

Abstract
The tumor microenvironment (TME) is a dynamic and complex matter shaped by heterogenous cancer and cancer‐associated cells present at the tumor site. Hyaluronan (HA) is a major TME component that plays pro‐tumorigenic and carcinogenic functions. These functions are mediated by different hyaladherins expressed by cancer and tumor‐associated cells triggering downstream signaling pathways that determine cell fate and contribute to TME progression toward a carcinogenic state. Here, the interaction of HA is reviewed with several cell‐surface hyaladherins—CD44, RHAMM, TLR2 and 4, LYVE‐1, HARE, and layilin. The signaling pathways activated by these interactions and the respective response of different cell populations within the TME, and the modulation of the TME, are discussed. Potential cancer therapies via targeting these interactions are also briefly discussed.
Keywords: CD44, HARE/Stab2, hyaladherins, layilin, LYVE‐1, RHAMM, TLR2/4
Hyaluronan is a major component of the tumor microenvironment (TME) that plays pro‐tumorigenic and carcinogenic functions. These functions are mediated by different hyaladherins, namely CD44, RHAMM, TLR2, and 4, LYVE‐1, HARE and layilin. The respective signaling pathways relevant to the modulation of the TME and potential cancer therapies are discussed via targeting the hyaladherins interactions.
1. Introduction
The tumor microenvironment (TME) is a complex and dynamic setting where tumorigenesis occurs. TME comprises multiple heterogeneous cellular populations, including cancer stem cells (CSC); tumor‐infiltrating immune, and inflammatory cells T and B lymphocytes, tumor‐associated macrophages, dendritic cells, natural killer cells, neutrophils, and myeloid‐derived suppressor cells; stromal cells – cancer‐associated fibroblasts, pericytes, mesenchymal stromal cells; blood and lymphatic vessels; and tissue‐specific cell types (Figure 1 ). These cells participate in autocrine and paracrine signaling that modify the surrounding extracellular matrix (ECM) through the secretion and deposition of bioactive molecules. The introduced changes modulate the cell‐cell and cell‐ECM interactions, contributing to tumor cells proliferation, invasion, metastasis, and determining the therapeutic response, as explained in detail by several excellent recent reviews.[ 1 , 2 , 3 , 4 , 5 , 6 ]
Figure 1.
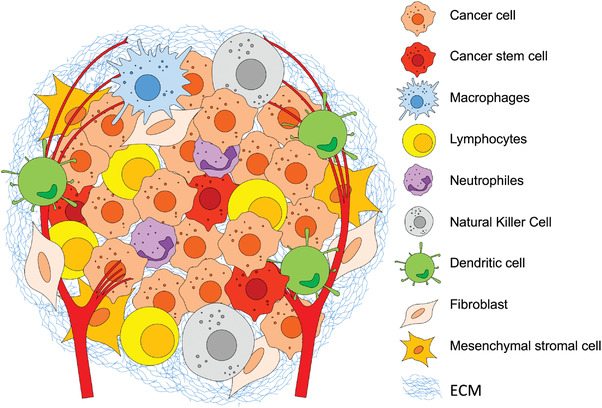
Schematic presentation of the tumor microenvironment complexity arising from the heterogeneous cellular populations that constantly remodel the surrounding extracellular matrix (ECM).
Among different signaling molecules in TME, hyaluronan (HA) is a well‐known multimodal player: it can regulate cell proliferation, apoptosis, multidrug resistance, and survival; induce cell invasion and metastases formation, and contribute to evading the immune response. HA homeostasis is altered since the early stages of tumorigenesis, and this alteration contributes to cancer initiation and progression (Figure 2 ).[ 7 , 8 , 9 , 10 , 11 ] HA is synthesized at the cell membrane by three hyaluronan synthases (HAS1‐3) through the alternate conjugation of D‐glucuronic acid and N‐acetyl‐D‐glucosamine (Figure 2A).[ 12 ] The synthesized linear polymer with a size of 3 to 4 MDa is secreted into the ECM, where it is degraded by hyaluronidases (HYAL) and reactive oxygen/nitrogen species (ROS/RNS) to shorter HA fragments and oligomers.[ 13 ] Both synthesis and degradation of HA are upregulated in most cancers, resulting in an accelerated turnover and accumulation of HA with different sizes in extra‐ and pericellular space (Figure 2B). Such HA accumulation is a hallmark of several cancers, e.g., pancreatic carcinomas, lung, breast, prostate, and colorectal cancers, and head to neck tumors, and a marker of poor prognosis.[ 5 , 14 , 15 ]
Figure 2.
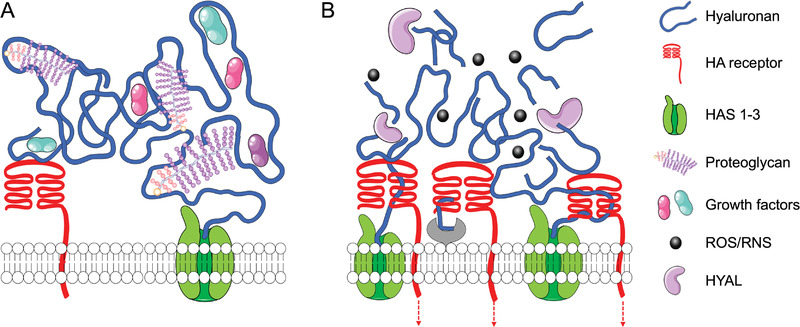
Hyaluronan synthesis and degradation. A) In healthy tissues, HA is synthesized at the cell membrane by HAS1‐3 into a high molecular weight polymer. HA is secreted and deposited into the pericellular and extracellular matrix, where it interacts with cell receptors and other ECM components, including proteoglycans and hyaladherins. B) In the tumor microenvironment, the increased expression and activity of HAS and HYAL, together with high redox potential and increased reactive oxygen/nitrogen species (ROS/RNS), lead to accelerated HA turnover and accumulation of HA fragments and oligomers.
The bioactivity of HA depends on its molecular weight: high molecular weight (HMW, molecular mass above 1 MDa) and low molecular weight HA (LMW, molecular mass up to 700 kDa) usually induce different and often opposite cell behavior (reviewed[ 16 , 17 ]). This divergent cell response is due to the different interactions between the available extra and pericellular HA and the specific cell receptors, namely cluster differentiation 44 (CD44), the receptor for hyaluronan‐mediated motility (RHAMM), toll‐like receptor 2 (TLR‐2), TLR4, lymphatic vessel endothelial hyaluronan receptor 1 (LYVE‐1), hyaluronan receptor for endocytosis (HARE), and layilin, which regulate the TME interactome and thus, the cancer aggressiveness and metastatic potential (Table 1 ).
Table 1.
Cell response induced by HA interactions with its receptors in different cancer models. Data from the past five years
| Receptor | Cell response | Model | Reference |
|---|---|---|---|
| CD44 | Proliferation | Cholangiocarcinoma cell lines; Non‐small cell lung cancer; Breast carcinoma cells; Colon cancer cells; Breast cancer stem cell‐like cells; Ovarian cancer cells; Pancreatic cancer. | [18, 19, 20, 21, 22, 23, 24] |
| Anti‐apoptosis/Survival | Non‐small cell lung cancer; Breast cancer cells; Colon cancer cells; Cancer stem cells of head and neck squamous cell carcinoma. | [19, 20, 21, 25, 26] | |
| Motility and Invasion | Cholangiocarcinoma cell lines; Breast carcinoma cells; Head and neck squamous cell carcinomas and its cancer stem cells; Ovarian cancer cells. | [18, 23, 26, 27, 28] | |
| Epithelial to mesenchymal transition | Head and neck squamous cell carcinomas; Breast cancer cells; Liver cancer cells. | [28, 29, 30] | |
| Stemness | Head and neck squamous cell carcinomas; Breast cancer stem cell‐like cells; Liver cancer cells. | [22, 28, 30] | |
| Radio/Chemotherapy resistance | Head and neck squamous cell carcinomas; Colorectal cancer; Breast cancer stem cell‐like cells. | [21, 22, 26, 28] | |
| Metastasis | Head and neck squamous cell carcinomas; Pancreatic cancer. | [24, 28] | |
| RHAMM | Proliferation | Lung adenocarcinoma cells; Ovarian cancer cells; Prostate cancer cells; Breast carcinoma cells; Adenocarcinoma of the breast duct; Non‐small cell lung cancer; Fibrosarcoma cells. | [19, 31, 32, 33] |
| Survival | Ovarian cancer cells; Prostate cancer cells; Breast carcinoma cells; Adenocarcinoma of the breast duct; Non‐small cell lung cancer. | [19, 32] | |
| Motility | Lung adenocarcinoma cells; choriocarcinoma cells. | [31, 34] | |
| Epithelial to mesenchymal transition; Chemotherapy resistance and stemness | Gastric cancer cells. | [35] | |
| TLR2,4 | Inflammation | Melanocytes. | [36] |
| Proliferation | Colorectal cancer; Colorectal cancer cells; Glioblastoma stem‐like cells. | [21, 37] | |
| Anti‐apoptosis/Survival | Colorectal cancer; Colorectal cancer cells. | [21] | |
| Radiotherapy resistance | Colorectal cancer. | [21] | |
| LYVE‐1 | (Lymph node) Metastasis | Breast cancer cells; Oral squamous cell carcinoma; Melanoma cells. | [38, 39, 40] |
| Proliferation | Lymphatic endothelial cells. | [41] | |
| Motility | Breast cancer cells; Lymphatic endothelial cells. | [38] | |
| Adhesion/Docking and migration (to TME) | Dendritic cells. | [42] | |
| Lymphangiogenesis | Lymphatic endothelial cells. | [38, 41] | |
| Tight junctions disruption | Lymphatic vessels endothelium. | [40] | |
| Soluble LYVE‐1 | Inhibits cell proliferation | Melanoma cells. | [43] |
| Inhibits lymphangiogenesis | Corneal lymphangiogenic models in mice. | [44] | |
| HARE | Proliferation, motility, and inflammation | Lymphatic endothelial cells. | [45] |
| Layilin | Immunomodulator | Immuno infiltrates in gastric and colon cancers; Infiltrating T cells in liver cancer; | [46, 47] |
| Invasion | Glioma cells; | [48] |
In this review, we discuss the interactions of HA with these receptors and the respective downstream signaling pathways leading to tumorigenic behavior of different cell populations within TME. We also tackle the potential of targeting HA/receptors interactions as a therapeutic approach.
2. Cluster of Differentiation 44 (CD44)
CD44 is the most studied HA receptor. It is involved in the normal homeostasis of different tissues and ubiquitously expressed on the surface of the cells of these tissues. It is overexpressed in several cancers and is a compelling marker of CSC.[ 28 ] CD44 is a non‐kinase transmembrane glycoprotein (P‐glycoprotein 1) that interacts with HA through two conserved HA‐binding regions (BX7B motifs – Link domain) at the N‐terminal region of the extracellular portion (Figure 3 ).[ 49 ] CD44 mediates HA signaling and HA internalization and degradation, thus contributing to HA local turnover.[ 50 ] CD44 is coded by a single gene but has different variants (CD44v) due to alternative splicing and post‐translational modifications, e.g., glycosylation, which are often deregulated in cancer. Alternative splicing of CD44 gene results in CD44 isoforms with variable exons – variant 2 to 10 (Figure 3), which provide new conformations and binding sites and have different/additional functions than standard CD44 (CD44s, 85–95 kDa), affecting tumor‐initiating and metastatic potential (reviewed[ 51 ]). Altered N‐ or O‐ glycosylation of CD44 also affects HA‐CD44 interactions and the metastatic potential of cancer cells.[ 52 , 53 ] As an example, truncated O‐glycosylation enhances the affinity of HA to CD44, thus, enhancing the tumorigenic signaling.[ 52 ]
Figure 3.
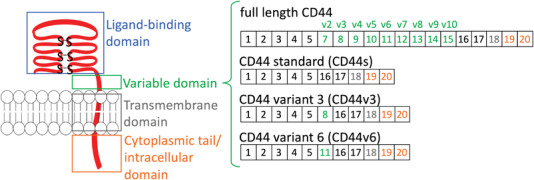
CD44 structural diversity is due to variable exons (v) that are inserted in the extracellular region next to the cell membrane. Some of the cancer‐associated variants are also shown.
2.1. CD44 as a Signaling Hub
In TME, CD44 acts as a signaling hub because it can trigger different signaling cascades in a function of the environment, thus, eliciting various cellular responses. The high expression of CD44 alone is not an indicator of its (pro)tumorogenic behavior, as the activation state of CD44 can be different.[ 54 ] CD44 is usually activated upon HA binding, which elicits protein conformational changes. The adopted conformations favor the formation of multimolecular complexes by additional binding of adaptor molecules, such as molecular switchers (e.g., Rho GTPase proteins RhoA, RhoC, Rac, and Cdc42) or cytoskeletal anchor proteins that trigger different downstream signaling.
There are also other mechanisms by which CD44 can affect the cell behavior in TME. One of these mechanisms implies the formation of clusters by multivalent interactions of HA with HAS and other receptors (e.g., Erbb2, Erbb3, EGFR, IGF1R‐b, PDGFR, and c‐MET) within lipid rafts.[ 55 , 56 , 57 , 58 , 59 , 60 , 61 , 62 ] Another mechanism involves sequential cleavage of the CD44 that is frequently observed in human tumors and associated with increased metastatic potential.[ 55 , 63 , 64 , 65 ] In this process, the ectodomain of CD44 is first cleaved by membrane‐type 1 matrix metalloproteinases (MT1‐MMP) and then presenilin‐1/γ‐secretase act to release the CD44 intracellular domain. CD44 intracellular domain translocates to the nucleus where is involved in the regulation of the transcription of genes related to survival, inflammation, glycolysis, and tumor invasion.
2.2. HA/CD44 Mediates Enhanced Cell Growth and Survival via RhoA/ROK Activation
HA binding to CD44 can activate transforming protein RhoA/Rho‐associated protein kinase (RhoA/ROK) downstream signaling via phosphorylation of proto‐oncogene tyrosine‐protein kinase (Src) (Figure 4A).[ 66 , 67 ] The activation of this signaling cascade is associated with enhanced cell growth, survival, and invasion in different cancers, including head and neck, pancreatic, colon, ovarian, and breast.[ 19 , 23 , 24 , 59 , 64 , 68 , 69 ]
Figure 4.
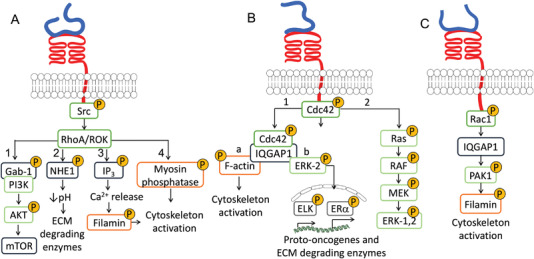
HA/CD44‐mediated RhoGTPase signaling pathways via A) RhoA/ROK, B) Cdc42, and C) Rac1 modulate cell proliferation, motility, and invasion.
The increased growth, survival and invasiveness of cancer cells result from the GRB2‐associated‐binding protein 1, phosphoinositide 3‐kinases and Protein kinase B pathway (Gab1‐PI3K/AKT, Figure 4A1)[ 67 , 68 , 71 ] that triggers activation of the transcriptor factor mammalian target of rapamycin (mTOR), downregulation of tumor suppressor proteins such as Homeobox D10 (HOXD10) and cyclin‐dependent kinase inhibitor 1 (p21), upregulation of survival proteins Baculoviral IAP repeat‐containing protein3 (cIAP‐2) and X‐linked inhibitor of apoptosis protein (XIAP), i.e., different factors that contribute to a (pro)tumorogenic profile.[ 18 , 26 , 70 , 71 , 72 , 73 ] As an example, in human metastatic breast tumor cells, RhoA/ROK activates the adaptor protein Gab‐1 linked to PI3K/AKT signaling, mediating cytokine macrophage‐colony stimulating factor (CSF‐1) production and enhancing cell growth, survival, and invasion.[ 70 ] In addition, the activation of AKT signaling can lead to HAS2 overexpression and induce a feedback loop: an enhanced HA synthesis leads to CD44 overexpression and sustain PI3K/AKT signaling.[ 25 ]
The HA/CD44‐mediated RhoA/ROK activation also affects the cytoskeletal function and cell motility of metastatic breast tumor cells and colon carcinoma cells (Figure 4A2–4).[ 74 , 75 , 76 ] RhoA/ROK activates Na+‐H+ exchanger‐1 (NHE1) (Figure 4A2), resulting in ECM acidification and activation of ECM‐degrading enzymes thus facilitating cell motility and invasion (Figure 4A2).[ 76 ] Another pathway involves RhoA/ROK triggered phosphorylation of inositol trisphosphate (IP3) receptors that induces internal Ca2+ release and activation of Ca2+‐dependent signaling (Figure 4A3) such as phosphorylation of filamin, which activates the cytoskeleton and enhances cell motility.[ 75 ] In addition, RhoA/ROK activates myosin phosphatase that is a direct regulator of cytoskeletal contraction (Figure 4A4).[ 67 , 74 ]
2.3. CD44 Is Involved in the Cytoskeleton Remodeling and Enhances Cell Motility
CD44 is involved in the formation and remodeling of the cell cytoskeleton via different signaling pathways. On one hand, structural and fundamental studies shows that CD44 can interact directly with cytoskeleton proteins, such as Ezrin/Radixin/Moesin (ERM), to facilitate microtubules and filamentous actin (F‐actin) binding.[ 77 , 78 , 79 , 80 ] On the other hand, HA/CD44 binding can also trigger different signaling pathways that affect the cytoskeleton formation and the related cellular motility. The RhoA/ROK pathway mentioned above is one example but other members of the RhoGTPase family (Ras, Rac1, and Cdc42) can also be phosphorylated upon HA/CD44 interaction and evoke cytoskeleton remodeling and increased cell migration of ovarian and breast tumor cells, lymphomas, neuroblastomas, melanomas and carcinomas.[ 81 , 82 , 83 , 84 , 85 ] HA/CD44‐dependent cell division control protein 42 (Cdc42) phosphorylation can activate the MAPK/ERK‐1,2 signaling pathway (Figure 4B2), which is involved in actin remodeling and cytoskeletal organization.[ 81 ] In ovarian cancer cells, the phosphorylated Cdc42 forms complex with the cytoskeletal adaptor protein IQGAP1 that regulates the cytoskeletal function via F‐actin (Figure 4B1a). Cdc42/IQGAP1 complex can be involved in the regulation of cell motility by association with extracellular‐signal‐regulated kinase 2 (ERK‐2), leading to ETS domain‐containing protein Elk‐1 (ELK‐1) and estrogen receptor‐α (ERα) mediated transcriptional up‐regulation and expression of proto‐oncogenes and ECM‐degrading enzymes (Figure 4B1b).[ 82 ]
HA/CD44‐dependent C3 botulinum toxin substrate 1 (Rac1) activation provides a fast response to changes in the TME. In breast tumor cells and invasive lymphoma cells, signaling through Rac1 activates P21‐Activated Kinase 1 (PAK1), IQGAP1, and filamin (Figure 4C), leading to actin assembly at membrane ruffling and pseudopod structures, mediating cell morphology alterations, adhesion, and motility.[ 83 , 84 , 85 ]
2.4. HA/CD44 Induces Epithelial to Mesenchymal Transition and Stem‐Like Phenotype
HA/CD44 interaction induces epithelial to mesenchymal transition (EMT), which together with the HA‐induced cell motility, supports tumor cell invasion and facilitates primary tumor extravasation.[ 86 ] In breast cancer, HA/CD44 interaction was associated with increased expression and activity of the EMT marker zinc finger E‐box‐binding homeobox 1 (ZEB1).[ 29 ] ZEB1 is involved in a feedback signaling cascade resulting in overexpression of HAS2 and CD44.[ 29 ] This autocrine signaling mechanism supports EMT and promotes the formation of metastasis. In CD44‐positive head and neck squamous cell carcinomas, the CD44‐mediated activation of Mitogen Activated Protein Kinases (MAPK)/ERK‐1,2/Homeobox protein NANOG (Nanog) induces stem‐like cells phenotypes (e.g., increased phosphorylation of ERK‐1,2 and Jun N‐terminal kinase 1,2 (JNK‐1,2) and ability to colony and form spheroids).[ 28 ] These cells show increased growth, migration, and invasion, associated with EMT‐characteristic markers and resistance to radiotherapy, which were reversed through Nanog knockdown.[ 28 ] CD44 also acts as a co‐receptor mediating HA‐dependent EMT (Table 2 ). In liver cancer cells, HA interaction with CD44 associated with transforming growth factor beta 1 (TGF‐β1) signaling plays a central role in EMT.[ 30 ] SNU‐368 hepatocarcinoma cells (CD44+ and TGF‐β1+) express EMT markers (increased N‐cadherin and decreased E‐cadherin) regulated by AKT/Glycogen synthase kinase‐3 (GSK‐3β)/β‐catenin pathway. The activation of this signaling pathway is dependent on both CD44 and TGF‐β1 and results in enhanced cell migration and ability to form spheres.
Table 2.
Hyaladherins as co‐receptors in TME signaling: activated pathways and cell response
| Hyaladherin | Co‐receptor | Model | Pathway | Cell response | Reference |
|---|---|---|---|---|---|
| CD44 | ErbB2 | Colon carcinoma cells | PI3K/AKT | Cell growth and survival | [90, 91] |
| EGFR | Head and neck cancer cells | MAPK/ERK‐1,2 | Cell growth, multidrug resistance, and cell motility | [59] | |
| Grb2‐HER2/Vav2 | Ovarian tumor cells | Ras/Rac | Cell growth and migration | [68] | |
| β1‐integrin | Breast cancer cells | Src/FAK – cortactin and paxillin phosphorylation | Cell motility | [86] | |
| TGFβ | Liver cancer cells | AKT/GSK‐3β/β‐catenin | EMT, stemness, and cell migration | [30] | |
| TLR‐2,4 | Breast tumor cells | AFAP‐110; MyD88/NF‐κB | Cell invasion and cytokine/ chemokine production | [97] | |
| RHAMM | CD44 and/or GFR | Breast cancer cells |
Ras/MAPK→ MEK/ERK‐1,2/RHAMM complex |
Rapid basal motility and invasion, cell cycle progression and inflammation | [27, 92, 113, 120] |
| GFR | Choriocarcinoma cells | PI3K/MEK1/Erl‐1,2 | Cell motility | [34] | |
| TGFβ | Gastric cancer | TGFβ/Smad‐2 | EMT, stemness and multifrug resistance | [35] | |
| LYVE‐1 | VEGF‐C and FGF‐2 | Lymphatic endothelial cells | Cell proliferation and lymphangiogenesis | [41] |
2.5. HA/CD44 Dependent Chemoresistance and Survival
HA/CD44 enhances cell survival by avoiding apoptosis and chemoresistance in several types of cancer. These events are regulated through CD44‐mediated signaling with diverse downstream effectors. One of the reported mechanisms is via protein kinase Cε (PKCε) activation. In breast and ovarian tumor cells, PKCε phosphorylates the transcription factor Nanog that at nuclear level enhances the expression of anti‐apoptotic proteins (IAPs) and multidrug‐resistant protein 1 (MDR1) and decreases tumor suppressor proteins, e.g., program cell death 4 (PDCD4) (Figure 5 ).[ 87 , 88 ] Nanog can also form complexes with STAT3 that have a similar effect.
Figure 5.
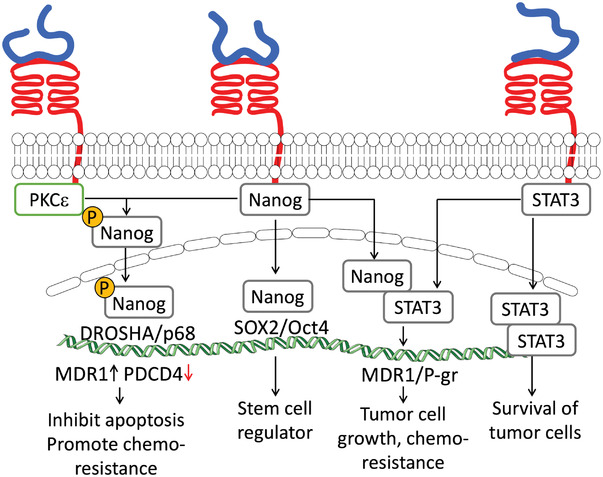
HA/CD44 binding activates different signaling pathways that promote chemoresistance and inhibit the apoptosis of tumor cells.
In head and neck CSC, HA/CD44 enhances cell survival and chemoresistance via DOT1‐like histone H3K79 methyltransferase (DOT1L)/miR‐10b‐dependent activation of RhoGTPase pathway that leads to the expression of survival proteins, e.g., cIAP‐2 and XIAP.[ 26 ] HA/CD44 binding can also indirectly regulate cell survival and chemoresistance. In CSC, HA/CD44 activates the nuclear factor erythroid 2‐like 2 (NFE2L2), a regulator of antioxidant genes, through p62 overexpression. As a result, CSC are protected from ROS‐rich microenvironment, and the tumor aggressiveness, growth, and chemoresistance are enhanced.[ 22 ] Recently, the role of HA/CD44 as a regulator of the Hippo signaling pathway (modulates cell proliferation and apoptosis) was reported.[ 20 ] CD44 clusters sequester the Hippo signaling‐inhibitor complex partitioning‐defective 1b‐Macrophage Stimulating (PAR1b‐MST). Upon declustering, this pathway is inhibited through yes‐associated protein (YAP) nuclear translocation and the expression of pro‐oncogenic and anti‐apoptotic genes.[ 20 , 89 ]
2.6. Co‐Association of CD44 with Other Receptors
As mentioned above, CD44 can act as a co‐receptor in signaling cascades, increasing pro‐tumorigenic behavior (Table 2). In colon and breast carcinoma cells, the formation of HA/CD44/ErbB2 (receptor tyrosine‐protein kinase) complex activates PI3K/AKT signaling pathway, increasing β‐catenin transcription factor activity and, consequently, the transcription of proliferative (cyclooxygenase‐2 (COX‐2)) and multidrug resistance (MDR1) genes, leading to cancer cell growth, survival and chemoresistance (Figure 6A).[ 90 , 91 ] In head and neck cancer cells, the HA/CD44/EGFR (epidermal growth factor receptor) complex increases cell growth, drug resistance, and motility through downstream MAPK/ERK‐1,2 signaling.[ 59 ] HA/CD44/RHAMM (receptor for hyaluronan‐mediated motility) forms a signaling complex with ERK‐1,2 to sustain rapid basal motility of invasive breast cancer cell lines.[ 27 , 92 ] In ovarian tumors, the association of HA/CD44 with growth factor receptor‐bound protein 2, human epidermal growth factor receptor 2 (Grb2‐HER2) and Guanine nucleotide exchange factor Vav2 (Vav2) protein activates Rac1 and Ras pathways, increasing cell growth and migration (Figure 6B).[ 68 ] HA/CD44 is also associated with the IP3 receptor in lipid rafts, promoting HA‐mediated Ca2+ signaling leading to nitric oxide generation and proliferation.[ 93 ]
Figure 6.
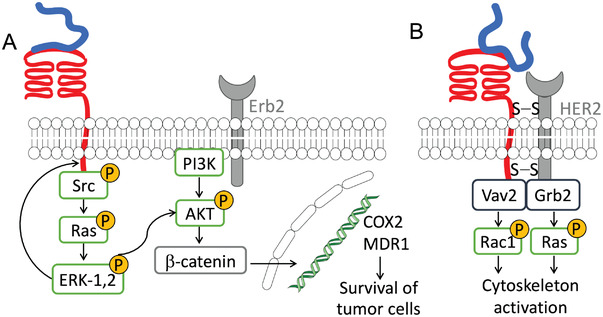
Co‐association of HA/CD44 signaling with A) Erb2 and B) HER2, supporting tumor cell survival and migration.
The supplementation of breast cancer cells with HA induces the association of CD44 and α5β1‐integrin via talin.[ 86 ] This signaling complex activates Src and focal adhesion kinase (FAK), inducing phosphorylation of cortactin and paxillin that promote cell motility during cancer cell extravasation from TME and cell adhesion to endothelial cells and fibronectin‐rich matrix of the metastatic environment.[ 86 ] Simultaneously, tumor cell motility and TME extravasation are facilitated through the upregulation and proteolytic activity of urokinase plasminogen activator (uPa), serin protease, collagen‐degrading enzymes, metalloproteinases (MMP), and cathepsin mediated by HA/CD44 interaction.[ 94 , 95 , 96 ]
The complex of HA, CD44 and toll‐like receptors (TLRs) modulate cytokine/chemokine production in breast tumor cells (Table 2).[ 97 ] Interaction of LMW HA with CD44 and TLR‐2,4 recruits actin filament‐associated protein (AFAP‐110) and Myeloid differentiation primary response 88 (MyD88) into signaling complexes (Figure 9B). Active AFAP‐110 binds to F‐actin and activates the cytoskeleton, while MyD88 binds to nuclear factor kappa light chain enhancer of activated B cells (NF‐κB) at the nuclear level and induces interleukin 1 beta (IL‐1β) and interleukin 8 (IL‐8) expression. These signaling events lead to the stimulation of cell invasion and production of cytokine/chemokine in breast tumor cells.[ 97 ]
Figure 9.

A) TLR structure and signaling pathways activated upon B) co‐association of TLR4 with HA/CD44 and C) homodimerization of TLR toward inflammation and cell survival in cancer.
3. Receptor for Hyaluronan Mediated Motility (RHAMM)
RHAMM, also known as intracellular hyaluronan receptor (IHABP), hyaluronan‐mediated motility receptor (HMMR), and the cluster of differentiation 168 (CD168) is not expressed in normal tissues. It is found transiently expressed during tissue repair[102, 103] and constitutively present in many carcinomas, e.g., breast, prostate, gastrointestinal tract, and the aggressive forms of multiple myeloma, leukemias, and lymphomas.[ 100 , 101 , 102 ] The secondary structure of RHAMM is largely helical (Figure 7A). It contains an HA‐binding domains at the C‐terminus that are rich in basic amino acids (Figure 7B) but structurally different from the BX7B motif of the CD44 HA‐binding domain.[ 11 , 103 ] Like CD44, RHAMM is coded by a single gene, HMMR, and different isoforms of RHAMM result from alternatively spliced transcript variants, alternate start codon, and post‐translational processes (Figure 7C).[ 104 ]
Figure 7.
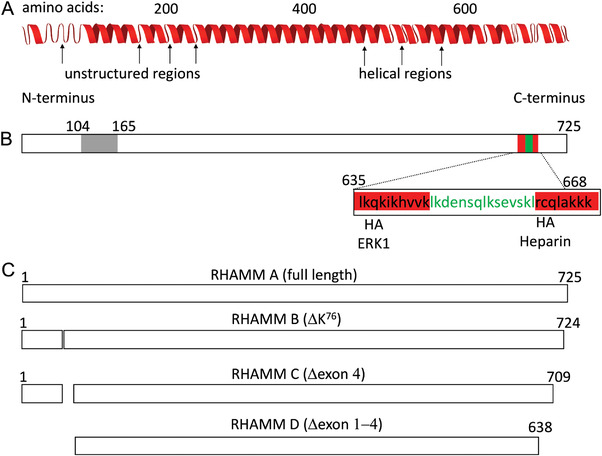
A) RHAMM exists as a helical protein containing short unstructured regions. B) It has several binding domains: at the N‐terminus a sequence that mediates binding to micritubules (in grey) and at the C‐terminus a leucine zipper (in green) which binds TPX2 and clusters of basic aminoacids (in red) that bind HA. C) Different isoforms of RHAMM in human. Adapted with permission under the terms of the CC‐BY 4.0 license.[ 105 ] Copyright 2021, the Author(s). Published by MDPI.
RHAMM does not contain a membrane‐spanning domain – it is soluble and, thus, can be localized in different cell compartments, including cytoplasm, nucleus, cell membrane, and/or in the ECM usually as a complex with two or more biomolecules.[ 106 , 107 ] The different localizations of RHAMM contribute to the intra‐/ extracellular exchange of information – a phenomenon called dynamic reciprocity.[ 100 , 108 ] Initially, intracellular RHAMM was identified (85 kDa in humans and 95 kDa in murine) and designated as RHAMMv5. It was associated with cell locomotion as indicated by its name. Currently, RHAMM is known as a multifunctional protein involved in different signaling processes at the cell surface and intracellularly. RHAMM variants have different functions in cancer. For example, overexpression of RHAMM isoform lacking N‐terminal microtubule‐binding domain[113] promotes pancreatic cancer in mouse and xenograft models. However, a recent study shows that RHAMM plays different roles in different cancer subtypes. While in malignant breast cancer, RHAMM overexpression correlates with aggressiveness and motility, overexpression of RHAMM on the luminal A subtype breast cancer inhibits cell migration via the AKT/GSK3β/Snail pathway.[ 110 ]
3.1. Cell Surface RHAMM
Cell surface RHAMM/HA complexes usually include other proteins, e.g., protein kinase receptors (RTKs) and non‐RTKs, such as CD44[ 19 , 27 , 92 , 111 , 112 ], platelet‐derived growth factor (PDGF)[ 113 ], TGFβ [ 35 , 112 ] or Recepteur d'origine nantais (RON)[ 114 ]. The composition of these different complexes modulates the downstream signaling by activating alternative molecular switchers, e.g., Src[ 115 ] and Ras.[ 113 , 115 , 116 , 117 , 118 ]
3.1.1. Cytoskeleton Dynamics and Cell Motility
The main cellular events arising from HA/RHAMM interaction are enhanced cell motility and invasion, which are regulated by different and complementary signaling pathways. HA/RHAMM interaction directly activates protein kinase Cς (PKCς) and FAK, recruiting cortactin and paxillin to focal adhesions (Figure 8A1).[ 119 ] Alternative signaling involves the association of HA/RHAMM either with CD44 or growth factor receptors (GFR) to transiently activate Src in cell lamellae, where it recruits and activates FAKs and cortactin, coordinating cell motility (Figure 8A2, Table 2).[ 115 ] In c‐H‐ras oncogene transformed fibroblasts, the activation of Ras signaling pathway promotes a rapid assembly and disassembly (turnover) of focal adhesions at cell lamellae, promoting high random cell motility (Figure 8A2).[ 116 , 117 ] In breast cancer cells, HA recognition by RHAMM activates Ras/MAPK pathway and requires both cell surface and intracellular RHAMM.[ 113 ] Cell surface RHAMM/HA complex, associated with CD44 and/or GFRs, activates Ras/MAPK, forming complexes between the intracellular RHAMM, Mitogen‐activated protein kinase kinase (MEK), and ERK‐1,2 (Figure 8A2). This complex binds to microtubules and promotes cytoskeleton dynamics required for cell motility and cell cycle progression.[ 92 , 111 , 113 , 120 ] RHAMM/MEK/ERK‐1,2 complex displays different functions at nuclear level, where it promotes the expression of plasminogen activator inhibitor‐1 (PAI‐1) and MMP‐9 that are involved in cell invasion and inflammation (Figure 8A3).[ 102 ] In choriocarcinoma cells (CD44−/RHAMM+ phenotype), HA/RHAMM/GFR‐induced motility results from MEK/ERK‐1,2 activation mediated by PI3K and evidence a crosstalk between PI3K/AKT and Ras/MAPK signaling pathways (Figure 8B, Table 2).[ 34 ]
Figure 8.
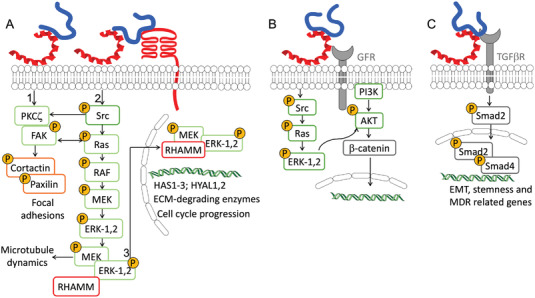
The HA/RHAMM interactions at the cell surface activate different downstream signaling in cancer that can lead to A) enhanced cell motility, B) proliferation, and C) epithelial to mesenchymal transition (EMT).
3.1.2. Cell Proliferation
Recently, cell proliferation has also been associated with HA/RHAMM interaction.[ 31 , 33 , 35 ] HT1080 fibrosarcoma cells treated with LMW HA showed RHAMM‐dependent growth induced via Ras/MAPK interaction with the β‐catenin pathway (Figure 8B).[ 33 ] This leads to β‐catenin overexpression and complexation with ERK‐1,2. The translocation of this complex to the nucleus upregulates c‐Myc transcription and alters cell proliferation.[ 33 , 121 , 122 ] HA/RHAMM interaction promotes the association of TPX2 Microtubule Nucleation Factor (TPX2) with RHAMM at the nucleus and the activation of Aurora Kinase A (AURKA), which directly regulates transcription of HAS1, HAS2 and HAS3, HYAL1, and HYAL2, cyclins and motility effectors, increasing cell proliferation, cell motility and enhancing tumorigenic HA signaling.[ 101 , 123 , 124 ] Other RHAMM‐dependent pathways might be involved in enhancing cell proliferation as HMMR silencing decreases both mRNA and protein expression of Cdc2 and CyclinB1 related to cell cycle progression.[ 31 ] Blocking HA/RHAMM interactions, either by abolishing HA synthesis or RHAMM expression, decreases cancer cell proliferation and survival. Silencing of HAS2 and HAS3 in lung cancer cells has such effect. Blocking of HA synthesis downregulates CD44, RHAMM, EGFR, AKT, and ERK‐1,2, and induces cleavage of caspase‐3 and poly(ADP‐ribose)polymerase (PARP), i.e., an apoptotic effect, which is reversed upon supplementation with HA.[ 19 ] RHAMM blockage with specifically designed peptides also inhibits proliferation and compromises cell viability, as demonstrated for ovarian cancer, prostate cancer, breast carcinoma, and adenocarcinoma of the breast duct cells, but not in non‐tumor fibroblasts and fibroblasts RHAMM(‐/‐).[ 32 ]
3.1.3. Epithelial to Mesenchymal Transition and Multidrug Resistance
Multidrug resistance, EMT, and stem cell‐like properties depend on RHAMM and Smad2 expression and activity (Figure 8C, Table 2).[ 35 ] As an example fluorouracil (5‐FU) resistant gastric cancer cells upregulate HMMR and its knocking down recovers the drug sensitivity. On the other hand, ectopic expression of RHAMM in parental SGC7901 and BGC823 gastric cancer cell lines resulted in 5‐FU resistance.[ 35 ] This effect was observed together with EMT confirmed by a reduced E‐cadherin expression and increased expression of vimentin, N‐cadherin, fibronectin, and pluripotency‐associated markers, including SOX2, NANOG, OCT4, and BMI—a pattern that was reversed with HMMR silencing.[ 35 ]
3.2. Intracellular RHAMM
Intracellularly, RHAMM is found in the cytoskeleton and nucleus. Because RHAMM interacts with several kinases, it has been suggested that the primary function of intracellular RHAMM (iRHAMM) is to connect the cytoskeleton to signaling complexes, similar to what happens at the cell membrane. Indeed, RHAMM can bind to different microstructures, like actin filaments, podosomes, centrosomes, microtubules, and mitotic spindle thus, affecting cell motility and proliferation.[ 106 , 125 , 126 ]
iRHAMM can transduce extracellular signaling activated upon interactions of cell surface RHAMM. Fundamental studies using fibroblast models show that iRHAMM binds and forms complexes with MEK1 and ERK‐1,2 in the cytoskeleton or nucleus.[ 111 , 127 ] These complexes regulate the cell's random motility, mitotic spindle integrity, cell cycle progression, and gene expression in breast cancer cells and fibroma cells. iRHAMM/ERK‐1,2 complexes are required for microtubule nucleation and link to centrosomal proteins, e.g., TPX2 and AURKA, thus, playing a fundamental role in centrosome function, including dynamic turnover of interphase microtubules and mitotic spindles.[ 128 , 129 , 130 , 131 ] Additionally, iRHAMM interacts with the cortical proteins, such as supervillin, to coordinate myosin II contraction and activate ERK‐1,2, required for cell migration and proliferation.[ 132 , 133 , 134 ]
4. Toll‐Like Receptors (TLR) 2 and 4
TLR are type I transmembrane receptors (700‐1100 amino acids) expressed by immune cells as macrophages and dendritic cells (Figure 9A). They have a crucial role in innate immunity and the induction of adaptive immune responses. TLR contain extracellular leucine‐rich sequences and a cytoplasmic domain (Figure 9A) that are responsible for the recognition of molecular patterns expressed by a wide variety of pathogens (i.e., PAMP) and damage‐associated molecular patterns (so‐called DAMP), as HA fragments, causing inflammatory responses.[ 135 ]
TLR act as dimers that can be either homodimers or heterodimers (either with other members of the TLR family or with different proteins), thus increasing the ligand diversity. In humans, ten TLR have been identified (TLR1‐10) and classified into two groups: cell‐surface TLR (TLR1, TLR2, TLR4, TLR5, and TLR6) that recognize bacterial cell‐surface components, and endosomal TLR (TLR3, TLR7, TLR8, and TLR9), which recognize microbial or viral nucleic acids. Among these TLR, TLR2 and TLR4 can recognize and bind HA.[ 136 ] A minimal size of 4 monosaccharide units (4‐mer) of HA is required to interact with TLR2 or TLR4[142] but only longer HA fragments (6‐mer) can engage heterodimer association of TLR4 and CD44, thus, enhancing intracellular signaling.[ 138 ]
TLR are expressed in different cancers, including hepatocellular carcinoma, melanoma, neuroblastoma, lung, colon, breast, ovarian, cervical, and prostate cancers.[ 139 ] In advanced stages of carcinogenesis, HA/TLR2,4 downstream signaling contributes to tumor cell invasion and expression of cytokines and chemokines, which are closely related to cancer progression. The specific effect of HA/TLR2,4 depends on the cell and tumor environment.
4.1. Effect of HA Molecular Weight on TLR Signaling
TLR/HA interactions and binding depend on HA molecular weight. The TLR complexes involving HMW HA and the respective downstream signaling in cancer are not well studied, and the reports on HMW HA/TLR2,4 involvement in inflammatory disorders are controversial. There is limited evidence that TLR2 and TLR4 binding to HMW HA attenuates inflammatory processes.[ 140 , 141 , 142 , 143 ] Recently, the interactions between HMW HA and TLR were associated with the formation of a physical barrier that limits access to the receptors instead of direct binding.[ 144 ] This is, however, not a common mechanism, as TLR activation/signaling in cancer is complex and depends on the pericellular coat dynamics.
In breast cancer cells, HA oligomers (3‐5 kDa) form a triple complex with CD44 and TLR2 or TLR4 that promotes the invasiveness and synthesis of IL‐1β.[ 97 ] The formation of the triple complex LMW HA/TLR2,4/CD44 activates two downstream signaling pathways: AFAP‐110 that binds to F‐actin is recruited and regulates cytoskeleton activation (Figure 9B1), increasing cell invasion; and downstream signaling via MyD88 (Figure 9B2) that promotes NF‐κB activation and nuclear translocation leading to transcription and release of pro‐inflammatory cytokines, including Tumor necrosis factor alpha (TNF‐α), major intrinsic protein (MIP), IL‐1β, IL‐6, IL‐8, and IL‐12, essential for cell recruitment and inflammatory state of the tumor microenvironment.[ 97 , 145 , 146 , 147 , 148 ] In human melanoma cells, the LMW HA/TLR4 also activates NF‐κB that triggers the overexpression of MMP2 and IL‐8, contributing to melanoma progression (Figure 9C1).[ 149 ] In UVB‐exposed melanocytes, in situ degradation of endogenous HA, by HYAL treatment, induces expression of the inflammatory cytokines IL‐6, IL‐8, C‐X‐C motif ligand 1 (CXCL1), and C‐X‐C motif chemokine ligand 10 (CXCL10) and activation of AKT pathway via TLR4 binding (Figure 9C2) that lead to increased cell proliferation and survival.[ 36 ]
Several in vivo studies using specific animal models have shown that HA/TLR4 binding drives tumor growth and suppresses apoptosis. As an example, significant inhibition of tumor growth was found in TLR4 deficient in vivo models.[ 21 ] The inhibition of tumor growth was more pronounced than observed for the respective CD44 deficiency models. Moreover, the systemic administration of a specifically designed peptide, PEP1, which binds to endogenous HA and blocks HA/TLR4 interaction, reduced the number of adenocarcinomas and inhibited tumor growth.[ 21 ] Signaling through TLR4 was confirmed using the MyD88 deficient model, which had a similar effect as PEP1 treatment. HA/TLR4 binding also supports proliferation and prevents apoptosis of colon carcinoma CT26 tumor isografts in mice models. Molecular analysis showed that HA/TLR4 activates β‐catenin through PI3K/AKT pathway leading to expression of proliferative proteins (Leucine Rich Repeat Containing G Protein‐Coupled (Lgr5), Cyclin D1, β‐catenin, R‐spondin) that increase the risk of carcinogenesis (Figure 9C3).[ 150 ] In addition, tumor survival was enhanced due to arrest to spontaneous apoptosis resulted from NF‐κB activation.[ 151 , 152 ]
4.2. TLR Expression as a Function of TME
During tumor development, the expression of TLR varies because of their different regulation by the cancer cell populations existing at each development stage. In glioblastoma stem cells (GSC), the low expression of TLR4 receptors allow them to survive regardless of inflammatory signals in the tumor microenvironment.[ 153 ] During GSC differentiation into cancer cells, TLR4 is upregulated through endogenous HA synthesis and autocrine signaling.[ 37 ] TLR4 activation induces proliferation and inflammation via the NF‐κB pathway while avoiding terminal differentiation and senescence, supporting tumor growth and recurrence.[ 37 ]
The macrophages population in tumors is also tuned by the “cancerization” state of TME. After recruitment, monocytes differentiate to M2 immunosuppressive macrophages taking a pro‐resolving role at the initial stages of cancer development.[ 154 ] At this point, the interaction of LMW HA, but not HMW, with CD44 and TLR4 inhibits the expression of pro‐inflammatory cytokines (TNF‐α and IL‐12) and induces secretion of anti‐inflammatory molecules (IL‐10).[ 155 , 156 ] Similar pro‐resolving behavior is also observed in dendritic cells in the TME. HA fragments, but not native HA, interact with TLR4 in dendritic cells.[ 137 ] This interaction results in the activation of p38/p42/44 MAPK signaling pathway, nuclear translocation of NF‐κB, and production of TNF‐α that lead to dendritic cells maturation and immuno‐mediated anti‐tumor response (Figure 9C2).[ 137 ]
With tumor progression, M0 macrophages and M2 macrophages are polarized into M1 pro‐inflammatory macrophages and TLR signaling changes.[ 157 ] In human primary monocytes and murine macrophages, LMW HA engages TLR4 independently of other HA‐receptors, through TLR4/MyD88/ERK‐1,2/p38/JNK pathway resulting in the activation of cytosolic phospholipase 2 (cPLA2 α), responsible for the hydrolysis of arachidonic acid, which contributes to inflammation, proliferation, and metastasis.[ 157 ] Similar pro‐inflammatory effects were observed as a consequence of the LMW HA/TLR2 interactions in macrophages. LMW HA/TLR2 activated NF‐κB via MyD88/IL‐1 receptor‐associated kinase 4 (IRAK)/TNF Receptor Associated Factor 6 (TRAF6), and PKC‐ζ pathway (Figure 9C1), stimulating the synthesis of pro‐inflammatory cytokines (IL‐2 and interferon γ (IFN‐γ)). Supplementation of HMW HA inhibited this effect.[ 146 ]
5. Lymphatic Vessel Endothelial Hyaluronan Receptor 1 (LYVE‐1)
LYVE‐1, also known as extracellular link domain containing 1 (XLKD1), is a CD44 homolog encoded in humans by the LYVE1 gene. This receptor is mainly expressed (but not limited to) by lymph vessels, and it is overexpressed in several cancers where it is considered an unfavorable prognostic marker.[ 38 , 39 , 40 , 158 , 159 ]
LYVE‐1 binds to HA through the prototypic HA‐binding domain (Figure 10A) conserved in the hyaladherins superfamily.[ 160 ] However, HA/LYVE‐1 differs from HA/CD44 binding – while HA binding to CD44 is mediated by H‐bonding and hydrophobic interaction, the HA/LYVE‐1 interactions are mainly electrostatic and thus, sensitive to ionic strength (IC50 of 150 mM NaCl as compared with IC50>2 M NaCl for HA/CD44).[ 160 ] LYVE‐1 binds either soluble LMW HA or HA immobilized within the pericellular coat.[ 42 , 161 ] Mechanistically, LYVE‐1 can act as a surface receptor,[ 42 , 161 ] a signal transducer,[ 162 ] or a decoy for HA.[ 43 ] This receptor is also involved in HA uptake and degradation – it conveys HA for catabolism within lymphatic endothelial cells and also mediates HA transport into the lumen of afferent lymphatic vessels for subsequent re‐uptake and degradation in lymph nodes.
Figure 10.
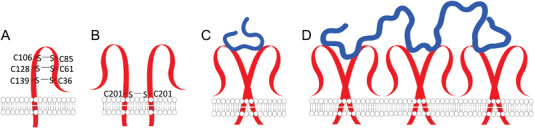
A) Schematic presentation of the structure of LYVE‐1 receptor and B) its homodimerization that occurs by formation of disulfide bond between the cysteines at C201 from the two receptors. C) The conformation of LYVE‐1 homodimers alters upon binding of low molecular weight HA. D) In the presence of high molecular weight HA, the dimers can form clusters.
The HA/LYVE‐1 interactions are favored at high LYVE‐1 density (focal clustering, Figure 10B,C) or in the presence of HMW HA that increases the binding avidity (Figure 10D)[ 165 , 166 ]. While the LYVE‐1 binding domain is active as a monomer, its binding affinity to HA (K d of 35.6 µM) is lower than the CD44 one (K d of 65.7 µM),[ 160 ] and usually, LYVE‐1 dimers bind HA with intermediate to high molecular weight and mediate its internalization.[ 163 , 164 , 165 ]
5.1. LYVE‐1 Role in Lymph Node Metastasis
The specific role of LYVE‐1 in cancer development and progression is yet unclear, but some data suggest LYVE‐1 involvement in lymphangiogenesis and lymph node metastasis formation. For example, LMW HA (generated in situ upon endogenous HA degradation) accumulates in interstitial tumor fluid (obtained from rat tumors and human colorectal tumors) and positively correlates with lymphatic invasion and lymph node metastasis.[ 159 ] LMW HA/LYVE‐1 interaction in lymph node endothelial cells stimulates proliferation, migration, and tube formation of lymphatic endothelial cells via tyrosine phosphorylation of protein kinase Cα/βII (PKCα/βII) and ERK‐1,2 (Figure 11A).[ 162 ] The LMW HA/LYVE‐1 complexes can also engage vascular endothelial growth factor C (VEGF‐C) and fibroblast growth factor 2 (FGF‐2) to induce lymphatic endothelial cell proliferation and lymphangiogenesis (Table 2).[ 41 ] Blocking the HA/LYVE‐1 interactions with antibodies inhibits cell proliferation and migration, reduces the number of lymphatic vessels and volume of primary tumors, and inhibits lymph node metastasis formation in breast cancer cells implanted in mice.[ 38 , 162 ] The process by which LMW HA potentiates the formation of lymph node metastasis is likely related to the altered permeability of lymph vessels.[ 40 ] LMW HA affects the lymphatic lumen integrity due to alterations on VE‐cadherin and β‐catenin at membrane junctions upon interaction with LYVE‐1 in human dermal lymphatic endothelial cells.
Figure 11.
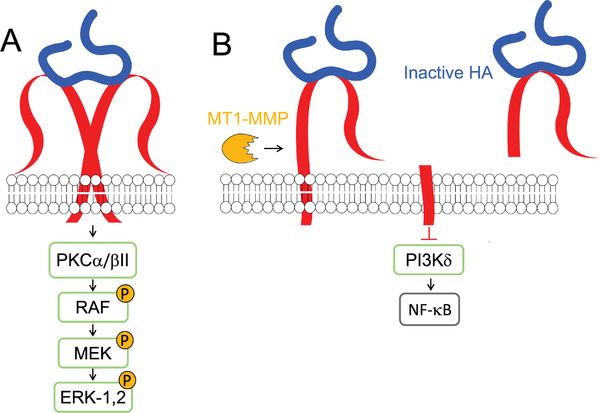
A) HA/LYVE‐1‐mediated intracellular signaling and B) function as a decoy receptor.
5.2. LYVE‐1 as a Decoy for HA
Contrary to the integral receptor, the shedded ectodomain of LYVE‐1 acts as a decoy receptor of LMW HA fragments, inhibiting cell proliferation, migration, and lymphangiogenesis (Figure 11B).[ 43 ] Soluble LYVE‐1 (sLYVE‐1) can be generated in situ, upon the action of MT1‐MMP, and released into the tumor interstitium.[ 44 ] sLYVE‐1 inhibits PI3Kδ signaling, as well NF‐κB activation and synthesis of VEGF‐C, and thus lymphangiogenesis (Figure 11B). On the other hand, sLYVE‐1 derived from M2‐like tumor‐associated macrophages (present at initial stages of cancer), has pro‐resolving functions via scavenging LMW HA from the TME. The generated sLYVE‐1/LMW HA complexes inhibit the cell proliferation of human and murine melanomas in the early tumor growth phase, but not in advanced stages.[ 43 ]
6. Hyaluronan Receptor for Endocytosis (HARE)
HARE, also known as Stabilin‐2 (Stab‐2), is coded by the STAB2 gene in humans (Figure 12A) and is mainly found in endocytic and recycling compartments of cells in lymph vessels and nodes, liver, and spleen.[ 166 ] HARE acts as a scavenger receptor for glycosaminoglycans, low‐density lipoprotein particles, phosphatidylserine, and other bioentities resulting from matrix degradation.[ 167 ] HARE receptor binds to HA through the Link domain, present in other hyaladherins, and it mediates HA internalization via clathrin‐mediated endocytosis, allowing a fast clearance of HA from biological fluids (Figure 12B).[ 168 ] HARE binding and endocytosis of HA with 40 to 400 kDa can activate MAPK, ERK‐1,2 and NF‐κB‐mediated gene expression (Figure 12C).[ 169 , 170 ] The exact downstream signaling(s) and its effect on cell behavior are yet unknown, but the sensitiveness to HA size might be related to ECM turnover.
Figure 12.

A) Human STAB2 domain organization and its involvement in tumor‐associated B) HA/HARE clathrin‐mediated endocytosis and C) HA/HARE signaling.
In cancer, inhibition or knockout of this receptor prevents HA uptake and metastasis formation.[ 168 , 171 ] Two possible mechanisms that drive toward this response have been proposed: (i) the resulting increase in HA fragments block or prevent the interaction of circulating cancer cells with surrounding tissues;[ 171 ] or (ii) possible actuation of HARE as an endothelial receptor for metastatic tumor cells with HA‐rich pericellular coats that promotes tissue penetration of tumor cells.[ 168 ]
The involvement of HARE in the interactions between invasive breast cancer cells and lymphatic endothelial cells has been demonstrated in co‐cultures.[ 45 ] Altered gene expression patterns in lymphatic endothelial cells were observed only when these cells were co‐cultured with highly metastatic breast and prostate cancer cells. The expression pattern changes included upregulation of metastasis‐related genes involved in the cell cycle (CDC6, CLSPN, kinases genes), cell adhesion, and motility (BST2, SELE, and HMMR), cytokines (CCL7, CXCL6, CXCL1, and CSF1), and factors of the complement system (C1R, C3, and CFB); which mediate lymphangiogenesis. Moreover, in these co‐cultures, HARE was downregulated, which might prevent HA uptake from the tumor microenvironment (via the above described clathrin‐mediated endocytosis), thus contributing to high levels of HA at TME.
7. Layilin
Layilin is a transmembrane homolog of C‐type lectins coded by the LAYN gene. Layilin recognizes LMW HA via the Link domain common to hyaladherins.[ 172 , 173 , 174 ] It participates in intracellular complexes with cytoskeletal‐membrane linker proteins, including talin, merlin, and radixin (Figure 13A).[ 172 , 175 ]
Figure 13.

HA/Layilin‐mediated intracellular signaling toward (A) motility and (B) invasion.
Although negative regulatory functions over the immune system and cancer cell motility have been identified, Layilin role in cancer is yet to be elucidated. Layilin is specifically and highly upregulated in tumor‐infiltrating Treg and CD8+ lymphocytes in human lung, colorectal, and hepatocellular carcinomas.[ 47 , 176 ] Layilin overexpression inhibits IFN‐γ production, a key cytokine in anti‐tumor immune responses.[ 47 ] As the Treg cells become suppressive, tumor‐infiltrating compromised CD8+ T cells increase, contributing to tumor immune escape and poor prognosis. A correlation between high layilin expression and the presence of immune infiltrating cells in TME, including CD8+ T cells, CD4+ T cells, macrophages, neutrophils, and dendritic cells, has been identified in several types of cancer, and it is suggested as a prognostic biomarker for colon and gastric cancers.[ 46 ]
Layilin ability to regulate cell motility was first observed in A549 human lung cancer cells. Knockdown of this HA receptor inhibited cell invasion and migration in vitro and lymphatic metastasis in vivo in the tumor‐bearing mice model.[ 177 ] Recently, the role of layilin was investigated in highly invasive malignant glioma cells (Figure 13B).[ 48 ] Knockdown experiments showed that layilin upregulates SNAI1 transcription activity by suppressing metastasis‐associated 1 family member 3 (MTA3, which is SNAI1 transcription repressor). At the nuclear level, SNAI1 causes overexpression of MMP2, MMP9, and collagen type I alpha 1 chain (COL1A1), potentiating the invasive ability of malignant glioma cells.[ 48 ]
Layilin might have other roles in cancer progression, including involvement in EMT. Previous studies have shown the importance of layilin in regulating intestinal epithelial tight junctions in inflammatory models[ 178 , 179 ] as well as its role TNF‐α induced EMT of renal tubular epithelial cells.[ 180 ]
8. Conclusions
In TME, HA homeostasis is altered by increased synthesis and degradation since the early stages of carcinogenesis. The HA receptors constitute a complex signaling hub that senses these changes and responds to them through clustering, formation of specific signaling complexes, and receptors cleavage. HA interaction with its receptors modulates several hallmarks of cancer, including proliferation (CD44, RHAMM, and TLR2,4), survival (CD44, RHAMM, and TLR2,4), invasion and metastasis (CD44, RHAMM, TLR2,4, LYVE‐1, HARE, and Layilin), immune response (CD44, TLR2,4, and Layilin) and lymphangiogenesis (LYVE‐1). In the TME, these interactions activate signaling cascades that affect different cell populations, including immune cells, fibroblasts, and vascular endothelial cells. HA and its receptors are also involved in the formation, protection, and sustained growth of stem cells niches and promote EMT of cancer cells, facilitating invasion and metastasis formation. HA receptors also participate in signaling cascades that lead to the upregulation and overexpression of several cytokines/chemokines and ECM‐degrading enzymes, which modify the TME and enhance carcinogenesis.
HA signaling is thus a potential target for cancer therapy. At the molecular level, targeting HA signaling can be achieved with different approaches: HA synthesis inhibition with 4‐methylumbelliferone or HAS silencing; HA inactivation with binding peptides; receptors inhibition with antibodies or specific peptides; and receptors silencing, knockdown, or knockout. Another strategy includes the use of inhibitors specific to the most transversal HA‐activated signaling pathways, as MAPK and PI3K/AKT pathways, blocking intracellular signaling. Nevertheless, given the role of HA and its signaling in normal tissue homeostasis, these strategies are not always applicable in a clinical scenario, and the development of specific systems for targeted and effective treatment remains a hot topic in the (bio)chemical, pharmaceutical, and medical sciences.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
The authors thank the Portuguese Fundação para a Ciência e a Tecnologia FCT (Grants no: SFRH/BD/114847/2016, PTDC/NAN‐MAT/28468/2017, and PTDC/CTM‐REF/0022/2020) for providing financial support to this project.
Biographies
Ana M. Carvalho graduated in applied biology and holds an M.Sc. in biophysics and bionanosystems from the University of Minho, Portugal. Recently, she obtained a Ph.D. in tissue engineering, regenerative medicine and stem cells from the same university. Her Ph.D. studies were performed at 3Bs Research Group, where she combined different biophysical and biological approaches to develop tools for studying the interactions of glycosaminoglycans with their receptors. Emphasis was given to hyaluronan and its receptors CD44 and RHAMM.

Rui L. Reis is a full professor of tissue engineering, regenerative medicine and stem cells at the University of Minho, Portugal where he is also leading 3Bs Research Group and the Research Institute on Biomaterials, Biodegradables, and Biomimetics. He has pioneered the development of functional biomaterials by non‐conventional processing of natural biomolecules such as proteins and carbohydrates. The developed biomaterials are used in different regeneration approaches and in the establishment of 3D disease models.

Iva Pashkuleva is a principal investigator at the 3Bs Research Group. She is working on the elucidation of glycan's role in cellular communication and explores the glycan's diversity in design and synthesis of functional biomaterials. Her team has developed new analytical methods and platforms for characterization and measurement of glycan−protein and glycan−cell interactions; glycan‐based delivery systems; selective cancer therapies based on glycan amphiphiles and biocatalytic self‐assembly; and extracellular supramolecular mimics.

Carvalho A. M., Reis R. L., Pashkuleva I., Hyaluronan Receptors as Mediators and Modulators of the Tumor Microenvironment. Adv. Healthcare Mater. 2023, 12, 2202118. 10.1002/adhm.202202118
References
- 1. Bejarano L., Jordāo M. J. C., Joyce J. A., Cancer Discovery 2021, 11, 933. [DOI] [PubMed] [Google Scholar]
- 2. Duan Q., Zhang H., Zheng J., Zhang L., Trends Cancer 2020, 6, 605. [DOI] [PubMed] [Google Scholar]
- 3. Mccarthy J. B., El‐Ashry D., Turley E. A., Front. Cell Dev. Biol. 2018, 6, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roma‐Rodrigues C., Mendes R., Baptista P., Fernandes A., Int. J. Mol. Sci. 2019, 20, 840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nikitovic D., Tzardi M., Berdiaki A., Tzanakakis G. N., Front. Immunol. 2015, 6, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brassart‐Pasco S., Brassart B., Ramont L., Oudart J.‐B., Monboisse J. C., Front. Oncol. 2020, 10, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caon I., Bartolini B., Parnigoni A., Moretto P., Viola M., Karousou E., Vigetti D., Passi A., Semin. Cancer Biol. 2020, 62, 9. [DOI] [PubMed] [Google Scholar]
- 8. Winkler J., Abisoye‐Ogunniyan A., Metcalf K. J., Werb Z., Nat. Commun. 2020, 11, 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cox T. R., Nat. Rev. Cancer 2021, 21, 217. [DOI] [PubMed] [Google Scholar]
- 10. Toole B. P., Nat. Rev. Cancer 2004, 4, 528. [DOI] [PubMed] [Google Scholar]
- 11. Turley E. A., Wood D. K., Mccarthy J. B., Cancer Res. 2016, 76, 2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Passi A., Vigetti D., Buraschi S., Iozzo R. V., FEBS J. 2019, 286, 2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nastase M.‐V., Janicova A., Wygrecka M., Schaefer L., Antioxid. Redox Signal. 2017, 27, 855. [DOI] [PubMed] [Google Scholar]
- 14. Henke E., Nandigama R., Ergün S., Front. Mol. Biosci. 2020, 6, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu M., Cao M., He Y., Liu Y., Yang C., Du Y., Wang W., Gao F., FASEB J. 2015, 29, 1290. [DOI] [PubMed] [Google Scholar]
- 16. Tavianatou A. G., Caon I., Franchi M., Piperigkou Z., Galesso D., Karamanos N. K., FEBS J. 2019, 286, 2883. [DOI] [PubMed] [Google Scholar]
- 17. Cyphert J. M., Trempus C. S., Garantziotis S., Int. J. Cell Biol. 2015, 2015, 563818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thanee M., Dokduang H., Kittirat Y., Phetcharaburanin J., Klanrit P., Titapun A., Namwat N., Khuntikeo N., Wangwiwatsin A., Saya H., Loilome W., PLoS One 2021, 16, e0245871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song J. M., Im J., Nho R. S., Han Y. H., Upadhyaya P., Kassie F., Mol. Carcinog. 2019, 58, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ooki T., Murata‐Kamiya N., Takahashi‐Kanemitsu A., Wu W., Hatakeyama M., Dev. Cell 2019, 49, 590. [DOI] [PubMed] [Google Scholar]
- 21. Makkar S., Riehl T. E., Chen B., Yan Y., Alvarado D. M., Ciorba M. A., Stenson W. F., Mol. Cancer Ther. 2019, 18, 2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ryoo I.‐G., Choi B.‐H., Ku S.‐K., Kwak M.‐K., Redox Biol. 2018, 17, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mao M., Zheng X., Jin B., Zhang F., Zhu L., Cui L., Exp. Ther. Med. 2017, 14, 5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matzke‐Ogi A., Jannasch K., Shatirishvili M., Fuchs B., Chiblak S., Morton J., Tawk B., Lindner T., Sansom O., Alves F., Warth A., Schwager C., Mier W., Ponta H., Abdollahi A., Gastroenterology 2016, 150, 513. [DOI] [PubMed] [Google Scholar]
- 25. Liu S., Cheng C., Cancer Res. 2017, 77, 3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bourguignon L. Y. W., Wong G., Shiina M., J. Biol. Chem. 2016, 291, 10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carvalho A. M., Soares Da Costa D., Paulo P. M. R., Reis R. L., Pashkuleva I., Acta Biomater. 2021, 119, 114. [DOI] [PubMed] [Google Scholar]
- 28. Huang C., Yoon C., Zhou X. H., Zhou Y. C., Zhou W. W., Liu H., Yang X., Lu J., Lee S. Y., Huang K., Cell Death Dis. 2020, 11, 266. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Preca B.‐T., Bajdak K., Mock K., Lehmann W., Sundararajan V., Bronsert P., Matzge‐Ogi A., Brabletz S., Brabletz T., Maurer J., Stemmler M. P., OncoTargets Ther. 2017, 8, 11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park N. R., Cha J. H., Jang J. W., Bae S. H., Jang B., Kim J.‐H., Hur W., Choi J. Y., Yoon S. K., Biochem. Biophys. Res. Commun. 2016, 477, 568. [DOI] [PubMed] [Google Scholar]
- 31. Chen F., Zhu X., Zheng J., Xu T., Wu K., Ru C., Math. Biosci. Eng. 2020, 17, 2150. [DOI] [PubMed] [Google Scholar]
- 32. Akentieva N. P., Topunov A. F., Biointerface Res. Appl. Chem. 2021, 11, 1225. [Google Scholar]
- 33. Kouvidi K., Berdiaki A., Tzardi M., Karousou E., Passi A., Nikitovic D., Tzanakakis G. N., Biochim. Biophys. Acta 2016, 1860, 814. [DOI] [PubMed] [Google Scholar]
- 34. Mascaro M., Pibuel M.A. A., Zotta E., Bianconi M. I., Otero S., Jankilevich G., Alvarez E., Hajos S. E., Histochem. Cell Biol. 2017, 148, 173. [DOI] [PubMed] [Google Scholar]
- 35. Zhang H., Ren L., Ding Y., Li F., Chen X., Ouyang Y., Zhang Y., Zhang D., FASEB J. 2019, 33, 6365. [DOI] [PubMed] [Google Scholar]
- 36. Takabe P., KäRn㤠R., Rauhala L., Tammi M., Tammi R., Pasonen‐Seppã¤Nen S., J. Invest. Dermatol. 2019, 139, 1993. [DOI] [PubMed] [Google Scholar]
- 37. Ferrandez E., Gutierrez O., Segundo D. S., Fernandez‐Luna J. L., Sci. Rep. 2018, 8, 6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hara Y., Torii R., Ueda S., Kurimoto E., Ueda E., Okura H., Tatano Y., Yagi H., Ohno Y., Tanaka T., Masuko K., Masuko T., Cancer Sci. 2018, 109, 3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arimoto S., Hasegawa T., Takeda D., Saito I., Amano R., Akashi M., Komori T., Anticancer Res. 2018, 38, 6157. [DOI] [PubMed] [Google Scholar]
- 40. Du Y., Cao M., Liu Y., He Y., Yang C., Wu M., Zhang G., Gao F., Oncoimmunology 2016, 5, e1232235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bauer J., Rothley M., Schmaus A., Quagliata L., Ehret M., Biskup M., Orian‐Rousseau V., Jackson D. G., Pettis R. J., Harvey A., Brã¤Se S., Thiele W., Sleeman J. P., J. Mol. Med. 2018, 96, 199. [DOI] [PubMed] [Google Scholar]
- 42. Johnson L. A., Banerji S., Lawrance W., Gileadi U., Prota G., Holder K. A., Roshorm Y. M., Cerundolo V., Gale N. W., Jackson D. G., Nat. Immunol. 2017, 18, 762. [DOI] [PubMed] [Google Scholar]
- 43. Dollt C., Becker K., Michel J., Melchers S., Weis C.‐A., Schledzewski K., Krewer A., Kloss L., Gebhardt C., Utikal J., Schmieder A., OncoTargets Ther. 2017, 8, 103682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong H. L. X., Jin G., Cao R., Zhang S., Cao Y., Zhou Z., Nat. Commun. 2016, 7, 10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oliveira‐Ferrer L., Milde‐Langosch K., Eylmann K., Rossberg M., Schmalfeldt B., Witzel I., Wellbrock J., Fiedler W., Int. J. Mol. Sci. 2020, 21, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pan J.‐H., Zhou H., Cooper L., Huang J.‐L., Zhu S.‐B., Zhao X.‐X., Ding H., Pan Y.‐L., Rong L., Front. Immunol. 2019, 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng C., Zheng L., Yoo J.‐K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J. Y., Zhang Q., Liu Z., Dong M., Hu X., Ouyang W., Peng J., Zhang Z., Cell 2017, 169, 1342. [DOI] [PubMed] [Google Scholar]
- 48. Kaji T., Arito M., Tsutiya A., Sase T., Onodera H., Sato T., Omoteyama K., Sato M., Suematsu N., Kurokawa M. S., Tanaka Y., Kato T., Brain Res. 2019, 1719, 140. [DOI] [PubMed] [Google Scholar]
- 49. Liao H. X., Lee D. M., Levesque M. C., Haynes B. F., J. Immunol. 1995, 155, 3938. [PubMed] [Google Scholar]
- 50. Knudson W., Chow G., Knudson C. B., Matrix Biol. 2002, 21, 15. [DOI] [PubMed] [Google Scholar]
- 51. Prochazka L., Tesarik R., Turanek J., Cell Signal 2014, 26, 2234. [DOI] [PubMed] [Google Scholar]
- 52. Mereiter S., Gomes C., Balmaã±A M., Macedo J. A., Polom K., Roviello F., Reis C. A., FEBS Lett. 2019, 593, 1675. [DOI] [PubMed] [Google Scholar]
- 53. Azevedo R., Gaiteiro C., Peixoto A., Relvas‐Santos M., Lima L. . S., Santos L. L., Clin. Proteomics 2018, 15, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lesley J., Kincade P. W., Hyman R., Eur. J. Immunol. 1993, 23, 1902. [DOI] [PubMed] [Google Scholar]
- 55. Yang C., Cao M., Liu H., Du Y., Liu Y., Wang W., Cui L., Hu J., Gao F., Bio. Chem. 2012, 287, 43094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Misra S., Hascall V. C., De Giovanni C., Markwald R. R., Ghatak S., J. Biol. Chem. 2009, 284, 12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Misra S., Ghatak S., Toole B. P., J. Biol. Chem. 2005, 280, 20310. [DOI] [PubMed] [Google Scholar]
- 58. Ghatak S., Misra S., Toole B. P., J. Biol. Chem. 2005, 280, 8875. [DOI] [PubMed] [Google Scholar]
- 59. Wang S. J., Bourguignon L. Y. W., Arch. Otolaryngol., Head Neck Surg. 2006, 132, 771. [DOI] [PubMed] [Google Scholar]
- 60. Sherman L. S., Rizvi T. A., Karyala S., Ratner N., J. Cell Biol. 2000, 150, 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Misra S., Toole B. P., Ghatak S., J. Biol. Chem. 2006, 281, 34936. [DOI] [PubMed] [Google Scholar]
- 62. Meran S., Luo D. D., Simpson R., Martin J., Wells A., Steadman R., Phillips A. O., J. Biol. Chem. 2011, 286, 17618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Okamoto I., Kawano Y., Tsuiki H., Sasaki J.‐I., Nakao M., Matsumoto M., Suga M., Ando M., Nakajima M., Saya H., Oncogene 1999, 18, 1435. [DOI] [PubMed] [Google Scholar]
- 64. Subramaniam V., Gardner H., Jothy S., Exp. Mol. Pathol. 2007, 83, 341. [DOI] [PubMed] [Google Scholar]
- 65. Senbanjo L. T., Chellaiah M. A., Front. Cell Dev. Biol. 2017, 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bourguignon L. Y. W., Wong G., Earle C., Krueger K., Spevak C. C., J. Biol. Chem. 2010, 285, 36721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bourguignon L. Y. W., Small GTPases 2012, 3, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bourguignon L. Y. W., Zhu H., Zhou B., Diedrich F., Singleton P. A., Hung M.‐C., J. Biol. Chem. 2001, 276, 48679. [DOI] [PubMed] [Google Scholar]
- 69. Li L., Qi L., Liang Z., Song W., Liu Y., Wang Y., Sun B., Zhang B., Cao W., Int. J. Mol. Med. 2015, 36, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bourguignon L. Y. W., Singleton P. A., Zhu H., Diedrich F., J. Biol. Chem. 2003, 278, 29420. [DOI] [PubMed] [Google Scholar]
- 71. Manning B. D., Toker A., Cell 2017, 169, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bourguignon L. Y. W., Semin. Cancer Biol. 2008, 18, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Faes S., Dormond O., Int. J. Mol. Sci. 2015, 16, 21138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K., J. Biol. Chem. 1996, 271, 20246. [DOI] [PubMed] [Google Scholar]
- 75. Bourguignon L. Y., Hyaluronan‐mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. in Seminars in cancer biology, Elsevier, Amsterdam, Netherlands, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bourguignon L Y. W., Singleton P. A., Diedrich F., Stern R., Gilad E., J. Biol. Chem. 2004, 279, 26991. [DOI] [PubMed] [Google Scholar]
- 77. Mori T., Kitano K., Terawaki S.‐I., Maesaki R., Fukami Y., Hakoshima T., J. Biol. Chem. 2008, 283, 29602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Duterme C., Mertens‐Strijthagen J., Tammi M., Flamion B., J. Biol. Chem. 2009, 284, 33495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Solinet S., Mahmud K., Stewman S. F., Ben El Kadhi K., Decelle B., Talje L., Ma A., Kwok B. H., Carreno S., J. Cell Biol. 2013, 202, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lokeshwar V. B., Fregien N., Bourguignon L. Y., J. Cell Biol. 1994, 126, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vigetti D., Viola M., Karousou E., Rizzi M., Moretto P., Genasetti A., Clerici M., Hascall V. C., De Luca G., Passi A., J. Biol. Chem. 2008, 283, 4448. [DOI] [PubMed] [Google Scholar]
- 82. Bourguignon L. Y. W., Gilad E., Rothman K., Peyrollier K., J. Biol. Chem. 2005, 280, 11961. [DOI] [PubMed] [Google Scholar]
- 83. Ridley A. J., Paterson H. F., Johnston C. L., Diekmann D., Hall A., Cell 1992, 70, 401. [DOI] [PubMed] [Google Scholar]
- 84. Habets G. G., Van der Kammen R. A., Stam J. C., Michiels F., Collard J. G., Oncogene 1995, 10, 1371. [PubMed] [Google Scholar]
- 85. Stam J. C., Michiels F., Van Der Kammen R. A., Moolenaar W. H., Collard J. G., EMBO J. 1998, 17, 4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mcfarlane S., Mcfarlane C., Montgomery N., Hill A., Waugh D. J. J., OncoTargets Ther. 2015, 6, 36762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bourguignon L. Y. W., Spevak C. C., Wong G., Xia W., Gilad E., J. Biol. Chem. 2009, 284, 26533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bourguignon L. Y. W., Peyrollier K., Xia W., Gilad E., J. Biol. Chem. 2008, 283, 17635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ooki T., Hatakeyama M., BioEssays 2020, 42, 2000005. [DOI] [PubMed] [Google Scholar]
- 90. Misra S., Obeid L. M., Hannun Y. A., Minamisawa S., Berger F. G., Markwald R. R., Toole B. P., Ghatak S., J. Biol. Chem. 2008, 283, 14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Misra S., Hascall V. C., Berger F. G., Markwald R. R., Ghatak S., Connect. Tissue Res. 2008, 49, 219. [DOI] [PubMed] [Google Scholar]
- 92. Hamilton S. R., Fard S. F., Paiwand F. F., Tolg C., Veiseh M., Wang C., Mccarthy J. B., Bissell M. J., Koropatnick J., Turley E. A., J. Biol. Chem. 2007, 282, 16667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Singleton P. A., Bourguignon L. Y. W., Exp. Cell Res. 2004, 295, 102. [DOI] [PubMed] [Google Scholar]
- 94. Montgomery N., Hill A., Mcfarlane S., Neisen J., O'grady A., Conlon S., Jirstrom K., Kay E. W., Waugh D. J., Breast Cancer Res. 2012, 14, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fieber C., Baumann P., Termeer C., Simon J. C., Hofmann M., Angel P., Herrlich P., Sleeman J. P., J. Cell Sci. 2004, 117, 359. [DOI] [PubMed] [Google Scholar]
- 96. Kung C.‐I., Chen C.‐Y., Yang C.‐C., Lin C.‐Y., Chen T.‐H., Wang H.‐S., Oncol. Rep. 2012, 28, 1808. [DOI] [PubMed] [Google Scholar]
- 97. Bourguignon L. Y. W., Wong G., Earle C. A., Xia W., Cytoskeleton 2011, 68, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Savani R. C., Modulators of inflammation in bronchopulmonary dysplasia. in Seminars in perinatology, Elsevier, Amsterdam, Netherlands, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Garantziotis S., Savani R. C., Matrix Biol. 2019, 78–79, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Telmer P. G., Tolg C., Mccarthy J. B., Turley E. A., Commun. Integr. Biol. 2011, 4, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Misra S., Hascall V. C., Markwald R. R., Ghatak S., Front. Immunol. 2015, 6, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tolg C., Mccarthy J. B., Yazdani A., Turley E. A., Biomed. Res. Int. 2014, 2014, 103923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Day A. J., Prestwich G. D., J. Biol. Chem. 2002, 277, 4585. [DOI] [PubMed] [Google Scholar]
- 104. Crainie M., Belch A. R., Mant M. J., Pilarski L. M., Blood 1999, 93, 1684. [PubMed] [Google Scholar]
- 105. Messam B. J., Tolg C., Mccarthy J. B., Nelson A. C., Turley E. A., Int. J. Mol. Sci. 2021, 22, 10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Assmann V., Marshall J. F., Fieber C., Hofmann M., Hart I. R., J. Cell Sci. 1998, 111, 1685. [DOI] [PubMed] [Google Scholar]
- 107. Entwistle J., Hall C. L., Turley E. A., J. Cell. Biochem. 1996, 61, 569. [DOI] [PubMed] [Google Scholar]
- 108. Xu R., Boudreau A., Bissell M. J., Cancer Metastasis Rev. 2009, 28, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Choi S., Wang D., Chen X., Tang L. H., Verma A., Chen Z., Kim B. J., Selesner L., Robzyk K., Zhang G., Pang S., Han T., Chan C. S., Fahey T. J., Elemento O., Du Y.‐C. N., Mol. Cancer 2019, 18, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang J., Li D., Shen W., Sun W., Gao R., Jiang P., Wang L., Liu Y., Chen Y., Zhou W., Wang R., Xiang R., Stupack D., Luo N., Anat. Rec. 2020, 303, 2344. [DOI] [PubMed] [Google Scholar]
- 111. Tolg C., Hamilton S. R., Nakrieko K.‐A., Kooshesh F., Walton P., Mccarthy J. B., Bissell M. J., Turley E. A., J. Cell Biol. 2006, 175, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Park D., Kim Y., Kim H., Kim K., Lee Y.‐S., Choe J., Hahn J.‐H., Lee H., Jeon J., Choi C., Kim Y.‐M., Jeoung D., Mol. Cells 2012, 33, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Choi S., Wang D., Chen X., Tang L. H., Verma A., Chen Z., Kim B. J., Selesner L., Robzyk K., Zhang G., Pang S., Han T., Chan C. S., Fahey T. J., Elemento O., Du Y.‐C. N., Mol. Cancer 2019, 18, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Manzanares D., Monzon M.‐E., Savani R. C., Salathe M., Am. J. Respir. Cell Mol. Biol. 2007, 37, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hall C. L., Lange L. A., Prober D. A., Zhang S., Turley E. A., Oncogene 1996, 13, 2213. [PubMed] [Google Scholar]
- 116. Hall C. L., Wang C., Lange L. A., Turley E. A., J. Cell Biol. 1994, 126, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hall C. L., Yang B., Yang X., Zhang S., Turley M., Samuel S., Lange L. A., Wang C., Curpen G. D., Savani R. C., Greenberg A. H., Turley E. A., Cell 1995, 82, 19. [DOI] [PubMed] [Google Scholar]
- 118. Wang C., Thor A. D., Moore D. H., Zhao Y., Kerschmann R., Stern R., Watson P. H., Turley E. A., Clin. Cancer Res. 1998, 4, 567. [PubMed] [Google Scholar]
- 119. Hall C. L., Collis L. A., Bo A J., Lange L., Mcnicol A., Gerrard J. M., Turley E. A., Matrix Biol. 2001, 20, 183. [DOI] [PubMed] [Google Scholar]
- 120. Mohapatra S., Yang X., Wright J. A., Turley E. A., Greenberg A. H., J. Exp. Med. 1996, 183, 1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nelson W. J., Nusse R., Science 2004, 303, 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Juan J., Muraguchi T., Iezza G., Sears R. C., Mcmahon M., Genes Dev. 2014, 28, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kouvidi K., Nikitovic D., Berdiaki A., Tzanakakis G. N., Adv. Cancer Res. 2014, 123, 319. [DOI] [PubMed] [Google Scholar]
- 124. Maxwell C. A., Mccarthy J., Turley E., J. Cell Sci. 2008, 121, 925. [DOI] [PubMed] [Google Scholar]
- 125. Maxwell C. A., Keats J. J., Belch A. R., Pilarski L. M., Reiman T., Cancer Res. 2005, 65, 850. [PubMed] [Google Scholar]
- 126. Assmann V., Jenkinson D., Marshall J. F., Hart I. R., J. Cell Sci. 1999, 112, 3943. [DOI] [PubMed] [Google Scholar]
- 127. Tolg C., Hamilton S. R., Morningstar L., Zhang J., Zhang S., Esguerra K. V., Telmer P. G., Luyt L. G., Harrison R., Mccarthy J. B., Turley E. A., J. Biol. Chem. 2010, 285, 26461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Maxwell C. A., Osorio A., Bonifaci N., Costes S. V., Chen H., Evans G. J. R., Mohan P., Petit A., Aguilar H., Villanueva A., Aytes A., Serra‐Musach J., Rennert G., Lejbkowicz F., Peterlongo P., Manoukian S., Peissel B., Ripamonti C. B., Bonanni B., Viel A., Allavena A., Bernard L., Radice P., Friedman E., Kaufman B., Laitman Y., Dubrovsky M., Milgrom R., Jakubowska A., Cybulski C., et al., PLoS Biol. 2011, 9, e1001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hatano H., Shigeishi H., Kudo Y., Higashikawa K., Tobiume K., Takata T., Kamata N., Lab. Invest. 2011, 91, 379. [DOI] [PubMed] [Google Scholar]
- 130. Chen H., Mohan P., Jiang J., Nemirovsky O., He D., Fleisch M. C., Niederacher D., Pilarski L. M., Lim C. J., Maxwell C. A., Cell Cycle 2014, 13, 2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Blanco I., Kuchenbaecker K., Cuadras D., Wang X., Barrowdale D., De Garibay G. R., Librado P., Rozas J., Bonifaci N., Mcguffog L., Pankratz V. S., Islam A., Mateo F., Berenguer A., Petit A., Brunet J., Osorio A., Nevanlinna H., Arun B. K., Toland A. E., Karlan B. Y., Walsh C., Lester J., Greene M. H., Mai P. L., Nussbaum R. L., Andrulis I. L., Domchek S. M., Nathanson K. L., Rebbeck T. R., et al., PLoS One 2015, 10, e0120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Smith T. C., Fridy P. C., Li Y., Basil S., Arjun S., Friesen R. M., Leszyk J., Chait B. T., Rout M. P., Luna E. J., Mol. Biol. Cell 2013, 24, 3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hasegawa H., Hyodo T., Asano E., Ito S., Maeda M., Kuribayashi H., Natsume A., Wakabayashi T., Hamaguchi M., Senga T., J. Cell Sci. 2013, 126, 3627. [DOI] [PubMed] [Google Scholar]
- 134. Gangopadhyay S. S., Takizawa N., Gallant C., Barber A. L., Je H.‐D., Smith T. C., Luna E. J., Morgan K. G., J. Cell Sci. 2004, 117, 5043. [DOI] [PubMed] [Google Scholar]
- 135. Jang G.‐Y., Lee J. W., Kim Y. S., Lee S. E., Han H. D., Hong K.‐J., Kang T. H., Park Y.‐M., Exp. Mol. Med. 2020, 52, 1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Beutler B. A., Blood 2009, 113, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Termeer C., Benedix F., Sleeman J., Fieber C., Voith U., Ahrens T., Miyake K., Freudenberg M., Galanos C., Simon J. C., J. Exp. Med. 2002, 195, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Campo G. M., Avenoso A., Campo S., Nastasi G., Calatroni A., Biochem. Pharmacol. 2010, 80, 480. [DOI] [PubMed] [Google Scholar]
- 139. Dajon M., Iribarren K., Cremer I., Immunobiology 2017, 222, 89. [DOI] [PubMed] [Google Scholar]
- 140. Zheng L., Riehl T. E., Stenson W. F., Gastroenterology 2009, 137, 2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Jiang D., Liang J., Fan J., Yu S., Chen S., Luo Y., Prestwich G. D., Mascarenhas M. M., Garg H. G., Quinn D. A., Homer R. J., Goldstein D. R., Bucala R., Lee P. J., Medzhitov R., Noble P. W., Nat. Med. 2005, 11, 1173. [DOI] [PubMed] [Google Scholar]
- 142. Termeer C., Benedix F., Sleeman J., Fieber C., Voith U., Ahrens T., Miyake K., Freudenberg M., Galanos C., Simon J. C., J. Exp. Med. 2002, 195, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Li F., Hao P., Liu G., Wang W., Han R., Jiang Z., Li X., Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 559. [DOI] [PubMed] [Google Scholar]
- 144. Ebid R., Lichtnekert J., Anders H.‐J., ISRN Nephrol. 2014, 2014, 714081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Taylor K. R., Yamasaki K., Radek K. A., Nardo A. D., Goodarzi H., Golenbock D., Beutler B., Gallo R. L., J. Biol. Chem. 2007, 282, 18265. [DOI] [PubMed] [Google Scholar]
- 146. Scheibner K. A., Lutz M. A., Boodoo S., Fenton M. J., Powell J. D., Horton M. R., J. Immunol. 2006, 177, 1272. [DOI] [PubMed] [Google Scholar]
- 147. Taylor K. R., Trowbridge J. M., Rudisill J. A., Termeer C. C., Simon J. C., Gallo R. L., J. Biol. Chem. 2004, 279, 17079. [DOI] [PubMed] [Google Scholar]
- 148. Kawasaki T., Kawai T., Front. Immunol. 2014, 5, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Voelcker V., Gebhardt C., Averbeck M., Saalbach A., Wolf V., Weih F., Sleeman J., Anderegg U., Simon J., Exp. Dermatol. 2008, 17, 100. [DOI] [PubMed] [Google Scholar]
- 150. Santaolalla R., Sussman D. A., Ruiz J. R., Davies J. M., Pastorini C., Sotolongo J., Burlingame O., Bejarano P. A., Philip S., Ahmed M. M., Ko J., Dirisina R., Barrett T. A., Shang L., Lira S. A., Fukata M., Abreu M. T., PLoS One 2013, 8, e63298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Rakoff‐Nahoum S., Medzhitov R., Nat. Rev. Cancer 2009, 9, 57. [DOI] [PubMed] [Google Scholar]
- 152. Mantovani A., Allavena P., Sica A., Balkwill F., Nature 2008, 454, 436. [DOI] [PubMed] [Google Scholar]
- 153. Alvarado A. G., Thiagarajan P. S., Mulkearns‐Hubert E. E., Silver D. J., Hale J. S., Alban T. J., Turaga S. M., Jarrar A., Reizes O., Longworth M. S., Vogelbaum M. A., Lathia J. D., Cell Stem Cell 2017, 20, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Duan Z., Luo Y., Signal Transduct. Target. Ther. 2021, 6, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Del Fresno C., Otero K., Soler‐Ranger L., Fuentes‐Prior P., Escoll P., Baos R., Caveda L., Arnalich F., J. Immunol. 2005, 174, 3032. [DOI] [PubMed] [Google Scholar]
- 156. Kuang D.‐M., Wu Y., Chen N., Cheng J., Zhuang S.‐M., Zheng L., Blood 2007, 110, 587. [DOI] [PubMed] [Google Scholar]
- 157. Sokolowska M., Chen L. Y., Eberlein M., Martinez‐Anton A., Liu Y., Alsaaty S., Qi H. Y., Logun C., Horton M., Shelhamer J. H., Clinical Trial 2014, 289, 4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Banerji S., Ni J., Wang S.‐X., Clasper S., Su J., Tammi R., Jones M., Jackson D. G., J. Cell Biol. 1999, 144, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Schmaus A., Klusmeier S., Rothley M., Dimmler A., Sipos B., Faller G., Thiele W., Allgayer H., Hohenberger P., Post S., Sleeman J. P., J. Cancer 2014, 111, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Banerji S., Hide B. R. S., James J. R., Noble M. E. M., Jackson D. G., J. Biol. Chem. 2010, 285, 10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lawrance W., Banerji S., Day A. J., Bhattacharjee S., Jackson D. G., J. Biol. Chem. 2016, 291, 8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wu M., Du Y., Liu Y., He Y., Yang C., Wang W., Gao F., PLoS One 2014, 9, e92857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Banerji S., Lawrance W., Metcalfe C., Briggs D. C., Yamauchi A., Dushek O., Van Der Merwe P. A., Day A. J., Jackson D. G., J. Biol. Chem. 2016, 291, 25004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Prevo R., Banerji S., Ferguson D. J. P., Clasper S., Jackson D. G., J. Biol. Chem. 2001, 276, 19420. [DOI] [PubMed] [Google Scholar]
- 165. Johnson L. A., Prevo R., Clasper S., Jackson D. G., J. Biol. Chem. 2007, 282, 33671. [DOI] [PubMed] [Google Scholar]
- 166. Harris E. N., Baker E., Int. J. Mol. Sci. 2020, 21, 3504.32429122 [Google Scholar]
- 167. Park S.‐Y., Jung M.‐Y., Kim H.‐J., Lee S.‐J., Kim S.‐Y., Lee B.‐H., Kwon T.‐H., Park R.‐W., Kim I.‐S., Cell Death Differ. 2008, 15, 192. [DOI] [PubMed] [Google Scholar]
- 168. Simpson M. A., Weigel J. A., Weigel P. H., Int. J. Cancer 2012, 131, E836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Pandey M. S., Weigel P. H., J. Biol. Chem. 2014, 289, 21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Pandey M. S., Baggenstoss B. A., Washburn J., Harris E. N., Weigel P. H., J. Biol. Chem. 2013, 288, 14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hirose Y., Saijou E., Sugano Y., Takeshita F., Nishimura S., Nonaka H., Chen Y.‐R., Sekine K., Kido T., Nakamura T., Kato S., Kanke T., Nakamura K., Nagai R., Ochiya T., Miyajima A., Proc. Natl. Acad. Sci. U S A 2012, 109, 4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Borowsky M. L., Hynes R. O., J. Cell Biol. 1998, 143, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Bono P., Rubin K., Higgins J. M. G., Hynes R. O., Mol. Biol. Cell 2001, 12, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Forteza R. M., Casalino‐Matsuda S. M., Falcon N. S., Valencia Gattas M., Monzon M. E., J. Biol. Chem. 2012, 287, 42288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Bono P., Cordero E., Johnson K., Borowsky M., Ramesh V., Jacks T., Hynes R. O., Exp. Cell Res. 2005, 308, 177. [DOI] [PubMed] [Google Scholar]
- 176. De Simone M., Arrigoni A., Rossetti G., Gruarin P., Ranzani V., Politano C., Bonnal R. J. P., Provasi E., Sarnicola M. L., Panzeri I., Moro M., Crosti M., Mazzara S., Vaira V., Bosari S., Palleschi A., Santambrogio L., Bovo G., Zucchini N., Totis M., Gianotti L., Cesana G., Perego R. A., Maroni N., Pisani Ceretti A., Opocher E., Geginat J., Stunnenberg H. G., Abrignani S., Pagani M., Immunity 2016, 45, 1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Chen Z., Zhuo W., Wang Y., Ao X., An J., Biotech. Appl. Biochem. 2008, 50, 89. [DOI] [PubMed] [Google Scholar]
- 178. Bellos D. A., Sharma D., Mcmullen M. R., Wat J., Saikia P., Motte C. A., Nagy L. E., Alcohol. Clin. Exp. Res. 2019, 43, 1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Kim Y., West G. A., Ray G., Kessler S. P., Petrey A. C., Fiocchi C., Mcdonald C., Longworth M. S., Nagy L. E., De La Motte C. A., Matrix Biol. 2018, 66, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Adachi T., Arito M., Suematsu N., Kamijo‐Ikemori A., Omoteyama K., Sato T., Kurokawa M. S., Okamoto K., Kimura K., Shibagaki Y., Kato T., Biochem. Biophys. Res. Commun. 2015, 467, 63. [DOI] [PubMed] [Google Scholar]