

Abstract
Candida albicans is the most common human fungal pathogen, able to reside in a broad range of niches within the human body. Even though C. albicans systemic infection is associated with high mortality, the fungus has historically received relatively little attention, resulting in a lack of optimized molecular and fluorescent tools. Over the last decade, some extra focus has been put on the optimization of fluorescent proteins (FPs) of C. albicans. However, as the FPs are GFP-type, they require an aerobic environment and a relatively long period to fully mature. Recently, we have shown the application of a novel type of fluorogen-based FP, with an improved version of fluorescence activating and absorption shifting tag (iFAST), in C. albicans. Due to the dynamic relation between iFAST and its fluorogens, the system has the advantage of being reversible in terms of fluorescence. Furthermore, the combination of iFAST with different fluorogens results in different spectral and cellular properties, allowing customization of the system.
Key features
• Genetic integration and tagging with the iFAST tag in Candida albicans.
• Imaging and localization of a protein of interest tagged with iFAST.
• Reversibility of fluorescence with iFAST.
Keywords: Candida albicans, iFAST, Fluorescence activating and absorption shifting tag, Reversible fluorescence, Protein tag, Multi-color, Fluorescence microscopy
Graphical overview
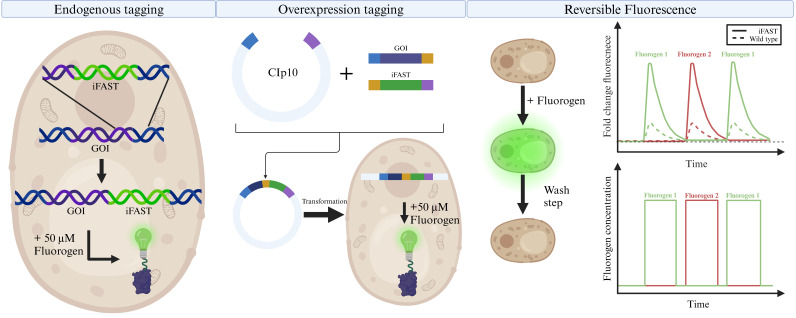
Background
The ubiquitous commensal Candida albicans is a fungus that resides in the gastrointestinal tract, blood, skin, and other mucosal surfaces [1–3]. In healthy individuals, the immune system is sufficiently capable of eradicating C. albicans, but upon opportunistic circumstances, the fungus can become pathogenic, often with a high morbidity rate [4]. As a species, C. albicans is closely related to the eukaryotic model organism Saccharomyces cerevisiae. However, despite this close relationship, C. albicans has an aberrant codon usage, translating the CUG codon to serine instead of leucine in 96% of cases [5,6]. Consequently, heterologous expression of C. albicans proteins requires codon optimization. Conversely, this also means that molecular tools and protein labeling techniques developed for use in S. cerevisiae cannot easily be translated to C. albicans, as these will suffer from the same mistranslation issues. Regarding this issue, several techniques for codon optimization in C. albicans have been reported [7,8], but studies have shown that these are not generally applicable [9].
In recent years, some fluorescent tools have been developed/optimized for use in C. albicans (and extensively reviewed in Van Genechten et al. [10]). In this protocol, we show the use of a novel type of fluorescent protein, an improved version of fluorescence activating and absorption shifting tag (iFAST), which requires the binding of a fluorogen, instead of chromophore maturation, to be fluorescently active. This novel system was developed by the group of Gautier for use in mammalian cells [11,12]. The iFAST protein is half the size of GFP and has the added benefit of reversibly binding its fluorogens, which are analogs of 4-hydroxybenzyliden-rhodanine (HBR). On their own, iFAST and HBR analogs are relatively non-fluorescent, but upon contact of iFAST protein with its fluorogen, fluorescence can be observed. An added benefit of this system is that the interaction is reversible and does not require oxygen, making this a reversible fluorescent system even in anaerobic conditions, where classical FPs like GFP do not work.
Materials and reagents
Biological materials
SC5314 Candida albicans wild-type strain [13]
SN152 Candida albicans arg4Δ/arg4Δ leu2Δ/leu2Δ his1Δ/his1Δ URA3/ura3Δ::imm434 IRO1/iro1Δ::imm434 [14]
Reagents
Granulated yeast extract (Merk, catalog number: 1.03753)
Bacterial peptone (Oxoid, catalog number: LP0037B)
Glucose (Fluka, catalog number: 49459)
Glycerol (Sigma, CAS number: 58-81-5)
Granulated bacteriological agar (Difco, catalog number: 214530)
Nourseothricin (Jena Bioscience, CAS number: 96736-11-7)
Complete synthetic mixture (CSM) (MP Biomedicals, catalog number: 114500022)
Yeast nitrogen base (YNB) without amino acids, ammonium sulfate, and riboflavin (Formedium, catalog number: YN6501)
Ammonium sulfate (VWR, CAS number: 7783-20-2)
Lithium acetate dihydrate (LiAc·2H2O) (Sigma-Aldrich, CAS number: 6108-17-4)
Polyethylene glycol (PEG) 3350 (Sigma-Aldrich, CAS number: 25322-68-3)
Trizma® base (Sigma-Aldrich, CAS number: 77-86-1)
HCl (Sigma-Aldrich, CAS number: 7647-01-0)
EDTA (Sigma-Aldrich, CAS number: 6381-92-6)
HMBR (Twinkle Factory, catalog number: 480541-250)
HBR-3,5DM (Twinkle Factory, catalog number: 499558-250)
HBR-3,5DOM (Twinkle Factory, catalog number: 516600-250)
HIFI Q5 polymerase (New England Biolabs, catalog number: NEB M0491L)
NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs, catalog number: NEB E2621X)
ssDNA, MB-grade from fish sperm (Merck, catalog number: 11467140001)
rCutsmart restriction buffer (New England Biolabs, catalog number: B6004S)
-
Restriction enzymes:
StuI (New England Biolabs, catalog number: R0187S)
NheI-HF (New England Biolabs, catalog number: R3131S)
PstI-HF (New England Biolabs, catalog number: R3140S)
Solutions
YPD (see Recipes)
YPD-NAT agar (see Recipes)
YPD-glycerol (see Recipes)
Low fluorescence medium (see Recipes)
1 M LiAc (see Recipes)
PEG 3350 (50%) (see Recipes)
TE (10×) (see Recipes)
LiAc/TE buffer (see Recipes)
LiAc/PEG buffer (see Recipes)
5 mM fluorogen stock solution (see Recipes)
Recipes
-
YPD (1 L)
Reagent Final concentration Quantity or Volume Yeast extract granulated 1% (w/v) 10 g Bacteriological peptone 2% (w/v) 20 g Glucose 2% (w/v) 20 g H2O (demineralized) n/a 1 L -
YPD-NAT agar (1 L)
*Note: Add the nourseothricin after heat sterilization and when the medium has cooled down to approximately 50 °C.
Reagent Final concentration Quantity or Volume Yeast extract granulated 1% (w/v) 10 g Bacteriological peptone 2% (w/v) 20 g Glucose 2% (w/v) 20 g Bacteriological agar 2% (w/v) 20 g Nourseothricin* 0.02% (w/v) 200 mg H2O (demineralized) n/a 1 L -
YPD-glycerol (100 mL)
Reagent Final concentration Quantity or Volume Yeast extract granulated 1% (w/v) 1 g Bacteriological peptone 2% (w/v) 2 g Glycerol 30% (v/v) 30 mL H2O (demineralized) n/a 70 mL -
Low fluorescence medium (1 L)
Reagent Final concentration Quantity or Volume CSM 0.079% (w/v) 0.79 g YNB (without amino acids, without ammonium sulfate, without riboflavin) 0.69% (w/v) 6.9 g Ammonium sulfate 0.5% (v/v) 5 g Glucose 2% (w/v) 20 g H2O (demineralized) n/a 1 L -
1 M LiAc (100 mL)
Reagent Final concentration Quantity or Volume LiAc·2H2O 1 M 10.2 g H2O (demineralized) n/a 100 mL -
PEG 3550 (50%) (100 mL)
*Note: First, add 50 mL of H2O and a stir bar. Let the PEG dissolve overnight and add water up to a total volume of 100 mL.
Reagent Final concentration Quantity or Volume PEG 3550 50% (w/v) 50 g H2O (demineralized) n/a Until 100 mL* -
TE (10×) (1 L)
*Note: After adding Tris, adjust the pH to 8 using HCl.
Reagent Final concentration Quantity or Volume Tris 10 mM 12.11 g* EDTA 1 mM 2.92 g H2O (demineralized) n/a 1 L -
LiAc/TE buffer (1 mL)
Reagent Final concentration Quantity or Volume LiAc (1M) 0.1 M 100 µL TE (10×) 1× 100 µL H2O (MiliQ) n/a 800 µL -
LiAc/PEG buffer (1 mL)
Reagent Final concentration Quantity or Volume LiAc (1 M) 0.1 M 100 µL TE (10×) 1× 100 µL PEG 3350 [50% (w/v)] 40% (w/v) 800 µL -
Fluorogen stock solution
*Note: The quantity is calculated to make a total of 100 mL. Store this stock solution in aliquots of 20 µL at -20 °C. Make a separate stock solution for each fluorogen; do not mix. Fluorogen stock solutions can withstand multiple freeze-thaw cycles; however, try not to defrost more than five times. Additionally, it is best to protect the fluorogens from light as much as possible when working.
Reagent Final concentration Quantity or Volume HMBR 5 mM 0.125 g* HBR-3,5DM 5 mM 0.133 g* HBR-3,5DOM 5 mM 0.149 g*
Laboratory supplies
Microtubes (1.5 mL) (Eppendorf, catalog number: 0030125150)
Micro pipettes (sizes 1,000, 200, and 10 µL)
Micro pipette tips (standard tips of 1,000, 200, and 10 µL)
Microscope slides (VWR, catalog number: 630-1985)
Glass covers (VWR, catalog number: 631-0122)
Glass culture tubes (10 mL) (Fisher Scientific, catalog number: 11557403)
Caps for culture tubes (VWR, catalog number: LUDI184010631)
Erlenmeyer flasks (300 mL) (VWR, catalog number: 391-0275)
Erlenmeyer flask caps (VWR, catalog number: 391-0950)
Clear cuvette (VWR, catalog number: 97000-590)
Vortex (VWR, catalog number: 444-1372)
NucleoSpinTM Gel and PCR Clean-up Kit (Macherey-Nagel, catalog number: 740611.50)
NucleoSpinTM Plasmid EasyPure Kit (Macherey-Nagel, catalog number: 740727.50)
Equipment
FV1000 microscope (Olympus, model: FV1000-IX81) with 60× UPlanSApo (NA1.35) objective lens (Olympus) or equivalent
ThermoMixer® F1.5 (Eppendorf, catalog number: 5384000020) or equivalent
Shaking incubator (New Brunswick Scientific, model: Innova40) or equivalent
Microtube centrifuge (Eppendorf, model: 5471C) or equivalent
BioPhotometer (Eppendorf, model: 6131)
NanoDropTM 1000 spectrophotometer (Thermo Scientific, model: ND-1000) or equivalent
Software and datasets
FV10-ASW 4.2 software package (Olympus)
ImageJ (v1.53t, 24/08/2022) (https://imagej.net/software/fiji/downloads)
RStudio (R version: 4.3.2, 31/10/2023) (https://cran.rstudio.com/; https://posit.co/download/rstudio-desktop/)
Procedure
Below we provide a stepwise overview of tagging a protein of interest with iFAST in C. albicans. We describe how to do this in order to obtain an endogenous tag or an overexpression construct. This protocol has been used to tag Ftr1 and Erg11, endogenously or as an overexpression construct, respectively [15]. Furthermore, we also describe a protocol to utilize the reversibility of the iFAST system to alter the absorption and excitation maxima of the tagged construct by exchanging the fluorogen. All steps of this protocol were performed at room temperature (22 °C) unless stated otherwise.
-
Transformation of Candida albicans (see General note 1)
Streak the wild-type C. albicans (SC5314) from the -80 °C stock and plate on YPD agar at 30 °C overnight (ON).
Transfer a single colony to 3 mL of YPD and grow ON at 30 °C in a shaking incubator (at 240 rpm).
Measure the cell density of the culture at 600 nm using the BioPhotometer. Dilute the ON culture to an OD600 of 0.2–0.4 in 50 mL of YPD in a 300 mL Erlenmeyer flask. Incubate again at 30 °C and grow the cells to a density of approximately 1.5; this will take 3–4 h.
Pellet the cells at 1,000× g for 5 min.
Remove the supernatant and resuspend the cells in 0.7 mL of LiAc/TE buffer.
While adding the LiAc/TE buffer, mix the cells well by pipetting up and down.
-
Pipette in a new Eppendorf tube (in the following order):
100 µL of the cell suspension in LiAc/TE buffer.
-
DNA (1 µg) that needs to be transformed.
i. In the case of a plasmid: First, linearize the plasmid via a restriction digestion. Then, use the unpurified restriction product.
ii. In case of PCR: Use 50 µL of unpurified linear PCR product.
20 µL of fish sperm ssDNA (10 mg/mL) (preheated to 98 °C for 5 min and cooled to 50 °C).
Leave at room temperature for 10 min.
0.7 mL of LiAc/PEG buffer.
Vortex shortly.
Incubate for 16 h at 30 °C while shaking (300 rpm).
Heat-shock the cell mixture using a heat block for 15 min at 44 °C.
Pellet cells at 5,200× g for 1 min.
Remove the supernatant and resuspend in 1 mL of YPD.
Let the cells recover for 4 h at 30 °C while shaking (300 rpm).
Pellet the cells at 5,200× g for 1 min.
Plate out on YPD agar containing 200 mg/L nourseothricin for both the pFA6-based endogenous tagging and the overexpression strains using the CIp10 plasmid. Incubate for 24 h at 30 °C and re-streak to single colonies on YPD-NAT agar. The plates should contain approximately 20 colonies for the pFA6 transformation and more than 100 colonies for the CIp10 transformation.
Colonies are verified based on the presence and localization of fluorescence under the confocal microscope. Alternatively, the transformation can be checked through colony PCR [16], targeting the tagged gene and iFAST (for iFAST reverse primer, see Table S3). Since the integration efficiency of the linearized CIp10 plasmid was around 90% in our case, testing five colonies proved to be sufficient. The integration efficiency of the PCR product for endogenous tagging of genes was less efficient (approximately 10%–20%), so at least 12 colonies were tested in that case.
-
Endogenous protein tagging with iFAST
-
The iFAST-SAT1 construct will be integrated between the end of the gene and the beginning of the terminator. For this, a forward and reverse primer need to be constructed with a sequence that overlaps with specific regions within the genome and binds on the pFA6-iFAST plasmid (available on AddGene: #209414). The genomic sequences of C. albicans genes can be accessed at http://www.candidagenome.org (see General note 2).
Design the forward primer to contain the last 50 bp of the gene of interest (GOI) excluding the stop codon, a double GGGGS linker (“GGAGGTGGAGGTTCTGGTGGAGGTGGTTCA”), and the first 20 bp of iFAST (“ATGGAACATGTTGCCTTTGG”).
The reverse primer consists of the first 50 complementary bp of the GOI terminator and the last 20 bp of the SAT1 gene (“TTAGGCGTCATCCTGTGCTC”).
Using the constructed primers, amplify the iFAST-SAT1 construct from the pFA6-iFAST plasmid using Q5 HiFi DNA polymerase (New England Biolabs) using the protocol in Table 1. The master mix was made following the manufacturer’s instructions.
Follow the protocol described in section A to transform the iFAST-SAT1 construct in C. albicans wild type. Select on medium containing 200 mg/L nourseothricin (see General note 3).
Pick a colony and re-streak again to obtain single colonies on YPD-NAT agar before continuing.
-
Colonies are verified based on fluorescence and its localization under the confocal microscope or by PCR, targeting the GOI and the iFAST gene (for iFAST reverse primer, see Table S3).
To check protein localization, grow a single colony overnight in low fluorescence medium at 30 °C. Dilute the culture in fresh medium to OD600 0.2–0.4 and grow for 4–5 h until the mid-exponential phase (~OD600 1). Incubate the strain for 15 min in low fluorescence medium with 50 µM of fluorogen. Afterward, add 1–2 µL of culture to a standard microscopy slide, cover with a coverslip, and check under the confocal microscope. Choose the settings of the microscope in accordance with the fluorogen (Table 2).
Transfer verified colonies to YPD-glycerol medium and place at -80 °C for long-term storage.
-
-
Overexpression tagging with iFAST
In silico Gibson cloning
The construction of the plasmid for overexpression tagging is based on Gibson assembly [17]. In the NEBuilder tool (https://nebuilder.neb.com/#!/) or a similar in silico cloning tool, insert the sequence of the plasmid backbone (CIp10-NAT1; AddGene: #225583) (Text S1). Process the plasmid for restriction digestion with PstI and NheI to allow the integration of two inserts.
As the first insert, add the full DNA sequence of the GOI, excluding the stop codon.
As the second fragment to insert, add the full iFAST sequence (Text S2). In between these fragments, add a double [GGGGS]2 linker.
In vitro cloning
-
Using the primers constructed by the in silico cloning tool, amplify the GOI and iFAST using Q5 HiFi DNA polymerase using the protocol in Table 1 (see General note 4).
Amplify the sequence of the GOI from isolated gDNA.
iFAST can be amplified from an iFAST-containing plasmid (pFA6a-iFAST available on AddGene: #209414) or gBlock.
Afterward, purify the PCR fragments using the NucleoSpinTM Gel and PCR Clean-up kit and measure the concentration using Nanodrop.
After the restriction of the backbone plasmid with PstI and NheI, purify the restriction mixture using the NucleoSpinTM Gel and PCR Clean-up kit. Measure the concentration of the purified, restricted plasmid using Nanodrop.
-
Construct the overexpression plasmid by ligating the linearized vector with the two PCR products (GOI and iFAST). The ligation mixture to do this consists of 50 ng of plasmid and a molar ratio of plasmid:fragments of 1:3 with 10 µL of NEBuilder® HiFi DNA Assembly Master Mix and the total volume adjusted to 20 µL. This mixture was incubated at 50 °C for up to 1 h (see General note 5).
Amplify the constructed plasmid using chemically competent E. coli [18]. Afterward, purify the plasmids using the NucleoSpinTM Plasmid EasyPure kit.
Before transformation into C. albicans, linearize the plasmid using restriction digestion by StuI. Transformation into C. albicans is done according to an adapted Gietz protocol (section A).
-
After transformation, select on YPD-NAT. Re-streak to single colonies and check successful tagging using fluorescence microscopy. Alternatively, colonies can be checked by PCR targeting the GOI and the iFAST gene. The FW primer within the GOI is unique, whilst the RV primer for the iFAST gene is “TTGCCAATCACTTGTTTGGG”.
To check protein localization, grow a single colony overnight in low fluorescence medium at 30 °C. Dilute the culture in fresh medium to OD600 0.2–0.4 and grow for 4–5 h until the mid-exponential phase (~OD600 1). Incubate the strain for 15 min in low fluorescence medium with 50 µM of fluorogen. Afterward, add 1–2 µL of culture to a standard microscopy slide, cover with a coverslip, and check under the confocal microscope. Choose the settings of the microscope in accordance with the fluorogen (Table 2).
Transfer verified colonies to YPD-glycerol medium and place at -80 °C for long-term storage.
-
Reversible fluorescence of iFAST
Grow the transformed cells overnight at 30°C in low fluorescence medium (Figure 1).
-
Dilute the transformed strain in fresh low fluorescence medium to OD600 of 0.2 and grow for 4–5 h until the mid-exponential phase (~OD600 1).
Before starting the experiment, predetermine the time points at which you want to image the cells (see General note 6).
Prior to adding any fluorogen, add 1–2 µL of culture on a standard microscopy slide, cover with a coverslip, and image using the fluorescent microscope (see section E). Use this during the analysis as the pre-treatment condition.
Add the fluorogen to the culture for a final concentration of 50 µM of fluorogen and a final volume of 1 mL.
-
For each predetermined time point repeat the following steps:
Add 1–2 µL of the culture on a standard microscopy slide and cover with a coverslip.
Let the rest of the culture incubate at 30 °C with gentle shaking (300 rpm) until the next time point.
Image the sample under the confocal microscope (see section E).
After the last time point, wash the cells twice by spinning down (1 min at 5,200× g), replacing the supernatant with medium lacking fluorogen. Image 1–2 µL of the culture on a standard microscopy slide after each of the washing steps (see General note 7).
If desired, the same culture can be used to repeat this experiment with another fluorogen to exploit the multicolor characteristic of the iFAST protein. For this, use the washed culture and repeat from step D4 (Figure 1).
-
Sample preparation and confocal microscopy
-
For handling the microscope, we used a previously published protocol [19]. For the selection of the laser lines, excitation filter, and appropriate emission filters for the different fluorogens, see Table 2.
Aside from the fluorescent channel, concurrent DIC or brightfield images are taken (see General note 8).
From the FV-10 software (or other user-specific software), images of both channels are exported to a (multi-)TIFF format (see General note 9).
-
Table 1. Thermocycling conditions for Q5 PCR reaction.
| Step | Temperature (°C) | Duration | Number of cycles |
|---|---|---|---|
| Initial denaturation | 98 | 30 s | 1 |
| Denaturation | 98 | 15 s |
30 Back to step 2 |
| Annealing | 64 | 25 s | |
| Extension | 72 | 30 s per kb | |
| Final extension | 72 | 5 min | 1 |
| Hold | 10 | ∞ | - |
Table 2. Overview of the validated fluorogens and their appropriate microscopy settings.
| Fluorogen | λexcmax/λemmmas | Excitation laser | Excitation filter | Emission filter |
|---|---|---|---|---|
| HMBR | 480/541 nm | 488 nm | DM405/488 | BA505–605 nm |
| HBR-3,5DM | 499/558 nm | 488 nm | DM405/488 | BA505–605 nm |
| HBR-3,5DOM | 516/600 nm | 515 nm | DM458/515 | BA575–620 nm |
Figure 1. Diagram of the experimental steps of section D.

All incubation steps are performed at 30 °C.
Data analysis
To evaluate the reversibility of fluorescence, the fluorescence intensity of the cells needs to be calculated and can be compared over time. All image analysis was done using the ImageJ2 software [20]. To do this, install and open the ImageJ2 software. Open a multi-TIFF image: File > Open…
Make a new macro to facilitate the image processing: Plugins > New>Macro. Past in the code written below.
run("Set Measurements...", "area mean display redirect=None decimal=3");
// This if statement clears out the ROI manager if it is not empty
if(roiManager("count")!= 0){
roiManager("Delete");
}
// The Min and Max numbers are chosen in accordance with our system, adjust these //if needed
setMinAndMax(0, 4095);
run("Stack to Images");
This macro makes sure that the correct measuring methods are set. Further, it will check if the ROI manager is empty; if not, it will clear the ROI manager. Additionally, it sets the minimal and maximal brightness of the image. Lastly, it separates the two channels of a multi-TIFF image (see General note 10). To run the macro, select the Run button.
Using the Freehand selection tool, select the regions of interest (ROIs). Select all C. albicans cells in the DIC or brightfield image and copy the ROIs to the ROI manager. To achieve this, select a cell and press the T button on the keyboard, and the ROI manager will open automatically.
In addition to the cells, select seven regions on the DIC image that only include background and no cells. These seven regions should be of similar size as the cells. These will be used to calculate the background fluorescence of the image.
Once all cells and background regions on the DIC or brightfield image have been selected and copied to the ROI manager, select the image of the fluorescent channel and measure via the ROI manager: Window > ROI manager > Measure. This will open a dialog box titled “Results.” Copy these results to an Excel file.
-
For the analysis of the fluorescence intensity at each timepoint, including the pre-treatment control, follow the following steps:
For each image, calculate the average intensity of the seven background ROI’s and subtract this value from the cell values.
To calculate the fold change in fluorescence, the data is normalized to the pre-treatment control (see General note 11). Calculate the average of the background-corrected cell measurement of the pre-treatment condition.
Divide every background-corrected cell measurement with this normalization value, including the cell measurements of the pre-treatment condition (see General note 12).
The fold change data of all cells is put into one big dataset, including information on which fluorogen was used and at which time point the data was collected (an example of such dataset can be found at https://github.com/Jonas-Ds/iFAST-RStudio-Figures).
For the visualization and statistical analysis, RStudio with the ggplot and stats packages were used, respectively. RStudio script with example data can be found here: https://github.com/Jonas-Ds/iFAST-RStudio-Figures/. At each time point, the two different strains (iFAST tagged strain vs. wild-type control) were compared using a Student's t-test.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Devos et al. [15]. A multi-colour fluorogenic tag and its application in Candida albicans. Microbiology (Figures 2–4; Figure S3).
General notes and troubleshooting
General notes
Transformation of C. albicans is based on a LiAc/PEG method, which promotes the uptake of DNA by the yeast cells.
-
Following are two example primers for the endogenous tagging of FTR1 in Candida albicans. The full genetic sequence including 1 kb up/downstream can be found on the Candida Genome Database (CGD) (www.candidagenome.org/). For the forward primer, the last 50 bp of the gene, excluding the stop codon, are in italics; the linker is underlined; and the first 20 bp of iFAST are in bold.
AAGTTGATGAAACTTCATCAAACAAATTGATCGAATCCAAAGAAAACAAA GGAGGTGGAGGTTCTGGTGGAGGTGGTTCA ATGGAACATGTTGCCTTTGG
For the reverse primer, the reverse complement of the first 50 bp of the terminator of FTR1 is in italics, and the reverse complement of last 20 bp of the SAT1 gene is in bold.
CACAGTCTCTTGCCTTATTCTTTTAGTTGTTGAATAATAATTAACTAAGT TTAGGCGTCATCCTGTGCTC (Table S3).
When using these primers for a different gene, only the italics part of the primers needs to be adjusted to the gene of interest.
No purification of the PCR product is performed, as this will lower the yield of transformation. C. albicans is not efficient in keeping plasmids even under selective pressure, therefore no purification is required.
The melting temperature for the PCRs is calculated using the Tm calculator tool of New England BioLabs (https://tmcalculator.neb.com/#!/main). In case a different DNA polymerase is used, the thermocycling conditions of Table 1 should be adjusted according to the DNA polymerase.
The NEBuilder® HiFi DNA Assembly Master Mix contains an exonuclease, a polymerase, and a DNA ligase. Due to the combination of these enzymes, no further processing of the PCR fragments is required after the restriction of the plasmid.
In the original publication of this protocol, a couple of different imaging and timing strategies can be found [15]. These include imaging at 2, 5, 10, and 15 min followed by two washing steps. This imaging and washing combination can then be followed by the addition of a different fluorogen and subsequent fluorescence imaging. The exact time points of imaging depend on the type of experiment and biological question. It is recommended that the imaging of the samples is done between timepoints and not to wait until the end of the experiment. For each time point, is it sufficient to take 1–2 µL of culture, so the same culture can be used for the entire experiment. Wash steps are performed on the whole culture.
It is recommended to image the culture after each wash step so that the decline in fluorescence due to the removal of the fluorogen can be followed and later represented during data analysis.
When the cells are in focus, it is recommended to take three pictures, each time of a different group of cells, to get more information about the cells at each time point. We recommend a minimum of 25 cells per strain per time point.
Here, we export as a multi-TIFF file, as this is our personal preference. The data analysis will also work fine if you export each channel as a separate TIFF file. If your software does not allow exporting to a TIFF file, you can use any other picture format that is compatible with image analysis in ImageJ2.
In case the channels of the fluorescent microscope were exported as separate TIFF files, remove the last line of the macro script.
In case of using multiple fluorogens in succession, the last image before the addition of the new fluorogen, of the second wash step, is used as the pre-treatment control for the new fluorogen.
The pre-treatment cell measurements are taken along to take into account the variation even before the addition of the fluorogen.
Troubleshooting
Problem 1: Failed C. albicans transformation
Possible cause: Expired nourseothricin, insufficient transformed DNA, PEG concentration changes, no fresh TE mixture.
Solution:
In the case of endogenous tagging, the selection of correct transformants is performed through the antibiotic nourseothricin. If you have too many incorrect transformants when transforming and selecting with nourseothricin, it may be that the nourseothricin is expired, added to the medium when it wasn’t cooled down sufficiently, or that your specific background strain is relatively resistant to nourseothricin. In these cases, we would advise using new nourseothricin, cooling the agar medium sufficiently, or increasing the nourseothricin concentration.
On the other hand, if no colonies are acquired after transformation, several adjustments could be made. First, prepare novel PEG and TE mixtures, since long-term evaporation might lead to concentration adjustments, as mentioned by Gietz and Schiestl [21]. Another possible explanation is insufficient PCR product or linearized plasmid. Check concentrations utilizing a Nanodrop spectrophotometer or equivalent instrument. If the concentration proved to be sufficient, you could still utilize more DNA, e.g., up to 5 µg. Using agar gel electrophoresis, it is also possible to verify whether the restriction digestion of the plasmid has worked.
In the case of endogenous construction, overhangs of 50 bp for homolog recombination might not be sufficient. It is possible to increase the overhang length; we have utilized overhangs of up to 100 bp.
Problem 2: Confocal microscopy
Possible cause: Insufficient fluorescence signal due to insufficient excitation or fluorogen.
For confocal microscopy, it is important to keep in mind that each genetically tagged strain will behave slightly differently, and this will require some optimization for each experiment. When experiencing low fluorescence from the tagged strains, we recommend adjusting the laser power and the voltage on the photomultiplier tube of the microscope, as this will influence the brightness and contrast of the images. However, keep in mind that higher laser power will also cause more photobleaching; in this protocol, we use an argon laser at 100 µW and the high voltage of the photomultiplier tube set to 580. Low fluorescence might also be caused by the fluorogen stock solution that has been through too many freeze-thawing cycles or has been exposed to light too much. For this, try to repeat the experiment with a new aliquot for the fluorogen stock. Increasing the fluorogen concentration might also solve the issue of low fluorescence; however, keep in mind that increasing the fluorogen concentration will also increase background fluorescence and can have a slight effect on the growth of the strains.
Additionally, we have not encountered that certain fluorogens are unable to reach specific organelles or are quenched at those organelle conditions. It is therefore a valid strategy to test other HMBR variants that are utilized in this protocol.
Acknowledgments
These protocols were used to acquire the data and results that were reported in our publication in Microbiology (Devos et al. [15], https://doi.org/10.1099/mic.0.001451). The protocol was developed using internal funding of the KU Leuven Research Council (grant # C14/22/075).
Competing interests
The authors declare that there are no competing interests.
Supplementary information
The following supporting information can be downloaded here:
Text S1. CIp10-NAT1plasmid map
Text S2. iFAST sequence
Table S3. Primers
References
- 1. Beigi R. H., Meyn L. A., Moore D. M., Krohn M. A. and Hillier S. L.(2004). Vaginal Yeast Colonization in Nonpregnant Women: A Longitudinal Study. Obstet Gynecol. 104: 926-930. [DOI] [PubMed] [Google Scholar]
- 2. Bougnoux M. E., Diogo D., N. Francois, Sendid B., Veirmeire S., Colombel J. F., Bouchier C., Van Kruiningen H., C. d'Enfert, Poulain D., et al.(2006). Multilocus Sequence Typing Reveals Intrafamilial Transmission and Microevolutions of Candida albicans Isolates from the Human Digestive Tract. J Clin Microbiol. 44(5): 1810-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Findley K., Oh J., Yang J., Conlan S., Deming C., Meyer J. A., Schoenfeld D., Nomicos E., Park M., et al.(2013). Topographic diversity of fungal and bacterial communities in human skin. Nature. 498(7454): 367-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown G. D., Denning D. W., Gow N. A. R., Levitz S. M., Netea M. G. and White T. C.(2012). Hidden Killers: Human Fungal Infections. Sci Transl Med. 4(165): e3004404. [DOI] [PubMed] [Google Scholar]
- 5. Gomes A. C., Miranda I., Silva R. M., Moura G. R., Thomas B., Akoulitchev A. and Santos M. A.(2007). A genetic code alteration generates a proteome of high diversity in the human pathogen Candida albicans . Genome Biol. 8(10): R206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Santos M. A. and Tuite M. F.(1995). The CUG codon is decoded in vivo as serine and not leucine in Candida albicans . Nucleic Acids Res. 23(9): 1481-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daniel E., Onwukwe G. U., Wierenga R. K., Quaggin S. E., Vainio S. J. and Krause M.(2015). ATGme: Open-source web application for rare codon identification and custom DNA sequence optimization. BMC Bioinf. 16(1): 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puigbo P., Guzman E., Romeu A. and Garcia-Vallve S.(2007). OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 35: W126-W131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Genechten W., Demuyser L., Dedecker P. and Van Dijck P.(2020). Presenting a codon-optimized palette of fluorescent proteins for use in Candida albicans . Sci Rep. 10(1): 6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Genechten W., Van Dijck P. and Demuyser L.(2021). Fluorescent toys‘n’ tools lighting the way in fungal research. FEMS Microbiol Rev. 45(5): 1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Plamont M. A., Billon-Denis E., Maurin S., Gauron C., Pimenta F. M., Specht C. G., Shi J., Quérard J., Pan B., Rossignol J., et al.(2015). Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc Natl Acad Sci USA. 113(3): 497-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tebo A. G., Pimenta F. M., Zhang Y. and Gautier A.(2018). Improved Chemical-Genetic Fluorescent Markers for Live Cell Microscopy. Biochemistry. 57(39): 5648-5653. [DOI] [PubMed] [Google Scholar]
- 13. Gillum A. M., Tsay E. Y. H. and Kirsch D. R.(1984). Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol General Genetics. 198(1): 179-182. [DOI] [PubMed] [Google Scholar]
- 14. Noble S. M. and Johnson A. D.(2005). Strains and Strategies for Large-Scale Gene Deletion Studies of the Diploid Human Fungal Pathogen Candida albicans . Eukaryotic Cell. 4(2): 298-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Devos J., Van Dijck P. and Van Genechten W.(2024). A multi-colour fluorogenic tag and its application in Candida albicans . Microbiology. 170(3): e001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mirhendi H, Diba K, Rezaei A, Jalalizand N, Hosseinpur L, and H Khodadadi (1970). Colony PCR Is a Rapid and Sensitive Method for DNA Amplification in Yeasts. Iran J Public Health. 36(1). [Google Scholar]
- 17. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. and Smith H. O.(2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6(5): 343-345. [DOI] [PubMed] [Google Scholar]
- 18. Froger A. and Hall J. E.(2007). Transformation of Plasmid DNA into E. coli Using the Heat Shock Method. J Visualized Exp. 6: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demuyser L., Van Genechten W. and Van Dijck P.(2022). Assessment of cAMP-PKA Signaling in Candida glabrata by FRET-Based Biosensors. In: Calderone, R.(Ed.). Candida Species(Vol. 2542, pp. 177–191). New York, NY: Springer US. [DOI] [PubMed] [Google Scholar]
- 20. Rueden C. T., Schindelin J., Hiner M. C., DeZonia B. E., Walter A. E., Arena E. T. and Eliceiri K. W.(2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinf. 18(1): 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gietz R. D. and Schiestl R. H.(2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2(1): 31-34. [DOI] [PubMed] [Google Scholar]