

Abstract
Tauopathies, including Alzheimer’s disease (AD), are a class of neurodegenerative diseases characterized by the presence of insoluble tau inclusions. Tau phosphorylation has traditionally been viewed as the dominant post-translational modification (PTM) controlling tau function and pathogenesis in tauopathies. However, we and others have identified tau acetylation as a primary PTM regulating both normal tau function as well as abnormal pathogenic features including aggregation. Prior work showed robust tau acetylation in aggregation hotspots located within the 2nd and 3rd repeat regions of tau (residues K280 and K311) in tauopathy brains, including AD, compared to non-tauopathy controls. By screening thousands of hybridoma clones, we generated site-specific and modification-specific monoclonal antibodies targeting acetylated tau at residues K280 or K311. To validate these antibodies in a bona fide neuronal system, we targeted the acetyltransferase CBP to the cytoplasm of neurons to promote tau acetylation. Several antibody clones specifically detected CBP-acetylated tau and co-localized with ac-tau in neurons. Additionally, our lead optimal anti-acetylated-tau monoclonal antibodies detected robust tau pathology in tangles and neuritic plaques of human AD brains. Given the now emerging interest in acetylated tau as critical regulator of tau functions, these sensitive and highly specific tools will allow us to further unravel the tau PTM code and, importantly, could be deployed as diagnostic or disease-modifying agents.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40478-024-01865-1.
Keywords: Alzheimer’s disease (AD), MAPT, Posttranslational modification (PTM), Acetylation, Tau, Neurodegeneration, Tauopathy, Monoclonal antibodies
Introduction
The microtubule-associated protein tau (MAPT) is one of the most highly abundant proteins in the brain. There are a total of six tau isoforms expressed in the adult brain that can form pathology, which result from alternative splicing of two variable N-terminal domains (exon 2, 3) and variable microtubule repeat region, R2 (exon 10). While tau normally functions to stabilize microtubules (MTs), various post-translational modifications (PTMs) can mediate tau’s ability to bind microtubules [1–3]. This loss of native function is linked to tau polymerization and aggregation, which is thought to underlie various tauopathies, including Alzheimer’s disease (AD), but also other tauopathies including frontotemporal dementia (FTD), Pick’s disease (PiD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD). While AD tau is comprised of both 3R and 4R tau, excluding or including the R2 region, respectively, several other tauopathies show tau-isoform specificity, in which the predominant tau isoforms are either 3R-tau (e.g. Pick’s disease [4]) or 4R-tau (e.g. corticobasal degeneration [5]).
It remains unclear exactly which tau species drive tau aggregation and pathology. While tau phosphorylation is classically associated with pathology, tau undergoes a variety of other post translational modifications including acetylation [1, 6, 7], methylation [8, 9], ubiquitination [10, 11], and SUMOylation [12, 13]. In particular, tau acetylation, unlike phosphorylation, occurs primarily in the microtubule binding region (MTBR) responsible for tau binding to MTs [14, 15]. Recent reports found tau acetylation favors tau beta strand stacking, consistent with a role for acetylation in defining the core tau protofilament, as shown by cryo-EM studies [16, 17]. Indeed, we and others found that tau acetylation can promote aberrant tau aggregation, proteolytic cleavage, and impair microtubule binding [15, 18, 19]. Further demonstrating the pathogenic effects of tau acetylation, loss of HDAC6, a tau deacetylase, exacerbated AD-like phenotypes in PS19 mice [18]. In human AD brains, two of the most prominent acetylated residues (K280 and K311) lie within homologous sequences within their respective 2nd and 3rd MTBR repeats [1, 18]. Intriguingly, these residues lie within two critical hexapeptide motifs (275VQIINK280 and 306VQIVYK311) that are responsible for tau’s ability to self-aggregate [20, 21]. Furthermore, acetylation at these two residues was observed in AD brains but not in non-tauopathy control brains, leading us to speculate that acetylation (at these two residues) occurs in a disease-specific manner (unlike tau phosphorylation), highlighting their exciting potential as therapeutic targets.
Given that tau acetylation can promote tau aggregation and prevent MT binding [14], these two particular sites (K280 and K311) emerged as potentially powerful drivers of tau pathology, though other sites of tau acetylation have also been documented (K163, K174, K274/K281, K369, and others), suggesting that a complex tau PTM profile exists. Among these other sites, acetylation at K174 and K274/K281 has been shown to promote tau pathology in several cellular and mouse models of tauopathy [22–24]. Furthermore, acetylated tau has been implicated in other non-AD tauopathies including traumatic brain injury (TBI), indicating broader significance of tau acetylation to brain health and disease [25].
Here, we developed, characterized, and validated a panel of several novel monoclonal antibodies detecting acetylated tau at residues K280 and K311, providing highly specific tools to unravel the tau PTM code that drives AD and other tauopathies. We anticipate that these antibodies will provide powerful tools for future biomarker assays or as immunotherapies.
Materials and methods
Plasmids and cell culture
The human tau constructs used were comprised of full-length tau with two N-terminal inserts and all four repeat domains (2N4R-tau). Site-directed mutagenesis (NEB) was used to insert the P301L and S320F mutations to generate 2N4R P301L/S320F tau. To generate plasmids pcDNA3.1-Flag-CBP CD-NES, pcDNA3.1-Flag-CBP CD-NLS, pcDNA3.1-Flag-CBP CD_LD-NES and pcDNA3.1-Flag-CBP CD_LD-NLS, pcDNA3.1-Flag-CBP CD and pcDNA3.1-Flag-CBP CD_LD were PCR amplified using CBP-BamHI-R and CBP-KpnI-F, NES_F and NES_R, or SV40-NLS-F and SV40_NLS_R were annealed and inserted. To generate lentiviral expression plasmids for pUltra-Flag-CBP CD-NES and pUltra-Flag-CBP CD-NLS, Flag-CBP CD-NES and Flag-CBP CD-NLS were amplified using primers 2xflag2pUltra F and 2xflag2pUltra R and inserted into AgeI and SalI linearized pUltra using NEB NEBuilder HiFi DNA Assembly Master Mix (Cat# E2621S). Similarly, for generating pUltra 2N4R wt, pUltra 2N4R PL, wild-type 2N4R and P301L 2N4R were amplified using primer Tau F and Tau R and inserted into AgeI and SalI linearized pUltra using T4 DNA ligase (NEB, M0202S). Tau and CBP plasmids were cloned into the pCDNA5/TO vector (Life Technologies). The K280R, K311R wild-type tau mutations were generated using site-directed mutagenesis (NEB). The primers used are listed in Supplemental Table 3.
QBI-293 cells (ThermoFisher, #R70507) are commercially available and maintained according to standard protocols. QBI-293 cells were cultured in DMEM media (supplemented with 10% fetal bovine serum, 1X L-glutamine, and 1X penicillin/streptomycin). FuGENE 6 transfection reagent (Promega) was used to transfect QBI-293 cells with 2N4R tau and CBP constructs for 48–72 h. Cells were lysed in 1X RIPA buffer [50 mM Tris pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholate, 1% Igepal CA-630, 0.1% SDS] supplemented with deacetylase, phosphatase and protease inhibitors: 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM Nicotinamide (NCA), 1 mM sodium orthovanadate (Na3VO4), 1 mM sodium fluoride (NaF), 0.5 M β-glycerophosphate, 2 μM Trichostatin A (TSA), and protease inhibitor cocktail composed of L-1-tosylamide-2-phenylethylchloromethyl, N-tosyl-L-lysine chloromethyl ketone, leupeptin, pepstatin, and soybean trypsin inhibitor, each at 1 μg/ml.
Primary cortical neurons
Mouse primary cortical culture were performed in accordance with the University of North Carolina (UNC) Institutional Animal Care and Use Committee (UNC IACUC protocol 21-257). Cortical neurons were isolated from C57Bl/6 mice (Charles River) at embryonic day 15–16 following lethal isoflurane anesthesia. Mouse embryos were removed from the pregnant abdominal cavity, washed with ice-cold 70% ethanol and placed in cold Hank’s-buffered Balanced Salt Solution (HBSS). Embryos in HBSS were transferred to a 10 cm dish. Embryos were then removed from the membrane and brains were dissected. The cortex was carefully isolated and transferred to Hibernate-E solution (BrainBits) with B27 (Gibco) and Glutamax (ThermoFisher). Cerebral cortices were minced with forceps and incubated for 30 min at 37°C in HBSS with 20 U/mL papain (Worthington Biomedical), 1 mM EDTA, 0.2 mg/mL L-Cysteine, and 5 U/mL DNAse (Promega). Cortical tissue was further dissociated in plating media [BrainPhys media (Stemcell), 5% fetal bovine serum, 1X B27 (Gibco), 1X penicillin/streptomycin (Gibco), and 1X Glutamax] using a P1000. The cell suspension was centrifuged for 5 min at 1.5 RCF. The resulting pellet was resuspended in plating media and passed through a 40 μm cell strainer. Cells were counted using a Countess 3 automated cell counter (Invitrogen) and plated in plating media at 600,000 cells/well in 6-well plates for western blot or 30,000 cells/well in 96-well plates for immunocytochemistry. The next day (DIV1), plating media was completely aspirated and fresh neuronal media [BrainPhys media (Stemcell), B27 (Gibco), 1X penicillin/streptomycin (Gibco), and 1X Glutamax] was added.
Lentiviral production
Lentiviral production was performed by co-transfecting 37.5 mg lentivirus plasmid with 25 mg psPAX2, 12.5 mg VSVG, and 6.25 mg REV plasmids per 15 cm culture plates of lenti-X 293 T cells (Takara), with three dishes of cells used for each lentiviral production. Two days after transfection, culture media was collected then centrifuged at 2000 g for 10 min. Lentiviral particles were purified using a double-sucrose gradient method. Supernatants were loaded onto a 70–60% to 30–20% sucrose gradient and centrifuged at 70,000 g for 2 h at 17 °C. The 30–60% fraction containing the viral particles was retrieved, resuspended in PBS, filtered with a 0.45 mm filter flask, loaded onto a 20% sucrose solution, and centrifuged a second time at 70,000 g for 2 h at 17 °C. The supernatants were carefully discarded, and the viral particles present in the pellet were resuspended in PBS, aliquoted and stored at − 80 °C.
Lentiviral neuron transductions and treatments
For lentiviral transductions, neurons were exposed to ~ 3.0 × 1012 viral particles per mL at DIV4 with the exception of the CBP-CD-NES and CBP-NLS constructs which were added at 7.5 × 1011 (1/4 dilution) to avoid CBP-induced toxicity. If multiple viruses were co-transduced, both were added at the same time at the above concentration. The following day, all media was aspirated and replaced with half conditioned media (from the same neuron culture) and half fresh neuronal media (BrainPhys with B27, Pen-strep, and Glutamax). Half of the media was discarded and replenished with fresh neuronal media every two days until the end of the experiment. For experiments where monoclonal antibodies were added to primary neurons, antibodies were added once at a final concentration of 26.6 nM based off previous work [26].
Immunoblotting and biochemical analysis
QBI-293 and primary cortical neuron RIPA lysates sonicated 20 times on ice, then centrifuged at 15,000 rpm for 20 min at 4 °C. Supernatant was removed from the pelleted material, mixed with 6X loading dye with DTT, then boiled at 98 °C for 10 min. Samples were resolved via SDS-PAGE on 4–20% gradient gels (BioRad), transferred to nitrocellulose membrane using the iBlot 2 (Invitrogen) using standard P0, 7-min settings. Membranes were blocked with 2% milk in 1X TBS for 30 min then incubated with primary antibody overnight at 4 °C. The next day, membranes were incubated with HRP-conjugated anti-mouse or anti-rabbit antibodies (1:1000) for 1 h at room temperature. Blots were developed with ECL Western Blotting substrate (Cytiva, Amsterdam) and imaged on an GE ImageQuant LAS4000. The immunoblots in Fig. 2 were analyzed on parallel blots to allow multiplexing of different antibodies with identical molecular weights. For sequential fractionations, transfected QBI-293 cells were collected in ice-cold PBS with all inhibitors, sonicated 20 times on ice, then centrifuged at 15,000 rpm for 20 min at 4 °C. The supernatant was removed and labeled as the “PBS fraction”. The pellet was washed 1X with cold PBS, centrifuged at 15,000 rpm for 20 min at 4 °C. This was repeated using hi-salt buffer (50 mM Tris, 10 mM EGTA, 5 mM MgSO4, 0.75 M NaCl, 0.02 M NaF, pH 7.6), then RIPA buffer, then 1% SDS buffer. Samples with SDS buffer were centrifuged at room temperature. Only PBS and SDS fractions are shown.
Fig. 2.

A neuronal tau acetylation assay using cytoplasmic targeted CBP to validate the specificity of ac-tau monoclonal antibodies. A QBI293 cells transfected with CBP catalytic domain (CD) with a nuclear exclusion sequence (NES) or nuclear localization sequence (NLS). CBP-FLAG labeled in 488, DAPI in blue. Scale bars, 50 μm. B Primary mouse cortical neurons transduced with lentivirus expressing CBP CD NES or CBP CD NLS. CBP-FLAG labeled in 488, DAPI in blue. Scale bars, 50 μm. C QBI293 cells transfected with wild-type 2N4R human tau with CBP-CD-NES, CBP-CD-NLS, or CBP-catalytically dead mutant (CBP-mut) NES probed with ac-K280 and ac-K311 antibodies. Each condition was performed in duplicate. D Primary mouse cortical neurons lentivirally transduced with empty vector, wild-type 2N4R human tau with or without CBP-CD-NES and CBP-CD-NLS probed with ac-K280 and ac-K311 antibodies. Each condition was performed in triplicate. Blots were probed with purified 19B6 or purified 15E8, rabbit polyclonal ac-K280 or rabbit polyclonal ac-K311, CBP (FLAG), and total tau (K9JA). E–F Quantification of ac-tau in primary neurons (ac-K280 (E) and ac-K311 (F) antibodies) comparing CBP-CD-NES and CBP-CD-NLS. N = 3; p-value: * < 0.05, ** < 0.01. * denotes ~ 100kd CBP-CD cross-reactive band. G Immunoprecipitation of ac-K280 tau in QBI293 cells with 19B6 (Input-left; IP-right) probed with polyclonal K280, 19B6, total tau (K9JA), and total ac-lysine antibodies. H QBI293 cells were transfected with wild-type 2N4R human tau or P301L/S320F 2N4R human tau with or without CBP-CD-NES. Lysates were processed by sequential fractionation through PBS, hi-salt buffer, RIPA buffer, then 1% SDS buffer and probed with 19B6 and 15E8 antibodies. PBS fraction (top) and 1% SDS buffer fraction (bottom) are shown
Immunoprecipitation
Following transfections, QBI-293 cells were collected via scraping in cold NETN buffer (20 mM Tris pH 8.0, 150 mM NaCl, 0.5mM EDTA, 0.5% NP-40/IGEPAL) plus deacetylase, phosphatase, and protease inhibitors. Protein levels in supernatants containing nuclear proteins were measured using Protein Assay Dye Reagent Concentrate (BioRad, 5000006). Roughly 10% of the lysate per sample was saved as Input samples, and equal protein concentrations were used in each IP reaction. Antibodies were conjugated to beads, then remaining lysate was added and incubated overnight, rotating at 4 °C. IP antibodies used: Mouse IgG1 kappa isotype control (Invitrogen, 14471485) and 19B6 (Ac-tau). 50 µL of Protein A/G magnetic beads (Pierce, 88803) were incubated with 8 µL antibody, per IP, for four hours prior to addition of lysates. The next day, beads were washed 3 × with cold NETN Buffer with all inhibitors. Protein complexes were eluted from beads in 100µL IgG Elution Buffer, pH 2.0 (Pierce, 21028) for 10 min, with continuous shaking at 190 rpm, at room temperature. 10 µL Tris buffer at pH 10.4 was added to neutralize each sample. Western blotting was performed on samples—15 µL of each IP was loaded and 2% Input was loaded.
Immunocytochemistry
Cell culture immunocytochemistry experiments were performed using QBI-293 cells or cortical primary neurons grown on poly-D-lysine (PDL)-coated 96-well plates. After transfection (48 h) or transduction (5–7 days unless otherwise stated), cells were rinsed in PBS then fixed in 4% paraformaldehyde for 15 min, then rinsed with PBS. Cells were then permeabilized with 0.2% Triton X-100 (Sigma) in PBS for 20 min then blocked in 4% NGS for 1 h. Primary antibodies were added in 2% milk and incubated overnight at 4°C. Cells were washed in PBS with 0.05% tween and incubated with Alexa 488, Alexa 594, or Alexa 647 conjugated secondary antibodies and DAPI for 1 h. Cells were imaged using the EVOS M7000 automated microscope or EVOS FL widefield microscope using EVOS 20X objective (Invitrogen AMEP4982) and 40X objectives (Invitrogen AMEP4754) with DAPI, GFP, Texas Red, and Cy5 light cubes. Note: Some representative images were artificially brightened in Microsoft PowerPoint or Celleste 5.0 software (Invitrogen) to aid visualization. Any panels assessing the same set of conditions (i.e. a specific monoclonal antibody across treatments) were brightened to identical levels.
Antibodies
The following primary antibodies were used in this study: mouse monoclonal 19B6 ac-K280 (large-scale purification) (Todd Cohen, 1:1000), mouse monoclonal 15E8 ac-K311 (large-scale purification) (Todd Cohen 1:1000), rabbit polyclonal ac-K280 (Todd Cohen, 1:500), rabbit polyclonal ac-K311 (Todd Cohen, 1:500), K9JA (DAKO, A0024 1:2000), Tau12 (Millipore, MAB2241, 1:1000), Ac-Lysine (CST, 9441, 1:1000), FLAG M2 (Sigma, F1804, 1:2000), HA (Cell Signaling, 3724, 1:1000), GAPDH (Millipore-Sigma, CB1001, 1:1000). Small-scale purifications of monoclonal antibodies from supernatant were used at 1:100 for SDS-PAGE and 1:25 for immunocytochemistry. For detection of 19B6 and 15E8 uptake, only mouse secondaries were used (no primary antibody incubation). Quantification of immunoblotting was performed using ImageStudio Lite Version 5.2.
Neuronal antibody uptake assay
For western blot, primary cortical neurons were transduced with lentivirus at DIV4 with P301L human tau with or without CBP-CD-NES. At DIV5, 19B6 or functional grade mouse IgG1 isotype control (Invitrogen, 16471482) antibodies were added directly to culture at a final concentration of 26.6 nM for seven days, replacing half of the media every three days with fresh neuronal media. Neurons were then washed 1X with PBS, then lysed in RIPA buffer with inhibitors and collected for SDS-PAGE analysis.
For immunocytochemistry, primary cortical neurons were transduced with lentivirus expressing P301L human tau with or without CBP-CD-NES at DIV10. Immediately prior to antibody application, 500ul of 19B6 and 15E8 antibodies were cleared of aggregates by centrifugation through a 0.1 um centrifugal filter unit (Millipore, UFC30VV25) for two minutes at 12,000 RCF. At DIV14 19B6 and 15E8 antibodies were directly applied to primary cortical neuron cultures at a final concentration of 26.6 nM and incubated at 37 °C, 5% CO2 for thirty minutes. A glycine wash step was then performed to remove antibody interacting with the cell surface but not completely internalized. Specifically, the pH was incrementally lowered by first adding one unit volume of 0.2 M glycine pH 2.0 to each well containing neurons (with neuronal media) for 30 s. All media was then removed and replaced with pure 0.2 M glycine pH 2.0 for an additional 30 s. The wash solution was removed and replaced with one well volume of 10 µM wheat-germ agglutinin conjugated to AlexaFluor 647 (Invitrogen, W32466) diluted in artificial cerebrospinal fluid (119 mM NaCl, 26.2 mM NaHCO3, 2.5 mM KCl, 1 mM NaH2PO4, 1.3 mM MgCl2, 10 mM glucose, and 2.5 mM CaCl2) for two minutes. WGA solution was removed, and cells were immediately fixed in 4% paraformaldehyde for ten minutes at room temperature. For experiments that evaluated clathrin-mediated endocytosis of monoclonal antibodies, primary neurons were pre-treated with each of the two inhibitors for 30 min prior to addition of the antibody. 15E8 antibody (8 ug/mL) was added into media containing inhibitors for another one hour. Inhibitors used were dynasore (Sigma D7693) and anti-mouse Fc Block (BD Pharmingen, 553,142). Cells were washed and fixed as described above. The number of ac-tau positive neurons and neurons displaying antibody uptake were quantified using Celleste 6 (Invitrogen).
Confocal imaging
Confocal micrographs were acquired using an inverted Zeiss 800 laser scanning confocal microscope equipped with 405 nm, 488 nm, 561 nm, and 640 nm diode lasers and two gallium arsenide phosphide (GaAsP) detectors. Micrographs were acquired using a Plan-Apo 40x/0.95 NA air objective lens and ZEN Blue software. Brightness and contrast were similarly adjusted between images using ImageJ (NIH).
Human monocyte-derived macrophage culture and differentiation
Primary hMDM were cultured from peripheral blood mononuclear cells (PBMCs), isolated from human buffy-coated leukopaks (New York Blood Center) as previously described. In all scenarios, blood products were screened against bacteremia and viremia, and the time from leukapheresis to culture was < 24h. Briefly, blood product was diluted 1:1 with sterile PBS then overlaid atop a Ficoll-Paque plus density matrix (Cytiva, GE17-1440-02) for differential centrifugation at 500 RCF for 20 min in a swinging-bucket centrifuge with no brake. Leukocytes were isolated, rinsed in Hank’s-balanced salt solution, and cleaned of erythrocytes by 10-min incubation in erythrocyte lysis solution (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) with agitation. Viable leukocytes were counted using trypan blue exclusion hemocytometry and plated in glass-bottom dishes (Nunc, 150680). Cells were maintained in complete media: Dulbecco’s modified eagle media with high glucose, 10% fetal bovine serum (Gibco, 160000–044), and 20 μg/ml gentamicin (Gibco 15750–60) at 37 °C, 5% CO2. Monocytes were allowed to adhere to the substrate for approximately 5–7 days in culture, then exchanged into differentiation media: complete media supplemented with 15 ng/mL granulocyte–macrophage colony stimulating factor (GM-CSF; RnD, 215-GM-010/CF). Differentiation media was replenished every other day by half-media changes for a total of two exchanges. Differentiated hMDM were then returned to complete media with half-media changes every two days.
pH-sensitive pHAb dye conjugation
Large-scale purified 19B6 (ac-K280) antibody was conjugated to pH-sensitive pHAb dye (Promega, G9841) per manufacturer protocol. Briefly, 100 µg of purified 19B6 antibody was buffer exchanged into amine conjugation buffer (10 mM sodium bicarbonate buffer, pH 8.5) using Zeba spin-desalting columns (Pierce, 89890). pHAb dye (10 mg/mL in DMSO) was added at a 20-molar excess to antibody (1.2 µL per 100 µg) and incubated at room temperature protected from light with orbital mixing at 200 RPM in a ThermoMixer C (Eppendorf, 2231001005). Unreacted dye was removed by passing the conjugation solution over a Zeba spin-desalting column equilibrated with sterile 1X PBS, pH 7.4 for a total of two exchanges. Antibody concentration was obtained by A280 and recovery efficiency determined to be approximately 70%. As a negative control, isovolumetric equivalents of buffers, free of 19B6, were subject to conjugation and exchange protocols in parallel to 19B6 reactions. As a positive control for uptake and cargo acidification, commercial DH5α E. coli were grown to stationary phase (OD600. > 1.0) and 1.0 mL of culture was heat-killed at 65 °C for 10 min. Bacteria were pelleted by centrifugation at 1,200 RCF for 5 min, washed twice in amine conjugation buffer, resuspended in 100 µL conjugation buffer with 1.2 µL of pHAb dye, and conjugated as above. Unreacted dye was removed by centrifugation with two wash cycles in sterile 1X PBS. To remove bacterial aggregates, the pellet was resuspended in 100 µL sterile PBS and sonicated 10-times at 25% amplitude using a QSonica hand-sonicator probe.
hMDM-K280 internalization and acidification assay
hMDM were pre-charged with 10 µM Chloroquine or isovolumetric equivalents of vehicle (sterile H2O) for 60 min at 37 °C, 5% CO2. Cells were then stimulated with 10 µL of either pHAB-19B6, pHAb-blank, or pHAb-E. coli, and returned to the incubator for 30 min. Cells were then imaged live using TAMRA filter settings on an EVOS widefield inverted microscope at 20X magnification with numerical aperture 0.40. Baseline fluorescence was set using the pHAb-blank stimulated condition.
Small-scale monoclonal antibody purification
For small-scale antibody purifications, Protein G Magnetic Beads (Pierce, 88848) were used to purify antibody from 500ul of hybridoma supernatant (provided by GenScript) as per manufacturer protocol. The bound antibody was eluted using 100 ul IgG Elution Buffer, pH 2.0 (Pierce, 21028) for ten minutes intermittently rotating tubes to mix, and then neutralized with 10 ul of Tris pH 10.5 buffer.
Mouse immunization to produce ac-K280 and ac-K311 monoclonal hybridomas
Acetylated peptide (QIIN {Lys-Ac} KLDLSNVQSC) and non-acetylated peptide (QIINKKLDLSNVQSC) were synthesized by the Peptide Department of GenScript. The acetylated peptide was conjugated to a carrier protein using the MBS method and mixed with Freund's adjuvant for immunization. Two groups of mice were immunized separately: BALB/C (Animal Number: #B1676 ~ 1680) and C57 BL/6 (Animal Number: #B1681 ~ 1685). Intraperitoneal injection method was performed with a dosage of 25 ug antigen/mice per injection. After three routine injections, the best two animals were selected for fusion based on the test results. The fusion partner used was SP2/0, and the fusion was carried out using electroporation. The successfully fused hybridoma cells were seeded onto a 96-well plate, and the cell supernatant was collected for detection and screening. Indirect ELISA was used to coat the antigen on the ELISA plate, followed by addition of the cell supernatant and secondary antibodies to promote the selection of specific clones. The subcloning was performed using a limiting dilution method to obtain a stable proliferating monoclonal cell line. Monoclonal cell lines can be used for large-scale production in a roller bottle. Mouse immunization and generation of hybridoma lines were performed by GenScript.
Indirect ELISA protocol for ac-tau monoclonal hybridomas
The antigen peptides (listed above) were diluted in coating buffer and incubated at 37 °C for 1h. Coating buffer with peptides was removed and washed with washing buffer. Plates were blocked with blocking buffer and incubated at 37 °C for 1h, then washed once with washing buffer. Hybridoma supernatant was diluted with blocking buffer and added to the plates with coated antigen peptides for 1h at 37 °C. Plates were then washed four times with washing buffer. HRP-conjugated secondary antibodies were diluted in blocking buffer and added to the plate for 30 min at 37 °C, then washed four times in washing buffer. 100 ul of TMB reagent was added to each well and incubated at room temperature until sufficient color development (15–20 min). Reaction was stopped with stop solution and absorbance read at 450 nm using a microplate reader. Indirect ELISAs were performed by GenScript.
Large-scale monoclonal antibody production and purification
Hybridomas were cultured in DMEM containing 10% FBS and incubated at 37 °C. When hybridomas reached > 70% confluence, cells were transferred to shake flask culture flask. When > 70% confluent, cells were transferred to a roller bottle and incubated in electro-thermal incubator for roughly 7 days. Cell suspension was centrifuged at 8000 rpm for 30 min to collect supernatant. Supernatant was then filtered with 0.22 um filter to remove debris. Protein A/G resin was bound to a purification column. Antibody sample was transferred to column, then washed several times. Antibody was then eluted with low pH elution buffer and then immediately neutralized the pH with neutralization buffer. Antibody eluate was dialyzed with a dialysis bag against PBS at 4 °C overnight. Large-scale monoclonal antibody production and purification was performed by GenScript.
Hybridoma cell culture
Prior to antibody sequencing, frozen vials of hybridoma cells from each clone (provided by GenScript) were thawed and transferred to a 15mL sterile centrifuge tube with 10 mLs of DMEM (Corning 10-013-CV) with 10% FBS and 1% penicillin/streptomycin. Cells were centrifuged at 800 rpm for 5 min. Supernatant was discarded and cells were suspended in 10 mL of media then transferred to a tissue-treated 10cm dish. Cells were incubated at 37 °C for 2–3 days until slightly adherent cells cover ~ 75% of the bottom of the dish. At this point, supernatant was collected from cells, and cells were gently washed off the bottom of the plate (cells are slightly adherent) with 10 mL of fresh media then divided into four 10cm dishes. Two days later, supernatant was aspirated from one plate and cells were scraped in Trizol then stored at − 80°C until sequencing protocol. The three other dishes were collected in FBS media containing 10% DMSO, then stored in liquid nitrogen.
Monoclonal antibody sequencing
Mouse monoclonal antibody sequencing was performed following previous work with additional modifications. Total RNA was extracted from hybridoma cell lines using TRIzol Reagent (Invitrogen), and reverse transcription polymerase chain reaction was applied using SMARTScribe Reverse Transcriptase (Takara cat# 639537) with primers in Supplemental Table 2. Antibody variable regions were amplified using touch-down PCR using Q5 High-Fidelity DNA Polymerase (98 °C for 30s; 10 cycles of 98 °C 15s, 63–57.5 °C for 30s (decrease 0.5 °C each cycle), and 72 °C 30s; 15 cycles of 98 °C 30s, 72 °C 30s; followed by 72 °C 2min and hold at 4 °C) with primers in Supplemental Table 3. Amplified antibody DNA fragments were gel extracted using QIAGEN QIAquick Gel Extraction Kit (Cat#28706) and inserted into NheI and BamHI linearized pcDNA3.1 using NEB NEBuilder HiFi DNA Assembly Master Mix (Cat# E2621S). Transformations were performed using NEB Stable Competent E. E. coli (Cat#C3040H), and DNA sequences were obtained by Sanger sequence using CMV forward primer.
Postmortem human immunohistochemistry
Human, Braak VI AD post-mortem brain was provided by the Center for Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania. Four AD cases and one Pick’s disease case were used for post-mortem human brain IHC. All tissues were ethanol fixed human tissue. Small-scale antibody purifications (from Protein G beads) were used at 1:25. Formic acid retrieval was used on 6μm tissue. We note that ethanol fixed tissue and formic acid retrieval are required for maximal detection sensitivity of ac-tau pathology in AD cases. On day 1 of IHC, slides were de-paraffinized with two five-minute incubations with Xylene. Slides were rehydrated in descending EtOH series for one minute each until 70% EtOH. Slides were then immersed in ddH20 for 1 min, then placed in 88% formic acid for five minutes. Slides were washed in H20, then incubated for 30 min in mixed methanol/H202. Sections were washed in H20 for another 10 min, then washed in 0.1 M Tris buffer pH 7.6 for five minutes. Slides were then blocked in blocking buffer (0.1 M Tris/2% FBS) for another five minutes. Primary antibodies were diluted in blocking buffer and added to each section. Slides were incubated at 4°C in humidified (Tris-soaked kimwipe) chamber overnight (16–20 h). On day 2, slides were washed in Tris three times for 2 min, then blocked in Tris/FBS for 5 min. Goat anti-mouse IgG1-HRP secondary antibody was applied at 1:1,000 in blocking buffer and incubated at room temperature for one hour. Slides were then washed in Tris twice for 3 min. Vector DAB solution was made with 1 drop of DAB per mL of stable DAB Buffer and inverted, not vortexed. DAB was applied to each slide using a transfer pipette and incubated for exactly 7 min per slide. Slides were rinsed with Tris buffer, then ddH20 for 5 min. Harris Hematoxylin (Thermo-Shandon) counterstain was applied for 20 s then washed twice in H20. Slides were washed in running tap water for 15 min, then dehydrated and cleared in xylenes (1 min each EtOH (70%, 80%, 95%, 95%, 100%, 100%) and two changes of Xylene for 5 min each. Slides were coverslipped with Cytoseal.
Statistical analyses
GraphPad Prism software was used to create all graphs and perform all statistical analyses. To compare ac-tau signal by immunoblotting, we performed separate two-tailed, unpaired student’s t-test from n = 3 replicates. For antibody uptake experiments with inhibitors, one-way ANOVA was performed using Dunnet’s multiple comparison test.
Results
Generation of anti-acetylated tau monoclonal antibodies
Using a hybridoma screening platform, we evaluated ~ 5,000 mouse monoclonal antibodies comprised of IgG1 and IgG2 isotypes that detected tau acetylated at either lysine K280 or K311 residues. Among these, 24 clones exhibited desirable specificity for their respective acetylated tau residue. The variable heavy and light-chain complementarity-determining regions (CDRs) determine the antibody’s ability to bind an antigen. To assess if any clones had identical or similar CDRs, we utilized a modified protocol to sequence heavy-chain and light-chain CDRs of all 24 clones [27]. Strikingly, several of these clones had either identical or highly overlapping CDRs (Table 1). Thus, CDR sequences were directly correlated with the optimal recognition of acetylated tau epitopes.
Table 1.
Heavy chain and light chain variable region CDRs of top 24 ac-K280 and ac-K311 hybridoma clones

Each ac-K280 (left) and ac-K311 (right) clone is listed with their respective complementarity determining regions (CDR1, CDR2, and CDR3). Heavy-chain CDRs are listed at the top, light-chain CDRs are listed at the bottom. Clones with identical sequences are listed together. An example sequence for the heavy-chain of 19B6 is shown with the location of each CDR depicted within the sequence.
Validation of ac-K280 and ac-K311 clones using engineered CBP variants
Creb-binding protein (CBP) was previously shown to act as a tau acetyltransferase [1, 28]. Therefore, we validated which of these 24 clones detected CBP-acetylated tau (ac-tau) in cells. Using antibodies purified from hybridoma supernatants, we found that several clones exhibited specificity for CBP-acetylated human tau, but not their respective acetylation-null K280R or K311R control mutants by western blot (Fig. 1A, B, Supplementary Fig. 1). As expected, the K280R and K311R mutants were acetylated at other lysine residues, as detected using a pan-acetylated lysine antibody (Fig. 1C). We also verified that most clones showed a high degree of specificity for ac-tau peptides vs. their comparable non-ac-tau control peptide by ELISA (Fig. 1D). Clones with similar CDRs were equally effective, while other less optimal clones that emerged from our screen lacked sensitivity or specificity when tested in the presence of the acetyltransferase CBP (Supplemental Fig. 1).
Fig. 1.

Molecular and biochemical validation of candidate ac-K280 and ac-K311 monoclonal antibodies. Representative immunoblots of QBI-293 cells transfected with wild-type 2N4R human tau or acetyl-null K280R/K311R 2N4R human tau and full length CBP. Small-scale purification of ac-tau monoclonal antibodies from hybridoma supernatants were applied to blots. A ac-K280 antibody clones. B ac-K311 antibody clones. C total acetylated lysine showing acetylation of K280R and K311R tau at other residues. D Quantification of ELISA OD450 signal from monoclonal ac-K280 and ac-K311 antibodies incubated with acetylated versus non-acetylated peptides. Peptides: Ac-K311-QIVY{Lys-Ac}PVDLSKVTSC, QIVYKPVDLSKVTSC; Ac-K280-QIIN{Lys-Ac}KLDLSNVQSC, QIINKKLDLSNVQSC
Since tau acetylation primarily occurs within the cytoplasm, we sought to develop an approach that would allow for cytoplasmic expression of CBP and avoid non-specific acetylation of nuclear CBP substrates. To achieve this, we generated lentiviruses expressing CBP’s catalytic domain (CD; residues 1088-1758) containing either a nuclear export signal (NES) or a nuclear localization signal (NLS). Unlike full-length ~ 265 kD CBP, the smaller active catalytic domain (~ 82 kD CBP-CD) is readily packaged and expressed via lentiviral-mediated transduction of neurons. CBP-CD-NES was successfully targeted to the cytoplasm of QBI-293 cells or primary cortical neurons (Fig. 2A, B). Co-transfection or co-transduction of CBP-CD with 2N4R tau was performed, followed by immunoblotting to assess the specificity of our two lead ac-K280 and ac-K311 clones, which we refer to as 19B6 and 15E8, respectively. In transfected QBI-293 cells, we found that 19B6 detected ac-K280 similar to a previously generated rabbit polyclonal antibody [1]. Similarly, 15E8 detected an ac-K311 banding pattern that was identical to a previously generated rabbit polyclonal K311 antibody [18] (Fig. 2C). We did not observe ac-tau when expressing a catalytically inactive CBP mutant (CBP-mut) lacking acetyltransferase activity (L1434A/D1435A), confirming CBP-dependent acetylation (Fig. 2C). In transduced primary cortical neurons, 19B6 and 15E8 detected acetylated tau in CBP-CD-NES expressing neurons (Fig. 2D). In these neurons, CBP-CD-NES induced ac-tau by ~ 2–5 fold compared to CBP-NLS, supporting ac-tau accumulation upon CBP targeting to the cytoplasm in transduced primary neurons (Fig. 2E, F). To further validate the specificity of 19B6, QBI-293 cells were co-transfected with wild-type tau or an acetylation-null K280R tau mutant in the presence or absence of CBP-CD-NES. We then immunoprecipitated tau using 19B6 and observed the expected ac-tau in the presence of CBP-CD-NES, but this was completely abrogated with the K280R mutant (Fig. 2G). This pulldown provides additional evidence that 19B6 demonstrates specificity for acetylated tau at residue K280.
We observed some differences in the extent of tau acetylation in QBI-293 and primary neurons. For example, nuclear targeted CBP-CD-NLS generated acetylated tau at low levels in neurons but more strongly in QBI-293 cells, the latter likely due to slight CBP leakage into the cytoplasm. We also noticed a non-tau cross-reactive 100 kD acetylated substrate that emerged only in the presence of the active truncated CBP-CD in either QBI-293 cells or primary neurons. This is a non-specific band resulting from expression of the constitutively active CBP-CD construct, since we did not observe this band with full length CBP (Supplemental Fig. 2A, B). Therefore, the 100 kD band likely represents an unknown acetylated substrate that is non-specifically detected by the 19B6 antibody. Any future immunoblotting performed with 19B6 should consider the reactivity of a 100 kD cross-reactive band. No other cross reactivity was observed. We reiterate that detection of the 100 kD cross-reactive band only occurs in the presence of the artificially truncated and active CBP construct but not full length CBP. Overall, our lead clones show highly desirable properties including acetylation-specificity and site-specificity in a neuronal tau acetylation assay.
To identify which tau species are recognized by the monoclonal ac-tau antibodies, we performed a sequential biochemical fractionation of QBI-293 cells in which we acetylated either wild-type soluble tau or a highly aggregate-prone tau mutant containing two FTD-causing familial mutations (P301L-S320F, termed PL-SF) [29]. In the soluble PBS extracted fractions, only monomeric ac-tau species were detected with 19B6 (ac-K280) and 15E8 (ac-K311) regardless of the tau variant. In the insoluble SDS-extracted fractions, the PL-SF tau variant formed more insoluble ac-tau than wild-type tau, as expected (Fig. 2H). Thus, we conclude that our monoclonal antibodies detect both soluble and insoluble acetylated tau species.
Immunocytochemistry validation of ac-K280 and ac-K311 clones in neurons
To determine if our candidate ac-tau antibodies detected acetylated tau by immunocytochemistry (ICC), we co-transduced neurons with 2N4R tau in the presence or absence of CBP-CD-NES and found that 19B6 selectively detected ac-tau with much greater sensitivity compared to other clones (e.g., 23B7 and 9G4) (Fig. 3A). We identified two ac-K311 clones that detected ac-tau to a similar degree (15E8, 14G8), though we note that 14G8 showed some non-specific background staining even in the absence of acetylated tau (Fig. 3B). Conversely, 13B12, 13H11, and 21D9 were either less sensitive or non-specific (Fig. 3B). We examined several other clones by ICC and found those with similar CDRs to be equally sensitive and specific while others either lacked detection sensitivity or were non-specific (Supplemental Fig. 3). Importantly, for scaling and production purposes, large-scale purifications of 19B6 and 15E8 maintained sensitivity and specificity in ICC assays employing acetylation of either wild-type or mutant P301L tau (Fig. 3C).
Fig. 3.

Immunocytochemistry validation of ac-tau monoclonal antibodies in primary neurons. Primary mouse cortical neurons were transduced for five days with lentiviruses for empty vector control, wild-type human tau, or wild-type tau and CBP-CD-NES. A Immunocytochemistry of neurons with rabbit polyclonal ac-K280 (RFP) and small-scale purification of 19B6, 23B7, and 9G4 (GFP). B Immunocytochemistry of neurons with rabbit polyclonal ac-K311 (RFP) and small-scale purification of 15E8, 14G8, 13B12, 13H11, and 21D9 (GFP). C Immunocytochemistry of large-scale purified 19B6 and 15E8 (GFP) and polyclonal ac-K280 and ac-K311 (RFP) after lentiviral transduction with wild-type or P301L human tau and CBP-CD-NES or CBP-CD-NLS. Scale bars, 125 μm. DAPI in blue
Clone 19B6 is internalized and shows target engagement
With the increasing interest in using anti-tau antibodies as immunotherapies, we sought to evaluate whether our most effective clones, 19B6 and 15E8, are internalized by primary cortical neurons and engage the desired target, which in this case is CBP-acetylated tau. Primary neurons were transduced with P301L tau in parallel with CBP-CD-NES to facilitate tau acetylation. After exposing cultures for seven days to 19B6, we found by immunoblotting that 19B6 more readily accumulated within neurons co-expressing tau and CBP-CD-NES compared to isotype control antibody, suggesting 19B6 uptake and affinity for ac-tau (Fig. 4A). We did not observe ac-tau degradation or clearance, suggesting antibody uptake may not be sufficient to target tau for degradation in this system (Fig. 4B). To confirm that 19B6 and 15E8 show target engagement, we allowed uptake of either 19B6 or 15E8 for one hour and subsequently performed double labeling to detect antibody engagement with ac-tau. Indeed, we observed a high degree of colocalization between the internalized antibodies and ac-tau, as both 19B6 and 15E8 labeled ac-tau positive cell bodies and neuronal processes. As expected, in the absence of CBP, we did not detect antibody engagement with non-acetylated tau (Fig. 4C, D). These data suggest that 19B6 and 15E8 are internalized by neurons and show desirable ac-tau target engagement.
Fig. 4.
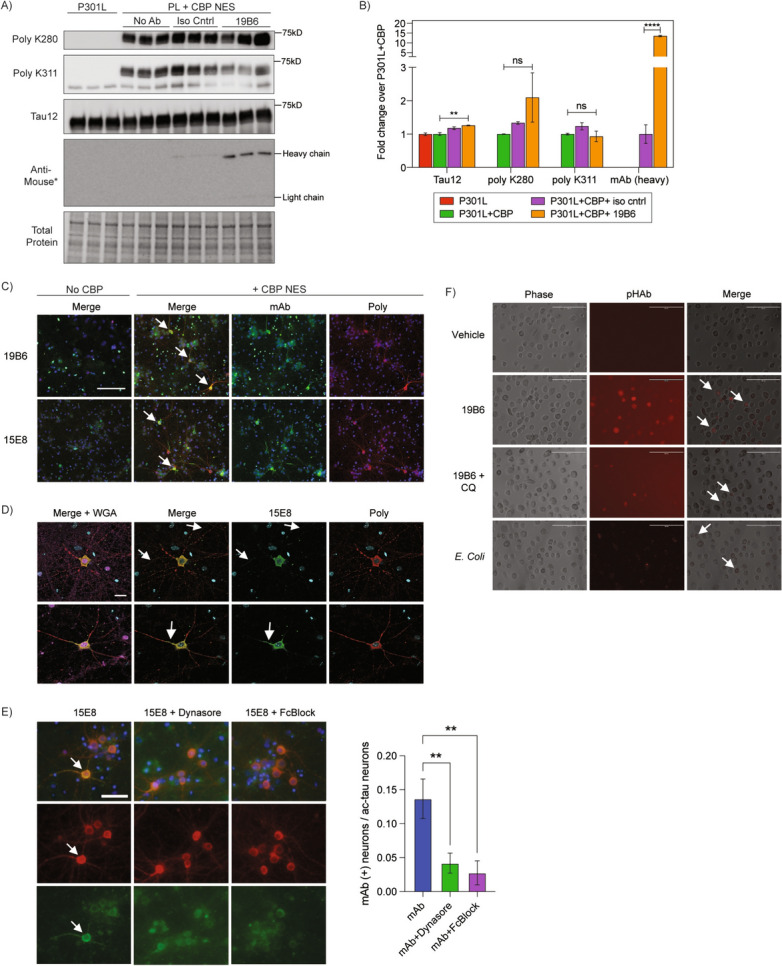
Ac-tau monoclonal antibodies 19B6 and 15E8 are internalized by primary neurons and microglia in an FcγII/III receptor-dependent manner. A Immunoblots of P301L transduced primary neurons with or without co-expression of CBP-CD-NES following antibody exposure. P301L + CBP-CD-NES neurons were exposed to 19B6 or IgG1 mouse isotype control antibody for seven days. Immunoblots were probed with polyclonal ac-K280, ac-K311, and total human tau (Tau12). The exact same lysates were run on a separate blot and probed with only anti-mouse secondary to assure maximal detection of the heavy and light chains. B Quantification of Tau12, polyclonal ac-K280, polyclonal ac-K311, and monoclonal antibody heavy chain signal from panel A. N = 3; p-value: *=< 0.05; ns = not significant. C Representative immunocytochemistry images of P301L transduced primary neurons with or without CBP-CD-NES after application of 19B6 and 15E8 antibodies. Neurons were labeled with polyclonal ac-K311 (RFP) and anti-mouse secondary (GFP) to label 19B6 and 15E8 uptake. White arrows label representative neurons exhibiting colocalization. Scale bars, 125 μm. DAPI in blue for C, D, and E. D Representative 40X confocal images from panel C highlighting co-localization of monoclonal 15E8 (GFP) and polyclonal ac-K311 (RFP). Neurons were stained with wheat germ agglutinin (magenta) to label the extracellular surface of neuronal processes. Regions of antibody uptake across ac-tau positive neuron processes and somas are labeled with white arrows. Scale bars, 20 μm. E Primary cortical neurons were pre-treated with dynasore (80 uM) or anti-mouse Fc Block (1 ug/16 cells) for 30 min, followed by the addition of 15E8 (8 ug/mL) for 1 h. Cells were fixed and stained with rabbit polyclonal ac-K311 and anti-mouse secondary (to detect antibody uptake). Quantification to the right shows the number of neurons positive for antibody uptake over the total number of ac-tau neurons per field. White arrows label neurons positive for antibody uptake. N = 15 fields per condition, across two separate biological replicates; p-value: ** < 0.05; Scale bars, 50 μm. F Cultured primary hMDMs were cultured and exposed to pHAb-vehicle, pHAb-19B6, pHAb-19B6 + chloroquine (10 uM), or E. coli as a positive control for uptake and cargo acidification. We note a low-level background fluorescence detected in apoptotic hMDMs after CQ treatment. Representative 20X images are shown. Cells exhibiting pHAb uptake are labeled with white arrows. Scale bars, 200 μm
Internalization of tau antibodies is primarily mediated by clathrin-mediated endocytosis (CME) involving FcγII/III receptor binding to IgGs [30]. To evaluate the mechanism by which ac-tau antibodies are internalized, we pre-treated primary cortical neurons with the CME inhibitor dynasore or anti-mouse Fc Block, which blocks IgG binding to FcγII/III receptors. The internalization of 15E8 was significantly reduced by both dynasore and mouse Fc Block confirming a CME and FcγII/III dependent mechanism of uptake (Fig. 4E).
Since microglia (brain-resident macrophages) can assist in clearing pathological tau as a response to immunotherapies [31, 32], we further assessed whether 19B6 could be internalized into immune cells. We adopted a primary human monocyte-derived macrophage model (hMDMs) that resemble human microglia based on transcriptional similarities [33, 34. Clone 19B6 was first pre-conjugated to pHAb, a pH-sensitive dye that fluoresces during endocytic-lysosomal processing and subsequent acidification [35], and then delivered 19B6-pHAb to cultured hMDMs. We found that 19B6 does indeed accumulate in hMDMs (Fig. 4F). Chloroquine (CQ), which inhibits lysosomal acidification, prevented most fluorescence of 19B6-pHAb, suggesting 19B6 is not only internalized but also processed by lysosomal acidification. As a positive control, E. coli, which are readily internalized for lysosomal degradation, were detected in the hMDMs (Fig. 4F). Thus, 19B6 is not only internalized by neurons but can also be processed by unrelated immune cell types.
Ac-tau monoclonal antibodies detect tau pathology in human AD brain
Finally, we evaluated whether ac-tau monoclonal antibodies have utility in detecting tau pathology in human AD brains. We performed histology by immunohistochemistry (IHC) on the frontal cortex of four AD patients. Both 19B6 and 15E8 detected neurofibrillary tangle (NFT) pathology in all four AD patients. Among ac-K280 clones, 17G8 shares an identical CDR with 19B6, while among ac-K311 clones, 14G8 shares a highly similar CDR with 15E8. All of these clones detected robust tau pathology by IHC. Only one of our candidate ac-K311 antibodies, 13B12, which detects ac-tau by western blot and ICC, was unable to detect ac-tau in human brain by IHC (data not shown). All of the ac-tau monoclonal antibodies, particularly the ac-K280 clones, recognized tau-positive neuritic plaque pathology in addition to NFTs (Fig. 5A, B). The ac-tau pathological lesions that we observed with ac-tau monoclonal antibodies were as robust as those observed with the AT8 antibody (phosphorylated S202/T205) (Fig. 5C). We conclude that our panel of highly specific antibodies have desirable biochemical and histological properties and therefore provide utility in the detection of pathogenic tau species in vitro, in primary neurons, and in human postmortem brain.
Fig. 5.

Ac-tau monoclonal antibodies detect AD pathological lesions in postmortem human brains. Sections were stained with small-scale purification of A monoclonal ac-tau K280 antibodies (clones 19B6 and 17G8) and B monoclonal ac-tau K311 antibodies (clones 14G8 and 15E8) and visualized using an avidin–biotin complex detection system. Sections were lightly counterstained with hematoxylin and then imaged. Cortical brain sections from four separate AD patients (across three different cortical regions) were stained with both sets of Ac-K280 and Ac-K311 clones. Patient 4 was only stained with 17G8 and 15E8. C Hippocampal brain tissue from the same AD cases were stained with AT8 (S202/T205) to label phosphorylated tau. Hippocampus from a non-diseased control patient is shown for comparison. The white square represents a magnified inset. Scale bars, 200 μm
Discussion
In this study we generated, validated, and characterized several new monoclonal antibodies targeting acetylated K280 and K311 residues of tau. We focused on these two residues since our previous mass spectrometry analysis found that they are most prominently acetylated in vitro and in human AD NFTs [1, 18]. Furthermore, the acetylation of these residues is robust by histology of AD brains [1, 18]. Interestingly, both residues are located in homologous positions within known hexapeptide aggregation motifs within the MTBR of tau and their acetylation is expected to dissociate tau from MTs via impaired electrostatic interactions, and hence further drive tau aggregation [1]. Indeed, our prior in vitro and rodent studies demonstrate a pathogenic role for these two residues in disease, which promotes tau dysfunction and drives tau aggregation, leading to their accumulation in a disease-specific manner in mice and humans [1, 18, 36–38]. While much of the literature supports aberrant tau acetylation as a driver of tau dysfunction, including more recent cryo-EM structural data [17], there are at least 15 ac-tau residues that have been identified in AD brains [39]. Some of these, such as K174, K274, and K281 have been found to promote tau dysfunction and exacerbate cognitive deficits in vivo [22–24, 40]. Though the majority of acetylated residues likely drive aberrant processes, others could potentially suppress pathogenic tau by preventing hyperphosphorylation at nearby phosphorylated epitopes (S252/356) [41, 42], however additional studies are needed to support any reported neuroprotective role for acetylated tau in vivo. These studies emphasize the complexity of the tau PTM code but nonetheless make a strong argument to develop acetylated tau-specific tools and assays to interrogate tau regulation and the impact of ac-tau on the brain.
While acetyl-mimic substitutions (K-to-Q substitutions) have been instrumental in studying lysine acetylation, these mutations do not always fully recapitulate genuine tau acetylation, partially because they are non-labile modifications. Instead, for this study, we developed assays to express the catalytic domain of CBP (CBP-CD) in the cytoplasm to induce tau acetylation in cell culture (Fig. 2). The future use of cytoplasmic CBP might allow us to interrogate reversible tau acetylation without the need for acetyl-mimic mutants. For example, tau can be deacetylated by HDAC6, a cytoplasmic-residing histone deacetylase. Our recent study revealed loss of HDAC6 potently exacerbated tau acetylation and subsequent pathology in PS19 mice [18]. SIRT1, another tau deacetylase, was found to deacetylate tau at K174, attenuating propagation of tau pathology across the brain [43]. Thus, cytoplasmic and/or nuclear CBP-CD can potentially be used as a discovery-based tool to delineate the signaling pathways that modulate tau acetylation/deacetylation. We note, however, in addition to enzymatic acetylation, tau can also undergo auto-acetylation in vitro, in the complete absence of CBP but in the presence of the cofactor acetyl-CoA [28, 44].
These new ac-K280 and ac-K311 monoclonal tools have numerous advantages. We previously generated rabbit polyclonal antibodies against ac-K280 and ac-K311 [1, 18], which detect multiple unresolved epitopes and cannot be traced back to a singular clone. However, these new monoclonal antibodies generated in this study are derived from an unlimited supply of immortalized hybridoma cells and can be engineered to refine and enhance specificity, activity, tissue uptake, or even humanized for translational immunotherapy studies. We were surprised to find that six of thirteen ac-K280 and two of thirteen ac-K311 antibodies had identical heavy-chain and light-chain CDRs (Table 1). This suggests strong sequence specificity conferred by specific CDRs that can be refined for optimal ac-tau recognition and could provide an excellent “backbone” for antibody engineering.
With the recent FDA approval of lecanemab and aducanumab [45, 46], immunotherapies have gained appreciable attention as a potential therapy for AD and other neurodegenerative diseases. This has spurred a flurry of pre-clinical and clinical trials targeting total tau and pathogenic strains of tau. Pre-clinical immunotherapy trials have found varying levels of success in alleviating tau pathology (hyperphosphorylation, insolubility, tau seeding and spread) and associated behavioral abnormalities (motor, memory) [47–52]. Five clinical trials have found evidence of target engagement and have progressed towards Phase 2 efficacy trials [53]. A future immunotherapy against ac-tau could provide a higher degree of tau sub-species specificity, thus leaving total normal tau intact by targeting only the pathogenic tau pool and avoiding potential off-target effects.
Tau immunotherapies could function by either (1) binding extracellular tau in the brain, thereby preventing neuronal uptake and propagation or (2) undergoing internalization into neurons and/or microglia and subsequently binding and promoting tau degradation by autophagy, proteosome, or microglial-dependent phagocytosis [31, 32, 54, 55]. We showed that 19B6 (ac-K280) can be internalized by both neurons and microglia (Fig. 4). Once internalized by neurons, 19B6 and 15E8 colocalized with ac-tau both in the cytoplasm and within ac-tau positive neuronal processes, supporting target engagement. The assumption is that, if taken up by neurons or microglia, one could conceivably block ac-tau pathogenesis as a potential treatment for AD and other tauopathies. Indeed, a recent study found that passive immunization of P301L mice using an ac-K280 antibody could rescue some aspects of cognitive decline by reducing the levels of tau oligomers [56]. These findings support the therapeutic potential of targeting ac-tau.
Given the striking disease-specificity for ac-K280 and ac-K311, with no apparent detection in non-tauopathy controls, ac-tau antibodies could also be employed in diagnostic biomarker assays. While phosphorylated tau (at residues T181, T205, T217, T231) is routinely measured in biofluids, it is worth noting that phosphorylated tau is detected even in healthy patients, albeit at lower levels, contrary to what we and others observed with ac-tau [39, 57–60]. It is certainly plausible that ac-tau biomarker assays could provide more accurate readouts of disease severity, disease progression, or as a distinguishing diagnostic feature among different but highly related tauopathies.
In summary, tau acetylation is a complex facet of an intriguing and evolving tau PTM profile that will require new tools and approaches to fully appreciate its role and regulation in health and disease. We generated a large panel of ac-tau monoclonal antibodies in this study, with two in particular (19B6 and 15E8) that show exceptional cellular, biochemical, and histological properties including high specificity and sensitivity for the detection of acetylated tau. We anticipate that future studies will extend these findings to other tauopathies beyond AD, which could inform more personalized and targeted therapeutic strategies.
Conclusions
In summary, we generated and characterized new monoclonal antibodies targeting tau acetylation at residues K280 and K311, two pathological acetylated residues which accumulate in AD. Furthermore, we validated the use of these antibodies using biochemical, cellular, and histological approaches. While some ac-tau antibodies have been generated in the field, their sensitivity and/or specificity is often times questionable or lacking, creating a need for more standardized ac-tau tools. Given the emerging evidence supporting tau acetylation in AD and other tauopathies, the antibodies developed here provide highly desirable new tools to unravel the complex tau PTM code and could be further deployed in biomarker assays and as novel anti-tau therapeutics.
Supplementary Information
Supplemental Figure 1. Immunoblot and ELISA validation of additional ac-K280 and ac-K311 clones. Representative immunoblots of QBI-293 cells transfected with wild-type 2N4R human tau or acetyl-null K280R/K311R 2N4R human tau and full length CBP. Small-scale purification of ac-tau monoclonal antibodies from hybridoma supernatant were applied to blots. A) ac-K280 antibody clones. B) ac-K311 antibody clones. Notes: 1A, clones in the top row have identical CDRs to 19B6; * or ** denote identical CDRs to 9G4 or 14G8 antibodies (listed at the bottom of the panel).
Supplemental Figure 2. A single non-specific ~100 kD band is detected by ac-tau monoclonal antibodies but only in response to constitutively active CBP-CD. A) Representative immunoblots of QBI-293 cells transfected with wild-type 2N4R human tau, tau and CBP-CD-NES/NLS, or CBP-CD-NES/NLS alone. Immunoblots were probed with 19B6, 15E8, FLAG (CBP-CD) and K9JA (total tau) antibodies. B) Representative immunoblots of QBI-293 cells transfected with wild-type 2N4R human tau with or without CBP-CD-NES or full length CBP. Cells were also transfected with catalytically dead versions (denoted LD) of CBP-CD-NES and full length CBP as negative controls. Immunoblots were probed with 19B6, 15E8, K9JA (total tau), FLAG (CBP-CD-NES), and HA tag (CBP full length) antibodies.
Supplemental Figure 3. Immunocytochemistry validation of additional ac-K280 and ac-K311 monoclonal antibodies in primary neurons. Primary mouse cortical neurons were transduced for five days with lentiviruses for empty vector control, wild-type human tau, or wild-type tau and CBP-CD-NES. A) Ac-K280 clones B) Ac-K311 clones. Note: * or ** denote identical CDRs to 19B6 or 14G8 antibodies (listed at the bottom of the panel).
Acknowledgements
We would like to thank the Center for Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania for providing post-mortem human AD brain sections. Monoclonal antibody hybridoma screening and large-scale antibody purifications were performed by Genscript.
Abbreviations
- AD
Alzheimer’s disease
- PTM
Post translational modification
- CBP
CREB binding protein
- MAPT
Microtubule associated protein tau
- MT
Microtubules
- FTD
Frontal temporal dementia
- PiD
Pick’s disease
- PSP
Supranuclear palsy
- CBD
Corticobasal degeneration
- MTBR
Microtubule binding region
- EM
Electron microscopy
- IgG
Immunoglobulin G
- CDR
Complementarity determining regions
- ELISA
Enzyme-linked immunosorbent assay
- CD
Catalytic domain
- NES
Nuclear exclusion sequence
- NLS
Nuclear localization sequence
- SDS
Sodium dodecyl sulfate
- PL-SF
P301L/S320F tau mutant
- ICC
Immunocytochemistry
- IHC
Immunohistochemistry
- CME
Clathrin mediated endocytosis
- hMDM
Human monocyte-derived macrophage
Author contributions
MRB designed and performed most experiments. XT performed all plasmid and lentiviral construct design and preparation. XT also performed all monoclonal antibody sequencing. JHT performed initial QBI293 and neuron experiments with CBP-CD-NLS and CBP-CD-NES and validation thereof. BAE and JVR performed hMDM uptake of monoclonal antibodies, aided in neuronal uptake of antibodies, and carried out confocal imaging of monoclonal uptake in primary neurons. AFB performed co-immunoprecipitation experiments and designed all figures. DI and WT performed all post-mortem human AD brain immunohistochemistry. MRB and TJC wrote the main manuscript text. TJC directed and supervised the study. All authors consented and contributed to the final version of the manuscript.
Funding
Support for this work was provided by National Institutes of Health (NIH) grants F32AG072826 (MRB) and RF1AG068063 (TJC), Duke/UNC Alzheimer’s Disease Research Center grant NIA-P30AG072958 (JHT), Alzheimer’s Association grants AARF-22-926617 (JHT) and AARG-22-924321 (TJC), and CurePSP grant 656-2018-06 (TJC). Human tissue from University of Pennsylvania were obtained as part of program project grants P01-AG-066597 and P30-AG-072979 (formerly AG010124) (DI/WT). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Availability of data and materials
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
TJC and MRB are inventors of the ac-tau monoclonal antibodies described in this study, own the copyright, and could receive royalties. These relationships have been disclosed and are under management by UNC-Chapel Hill.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cohen TJ et al (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2:252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bramblett GT et al (1993) Abnormal tau phosphorylation at Ser396 in alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 10:1089–1099 [DOI] [PubMed] [Google Scholar]
- 3.Butner KA, Kirschner MW (1991) Tau protein binds to microtubules through a flexible array of distributed weak sites. J cell Biol 115:717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y (1998) Vulnerable neuronal subsets in Alzheimer’s and Pick’s disease are distinguished by their τ isoform distribution and phosphorylation. Ann Neurol 43:193–204 [DOI] [PubMed] [Google Scholar]
- 5.Sergeant N, Wattez A, Delacourte A (1999) Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neurochem 72:1243–1249 [DOI] [PubMed] [Google Scholar]
- 6.Klein H-U et al (2019) Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat Neurosci 22:37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caballero B et al (2021) Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat Commun 12:2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huseby CJ et al (2019) Quantification of tau protein lysine methylation in aging and Alzheimer’s disease. J Alzheimer’s Dis 71:979–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balmik AA, Chinnathambi S (2021) Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun Signal 19:51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cripps D et al (2006) Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation*. J Biol Chem 281:10825–10838 [DOI] [PubMed] [Google Scholar]
- 11.Morishima-Kawashima M et al (1993) Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 10:1151–1160 [DOI] [PubMed] [Google Scholar]
- 12.Martins WC, Tasca CI, Cimarosti H (2016) Battling Alzheimer’s disease: targeting SUMOylation-mediated pathways. Neurochem Res 41:568–578 [DOI] [PubMed] [Google Scholar]
- 13.Qin M et al (2019) SET SUMOylation promotes its cytoplasmic retention and induces tau pathology and cognitive impairments. Acta Neuropathol Commun 7:21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goode B, Feinstein S (1994) Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol 124:769–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris M et al (2015) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci 18:1183–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fitzpatrick AWP et al (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547:185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arakhamia T et al (2020) Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell. 10.1016/j.cell.2020.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trzeciakiewicz H et al (2020) An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat Commun 11:5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ajit D et al (2019) A unique tau conformation generated by an acetylation-mimic substitution modulates P301S-dependent tau pathology and hyperphosphorylation. J Biol Chem 294:16698–16711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Lee VM-Y (2006) Characterization of two VQIXXK motifs for tau fibrillization in vitro †. Biochemistry 45:15692–15701 [DOI] [PubMed] [Google Scholar]
- 21.Seidler PM et al (2018) Structure-based inhibitors of tau aggregation. Nat Chem 10:170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sohn PD et al (2016) Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener 11:47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tracy TE et al (2016) Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90:245–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Min S-W et al (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21:1154–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin M-K et al (2021) Reducing acetylated tau is neuroprotective in brain injury. Cell. 10.1016/j.cell.2021.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbons GS et al (2020) Conformation-selective tau monoclonal antibodies inhibit tau pathology in primary neurons and a mouse model of Alzheimer’s disease. Mol Neurodegener 15:64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer L et al (2019) A simplified workflow for monoclonal antibody sequencing. PLoS One 14:e0218717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Min S-W et al (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67:953–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strang KH et al (2017) Distinct differences in prion-like seeding and aggregation between tau protein variants provide mechanistic insights into tauopathies. J Biol Chem 293:2408–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Congdon EE, Gu J, Sait HBR, Sigurdsson EM (2013) Antibody uptake into neurons occurs primarily via clathrin-dependent Fcγ receptor endocytosis and is a prerequisite for acute tau protein clearance. J Biol Chem 288:35452–35465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funk KE, Mirbaha H, Jiang H, Holtzman DM, Diamond MI (2015) Distinct therapeutic mechanisms of tau antibodies: promoting microglial clearance versus blocking neuronal uptake. J Biol Chem 290:21652–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo W et al (2015) Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 5:11161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quiroga IY et al (2022) Synthetic amyloid beta does not induce a robust transcriptional response in innate immune cell culture systems. J Neuroinflamm 19:99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Killebrew DA, Williams KS, Xie Y, Longo F, Meeker RB (2022) Suppression of HIV-associated macrophage activation by a p75 neurotrophin receptor ligand. J Neuroimmune Pharmacol 17:242–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nath N et al (2016) Homogeneous plate based antibody internalization assay using pH sensor fluorescent dye. J Immunol Methods 431:11–21 [DOI] [PubMed] [Google Scholar]
- 36.Trzeciakiewicz H et al (2017) A dual pathogenic mechanism links tau acetylation to sporadic tauopathy. Sci Rep 7:44102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng J-H et al (2017) The deacetylase HDAC6 mediates endogenous neuritic tau pathology. Cell Rep 20:2169–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irwin DJ et al (2012) Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain J Neurol 135:807–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wesseling H et al (2020) Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell. 10.1016/j.cell.2020.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Q et al (2023) Acetylated tau exacerbates learning and memory impairment by disturbing with mitochondrial homeostasis. Redox Biol 62:102697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cook C et al (2014) Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet 23:104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xia Y et al (2022) Pathogenic tau recruits wild-type tau into brain inclusions and induces gut degeneration in transgenic SPAM mice. Commun Biol 5:446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Min S-W et al (2018) SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J Neurosci 38:3680–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen TJ, Constance BH, Hwang AW, James M, Yuan C-X (2016) Intrinsic tau acetylation is coupled to auto-proteolytic tau fragmentation. PLoS One 11:e0158470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haeberlein SB et al (2022) Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimer’s Dis 9:197–210 [DOI] [PubMed] [Google Scholar]
- 46.van Dyck CH et al (2022) Lecanemab in early Alzheimer’s disease. New Engl J Med 388:9–21 [DOI] [PubMed] [Google Scholar]
- 47.Vitale F et al (2018) Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol Commun 6:82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM (2007) Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 27:9115–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walls KC et al (2014) p-Tau immunotherapy reduces soluble and insoluble tau in aged 3xTg-AD mice. Neurosci Lett 575:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chai X et al (2011) Passive immunization with anti-tau antibodies in two transgenic models: reduction of tau pathology and delay of disease progression. J Biol Chem 286:34457–34467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castillo-Carranza DL et al (2015) Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J Neurosci 35:4857–4868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai C et al (2018) Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 × Tg-AD mice. Alzheimer’s Res Ther 10:13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sandusky-Beltran LA, Sigurdsson EM (2020) Tau immunotherapies: lessons learned, current status and future considerations. Neuropharmacology. 10.1016/j.neuropharm.2020.108104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee S-H et al (2016) Antibody-mediated targeting of tau in vivo does not require effector function and microglial engagement. Cell Rep 16:1690–1700 [DOI] [PubMed] [Google Scholar]
- 55.Sigurdsson EM (2008) Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimer’s Dis 15:157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song H-L et al (2023) Monoclonal antibody Y01 prevents tauopathy progression induced by lysine280-acetylated tau in cell and mouse models. J Clin Invest. 10.1172/jci156537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanger DP et al (2007) Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis*. J Biol Chem 282:23645–23654 [DOI] [PubMed] [Google Scholar]
- 58.Morishima-Kawashima M et al (1995) Proline-directed and Non-proline-directed Phosphorylation of PHF-tau (∗). J Biol Chem 270:823–829 [DOI] [PubMed] [Google Scholar]
- 59.Xia Y, Prokop S, Giasson BI (2021) “Don’t Phos Over Tau”: recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol Neurodegener 16:37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsuo ES et al (1994) Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 13:989–1002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Immunoblot and ELISA validation of additional ac-K280 and ac-K311 clones. Representative immunoblots of QBI-293 cells transfected with wild-type 2N4R human tau or acetyl-null K280R/K311R 2N4R human tau and full length CBP. Small-scale purification of ac-tau monoclonal antibodies from hybridoma supernatant were applied to blots. A) ac-K280 antibody clones. B) ac-K311 antibody clones. Notes: 1A, clones in the top row have identical CDRs to 19B6; * or ** denote identical CDRs to 9G4 or 14G8 antibodies (listed at the bottom of the panel).
Supplemental Figure 2. A single non-specific ~100 kD band is detected by ac-tau monoclonal antibodies but only in response to constitutively active CBP-CD. A) Representative immunoblots of QBI-293 cells transfected with wild-type 2N4R human tau, tau and CBP-CD-NES/NLS, or CBP-CD-NES/NLS alone. Immunoblots were probed with 19B6, 15E8, FLAG (CBP-CD) and K9JA (total tau) antibodies. B) Representative immunoblots of QBI-293 cells transfected with wild-type 2N4R human tau with or without CBP-CD-NES or full length CBP. Cells were also transfected with catalytically dead versions (denoted LD) of CBP-CD-NES and full length CBP as negative controls. Immunoblots were probed with 19B6, 15E8, K9JA (total tau), FLAG (CBP-CD-NES), and HA tag (CBP full length) antibodies.
Supplemental Figure 3. Immunocytochemistry validation of additional ac-K280 and ac-K311 monoclonal antibodies in primary neurons. Primary mouse cortical neurons were transduced for five days with lentiviruses for empty vector control, wild-type human tau, or wild-type tau and CBP-CD-NES. A) Ac-K280 clones B) Ac-K311 clones. Note: * or ** denote identical CDRs to 19B6 or 14G8 antibodies (listed at the bottom of the panel).
Data Availability Statement
No datasets were generated or analysed during the current study.