

Abstract
The process of T-lymphocyte migration involves a complex interplay of chemical and mechanical signals. Mechanotransduction mechanisms in T lymphocytes enable them to efficiently navigate through diverse architectural and topographical features of the dynamic tissue macro- and micro-niches encountered during immune responses. Piezo1 mechanosensors are crucial for driving optimal T-cell migration by driving actin-cytoskeletal remodeling. Chemokine-stimulated T lymphocytes demonstrate significant asymmetry or polarity of Piezo1 and actin along the cell axis. The establishment and maintenance of polarity in migrating cells are paramount for facilitating coordinated and directional movements along gradients of chemokine signals. Live-cell imaging techniques are widely employed to study the trajectories of migrating cells. Our approach expands upon current methodologies by not only tracking migrating cells but also imaging fluorescently labeled cellular components. Specifically, our method enables measurement of protein enrichment in the front and rear halves of the moving cell by analyzing the temporal direction of cell trajectories, subsequently bisecting the cell into front-back halves, and measuring the intensities of the fluorescent signals in each cell half at each time frame. Our protocol also facilitates the quantification of the angular distribution of fluorescent signals, enabling visualization of the spatial distribution of signals relative to the direction of cell migration. The protocol describes the examination of polarity in chemokine-treated Jurkat cells transfected with Piezo1-mCherry and actin-GFP constructs. This approach can be extended to live-cell imaging and polarity assessment of other fluorescently labeled proteins.
Key features
• This experimental protocol allows real-time imaging of Jurkat cells expressing two fluorescent proteins (Piezo1 mCherry and actin-GFP).
• Measures cell polarity by examining spatial enrichment of Piezo1 and actin proteins within the front and rear halves of a moving Jurkat cell.
• The protocol enables analysis of cell polarity in 2D tracks of moving cells.
• Polarity analysis includes measuring fluorescent signal intensities in front-rear halves of a moving cell and calculation of signal polarization angles relative to the cell trajectory.
Keywords: Time-lapse confocal imaging, Jurkat cell line, Piezo1 mCherry, Actin-GFP imaging, Cell polarity, Angular distribution
Graphical overview
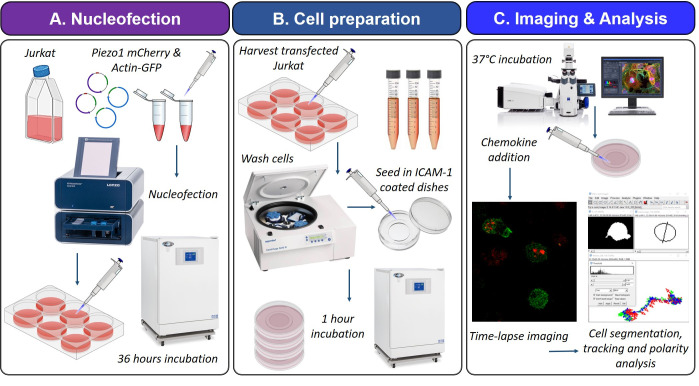
Schematic illustration of the experimental and analysis workflow. A. Jurkat cells were nucleofected with plasmids expressing Piezo1 mCherry and actin-GFP. The pulsed cells were cultured in complete RPMI medium for 36 h at 37 °C/5% CO2 to allow protein expression. B. Transfected Jurkat cells were collected, washed, and resuspended in imaging medium. Cells were seeded in ICAM-1-coated glass bottom confocal dishes and incubated for 1 h at 37 °C/5% CO2 to facilitate cell attachment. C. Live-cell tracking of transfected cells was performed in a confocal incubation setup prewarmed to 37 °C. Chemokine SDF1α was added 10–15 min prior to image acquisition. Images were acquired for a total duration of 5 min at intervals of 30 s. Z-stacks of 1 µm were captured at each frame. Fiji/ImageJ was used for cell segmentation by thresholding, ROI detection, trajectory, and polarity analysis.
Background
Migration of immune cells is finely orchestrated by an intricate interplay of molecular and physical signals [1–3]. These signals efficiently navigate the cells toward inflamed tissues and secondary lymphoid organs, where they mount the onset of the vital adaptive immune response. Migration of immune cells including T lymphocytes is mechanically intensive [4–6]. Motile T lymphocytes actively engage in continuous sensing and adaptation to mechanical cues within their surroundings. These signals encompass various parameters such as local tissue structure and topography, mechanical rigidity, confinement, hydrostatic pressure, and shear stress from interstitial fluid and circulating blood [5–7]. In addition, T lymphocytes also sense cell-intrinsic mechanical forces that stem from numerous cellular processes including tethering interactions with the extracellular matrix (ECM), actomyosin cytoskeletal remodeling, generation of traction forces, consequent exertion of forces on the tethered membrane, and changes in membrane tension [4–7]. Forces produced by cells facilitate unimpeded movement by overcoming resistance stress generated from local tissue deformation during migration [8–10].
The mechanosensitive ion channel Piezo1 has been widely implicated in numerous physiological processes that are subject to intricate mechanical regulation [11–13]. These processes include vascular remodeling, stem cell differentiation, epithelial homeostasis, and regulation of red blood cell volume [14–19]. Key research has also delved into the function of these mechanosensors in regulating the activation and function of T and B lymphocytes, dendritic cells, and macrophages [20–27]. The homo-trimeric subunits arranged in a propeller blade-like fashion facilitate Piezo1 to sense changes in membrane tension caused by applied force. Piezo1 subsequently undergoes a change in conformation allowing the opening of the central ion pore and passage of extracellular cations into the cell [28,29].
A pivotal investigation conducted by our team discovered the crucial role of Piezo1 in driving optimal human T-lymphocyte activation in response to T-cell receptor (TCR) triggering [20,30]. Further examination revealed that Piezo1-driven calcium response activates downstream calpain-dependent polymerization of the actin cytoskeleton, resulting in the formation of stable immunological synapses [20]. Subsequent deeper examination enabled us to identify a similar mechanism operating during chemokine-driven, ICAM-1-dependent human CD4+ T-lymphocyte migration [31]. Utilizing interference reflection microscopy (IRM) and confocal imaging, the study observed an increase in membrane tension in chemokine-stimulated T lymphocytes at focal adhesion sites. Polarized Piezo1 recruitment to these high-tension regions at the leading edge was accompanied by localized calcium signaling and calpain-driven F-actin polymerization. Local cytoskeletal rearrangement facilitated the recruitment of CD11a/LFA-1, triggering downstream PI3-kinase/Akt signaling that promoted further actin polymerization and flow, thus driving cell movement [31]. A notable finding of this study was the enrichment of Piezo1 at the leading edges of the cell, accompanied by a dynamic distribution of actin. In order to examine the spatial polarity of Piezo1 and actin in moving cells, we used the Jurkat cell line expressing Piezo1 mCherry and actin-GFP. Time-lapse imaging was conducted. Using a custom-designed code, the trajectories of the transfected cells were analyzed to determine the front-back polarity of the fluorescent signals, along with the distribution of their polarization angles.
Migrating cells exhibit distinct polarity characterized by an asymmetric distribution of cellular components along their migratory axis [32–34]. Generation and maintenance of cell polarity are essential for driving coordinated and directed movement of cells, in response to chemical (such as chemotaxis) and physical cues (such as haptotaxis) [35,36]. The protocol outlined below offers a comprehensive, step-by-step account of the experimental methodology and analytical approach employed to assess the polarity of Piezo1 mCherry and actin-GFP in chemokine-stimulated Jurkat cells. The methodologies described herein can be readily adapted for the evaluation of other proteins and in various experimental contexts.
Materials and reagents
Biological materials
Jurkat cell line [from Dr. Santusabuj Das at National Institute of Cholera and Enteric Diseases (NICED), Kolkata, India] (ATCC, catalog number: TIB-152)
Piezo1 mCherry plasmid (from Dr. Charles Cox at Victor Chang Cardiac Research Laboratory, Darlinghurst, Australia)
pCAG-mGFP-Actin plasmid (Addgene, catalog number: 21948)
Reagents
Recombinant human SDF1α/CXCL12 (PeproTech, catalog number: 300-28A)
Recombinant human ICAM-1 (PeproTech, catalog number: 150-05)
SE Cell Line 4D-NucleofectorTM X Kit L (Lonza, catalog number: V4XC-1024)
RPMI 1640 medium (Gibco, catalog number: 11875093)
RPMI 1640 medium, no phenol red (Gibco, catalog number: 11835030)
Fetal bovine serum (FBS) (Gibco, catalog number: 16000044)
Sodium pyruvate, 100 mM (Gibco, catalog number: 11360070)
MEM non-essential amino acids solution, 100× (Gibco, catalog number: 11140050)
Penicillin-streptomycin solution, 100× (Gibco, catalog number: 15140122)
Antibiotic-antimycotic, 100× (Gibco, catalog number: 15240062)
HEPES (Sigma-Aldrich, catalog number: H3375)
Phosphate-buffered saline, pH 7.4 (Himedia, catalog number: M1866)
Sodium hydroxide (Himedia, catalog number: PCT1325), 10 N solution in double-distilled water
Endotoxin-free water (InvivoGen, catalog number: h2olal-1.5)
UltraPureTM DNase/RNase-free distilled water (Invitrogen, catalog number: 10977015)
Solutions
Complete RPMI medium (see Recipes)
Post-nucleofection RPMI medium (see Recipes)
1 M HEPES solution, pH 7.4 (see Recipes)
RPMI medium without phenol red for imaging (see Recipes)
Phosphate-buffered saline (1×) (see Recipes)
Nucleofection solution (see Recipes)
Recombinant human ICAM-1 stock solution (see Recipes)
Recombinant human SDF1α stock solution (see Recipes)
Recipes
-
Complete RPMI medium
Reagent Final concentration Quantity or Volume RPMI 1640 500 mL Fetal bovine serum 10% 50 mL Sodium pyruvate (100×) 1× 6 mL MEM Non-essential amino acid (100×) 1× 6 mL Penicillin-streptomycin (100×) 1× 6 mL Antibiotic-antimycotic (100×) 1× 6 mL * Filter final media using a 0.22 µm bottle-top vacuum filter system and store at 4 °C.
-
Post-nucleofection RPMI medium
Reagent Final concentration Quantity or Volume RPMI 1640 40 mL Fetal bovine serum 20% 10 mL Sodium pyruvate (100×) 1× 500 µL MEM Non-essential amino acid (100×) 1× 500 µL * Filter final media using a 0.22 µm bottle-top vacuum filter system and store at 4 °C.
-
1 M HEPES solution, pH 7.4
Reagent Final concentration Quantity or Volume HEPES 1 M 11.9 g 10 N sodium hydroxide as needed for pH = 7.5 Ultrapure distilled water 50 mL * Adjust the pH to 7.5 using sodium hydroxide and make up the volume with ultrapure distilled water to 50 mL. Filter the solution using a 0.22 µm syringe filter and store at 4 °C.
-
RPMI medium without phenol red
Reagent Final concentration Quantity or volume RPMI 1640 medium, no phenol red 500 mL Fetal bovine serum 10% 50 mL Sodium pyruvate (100×) 1× 6 mL MEM Non-essential amino acid (100×) 1× 6 mL Penicillin-streptomycin (100×) 1× 6 mL Antibiotic-antimycotic (100×) 1× 6 mL HEPES (100 mM), pH 7.5 25 mM 1.25 mL * Filter the final media using a 0.22 µm bottle-top vacuum filter system and store at 4 °C.
-
Phosphate-buffered saline (PBS), 1×
Reagent Final concentration Quantity or volume Phosphate-buffered saline 1× 9.9 g Milli-Q water 1 L * Autoclave the buffer and filter using a 0.22µm vacuum filter system. Store at 4 °C.
-
Nucleofection solution
Reagent Final concentration Quantity or volume SE Cell Line NucleofectorTM solution (Lonza kit) 2.25 mL Supplement buffer (Lonza kit) 0.5 mL * Store the solution at 4 °C.
-
ICAM-1 stock solution
Reagent Final concentration Quantity or volume Recombinant human ICAM-1 0.5 mg/mL 50 µg LAL endotoxin-free water 100 µL * Make small aliquots of the reconstituted stock solution and store them at -80 °C.
-
SDF1α stock solution
Reagent Final concentration Quantity or volume Recombinant human SDF1α/CXCL12 0.1 mg/mL 10 µg Endotoxin-free water 100 µL * Make small aliquots of the reconstituted stock solution and store them at -80 °C.
Laboratory supplies
NuncTM glass-bottom confocal dishes (Thermo Scientific, catalog number: 150680)
T25 cell culture flasks (Thermo Scientific, catalog number: 156340)
1.5 mL microcentrifuge tubes (Tarsons, catalog number: 500010)
15 mL centrifuge tubes (Tarsons, catalog number: 546021)
Corning® Costar® tissue culture-treated 6-well plates (Merck, catalog number: CLS3516)
Corning® bottle-top vacuum filter system (Merck, catalog number: CLS431097)
Millex® PVDF syringe filter, pore size: 0.22 µm, diameter: 33 mm (Merck, catalog number: SLGVR33RS)
Nitrile gloves (Kimtech, catalog number: 55081)
Neubauer chamber
Equipment
AmaxaTM 4D-Nucleofector (Lonza)
Zeiss LSM 980 confocal microscope with 37 °C heating equipment and chamber
Laminar flow hood
37 °C, 5% CO2 incubator
pH meter
Refrigerated centrifuge
Software and datasets
Zen blue v3.3 (for imaging)
Fiji (for image analysis, https://fiji.sc/)
GraphPad Prism 8.0
Origin 2019b
Procedure
-
Preparation of Jurkat cell line for plasmid nucleofection
Passage approximately 0.1 × 106/mL Jurkat cells in T-25 flasks containing 5 mL of complete RPMI medium (see Recipe 1) between 36 and 48 h before transfection.
Do not allow confluency to reach more than 75% on the day of nucleofection.
Avoid using cells that have undergone more than 10 passages since revival.
-
Nucleofection of Piezo1-mCherry and actin-GFP plasmids
Pre-equilibrate post-nucleofection RPMI media (see Recipe 2) at 37 °C/5% CO2 in tissue-culture-treated 6-well plates. Add 2 mL of media per well.
Prewarm an aliquot of post-nucleofection RPMI media at 37 °C.
Switch on the Amaxa 4D-Nucleofector system at least 30 min before nucleofection.
Incubate the nucleofection solution at room temperature prior to nucleofection.
Count Jurkat cells using a Neubauer chamber.
-
Centrifuge Jurkat cells at 165× g for 3 min and remove media completely. Resuspend 2 × 106 cells in 100 µL of nucleofection solution.
Caution: Avoid leaving the cells in nucleofection solution for more than 5 min. Prolonged incubation of cells in the nucleofection solution causes a drastic reduction in cell viability.
Add Piezo1 mCherry-expressing plasmid and actin-GFP plasmid at a concentration of 2–3 µg/mL each into the Jurkat cell suspension. Mix gently. Ensure that the volume of plasmids does not exceed 10% of the final sample volume.
Transfer the plasmid–cell mix into nucleofection cuvettes and nucleofect using program CL-120, specific for the Jurkat cell line, in the Amaxa 4D-Nucleofector system.
Add 1 mL of prewarmed post-nucleofection RPMI medium to the cuvettes containing nucleofected cells and let it stand for 2 min.
Mix the cells by gentle pipetting and transfer the cells to culture plates containing equilibrated post-nucleofection RPMI medium. Seed the transfected cells at a density of 1 × 106 cells per well.
Incubate the cells at 37 °C/5% CO2 for 12 h. Wash the cells with 1× PBS at 165× g for 3 min and add fresh post-nucleofection RPMI medium for approximately an additional 24 h before imaging.
-
Preparation of cell chambers for live-cell imaging
Prepare 4 µg/mL of ICAM-1 solution in sterile and filtered 1× PBS from stock solution (see Recipes).
Coat glass-bottom confocal dishes (12 mm diameter) with 100 µL of ICAM-1 solution overnight at 4 °C.
Remove ICAM-1 solution. Wash the dish five times with 1× PBS (200 µL, 5 min each).
Remove PBS completely and allow the dishes to dry at room temperature for 2–3 h. The coated dishes can be stored at 4 °C in sealed conditions for approximately four weeks.
-
Preparation of Piezo1 mCherry/actin-GFP nucleofected Jurkat cells for live-cell imaging
Prewarm RPMI medium without phenol red at 37 °C (see Recipes).
Wash transfected Jurkat cells twice in 1× PBS at 165× g for 3 min. Resuspend cells in RPMI medium without phenol red for imaging. Measure transfection efficiency using flow cytometry (cells showing at least 30% transfection efficiency were used for downstream imaging).
Count cells using a Neubauer chamber.
Seed approximately 50,000 transfected Jurkat cells per 12 mm ICAM-1-coated confocal dish in 100 µL of imaging medium.
Allow the cells to adhere to the coated dishes for 1 h at 37 °C/5% CO2.
Note: The imaging medium contains 25 mM HEPES (pH 7.5). The buffering provided by HEPES operates independently of CO2/bicarbonate buffering system. Since our imaging system lacked CO2 supply, the addition of HEPES ensured the stability of medium pH during imaging.
-
Live-cell tracking of Piezo1 mCherry/actin-GFP expressing Jurkat cells
Prewarm the incubation chamber of the confocal microscope to 37 °C at least 30 min prior to imaging.
-
Transfer the confocal dish into the 37 °C incubation chamber of the confocal imaging setup. Incubate for an additional 15 min before tracking.
Note: Adequate prewarming and pre-incubation of cells within the microscope’s incubation chamber are critical to ensure the stability of temperatures and prevent thermal drift, which could otherwise cause gradual loss of focus or focal drift during imaging.
Add 0.1 µg/mL of recombinant SDF1α (diluted in imaging medium, see Recipe 8) to the dish 10–15 min prior to acquisition during incubation.
Duration and frequency of imaging for a time-lapse video depends on the nature of the events under examination. The time-lapse imaging of the representative cell was performed at intervals of 30 s for a total duration of 5 min. Z-stacks of 1 µm were acquired (total 12 stacks) at each frame. Images were acquired at 63×/1.4 magnification. For higher-speed imaging, 512 × 512 frames at a bi-directional scanning speed of 8–9 (maximum) was used.
Live-cell imaging without CO2 supplementation and adequate humidification should be limited to shorter durations of no more than 30–40 min.
-
Imaging sessions should be kept brief to prevent photo-bleaching of fluorescent proteins. In our study, we have restricted the maximum acquisition duration to 15–20 min, with intervals of 30 s between captures, and limited the total number of Z-stacks to 12–14. This approach helps minimize prolonged illumination and consequent photo-bleaching.
Note: The confocal dishes were sealed with parafilm to avoid the effect of air currents generated as a result of the heating mechanism and consequent drift.
Data analysis
Measuring Piezo1 and actin polarity in moving cells
Import the exported imaging files into Fiji (https://fiji.sc/).
In the Bio-Formats Import Options window, view the image as Hyperstack and in Composite color mode.
Duplicate the hyperstack. Specify the channels (fluorescence signals for Piezo1 mCherry and actin-GFP) and delete the stacks (or slices) and time frames that are not essential for the analysis from either/both ends of the range. Use this duplicated hyperstack for further analysis.
-
Select Image > Stacks > Z Projection > Max intensity (or other suitable method of Z projection). This will produce a time-lapsed 2D projection of your hyperstack. Use this resulting 2D video for further tracking and polarity analyses. The hyperstack is split into its constituent channels—Piezo1-mCherry (red) and actin-GFP (green)—for subsequent analysis (Image > Color > Split Channels) (Figure 1).
The analysis described below employs a custom code designed for the study by Liu et al. [31] to facilitate automated image processing of multiple regions of interest (ROIs) across multiple time frames of a single time-lapse video.
Optional: Gamma adjustment can be performed to control brightness and contrast of the images to aid the intensity thresholding. It employs a nonlinear mapping of the pixel intensity values. Gamma < 1 amplifies the faint pixels while reducing the intensities of the bright pixels, thereby making the faint pixels more visible. Conversely, Gamma >1 amplifies the bright pixels while further lowering the intensities of the faint pixels. Linear mapping is performed for gamma values equal to 1 (Figure 2A).
Select an ROI to measure background intensity and execute subtraction of background noise (Figure 2B).
Merge the two fluorescence channels. Perform a maximum intensity Z projection along the time axis to obtain a 2D image depicting the total extent of all the cell trajectories within the x-y frame. Use the freehand selection tool to manually draw ROIs encompassing the cell trajectories suitable for downstream analysis. Each ROI should contain a single-cell trajectory exhibiting signals in both fluorescence channels. Save these ROIs, as cells contained within will be used for tracking and polarity analyses across all time frames in the 2D time-lapse video (Figure 2C).
Create binary masks for each cell across all time frames within each delineated trajectory ROI, as specified above. Smooth the time stacks to blur the time-lapsed images and diminish speckled background noise. This facilitates image thresholding, ensuring accurate delineation of the specific cell ROI. Execute auto threshold by appending dark stack to the thresholding method, indicating that thresholding is performed on an image stack where the object of interest appears darker than the background. Utilize Covert to Mask and background = dark to generate binary masks of the cells, where darker regions (object of interest/cells) are interpreted as foreground while brighter regions are designated as image background (pixel values: 0 for background, 255 for foreground, for an 8-bit image) (Figure 3). These settings ensure precise segmentation of cells across all time frames. Utilize Fill Holes and Analyze Particles while specifying the maximum and minimum cell area to generate outlines of ROIs corresponding to each cell mask. Save these ROIs (Figure 3).
-
Using Set Measurements, analyze the following parameters of each cell ROI:
Area
Center of Mass (intensity-weighted average of x and y of all pixels) = XM and YM
Centroid (average of x and y coordinates of all pixels) = X and Y
Shape descriptors = Circularity, Aspect Ratio, Round, and Solidity
Fit ellipse = lengths of Major (primary) and Minor (secondary) axis, and Angle between the primary axis of the cell (the major axis of the fitted ellipse) and a line parallel to the x-axis of the image.
Store these values corresponding to each cell in every time frame as separate arrays.
-
Convert image stacks to 32-bit. Perform Fit Ellipse on the cell ROI to extract the shape parameters, then bisect each cell ROI into equal parts along the minor axis using the following calculations:
-
From the Angle value given by ImageJ, calculate the angle subtended by the minor axis of the fitted ellipse on the x-axis of the image (Figure 4A).
θ = angle perpendicular to the direction of the major axis of ellipse, i.e., the minor axis
-
Draw the minor axis from the ellipse’s centroid to one edge of the ROI by specifying coordinates of the edge (xbl1, ybl1) (Figure 4A).
-
Draw minor axis from the centroid to the other edge of the ROI by specifying coordinates of the other edge (xbl2, ybl2) (Figure 4A).
widthPxl and heightPxl = width (along x-axis) and height (along y-axis) of the ROI.
Xc[j] and Yc[j] = centroids of the ROIs (no. of ROIs = j).
Major = length of the major axis of the ellipse.
Draw a line connecting xbl1, ybl1 and xbl2, ybl2, which will bisect the ellipse along the minor axis of the ROI ellipse. The length of the bisecting line along the minor axis is equal to the length of the major axis so that the straight line completely passes over the ellipse’s edges. Without the overlap, gaps remain between the endpoints of the line and the ellipse’s edges, which does not allow selecting either half of the ellipse using the Wand tool later. Do this for all ROIs corresponding to cell masks in each time frame (Figure 4A, B).
-
-
To define the front and back halves of the bisected moving cell
-
Measure the coordinates of the half-centers of each half of the cell. The half-center of each cell-half lies at the mid-point between the centroid of the whole cell and the centers of each half-cell (Figure 4A):
xh1, yh1 and xh2, yh2 are half-center coordinates of each half of the ROI.
Note: Ideally, any point within each bisected ROI would enable Fiji to use the Wand tool and detect each bisected ROI. To avoid randomness, we chose the “half-center” that lies closer to the bisection line, thereby increasing the probability of finding positive fluorescence pixels that will facilitate detection (Figure 4A).
-
Calculate the distance of the half-center points of each of the above-calculated half-ellipses from the ellipse’s centroid in the preceding frame in the trajectory. The closest half is the back half of the cell while the farthest half is the front half of the cell (Figure 4A).
-
Using the whole cell masks or ROIs stored earlier (step 8) and the calculated half-center coordinates of each half of the ellipse (xh1, yh1 and xh2, yh2), measure the ROIs corresponding to each half of the ellipse using the Wand tool. Measure the raw integrated density (RawIntDen) of both front and back halves of the ellipse.
-
Measure the polarity index by determining the ratio of the raw integrated density of the front and back halves of the cell (F/B). File S1 contains the results of the analysis. A polarity index greater than 1 signifies skewed frontal polarization and distribution of the fluorescent signals (Figure 5A). A higher Piezo1 F/B polarity compared to actin aligns with the leading-edge distribution of Piezo1, as observed in Liu et al. [31].
Note: Perform the above analysis steps for both fluorescent channels to calculate F/B polarity of both Piezo1 mCherry and actin-GFP.
-
The relative angle of polarity or angular distribution of fluorescent signals with respect to displacement of cell can also be measured. Perform the calculation below for all the ROIs (no. of ROIs = j), corresponding to varying cell positions across different time frames.
-
Using the atan2 function as below, the polarization angle of fluorescent signals can be calculated with respect to the cell center. Convert the angles to degrees ( .
XcMA, YcMA = center of mass (intensity-weighted spatial coordinates)
Xc, Yc = centroid (spatial coordinates of center of cell area)
-
Similarly, the displacement magnitude and the displacement angle (direction of trajectory) can also be calculated. Convert the angles to degrees.
j-1, j = two subsequent time frames.
-
Calculate the relative angle of polarization of fluorescence intensity with respect to the trajectory of displacement by subtracting the displacement angle from the polarization angle (Figure 5B).
Note: Perform the above analysis steps for both fluorescent channels to obtain polarization angles of both Piezo1 mCherry and actin-GFP across all time frames.
Utilize the Arrow tool alongside a suitable scaling method (bilinear interpolation) to depict vectors representing the polarization angles for all the ROIs across time frames (Figure 5C).
Visualization software such as GraphPad Prism 8 and Origin 2019b can be used to plot the F/B ratios along with polarization angles of both Piezo1 mCherry and actin-GFP signals. Polar plots can be generated to examine the distribution of Piezo1 and actin relative angles. The relative angle values calculated for all cells at each time frame can be binned into regular intervals ranging from -180° to +180°. The frequency distribution can then be subsequently plotted.
-
Figure 1. Generation of 2D time lapse videos by performing maximum intensity Z-projection of the multi-channels/multi-time-points hyperstacks.
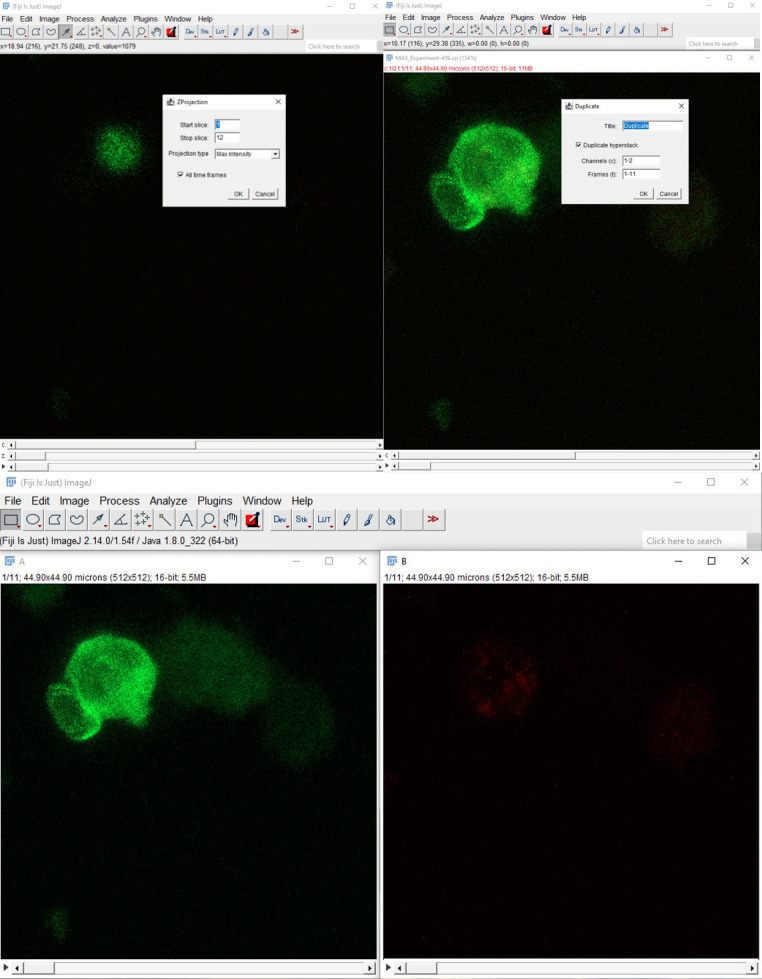
The multichannel 2D time hyperstack was split into two time-lapse videos corresponding to actin-GFP (green) and Piezo1 mCherry (red).
Figure 2. Preprocessing of 2D time-lapse stack before ROI detection.

A. Gamma adjustment enhances the contrast between the object of interest and background signal. B. Background signal correction involves selecting an ROI in the background region, measuring its average intensity, and subtracting the signal from the original image. C. The maximum intensity projection along the time axis generates a comprehensive view of the cell’s overall trajectory over the course of imaging. Defining this ROI facilitates easier detection and analysis of the cell within it across all time points.
Figure 3. Mapping out cell ROIs.

A. A generalized analysis scheme for ROI detection using thresholding and creation of binary masks (0–255). B. Resulting binary masks and outline of analyzed cell ROI at each time frame. Both actin-GFP and Piezo1-mCherry channels have been shown for the detected ROI.
Figure 4. Bisecting cell ROI into front and back halves.

A. A cell ROI is fitted into an ellipse and its shape parameters are measured. The centroids (XC, YC) of the cell ROI at subsequent time frames (Tj-1 and Tj) are used to calculate the cell displacement. The ROI is split into two halves along the minor axis (perpendicular to the major axis). θ = angle perpendicular to the major axis, φ = angle (radians) subtended by the minor axis. To determine the back and front half of the bisected ROI, dist1 and dist2 are calculated. dist1 < dist2. dist1 corresponds to the back half and dist2 corresponds to the front half of the bisected ROI. (xh1, yh1): half-centers of front half; (xh2, yh2): half-centers of back half. B. The cell ROIs across time frames (F0–F10) were fitted into an ellipse. The ellipse was bisected along the minor axis (perpendicular to the major axis) to generate two halves of the moving cell.
Figure 5. Measuring live-cell polarity of Piezo1 mCherry and actin-GFP in chemokine-stimulated Jurkat cells.
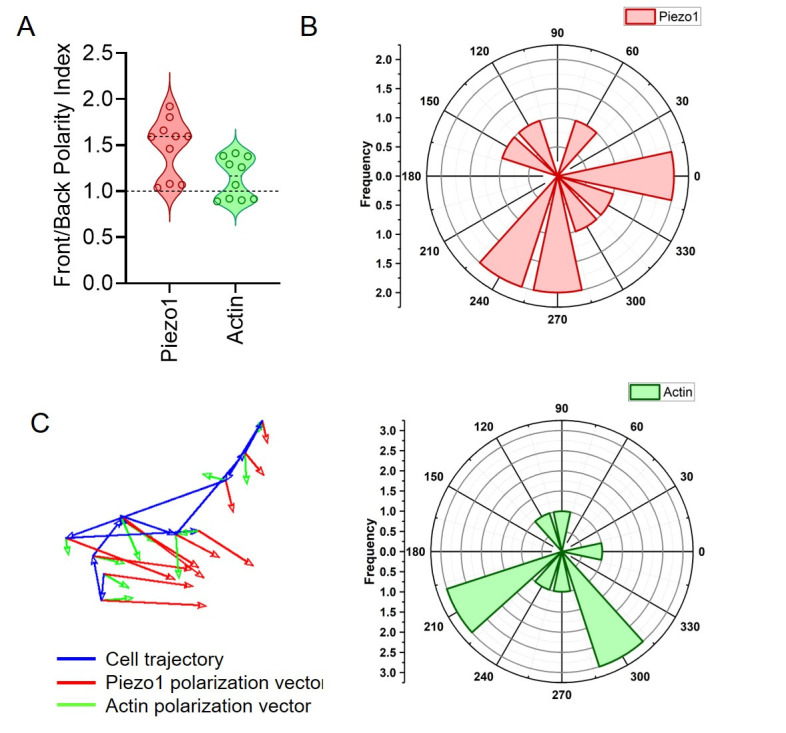
A. Front/back (F/B) polarity of Piezo1 and actin of a single cell. Each point corresponds to F/B value at a specific time frame of a single moving cell. B. Polar plots depicting relative angles of Piezo1 (upper panel) and actin (lower panel). The relative angles correspond to a single cell against different time frames. C. Vector plot of a single cell depicting the direction of cell trajectory (blue), Piezo1 polarization (red), and actin polarization (green) at each time frame.
Validation of protocol
The protocol was used and published in the following research article:
Liu et al. [31]. Piezo1 mechanosensing regulates integrin-dependent chemotactic migration in human T cells. Experimental setup and analysis were used for figures 6D, E, and figure 6 – figure supplement 1B and C.
General notes and troubleshooting
General notes
Nucleofection often leads to significant cell death. To enhance transfection efficiencies, ensure that the cell line is healthy and has undergone no more than 10 passages. Increase cell viability by using prewarmed and CO2-equilibrated medium. Avoid using confluent cells for transfection, as this can lead to decreased viability and transfection efficiencies. Additionally, minimizing cell death can be achieved by increasing FBS content and removing antibiotics in the post-nucleofection media.
All media and buffers used for cell preparation and imaging should be filtered using 0.22 µm filters. This step helps reduce debris contaminants, which can otherwise compromise imaging quality by contributing to elevated background noise and signal artifacts.
It is crucial to maintain stable temperatures throughout the imaging process. Temperature fluctuations and other mechanical disturbances can cause substantial focal drift during imaging, rendering the resulting images unsuitable for subsequent analysis. While there are tools and algorithms available to compensate for manual focal drift, it is advisable to minimize disturbances to capture high-quality time-lapse images. Prior heating of the enclosed imaging chamber minimizes such temperature- and mechanical-induced variations.
Images should be carefully monitored for any evidence of photobleaching and consequent loss of signal during the course of live-cell imaging. Minimizing the total duration of acquisition, along with the number of frames and/or Z-stacks, reduces the frequency of illumination of the sample and mitigates resulting photobleaching.
Additionally, given the dynamic nature of live-cell imaging, it is crucial to capture each frame as quickly as possible to minimize significant cell movement within a single frame. This can be achieved by employing bi-directional high-speed scanning at a moderate resolution of 512 × 512 pixels and enhancing magnification for faster scanning of smaller areas.
Efficient segmentation of cells to determine ROIs for subsequent analysis relies on robust pre-processing of the images, including enhancing image contrast, smoothing for background noise, and correcting background intensities. There is no singular method that can be considered ideal for achieving optimal results in these steps. The choice of method depends on various parameters, including image quality, signal strength, and background noise level. Numerous tools and algorithms are available for the purposes of reducing background and thresholding (delineation of cell ROIs), which should be carefully tried and implemented to obtain accurate results.
Acknowledgments
We express our sincere gratitude towards all co-authors of the corresponding study [31] for their significant contributions and support. The study was funded by the Council of Scientific and Industrial Research, India (Grant no. FBR MLP-140). D.G. was additionally supported by the Swarnajayanti Fellowship award granted by the Department of Science and Technology, Government of India. Special thanks are extended to Dr. Charles Cox at the Victor Chang Institute of Cardiac Research, Australia, for generously providing the Piezo1 constructs.
Competing interests
The authors declare no competing financial interests.
Ethical considerations
There are no ethical considerations associated with this protocol.
Supplementary information
The following supporting information can be downloaded here:
-
File S1
File S1.xlsx contains source data for Figures 6A–B. It contains the Fiji analysis outputs: ROI parameters(area, circularity), trajectory analysis(displacement magnitude, displacement angle), polarization angles and relative polarization angles for Piezo1(P) and actin(A), raw integrated densities of front and back-halves of the ROI, along with front/back(F/B) ratios of Piezo1 and actin. Each data point corresponds to a single cell at different time frames. Calculation of polar angular distribution has also been added to the file.
References
- 1. Moreau H. D., Piel M., Voituriez R. and Lennon-Duménil A. M.(2018). Integrating Physical and Molecular Insights on Immune Cell Migration. Trends Immunol. 39(8): 632-643. [DOI] [PubMed] [Google Scholar]
- 2. Kameritsch P. and Renkawitz J.(2020). Principles of Leukocyte Migration Strategies. Trends Cell Biol. 30(10): 818-832. [DOI] [PubMed] [Google Scholar]
- 3. Delgado M. and Lennon-Duménil A. M.(2022). How cell migration helps immune sentinels. Front Cell Dev Biol. 10: e932472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rossy J., Laufer J. M. and Legler D. F.(2018). Role of Mechanotransduction and Tension in T Cell Function. Front Immunol. 9: e02638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fowell D. J. and Kim M.(2021). The spatio-temporal control of effector T cell migration. Nat Rev Immunol. 21(9): 582-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Du H., Bartleson J. M., Butenko S., Alonso V., Liu W. F., Winer D. A. and Butte M. J.(2022). Tuning immunity through tissue mechanotransduction. Nat Rev Immunol. 23(3): 174-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tabdanov E. D., Rodríguez-Merced N. J., Cartagena-Rivera A. X., Puram V. V., Callaway M. K., Ensminger E. A., Pomeroy E. J., Yamamoto K., Lahr W. S., Webber B. R., et al.(2021). Engineering T cells to enhance 3D migration through structurally and mechanically complex tumor microenvironments. Nat Commun. 12(1): 2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lange J. R. and Fabry B.(2013). Cell and tissue mechanics in cell migration. Exp Cell Res. 319(16): 2418-2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yao L. and Li Y.(2022). Effective Force Generation During Mammalian Cell Migration Under Different Molecular and Physical Mechanisms. Front Cell Dev Biol. 10: e903234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J., Yan D. and Chen Y.(2023). Understanding the driving force for cell migration plasticity. Biophys J. 122(18): 3570-3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coste B., Mathur J., Schmidt M., Earley T. J., Ranade S., Petrus M. J., Dubin A. E. and Patapoutian A.(2010). Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science. 330(6000): 55-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murthy S. E., Dubin A. E. and Patapoutian A.(2017). Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat Rev Mol Cell Biol. 18(12): 771-783. [DOI] [PubMed] [Google Scholar]
- 13. Ridone P., Vassalli M. and Martinac B.(2019). Piezo1 mechanosensitive channels: what are they and why are they important. Biophys Rev. 11(5): 795-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ranade S. S., Qiu Z., Woo S. H., Hur S. S., Murthy S. E., Cahalan S. M., Xu J., Mathur J., Bandell M., Coste B., et al.(2014). Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA. 111(28): 10347-10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pathak M. M., Nourse J. L., Tran T., Hwe J., Arulmoli J., Le D. T. T., Bernardis E., Flanagan L. A. and Tombola F.(2014). Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc Natl Acad Sci USA. 111(45): 16148-16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cahalan S. M., Lukacs V., Ranade S. S., Chien S., Bandell M. and Patapoutian A.(2015). Piezo1 links mechanical forces to red blood cell volume. eLife. 4: e07370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Retailleau K., Duprat F., Arhatte M., Ranade S. S., Peyronnet R., Martins J. R., Jodar M., Moro C., Offermanns S., Feng Y., et al.(2015). Piezo1 in Smooth Muscle Cells Is Involved in Hypertension-Dependent Arterial Remodeling. Cell Rep. 13(6): 1161-1171. [DOI] [PubMed] [Google Scholar]
- 18. Ma N., Chen D., Lee J. H., Kuri P., Hernandez E. B., Kocan J., Mahmood H., Tichy E. D., Rompolas P., Mourkioti F., et al.(2022). Piezo1 regulates the regenerative capacity of skeletal muscles via orchestration of stem cell morphological states. Sci Adv. 8(11): eabn0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gudipaty S. A., Lindblom J., Loftus P. D., Redd M. J., Edes K., Davey C. F., Krishnegowda V. and Rosenblatt J.(2017). Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. 543(7643): 118-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu C. S. C., Raychaudhuri D., Paul B., Chakrabarty Y., Ghosh A. R., Rahaman O., Talukdar A. and Ganguly D.(2018). Cutting Edge: Piezo1 Mechanosensors Optimize Human T Cell Activation. J Immunol. 200(4): 1255-1260. [DOI] [PubMed] [Google Scholar]
- 21. Chakraborty M., Chu K., Shrestha A., Revelo X. S., Zhang X., Gold M. J., Khan S., Lee M., Huang C., Akbari M., et al.(2021). Mechanical Stiffness Controls Dendritic Cell Metabolism and Function. Cell Rep. 34(2): 108609. [DOI] [PubMed] [Google Scholar]
- 22. Atcha H., Jairaman A., Holt J. R., Meli V. S., Nagalla R. R., Veerasubramanian P. K., Brumm K. T., Lim H. E., Othy S., Cahalan M. D., et al.(2021). Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat Commun. 12(1): 3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geng J., Shi Y., Zhang J., Yang B., Wang P., Yuan W., Zhao H., Li J., Qin F., Hong L., et al.(2021). TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat Commun. 12(1): 3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Yang H., Jia A., Wang Y., Yang Q., Dong Y., Hou Y., Cao Y., Dong L., Bi Y., et al.(2022). Dendritic cell Piezo1 directs the differentiation of TH1 and Treg cells in cancer. eLife. 11: e79957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abiff M., Alshebremi M., Bonner M., Myers J. T., Kim B. G., Tomchuck S. L., Santin A., Kingsley D., Choi S. H., Huang A. Y., et al.(2023). Piezo1 facilitates optimal T cell activation during tumor challenge. Oncoimmunology. 12(1): e2281179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwak K., Sohn H., George R., Torgbor C., Manzella-Lapeira J., Brzostowski J. and Pierce S. K.(2023). B cell responses to membrane-presented antigens require the function of the mechanosensitive cation channel Piezo1. Sci Signaling. 16(804): eabq5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pang R., Sun W., Yang Y., Wen D., Lin F., Wang D., Li K., Zhang N., Liang J., Xiong C., et al.(2024). PIEZO1 mechanically regulates the antitumour cytotoxicity of T lymphocytes. Nat Biomed Eng. doi: 10.1038/s41551-024-01188-5. [DOI] [PubMed] [Google Scholar]
- 28. Zhao Q., Zhou H., Chi S., Wang Y., Wang J., Geng J., Wu K., Liu W., Zhang T., Dong M. Q., et al.(2018). Structure and mechanogating mechanism of the Piezo1 channel. Nature. 554(7693): 487-492. [DOI] [PubMed] [Google Scholar]
- 29. Vasileva V. and Chubinskiy‐Nadezhdin V.(2023). Regulation of PIEZO1 channels by lipids and the structural components of extracellular matrix/cell cytoskeleton. J Cell Physiol. 238(5): 918-930. [DOI] [PubMed] [Google Scholar]
- 30. Liu C. S. C. and Ganguly D.(2019). Mechanical Cues for T Cell Activation: Role of Piezo1 Mechanosensors. Crit Rev Immunol. 39(1): 15-38. [DOI] [PubMed] [Google Scholar]
- 31. Liu, C. S. C., Mandal, T., Biswas, P., Hoque, M. A., Bandopadhyay, P., Sinha, B. P., Sarif, J., D'Rozario, R., Sinha, D. K., Sinha, B. et al.(2024). Piezo1 mechanosensing regulates integrin-dependent chemotactic migration in human T cells. eLife. 12: e91903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ludford-Menting M. J., Oliaro J., Sacirbegovic F., Cheah E. Y., Pedersen N., Thomas S. J., Pasam A., Iazzolino R., Dow L. E., Waterhouse N. J., et al.(2005). A Network of PDZ-Containing Proteins Regulates T Cell Polarity and Morphology during Migration and Immunological Synapse Formation. Immunity. 22(6): 737-748. [DOI] [PubMed] [Google Scholar]
- 33. SenGupta S., Parent C. A. and Bear J. E.(2021). The principles of directed cell migration. Nat Rev Mol Cell Biol. 22(8): 529-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Banerjee T., Biswas D., Pal D. S., Miao Y., Iglesias P. A. and Devreotes P. N.(2022). Spatiotemporal dynamics of membrane surface charge regulates cell polarity and migration. Nat Cell Biol. 24(10): 1499-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Autenrieth T. J., Frank S. C., Greiner A. M., Klumpp D., Richter B., Hauser M., Lee S. I., Levine J. and Bastmeyer M.(2016). Actomyosin contractility and RhoGTPases affect cell-polarity and directional migration during haptotaxis. Integr Biol. 8(10): 1067-1078. [DOI] [PubMed] [Google Scholar]
- 36. Ghose D., Elston T. and Lew D.(2022). Orientation of Cell Polarity by Chemical Gradients. Annu Rev Biophys. 51(1): 431-451. [DOI] [PMC free article] [PubMed] [Google Scholar]