 ] Enzymes such as catalase can be encapsulated with reasonable efficiency when exosomes are subjected to saponin treatment, freeze‐thawing, or sonication.[ 23 ] A comparison of the loading methods’ efficiencies between different studies is, however, severely restricted due to inconsistencies in the declaration of drug loading levels (e.g., drug molecules per vesicles,[ 20 ] drug concentration per vesicles,[ 17 ] and total encapsulated drug concentration[ 18 ]), and in the purification protocols used to remove excess drug (e.g., ultracentrifugation,[ 21 ] size exclusion chromatography[ 20 ]). Apart from nucleic acids, mainly classical drugs with a hydrophobic to moderate hydrophilic nature have hitherto been incorporated into EV,[ 1 , 15 ] leaving the capacity of current loading methods to encapsulate highly hydrophilic classical drugs largely unaddressed.
Particularly relevant for the applicability of EV in drug delivery, analyses of their physicochemical features, morphological appearance, and cellular uptake revealed that the loading methods can partially affect the vesicle integrity.[ 19 , 20 , 23 , 24 ] In a study of macrophage‐derived exosomes, sonication induced a significant size increase and re‐shaping of the vesicle structure, while freeze‐thawing led to aggregation.[ 23 ] Treatment with the surfactant saponin, by contrast, preserved the vesicle characteristics,[ 23 ] as independently corroborated by another study applying this method on cancer cell‐derived EV.[ 20 ] Originally described for erythrocyte loading,[ 25 ] hypotonic dialysis has been used for encapsulating intermediately hydrophilic porphyrins into EV.[ 20 ] Although the loading efficiency was reported to be relatively high, the vesicles’ size profile was significantly altered.[ 20 ] Interestingly, the investigated loading methods seemed to not impair the cellular uptake of EV, which was interpreted as indicative of preserved biological functions.[ 20 , 23 ] The latter has in most cases been corroborated by the pharmacological response induced by the encapsulated drug within the target cells.[ 20 , 21 , 23 ] However, little is known about the potential impact of the loading methods on the intrinsic enzymatic and biological activity of the EV themselves.
In this work, several loading methods are investigated with the aim of identifying the optimal one(s) in terms of loading efficiency of hydrophilic compounds, and minimal impact on the integrity and bioactivity of small EV. The pH‐sensing probe pyranine and the phosphodiesterase inhibitor pentoxifylline were selected as model hydrophilic candidates to determine the loading capacity of the investigated encapsulation processes. The impact of the loading methods on the vesicle integrity was subsequently investigated, thereby focusing on the intrinsic biological functions of small EV derived from HPV‐16 E6/E7 transformed human bone marrow mesenchymal stromal cells (HS‐5).
2. Results and Discussion
2.1. HS‐5 Cell‐Derived Small EV
As reported in our previous work,[ 26 ] small EV, including exosomes, were isolated from HS‐5 cells by differential ultracentrifugation and extensively characterized following the recommendations of the International Society for Extracellular Vesicles.[ 27 , 28 ] In brief, nanoparticle tracking analysis (NTA) revealed that the diameters of the isolated vesicles were generally below 200 nm, with the main population having a mode of 101 ± 12 nm (Figure S1A, Supporting Information). Vesicles displayed a characteristic, though artificially created, cup‐shaped appearance when analyzed by transmission electron microscopy (TEM), and expressed broadly accepted EV marker proteins (Figure S1B,C, Supporting Information). Importantly, depletion of the representative contaminant calregulin indicated purity (Figure S1C, Supporting Information).
For subsequent drug loading studies of the obtained HS‐5 cell‐derived small EV, different physical principles were assessed for their efficiency in promoting a temporary destabilization of the vesicle membrane, including liposomes fusion,[ 29 ] freeze‐thawing,[ 29 ] sonication,[ 23 ] osmotic shock,[ 20 , 25 ] and saponin treatment[ 23 ] (Figure 1 ). For the fusion of HS‐5 cell‐derived small EV with drug‐loaded liposomes, the optimal liposomal formulation was first determined.
Figure 1.
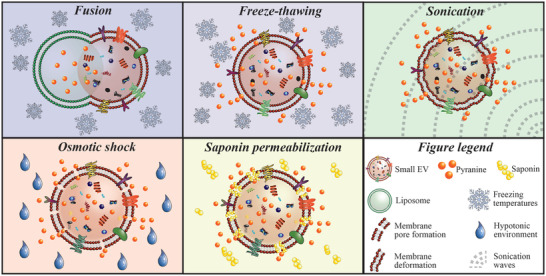
Schematic snapshot view of investigated loading approaches. The hydrophilic low molecular weight fluorophore pyranine was incorporated into small EV by the depicted loading strategies, i.e., fusion with preloaded liposomes, freeze‐thawing, sonication, osmotic shock‐controlled loading, and saponin permeabilization under isotonic conditions.
2.2. HS‐5 Cell‐derived Small EV Efficiently Fuse with Cationic Liposomes to form Hybrid Vesicles
The neutral lipid 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) was selected as base component of the screened liposomal formulations (Figure 2A) as it is abundantly present in HS‐5 exosomes[ 26 ] and commonly used in drug delivery.[ 30 , 31 ] To increase the liposomes’ fusion propensity with the negatively‐charged HS‐5 cell‐derived small EV, the cationic lipid 1,2‐dioleoyl‐3‐trimethylammonium‐propane (DOTAP) was incorporated at varying molar ratios.[ 26 , 32 , 33 ] Phosphoethanolamines (i.e., 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE) and 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[carboxy(polyethylene glycol)‐2000] (DSPE‐PEG)) were additionally incorporated into the liposome formulations to modulate their fusion efficiency.[ 29 , 33 , 34 ] Overall, positively‐charged liposomes with similar modal diameters as native HS‐5 cell‐derived small EV were generated (Figure S2A,B, Supporting Information). Liposomes fusion with these EV was subsequently induced by freeze‐thawing to spare the need for contaminating fusion‐promoting chemicals (e.g., polyethylene glycol or calcium).[ 29 , 33 ] The fusion process was traced by a fluorescence resonance energy transfer (FRET) assay, employing liposomes containing the fluorescent probes nitrobenzoxadiazol (NBD) and rhodamine.[ 29 , 33 , 35 ] Fusion efficiencies of labeled liposomes with unlabeled small EV were detected as an increase of the NBD fluorescence.[ 35 ]
Figure 2.
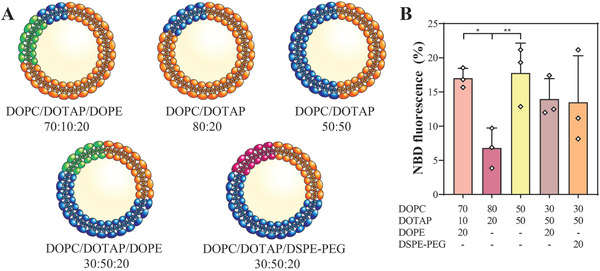
HS‐5 cell‐derived small EV can be fused with cationic liposomes. A) Schematic overview of the different liposomal formulations (in mol%) tested for fusion with HS‐5 cell‐derived small EV. B) Fusion efficiency as a function of the liposome composition. Fusion efficiency is expressed as percentage of the NBD fluorescence (y‐axis). Lipid concentrations are indicated as mol% (x‐axis). Data represent mean + SD, n = 3. Significance was calculated with an ordinary two‐way ANOVA followed by a post‐hoc Tukey's multiple comparison test, * p < 0.05, ** p < 0.01.
Here, it was found that DOPC liposomes fused to a significantly higher extent with HS‐5 cell‐derived small EV at a DOTAP content of 50 mol% compared to 20 mol% (Figure 2B). The fusogenicity of liposomes formulated with 50 mol% DOTAP remained unaffected upon incorporation of either DSPE‐PEG or DOPE at 20 mol% (Figure 2B). Yet in presence of DOPE, the liposomes’ fusion efficiency seemed independent of the DOTAP content (Figure 2B). Interestingly, liposomes containing DOPC/DOTAP/DOPE at a molar ratio of 70:10:20 fused to a significantly higher degree than those composed of DOPC/DOTAP 80:20 mol%, indicating that DOPE may promote fusion at low DOTAP contents.[ 34 ]
Overall, liposomes formulated with 20 mol% DOPE and either 10 or 50 mol% DOTAP had the highest fusion efficiencies, although the latter remained generally modest (< 20%). Since cationic lipids are associated with toxicity issues,[ 36 ] the formulation containing the lowest content thereof (i.e., DOPC/DOTAP/DOPE at 70:10:20 mol%) was used for subsequent drug loading studies.
2.3. Hydrophilic Compounds can be Incorporated into Small EV
To compare different loading methods in terms of their loading capacity for hydrophilic low molecular weight compounds, the fluorophore pyranine (524.4 g mol−1) was selected as model cargo (Figure 3A) as its three negative charges at pH 7.4 render it non‐permeable across lipid bilayers.[ 37 ] To account for potential passive incorporation of pyranine within the vesicle structure, coincubation at isotonic conditions was performed as control. Of note, the pyranine concentration was similar between the different loading methods to ensure comparability. Taking advantage of the strong intrinsic pyranine fluorescence at neutral pH,[ 38 ] a rapid qualitative screening by fluorescence microscopy was first performed. Highest fluorescence signals were observed with the osmotic shock and freeze‐thawing methods, whereas no fluorescence was detected for sonicated and saponin‐treated EV (Figure S3A–F, Supporting Information). Fusion and coincubation control revealed comparable fluorescence signals, which were both below those of the osmotic shock and freeze‐thawing (Figure S3G,H, Supporting Information).
Figure 3.
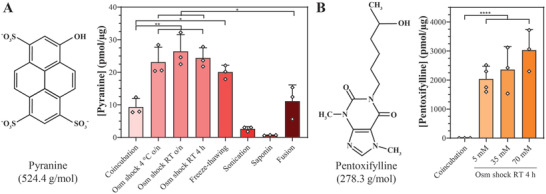
Osmotic shock‐controlled loading achieves highest loading capacities. A) Pyranine loading into HS‐5 cell‐derived small EV. Small EV were loaded with pyranine by osmotic (osm) shock (performed at 4 °C or room temperature (RT), for overnight (o/n) or 4 h), freeze‐thawing, sonication, saponin permeabilization, or fusion with pyranine‐loaded liposomes (DOPC/DOTAP/DOPE 70:10:20 mol%). Coincubation served as loading control. B) Pentoxifylline loading into HS‐5 cell‐derived small EV. Pentoxifylline was incorporated into small EV by a 4‐h osmotic shock at RT at varying drug concentrations. Coincubation, representatively shown for 70 mm pentoxifylline, served as loading control. All data represent mean + SD, n = 3. Significance was calculated with an ordinary two‐way ANOVA followed by a post‐hoc Tukey's multiple comparison test, * p < 0.05, ** p < 0.01, **** p < 0.0001.
Confirming the qualitative results, quantification of encapsulated pyranine levels revealed that permeabilization of the vesicle membrane through osmotic shock encapsulated significantly higher pyranine quantities than simple coincubation, reaching levels around 25 pmol µg−1 independently of the temperature and incubation time (Figure 3A). These results were in line with previous reports of efficient encapsulation of highly water‐soluble glucocorticoid‐analogues and intermediately hydrophilic porphyrin analogues by osmotic shock at room temperature (RT) for 30 min into erythrocytes and at 4 °C for 4 h into EV, respectively.[ 20 , 25 ] The apparent loading capacity reached following coincubation might have resulted from unspecific dye binding to membrane surface structures (e.g., heparan sulfate proteoglycans, surface proteins, or lipid head groups) as pyranine is not permeable across lipid bilayers.[ 37 ] When small EV were subjected to repeated freeze‐thawing, pyranine contents were comparable to those obtained with the osmotic shock, while those after the fusion approach were approximately two‐fold lower (Figure 3A). Sonication and saponin treatment, on the other hand, were ineffective in loading pyranine (Figure 3A), which was contrary to a former study reporting that these two methods performed under similar experimental conditions were efficient in encapsulating the enzyme catalase into exosomes.[ 23 ] Interestingly, pyranine contents following sonication and saponin methods were even below those of the coincubation control (Figure 3A). Possibly, the hydro‐mechanical shear forces induced by sonication disrupted non‐specific pyranine interactions with the vesicle membrane. Likewise, the detergent saponin may have displaced surface‐bound pyranine given its use in immunofluorescence stainings to block non‐specific antibody binding.[ 39 ]
Overall, osmotic shock and freeze‐thawing were identified as the methods with the highest loading capacity, while fusion resulted in intermediate values. Sonication and saponin treatment, on the other hand, failed to load the hydrophilic fluorophore pyranine. The same trends applied to the estimated transfer efficiency of the investigated encapsulation processes (Table S1, Supporting Information).
2.4. Osmotic Shock can Encapsulate the Hydrophilic Model Drug Pentoxifylline
Since the osmotic shock procedure performed at RT for 4 h yielded one of the highest loading capacities, it was representatively selected for encapsulating the hydrophilic model drug pentoxifylline (278.3 g mol−1) (Figure 3B). Unlike pyranine, pentoxifylline did not associate with the vesicles upon coincubation, even when used at significantly higher concentrations than pyranine (70 mm pentoxifylline vs. 5 mm pyranine) (Figure 3A,B). Following osmotic shock, relatively high amounts of pentoxifylline (2000–3000 pmol µg−1 on average) could be incorporated, whereby an increase of the pentoxifylline concentration from 5 to 70 mm produced a non‐significant trend toward higher encapsulated quantities (Figure 3B). Compared to pyranine, a 100‐fold higher concentration of pentoxifylline could be loaded into small EV. We hypothesized that, compared to pyranine, the smaller molecular weight and largely uncharged state of pentoxifylline may have facilitated its diffusion across the vesicle membrane under permeabilizing conditions.
2.5. Loading Methods can Modify the Size and Zeta Potential of Small EV
To track potential alterations of the vesicles’ size and surface charge after being subjected to the loading methods, EV were characterized by NTA. When compared to native HS‐5 cell‐derived small EV, the modal diameters and size distribution profiles remained relatively unchanged irrespective of the loading procedure (Figure 4A; Figure S4A–H, Supporting Information). Only the EV‐liposome hybrid vesicles obtained via the fusion approach displayed a small, but significant increase of the modal diameter compared to native EV (Figure 4A;Figure S4H, Supporting Information), which corresponded to previous findings.[ 29 , 33 ] This size increase was expected as the fusion process of two vesicles should result in a larger hybrid vesicle. Sonication, on the other hand, was the only method with which additional vesicle populations between 200 and 700 nm were clearly noted, albeit in low portions (Figure S4F, Supporting Information). These results were in direct contrast with previous reports showing significant size increases and/or massive vesicle aggregates formation following the different methods, even when performed under similar experimental conditions.[ 20 , 23 , 24 , 29 , 33 ]
Figure 4.
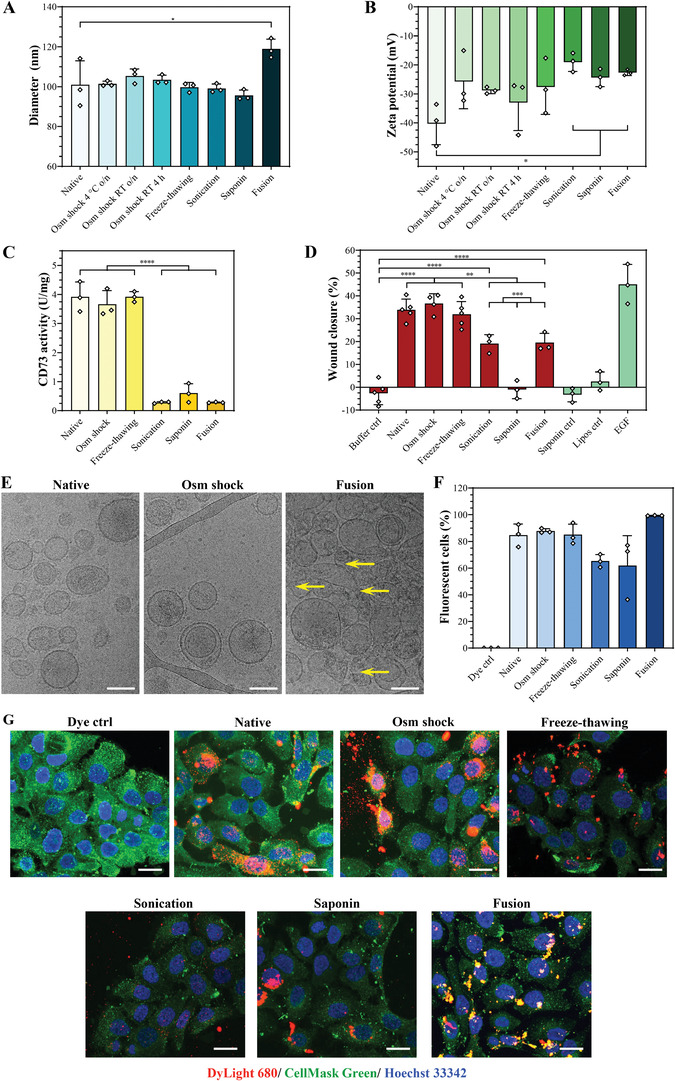
The loading methods can affect the vesicle characteristics. A,B) Modal diameters and zeta potentials of HS‐5 cell‐derived small EV, as characterized by NTA. C) CD73 activity of HS‐5 cell‐derived small EV was determined by a malachite green assay after applying the different loading methods. D) Scratch wound healing assay with HaCaT cells. Cells were treated with small EV (13.5 µg protein) subjected to the different loading procedures, negative controls (buffer, saponin, or liposomes formulated with DOPC/DOTAP/DOPE 70:10:20 mol%), or the positive control epidermal growth factor (EGF, 50 ng mL−1). Wound closure was analyzed after 24 h. E) Morphology of HS‐5 cell‐derived small EV. The vesicles’ native‐like state was visualized by cryo‐TEM. Yellow arrows mark potentially damaged membranes. Representative images are shown, scale bars = 100 nm. F) Cellular uptake studies in HaCaT cells. Cells were treated for 24 h with DyLight 680‐labeled small EV (10 µg protein) after being subjected to the different loading methods, and the percentage of fluorescent cells was determined by flow cytometry. G) Visualization of cellular uptake in HaCaT cells. Confocal images of HaCaT cells that were treated for 24 h with DyLight 680‐labeled small EV (10 µg protein, red) subjected to the indicated loading procedures. The cellular plasma membrane was stained with CellMask Green (green) and nuclei with Hoechst 33342 (blue). Representative images are shown, scale bars = 20 µm. All data represent mean + SD, n = 3. Significance was calculated with an ordinary two‐way ANOVA followed by a post‐hoc Tukey's multiple comparison test, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Regarding the zeta potential, EV subjected to osmotic shock or freeze‐thawing presented similar mean values as native vesicles (Figure 4B), which confirmed previous findings.[ 20 , 24 ] In contrast, fusion with cationic liposomes, sonication, and saponin treatment significantly increased the zeta potential vs. native vesicles (Figure 4B). Regarding the fusion approach, similar trends have been previously shown for the zeta potential of hybrid vesicles when exosomes were fused with cationic liposomes.[ 40 ] Following sonication and saponin treatment, on the other hand, it was formerly reported that the vesicles’ zeta potential remained unchanged,[ 20 , 24 ] a difference that we hypothesized could be due to the specific lipid composition of HS‐5 cell‐derived small EV,[ 26 , 41 ] which could render them more susceptible to alterations of their membrane integrity in comparison to vesicles of other cell sources. Membrane remodeling processes experienced upon sonication and saponin treatment may have led to changes in the surface‐exposed membrane proteins and/or lipids, hence potentially affecting the zeta potential of the EV. This phenomenon was previously presumed to account for an increased cellular uptake of sonicated vs. native exosomes.[ 23 ] It is conceivable that the degree of such remodeling processes might, apart from the membrane lipid composition, also depend on the applied saponin concentration and sonication power, respectively, which differed across studies.[ 20 , 23 , 24 ]
Taken together, freeze‐thawing and osmotic shock methods preserved both the size and zeta potential of HS‐5 cell‐derived small EV. In contrast, fusion with cationic liposomes, sonication, and saponin treatment substantially affected the surface charge of the vesicles, and sonication further clearly induced the formation of larger vesicle and/or aggregate populations.
2.6. Osmotic Shock and Freeze‐Thawing Preserve the Biological Functions of HS‐5 Cell‐Derived Small EV
The impact of the drug loading methods on the EV integrity is primarily studied as a function of the physicochemical properties, morphology, and cellular uptake of the vesicles,[ 20 , 23 ] excluding potential effects on their bioactivity. HS‐5 cell‐derived small EV are particularly useful to monitor the biological functionality post‐loading because they abundantly express the transmembrane enzyme CD73, which can serve as a proxy for the vesicle functionality.[ 26 ] Here, osmotic shock and freeze‐thawing were both found to preserve the CD73 activity of HS‐5 cell‐derived small EV (Figure 4C). Interestingly, sonication, saponin permeabilization, and fusion significantly reduced this enzymatic activity by approximately ten‐fold on average (Figure 4C).
To further assess whether the loading process impacted the vesicles’ biological activity, an in vitro scratch wound healing assay was performed on human immortalized keratinocytes (HaCaT cells[ 42 ]). We previously employed this assay to investigate the efficacy of HS‐5 exosomes and concurrently monitor their CD73 activity.[ 26 ] As with the CD73 activity, HS‐5 cell‐derived small EV subjected to osmotic shock or freeze‐thawing displayed similar wound closure rates as native vesicles (Figure 4D), indicating that their bioactivity was unaffected by these methods. In contrast, sonication and fusion significantly reduced the wound closure rates of HS‐5 cell‐derived EV by half, which might be associated to the near‐complete loss of their CD73 activity.[ 26 ] Remarkably, saponin permeabilization fully abrogated the wound closure‐promoting activity of the EV (Figure 4D), which could be a combination of the ablated CD73 activity and a potential loss of intraluminal bioactive EV components (e.g., nucleic acids, proteins), which are essential for the bioactivity of HS‐5 cell‐derived EV.[ 26 ] Importantly, control treatments with saponin and liposomes used for fusion did not affect the scratch wound closure rate (Figure 4D), confirming the method's suitability for an unbiased tracing of the functions of EV.
These results were subsequently verified by cryo‐TEM, which was representatively performed for the osmotic shock and fusion as their CD73 and wound closure‐promoting activity suggested a preserved and altered integrity, respectively. Indeed, vesicle membranes remained morphologically intact following the osmotic shock procedure, while after fusion the membranes appeared to be damaged (Figure 4E; Figure S5A–C, Supporting Information). Interestingly, only few membranes seemed to be damaged when native EV were subjected to repeated freeze‐thawing cycles (Figure S5D, Supporting Information), suggesting that alterations in the lipid composition of the EV‐liposome hybrid vesicles might reduce their resistance against freeze‐thawing induced stress. Of note, since liposomes subjected to the fusion conditions remained clearly morphologically intact and could be distinguished from EV by their smoother membrane and more transparent vesicle core (Figure S5E, Supporting Information), the damaged membranes observed after the fusion process likely belonged to (un)fused EV. These findings correlated well with the measured activity of the transmembrane enzyme CD73, which would likely lose its activity upon membrane damage.
Overall, from the investigated loading approaches, the osmotic shock and freeze‐thawing methods were able to preserve the morphological integrity as well as the CD73 enzymatic and in vitro wound closure‐accelerating activity of HS‐5 cell‐derived small EV.
2.7. Cellular Uptake of Small EV is Only Minimally Affected by the Loading Procedures
Since changes of the vesicles’ biological functionality and membrane integrity may also affect their cellular uptake, the latter was subsequently investigated in HaCaT cells using DyLight 680‐labeled vesicles. As determined by flow cytometric analysis, osmotic shock and freeze‐thawing had no effect on the cellular uptake of HS‐5 cell‐derived small EV, whereas saponin treatment and sonication non‐significantly diminished it (Figure 4F). The decrease observed with sonicated small EV contradicted a former study reporting an increased uptake efficiency of these exosomes compared to freeze‐thawed and native vesicles.[ 23 ] We hypothesized that the different membrane composition (i.e., lipids and proteins) of EV obtained from varying cell sources (HS‐5 cells in our study vs. macrophages[ 23 ]) may have resulted in the observed differences. Following fusion with cationic liposomes, the uptake of EV‐liposome hybrid vesicles was enhanced by approximately 10% after a 24‐h incubation time (Figure 4F). Confocal imaging confirmed a general uptake of native DyLight 680‐labeled EV and those being subjected to the loading methods by HaCaT cells (Figure 4G). After the osmotic shock, freeze‐thawing, saponin, and fusion methods, the vesicles' location appeared as smaller or larger red dots distributed throughout the whole cell, whereas only weak fluorescent signals of sonicated vesicles could be observed (Figure 4G). Interestingly, internalization of EV‐liposome hybrid vesicles seemed to be higher compared to native small EV and those after the other loading approaches (Figure 4G). Similar results have been obtained in a previous work when exosomes were modified with cationic lipids.[ 43 ] Whether this observation may result from a different uptake route and/or intracellular trafficking of the EV‐liposome hybrid vesicles remains to be determined. Moreover, given the fact that cationic lipids have been reported to show a concentration‐dependent toxicity and enhanced uptake by the mononuclear phagocyte system,[ 36 , 44 ] the safety as well as the pharmacokinetic and biodistribution profiles of the EV‐liposome hybrid vesicles have to be examined in future in vivo studies.
Taken together, the investigated loading methods only minimally impacted the extent of the cellular uptake of small EV, suggesting that this parameter by itself might not accurately reflect potential modifications of the intrinsic vesicle functions.
3. Conclusion
In this study, different loading methods (i.e., fusion with liposomes, freeze‐thawing, sonication, osmotic shock, and saponin treatment) were investigated for their capacity to encapsulate hydrophilic low molecular weight compounds into HS‐5 cell‐derived small EV. Moreover, their impact on both the physicochemical properties and intrinsic bioactivity of HS‐5 cell‐derived small EV was monitored.
Freeze‐thawing and particularly osmotic shock proved to be the only methods able to encapsulate hydrophilic low molecular weight probes (i.e., the fluorophore pyranine and the drug pentoxifylline), while fully preserving the vesicle functions. Moreover, the importance of thoroughly characterizing the vesicle functions post‐loading was demonstrated, not only in terms of their physicochemical characteristics and cellular uptake, but particularly their intrinsic biological activity. Remarkably, the latter was found to be a critical parameter allowing to probe alterations of the EV functions that cannot be unambiguously captured by physicochemical characterization tools and cellular uptake studies. We strongly believe that future biomedical investigations may benefit from our results as they provide first insights on the loading capacity of certain methods for hydrophilic drugs and on the impact of the loading step on the intrinsic bioactivity of EV.
4. Experimental Section
Small EV Preparation and Characterization
Small EV were isolated from the HPV‐16 E6/E7 transformed human bone marrow mesenchymal stromal cell line HS‐5 (ATCC, Manassas, VA, USA) as previously described.[ 26 ] Prior EV production, cells were tested for mycoplasma using the MycoAlert Mycoplasma Detection kit (Lonza Group AG, Basel, Switzerland). Briefly, 27 × 105 cells cm−2 were seeded in 15 mL complete growth medium (Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 100 U mL−1 penicillin, 100 µg mL−1 streptomycin, and 10% v/v fetal bovine serum (FBS) (all from Thermo Fisher Scientific, Waltham, MA, USA)) and grown for 40 h. For EV production, cells were washed twice with phosphate‐buffered saline (PBS, Thermo Fisher Scientific) and cultured in 18 mL fresh serum‐free DMEM for 48 h. The supernatant (i.e., conditioned medium (CM)) was clarified by centrifugation at 4 °C and 2000 × g for 5 min, followed by 10 000 × g for 15 min, and subsequent 0.22‐µm filtration. Small EV were then enriched from 396 mL of the obtained clarified CM by ultracentrifugation at 4 °C and 100 000 × g for 70 min with an Optima XE‐90 ultracentrifuge equipped with a Type 45 Ti Fixed‐Angle Titanium Rotor (Beckman Coulter Life Sciences, Indianapolis, IN, USA). Small EV pellets were resuspended in PBS, pooled, and re‐centrifuged under identical conditions. The purified small EV pellet was then resuspended in either 200 µL PBS or, for CD73 activity measurements, HEPES‐buffered saline (20 mm HEPES (Thermo Fisher Scientific), 150 mm sodium chloride (Sigma Aldrich, St. Louis, MO, USA), pH 7.4) and stored at −20 °C. The vesicles’ size profile was determined by NTA (see section NTA). For analyzing the vesicle morphology, small EV were fixed with 1% v/v glutaraldehyde (Sigma Aldrich) on carbon‐coated grids (Quantifoil Micro Tools GmbH, Grosslöbichau, Germany), stained with uranyl oxalate (Sigma Aldrich), and visualized with a FEI Morgagni 268 microscope (Field Electron and Ion Company, Hillsboro, OR, USA) at 100 kV.[ 45 ] EV protein and contamination markers were detected by Western blot analysis.[ 46 , 47 ] Briefly, after HS‐5 cell and EV lysis, proteins were separated on 12% sodium dodecyl sulfate polyacrylamide gels and subsequently transferred on Immun‐Blot PVDF membranes (Bio‐Rad Laboratories, Hercules, CA, USA). The membrane was blocked in 5% w/v skim milk in Tris buffered saline containing 1% v/v polysorbate 20 (Sigma Aldrich), followed by incubation with CD73, CD63, CD9, TSG101, calregulin, and GAPDH primary antibodies (Santa Cruz Biotechnology Inc., Dallas, TX, USA), and secondary HRP‐conjugated antibody (Dako Denmark A/S, Glostrup, Denmark). Protein bands were visualized with Western Blotting Luminol Reagent (Santa Cruz Biotechnology Inc.), and developed on Fuji medical X‐ray films (FUJIFILM Europe GmbH, Düsseldorf, Germany).
Liposome Preparation
Liposomes composed of varying molar concentrations of DOPC, DOTAP, DOPE, and DSPE‐PEG (PEG M w 2000 g mol−1) (all from Avanti Polar Lipids, Alabaster, AL, USA) were prepared by the thin‐film hydration method.[ 48 ] Lipids dissolved in chloroform ReagentPlus (Sigma Aldrich) were mixed at the corresponding molar ratios indicated in the Results and Discussion section, and dried in vacuo for overnight to form a thin lipid film. Liposomes at a final lipid concentration of 2 mm were obtained by rehydration in PBS with or without 25 mm pyranine (Thermo Fisher Scientific) at 55 °C and subsequent extrusion through two‐stacked 100‐nm pore‐sized polycarbonate filter membranes at 55 °C. Liposomes were purified from non‐encapsulated pyranine by size exclusion chromatography using PD MidiTrap columns with a Sephadex G‐25 resin (GE Healthcare, Chicago, IL, USA) and subsequently stored at 4 °C.
Fusion Efficiency
The fusion efficiency between small EV and liposomes containing 1.5 mol% each of NBD‐ and rhodamine‐labeled 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphoethanolamine (Avanti Polar Lipids) was evaluated by a FRET assay as previously described.[ 29 , 35 ] Small EV were fused with liposomes by freeze‐thawing (see section Loading procedures) and the NBD fluorescence was monitored at an excitation and emission wavelength of 470 and 530 nm, respectively, using a Tecan Infinite M200 plate reader (Tecan Group Ltd., Männedorf, Switzerland). The percentage of the NBD fluorescence, indicative of the fusion efficiency, was calculated as ratio of the NBD fluorescence of the samples and the NBD fluorescence obtained upon lysing the vesicles with 2.5% w/v n‐dodecyl‐β‐D‐maltoside (Sigma Aldrich).[ 35 ] The percentage of the NBD fluorescence obtained for small EV and liposomes incubated under non‐fusion conditions served as control to account for spontaneous dye transfer, and was subtracted from the value yielded under fusion conditions.
Loading Procedures
Pyranine encapsulation into HS‐5 cell‐derived small EV was achieved by either sonication,[ 23 ] saponin permeabilization under isotonic conditions,[ 23 ] freeze‐thawing,[ 29 ] osmotic shock induction,[ 20 , 25 ] or fusion with pre‐loaded liposomes. Coincubation[ 20 ] served as a loading control. Unless indicated otherwise, 20 µg EV proteins and 5 mm pyranine were mixed in a sample volume of 100 µL and subjected to the different drug loading procedures.
For control loading, samples were incubated under isotonic conditions at RT for 4 h. For saponin permeabilization, 2 mg mL−1 saponin (Sigma Aldrich) was added to the EV‐pyranine mixture and incubated at RT for 20 min under agitation. In case of freeze‐thawing, the EV‐pyranine mixture was subjected to ten cycles of freezing in liquid nitrogen and thawing in a water bath at 30 °C. For sonication, the EV‐pyranine mixture was cooled on an ice bath and sonicated for 24 s at a 20% amplitude (700 W, 50/60 Hz, 6 cycles of 4 s pulse and 2 s pause) using a FB‐705 Sonic Dismembrator (Fisher Scientific, Reinach, Switzerland). After a 2‐min cooling period on ice, the sample was re‐sonicated under identical conditions. In case of the osmotic shock procedure, EV were loaded in 3.5 kDa molecular weight cut‐off dialysis units (Thermo Fisher Scientific) and first dialyzed against 2 mL water at RT for 3 h. Then, pyranine was carefully added to the hypotonic EV and the mixture was dialyzed against 1 mL water containing 5 mm pyranine for either 4 h at RT or 18 h at RT or 18 h at 4 °C. EV were recovered by diluting the mixture in hypertonic buffer to reach a final osmolality of 300 mOsmol kg−1 and dialyzed against an isotonic buffer at RT for 2.5 h. In case of fusion with pyranine‐loaded liposomes, 20 µg EV proteins and 100 µm liposomes were subjected to ten freeze‐thawing cycles and non‐fused liposomes were removed by cation exchange chromatography using Dowex Monosphere 650C resin (Merck KGaA, Darmstadt, Germany). To account for pyranine‐loaded liposomes that could not be removed from fused vesicles, 100 µm pyranine‐loaded liposomes were subjected to ten freeze‐thawing cycles and purified by cation exchange chromatography as control. Control pyranine concentrations were subsequently subtracted from the ones obtained for fused vesicles.
HS‐5 cell‐derived small EV were additionally loaded with the hydrophilic drug pentoxifylline (Sigma Aldrich) at varying concentrations (5, 35 or 70 mm) via the osmotic shock procedure for 4 h at RT. Coincubation served as a loading control, and was performed with 5 and 70 mm pentoxifylline.
For all loading procedures, except the fusion approach, free compound was removed by size exclusion chromatography using PD MidiTrap columns with a Sephadex G‐25 resin.
Quantification of Loading Levels
Lyophilized small EV loaded with either pyranine or pentoxifylline were resuspended in 65 µL 4 m hydrochloric acid (Sigma Aldrich) and incubated at 95 °C for 1 h under agitation to lyze the vesicles. For neutralization, 65 µL 4 m sodium hydroxide (Sigma Aldrich) was added and re‐incubated at 95 °C for 30 min under agitation. Following centrifugation at 10 000 × g for 5 min, 60 µL aliquots were analyzed in a randomized order on a XBridge BEH C18 column (Waters Corporation, Milford, MA, USA) using a VWR‐Hitachi Chromaster HPLC (VWR International LLC, Radnor, PA, USA) as follows.
For pyranine quantification, samples were separated by a 10 mm glycine and 2 mm tetrabutyl ammonium hydroxide (Sigma Aldrich) in water pH 10 (solvent A) and acetonitrile (solvent B, Sigma Aldrich) gradient (from 20% to 60% B in 5 min, 60% B for 1 min, 50% B in 1 min and for 5 min, 20% B in 1 min and for 7 min) at 35 °C and a flow rate of 0.7 mL min−1. Pyranine was detected at a 454 nm excitation and 511 nm emission wavelength using the fluorescence system detector (VWR International LLC) and quantified based on a pyranine calibration curve ranging from 0.4 to 50 nm.
For pentoxifylline quantification, samples were separated by a 10 mm sodium acetate (VWR International LLC) in water pH 4.0 (solvent A) and acetonitrile (solvent B) gradient (10% B for 3 min, 30% B in 3 min and for 7 min, 50% B in 1 min and for 3 min, and 10% B in 2 min and for 6 min) at 30 °C and a flow rate of 0.8 mL min−1. Pentoxifylline was detected at 275 nm using the UV absorbance system detector (VWR International LLC) and quantified based on a pentoxifylline calibration curve ranging from 4 to 90 μm.
The loading capacity was calculated as pmol compound per µg EV protein. To obtain the estimated transfer efficiency, the encapsulated compound concentration was calculated assuming the vesicles as a perfect water‐filled sphere with a mean diameter of 101 nm, and divided by the initial compound concentration. A 100% transfer efficiency would correspond to an internal EV compound concentration being equivalent to the initial one in the bulk solution.
NTA
Size, concentration and zeta potential of small EV and liposomes were analyzed with a ZetaView PMX 120‐Z instrument equipped with a CMOS camera and a 405‐nm laser source (Particle Metrix GmbH, Meerbusch, Germany). Samples were diluted to 107–108 vesicles mL−1 (50–200 vesicles per frame), and measured in a randomized order with a sensitivity of 85, shutter of 150 and frame rate of 30 s−1 at 11 positions with two readings per position. Zeta potential measurements were performed in the pulsed mode applying similar acquisition settings. Data were analyzed with the ZetaView software package (version 8.05.12 SP1, Particle Metrix GmbH), applying the instrument's default post‐acquisition parameter settings with the width of the bin class set to 5 nm.
Protein Quantification
EV protein contents post‐loading were quantified by the Micro BCA assay following the manufacturer's instructions (Thermo Fisher Scientific). Briefly, 80 µL of the EV suspension were diluted with nanopure water ad 150 µL and incubated with 150 µL Micro BCA reagent at 37 °C for 2 h. The absorbance at 562 nm was measured using a Tecan Infinite M200 plate reader.
CD73 Enzymatic Assay
The CD73 activity of small EV was determined as reported before.[ 26 ] In brief, 0.1 µg EV protein and 24 µmol adenosine‐5′‐monophosphate (Acros Organics, Geel, Belgium) were incubated in a reaction volume of 60 µL at RT for 10 min. Following incubation with 40 µL color reagent (0.034% w/v malachite green (Bender & Hobein GmbH, Munich, Germany), 1.55% w/v ammonium molybdate tetrahydrate (abcr GmbH, Karlsruhe, Germany), 0.0625% v/v polysorbate 20 (Sigma Aldrich)) at RT for 1 h, the absorbance at 620 nm was measured with a Tecan Infinite M200 plate reader.
Cryo‐TEM
Small EV (3 µL) were added on 300‐mesh lacey carbon‐coated copper grids (Quantifoil Micro Tools GmbH) previously glow‐discharged for 30 s in an Emitech K100X glow discharge device (Quorum Technologies Ltd., Lewes, UK), and vitrified in liquid ethane/propane using a Vitrobot Mark II (Field Electron and Ion Company). Micrographs were subsequently recorded with a Tecnai F20 Cryo transmission electron microscope equipped with a Falcon II 4K Direct Electron Detector (Field Electron and Ion Company). The cryo‐microscope was operated at −180 °C in the bright field mode at a 200 kV acceleration voltage and <500 electrons nm−2.
In vitro Scratch Wound Healing Assay
The scratch wound healing assay was performed as described previously.[ 26 ] In brief, human immortalized keratinocytes (HaCaT cells,[ 42 ] kindly provided by Dr. P. Boukamp (Leibniz Institute for Environmental Medicine, Düsseldorf, Germany)) were seeded in a 48‐well plate (TPP Techno Plastic Products AG, Trasadingen, Switzerland) at a density of 105 000 cells per well in DMEM supplemented with 100 U mL−1 penicillin, 100 µg mL−1 streptomycin, and 10% v/v FBS. When confluency was reached, a sterile 200 µL pipette tip was used to scrape a cross‐shaped wound into the cell monolayer. Cells were then washed with PBS and treated with complete growth medium containing 13.5 µg HS‐5 cell‐derived small EV proteins previously subjected to the investigated drug loading methods, or negative controls. Directly after scratching and after a 24‐h stimulation time, the wound was imaged at a 2.5× magnification using a Leica DMI6000 B epifluorescence microscope (Leica Microsystems, Wetzlar, Germany). The wound closure rate was analyzed randomly in a blinded fashion with FIJI software[ 49 ] and normalized to medium controls. HaCaT cells were regularly tested for mycoplasma using the MycoAlert Mycoplasma Detection kit (Lonza Group AG).
Labeling of HS‐5 Cell‐Derived Small EV
For uptake studies and confocal microscopy measurements, small EV were labeled with the near‐infrared dye DyLightTM 680 N‐succinimidyl (NHS) ester, mainly adhering to the manufacturer's instructions (Thermo Fisher Scientific). Briefly, small EV and dye (20:1 w/w, whereby the EV quantity refers to µg of EV proteins) were incubated for 2 h at RT, followed by 2 h at 4 °C. Excess dye was subsequently removed by ultracentrifugation at 4 °C and 100 000 × g for 70 min, and the labeled small EV pellet was resuspended in 150 µL PBS. In parallel, an equal dye quantity was purified in a similar way and used as dye control to assess the purification efficiency.
Flow Cytometry
HaCaT cells were seeded at 80 000 cells per well in a 24‐well plate (TPP Techno Plastic Products AG) and grown over 24 h. Cells were then incubated with 10 µg DyLight 680‐labeled HS‐5 cell‐derived small EV proteins previously subjected to the investigated drug loading methods, or controls for 24 h. For flow cytometry measurements, cells were detached, centrifuged at 4 °C and 300 × g for 10 min and resuspended in 100–150 µL ice‐cold PBS containing 2 mm ethylenediaminetetraacetic acid (Sigma Aldrich) and 0.5% w/v bovine serum albumin (Sigma Aldrich). Data of 10 000 cells per sample were subsequently acquired with a CytoFLEX S flow cytometer (Beckman Coulter Life Sciences) and analyzed with FlowJo software (BD Biosciences, Franklin Lakes, NJ, USA).
Confocal Microscopy
In 24‐well plates, HaCaT cells were seeded on sterile 12 mm round coverslips (VWR International LLC) at a density of 80 000 cells per well, grown over 24 h and treated with 10 µg DyLight 680‐labeled HS‐5 cell‐derived small EV proteins, either native or after the investigated drug loading methods, or dye control for 24 h. After PBS‐washing, cell plasma membranes were stained with CellMaskTM Green according to the manufacturer's instructions (Thermo Fisher Scientific). Cells were subsequently fixed with 4% v/v PBS‐buffered paraformaldehyde (Sigma Aldrich) for 15 min at RT, washed with PBS and counter‐stained with Hoechst 33342 (Sigma Aldrich). Coverslips were subsequently mounted in ProlongTM Diamond Antifade Mountant (Thermo Fisher Scientific) and imaged on a Leica TCS SP8 STED 3× confocal microscope (Leica Microsystems).
Statistical Analysis
Data represent mean + SD, n = 3, unless stated otherwise. Assuming a normal distribution, significance between groups was calculated with an ordinary two‐way ANOVA followed by a post‐hoc Tukey's multiple comparison test (α = 0.05, p‐value calculated) using GraphPad Prism software (version 8.2.0, GraphPad Software Inc., San Diego, CA, USA).
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
J.‐C.L. and B.F.H. designed the overall study and wrote the manuscript. B.F.H. performed most experiments and analyzed all data. J.J.B. was involved in the EV production and cellular uptake measurements.
Supporting information
Supporting Information
Acknowledgements
The authors acknowledge Stephan Handschin (ScopeM, ETH Zurich, Switzerland) for performing the cryo‐TEM measurements, and Justine Kusch‐Wieser (ScopeM, ETH Zurich, Switzerland) for performing the confocal imaging studies. The authors further gratefully acknowledge Dr. Jong‐Ah Kim for her careful proof‐reading of the manuscript. Lastly, the authors thank the ETH research grant (ETH‐10 16‐1) and the Phospholipid Research Center (Heidelberg, Germany; JCL‐2018‐065/2‐1) for generous funding.
Hettich B. F., Bader J. J., Leroux J.‐C., Encapsulation of Hydrophilic Compounds in Small Extracellular Vesicles: Loading Capacity and Impact on Vesicle Functions. Adv. Healthcare Mater. 2022, 11, 2100047. 10.1002/adhm.202100047
Data Availability Statement
Research data are not shared.
References
- 1. Elsharkasy O. M., Nordin J. Z., Hagey D. W., de Jong O. G., Schiffelers R. M., Andaloussi S. E., Vader P., Adv. Drug Deliv. Rev. 2020, 159, 332. [DOI] [PubMed] [Google Scholar]
- 2. Patra J. K., Das G., Fraceto L. F., Campos E. V. R., d. P. Rodriguez‐Torres M., Acosta‐Torres L. S., Diaz‐Torres L. A., Grillo R., Swamy M. K., Sharma S., Habtemariam S., Shin H.‐S., J. Nanobiotechnol. 2018, 16, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bisso S., Leroux J. C., Int. J. Pharm. 2020, 578, 119098. [DOI] [PubMed] [Google Scholar]
- 4. Batrakova E. V., Kim M. S., J. Control. Release 2015, 219, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Armstrong J. P., Stevens M. M., Adv. Drug Deliv. Rev. 2018, 130, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang M., Zang X., Wang M., Li Z., Qiao M., Hu H., Chen D., J. Mater. Chem. B 2019, 7, 2421. [DOI] [PubMed] [Google Scholar]
- 7. Colombo M., Raposo G., Théry C., Annu. Rev. Cell Dev. Biol. 2014, 30, 255. [DOI] [PubMed] [Google Scholar]
- 8. Théry C., Zitvogel L., Amigorena S., Nat. Rev. Immunol. 2002, 2, 569. [DOI] [PubMed] [Google Scholar]
- 9. Ferguson S. W., Nguyen J., J. Control. Release 2016, 228, 179. [DOI] [PubMed] [Google Scholar]
- 10. Raposo G., Stoorvogel W., J. Cell Biol. 2013, 200, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Phinney D. G., Pittenger M. F., Stem Cells 2017, 35, 851. [DOI] [PubMed] [Google Scholar]
- 12. Andrzejewska A., Lukomska B., Janowski M., Stem Cells 2019, 37, 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. György B., Hung M. E., Breakefield X. O., Leonard J. N., Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kordelas L., Rebmann V., Ludwig A., Radtke S., Ruesing J., Doeppner T., Epple M., Horn P., Beelen D., Giebel B., Leukemia 2014, 28, 970. [DOI] [PubMed] [Google Scholar]
- 15. Armstrong J. P. K., Holme M. N., Stevens M. M., ACS Nano 2017, 11, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Donoso‐Quezada J., Ayala‐Mar S., González‐Valdez J., Crit. Rev. Biotechnol. 2020, 40, 804. [DOI] [PubMed] [Google Scholar]
- 17. Smyth T., Kullberg M., Malik N., Smith‐Jones P., Graner M. W., Anchordoquy T. J., J. Control. Release 2015, 199, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saari H., Lázaro‐Ibáñez E., Viitala T., Vuorimaa‐Laukkanen E., Siljander P., Yliperttula M., J. Control. Release 2015, 220, 727. [DOI] [PubMed] [Google Scholar]
- 19. Wang J., Chen D., Ho E. A., J. Control. Release 2020, 329, 894. [DOI] [PubMed] [Google Scholar]
- 20. Fuhrmann G., Serio A., Mazo M., Nair R., Stevens M. M., J. Control. Release 2015, 205, 35. [DOI] [PubMed] [Google Scholar]
- 21. Alvarez‐Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. J., Nat. Biotechnol. 2011, 29, 341. [DOI] [PubMed] [Google Scholar]
- 22. Kooijmans S. A. A., Stremersch S., Braeckmans K., de Smedt S. C., Hendrix A., Wood M. J. A., Schiffelers R. M., Raemdonck K., Vader P., J. Control. Release 2013, 172, 229. [DOI] [PubMed] [Google Scholar]
- 23. Haney M. J., Klyachko N. L., Zhao Y., Gupta R., Plotnikova E. G., He Z., Patel T., Piroyan A., Sokolsky M., Kabanov A. V., J. Control. Release 2015, 207, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim M. S., Haney M. J., Zhao Y., Mahajan V., Deygen I., Klyachko N. L., Inskoe E., Piroyan A., Sokolsky M., Okolie O., Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Magnani M., Rossi L., D'ascenzo M., Panzani I., Bigi L., Zanella A., Biotechnol. Appl. Biochem. 1998, 28, 1. [PubMed] [Google Scholar]
- 26. Hettich B. F., Ben‐Yehuda Greenwald M., Werner S., Leroux J.‐C., Adv. Sci. 2020, 7, 2002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lötvall J., Hill A. F., Hochberg F., Buzás E. I., Di Vizio D., Gardiner C., Gho Y. S., Kurochkin I. V., Mathivanan S., Quesenberry P., Sahoo S., Tahara H., Wauben M. H., Witwer K. W., Théry C., J. Extracell. Vesicles 2014, 3, 26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Théry C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin‐Smith G. K., J. Extracell. Vesicles 2018, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato Y. T., Umezaki K., Sawada S., Mukai S.‐a., Sasaki Y., Harada N., Shiku H., Akiyoshi K., Sci. Rep. 2016, 6, 21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu M., Zhao X., Xing H., Xun Z., Zhu S., Lang L., Yang T., Cai C., Wang D., Ding P., Int. J. Pharm. 2018, 550, 100. [DOI] [PubMed] [Google Scholar]
- 31. Briuglia M.‐L., Rotella C., McFarlane A., Lamprou D. A., Drug Delivery Transl. Res. 2015, 5, 231. [DOI] [PubMed] [Google Scholar]
- 32. Kolašinac R., Kleusch C., Braun T., Merkel R., Csiszár A., Int. J. Mol. Sci. 2018, 19, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Piffoux M., Silva A. K. A., Wilhelm C., Gazeau F., Tareste D., ACS Nano 2018, 12, 6830. [DOI] [PubMed] [Google Scholar]
- 34. Du Z., Munye M. M., Tagalakis A. D., Manunta M. D. I., Hart S. L., Sci. Rep. 2014, 4, 7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parlati F., Weber T., McNew J. A., Westermann B., Söllner T. H., Rothman J. E., Proc. Natl. Acad. Sci. USA 1999, 96, 12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lv H., Zhang S., Wang B., Cui S., Yan J., J. Controlled Release 2006, 114, 100. [DOI] [PubMed] [Google Scholar]
- 37. Straubinger R. M., Papahadjopoulos D., Hong K., J. Biochem. 1990, 29, 4929. [DOI] [PubMed] [Google Scholar]
- 38. Overly C. C., Lee K.‐D., Berthiaume E., Hollenbeck P. J., Proc. Natl. Acad. Sci. USA 1995, 92, 3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donaldson J. G., Curr. Protoc. Cell Biol. 2015, 69, 4.4.1. [DOI] [PubMed] [Google Scholar]
- 40. Yang Y., Shen G., Wang H., Li H., Zhang T., Tao N., Ding X., Yu H., Proc. Natl. Acad. Sci. USA 2018, 115, 10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Skotland T., Sagini K., Sandvig K., Llorente A., Adv. Drug Deliv. Rev. 2020, 159, 308. [DOI] [PubMed] [Google Scholar]
- 42. Boukamp P., Petrussevska R. T., Breitkreutz D., Hornung J., Markham A., Fusenig N. E., J. Cell Biol. 1988, 106, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nakase I., Futaki S., Sci. Rep. 2015, 5, 10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Knudsen K. B., Northeved H., Kumar Ek P., Permin A., Gjetting T., Andresen T. L., Larsen S., Wegener K. M., Lykkesfeldt J., Jantzen K., Loft S., Møller P., Roursgaard M., Nanomed. Nanotechnol. Biol. Med. 2015, 11, 467. [DOI] [PubMed] [Google Scholar]
- 45. Théry C., Amigorena S., Raposo G., Clayton A., Curr. Protoc. Cell Biol. 2006, 30, 3.22. 1. [DOI] [PubMed] [Google Scholar]
- 46. Laemmli U. K., Nature 1970, 227, 680. [DOI] [PubMed] [Google Scholar]
- 47. Towbin H., Staehelin T., Gordon J., Proc. Natl. Acad. Sci. USA 1979, 76, 4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hope M., Bally M., Webb G., Cullis P., Biochim. Biophys. Acta, Biomembr. 1985, 812, 55. [DOI] [PubMed] [Google Scholar]
- 49. Schindelin J., Arganda‐Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Nat. Methods 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.