

Abstract
Effective and accessible treatments for Alzheimer's disease (AD) are urgently needed. Soluble Aβ oligomers are identified as neurotoxic species in AD and targeted in antibody‐based drug development to mitigate cognitive decline. However, controversy exists concerning their efficacy and safety. In this study, an alternative strategy is proposed to inhibit the formation of Aβ oligomers by selectively oxidizing specific amino acids in the Aβ sequence, thereby preventing its aggregation. Targeted oxidation is achieved using biocompatible and blood‐brain barrier‐permeable multicomponent nanoscintillators that generate singlet oxygen upon X‐ray interaction. Surface‐modified scintillators interact selectively with Aβ and, upon X‐ray irradiation, inhibit the formation of neurotoxic aggregates both in vitro and in vivo. Feeding transgenic Caenorhabditis elegans expressing human Aβ with the nanoscintillators and subsequent irradiation with soft X‐ray reduces Aβ oligomer levels, extends lifespan, and restores memory and behavioral deficits. These findings support the potential of X‐ray‐based therapy for AD and warrant further development.
Keywords: Aβ amyloids, Alzheimer's disease, hybrid materials, nanoscintillators, singlet oxygen, X‐rays
It proposes to inhibit formation of neurotoxic Aβ oligomers in Alzheimer's disease through targeted oxidation of specific amino acids by reactive singlet oxygen produced locally by surface‐modified nanoscintillators following X‐ray irradiation. The nanoscintillators interact selectively with Aβ and upon irradiation inhibit neurotoxic Aβ aggregation both in vitro and in vivo.
1. Introduction
Aggregation of misfolded proteins is the fundamental cause of many degenerative conditions including Alzheimer's and Parkinson's diseases (AD and PD) as well as type II diabetes.[ 1 ] Misfolded proteins are usually inactive, however, the dynamic equilibrium between monomeric and oligomeric aggregates may lead to cytotoxic states that induce degeneration.[ 2 ] In AD, amyloid‐β (Aβ) and tau proteins misfold and form oligomers and fibrils that accumulate in the brain in pathogenic pathways leading to synaptic loss and selective neuronal death (Figure 1a).[ 1 ]
Figure 1.
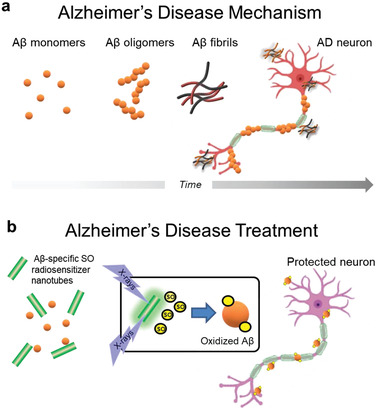
Proposed principle of X‐ray mediated treatment of Alzheimer's disease (AD). a) Amyloid‐β (Aβ) misfolds and forms soluble toxic oligomers and fibrils that accumulate in the brain leading to synaptic loss and selective neuronal death. b) X‐ray irradiation of Aβ‐specific radiosensitizing nanotubes generates singlet oxygen (SO) that selectively oxidizes Aβ to inhibit aggregation and neurotoxicity, thereby preventing AD symptoms.
Rich in cross β‐sheet conformations, soluble oligomers of Aβ are suspected to be more deleterious than fibrils and plaques and appear prior to tau accumulation and the clinical symptoms.[ 2 ] Therefore, targeting soluble Aβ oligomers has emerged as a promising approach for developing AD‐specific drugs to prevent cognitive decline. Translation from mouse to man has been demonstrated with antibody‐based drugs,[ 3 ] and the monoclonal antibodies Aducanumab and Lecanemab were recently approved for early AD treatment by the Food and Drug Administration.[ 4 ] Controversy over efficacy and side effects, such as micro‐hemorrhage and brain swelling highlight the urgent need for improved therapies and novel diagnostic tools. As an alternative and noninvasive therapy, singlet oxygen (SO) generated by suitable photosensitizers and visible light has shown inhibitory effects on Aβ aggregation and toxicity achieving a peculiar photodynamic therapeutic effect.[ 5 ] However, this strategy is largely impractical for AD treatment due to limited penetration of UV–vis photons through deep tissues and the skull.
Here, we present a revised strategy to overcome this limitation by using a combination of multicomponent hybrid scintillating nanotubes (NTs) and low doses of tissue‐penetrating X‐rays (Figure 1b). The NTs are functionalized with a SO photosensitizer that preferentially targets and interacts with Aβ oligomers, along with polyethylene glycol (PEG) molecules to increase water solubility and colloidal dispersion by reducing the NT's aggregation and minimize nonspecific interactions with other biomolecules. The dense NTs facilitate localized deposition of the ionizing radiation energy and activation of the photosensitizers, boosting the production of SO in the deep brain in close proximity to Aβ thus significantly reducing their aggregation.
2. Results and Discussion
2.1. Design and Synthesis of Multicomponent Scintillating NTs as SO Radiosensitizers
Hydrated magnesium silicate (Mg3(Si2O5)(OH)4) nanotubes (NTs) of ≈100 nm height and 50 nm diameter were chosen as scintillating dense nanomaterial to carry the SO sensitizers (Figure 2a; Figure S1a, Supporting Information).[ 6 ] Prepared hydrothermally, these biocompatible NTs are taken up by cells and can penetrate the blood–brain barrier in pathological conditions.[ 6 ] The X‐ray diffractometric (XRD) spectrum (Figure S1b, Supporting Information) shows that the NTs comprise alternating and wrapped layers of silica and brucite, giving rise to the tubular arrangement. The external surface of NTs is brucitic with positive ζ‐potential, enabling its modification by ionic self‐assembly (ISA) in an aqueous environment. This approach utilizes Coulombic attraction between positively charged NTs and negatively charged components, providing a simple bottom‐up approach with broad applicability.[ 7 ]
Figure 2.
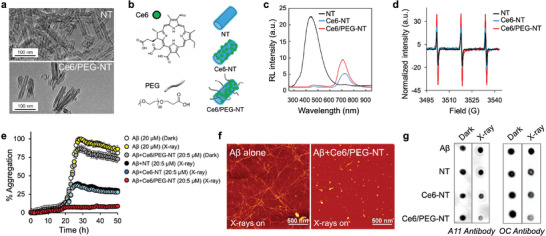
Multicomponent scintillating nanotubes (NTs) for singlet oxygen (SO) radiosensitization and inhibition of Aβ aggregation. a) TEM images of bare and functionalized Ce6/PEG‐NT scintillators. b) Molecular structures of Ce6 and the PEG ligand with sketches of single and double decorated NTs. c) Scintillation spectra of NT, Ce6‐NT and Ce6/PEG‐NT powders under soft X‐ray excitation (2 Gy). d) Comparative EPR spectra of NT (5 µm) and TEMP (40 µm) dispersions in PBS/D2O (1:9) after irradiation. e) Effect of irradiated NTs on Aβ40 aggregation using Thioflavin T (ThT) assay. Monomeric Aβ40 (20 µm) was incubated in absence or presence of various NTs (5 µm) in PBS (50 mm, pH7.2) with and without exposure to X‐rays (100% aggregation is taken as ThT fluorescence of Aβ in dark). Data are mean ± SD of experiments carried out in triplicate and repeated twice. f) AFM images of Aβ alone (20 µm, left) and treated with Ce6/PEG‐NTs (1 µm, right) following irradiation and 72 h incubation at 37 °C. g) Effect of NTs on Aβ40 oligomer and fibril formation in the dark and after irradiation. Aβ40 (20 µm) was aged for 48 h in the absence or presence of 1 µm or equivalent amount of NTs. Samples were spotted onto nitrocellulose membranes and probed with (left) A11 or (right) OC antibodies.
We chose to functionalize the NTs with Chlorin e6 (Ce6) as a negatively charged SO photosensitizer (Figure 2b). Clinically approved for photodynamic therapy of lung cancer,[ 8 , 9 ] Ce6 exhibits minimal toxicity, high stability, and excellent aqueous solubility. Moreover, it shows remarkable selectivity to Aβ and inhibits its aggregation and toxicity upon excitation with visible light.[ 5b ] We have demonstrated that SO produced by photoexcited Ce6 selectively oxidizes the Histidine residues in Aβ, generating cross‐linked aggregates that impede further aggregation.[ 5b ] Ce6 was coupled to the NTs surface (Ce6‐NTs) by ISA to simultaneously target early Aβ species and block further aggregation through the SO radiosensitization of the dense NTs with X‐rays. To improve colloidal stability and prevent aggregation in an aqueous environment, the NTs were also decorated by Food and Drug Administration‐approved m‐dPEG37acid (Ce6/PEG‐NTs, Figure 2a,b).[ 10 ] Modification of the NTs surface by PEG is expected to reduce toxicity and immunogenicity by preventing NTs agglomeration and opsonization by the immune system and increase the in vivo circulation time.[ 11 ] Furthermore, it has been demonstrated that PEGylation of Porphyrin‐coated NTs preserves the original luminescence properties of the single photosensitizer molecule.[ 7 ]
The successful decoration of Ce6 and PEG on the NTs is demonstrated by attenuated total reflection Fourier transform infrared spectroscopy (Figure S1c, Supporting Information). Steady‐state and time‐resolved photoluminescence experiments demonstrate that the electronic properties of Ce6 are fully preserved upon binding to the NT surface (Figure S2, Supporting Information). Figure 2c shows the scintillation emission of bare and functionalized NTs powders (Experimental Section). Upon functionalization with Ce6, the broad emission of bare NTs at ≈450 nm is quenched, while a dominant emission from the dye emerges at ≈710 nm. The blue emission quenching suggests that in addition to direct Ce6 excitation during scintillation by recombination of diffusing charges on the dye molecules, there is partial reabsorption of the NT emission, with a portion of energy being transferred from the NTs to the sensitizers activating their fluorescence (Figure S3, Supporting Information).[ 7 , 12 ] The decoration with PEG had a similar quenching effect; however, the emission at 710 nm became more intense, suggesting that PEG improves the scintillation of the NTs.
The X‐ray‐induced SO sensitization ability of the NTs was confirmed by electron paramagnetic resonance (EPR) spectroscopy in combination with 2,2,6,6‐tetramethylpiperidine (TEMP) as the SO‐specific spin trap (Experimental Section).[ 13 ] As shown in Figure 2d, upon irradiation, the NTs produce a clear TEMPO triplet indicative of SO generation. In control experiments with nonirradiated samples, no EPR signals were detected (Figure S4, Supporting Information). Consistent with the scintillation experiments (Figure 2c) the signal intensity progressively increases with the NTs, Ce6‐NTs, and Ce6/PEG‐NTs, respectively, indicating that the latter composition is more efficient, correlating with the improved activation of Ce6 molecules.
2.2. In Vitro Inhibition of Aβ Aggregation by Irradiated NTs
The ability of NTs to disrupt the Aβ aggregation under X‐ray irradiation was investigated using Thioflavin T (ThT), which emits fluorescence upon binding to cross‐β‐sheets of amyloid fibrils (Experimental Section).[ 14 ] Specifically, we targeted the most abundant Aβ isoform found in amyloid deposits in AD, that is the 40‐residue peptide (Aβ40), as a validated AD‐related species. Figure 2e shows that Aβ aggregation is insensitive to X‐rays in absence of radiosensitizers. In sharp contrast, Ce6‐NTs and Ce6/PEG‐NTs dose‐dependently reduced Aβ aggregation when irradiated with X‐rays. At the lowest concentration tested (1 µm), Ce6‐NTs and Ce6/PEG‐NTs reduced Aβ aggregation by 67% and 85%, respectively (Figure 2e; Figure S5, Supporting Information). The enhanced performance of Ce6/PEG‐NTs can be attributed to both the improved scintillation properties of the PEGylated system and its superior solubility in an aqueous environment.
The inhibitory effect of the radiosensitizers on Aβ aggregation was further confirmed through atomic force microscopy (AFM). The AFM image of untreated Aβ shows long fibril structures after 72 h of aging in dark (Figure S6, Supporting Information, left) and after exposure to X‐rays (Figure 2f, left). Consistent with the ThT experiments, Ce6/PEG‐NTs did not affect fibril formation when incubated in the dark (Figure S6, Supporting Information, right), while completely prevented fibril formation upon irradiation (Figure 2f, right).
Dot blot assays were performed to investigate in more detail the effect of the radiosensitizers on formation of Aβ aggregates. Conformation‐specific antibodies A11 and OC were used to identify oligomers and fibrils, respectively (Experimental Section).[ 15 ] The results, as shown in Figure 2g, reveal that without irradiation, the presence of radiosensitizers does not affect Aβ aggregation into oligomers and fibrils. Similarly, in the absence of radiosensitizers, the aggregation is unaffected by irradiation. In sharp contrast, a clear reduction of both oligomers and fibrils is observed upon X‐ray irradiation. Notably, the reduction of oligomers and fibrils is significantly more efficient when using Ce6/PEG‐NTs, which agrees with their better ability to sensitize SO. This finding is further supported by circular dichroism experiments demonstrating that the largest effect on the transition of Aβ from random coil to β‐sheet‐rich conformation is induced by Ce6‐PEG‐NTs after X‐ray irradiation (Figure S7, Supporting Information).
Next, electrospray ionization mass spectrometry (ESI‐MS, Experimental Section) was utilized to investigate the molecular mechanism underlying X‐ray‐induced oxidation that prevents Aβ aggregation. The ESI‐MS spectra of Aβ40 before and after irradiation revealed the formation of the same quadruple charged ion ((Aβ40 + 4H)4+; m/z 1083.29) corresponding to the molecular mass of Aβ40 (m/z 4329.16, Figure 3a; Figure S8 and Table S1, Supporting Information).[ 16 ] Bare NTs have no significant effect on Aβ mass even after irradiation (Figure 3b) in agreement with their weak radiosensitization. In the presence of Ce6‐NTs, irradiation led to the appearance of a prominent Aβ oxidized species at m/z = 1086.79, likely corresponding to the oxidation of one histidine residue in the Aβ chain ((Aβ40 + 1 His ox.)4+; Figure 3c and Table S1, Supporting Information).[ 5 , 16 ] On the other hand, irradiation of Aβ with the best sensitizer (Ce6/PEG‐NTs) generated a diverse mixture of oxidized Aβ species in the ESI‐MS spectrum (Figure 3d), likely associated with the oxidation of His6/13/14, Tyr 10 and Met35. This indicates a strong interaction between the dual‐functionalized NTs and Aβ, leading to efficient oxidation of the latter (Table S1, Supporting Information).[ 17 , 18 ] Previous studies have demonstrated that His13 and His14 are associated with Aβ‐mediated ion channel formation and cytotoxicity, and blocking these channels with small peptides or molecules can prevent Ca2+ influx and neurotoxicity.[ 19 ] Notably, photodynamic treatment of Ce6 with visible light in our previous studies did not cause oxidation of Tyr and Met residues[ 5b ] suggesting that X‐ray excited Ce6/PEG‐NTs are highly effective in generating SO.
Figure 3.
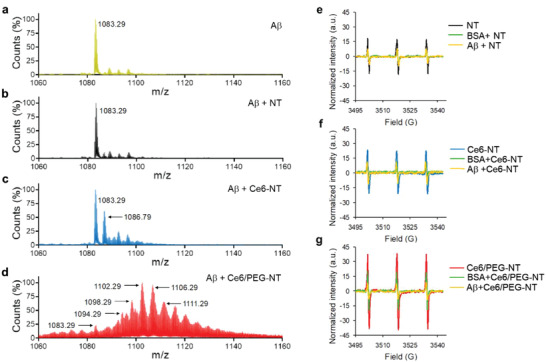
Mechanism of Aβ oxidation by X‐ray irradiated multicomponent NTs. a–d) ESI‐MS spectra obtained from incubation of Aβ40 a) without and with b) NTs, c) Ce6‐NTs, and d) Ce6/PEG‐NTs after irradiation. Aβ40 (40 µm) was treated with 2 µm or equivalent amount of NTs irradiated with 2 Gy X‐rays in PBS (50 mm, pH7.4) and analyzed by ESI–MS. e–g) Effect of Aβ40 and BSA on singlet oxygen generated from X‐ray excited NTs. Comparative TEMPO EPR spectra formed from incubation of TEMP (40 µm) and 5 µm dispersions of e) NTs, f) Ce6‐NTs, and g) Ce6/PEG‐NTs in PBS/D2O (1:9) following X‐ray exposure with or without Aβ40 and BSA (40 µm each).
To assess the specificity of Ce6/PEG‐NTs as inhibitors of Aβ aggregation, a competitive assay was conducted between Aβ monomers and bovine serum albumin (BSA) in their ability to react with X‐ray‐induced SO, using EPR/TEMP spin trap (Figure 3g). Both Aβ and BSA equally reduced the intensity of the TEMPO spin adduct generated by X‐ray‐excited NTs and Ce6‐NTs, indicating that they were unable to differentiate between Aβ and BSA (Figure 3e,f). In contrast, the SO signal generated by X‐ray‐irradiated Ce6/PEG‐NTs was more effectively reduced by Aβ compared to BSA, suggesting that Ce6/PEG‐NTs exhibit greater selectivity toward Aβ, possibly due to the improved colloidal stability conferred by PEG and its ability to reduce nonspecific protein binding.[ 20 ]
2.3. In Vivo Effect of Irradiated NTs on Aβ Toxicity and Symptoms
The biocompatibility and efficacy of NTs against Aβ toxicity were evaluated in C. elegans models expressing human Aβ42. Transgenic C. elegans are largely used as validated models for AD and other age‐related neurodegenerative diseases due to their age‐dependent humanlike physiological changes at tissue, cellular and molecular levels. Moreover, the genome sequence of C. elegans is similar to the human one with roughly 38% of worm genes having a human ortholog (e.g., APP, tau).[ 21 ]
Considering that aging plays a crucial role in AD pathology and Aβ‐mediated neurotoxicity, the effects of irradiated NTs were first investigated by monitoring the lifespan of wild type (WT) CL802 and transgenic CL2006 C. elegans, which expresses Aβ3–42 in the body‐wall muscles causing progressive paralysis and premature death.[ 22 ] Prior to these experiments, the biocompatibility and safety of the NTs as well as the tolerated dose of X‐ray irradiation, were assessed in human neuroblastoma SH‐SY5Y cells and WT C. elegans (Figures S9–S11, Supporting Information). These studies collectively demonstrated that NTs are safe even at the highest concentration tested. Furthermore, X‐ray irradiation up to 10 Gy was found to be well‐tolerable by the worms (Figure S11, Supporting Information). Figure 4a,b shows that X‐ray exposure has no negative effect on the mean lifespan of WT worms treated with different NTs. It also shows that mutant worms live significantly shorter than WT worms (14.5 vs 12.9 days, p < 0.0001) and that NTs and Ce6‐NTs have no significant protecting effect on the mutant worms upon irradiation. In agreement with their greater ability to modify Aβ and inhibit its aggregation, Ce6/PEG‐NTs also increase the longevity of transgenic worms upon irradiation (12.9 vs 14.1 days, p < 0.0001) and negate the effect of overexpressed Aβ.
Figure 4.
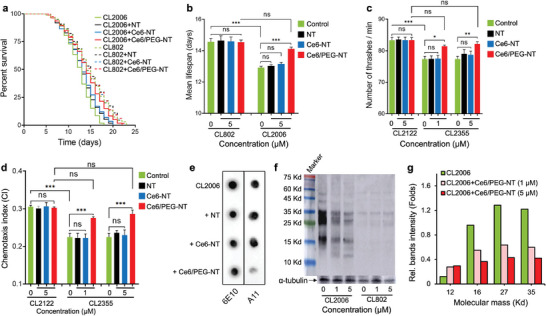
Effect of multicomponent scintillating nanotubes (NTs) on transgenic C. elegans AD models. a) Kaplan–Maier survival plots and b) median lifespan of transgenic CL2006 and control WT CL802 worms fed with 5 µm of NTs, Ce6‐NTs and Ce6/PEG‐NTs in PBS and irradiated with soft X‐rays (2 Gy). Data are presented as mean ± SD from three experiments and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test (n = 100 each; *** p < 0.0001, ns = not significant). c) Effect of NT treatment on mobility (thrashes min−1) of transgenic CL2355 and WT CL2122 worms following irradiation. Results are mean ± SD from three experiments with 20 worms per group. Statistical significance was determined as described above (* p < 0.05. ** p < 0.005, *** p < 0.0001, ns = not significant). d) Effect of NTs and X‐rays on chemotaxis of transgenic CL2355 and WT CL2122 worms. The chemotactic index CI = (number of worms at attractant sites – number of worms at control sites)/total number of worms. Results are reported as described above (n = 60 each; *** p < 0.0001, ns = not significant). e) Representative dot‐blot analysis of equal amounts of the proteins extracted from transgenic worms treated with NTs or vehicle after X‐ray irradiation and probed with sequence specific 6E10 and oligomer specific A11 antibodies. f) Representative Western‐blot analysis of Aβ species in transgenic CL2006 and control WT CL2122 strains untreated and treated with Ce6/PEG‐NTs (1 and 5 µm) following irradiation. Equal amounts of extracted proteins were loaded onto each lane and blotted with an anti‐Aβ antibody (6E10) or α‐tubulin. g) Quantification of major Aβ species in transgenic CL2006 animals treated with different amounts of Ce6/PEG‐NTs following irradiation using ImageJ software.
The effect of NTs on neuronal functionalities were investigated on temperature sensitive CL2355 transgenic C. elegans, which express pan‐neuronal human Aβ1–42 when the temperature up‐shifted to 23–25 °C leading to difficulties in associated learning, chemotaxis, and thrashing.[ 23 ] The transgenic and WT worms were fed with increasing concentrations of NTs and kept at 16 °C for 36 h. The temperature was then elevated to 23 °C to induce Aβ expression. Figure 4c demonstrates that mutant and WT worms bend, respectively, ≈77 and 83 times per minute at an elevated temperature (23 vs 16 °C) (p < 0.0001). In control experiments, the WT worms were not affected when fed with NTs and irradiated with 2 Gy of X‐rays, suggesting the lack of toxicity of the treatment. Consistent with the lower in vitro activity of bare and Ce6‐conjugated NTs, no significant improvement in thrashing performance was noticed when mutant worms were fed with NTs or Ce6‐NTs and irradiated. However, on feeding with increasing concentrations of Ce6/PEG‐NTs and irradiation, the AD worms showed a significant improvement in trashing ability to levels exhibited by untreated WT worms indicating that the treatment enables full recovery of motility in AD worms.
In C. elegans, activation of several sensory neurons is required to stimulate motor neurons and chemotaxis behavior.[ 24 ] To demonstrate further the therapeutic effect of irradiated NTs, we investigated their impact on chemotaxis. The chemotaxis index (CI) measures the fraction of animals that reach the attractant region using their chemosensory detection system.[ 24 ] As shown in Figure 4d, the CI of the control WT and transgenic strains was respectively 0.300 ± 0.003 and 0.22 ± 0.01, indicating substantial chemotactic dysfunction induced by Aβ expression in the worm's neurons. Treatment with NTs and Ce6‐NTs followed by irradiation had no significant effect on WT animals, and they were also ineffective in reducing the chemotactic dysfunction of transgenic worms. In contrast, feeding the worms with Ce6/PEG‐NTs and subsequent irradiation led to a significant improvement in the CI to 0.270 ± 0.003 (p < 0.0001) at 1 µm concentration and further to 0.290 ± 0.003 (p < 0.0001) at 5 µm. These results indicate that the chemotaxis ability of the mutant worms was fully recovered after treatment with Ce6/PEG‐NTs and X‐ray irradiation.
To gain insights into the mechanism by which irradiated Ce6/PEG‐NTs improve pathological behavior in transgenic animals, we analyzed the levels of Aβ species in equal amounts of extracted proteins using immunochemical dot‐blot analyses with sequence and oligomer‐specific 6E10 and A11 antibodies.[ 25 ] The results, as shown in Figure 4e, indicate that irradiated NTs and Ce6‐NTs have no effect on the total amount of Aβ or A11‐reactive oligomers. Conversely, Ce6/PEG‐NTs dramatically reduce the overall levels of the oligomers, in agreement with their superior radiosensitization ability.
Western blot analysis of the extracted proteins suggested that the decrease in Aβ oligomers is associated with decreased levels of Aβ species with molecular mass range of ≈12 to 60 kDa. More specifically, the levels of Aβ tetramers (≈18 kDa), hexamers (≈27 kDa) and heptamers (≈31 kDa) decreased with increasing the feeding of transgenic CL2006 worms with Ce6/PEG‐NTs and irradiation with X‐rays (Figure 4f,g). Hexamers of Aβ have been suggested to serve as building blocks for toxic aggregates, including Aβ‐derived diffusible ligands, dodecamers and globulomers.[ 26 ] Aβ hexamers were also associated with pore‐induced toxicity by organizing into β‐barrel‐shaped structures.[ 27 ] In control WT worms, as expected, no significant amount of Aβ and its aggregates were observed.
3. Conclusion
In the search for an effective treatment strategy for AD, we propose to inhibit the formation of neurotoxic Aβ oligomers by oxidation of specific amino acids. Such oxidation is achieved in vivo by exploiting highly reactive singlet oxygen (SO) locally produced by interaction of engineered biocompatible multicomponent nanoscintillators with low and safe doses of X‐ray irradiation. Our findings demonstrate that the scintillators interact preferentially with Aβ, effectively preventing the formation of neurotoxic aggregates both in vitro and in transgenic C. elegans AD models. Animals fed with the nanoscintillators and irradiated with X‐rays completely recovered from neuronal dysfunction and associated symptoms.
Considering the emerging pre‐symptomatic diagnostic methods for AD[ 28 ] that enable early detection and precise localization of Aβ oligomers and fibrils, along with the highly advanced and accurate X‐ray irradiation methods developed for cancer radiotherapy, our results strongly support the further development of low dose X‐ray therapies that exploit Aβ‐targeting radiosensitizers to treat AD and impede its progression.
By oxidizing Aβ monomers and early oligomers, we successfully prevent their aggregation into toxic forms, achieving a reduction in aggregation of more than 80%. In transgenic C. elegans models, the combination of NTs and X‐rays dramatically reduced the levels of Aβ oligomers and restored functional and behavioral symptoms associated with Aβ aggregation and toxicity. These results highlight the potential of Aβ‐targeting radiosensitizers as a revolutionary and unexplored strategy to treat AD and hinder its advancement.
4. Experimental Section
Unless otherwise mentioned reagents were used as received. Ce6 was obtained from Frontier Scientific (Logan, Utah). Aβ40 was acquired from Hanhong Scientific (Shanghai, China), purified to homogeneity (>95%) by RP‐HPLC and stored until use as a lyophilized powder at −20 °C. The monomeric state of the peptide was ensured by triple evaporation from high‐grade 1,1,1,3,3,3‐hexafluoroisopropanol using a stream of nitrogen.[ 26 ] A11 and OC antibodies were generously provided by Dr. Rakez Kayed (Department of Neurology, University of Texas).[ 15 , 25 ]
Synthesis of Stoichiometric Hydrated Magnesium Silicate Nanotubes (NTs)
NTs were synthesized according to previously used synthetic method.[ 6 ] A hydrothermal reactor with a 100 cm3 polypropylene vessel was used to carry the hydrothermal reaction of 1.522 g of Na2SiO3 and 0.764 g of MgCl2 in an aqueous solution of NaOH (220 mL, 0.4 m) at 250 °C for 16 h. The precipitate removed from the solution was repeatedly washed with deionized water (DW) before drying for 3 h at 110 °C.
Functionalization of NTs with Chlorin e6 (Ce6)
Dry NT powder (30 mg) was suspended in 5 mL of NaOH solution (0.4 m) and 2.5 mL of Ce6 solution (650 µm in 0.4 m NaOH) was added slowly under stirring (Ce6‐NTs). Ce6 molecules adsorbed on the surface of NTs were centrifuged and dried at 50 °C in a drying stove for 3 h.
Pegylation of Functionalized Ce6‐NTs
Ce6‐NTs (40 mg) were dispersed in 20 mL of Tris buffer (100 mm, pH 7.4) and sonicated for 30 min. To this suspension, m‐PEG37‐acid (10 mg, Sigma) was added under stirring. The product was washed and centrifuged three times with DW and dried as described above.
Transmission Electron Microscope (TEM) Analysis
Samples were prepared by dispersing a few mg of the NTs in 2 mL of DW and dropping 3 µL of the suspension on carbon‐coated copper grids. The samples were analyzed by a JEOL JEM1220 TEM operated at 120 kV.
Radioluminescence and Irradiation Experiments
Steady–state scintillation measurements were performed by irradiating the samples at room temperature (RT) with a Philips 2274 X‐ray tube with a tungsten target equipped with a beryllium window operated at 20 kV (steady–state radioluminescence spectroscopy, RL). At this operating voltage, X‐rays were produced by the Bremsstrahlung mechanism superimposed to the L and M transition lines of tungsten due to the impact of electrons generated through a thermionic effect and accelerated onto the tungsten target. No beam filtering was applied. RL spectra were recorded using a homemade apparatus featuring a liquid nitrogen‐cooled charge‐coupled device (CCD, Jobin‐Yvon Symphony II) coupled to a monochromator (Jobin‐Yvon Triax 180) with a 100 grooves/mm grating as a detection system. Spectra were corrected for the setup optical response. For the irradiation experiments, samples received unfiltered X‐ray radiation using a Machlett OEG 50 tube with a tungsten target. Dose values were evaluated in air with an ionization chamber.
EPR Spectroscopy
Continuous wave EPR (CW‐EPR) spectra were recorded with an Elexsys Bruker E500 X‐band spectrometer operating at 9.0–9.5 GHz, 20 mW of microwave power, 100 G scan range, and 1 G field modulation. The specific spin trap 2,2,6,6‐tetramethylpiperidine (TEMP) was used to detect and characterize the generated 1O2. A solution of different NTs (25 µM, 20 µL) and TEMP (400 µM, 10 µL) in D2O was incubated in absence or presence of Aβ (200 µM, 20 µL) or BSA (200 µM, 20 µL) in D2O. The total volume was adjusted to 100 µL using PBS/D2O (1:9). The final concentrations were 40 µm for Aβ, BSA, and TEMP and 5 µm for the NTs. The mixture was irradiated with 2 Gy of X‐rays and placed in capillary quartz tubes (Vitrocom, Mountain Lakes, NJ, USA) for measurements.
Thioflavin T (ThT) Aggregation Assay
Anti‐amyloidogenic activity was assessed by the ThT assay.[ 29 ] Briefly, monomerized Aβ40 solution (67 µM, 30 µL) was mixed with increasing concentrations of NTs (10 µL in DDW) and ThT solution (500 µm, 4 µL in H2O) to afford final concentrations of 20 µm (Aβ40), 1 and 5 µm (NTs) and 20 µm (ThT) in 100 µL total volume (see Supporting Information). The solution was irradiated with 2 Gy X‐rays at 14 keV and placed immediately in a 96 well flat‐bottom black plate. The plate was covered with a transparent polyolefin film and placed in a microplate reader (Synergy H1 microplate reader, BioTek, US). Fluorescence intensity of amyloid bound ThT samples was examined hourly over 65 h using excitation/emission wavelengths of 430/492 nm at 37 °C. Before each reading, the plate was shaken for 2 min. Hundred percent aggregation was determined by ThT fluorescence of Aβ alone and presented as mean ± SD of three experiments.
Atomic Force Microscopy (AFM)
Samples were prepared as described for the ThT assay, but without addition of ThT. After irradiation, the samples were incubated for 72 h at 37 °C. A volume of 5 µL of the sample was deposited onto a cleaved mica substrate and dried at RT. AFM images were acquired in tapping mode using a Bio FastScan scanning probe microscope (Bruker AXS) with a silicon probe (spring constant of 18 N m−1) equipped with a cantilever (≈1400 kHz resonance frequency).
Immunoblot Assay
Monomerized Aβ40 solution (20 µm) was treated without or with 1 µm or equivalent amount of NTs, irradiated with 2 Gy of X‐rays and incubated for 48 h at 37 °C. Samples were stored at −80 °C until analyzed. For dot blot studies, 2 µL of each sample was spotted onto a nitrocellulose membrane (Whatman) and dried at RT. The blots were blocked for 1 h with 5% BSA solution in Tris‐buffered saline (TBS, 10 mm) containing 0.1% Tween 20 (TBST). Membranes were washed three times (10 min each) with TBST and incubated at ≈4 °C overnight with antioligomer Aβ antibody (A11, 1:5000 dilution) and antifibril antibody (OC, 1:5000 dilution) in 0.5% BSA in TBST. Membranes were washed three times with TBST and incubated further with horseradish peroxidase (HRP) conjugated antirabbit IgG at 1:5000 in 0.5% BSA solution at RT for 1 h. Finally, membranes were washed four times with TBST and developed using the ECL reagent kit (Bio‐rad). Chemiluminescence was measured with ImageQuant LAS 4000 imaging system.
Mass Spectrometry
Aβ40 (40 µm) solutions were prepared in the absence or presence of 2 µm or equivalent amount of NTs and irradiated with 2 Gy X‐rays or kept in dark. Samples were then centrifuged at 4000 rpm for 15 min to precipitate the NTs, and supernatants were analyzed by an electrospray ionization mass spectrometer (ESI‐Q‐TOF, Agilent Technologies, USA).
C. Elegans Strains, Maintenance, and Treatment
Transgenic CL2355 (dvIs50, pCL45 (snb‐1::Aβ 1–42::3′ UTR(long) + mtl‐2::GFP] and CL2006 (dvIs2, pCL12(unc‐54/human Aβ 1–42 minigene) + rol‐6(su1006)) and control CL2122 (dvIs15, (pPD30.38) unc‐54(vector) + (pCL26) mtl‐2::GFP) and CL802 (smg‐1(cc546) I; rol‐6(su1006) II) strains were obtained from Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN). All strains were propagated at 16 °C on nematode growth medium (NGM) plates seeded with Escherichia coli strain OP50 (100 µL) as food source. Worms were age‐synchronized for all assays by hypochlorite bleaching (0.1 m KOH, 2.6% NaClO). The observed eggs were hatched overnight (L1) in M9 buffer and cultivated on NGM plates as described.[ 30 ]
C. Elegans Lifespan
Age‐synchronized transgenic (CL2006) and WT (CL802) animals were cultured on 60 mm NGM plates at 16 °C. For each treatment group, a total of 100 synchronized L4 worms were seeded (25 worms/plate) and fed with heat inactivated OP50 solution containing the vehicle (PBS) or 5 µm of NTs. After overnight feeding, worms were washed with M9 three times, exposed to 2 Gy X‐rays, or kept in dark. Animals were transferred to new plates every third day and exposed again to X‐rays or kept in dark. Worms with internal hatching were removed and scored each day. The experiment was terminated when all worms were scored as dead or censored. Survival plots were obtained from Kaplan–Meier survival analysis and lifespan data were analyzed by a one‐way ANOVA followed by Tukey's multiple comparison test.
C. Elegans Locomotion Assay
Synchronized L4 worms (CL2355) and control (CL2122) strains were seeded on NGM plates (60 mm) and fed overnight with vehicle (PBS) or increasing concentrations of NTs. The worms were washed with M9 three times, exposed to 2 Gy X‐rays or kept in dark and incubated at 16 °C for 36 h. Animals were transferred to new plates, incubated at 23 °C and fed with the samples. After feeding overnight, animals were washed with M9 and irradiated again or kept in dark and incubated for 36 h at 23 °C. Worms were individually placed in a 12‐wells plate containing 1 mL of M9 buffer. After a recovery time of 2 min, worm thrashes in which the body bends from one side to the other and back were counted for 1 min for each animal (20 animals per group). Data were mean ± SD from three independent experiments and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test.
C. Elegans Chemotaxis Assay
Chemotaxis assays were carried out as described previously.[ 31 ] Briefly, synchronized transgenic C. elegans CL2355 and its control strain CL2122 were fed and handled as described in locomotion assay. After treatment, worms were washed twice with M9 buffer. Sodium azide (1 µL, 0.25 m) with 0.1% benzaldehyde in absolute ethanol (1 µL) as odorant were added to two opposite “attractant” corners of a 100 mm agar plate. On the control spots, sodium azide (1 µL) and absolute ethanol (1 µL) solutions were spotted. A suspension of worms (5 µL, ≈60 worms) was immediately applied to the center of each plate. The plates were incubated at 23 °C for 1 h and the number of worms in each quadrant was scored. The chemotaxis index (CI) was calculated by subtracting the number of worms in both control quadrants from the number of worms in both attractant quadrants and dividing by the total number of scored worms. Data were mean ± SD from three independent experiments analyzed as described above.
Immunoblotting of Aβ Species
After experimental treatment, transgenic CL2006 and WT CL802 worms were washed with M9 buffer, centrifuged, collected, frozen quickly in liquid nitrogen, and stored at −80 °C until analyzed. The worm pellets were immersed in a lysis buffer (62 mm Tris‐HCl, pH 6.8, 10% glycerol, 2% SDS, 4% β‐marcaptoethanol and 1X protease inhibitor cocktail), sonicated for 3 min, and boiled for 5 min. Total protein amounts were quantified by using the Bradford assay (BioRad) or by NanoDrop absorbance quantification at 280 nm. Equal amounts of proteins were spotted onto a nitrocellulose membrane (0.2 µm) in duplicate and blocked with 5% BSA in PBS (pH7.4) containing 0.1% (v/v) Tween 20 (PBS‐T). The membranes were incubated overnight with either 6E10 monoclonal antibody (1:2000 dilution, Covance) or a rabbit polyclonal antibody (A11, 1:1000 dilution) in PBS‐T containing 0.5% BSA. The blots were washed with PBST and incubated for 1 h at RT with horseradish peroxidase (HRP) conjugated to either an anti‐mouse IgG (1:10000 dilution in 0.5% BSA in PBST) or anti‐rabbit IgG (1:5000 in 0.5% BSA in PBST). The blots were washed four times with PBST and developed using the ECL kit as described. For Western blot assay, equal amounts of protein lysate were heated for 3 min at 95 °C with sample buffer containing 5% β‐mercaptoethanol. Proteins were separated by SDS‐PAGE gel using 15% Tricine gel and transferred to a nitrocellulose membrane using transfer buffer containing 10% methanol at 70 V for 75 min. The blots were blocked with 5% BSA in TBS plus 0.01% (v/v) Tween 20 for 1 h at RT, incubated with 6E10 antibody, and developed as above. Identical blots were reacted with anti‐α‐tubulin antibody and developed. The mean densities of β‐amyloid reactive bands were analyzed by ImageJ (National Institutes of Health, USA).
Statistics
Data were analyzed either by one‐way ANOVA followed by post hoc analysis where appropriate, or by two‐tailed, unpaired Student's t‐test unless indicated otherwise using GraphPad Prism 9 for Mac OS. Statistical details were provided in respective figure legends.
Conflict of Interest
There is a patent associated with the work in this article, patent application no. 102023000022626, IT0842‐23‐PA103305IT01 ‐ Università degli Studi Milano Bicocca‐ HS.
Author Contributions
S.S., V.S., S.R., and A.M. conceived the project idea. S.S., M.R., R.Y., and Y.S. performed in vitro, in vivo, and ex vivo experiments under the supervision of S.R., V.S., and M.C. A.M. designed and synthesized the multicomponent NTs. V.S. performed the experiments for material structural characterization. F.C. and I.V. performed scintillation and photoluminescence spectroscopy experiments under supervision of A.M. All authors contributed to the manuscript preparation.
Supporting information
Supporting Information
Acknowledgements
This study was supported by a collaborative research fund from the Italian Ministero degli Affari Esteri e della Cooperazione Internazionale (MAECI) and Israel Ministry of Science and Technology (MOST) # 2020‐H45H19000070001 and 3–17479 “X‐ray activated photodynamic therapy for targeted treatment of Alzheimer's disease”. A.M. also acknowledges funding from the Italian Ministry of Health (project code RF‐2016‐02362263, NanoTrack‐EXO). The authors thank Dr. Ronit Lavi and Dr. Anat Haviv‐Chesner for their support in conducting EPR and C. elegans studies respectively. S.S. thanks the Planning and Budgeting Committee (PBC) of the Council for Higher Education, Israel for providing the postdoctoral fellowship.
Senapati S., Secchi V., Cova F., Richman M., Villa I., Yehuda R., Shenberger Y., Campione M., Rahimipour S., Monguzzi A., Noninvasive Treatment of Alzheimer's Disease with Scintillating Nanotubes. Adv. Healthcare Mater. 2023, 12, 2301527. 10.1002/adhm.202301527
Contributor Information
Shai Rahimipour, Email: Shai.Rahimipour@biu.ac.il.
Angelo Monguzzi, Email: angelo.monguzzi@unimib.it.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Karran E., Mercken M., Strooper B. D., Nat. Rev. Drug Discovery 2011, 10, 698. [DOI] [PubMed] [Google Scholar]
- 2.a) Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., Klein W. L., Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 6448; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fändrich M., J. Mol. Biol. 2012, 421, 427; [DOI] [PubMed] [Google Scholar]; c) Hyman J. M., Firestone A. J., Heine V. M., Zhao Y., Ocasio C. A., Han K., Sun M., Rack P. G., Sinha S., Wu J. J., Solow‐Cordero D. E., Jiang J., Rowitch D. H., Chen J. K., Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14132; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cline E. N., Bicca M. A., Viola K. L., Klein W. L., J. Alzheimers Dis. 2018, 64, S567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Sevigny J., Chiao P., Bussière T., Weinreb P. H., Williams L., Maier M., Dunstan R., Salloway S., Chen T., Ling Y., O'Gorman J., Qian F., Arastu M., Li M., Chollate S., Brennan M. S., Quintero‐Monzon O., Scannevin R. H., Arnold H. M., Engber T., Rhodes K., Ferrero J., Hang Y., Mikulskis A., Grimm J., Hock C., Nitsch R. M., Sandrock A., Nature 2016, 537, 50; [DOI] [PubMed] [Google Scholar]; b) Panza F., Lozupone M., Dibello V., Greco A., Daniele A., Seripa D., Logroscino G., Imbimbo B. P., Immunotherapy 2019, 11, 3. [DOI] [PubMed] [Google Scholar]
- 4.a) Reiman E. M., Nature 2023, 615, 42; [DOI] [PubMed] [Google Scholar]; b) van Dyck C. H., Swanson C. J., Aisen P., Bateman R. J., Chen C., Gee M., Kanekiyo M., Li D., Reyderman L., Cohen S., Froelich L., Katayama S., Sabbagh M., Vellas B., Watson D., Dhadda S., Irizarry M., Kramer L. D., Iwatsubo T., N. Engl. J. Med. 2022, 388, 9. [DOI] [PubMed] [Google Scholar]
- 5.a) Li C., Wang J., Liu L., Front. Chem. 2020, 8, 509; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Leshem G., Richman M., Lisniansky E., Antman‐Passig M., Habashi M., Gräslund A., Wärmländer S. K. T. S., Rahimipour S., Chem. Sci. 2019, 10, 208; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hirabayashi A., Shindo Y., Oka K., Takahashi D., Toshima K., Chem. Commun. 2014, 50, 9543; [DOI] [PubMed] [Google Scholar]; d) Ishida Y., Fujii T., Oka K., Takahashi D., Toshima K., Chem. Asian J. 2011, 6, 2312; [DOI] [PubMed] [Google Scholar]; e) Taniguchi A., Shimizu Y., Oisaki K., Sohma Y., Kanai M., Nat. Chem. 2016, 8, 974. [DOI] [PubMed] [Google Scholar]
- 6. Villa C., Campione M., Santiago‐González B., Alessandrini F., Erratico S., Zucca I., Bruzzone M. G., Forzenigo L., Malatesta P., Mauri M., Trombetta E., Brovelli S., Torrente Y., Meinardi F., Monguzzi A., Adv. Funct. Mater. 2018, 28, 1707582. [Google Scholar]
- 7. Villa I., Villa C., Crapanzano R., Secchi V., Tawfilas M., Trombetta E., Porretti L., Brambilla A., Campione M., Torrente Y., Vedda A., Monguzzi A., ACS Appl. Mater. Interfaces 2021, 13, 12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mojzisova H., Bonneau S., Maillard P., Berg K., Brault D., Photochem. Photobiol. Sci. 2009, 8, 778. [DOI] [PubMed] [Google Scholar]
- 9. Kim T. E., Chang J. E., Pharmaceutics 2023, 15, 2257.37765226 [Google Scholar]
- 10. Gref R., Domb A., Quellec P., Blunk T., Müller R., Verbavatz J.‐M., Langer R., Adv. Drug Del. Rev. 1995, 16, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Klibanov A. L., Maruyama K., Torchilin V. P., Huang L., FEBS Lett. 1990, 268, 235; [DOI] [PubMed] [Google Scholar]; b) Dufort S., Sancey L., Coll J.‐L., Adv. Drug Del. Rev. 2012, 64, 179; [DOI] [PubMed] [Google Scholar]; c) Suk J. S., Xu Q., Kim N., Hanes J., Ensign L. M., Adv. Drug Del. Rev. 2016, 99, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Secchi V., Monguzzi A., Villa I., Int. J. Mol. Sci. 2022, 23, 8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Han S. K., Hwang T.‐M., Yoon Y., Kang J.‐W., Chemosphere 2011, 84, 1095; [DOI] [PubMed] [Google Scholar]; b) Rahimipour S., Gescheidt G., Bilkis I., Fridkin M., Weiner L., J. Appl. Magn. Reson. 2010, 37, 629. [Google Scholar]
- 14. LeVine H. 3rd, Protein Sci. 1993, 2, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Shea D., Hsu C.‐C., Bi T. M., Paranjapye N., Childers M. C., Cochran J., Tomberlin C. P., Wang L., Paris D., Zonderman J., Varani G., Link C. D., Mullan M., Daggett V., Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8895; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G., Mol. Neurodegener. 2007, 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fanni A. M., Okoye D., Monge F. A., Hammond J., Maghsoodi F., Martin T. D., Brinkley G., Phipps M. L., Evans D. G., Martinez J. S., Whitten D. G., Chi E. Y., ACS Appl. Mater. Interfaces 2022, 14, 14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Lee B. I., Suh Y. S., Chung Y. J., Yu K., Park C. B., Sci. Rep. 2017, 7, 7523; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pattison D. I., Rahmanto A. S., Davies M. J., Photochem. Photobiol. Sci. 2012, 11, 38. [DOI] [PubMed] [Google Scholar]
- 18. Taniguchi A., Sasaki D., Shiohara A., Iwatsubo T., Tomita T., Sohma Y., Kanai M., Angew. Chem., Int. Ed. 2014, 53, 1382. [DOI] [PubMed] [Google Scholar]
- 19.a) Diaz J. C., Linnehan J., Pollard H., Arispe N., Biol. Res. 2006, 39, 447; [DOI] [PubMed] [Google Scholar]; b) Arispe N., Diaz J. C., Flora M., Biophys. J. 2008, 95, 4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kingshott P., Griesser H. J., Curr. Opin. Solid State Mater. Sci. 1999, 4, 403. [Google Scholar]
- 21.a) Wu Y., Luo Y., Curr. Alzheimer Res. 2005, 2, 37; [DOI] [PubMed] [Google Scholar]; b) Alexander A. G., Marfil V., Li C., Front. Genet. 2014, 5, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Link C. D., Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 9368; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guglielmotto M., Giliberto L., Tamagno E., Tabaton M., Front. Aging Neurosci. 2010, 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Y., Wu Z., Butko P., Christen Y., Lambert M. P., Klein W. L., Link C. D., Luo Y., J. Neurosci. 2006, 26, 13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hobert O., J. Neurobiol. 2003, 54, 203. [DOI] [PubMed] [Google Scholar]
- 25.a) Baghallab I., Reyes‐Ruiz J. M., Abulnaja K., Huwait E., Glabe C., J. Alzheimers Dis. 2018, 66, 1235; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kayed R., Head E., Thompson J. L., Mcintire T. M., Milton S. C., Cotman C. W., Glabe C. G., Science 2003, 300, 486. [DOI] [PubMed] [Google Scholar]
- 26.a) Bitan G., Kirkitadze M. D., Lomakin A., Vollers S. S., Benedek G. B., Teplow D. B., Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 330; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bernstein S. L., Dupuis N. F., Lazo N. D., Wyttenbach T., Condron M. M., Bitan G., Teplow D. B., Shea J.‐E., Ruotolo B. T., Robinson C. V., Bowers M. T., Nat. Chem. 2009, 1, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Do T. D., Lapointe N. E., Nelson R., Krotee P., Hayden E. Y., Ulrich B., Quan S., Feinstein S. C., Teplow D. B., Eisenberg D., Shea J.‐E., Bowers M. T., J. Am. Chem. Soc. 2016, 138, 549; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Österlund N., Moons R., Ilag L. L., Sobott F., Gräslund A., J. Am. Chem. Soc. 2019, 141, 10440. [DOI] [PubMed] [Google Scholar]
- 28. Habashi M., Vutla S., Tripathi K., Senapati S., Chauhan P. S., Haviv‐Chesner A., Richman M., Mohand S. A., Dumulon‐Perreault V., Mulamreddy R., Okun E., Chill J. H., Guerin B., Lubell W. D., Rahimipour S., Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2210766119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richman M., Wilk S., Chemerovski M., Wärmländer S. K. T. S., Wahlström A., Gräslund A., Rahimipour S., J. Am. Chem. Soc. 2013, 135, 3474. [DOI] [PubMed] [Google Scholar]
- 30. Stiernagle T., in WormBook: The Online Review of C. elegans Biology, Oxford Univeristy Press, New York (NY) 2006, pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Habashi M., Chauhan P. S., Vutla S., Senapati S., Diachkov M., El‐Husseini A., Guerin B., Lubell W. D., Rahimipour S., J. Med. Chem. 2023, 66, 3058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.