

Abstract
Load-bearing soft tissues are soft but strong, strong yet tough. These properties can only be replicated in synthetic hydrogels, which do not have the biocomplexity required by many biomedical applications. By contrast, natural hydrogels, although retaining the native complexity, are weak and fragile. Here we present a thermomechanical casting method to achieve the mechanical capabilities of synthetic materials in biopolymer hydrogels. The thermomechanical cast and chemically crosslinked biopolymer chains form a short-range disordered but long-range ordered structure in water. Upon stretch, the disordered structure transforms to a hierarchically ordered structure. This disorder-order transformation resembles the synergy of the disordered elastin and ordered collagen in load-bearing soft tissues. As entropy drives a reverse order-disorder transformation, the hydrogels can resist repeated cycles of loads without deterioration in mechanical properties. Gelatin hydrogels produced by this method combine tissue-like tunable mechanical properties that outperform the gelatin prepared by synthetic approaches, and in vivo biocomplexity beyond current natural systems. Unlike polymer engineering approaches, which rely on specific crosslinks provided by special polymers, this strategy utilizes the entropy of swollen chains and is generalizable to many other biopolymers. It could thus significantly accelerate translational success of biomaterials.
Keywords: fatigue resistant hydrogels, biomaterials, gelatin
Graphical Abstract
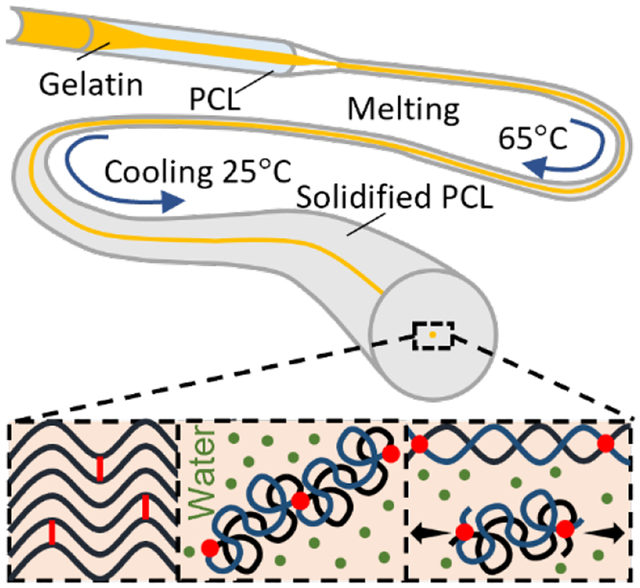
1. Introduction
Advances in biomaterials have helped launch emerging fields such as tissue engineering and regenerative medicine,[1] and facilitate fundamental research such as mechanobiology.[2] Synthetic biomaterials such as polyacrylamide hydrogels have tunable mechanical properties but lack biochemical, structural, and cell adhesive properties. While naturally derived materials such as reconstituted native extracellular matrix (ECM) components retain native biochemical properties but lose the bulk material mechanical properties. Integration of the mechanical capabilities of synthetic materials and the biochemical and architectural complexity of natural materials is highly demanded for next generation biomaterials but remains extremely challenging.
Innovations in polymer engineering have led to a leap in mechanical properties of synthetic hydrogels, especially the combination of freeze casting and salting-out, which produced the by far best-performing hydrogels.[3] However, the polymer engineering approaches are often limited to specific polymers that are biologically harsh. When applied to naturally derived materials, they either do not or only moderately improve the mechanical properties. For example, the success of freeze casting-assisted salting-out relies on strong nanocrystalline crosslinks, which is uniquely formed in polyvinyl alcohol.[4] By contrast, natural hydrogels such as gelatin form hydrophobic aggregates by the salting-out process.[5] The aggregates dissociate in water and gelatin melts at 37°C. This greatly undermines the usefulness of polymer engineering methods in biomaterial applications. Therefore, a new strategy designed for biopolymers is imperative.
Tissues’ mechanical properties originate from the hierarchical architectures of ECM components,[6] but most ECM derived hydrogels lose their hierarchical architectures. Restoring their native structure, especially the fiber structure, in principle should improve mechanical properties.[7] However, this strategy again only works for synthetic materials,[8] electrospun collagen and gelatin fibers and gelatin fiber composites are still weak and brittle like their homogeneous hydrogels.[9] This shows the importance of replicating the multiscale characteristics of ECM structures.
Collagen and elastin are primary load-bearing ECM components.[10] Collagen has an ordered triple helix structure, which is strong,[11] while elastin is soft and stretchable due to its disordered random coil structure.[12] Their synergy makes loading-bearing soft tissues soft but strong, and strong yet tough, especially in cardiovascular tissues, which are compliant when elastin bears most of the load at small strains but become exponentially stronger when collagen gradually straightens to take the load at large strains.[13] Here, we mimic this synergy by creating an elastin-like structure that can transform to a collagen-like structure under stretch.
To realize such structure, we adopted a philosophy that is opposite to both synthetic engineering and natural abstraction approaches – we used thermomechanical casting to intentionally create denatured biopolymers. The denatured biopolymer chains randomly swell and coil into a short-range elastin-like structure, but chemical crosslinks preserve their long-range ordered structure. Upon stretch, the elastin-like structure transforms to a collagen-like structure. This disorder-order transformation resembles the synergy of collagen and elastin in load-bearing soft tissues. As a demonstration, we fabricated metastable high-entropy gelatin (MHEG) hydrogels. To show their translational usefulness, we compared the mechanical properties of MHEG with those of the gelatin hydrogels prepared by the best-performing polymer engineering approaches. We also compared the cell remodeling in MHEG with current naturally derived hydrogel systems. Since entropy drives an order-disorder transformation, we also semi-quantified the role of chain entropy in the durability of MHEG.
2. Results
2.1. Inspiration from durable tissues
Deeper understanding of how collagen and elastin utilize their structural energy inspired our strategy. More importantly, an engineering definition of durability is in need, as many applications require biomaterials to sustain millions of fatigue loads and maintain original mechanical properties. This property, in polymer engineering, is fatigue threshold, which is defined as the resistance of hydrogels to propagation of an existing crack.[14] However, crack propagation is not the primary concern for biological tissues and related applications, as tissues consist of fibers that are insulated from cracks. Instead, maintenance of mechanical properties under prolonged loading is highly desirable. To better quantify this durability for biomaterial design, we introduced two properties: maximum energy density W (detailed measurements in Figure S1) and toughness consumption ratio ΔU/U. The maximum energy density is the area under stress-strain curve when samples were loaded in creep-fatigue at their critical creep stress, it describes the maximum energy a material can withstand under creep-fatigue loading for infinite cycles. The toughness consumption ratio is the ratio of areas under the stress-strain curves of the fatigue loaded and unloaded samples, it describes the capability of this material to maintain its mechanical properties and intrinsic energy.
Here we used the elastin and collagen isolated from porcine aortas as the model to establish the definition, measurement, and baseline of these two properties. To minimize the inconsistency in mechanical tests of aortas,[15] we only used the media of ascending aortas and dissected in circumferential direction (Figure 1A). Elastin was purified by autoclaving, a routine protocol to isolate insoluble elastin.[16] Collagen was isolated by elastase treatment, which has been widely used to decouple collagen and elastin in various tissues including human aortas,[17] When loaded in creep-fatigue to 40% of their ultimate tensile strength (UTS), collagen failed within minutes while elastin appeared to infinitely sustain the loading (Figure 1B). During the loading, collagen exhibited creep behaviors, with increasing strains after each cycle (Figure1C). By contrast, elastin maintained its strain even when loaded to 50% of its UTS. Elastin sets a baseline for durable materials, as it has a large We = 0.12 ± 0.02 MJ m−3 and small ΔUe/Ue = 1.8 ± 0.3% (Figure 1D). Whereas collagen is a typical disposable material with a small Wc = 0.03 ± 0.01 MJ m−3 and large ΔUc/Uc = 69 ± 5% (Figure 1E). Fourier Transform Infrared (FTIR) further showed that the fatigue loads did not affect the random coil structure of elastin but denatured almost half of triple helices in collagen (Figure S2). The triple helices in collagen fibers failed in two modes: simultaneous rupture of three helices or pulling a single α-chain out of the triple helix.[18] When subjected to cyclic stretch, the triple helices in collagen underwent the second mode, as it only required 20% of the stress required by the first mode.[19] The unfolding of helices accumulated into a permanent elongation. When the permanent elongation was large enough, samples ruptured. Unlike collagen, the coiled coils in elastin fibers can reversibly extend and recoil under the repeated cycles of loading. Therefore, after loading, the surviving collagen samples did not take any load when their strain was within their permanent elongation. Upon further stretch, their stress-strain curves ramped up steeply to a lower ultimate stress within a small stretchability. This study showed that the coil structure is the primary fatigue resistant component.
Figure 1. Inspiration from aortas.
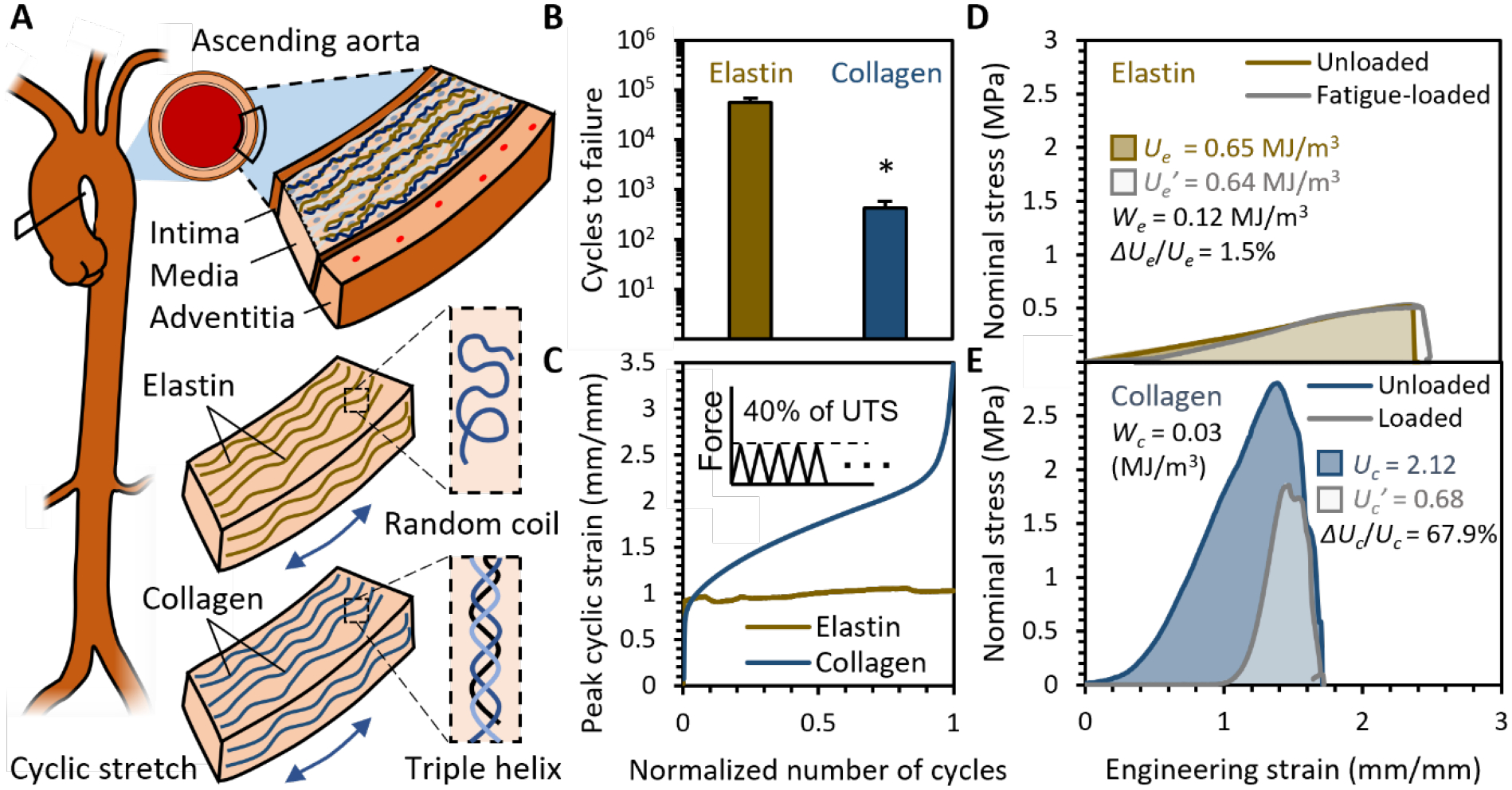
(A) Elastin and collagen isolated from porcine ascending aortas were used as the model to study and define durability for biomaterials and set baselines for maximum fatigue energy density We and toughness consumption ratio ΔU/U. (B) Samples were loaded in creep-fatigue to 40% of their UTS at 1 Hz until tissue failure or 20 hrs. Elastin sustained the loading, while collagen failed within limited cycles. n = 15, *p < 0.05, two tailed student T test. (C) Collagen exhibited creep behaviors, as their peak strain increased with cycles, while elastin did not show creep behavior. (D) Elastin has a small ΔUe/Ue, as its toughness remained the same even after having been loaded to We. (E) Collagen has a large ΔUc/Uc, as its toughness was significantly consumed after having been loaded to Wc.
2.2. Thermomechanical casting and chemical crosslinking
Inspired by the synergy of collagen and elastin, and the durability of disordered structures, we proposed a method combining thermomechanical casting and chemical crosslinking to make full use of the coiled biopolymer chains.
Thermomechanical casting induced a long-range ordered and short-range metastable structure. Highly concentrated gelatin (50 wt%) was encapsulated in polycaprolactone (PCL) tubes (Figure 2A). When heated to 65 °C, both polymers melted, and stretching the plastic PCL also applied a shear thinning effect on gelatin, which gradually aligned gelatin chains. After cooling to room temperature, the alignment was fixed as PCL solidified. This alignment is a metastable state, the gelatin chains will coil upon hydrated, due to the high content of hydrophobic glycine and proline in gelatin.[20] The gelatin was retrieved by dissolving PCL in acetone, which also dehydrated the gelatin to maintain the metastable alignment.
Figure 2. Fabrication of metastable gelatin that can transform between elastin-like and collagen-like structures.
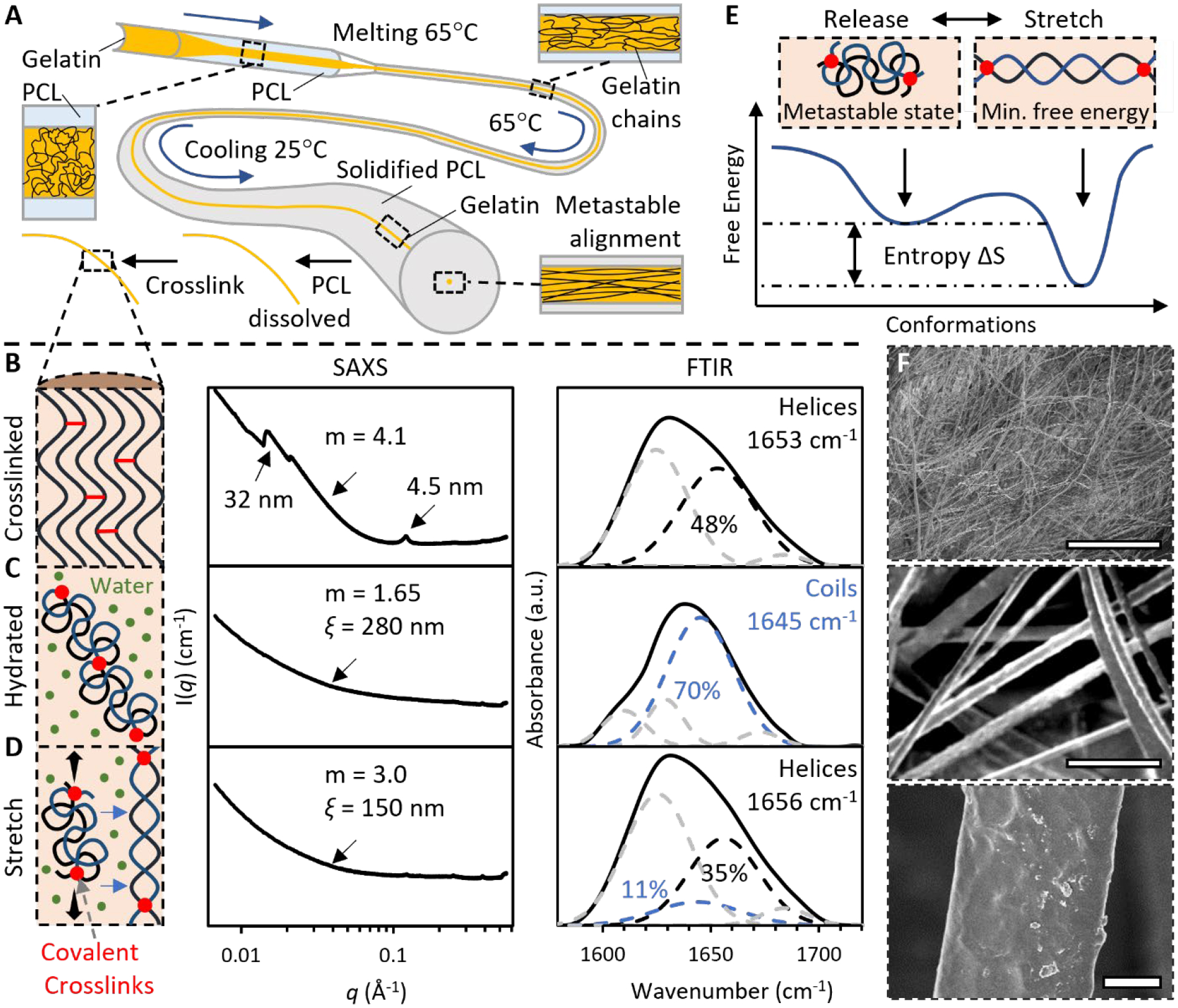
(A) Thermomechanical casting gradually aligned gelatin chains in polycaprolactone (PCL). The metastable alignment maintained when PCL was dissolved by acetone and gelatin was crosslinked by glutaraldehyde in methanol. (B) The fully crosslinked gelatin had a collagen-like structure. (C) When forming hydrogels, the gelatin had an elastin-like structure. (D) Under stretch, the hydrogel adapted a collagen-like structure. The nanostructures were estimated by fitting SAXS curve for the correlation length model, with Porod exponent m and correlation length ξ. Deconvolved FTIR amide I spectra estimated composition of secondary structures. (E) Stretch induces coil-to-helix transition while entropy drives helix-to-coil transition. (F) SEM images show that the microstructure of the gelatin hydrogels is tunable within ECM length scales. Scale bar: 10 μm.
Chemical crosslinking at the metastable state by glutaraldehyde in methanol introduced covalent bonding between aligned gelatin chains (Figure 2B). At this stage, the gelatin had a generally collagen-like ordered structure, as indicated by the peaks on the curve of Small Angle X-ray Scattering (SAXS), and the deconvoluted FTIR amide I spectra. The amide I band is mainly associated with C = O stretch of amides and is the most informative band on the secondary structure of protein, and the deconvoluted peaks around 1655 cm−1 and 1645 cm−1 were assigned to helices and random coils, respectively.[21]
2.3. Transforming between elastin-like and collagen-like structures
When forming hydrogels, the short-range metastable structure swelled and coiled into an elastin-like structure (Figure 2C). The FTIR showed that the fully crosslinked gelatin had a predominantly coil structure with 70 ± 8% random coils when hydrated, the highly crosslinked random coils is a characteristic structure of elastin. SAXS further elucidated the nanostructure of the hydrated gelatin, with data fitting for the correlation length model.[22] In the model, Porod exponent m describes polymer nanostructures,[23] and correlation length ξ represents the size of growing chemical crosslinks.[24] The hydrated gelatin had a ξ = 300 ± 19 nm, and m = 1.65 ± 0.1, which indicates the fully swollen polymer coil. Together these data revealed that the gelatin chains were crosslinked in clusters with a length of around 300 nm, close to the persistence length of collagen triple helices. Within clusters, the chains were fully swollen and coiled, and there were no small crosslinks between them.
Upon stretch, the disordered elastin-like structure transformed to the ordered collagen-like structure due to the covalent bonding between chains (Figure 2D). The Porod exponent sharply increased from m = 1.65 ± 0.1 to m = 3.0 ± 0.2 which stands for a rough surface. This echoes the coil-helix transition of DNA, in which m varies between 1.7 in coil phase and 3.7 in helix phase.[25] The FTIR amide I spectra further corroborated the stretch induced coil-helix, The content of coil structure dropped to 10 ± 5%, while helical structures with a content of 35 ± 5%, became the main structure under stretch. This extension-generated coil-helix transition is very common in biopolymers.[26]
The transformation between the elastin-like and collagen-like structures is reversible by entropy. In conformational energy profile, the coil is a metastable state while the helix is the global minimum (Figure 2E). When stretch is released, entropy drives a helix-coil transition.
The thermomechanical cast gelatin hydrogel resembles the architectural characteristics of native ECM (Figure 2F). It comprises fibers with diameters tunable from 200 nm to 200 μm, covering the typical length scales of collagen fibers and elastin fibers.
2.4. Energy state – structure – mechanical property correlation
The thermomechanical stretch has a profound impact on nanostructures of the gelatin hydrogels. Insufficient treatment did not give the metastable alignment of gelatin chains, instead, the chains stabilized as aggregates (Figure 3A). When fully crosslinked, the aggregates were so dense that electrons of Transmission Electron Microscopes (TEM) could not pass through. Their m = 2.2 ± 0.1 showed a branch structure with crosslinks between chains, and the small ξ = 9.5 ± 1.1 nm showed that those crosslinked chains were densely packed. Such structure restricts the extension of polymer chains and leads to a brittle hydrogel, as covalent crosslinks break even at a small stretch. Overmuch treatment, on the other hand, broke the polymer network (Figure 3C). The dissociated chains formed disconnected nanofibrils instead of an integrated structure, as unveiled by the TEM images of the inside structure of the gelatin hydrogels. The m = 3.4 ± 0.1 confirmed the discrete nanostructure as it points to a very rough surface, and the ξ = 10.1 ± 2.1 nm was in concert with length of nanofibrils imaged by TEM. Such disconnected structures cannot bear loads. Only chains at the metastable alignment can form the “spring” structure (Figure 3B). As a result, both hydrogels made by stabilized and unstable structures were weak and brittle, only the metastable structure fundamentally refined the mechanical properties (Figure S3). To obtain the metastable structure, the thermomechanical stretch should be kept within 103 times to 104 times.
Figure 3. Energy state – structure relation.
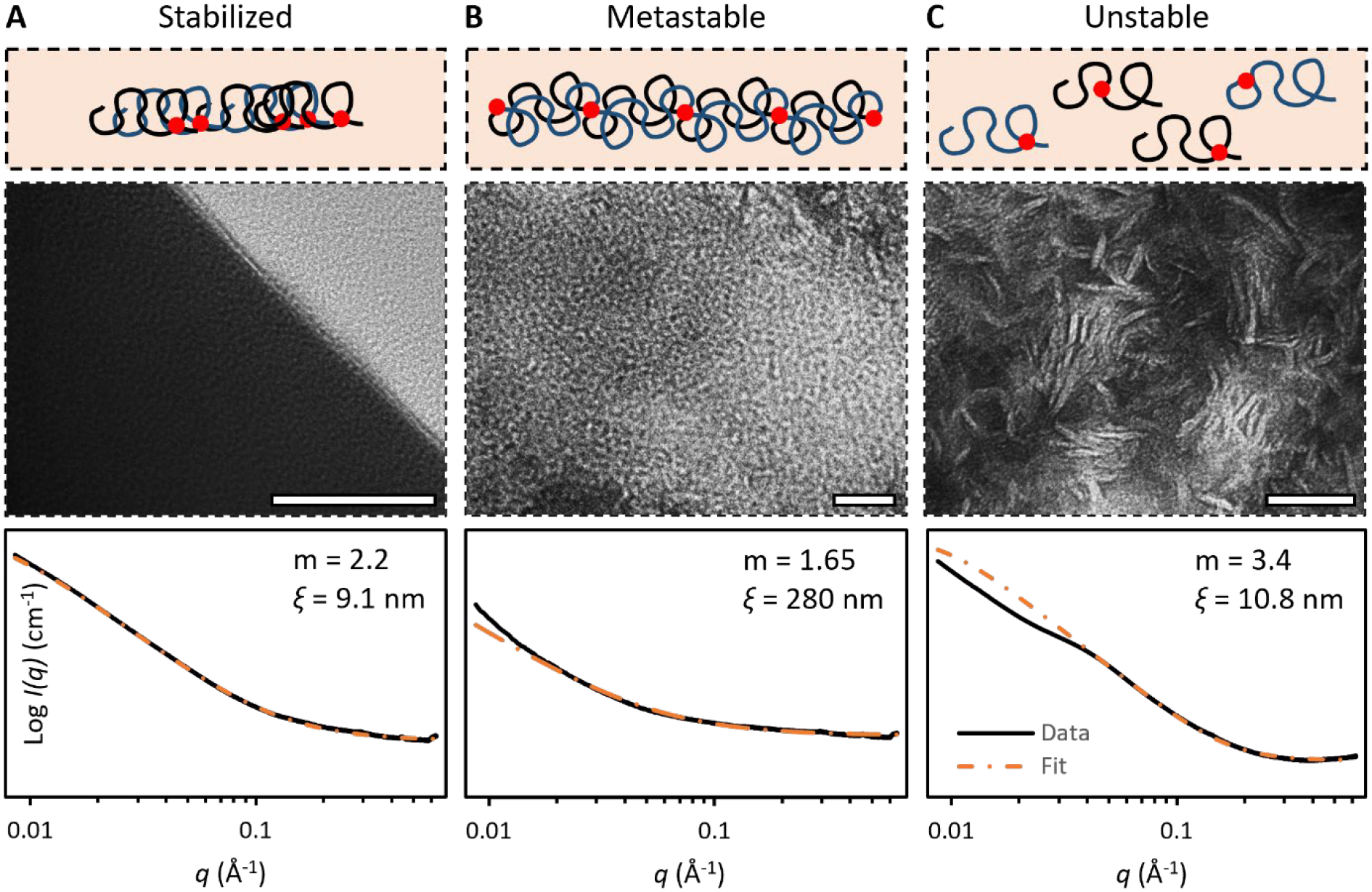
TEM images and SAXS of gelatin hydrogels comprising networks at (A) stabilized state, (B) metastable state, and (C) unstable state. Porod exponent m and correlation length ξ were measured by fitting SAXS curve for the correlation length model. Scale bar: 50 nm.
2.5. Tissue-like tunable mechanical properties
The 20% metastable high-entropy gelatin (MHEG) made by hydrating highly entangled fiber bundles were very soft, with a Young’s modulus of 0.3 – 0.5 MPa and strength of 2.0 ± 0.2 MPa, an extensibility of over 600% (Figure 4A). These properties match those of porcine aortas. In this hydrogel, dense entanglement and sparse crosslinks between fibers enabled tension to transmit from single fibers to many others, resulting in the extreme extensibility. The mechanical properties are tunable by fiber alignment and welding to match other tissues. Aligned fibers increased the modulus of MHEG by 4 times and strength by 1.5 times but also significantly decreased the extensibility by 3 times. These properties are comparable to heart valves. In this hydrogel, fibers simultaneously strengthened and toughened, thus making the hydrogel stronger. In the meantime, they were allowed to slide against each other, reserving the extensibility. The aligned fibers could be covalently welded to further increase the modulus and strength, at a cost of significantly reducing the water content and extensibility. The strength increased to up to 4 MPa and the extensibility decreased to 20%, mimicking the mechanical properties of tendons. In this hydrogel, aligned fibers were first glued together by drying from 75% ethanol and then welded by crosslinking. This process significantly restricted the transform from metastable collagen-like structure to elastin-like structure, eliminating the extensibility. Compared to crosslinked and homogenous 20% gelatin hydrogels, these MHEG hydrogels were 100-fold stronger and 1000-fold tougher.
Figure 4. Tissue-like tunable mechanical properties and durability.
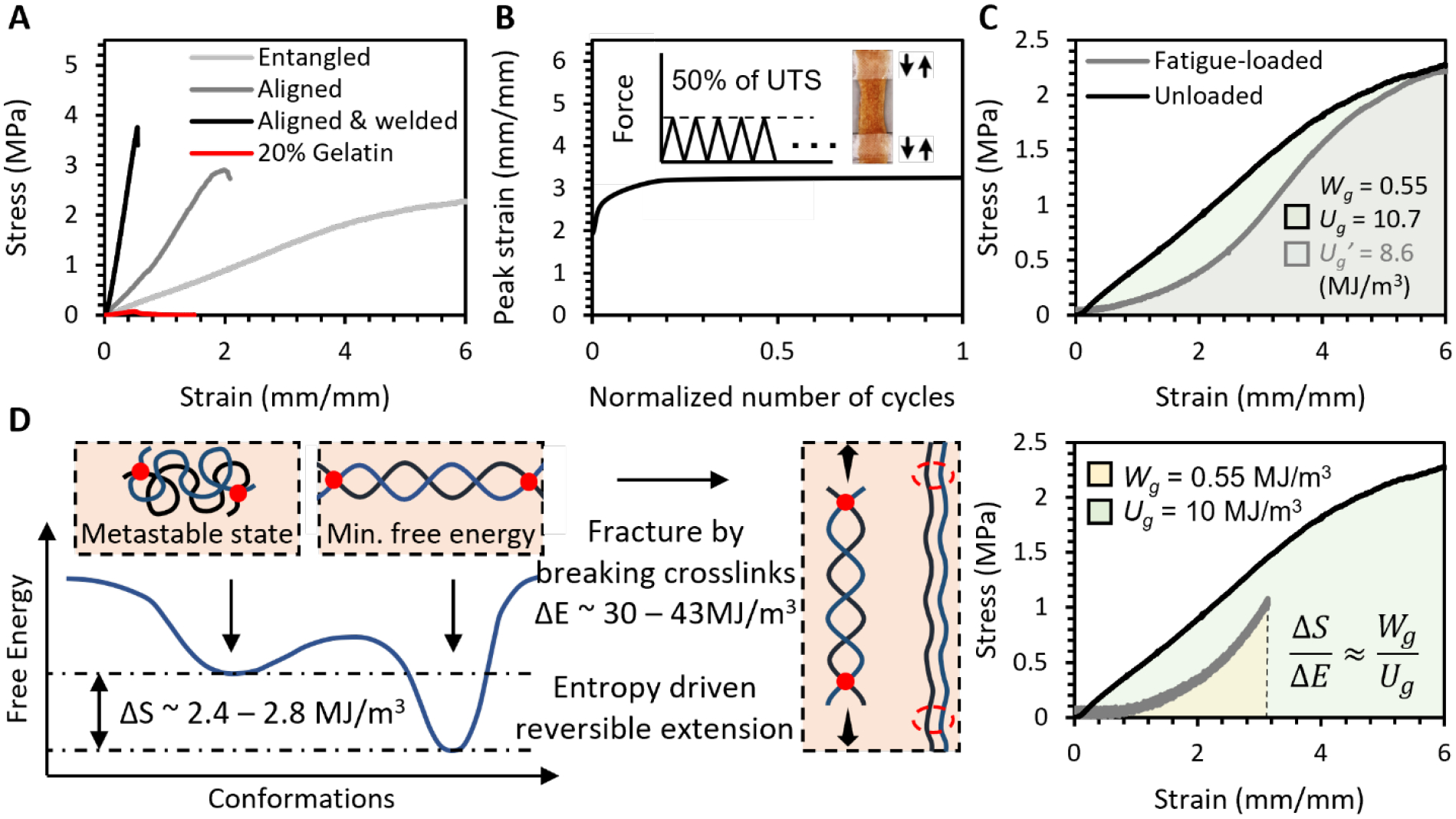
A. Stress-strain curves of hierarchically structured gelatin fiber hydrogels that resemble various soft tissues. B. The metastable high-entropy gelatin (MHEG) sustained the fatigue loading of 50% UTS at 1 Hz for 20 hours and did not exhibit creep behavior. C. Durability of the MHEG. Wg: fatigue energy density, Ug: toughness D. Entropy is the durability. ΔS is the estimated entropy difference between the elastin-like and collagen-like structures. ΔE is the energy required to break covalent bonds in fully crosslinked gelatin. ΔS/ΔE ≈ Wg/Ug.
We followed our newly defined properties to evaluate the durability of MHEG. When loaded in creep-fatigue to 50% of their UTS, it did not show creep behaviors (Figure 4B), giving Wg = 0.55 ± 0.07 MJ/m3 which is almost 5 times that of elastin. The fatigue loads only consumed less than 20% of its toughness with a ΔUg/Ug = 18.5 ± 1.5% that is about 4-fold better than that of collagen (Figure 4C). These showed MHEG had an excellent durability.
2.6. Chain entropy is durability
To bridge the molecular energy and bulk material properties, we compared ΔS/ΔE and Wg/Ug. ΔS is the entropy difference between the elastin-like and collagen-like structures, ΔE is the energy required to fracture the fully crosslinked gelatin by breaking its covalent bonds (Figure 4D). Wg is the energy of the bulk gelatin hydrogel to resist repeated cycles of loads, and Ug is the energy to resist a single cycle of load that fractures the hydrogel. By comparing ΔS/ΔE and Wg/Ug, we can determine the role of chain entropy in the durability of MHEG.
The ΔS was estimated by helical propensities.[27] Glycine and Proline are the two most abundant amino acids in gelatin, their helical propensities Δ(ΔG)(Gly) ≈ 4.18 KJ mol−1, Δ(ΔG)(Pro) ≈ 13.22 KJ mol−1. The 20 w/v% gelatin hydrogel had 200 kg gelatin molecules in 1 m−3. With an average molar mass = 80 g mol−1 per residue, the number of amino acids in the hydrogel = 200 kg (80 g mol−1)−1 = 2500 mol. The type A gelatin with a bloom number of 300g that we used typically contains 21% Gly and 12% Pro, this yields a reasonable estimate of the energy difference between coils and helices ΔS(ideal) ≈ Δ(ΔG)(Gly) × 2500 mol × 21% + Δ(ΔG)(Pro) × 2500 mol × 12% + … (the rest residues) ≈ 8 MJ. However, based on FTIR measurement, only 30 – 35% of all amino acids underwent the coil-helix transition, this gives the metastable entropy in the gelatin hydrogels ΔS ≈ 2.4 – 2.8 MJ m−3. The ΔE ≈ 300 – 430 KJ/mol × 2500 mol × 4% ≈ 30 – 43 MJ, where 300 – 430 KJ mol−1 is the energy of C-N, 4% is content of unprotonated d ε-amino groups, through which gelatin is crosslinked by glutaraldehyde.
The ΔS/ΔE ≈ 5.3 – 8.5% and Wg/Ug = 4.2 – 6.8%. The values are surprisingly close, especially given the huge crossing in length scales. This showed that entropy is enough to resist fatigue loading and protect structures from deteriorating.
2.8. Comparison with polymer engineering approaches
Double-network is one of the most widely adopted strategy for both synthetic polymers and natural biomaterials, reported naturally derived double-network hydrogels include gellan gum methacrylate/gelatin methacrylate (GelMA),[29] methacrylated tropoelastin/GelMA,[30] GelMA/tannic acid.[31] When compared to those hydrogels and salting-out gelatin, MHEG was both stronger and much tougher (Figure 5A). It showed a superior durability that is 1000-fold better (Figure 5B), and its toughness and durability are even comparable to metals (Figure 5C).
Figure 5. Comparison between metastable high-entropy gelatin and gelatin prepared by polymer engineering approaches.
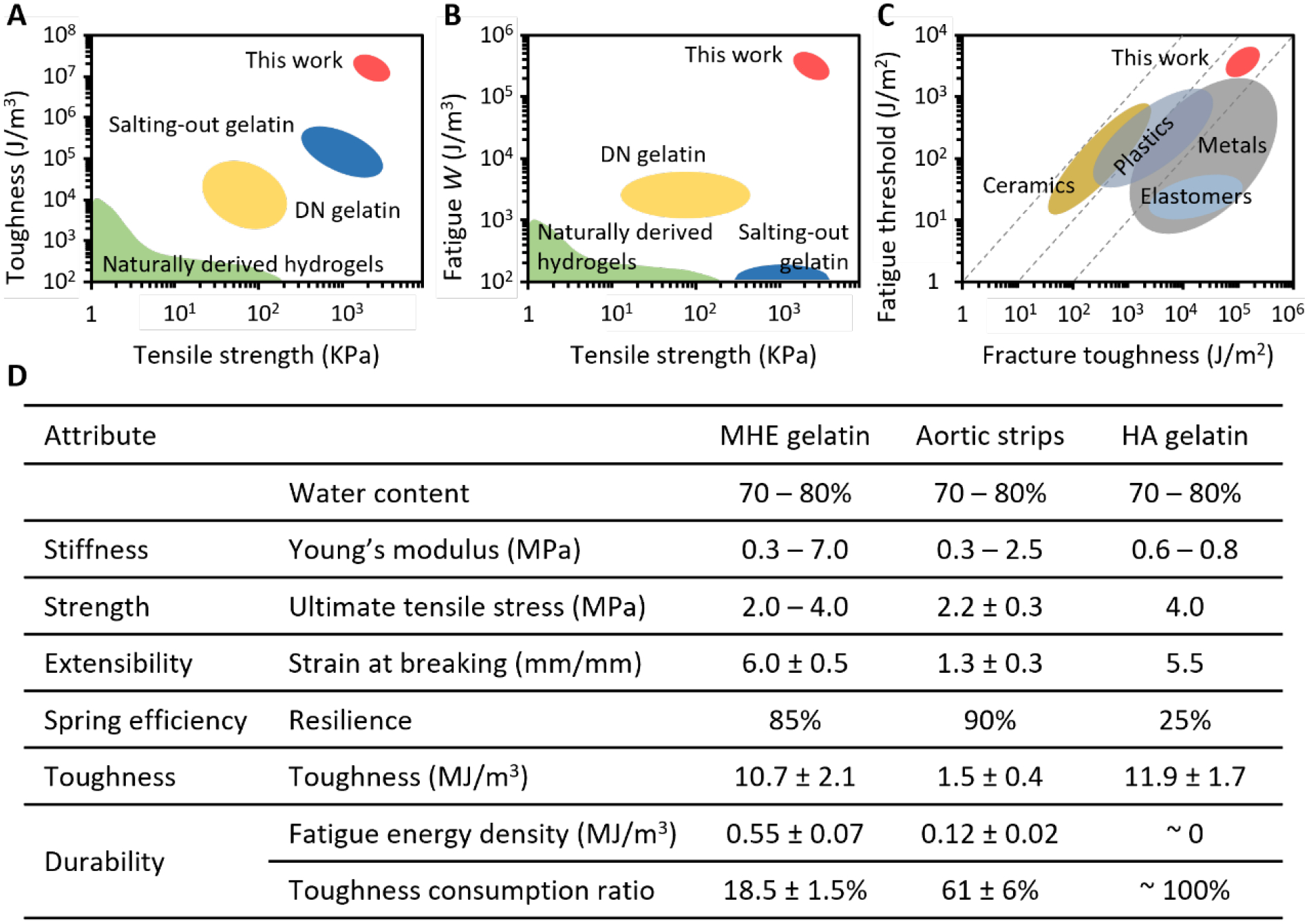
A. Toughness-strength chart compares MHEG to conventionally prepared naturally derived hydrogels,[6b, 36b] various salting-out gelatin[3, 5], various double network gelatin such as GelMA – methacrylated gellan gum,[28] GelMA – methacrylated tropoelastin,[29] and tannic acid treated GelMA.[30] B. Fatigue energy density-strength chart compares MHEG to those hydrogels, whose fatigue resistance is generally on or below 103 J/m3.[14] C. Location of MHEG among various classes of materials,[28] in durability-toughness chart. D. Detailed comparison of mechanical properties between MHEG and hierarchically anisotropic gelatin (HAG), which was produced by freeze casting-assisted salting-out.[3] This method has produced by far the best-performance hydrogels. Circumferential aortic media strips are a reference.
Freeze casting-assisted salting-out produced by far the best-performing hydrogels. Here we side-by-side compared our MHEG and the hierarchically anisotropic gelatin (HAG) produced by the freeze casting-assisted salting-out (Figure 5D). The circumferential porcine aortic media strips were used as the reference. MHEG reached the same strength, extensibility, and toughness of HAG, but HAG did not have the aorta-like resilience and durability as MHEG did.
2.9. Biocomplexity beyond current natural systems
Native ECM comprises collagen and elastin fibers with diameters typically of the order of micrometers. We produced bundles of gelatin fibers with the same dimensions. The crosslinked fibers were neutralized by glycine and dialyzed to remove possible chemical residuals. The clean and sterile fiber hydrogels were then lyophilized to obtain cotton-like dry fibrous scaffolds. The scaffolds can absorb water up to ten times their weight to form hydrogels due to the capillary action. This enabled a uniform 3D cellularization even when seeding cells on top of the scaffolds. The resultant hydrogels resemble the fibrous architecture of ECM and provide the native cell adhesion properties. These properties facilitated a fast formation of interconnected 3D cell meshwork (Figure 6A). Inside the hydrogel, 2D cell density was roughly constant throughout micrometer-scale thickness (Figure 6B), and 3D cell density was also consistent throughout millimeter-scale thickness and close to the seeding density (Figure 6C).
Figure 6. ECM-like cell functional property.
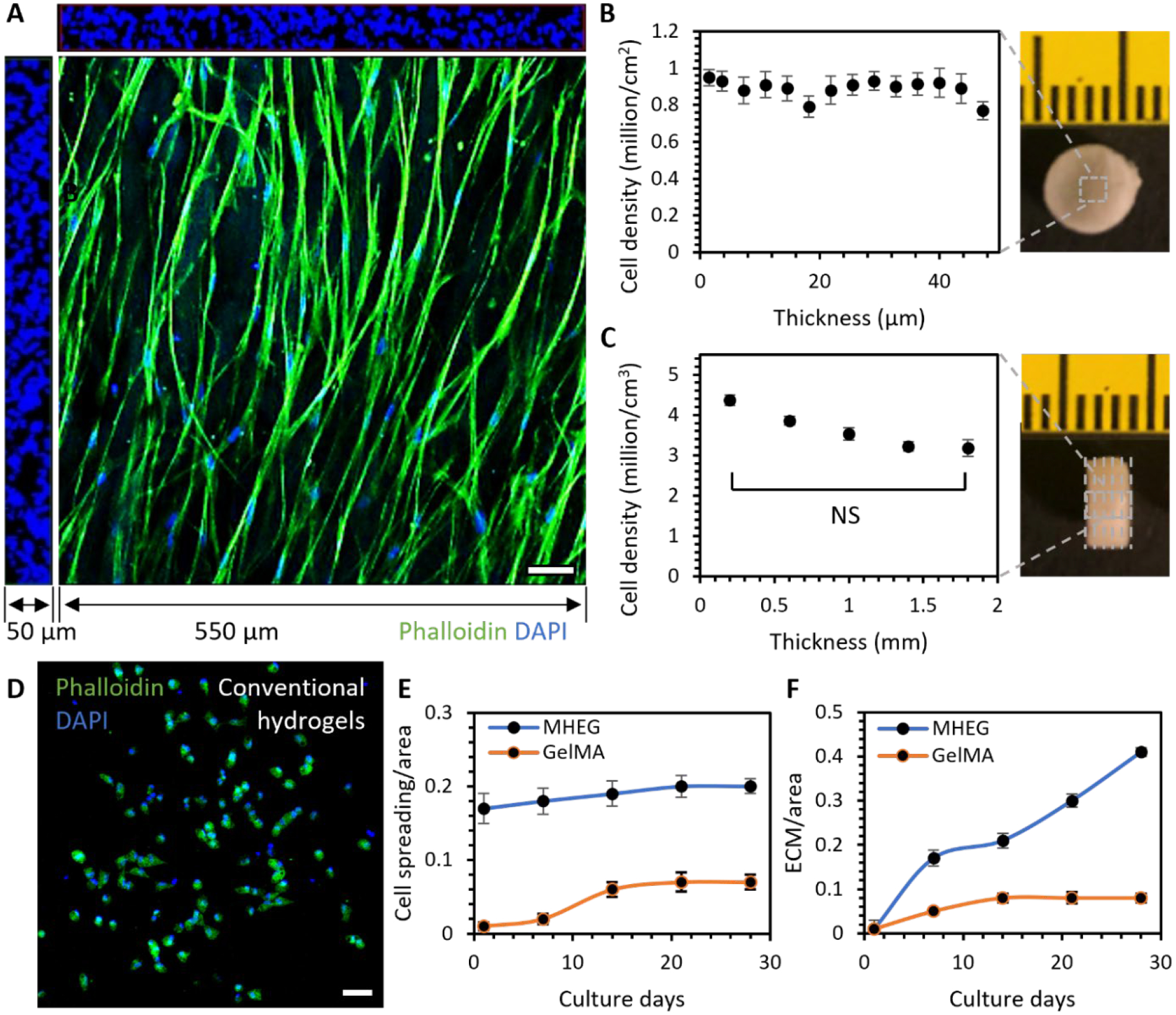
A. The 3D rendering of confocal stacks with dimensions of 550 × 550 × 50 μm showed that cells formed an interconnected 3D meshwork in MHEG. B. Cell density in each 2D stack throughout the 50 μm thickness. C. Cell density in each 3D volume at different depths of a sample with a thickness of 2 mm. D. Cells were normally isolated inside conventional GelMA hydrogels. E. Comparison of cell spreading area/image area in MHEG and GelMA with increasing culture time. F. Comparison of newly deposited ECM area/image area in MHEG and GelMA with increasing culture time. n = cells in a volume of 3 × 550 μm × 550 μm × 50 μm, two tailed student T test. Scale bar: 50 μm.
We also compared the cell-culture properties of MHEG and one of the most widely used naturally derived materials, GelMA. MHEG and GelMA have different chemistry and architecture, which lead to different properties that can affect cell responses. Chemically, MHEG is crosslinked by glutaraldehyde, which binds two free amine groups together. GelMA is crosslinked via binding between functionalized amine (methacrylamide) and functionalized hydroxyl (methacrylate) groups. Despite this difference, studies have shown that both crosslinking mechanisms only target free amine groups and do not affect the ell-binding motifs such as the Arg-Gly-Asp, or RGD sequence in gelatin.[32] However, functionalization of amine and hydroxyl groups also makes gelatin more hydrophilic than glutaraldehyde crosslinked gelatin and gives GelMA a higher wettability. These properties could influence cell adhesion onto the materials. Mechanically, although MHEG has an elasticity almost one order of magnitude higher than fully functionalized and crosslinked GelMA, the difference in local elasticity that cells can sense is much smaller. At the same crosslinking density, the glutaraldehyde crosslinked gelatin only has slightly higher elasticity than GelMA. This difference in elasticity could have a great impact on cell spreading. Architecturally, MHEG has a well-defined fibrous structure, which promotes cell spreading, migration and proliferation. In addition, MHEG has a pore size on the order of a hundred micrometers. In comparison, GelMA is a homogeneous hydrogel with nanometer scale pore size. All these differences led to a strikingly different cell behavior in MHEG and GelMA. Cells cultured inside GelMA were normally isolated from each other (Figure 6D). They had to degrade the surrounding matrix to spread, leaving a much slower cell spreading and an extra-long culture time (Figure 6E). This led to a striking difference in cell remodeling of the materials, as cells steadily deposited ECM in MHEG while they were still confined in the nanometer-scale architecture of GelMA (Figure 6F).
3. Discussion
Thermomechanical treatment such as melting and rolling are often used in casting metals but has been avoided in processing natural materials such as proteins, lipids, and carbohydrates. The natural materials often denature and lose architectural properties under the melting and rolling. The denatured chains randomly coil in water and form hopelessly weak and brittle hydrogels. However, the random coils are also a structural characteristic of elastin. We make use of this coiled chains by introducing chemical crosslinks at a metastable state when biopolymer chains are thermomechanically aligned. Upon hydration, those chains swell and coil into short-range disordered structure. Nevertheless, the chemical crosslinks preserve the long-range alignment, so that the structure can undergo the disorder-order transformation to combine the stretchability of elastin and strength of collagen. This strategy is also opposite to the philosophy of polymer engineering, which takes advantage of special synthetic polymer chains that can form specific crosslink. However, naturally derived materials biopolymers do not form such specific crosslinks, instead they have swollen and coiled chains. The philosophy that makes use of those “hopelessly useless” chains could extend to many other bio-hydrogels that cannot be enhanced by polymer engineering approaches.
The thermomechanical process is versatile and scalable. The whole process can even be done on a kitchen table by repeatedly stretching and folding a gelatin-loaded PCL rods at the melting temperature of PCL (Figure S4). This simple and no-need-to-setup technique would encourage the accessibility of biomaterial products.
The fiber architecture can regulate how cells sense and respond to biomaterials,[33] and thus control stem cell differentiation.[34] Providing an ECM-like microenvironment can maximize the efficacy of cell therapy by minimizing the maladaptive phenotype change, such as chondrocyte-fibrochondrocyte,[35] and fibroblast-myofibroblast differentiation.[36] By providing a 3D interconnected cell meshwork, MHEG is also a scarce platform for applications and studies that require mechanical or electrical connections between cells, such as cardiac and neural engineering.[37] Gelatin-based hydrogels are perhaps the most popular biomaterials by numbers of publications,[38] this work would significantly advance many emerging biomedical fields such as tissue engineering and regenerative medicine.
We also show that the chain entropy is sufficient to resist repeated loading while maintaining mechanical properties of hydrogels. The discovery of the high-entropy, structures, instead of the ordered collagen-like structures, being the primary durability component is perhaps surprising to people who focus on replicating the aligned collagen structures in hydrogels. Entropy is the energy that is unavailable to do work in a closed system. It inevitably increases in ordered structures under loading, reducing the available energy to do work, and eventually resulting in creep fatigue. Conversely, entropy maintains the state of disorder of disordered structures. Therefore, the high-entropy structures would not fatigue unless the input work overturns their entropy.
Loss of compliance is a common feature in aging tissues and injured cardiovascular tissues.[39] The “hydrogel therapy” has been proposed and shown great promise. For example, most patients who suffer from heart attacks survive and initially have adequate cardiac function. Only with time do patients experience progressive ventricular dilatation and symptoms of heart failure.[40] To slow down the maladaptive dilatation, hydrogel injections and patches have been developed,[41] but widespread efficacy has not been achieved. The “therapeutic hydrogels” should be soft enough to match the surrounding tissues and durable to accommodate long-term performance. Combination of these properties has not been realized in biologically friendly materials. The metastable high-entropy strategy could significantly accelerate translational success of “hydrogel therapy”.
4. Conclusion
In summary, we used a thermomechanochemical process to combine the structures of two primary load-bearing ECM, collagen, and elastin, in one hydrogel. Unlike the popular polymer engineering approaches, which rely on the inter-/intra-network crosslinks provided by specific polymers, our metastable high-entropy strategy makes best use of the entropy of swelling chains and thus is generalizable to many biopolymers. MHEG exhibited a combination of mechanical properties better than most hydrogels prepared by synthetic methods, and biocomplexities better than most biomaterials. This work, combining synthetic mechanical properties and natural biological complexity, sets a paradigm for next generation biomaterials.
5. Experimental Section/Methods
Aorta preparation and biomechanical tests:
Healthy porcine hearts with ascending aorta were obtained from a local slaughterhouse. First, the aortas were dissected and cleaned. Next, the intima was removed, and circumferential strips (30 mm × 10 mm) were cut out from the media-adventitia composite. Finally, the strips were sectioned into 1 mm thickness using a vibratome. Only media stripes were used and cut into a dog-bone shape with a gauge width of 10 mm. Elastin was purified by the autoclaving method. In brief, the strips were autoclaved in 20 volumes distilled water at 1 atm for 45 min. The autoclave step was repeated with fresh DI water for 3 times until no further protein was detected in the supernatant. The residue was dried by lyophilization and stored in −20 °C before tested. Collagen was prepared via digesting elastin by elastase (EC134, Elastase-High Purity, porcine, Elastin Products Company, Inc). In brief, the strips were incubated in the PBS containing 100 U/ml elastase and 0.1 mg/ml soybean trypsin inhibitor (SBTI) solution for 16 hours at room temperature with gentle agitation. The elastase treated samples were washed with fresh DI water for 3 times, lyophilized and stored in – 20 °C. A Mark-10 F105 with a 250 N load sensor was used for biomechanical tests. Dry samples were rehydrated in PBS overnight and mounted in a customized water chamber filled with PBS to prevent drying out. The stress-strain curves were measured by uniaxial tensile tests a strain-rate of 100% /s. The durability was evaluated by creep-fatigue tests. For all tests, n = 12 per group from 6 aortas. The nominal stress and strain were used.
Fiber fabrication and hydrogel preparation:
Gelatin (50 wt%, Sigma) was injected into a PCL tube (Fig.S5), which was made from moldable plastic pellets (Amazon). The tube was then sealed and heated to 65°C to melt both PCL and gelatin. The melting tube was then repeatedly stretched and folded. When cooling at room temperature, the PCL tube solidified, and the gelatin solution became strands of fibers. The fibers were retrieved by dissolving the PCL in acetone. The collected fibers were crosslinked by 1 v/v% glutaraldehyde in methanol overnight. To prepare hydrogels, fiber bundles of a g were rehydrated by dropping 3 × a cm3 water onto fibers. For cell applications, fibers were first washed in water containing 2 w/v% glycine to neutralize glutaraldehyde residues and then rinsed to remove glycine residues. The rinsed fiber bundles were dialyzed for 3 days and lyophilized for 1 day.
Small-angle X-ray scattering (SAXS):
The nanostructure analysis was based on the SAXS measurement, which was performed at the beamline ID-7A1 of Cornell High Energy Synchrotron Source using an Eiger 4M detector (Dectris, Switzerland) in vacuum. Samples were loaded into the bottom of a quartz capillary (Hampton Research, USA). Measurements were conducted at 20 °C with a sample-to-detector distance of 1648 mm. The photon energy of the X-ray was 11.3 keV, and the beam diameter was 0.25 × 0.25 mm. The SAXS detector covered a collected q range of 0.009 Å−1 to 0.56 Å−1. Each measurement was made by exposing the sample to the beam ten times for one second per exposure. Porod exponent m and correlation length ξ were determined through fitting with correlation length model at q range of 0.02 – 0.5 Å−1
where I(q) is the scattering intensity, q is the scattering vector, C is the scaling factor, B is the incoherent background scattering.
Attenuated Total Reflectance Fourier Transform Infrared spectroscopy (ATR-FTIR):
Secondary structures were estimated by FTIR, using a Bruker Vertex V80V Vacuum system with single reflection diamond crystal mount (Bruker Optik GmbH, Ettlingen, Germany). Spectra were measured at a scanner velocity of 40 kHz and a resolution of 4.0 cm−1. Data was collected over the wavenumber 3900 – 400 cm−1. The amide I band (1720 – 1580 cm−1) was deconvoluted by taking second derivatives with a 9-point smoothing Savitzky–Golay algorithm. The ratio of areas under deconvoluted peaks were used to estimate the content for each secondary structure.
Scanning Electron Microscopy (SEM):
The fiber morphology was imaged by Zeiss Gemini 500. Fibers were first coated with Gold-Platinum and then imaged under an electron beam intensity at 2 kV.
Negative Stain Transmission Electron Microscopy (NS-TEM):
The internal nanostructures of hydrated fibers were imaged by negative stains. Samples were sonicated and dispersed in water, which was broken into microscale fiber to perform TEM. Formvar/carbon 200 mesh copper grids (Electron Microscopy Sciences) was plasma cleaned by the easiGlow glow discharge system (PELCO). Fiber suspension of 10 μL was applied onto the grid, incubated for two minutes, and the excess solution was blotted with filter paper. Uranyl acetate 2% of 10 μl was then applied to the grid twice for 30-second incubation followed by blotting. The grid was air-dried for 5 minutes and visualized by Thermo Fisher F200i Transmission Electron Microscope at 120 keV.
Mechanical tests of hydrogels:
A Mark-10 F105 with a 250 N load sensor was used for mechanical tests. Sample dimensions: width 10 mm, length between two grippers 20 mm, thickness around 1 mm. The stress-strain curves were measured by uniaxial tensile tests a strain-rate of 2% s−1 until fracture. The durability (maximum fatigue energy density and toughness consumption ratio) was evaluated by creep-fatigue tests (Figure S1). Samples were loaded in creep-fatigue to 10 – 50% of their ultimate tensile stress at a frequency of 1 Hz until sample rupture or 20 hours. After the creep-fatigue tests, the stress-strain curves of surviving samples were measured again to calculate the toughness consumption ratio. For all tests, n = 5 per group.
Cell culture:
hMSCs were purchased from Lonza and Passage-5 cells were used. Cells were dropped onto dry scaffolds at a seeding density of 5 million cm−3. The scaffolds absorbed the medium and cells by capillary action. The scaffolds were incubated for 1 – 3 hours to allow cell adhesion before adding culture medium, which was prepared by supplementing Dulbecco’s modified Eagle medium (Invitrogen) with 10% fetal bovine serum (FBS, Invitrogen) and 1% penicillin/streptomycin (P/S, Invitrogen).
Cell imaging and analysis:
Scaffolds were fixed in 4% paraformaldehyde in PBS for 15 min at room temperature and permeabilized with 0.05% Triton X-100 in PBS for 15 min. The cytoskeleton was stained by phalloidin (Invitrogen) and cell nuclei were labeled by DAPI (Invitrogen). Confocal images were taken by Zeiss LSM 710. ImageJ was used for image analysis. Nuclei were counted in each stack of confocal images for estimation of the 2D cell density. The 3D object algorithm was used to count nuclei in 3D stacks. For significance analysis between two groups, the two-tailed Student T-Tests were used.
Supplementary Material
Acknowledgements
This work was supported by American Heart Association grant 821615 (MW), National Science Foundation award DMR-1829070 (Cornell High Energy Synchrotron Source), National Institute of General Medical Sciences award GM-124166 (Macromolecular Diffraction at Cornell High Energy Synchrotron Source), National Science Foundation award DMR-1719875 (Cornell Center for Materials Research Shared Facilities)
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dani Liu, School of Aeronautic Science and Engineering, Beihang University, Beijing, 100191, China.
Shi Feng, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY, 14853, USA.
Qingqiu Huang, Cornell High Energy Synchrotron Source, Cornell University, Ithaca, NY, 14853, USA.
Shuofei Sun, Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY, 14853, USA.
Gening Dong, Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY, 14853, USA.
Feifei Long, Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY, 14853, USA.
Mario Milazzo, Department of Civil and Industrial Engineering, University of Pisa, Pisa, 56122 Italy; Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA.
Mingkun Wang, School of Aeronautic Science and Engineering, Beihang University, Beijing, 100191, China; Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY, 14853, USA.
References
- [1].a) Speidel AT, Grigsby CL, Stevens MM, Nature Materials 2022, 21, 989; [DOI] [PubMed] [Google Scholar]; b) Han X, Alu A, Liu H, Shi Y, Wei X, Cai L, Wei Y, Bioactive Materials 2022, 17, 29; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Facklam AL, Volpatti LR, Anderson DG, Advanced Materials 2020, 32, 1902005. [DOI] [PubMed] [Google Scholar]
- [2].a) Brusatin G, Panciera T, Gandin A, Citron A, Piccolo S, Nature Materials 2018, 17, 1063; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li L, Eyckmans J, Chen CS, Nature Materials 2017, 16, 1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hua M, Wu S, Ma Y, Zhao Y, Chen Z, Frenkel I, Strzalka J, Zhou H, Zhu X, He X, Nature 2021, 590, 594. [DOI] [PubMed] [Google Scholar]
- [4].a) Lin S, Liu X, Liu J, Yuk H, Loh H-C, Parada GA, Settens C, Song J, Masic A, McKinley GH, Zhao X, Science Advances 2019, 5, eaau8528; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin S, Liu J, Liu X, Zhao X, Proceedings of the National Academy of Sciences 2019, 116, 10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].He Q, Huang Y, Wang S, Advanced Functional Materials 2018, 28, 1705069. [Google Scholar]
- [6].a) Burla F, Mulla Y, Vos BE, Aufderhorst-Roberts A, Koenderink GH, Nature Reviews Physics 2019, 1, 249; [Google Scholar]; b) Zhao X, Chen X, Yuk H, Lin S, Liu X, Parada G, Chemical Reviews 2021, 121, 4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wegst UGK, Bai H, Saiz E, Tomsia AP, Ritchie RO, Nature Materials 2015, 14, 23. [DOI] [PubMed] [Google Scholar]
- [8].Ni J, Lin S, Qin Z, Veysset D, Liu X, Sun Y, Hsieh AJ, Radovitzky R, Nelson KA, Zhao X, Matter 2021, 4, 1919. [Google Scholar]
- [9].Kong B, Liu R, Guo J, Lu L, Zhou Q, Zhao Y, Bioactive Materials 2023, 19, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Harrington MJ, Fratzl P, Progress in Materials Science 2021, 120, 100767. [Google Scholar]
- [11].Bailey AJ, Macmillan J, Shrewry PR, Tatham AS, Puxkandl R, Zizak I, Paris O, Keckes J, Tesch W, Bernstorff S, Purslow P, Fratzl P, Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 2002, 357, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bailey AJ, Macmillan J, Shrewry PR, Tatham AS, Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K, Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 2002, 357, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fomovsky GM, Thomopoulos S, Holmes JW, Journal of Molecular and Cellular Cardiology 2010, 48, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bai R, Yang J, Suo Z, European Journal of Mechanics - A/Solids 2019, 74, 337. [Google Scholar]
- [15].Pei M, Zou D, Gao Y, Zhang J, Huang P, Wang J, Huang J, Li Z, Chen Y, PLOS ONE 2021, 16, e0244390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Halabi CM, Mecham RP, in Methods in Cell Biology, Vol. 143 (Ed: Mecham RP), Academic Press, 2018. [Google Scholar]
- [17].a) Schriefl AJ, Schmidt T, Balzani D, Sommer G, Holzapfel GA, Acta Biomaterialia 2015, 17, 125; [DOI] [PubMed] [Google Scholar]; b) Ross CJ, Laurence DW, Echols AL, Babu AR, Gu T, Duginski GA, Johns CH, Mullins BT, Casey KM, Laurence KA, Zhao YD, Amini R, Fung K-M, Mir A, Burkhart HM, Wu Y, Holzapfel GA, Lee C-H, Acta Biomaterialia 2021, 135, 425; [DOI] [PubMed] [Google Scholar]; c) Godinho MS, Thorpe CT, Greenwald SE, Screen HRC, Acta Biomaterialia 2021, 123, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zitnay JL, Li Y, Qin Z, San BH, Depalle B, Reese SP, Buehler MJ, Yu SM, Weiss JA, Nature Communications 2017, 8, 14913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zitnay JL, Jung GS, Lin AH, Qin Z, Li Y, Yu SM, Buehler MJ, Weiss JA, Science Advances 2020, 6, eaba2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rauscher S, Pomès R, eLife 2017, 6, e26526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Yang H, Yang S, Kong J, Dong A, Yu S, Nature Protocols 2015, 10, 382; [DOI] [PubMed] [Google Scholar]; b) Staroszczyk H, Pielichowska J, Sztuka K, Stangret J, Kołodziejska I, Food Chemistry 2012, 130, 335. [Google Scholar]
- [22].a) Boyd-Moss M, Firipis K, O’Connell CD, Rifai A, Quigley A, Boer G, Long BM, Nisbet DR, Williams RJ, Materials Chemistry Frontiers 2021, 5, 8025; [Google Scholar]; b) Wisotzki EI, Tempesti P, Fratini E, Mayr SG, Physical Chemistry Chemical Physics 2017, 19, 12064. [DOI] [PubMed] [Google Scholar]
- [23].Yang Z, Hemar Y, Hilliou L, Gilbert EP, McGillivray DJ, Williams MAK, Chaieb S, Biomacromolecules 2016, 17, 590. [DOI] [PubMed] [Google Scholar]
- [24].Bode F, da Silva MA, Smith P, Lorenz CD, McCullen S, Stevens MM, Dreiss CA, Soft Matter 2013, 9, 6986. [DOI] [PubMed] [Google Scholar]
- [25].Hammouda B, 2016.
- [26].Courty S, Gornall JL, Terentjev EM, Proceedings of the National Academy of Sciences 2005, 102, 13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nick Pace C, Martin Scholtz J, Biophys J 1998, 75, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xiang C, Wang Z, Yang C, Yao X, Wang Y, Suo Z, Materials Today 2020, 34, 7. [Google Scholar]
- [29].Shin H, Olsen BD, Khademhosseini A, Biomaterials 2012, 33, 3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Annabi N, Mithieux SM, Zorlutuna P, Camci-Unal G, Weiss AS, Khademhosseini A, Biomaterials 2013, 34, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu B, Wang Y, Miao Y, Zhang X, Fan Z, Singh G, Zhang X, Xu K, Li B, Hu Z, Xing M, Biomaterials 2018, 171, 83. [DOI] [PubMed] [Google Scholar]
- [32].Yang G, Xiao Z, Long H, Ma K, Zhang J, Ren X, Zhang J, Scientific Reports 2018, 8, 1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Baker BM, Trappmann B, Wang WY, Sakar MS, Kim IL, Shenoy VB, Burdick JA, Chen CS, Nature Materials 2015, 14, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang M, Cui C, Ibrahim MM, Han B, Li Q, Pacifici M, Lawrence JTR, Han L, Han L-H, Advanced Functional Materials 2019, 29, 1808967. [Google Scholar]
- [35].Lee H.-p., Gu L, Mooney DJ, Levenston ME, Chaudhuri O, Nature Materials 2017, 16, 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fioretta ES, Motta SE, Lintas V, Loerakker S, Parker KK, Baaijens FPT, Falk V, Hoerstrup SP, Emmert MY, Nature Reviews Cardiology 2021, 18, 92. [DOI] [PubMed] [Google Scholar]
- [37].Elkhoury K, Morsink M, Sanchez-Gonzalez L, Kahn C, Tamayol A, Arab-Tehrany E, Bioactive Materials 2021, 6, 3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].a) Kurian AG, Singh RK, Patel KD, Lee J-H, Kim H-W, Bioactive Materials 2022, 8, 267; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A, Biomaterials 2015, 73, 254; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yue K, Li X, Schrobback K, Sheikhi A, Annabi N, Leijten J, Zhang W, Zhang YS, Hutmacher DW, Klein TJ, Khademhosseini A, Biomaterials 2017, 139, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Villalobos Lizardi JC, Baranger J, Nguyen MB, Asnacios A, Malik A, Lumens J, Mertens L, Friedberg MK, Simmons CA, Pernot M, Villemain O, Nature Cardiovascular Research 2022, 1, 8. [DOI] [PubMed] [Google Scholar]
- [40].Burdick JA, Mauck RL, Gorman JH, Gorman RC, Science Translational Medicine 2013, 5, 176ps4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lin X, Liu Y, Bai A, Cai H, Bai Y, Jiang W, Yang H, Wang X, Yang L, Sun N, Gao H, Nature Biomedical Engineering 2019, 3, 632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.